

Abstract
Cancer cells develop mechanisms to escape immunosurveillance, among which modulating the expression of immune suppressive messenger RNAs is most well-documented. However, how this is molecularly achieved remains largely unresolved. Here, we develop an in vivo mouse model of liver cancer to study oncogene cooperation in immunosurveillance. We show that MYC overexpression (MYCTg) synergizes with KRASG12D to induce an aggressive liver tumor leading to metastasis formation and reduced mouse survival compared with KRASG12D alone. Genome-wide ribosomal footprinting of MYCTg;KRASG12 tumors compared with KRASG12D revealed potential alterations in translation of mRNAs, including programmed-death-ligand 1 (PD-L1). Further analysis revealed that PD-L1 translation is repressed in KRASG12D tumors by functional, non-canonical upstream open reading frames in its 5′ untranslated region, which is bypassed in MYCTg;KRASG12D tumors to evade immune attack. We show that this mechanism of PD-L1 translational upregulation was effectively targeted by a potent, clinical compound that inhibits eIF4E phosphorylation, eFT508, which reverses the aggressive and metastatic characteristics of MYCT9;KRASG12D tumors. Together, these studies reveal how immune-checkpoint proteins are manipulated by distinct oncogenes at the level of mRNA translation, which can be exploited for new immunotherapies.
Reporting Summary.
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
The concept that the immune system can control tumor growth can be traced back to 1893 when William Coley used live bacteria as an immune stimulant to treat cancer. This natural protective mechanism of the human body to control cancer is now known to be elicited by key immune-checkpoint molecules, often present on cytotoxic T cells1. However, tumor cells can exploit these immune-checkpoint pathways as a mechanism to evade detection by the immune response, for example by altering the expression of key ligands on tumor cells such as PD-L1 and CD80/86, to inhibit the cytotoxicity activity of immune cells. Understanding the underlying mechanism by which cancer cells can ‘molecularly cloak’ themselves and remain hidden to immune surveillance is of great interest to cancer biology and provides the rationale design of therapeutic interventions. Although direct transcriptional control of inhibitory immune-checkpoint molecules by specific oncogenic pathways has been reported2,3, post-transcriptional control may offer a fast, highly tunable mechanism to control the abundance of effector proteins guiding immune evasion of cancer cells. Yet, this level of regulation of the immune-checkpoint molecules remains largely uncharacterized.
To study how oncogene cooperation directs immune-checkpoint deregulation, we focused on primary liver cancer, the second leading cause of cancer deaths worldwide4. Hepatocellular carcinoma (HCC) lacks effective cures except for early surgical intervention or liver transplantation, highlighting the urgent need for new therapeutic strategies. c-MYC, overexpression of which is commonly caused by genomic amplification, is widely amplified5 and present in 70% of viral and alcohol-related HCC6 and has a critical role in the malignant transformation of hepatocytes into HCC7,8. Of all genetic alternations in HCC patients, there is an over-representation of pathways related to receptor tyrosine kinase (RTK) signaling, with ~22–37% of HCC patients having at least one alternation in genes associated to RTK/RAS/PI3K pathway5. Here, we discovered that MYC, when cooperating with RTK/RAS signaling, markedly increases HCC formation and metastasis development. We further characterize the importance of evasion from immune attack as a key driver of primary and metastatic cancer development and define an unexpected post-transcriptional control of gene expression that drives this process. We also show that a potent clinical compound inhibiting the phosphorylation of the major cap-binding protein, eIF4E, can restore cancer immunosurveillance.
Results
MYC and KRAS cooperate in inducing liver cancer with metastatic potential and an altered immune microenvironment.
We developed a genetic engineered mouse (GEM) model of liver cancer with enhanced MYC and RTK signaling. We first generated an HA-tagged, c-MYC transgene, whereby GFP is co-translated via a viral internal ribosome entry site element that is driven in a CRE- dependent manner (referred to here as: MYCTg; Supplementary Fig. 1a–c).We then crossed this c-MYC transgenic mouse to a well-established Cre-inducible, constitutively active KRASG12D mouse that mimics notably enhanced downstream signaling of RTK9,10, whereby liver-specific expression is achieved through Albumin-Cre- mediated recombination (Supplementary Fig. 1d,e). In this setting, MYC overexpression in the liver was not sufficient to drive cancer in vivo, whereas KRASG12D and MYC (referred to here as MYCTg;KRASG12D) synergized to provoke a more aggressive tumor compared with the KRASG12D alone (Fig. 1a). This aggressive tumor was characterized by decreased tumor latency (Fig. 1a), the simultaneous presence of intrahepatic cholangiocarcinoma and HCC (Fig. 1b), as well as decreased survival. Notably, MYCTg;KRASG12D mice showed a marked increase in metastasis in 90% of animals, compared with only 10% in KRASG12D alone (Fig. 1c,d). Metastasis occurred primarily in the lungs (Fig. 1d), but could also be identified in lymph nodes (Supplementary Fig. 1f). These findings suggest that the barrier for primary and metastatic tumor development in KRASG12D tumors is broken by MYC hyperactivation.
Fig. 1 |. MYC and KRAS cooperate to promote liver cancer with metastatic potential and induce marked alterations of immune populations compared with KRAS alone.
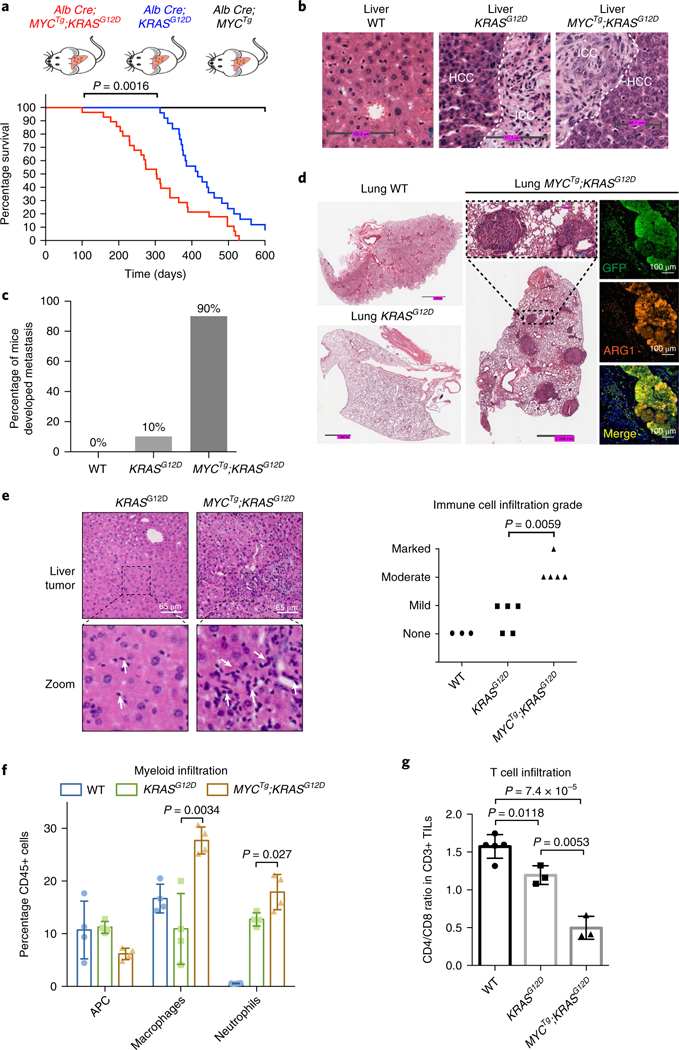
a, Kaplan–Meier curves showing tumor-free survival of MYCTg;KRASG12D, KRASG12D, and MYCTg mice. n = 22 per genotype (log-rank test). b, Representative wild-type (WT), KRASG12D, or MYCTg;KRASG12D mice liver histology, n = 3 independent experiments is shown. ICC: intrahepatic cholangiocarcinoma (scale bars, WT: 103.3 μm, KRASG12D: 111.3 μm, MYCTg;KRASG12D: 65.7 μm). c, Percentage of wild-type (n = 3) KRASG12D (n = 10) or MYCTg;KRASG12D (n = 10) mice developed lung metastasis during cancer development. d, Representative wild-type, KRASG12D and MYCTg;KRASG12D mice lung histology, n = 5 independent experiments are shown. Immunofluorescence staining for GFP (indication of MYC overexpression), ARG1 (arginase-1, HCC marker, not expressed in the lung), and DAPI in MYCTg;KRASG12D lung section. e, Left: representative HCC histology in KRASG12D and MYCTg;KRASG12D mice showing different grades of immune infiltration, n = 3 independent experiments (scale bars, 65 μm). White arrows represent inflammatory cell infiltration. Right: clinical evaluation of the grade of immune cells infiltration of the liver histology of wild-type (n = 3), KRASG12D (n = 5), and MYCTg;KRASG12D (n = 5) mice (none: 0/HPF, mild: 1–3 per HPF, moderate 4–10 per HPF, marked >10 per HPF). Two-sided t test. f, Quantifications by flow cytometry of antigen-presenting cell (APC) (CD45+, CD11b–, CD11c + ), macrophages (CD45 + , CD11b + , CD11c–, Ly6G/C–), and neutrophils (CD45+, CD11b+, CD11c–, Ly6G/C+) infiltrations into the liver tumors of wild-type, KRASG12D, or MYCTg;KRASG12D mice. n = 4. Two-sided t test. g, CD4/CD8 T cell ratio in wild-type liver (n = 5), KRASG12D (n = 3), and MYCTg;KRASG12D (n = 3) mice liver tumors. Two-sided t test. TILs: tumor-infiltrating lymphocytes. All values represent the mean ± s.d.
By characterizing the microenvironment of MYCTg;KRASG12D tumors, we observed a robust invasion of inflammatory cells in comparison to KRASG12D alone (Fig. 1e). Notably, MYCTg;KRASG12D-driven inflammatory cell infiltration is characterized by increased CD45+ leukocytes including neutrophils and macrophages (Fig. 1f and Supplementary Fig. 1g–i). The infiltration of inflammatory cells has been shown to enhance tumor angiogenesis, proliferation, metastasis, and therapy resistance11, and therefore may contribute to the aggressive phenotype of MYCTg;KRASG12D-driven cancer. Notably, we also detected an increased infiltration of a cytotoxic, CD8+ T cell biased tumor-infiltrating lymphocytes (Fig. 1g and Supplementary Fig. 1j), which has a central role in anti-tumor immunity and tumor regression. However, despite the increased CD8+ T cell population that has been associated with an improved prognosis12–14, MYCTg;KRASG12D-driven liver cancer progresses more aggressively. Based on these, we proposed that cancer cells with MYC and KRAS activation may escape immune surveillance and suppress the cytotoxicity of CD8+ T cells in the tumor microenvironment15–18. This escape from immune surveillance may be a broader characteristic of MYC and KRAS-driven cancers19.
MYC and KRAS cooperation selectively alters gene expression at the translation level.
We proposed that MYC and KRAS cooperate to drive unique gene expression programs in cancer cells to evade immune-destruction and develop metastasis. To survey the genome-wide gene expression at both the transcription and translation level in an unbiased manner, we first carefully micro-dissected primary liver tumor nodules in MYCTg;KRASG12D and KRASG12D mice as well as wild-type livers as controls. We deep-sequenced total mRNAs and ribosome-protected mRNA fragments using ribosome profiling20 to generate genome-wide transcriptional and translational landscapes, respectively (Supplementary Fig. 2). Applying the framework of the generalized linear model, a linear regression was performed to the normalized read counts, as a function of sample type variables (‘WT’, ‘KRASG12D’, or ‘MYCTg;KRASG12D’). Here, the coefficient of sample type variables (that is, ‘MYCTg;KRASG12D’ over ‘KRASG12D’) is a measurement of corresponding differential gene expression. The transcriptional expression profile in the MYCTg;KRASG12D murine model of HCC exhibits significant similarities to human HCC (Supplementary Fig. 3). Human HCC samples harboring both MYC amplification and RTK signaling activation (Supplementary Fig. 3a) possess a gene expression pattern that strongly correlated with that of our MYCTg;KRASG12D murine HCC (Supplementary Fig. 3b,c). These findings support that our GEM model is a highly relevant and suitable model for studying human HCC. When the global transcriptome (RNA-Seq) and translatome (Ribo-Seq) profiles were directly compared, we observed that activation of KRAS alone results in a coordinated change in RNA-Seq and Ribo-seq compared with wild-type hepatocytes (Fig. 2a, Supplementary Fig. 4, and Supplementary Table 1). In the context of both MYC and KRAS activation, we detected few statistically significant changes at the mRNA level compared with KRAS activation alone (11 down- and 13 upregulated transcripts with Padj < 0.1 and |log2FC| > 2 in RNA-Seq) (Fig. 2a and Supplementary Table 2), but we observed changes in ribosome footprints for a subset of transcripts (130 down- and 339 upregulated transcripts with Padj < 0.1 and |log2FC| > 2 in Ribo-Seq) (Fig. 2a, Supplementary Fig. 4, and Supplementary Table 2). Further validation will be required to determine whether these Ribo-Seq changes are a result of a combination of changes in transcription and translation or translation/transcription alone (Supplementary Fig. 4). Nevertheless, these findings raise the possibility that in the context of two cooperating oncogenes, the translational profile of gene expression is distinct.
Fig. 2 |. A dichotomy in gene regulation between transcription versus translational control in KRASG12D versus MYCTg;KRASG12D tumors.
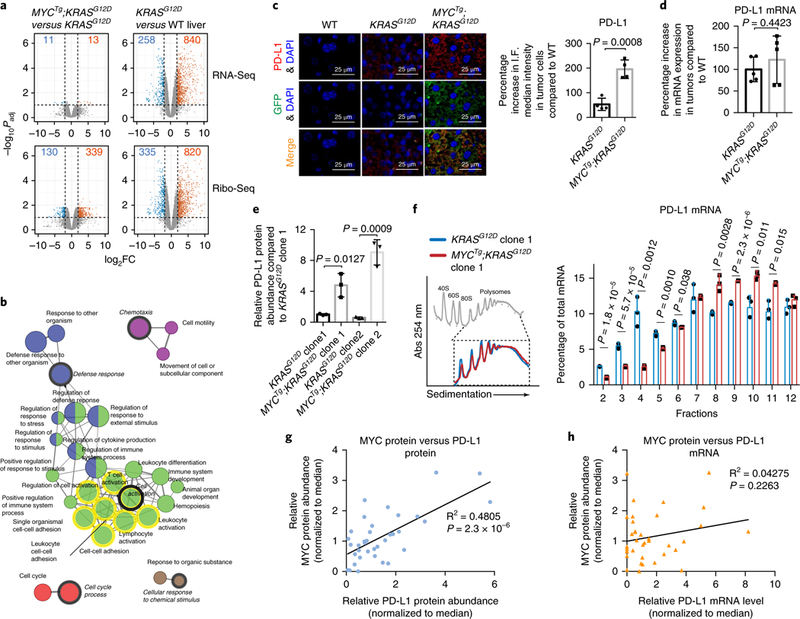
a, Differential gene expression analysis shown as volcano plots, comparing MYCTg;KRASG12D (n = 2 animals) to KRASG12D (n = 2 animals) (left panel), and comparing KRASG12D (n = 2 animals) to wild type (n = 3 animals) (right panel) with RNA-Seq on the top and Ribo-Seq on the bottom. P values (two-sided t test) were adjusted using the Benjamini–Hochberg procedure for multi-testing. Dashed lines mark the threshold of significance in this study: the horizontal line marks the statistical significance (Padj< 0.1) and the vertical lines mark the fold change (FC) (|log2FC| > 2). The numbers of genes that pass this threshold are shown, with downregulated genes in blue and upregulated genes in orange. b, The network of enriched gene ontology categories (biological process) among 339 genes that are upregulated in Ribo-Seq comparing MYCTg;KRASG12D to KRASG12D (Padj< 0.1 and log2FC > 2). The gene ontology network reflects the relationships between the gene ontology categories on the basis of the similarity of their associated genes, generated by ClueGO. Each node represents one enriched gene ontology category, with the size of the node representing the term enrichment significance (right-sided hypergeometric test). Edges indicate similarity (Kappa score > 0.4) between the two connected gene ontology categories. Gene ontology categories are clustered into functional groups represented by different color codes. The mixed color coded nodes are shared between two functional groups. The most significantly enriched gene ontology category from each group is designated as the leading group term and highlighted in italics. The gene ontology terms that PD-L1 belongs to are highlighted in yellow circles. c, Left: representative PD-L1, GFP (indication of MYC overexpression) and DAPI immunofluorescence staining of wild-type mice livers, KRASG12D, or MYCTg;KRASG12D mice liver tumors (scale bars, 25 μm). Right: quantification of percentage increase in PD-L1 median intensity in KRASG12D and MYCTg;KRASG12D tumor cells relative to the wild type. n = 4 independent experiments, two-sided t test. d, Percentage increase in PD-L1 mRNA expression levels in KRASG12D or MYCTg;KRASG12D mice liver tumors relative to wild type, normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA. n = 5. e, Flow cytometry assessment of PD-L1 protein abundance in the KRASG12D and MYCTg;KRASG12D HCC cell lines, n = 3 independent experiments, two-sided t test. f, Left: representative polysome trace of KRASG12D and MYCTg;KRASG12D cell lines, n = 3 independent experiments. Inset highlights the polysomal fractions (7–12). Right: qualitative PCR with reverse transcription (RT-qPCR) analysis of PD-L1 mRNA levels in fractions, percentages of PD-L1 mRNA distributed in each fraction against total PD-L1 mRNA are shown. Two-sided t test. g, Comparison of relative MYC and PD-L1 protein abundance (normalized to median) in 36 fresh frozen human HCC patient samples by western blot. The 95% confidence interval for the Pearson coefficients is 0.4722–0.8322 (Pearson’s correlation test, two-sided P = 2.3×10–6). h, Comparison of relative MYC protein abundance and relative PD-L1 mRNA expression (normalized to median) in 36 fresh frozen human HCC patient samples. The 95% confidence interval for the Pearson coefficients is –0.1307 to 0.5012 (Pearson’s correlation test, two-sided P = 0.2263). All values represent the mean± s.d.
MYC reprograms PD-L1 expression at the translation level.
To further investigate possible translational regulation in MYCTg;KRASG12D tumors, we first performed gene ontology analysis on the 339 transcripts with significantly upregulated ribosome footprints in MYCTg;KRASG12D tumors compared with KRASG12D (Padj < 0.1 and log2FC > 2 in Ribo-Seq). This revealed several significantly enriched gene ontology categories (Padj < 0.05) that were grouped into major functional clusters including: defense response, cell activation, chemotaxis, cell cycle process, and cellular response to chemical stimulus (Fig. 2b and Supplementary Table 3). Notably, in the cell activation cluster, we found the immune-checkpoint gene Cd274 encoding the mouse ortholog of PD-L1 (Fig. 2b and Supplementary Fig. 5). Despite PD-L1 mRNA being relatively equally induced in both KRASG12D and MYCTg;KRASG12D tumors, PD-L1 ribosome foot-prints are notably upregulated in MYCTg;KRASG12D (Supplementary Table 2), suggesting possible translational upregulation of the gene. PD-L1 is a transmembrane protein expressed on tumor cells that is a ligand of the immune-checkpoint receptor programmed death-1 (PD-1) on T cells. PD-L1/PD-1 interaction leads to the negative regulation of T cell proliferation, migration and activation, allowing tumor cells to evade the anti-tumor immune response21, which matches the phenotype of the MYCTg;KRASG12D tumor. Thus, we suggested that the translational upregulation of PD-L1 by MYC and RAS cooperation may lead to immune evasion and the aggressive cancer phenotype in MYCTg;KRASG12D mice. As PD-L1 is also widely expressed on tumor-infiltrating immune cells such as T cells and some myeloid cells22,23, we first confirmed HCC tumor-specific expression of PD-L1 using immunofluorescence-based co-staining of MYCTg-expressing liver cells with PD-L1 (Fig. 2c). Consistent with our ribosome-profiling analysis showing an approximate fivefold increase in ribosome footprints at the PD-L1 mRNA in MYCTg;KRASG12D compared with KRASG12D (Supplementary Fig. 5), there is a corresponding fourfold increase of PD-L1 protein abundance observed in MYCTg;KRASG12D tumors by immunofluorescence (Fig. 2c), and this occurred independently of changes in mRNA amounts (Fig. 2d).
To validate that PD-L1 is regulated at the translation level in MYCTg;KRASG12D liver cancer cells, we derived ex vivo cultures of primary, single-clone cell lines from individual liver tumors from KRASG12D or MYCTg;KRASG12D mice (Supplementary Fig. 6a–c). Noteworthy and consistent with previous studies, PD-L1 protein expression is typically assessed by flow cytometry24–27. Using the in vitro culturing of these cell lines, we specifically observed an increase in protein abundance of PD-L1 by flow cytometry in the MYCTg;KRASG12D background with no significant difference at the mRNA level (Fig. 2e and Supplementary Fig. 6d), which also demonstrates that the upregulation of PD-L1 in MYCTg;KRASG12D tumor is a result of primary oncogenic cooperation, rather than a reaction to anti-tumor immunity from the tumor microenvironment.Besides PD-L1, we also validated other translationally regulated mRNAs identified by ribosome profiling in the MYCTg;KRASG12D tumor setting. We confirmed that cytokines involved in immune cell attraction and inflammation (CCL2 and CXCL9), proteins involved in the cell cycle process (PLK1, CDC20), and proteins involved in cell motility (SNAIL and CXCR6) are also regulated by MYC at the protein level (Supplementary Fig. 6e–g) without affecting their mRNA expression (Supplementary Fig. 6h).
Although MYC itself is a regulator of global protein synthesis, no noticeable increase in global protein synthesis rates in MYCTg;KRASG12D compared with KRASG12D cells was evident (Fig. 2f). To further characterize translational control of the PD-L1 mRNA in MYCTg;KRASG12D liver tumor cells, we examined its distribution in polysomes (translationally active ribosome fractions) on sucrose gradient fractionation (Fig. 2f). There is a higher percentage of PD-L1 mRNA accumulated in the translationally inactive free subunit and monosome fractions (fractions 2–6) in KRASG12D liver tumor cells (Fig. 2f and Supplementary Fig. 7a), revealing that the translation of PD-L1 mRNA is relatively suppressed in KRASG12D cells. However, in MYCTg;KRASG12D cells there was a significant shift of PD-L1 mRNA, but not the GAPDH control mRNA, towards the translationally active, polysome fractions (fractions 8–11) (Fig. 2f and Supplementary Fig. 7a,b), showing that the control of PD-L1 expression is at the level of mRNA translation in MYCTg and KRASG12D cooperation-driven cancer. This is consistent with the observation made in fresh frozen human HCC tumor samples (Supplementary Fig. 8a,b), where we observed a significant correlation between MYC amounts and PD-L1 protein abundances (Fig. 2g), whereas PD-L1 mRNA expression was not correlated (Fig. 2h).
MYC enhances PD-L1 translation through bypassing upstream open reading frame (uORF)-mediated translational repression.
We next sought to address the mechanism by which PD-L1 mRNA is inefficiently translated in KRASG12D liver tumor cells, but becomes translationally activated in MYCTg;KRASG12D. The translation of mRNAs is highly controlled by the 5′ untranslated region (5′UTR), which can contain several regulatory elements including complex structures as well as sequence-specific RNA elements28. To explore how PD-L1 is translationally regulated, we analyzed the 5′UtR sequence of the PD-L1 mRNA. Notably, the mouse PD-L1 mRNA contained three putative uORFs, one of which initiates from an upstream AUG (uAUG), and the other two from non-canonical upstream CUG start codons (uCUG) that partially overlaps with the main PD-L1 ORF (Fig. 3a). To molecularly validate the engagement of the ribosome on these uORFs, we performed a toeprint assay in KRASG12D and MYCTg;KRASG12D cell lysates to directly visualize ribosome footprints on the upstream initiation codons of an in-vitro-transcribed PD-L1 5′UTR reporter. We detected that, in KRASG12D cell lysate, ribosome footprints are preferably observed at the uAUG and the proximal uCUG (the uCUG near the main ORF), rather than the main AUG (Fig. 3b, RNA loading controls in Supplementary Fig. 9a). Mutating the uCUG in this reporter significantly abolished ribosome recognition of uCUG upstream start codons (Supplementary Fig. 9b). On the contrary, in MYCTg;KRASG12D cell lysates, the major ribosome footprints occur at the main AUG, compared to the uAUG or uCUG (Fig. 3b).
Fig. 3 |. PD-L1 expression is upregulated in MYCTg;KRASG12D cells through a bypass in uORF-mediated translational repression.
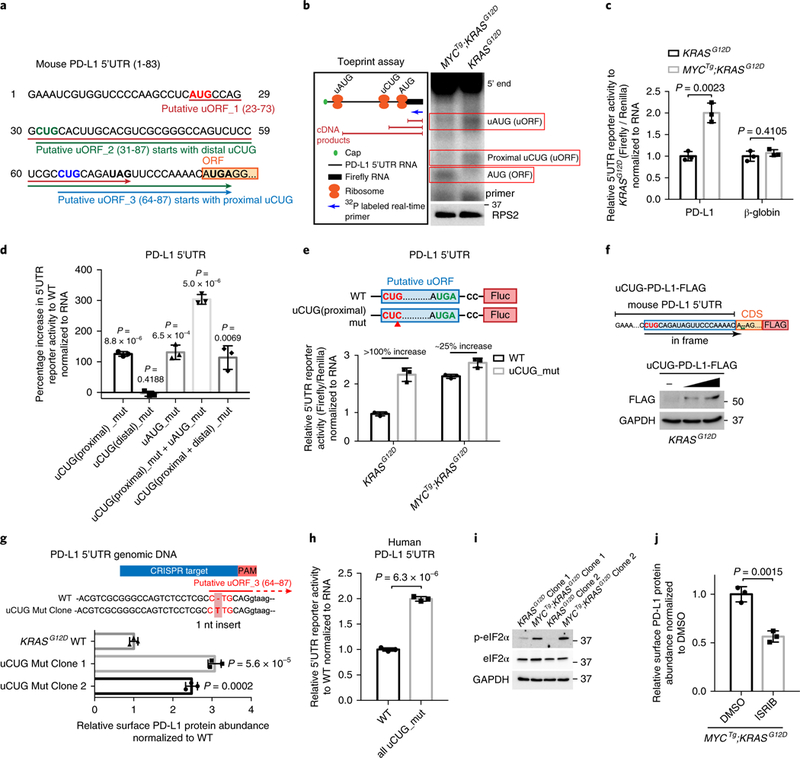
a, The sequence of the 5′UTR of mouse PD-L1 mRNA containing three putative uORFs initiating from uAUG, uCUG(distal), and uCUG(proximal), respectively. Stop codons for each putative uORF are in bold. b, Left: schematics of the toeprint assay. mRNAs composed of mouse PD-L1 5′UTR linked with firefly luciferase were used as the toeprint assay template. Right: autoradiography of the complementary DNA products from the toeprint assay using cytoplasmic lysate from KRASG12D and MYCTg;KRASG12D cell lines. Red boxes highlight the bands indicating ribosome recognitions of the uAUG, uCUG(proximal), and AUG of the main ORF. RPS2 western blot is shown as controls for the amount of ribosomes supplied in each reaction. c, 5′UTR reporter activity of mouse PD-L1 uORF-luciferase and β-globin 5′UTR luciferase (control) in KRASG12D and MYCTg;KRASG12D cell lines. Two-sided t test, n = 3 independent experiments. d, Percentage increase in the 5′UTR reporter activity of mouse PD-L1 uORF-luciferase with point mutations (CU’G’ to CU’C’ or AU’G’ to AU’C’) in the three upstream start codons over that with wild type, in KRASG12D cells. Two-sided t test, n = 3. e, 5′UTR reporter activity of mouse PD-L1 uORF-luciferase with and without a point mutation (CU’G’ to CU’C’) in the proximal uCUG start codon, in KRASG12D and MYCTg;KRASG12D cells. n = 3 independent experiments. Percentages of increase (100% in KRASG12D and 25% in MYCTg;KRASG12D) in the luciferase activity of the uCUG mutation compared with wild type (WT) were shown. f, Upper: constructs for expressing a FLAG-tagged PD-L1 with ‘AUG’ start codon mutation (A’UGA’ to A’A’) (uCUG-PD-L1-FLAG). Lower: western blot of FLAG and GAPDH in KRASG12D cell lines after 48-h transfection, n = 3 independent experiments. g, Upper: mouse PD-L1 5′UTR exon and intron DNA sequence of the wild-type (WT) and uCUG mutation KRASG12D clonal cell line are shown. PAM region (AGG) is highlighted in red, which spans the 5′UTR exon and intron; CRISPR target region that against the 5′UTR exon is labeled in blue. 1 nucleotide (nt) insertion (CTG to C”T”TG) is detected in the uCUG start codon after the CRISPR/Cas9 editing. Lower: relative PD-L1 abundance in wild-type and two-mutation clonal cell lines determined by flow cytometry. Two-sided t test, n = 3 independent experiments. h, 5′UTR reporter activity of human PD-L1 uORF-luciferase with and without point mutations (CU’G’ to CU’C’) in the uCUG start codons in SNU-449 human HCC cell line (wild type, WT). Two-sided t test, n = 3 independent experiments. i, Western blot of p-eIF2α, eIF2α and GAPDH in KRASG12D and MYCTg;KRASG12D cell lines. j, Relative PD-L1 protein abundance assessed by flow cytometry in MYCTg;KRASG12D cell lines treated with DMSO or 0.5 μM ISRIB for 72h. Two-sided t test, n = 3 independent experiments. All values represent the mean± s.d. Uncropped blots are provided in Supplementary Fig 15.
Although recent studies have identified putative uORFs genome-wide29, little is known about the functional contribution of uORF-mediated regulation, particularly in the context of cancer development and the regulation of the immune-checkpoint molecules. To understand whether the uORFs in the 5′UtR of PD-L1 have a significant role in PD-L1 translational control, we transfected the full-length PD-L1 5′UTR upstream of a luciferase reporter in KRASG12D and MYCTg;KRASG12D cells. We observed that the reporter activity of wild-type PD-L1 5′UTR in MYCTg;KRASG12D cells was significantly higher compared with that in KRASG12D cells (Fig. 3c). To characterize the functionality of these uORFs, we mutated the start codons of individual uORFs (Fig. 3d). Notably, disrupting either the uCUG near the main ORF (uCUG(proximal)) or the uAUG in KRASG12D cells increased the reporter activity by >100%, whereas mutating the uCUG apart from the main ORF (uCUG(distal)) had no effect (Fig. 3d). This increase in the reporter activity on proximal uCUG mutation was less significant (~25%) in MYCTg;KRASG12D cells (Fig. 3e). In addition, mutating the stop codon of the uORF, which placed the proximal uCUG in frame with the main ORF, led to a significant increase in reporter activity in KRASG12D cells (Supplementary Fig. 9c). We also designed a vector where the uCUG is the only initiating codon that can produce a full-length flag-tagged PD-L1 protein (uCUG-PD-Ll-FLAG). In this setting, we observed the expression of a full-length flagged-PD-Ll protein in KRASG12D cells (Fig. 3f), further validating the functionality of the uCUG. Moreover, mutating both the uCUG(proximal) and the uAUG led to further enhanced reporter activity (300% increase) (Fig. 3d). These results indicate that there are two functional uORFs (uORF_1 and uORF_3 mediated by uAUG and the proximal uCUG, respectively) in mouse PD-L1 5′UTR that act in an additive manner to suppress downstream main ORF translation.
To further characterize the functional significance of these two uORFs in regulating endogenous PD-L1 translation, we used CRISPR/Cas9 genome editing to generate KRASG12D-derived tumor cell lines in which the proximal uCUG (Fig. 3g) or the uAUG (Supplementary Fig. 9d) in the PD-L1 mRNA 5′UTR is disrupted. Disruption of the uCUG or the uAUG start codon led to significantly enhanced protein abundance of endogenous PD-L1 compared to the non-mutated parental clone (Fig. 3g and Supplementary Fig. 9d), with no effect on PD-L1 mRNA amounts (Supplementary Fig. 9e). Dependence of these non-canonical uORFs highlights their key role in control of PD-L1 expression at the translation level. Similarly, the human PD-L1 5′UTR also contains uCUG start codon uORFs, where the initiation ribosome footprints on these uORFs can be detected in previously published ribosome-profiling datasets carried out in the presence of lactimidomycin30, which stalls ribosomes on initiating codons (Supplementary Fig. 9f). By mutating the uCUGs in the human PD-L1 5′UTR, we observed an 100% increase in the translation of the downstream main ORF (Fig. 3h), suggesting that similar to mouse, human PD-L1 translation is also controlled by uORFs.
An outstanding question is how PD-L1 uORF-mediated translation repression is bypassed in MYCTg;KRASG12D cells. We observed that MYCTg;KRASG12D clones had significantly higher eIF2α phosphorylation amounts compared with our KRASG12D clones (Fig. 3i). eIF2α is a key component of the eIF2 ternary complex31–33 that when phosphorylated facilitates the bypass of uORFs, thereby favoring translation of the main ORF. When we treated MYCTg;KRASG12D cells with integrated stress response inhibitor (ISRIB), a compound selectively reverses the effects of eIF2α phosphorylation34,35, the abundance of PD-L1 protein in MYCTg;KRASG12D cells was markedly downregulated (Fig. 3j), whereas PD-L1 mRNA remained unchanged (Supplementary Fig. 10). These results suggest, at least in part, a functional role for eIF2α phosphorylation in bypass of uORFs. Altogether, several orthogonal lines of evidence demonstrate that MYC elevates PD-L1 protein abundance through relieving the inhibition of PD-L1 mRNA translation mediated by functional, non-canonical uORFs.
The metastatic potential of MYCTg;KRASG12D tumors is dependent on PD-L1 mediated immune evasion.
Given that liver tumors that developed from MYCTg;KRASG12D cells expressed significantly higher PD-L1 protein abundance than KRASG12D (Fig. 2c), whether the increased PD-L1 expression and subsequent immune evasion are responsible for the cancer metastatic potential remains unclear. To this end, we orthotopically injected two individual uCUG mutant KRASG12D clones containing a mutation of the uORF that stimulates PD-L1 translation (KRASG12D Mut clone 1 and 2), into the subcapsular region of the median liver lobe of mice (Supplementary Fig. 11a). Noteworthy, we confirmed that this immunocompetent C57BL/6 orthotopic model was able to recapitulate disease progression in a similar manner as our GEM models: C57BL/6 mice injected with tumor cells coexpressing MYCTg and KRASG12D had a reduced survival time (Fig. 4a), aggressive immune infiltration in the liver (Fig. 4b and Supplementary Fig. 11b), increased PD-L1 abundance (Supplementary Fig. 11c), and increased metastatic potential (Fig. 4b,c and Supplementary Fig. 11d) when compared with those injected with KRASG12D cells. In this system, KRASG12D cells only developed primary tumors with no metastasis (Fig. 4c and Supplementary Fig. 11 d). Notably, 44.4 and 50% of animals injected with the two KRASG12D uORF mutant clones, respectively, developed metastasis (Fig. 4c,d). These results show that increasing the translation of PD-L1 in KRASG12D uORF mutant tumor cells is sufficient to facilitate metastasis, suggesting that the activity of PD-L1 on inhibiting immune surveillance is a key determinant of metastatic potential and cancer aggressiveness. Therefore, to delineate the contribution of T cell surveillance on metastases and survival, we injected MYCTg;KRASG12D and KRASG12D cells into athymic nude mice that lack functional T cells. Ablation of T cells in nude mice ameliorated the difference in the survival rates of KRASG12D and MYCTg;KRASG12D HCC-injected mice (Fig. 4e). Both KRASG12D and MYCTg;KRASG12D HCC-injected athymic nude mice were able to develop pulmonary metastases (Fig. 4f), suggesting that the T cell immune surveillance normally restrains metastasis formation in KRASG12D-derived tumors.
Fig. 4 |. The metastatic potential of MYCTg;KRASG12D tumors is dependent on PD-L1-mediated immune suppression.
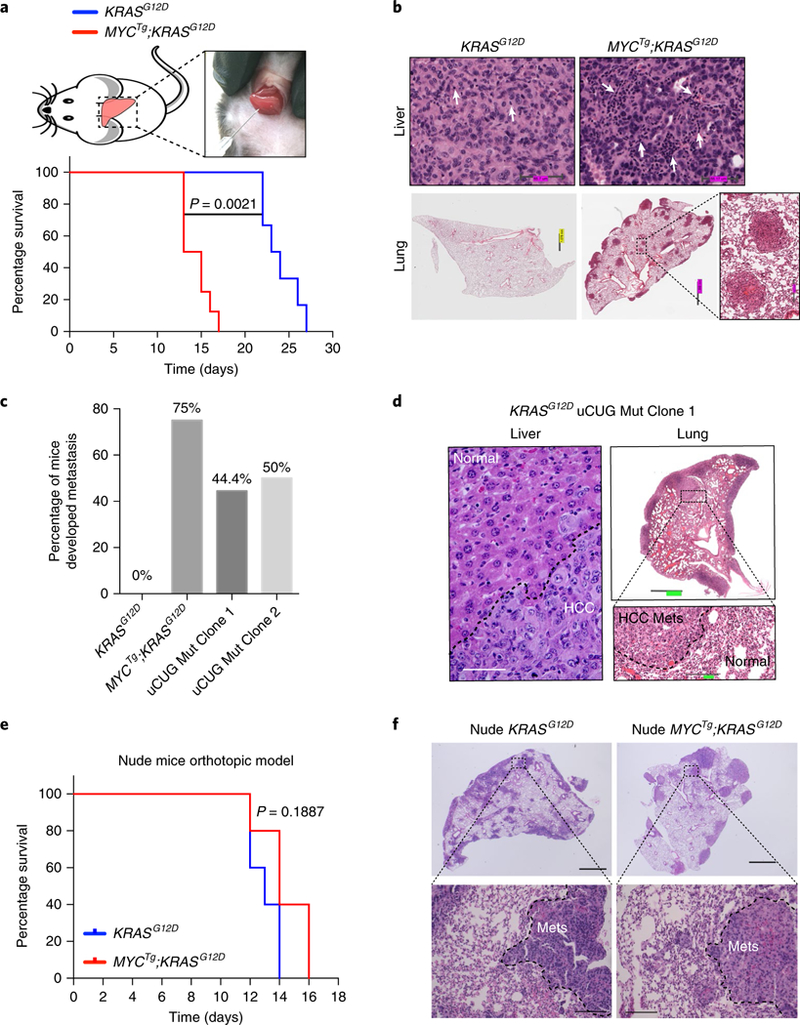
a, Kaplan–Meier curves showing the survival of male C57/BL6 mice with KRASG12D (n = 6) and MYCTg;KRASG12D (n = 8) cells orthotopically injected into livers. Wilcoxon rank sum test. b, Representative histology of livers and lungs of orthotopic KRASG12D (n = 5) and MYCTg;KRASG12D (n = 5) C57BL/6 mice. White arrows represent inflammatory cell infiltration. c, Percentage of C57BL/6 mice injected with KRASG12D (n = 6), MYCTg;KRASG12D (n = 8), KRASG12D Mut Clone 1 (n = 9), or KRASG12D Mut Clone 2 (n = 8) cells developed metastasis. d, Representative histology of livers and lungs of orthotopic KRASG12D Mut Clone 1 mice (n = 5). Scale bars: left, 50 μm; upper right, 909.6 μm; lower right, 151.4 μm. e, Kaplan–Meier curves showing the survival of male athymic nude mice with KRASG12D (n = 5) and MYCTg;KRASG12D (n = 5) cells orthotopically injected into the livers of nude mice. Wilcoxon rank sum test. f, Representative histology of lungs of orthotopic KRASG12D (n = 5) and MYCTg;KRASG12D (n = 5) nude mice. Upper images: scale bar, 1,000 μm. Lower images: scale bar, 100 μm. Mets, metastases.
eFT508, a potent clinical translation inhibitor, dramatically downregulates PD-L1 protein abundance.
Given the finding that a new layer of translational control guides PD-L1 expression, which is important for tumor development, we sought to design a rational pharmacological approach to target PD-L1 translation. Although pharmacological agents that target the core translation machinery itself may induce unwanted toxicity, targeting their post-translational modifications may offer a more favorable therapeutic window. In this respect, it is known that phosphorylation of the main cap-binding protein, eIF4E at Serine 209, positively regulates the oncogenic activity of eIF4E and promotes specific mRNA translation36,37, whereas mutating the eIF4E phosphorylation site in knock-in mice has no effect on organismal development or physiology36. Similarly, ablation of the kinases responsible for phosphorylating eIF4E, mitogen-activated protein kinase interacting kinase 1 and 2 (MNK1/2), has proven to be dispensable for organismal life and the resulting mice do not show any discernible phenotypes38. MYCTg;KRASG12D cells or liver tumors do not possess higher eIF4E phosphorylation than KRASG12D alone (Supplementary Fig. 12a, b), yet given the selective increase in the translational oncogenic program in MYCTg;KRASG12D cells, we reasoned that these tumors may be more sensitive to inhibitors of translation. Thus, to determine whether increased PD-L1 translation can be exploited therapeutically, we used eFT508: a potent and highly selective dual MNK1/2 inhibitor39, now in phase 2 clinical trials (NCT02937675, NCT02605083 and NCT03258398). eFT508 is considerably more potent than other reported MNK inhibitors both biochemically (MNK1 and MNK2, 2.4 and 1 nM, respectively) and in cells inhibiting phosphorylation of eIF4E (6.5 nM). Moreover, eFT508 has exceptional kinome selectivity (tested against >400 kinases) relative to reported MNK inhibitors (the nearest kinase is inhibited approximately 100-fold weaker than MNK1 or MNK2). In addition, eFT508 has excellent drug-like properties (MW 340, protein binding ~50%, logD = 2.7, Caco2 A-B/B-A 15/5, excellent multi-species oral PK)39. In liver tumors, eFT508 efficiently eliminated eIF4E phosphorylation (Supplementary Fig. 12a) without affecting global protein synthesis (Supplementary Fig. 12c). At first, we tested the effects of eFT508 on PD-L1 expression in MYCTg;KRASG12D cells. Notably, inhibiting eIF4E phosphorylation by eFT508 resulted in significant and selective downregulation of MYCTg;KRASG12D-induced PD-L1 protein abundance (Fig. 5a), but not mRNA expression (Fig. 5b); similar effects were also detected for several other translational targets upregulated in MYCTg;KRASG12D cells (Supplementary Fig. 12d–f). On the contrary, eFT508 had limited additional effects in reducing PD-L1 protein abundance in KRASG12D cells where PD-L1 translation is suppressed (Supplementary Fig. 12g). We further found that KRASG12D cells harboring mutations in the uAUG and uCUG uORFs of PD-L1 (KRASG12D uAUG + uCUG_mut) exhibited reduced PD-L1 protein expression by eFT508 (Supplementary Fig. 12h), suggesting that the enhanced translation of PD-L1 can sensitize cells to this translation inhibitor. However, we cannot at present exclude a potentially more direct effect of eIF4E phosphorylation on uORF-mediated translational repression. Moreover, the effects of eFT508 on PD-L1 translation can be mirrored genetically in eIF4E phosphorylation-ablated (Eif4eS209A/S209A) mice36 that show a 50% decrease in PD-L1 protein expression compared with wild type when livers of these animals are transfected with MYC and KRASG12D in vivo (Fig. 5c and Supplementary Fig. 12i).
Fig. 5 |. p-eIF4E inhibition by eFT508 reduces PD-L1 abundance and prevents liver cancer progression and metastasis in vivo.
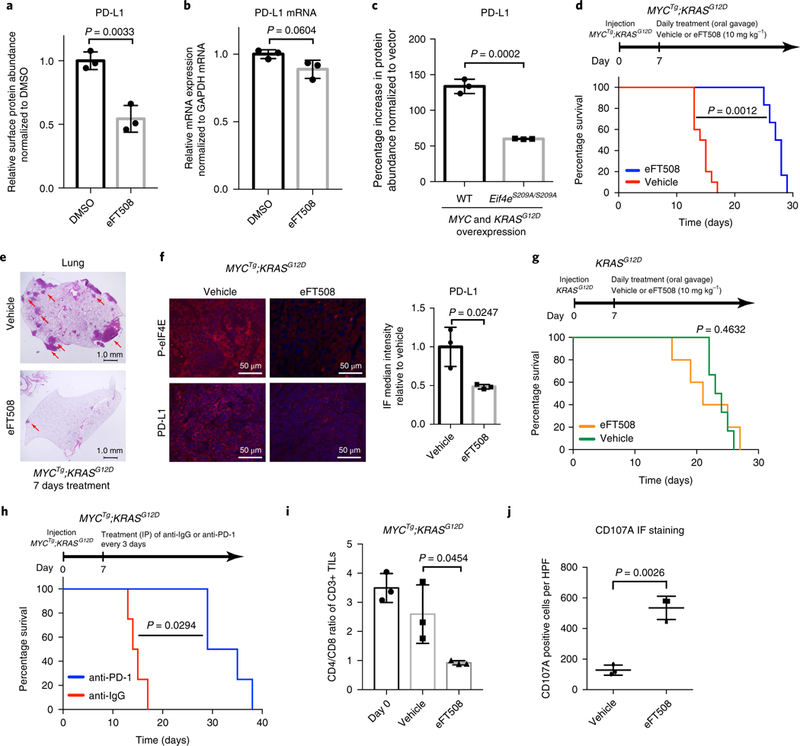
a, Relative surface PD-L1 abundance assessed by flow cytometry in MYCTg;KRASG12D cell lines treated with DMSO or 0.5 μM eFT508 for 72 h. n = 3 independent experiments, two-sided t test. b, Relative PD-L1 mRNA expression in KRASG12D cell lines treated with DMSO or eFT508 for 48h. n = 3 independent experiments, two-sided t test. c, Percentage increase in the PD-L1 abundance in MYC and KRASG12D overexpressed hepatocytes from wild type and Eif4eS209A/S209A, compared with control vector transfected wild-type (WT) and Eif4eS209A/S209A hepatocyte, respectively, assessed by flow cytometry. n = 3 independent experiments, two-sided t test. d, Kaplan–Meier curves showing the survival of male C57/B6 mice with MYCTg;KRASG12D cells orthotopically injected into the livers. Vehicle (n = 10 animals) or eFT508 (10 mg kg–1) (n = 6 animals) was orally administered once a day starting 7 d post-tumor implantation. Wilcoxon rank sum test. e, Representative histology of lungs of MYCTg;KRASG12D injected mice after 7 d vehicle or eFT508 treatments (scale bars, 1.0 mm). Red arrows present metastases in the lungs. n = 5 animals. f, Immunofluorescence staining (IF) for p-eIF4E, PD-L1, and DAPI in MYCTg;KRASG12D liver section on 7d vehicle or eFT508 treatments, n = 3 independent experiments, two-sided t test (scale bars, 50 μm). g, Kaplan–Meier curves showing the survival of male C57/B6 mice with KRASG12D cells orthotopically injected into the livers. Vehicle (n = 6 animals) or eFT508 (10 mgkg–1) (n = 5 animals) was orally administered once a day starting 7 d post tumor implantation. Wilcoxon rank sum test. h, Kaplan–Meier curves showing the survival of male C57/B6 mice with MYCTg;KRASG12D cells orthotopically injected into the livers. Monoclonal antibodies against IgG (n = 4 animals) or PD-1 (200 μg per mouse) (n = 4 animals) were administered by intraperitoneal (IP) injection every 3 d starting 7 d post-implantation. Wilcoxon rank sum test. i, CD4/CD8 T cell ratio in the livers of mice injected with MYCTg;KRASG12D cells on 7-d vehicle (n = 3 animals) or eFT508 (n = 3 animals) treatment. Two-sided t test. j, Quantification of CD107A immunofluorescence intensity in MYCTg;KRASG12D liver section on 7-d vehicle (n = 3 animals) or eFT508 (n = 3 animals) treatments. Two-sided t test. All values represent the mean± s.d.
eFT508 prevents liver cancer progression and metastasis in vivo.
Given that there was a marked inhibitory effect on PD-L1 by eFT508 treatment in MYCTg;KRASG12D HCC cells, but not in KRASG12D cells, we next examined whether eFT508 could prevent tumor progression in the MYCTg;KRASG12D orthotopic preclinical model. Daily oral treatment of eFT508 reduced primary tumor growth (Supplementary Fig. 12j), doubled the survival time of C57BL/6 mice bearing MYCTg;KRASG12D tumors (Fig. 5d), and prevented lung metastasis formation (Fig. 5e). Moreover, eFT508 led to a marked inhibition of eIF4E phosphorylation and 50% reduction in PD-L1 abundance in vivo (Fig. 5f), which recapitulated our in vitro findings. Of note, eFT508 had no effect in mice bearing KRASG12D tumors (Fig. 5g), suggesting that although more aggressive, MYCTg;KRASG12D tumors are more sensitive to eFT508 treatment, as they rely on PD-L1 translation for their progression. The possibility of the presence of an HA model antigen derived from the HA-tag linked to MYC transgene does not influence the immune microenvironment (Supplementary Fig. 13a,b) nor the sensitivity to eFT508 treatment (Supplementary Fig. 13c).
To further address whether inhibiting the PD-1/ PD-L1 functional complex could mimic the anti-tumor effect of eFT508, we treated the MYCTg;KRASG12D orthotopic model with monoclonal antibodies against PD-1. Treatment with anti-PD-1 resulted in a significant increase in survival (Fig. 5h), suggesting that targeting the PD-1/PD-L1 axis can markedly reduce the aggressiveness of MYCTg;KRASG12D tumors. However, unlike the monoclonal antibodies against PD-L1 that also target and may kill immune cells in the tumor microenvironment that express PD-L140,41, eFT508 has a high selectivity against the PD-L1 protein on tumor cells, but not the infiltrated myeloid cells, liver-resident macrophages, and T cells (where PD-L1 expression is relatively low in our tumor model) (Supplementary Fig. 14a,b). Furthermore, eFT508 treatment resulted in a significant increase in cytotoxic CD8+ T cells compared with untreated tumors (Fig. 5i) that were positive in cytotoxic anti-tumor activity as assessed by surface CD107A abundance42 (Fig. 5j). Together, these findings reveal a key molecular underpinning of the immune response in acquiring metastatic potential, the leading cause of cancer lethality, and highlight that oncogene-mediated translational control of the immune checkpoint is effectively targeted with eFT508 specifically in tumor cells without affecting immune cells, maximizing the anti-tumor effect.
Discussion
Oncogenes widely manipulate many diverse gene expression programs to direct cancer progression. Given the fact that we have profiled two end-stage tumor types (KRASG12D and MYCTg;KRASG12D tumors), during the course of their evolution, similar transcriptome profiles may have been co-opted and are consistent with Myc hyperactivation in fully developed Ras tumors. Instead, a more profound remodeling of gene expression at the step of translation control offers unique gene regulatory programs to selectively synthesize effector proteins in unique stages of cancer development, for example, to shape the tumor microenvironment (Fig. 2b). The broader implication of our data is that translational regulation of the immune regulators facilitates tumor cells evasion from immune attack to promote tumor progression and metastasis, and this regulation is not typically detected by standard methodologies that profile cancer cells (that is, genomics and transcriptomics). In particular, although a potent uORF embedded in the PD-L1 5′ UTR serves as the main obstacle for the efficient translation of PD-L1 mRNA, this mechanism leaves a ‘backdoor’ for specific oncogenes, such as MYC, to re-activate PD-L1 translation at least in part by hijacking the phospho-eIF2α dependent adaptive stress pathway, which has been previously linked to the oncogenic activity of MYC43. Cancer cells may use this unique mechanism to more rapidly synthesize PD-L1 protein (compared with transcriptional control), in response to tumor immune environment changes, allowing for tumor progression and metastasis. It is also important to note that genetic alterations in regulatory elements in the 3′ untranslated region of PD-L1 increase PD-L1 expression44 and it will be interesting to determine the interplay between translation control and mRNA stability of this key checkpoint protein. In this respect, a selective, clinically relevant translational inhibitor, eFT508, exhibits remarkable sensitivity when the translation of the immune-check-point protein is hyperactive. In addition, while this manuscript was in press, a recent study has shown that components of the translation initiation complex, eIF4F, can also affect PD-L1 expression through the translational upregulation of STAT1, a transcription factor controlling PD-L1 expression45. Together with our work, this suggests that key immune-checkpoint proteins can be regulated by translation factors at many levels and that translation inhibitors may serve as important drugs for immunotherapies. In the context of liver cancer, our findings are significant as HCC is a major cause of cancer death worldwide with surgical resection as the only means to manage the disease, yet most patients are not suitable for surgery because of poor hepatic reserve and metastatic lesions. Furthermore, as the amplification and activation of MYC and RAS are among the most frequent lesions that cooperate in many human cancers19,46,47, translational control of the immune checkpoint is likely to be more widespread in many cancer types.
It remains poorly understood why only a fraction of cancer patients respond to immune-checkpoint antibody blockade, and resistance to immune-checkpoint inhibitors has been reported in cancer patients48. With respect to our ribosome-profiling data, besides PD-L1, there are several other key factors or pathways involved in cell cycle regulation, tumor cell motility and immune response that are upregulated by Ribo-Seq in tumors with MYC hyperactivation (Supplementary Figs. 5 and 6), which are also effectively targeted by the translation inhibitor eFT508 (Supplementary Fig. 12). Therefore, selective inhibitors of mRNA translation, such as eFT508, may have the potential to both enhance anti-tumor immune response and inhibit cancer progression and metastasis, offering a therapeutic approach that could improve therapeutic efficacy over existing checkpoint inhibitors and could be further combined to achieve beneficial outcomes.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, statements of data availability and associated accession codes are available at https://doi.org/10.1038/s41591–018-0321–2.
Methods
Mice.
A modified pCAGEN vector49 containing PmeI sites flanking the CAG promoter50 and rabbit beta-1 globin gene polyadenylation sites was used to as the backbone of the transgenic vector. Cloning of a Lox-STOP-Lox element51, N-terminal HA-epitope tagged c-MYC (human), encephalomyocarditis virus internal ribosome entry site52, and palmitoylated-eGFP53 was performed to insert these sequences in the multi cloning site located between the CAG promoter and the downstream beta-1 globin gene untranslated sequences containing polyadenylation sites to generate a Cre-activateable bicistronic mRNA capable of simultaneous expression of HA-tagged MYC and palmitoylated GFP in a wide array of cells and tissues. MYCTg mice were created via pronuclear injection of the purified PmeI digested fragment containing the transgene sequence into C57Bl/6 zygotes by the Gladstone Institutes transgenic core facility. Our MYCTg mice has a similar MYC overexpression range to human HCC. The LSL-KRASG12D mouse (a point mutation endogenous KRAS locus) was purchased from The Jackson Laboratory, and mice with liver-specific KRASG12D expression (Alb Cre; KRASG12D) were generated. To generate Alb Cre;MYCTg;KRASG12D mice, we intercrossed the MYCTg mouse to the Alb Cre; KRASG12D mouse. The University of California San Francisco Institutional Animal Care and Use Committee approved all studies involving live mice.
In vivo sample preparation.
Mouse liver tumors were micro-dissected immediately after animal euthanization. Liver perfusion (with ice-cold PBS containing 10 mg ml–1 cycloheximide). Wild-type liver or the micro-dissected tumors were snap frozen in liquid nitrogen, followed by grounding with mortar and pestle. Tumor was resuspended in the lysis buffer (20 mM Tris pH 7.5, 150 mM NaCl, 15 mM MgCl2, 100 μg ml–1 cycloheximide, 1 mM DTT, 0.5% Triton X-100, 0.1 mg ml–1 heparin, 8% glycerol, 20 U per ml TURBO DNase, ×1 Combined Protease and Phosphatase Inhibitor).
Deep-sequencing library preparation.
The ribosome-profiling analysis was carried out as described previously54. Briefly, after removing the nuclei and mitochondria, lysate was treated with RNase A/T1 mix (0.5 mg RNase A (Ambion, AM2272) and 1,000 U RNase T1 (Life Technologies, 2280)) to digest mRNAs that are not protected by the ribosome. The digestion was stopped by adding SUPERase-In RNase Inhibitor (20 U per μl, Ambion, AM2696), followed by centrifuging to pellet ribosomes. Ribosome-protected fragments were purified by TRIzol, and two biological replicates were performed for each type of Ribo-Seq and all Ribo-Seq libraries (three biological replicates for wild-type liver).
Analysis of RNA-Seq and Ribo-Seq libraries.
A layered alignment was performed to first discard reads mapping to mouse ribosomal RNA (rRNA) sequences using Burrows–Wheeler Aligner55. Non-rRNA reads were then aligned against the mouse transcriptome (GRCm38 build 76) using RSEM56 and bowtie2 57. Reads were aligned strand-specifically to exclude anti-sense or other non-transcript reads. For each gene, total number of ribosome-protected fragments and RNA reads were counted and fed into the downstream analysis. A minimum of two counts per million in at least two Ribo-Seq libraries was used as a cutoff to filter out lowly expressed genes, following the guideline58. Differential gene expression analyses were performed using the limma package59 with voom method60. Briefly, the effective library size of each deep-sequencing library was calculated and raw read count data were normalized by the trimmed mean of M values method61. Normalized read counts were reported in Supplementary Tables 1 and 2. Variance-mean relationship in count data was estimated using the voom method60 and differential gene expression was tested using a model of negative binomial distribution. A linear regression was performed to the normalized read counts, as a function of sample type variables (‘WT’, ‘KRASG12D’ or ‘MYCTg;KRASG12D’). Here, the coefficient of sample type variables (that is, ‘MYCTg;KRASG12D’ over ‘KRASG12D’) is a measurement of corresponding differential gene expressions. P values were adjusted using the Benjamini–Hochberg procedure for multi-testing, and these adjusted P values (Padj or false discovery rate) were reported for each gene in Supplementary Data. To visualize the deep-sequencing datasets, sequencing reads were aligned to the mouse genome (GRCm38) using the STAR RNA-Seq aligner62, and imported into the Integrative Genomics Viewer63.
For the comparison of the similarity of human liver cancer and mouse liver cancer: human liver hepatocellular carcinoma and normal liver samples were based on data generated by The Cancer Genome Atlas Research Network (http://cancergenome.nih.gov/). Raw estimated number of reads that aligned to each transcript were obtained for 370 primary tumors and 50 normal liver samples from FireBrowse (http://firebrowse.org/). Differential gene expression analyses were performed similar as for the mouse samples. Here, a linear regression was performed to the normalized read counts, as a function of sample type variables (‘primary tumor’ or ‘normal’). The coefficient of sample type variables (‘primary tumor’ over ‘normal’) is a measurement of differential gene expressions. P values were adjusted using the Benjamini–Hochberg procedure for multi-testing.
Gene functional analyses and visualization.
Gene Ontology enrichment and clustering analysis was carried out using ClueGO64 for genes that are upregulated in Ribo-Seq comparing MYCTg;KRASG12D to KRASG12D (Padj < 0.1 and log2FC > 2). The following parameters were used: Reference set: all genes analyzed; Enrichment: right-sided hypergeometric test, P value adjustment: Benjamini–Hochberg adjustment. Enriched gene ontology terms were then grouped using Kappa statistics with leading group term on the basis of the highest statistical significance. Protein interaction networks were retrieved from STRING v.10.0 database65 and imported into Cytoscape66 for visualization. Each node represents a gene and edges show protein-protein association with width proportional to the association score. Disconnected nodes were removed from the plot for simplicity.
Plasmids.
PD-L1 5′UTR was synthesized and cloned into the HindIII and NcoI sites of pGL3-SV40 control vector (Promega). CUG and STOP mutations were carried out using site-direct mutagenesis. PD-L1 5′UTR, CDS and 3′UTR fused with a FLAG sequence were amplified and cloned into murine stem cell virus backbone vector. CDS start codon ATG deletion and frame shift (‘TG’ removal from ‘ATG’) were generated by site-direct mutagenesis. Human MYC, a T2A peptide sequence and KRASG12D were together cloned into a murine stem cell virus backbone vector. Sequences were based off the NCBI database.
Immunofluorescence.
Liver and lung tissues were dissected from mice and fixed in 10% formalin overnight at 4 °C. Tissues were subsequently dehydrated in ethanol at room temperature, mounted into paraffin blocks and sectioned at 5 μm. Specimens were de-paraffinized, rehydrated, and antigen unmasking was performed as previously described54. The sections were then incubated in 5% goat serum, 1% BSA in tris-buffered saline for 1 h at room temperature. Primary antibodies were diluted 1:50–1:500 in blocking solution and incubated on sections overnight at 4 °C. Specimens were incubated with the appropriate Alexa 488 and 594 labeled secondary (Invitrogen) at 1:500 for 2 h at room temperature after washing, followed by mounting with DAPI Hardset Mounting Medium (Vector Lab). Zeiss AxioImager M1 microscope was used to image. Individual cells were quantified for mean fluorescence intensity using the Axiovision (Zeiss, Release 4.8) densitometric tool.
H&E staining.
Paraffin-embedded liver and lung specimens were de-paraffinized and rehydrated, and stained with hematoxylin (Thermo Scientific), followed with washes with water. An incubation in differentiation RTU (VWR) and two washes with water followed by two 70% ethanol washes were performed. The samples were then stained with eosin (Thermo Scientific) then dehydrated with ethanol followed by CitriSolv (Fisher). Slides were mounted with Cytoseal XYL (Richard Allan Scientific).
Luciferase assay.
KRASG12D and MYCTg;KRASG12D cells were transfected in 12-well plates with 200 ng of pGL3 (Firelfy luciferase) constructs containing full-length or mutant 5′UTR of PD-L1 and 40 ng of pRL (Renilla luciferase) plasmid using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Cells were collected 24 h post-transfection and half of the cells were assayed using Dual luciferase kit (Promega), the other half were proceeded for TRIzol (Invitrogen) purification of RNA. Firefly luciferase activity was normalized to Renilla activity, and further normalized to Firefly and Renilla luciferase RNA amounts quantified by RT-qPCR.
Western blotting.
Western blots were performed with antibodies against of GFP (Abcam no. ab290, 1:2,000), MYC (Cell Signaling no. 5605, 1:1,000), β-actin (Sigma no. A5316, 1:10,000), ARG1 (Sigma no. HPA024006, 0.4 μg ml–1 for western blot), RASG12D (Cell Signaling no. 14429, 1:1,000), p-eIF2α Ser51 (Cell Signaling no. 3597, 1:500), eIF2α (Cell Signaling no. 9722, 1:1,000), p-eIF4E Ser209 (Cell Signaling no. 9721, 1:1,000), eIF4E (BD no. 610270, 1:1,000), GAPDH (Cell Signaling no. 2118, 1:1,000), p-ERK1/2 (Cell Signaling no. 4370, 1:1,000), SNAIL (Proteintech, 13099–1-AP, 1:500), CDC20 (Proteintech, 10252–1-AP, 1:1,000), PLK1 (Proteintech, 10305–1-AP, 1:1,000), ADAM8 (Proteintech, 23778–1-AP, 1:1,000), RPS2 (Abcam, ab58341, 1:1,000), HA-HRP (Cell Signaling no. 2999, 1:1,000), HSP90 (Cell Signaling, no. 4877, 1:1,000), and PD-L1 (Cell Signaling, no. 13684, 1:1,000). Blots are developed using ChemiDoc Imaging Systems (BioRad).
Sucrose gradient fractionation.
KRASG12D and MYCTg;KRASG12D cells were treated with 0.1 mg ml–1 Cycloheximide for 5 min, before lysing in 300 μl of lysis buffer (20 mM Tris pH 7.5, 200 mM NaCl, 15 mM MgCl2, 1 mM DTT, 1% Triton X-100, 0.1 mg ml–1 cycloheximide, 200 U per ml RNasin (Promega)). Nuclei and membrane debris was then removed by centrifuging at 10,000g, for 5 min. The lysate was loaded onto a sucrose gradient (10–50% sucrose(w/v), 20 mM Tris pH 7.5, 100 mM NaCl, 15 mM MgCl2 and centrifuged in a SW41Ti rotor (Beckman) for 2.5 h at 38,000 r.p.m. at 4 °C. Fractions were collected by density gradient fractionation system (Teledyne ISCO).
qPCR.
RNA was isolated using TRIzol (Invitrogen) purification using PureLink RNA Mini Kit (Thermo Fisher, 12183018). RNAs were converted into cDNAs using High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). cDNAs were diluted 1 to 10, and used for SYBR green detection RT-qPCR assay. Primers were used at 250 nM per reaction. Cd274 F 5′-AGTCTCCTCGCCTG CAGATA, Cd274 R 5′-AGTAAACGCCCGTAGCAAGT. Gapdh F 5′- C A ATG A ATACGGCTACAGCAA, Gapdh R 5′-AGGGAGATGCTCAGTGTTGG. Fluc F 5′ -CAACTGCATAAG GCTATGAAGAGA, Fluc R 5′-ATTTGTATTCAGCCCATATCGTTT. Rluc F 5′- CAAAGAG AAAGGTGAAGTTCGTC, Rluc R 5′- TGTACAACGTCAGGTTTACCAC.
Hydrodynamic tail vein injection.
Plasmid DNA was injected by hydrodynamic technique as previously described67. Briefly, endotoxin-free plasmid DNA was prepared using Qiagen Endo Free Maxi Kit and dissolved in Saline solution to a final volume of 8% of body weight at room temperature. 8–10-week-old gender-matched mice were used for comparison. They were restrained and the lateral tail vein was accessed with a 26 G needle. Each mouse was administrated with 10 μg of dissolved DNA and the administration of the solution was performed in 10 s or less without extravasation.
Flow cytometry.
Cultured cell lines were detached and gathered with ×1 citric saline. Primary hepatocytes were collected from mice and prepared with liver dissociation kit (Miltenyi Biotech, 130–105-807) according to the manufacturer instructions. Mouse anti-PD-L1 antibody (BioLegend, 124314) were then added to single cell suspension, followed by incubation for 30 min at 4 °C. Data were acquired through a BD FACSVerse and were analyzed with FlowJo software (Tree Star).
Immune profiling.
Mice were euthanized at described post-natal ages and when clinically indicated due to terminal burden of disease. After euthanasia, the thoracic cavity was revealed and 24 G butterfly needle was inserted into the left ventricle. Distal tissues were perfused using instillation of 20 ml of PBS until blanching of the liver is observed. Harvested livers were minced with sterile scalpels before transfer into GentleMACS C Tubes (Miltenyi Biotec). Digestion media with enzyme composition per Miltenyi Liver Dissociation Kit (5 ml) was added to each tube and digestion to single cell suspension performed using GentleMACS on program ‘37_m_LIDK_1’. Digestion was then filtered through 70 μm mesh and washed with 20 ml PBS followed by centrifugation at 450g for 8 min at 4 °C. Red blood cells were lysed by resuspension of the pellet in 5 ml of ACK lysis buffer (ThermoFisher). Lysis quenched after 3 min with 25 ml of ice cold FACS (PBS with 0.5% BSA and 5 mM EDTA) buffer. Suspension again pelleted by centrifugation at 450 g for 8 min at 4 °C. Resulting pellet was resuspended in 5 ml of RPMI-L (RPMI supplemented with 5% FBS and 5 mM EDTA) and overlayed onto 10 ml of Lymphoprep (StemCell Technologies) followed by centrifugation at 800 g for 25 min at room temperature. Isolated aqueous- and interphases were collected and washed with FACS buffer by spinning down at 300g for 5 min at 4 °C. Mononuclear cells were counted and aliquoted to 1 × 107 cells before staining for flow cytometry.
For analysis of infiltrating mononuclear cells, isolated cells were blocked with CD16/32 FcB (Cell Culture Facility, UCSF) for 15 min on ice. Samples were then divided into two fractions for staining of either Lymphoid or Myeloid cell markers for 45 min on ice in dark. Lymphoid markers were analyzed by staining APC-CD45, PECy7-CD3, PB-CD19, PE-CD4, and PerCPCy5.5-CD8. Myeloid markers were analyzed by staining APC-CD45, PECy7-Ly6G/C, PE-CD11c, PerCPCy5.5-CD14, and Bv510-CD11b. All antibodies were purchased from eBioscience. Samples were then acquired by BD LSR II (Becton Dickinson) and analyzed with FlowJo (FlowJo, LLC).
Generating mice liver cancer cell lines.
Alb-Cre; KRASG12D and Alb-Cre; MYCTg;KRASG12D mice were anesthetized and disinfected with 70% ethanol. Abdomen and chest were exposed and then portal vein was cut, followed immediately by cannulating the thoracic SVC via atrium to clear the blood using a breathing apparatus. The surface of the liver was wet with L-15 solution. The liver was then rinsed with the Liver Perfusion Media (GIBCO cat. no. 17701–038) for 4 min, followed by perfusion with the liver digest media (GIBCO cat. no. 17703–034) for 4–8 min. After a sufficient digestion, the liver was isolated and transferred to a sterile plastic petri dish in a cell culture hood. The liver was then minced in the hepatocyte plating medium (DMEM H21 high glucose medium supplied with ×1 Pen/Strep solution, ×1 insulin-transferrin-selenium solution (GIBCO no. 41400–045) and 5% FBS) and filtered through a sterile gauze into a 50 ml conical tube. Cells were washed with and resuspended in hepatocyte plating medium and plated on a sterile plastic petri dish for culturing. Cells were then plated into 96-well plates for single cell selection. KRASG12D and MYCTg;KRASG12D single clonal cancer cells were confirmed by western blot and immunofluorescence with MYC, KRASG12D, ARG1 and HA antibodies.
Toeprint assay.
Mouse PD-L1 5′UTR sequence was cloned into pGL3 (Firefly luciferase) vector that contains a T7 promoter. mRNA was in vitro transcribed using mMESSAGE mMACHINE T7 Transcription Kit (Thermo Fisher Scientific). The reverse transcription oligonucleotide (TATCTCTTCATAGCCTTATG) was [γ−32P]ATP labeled, and pre-annealed to the mRNA by heating for 1 min at 65 °C followed by 37 °C for 8 min in 40 mM Tris-HCl (pH 7.5) and 0.2 mM EDTA. Cytoplasmic cell lysates from KRASG12D and MYCTg;KRASG12D cells were extract and incubated with the oligo-mRNA complex, followed by reverse transcription as previously described68. cDNA products were extracted with phenol and analyzed on 8% TBE gels.
CRISPR in the Alb-Cre; KRASG12D cell line.
A guide RNA (sgRNA) against Cd274 uORF sequence was designed from the F. Zhang laboratory database (crispr.mit.edu)69, and cloned into pSpCas9(BB)-2A-GFP (PX458) vector (Addgene no. 48138)69. Non-targeting control sgRNA was used side-by-side with Cd274 sgRNA to rule out phenotypes result from nonspecific editing. To obtain single clonal cell lines, GFP-positive cells were sorted into 96-well plates. Genomic DNAs were isolated, PCR amplified and sequenced to confirm editing. PD-L1 protein changes were detected by flow cytometry. Cd274 sgRNA (targeting uCUG): 5′-GGGCCAGTCTCCTCGCCTGC-3′. Cd274 sgRNA (targeting uAUG): 5′-TGGTCCCCAAGCCTCATGCC-3′.
Intrahepatic metastatic HCC graft implantation and drug treatment.
Ex vivo cultures of primary, single-clone cell lines from individual liver tumors were derived from one Alb-Cre; KRASG12D and one Alb-Cre; MYCTs;KRASG12D mice. HCC cells described above were trypsinized, counted and 5 ×105 of cells were injected into the subcapsular region of the median liver lobe of C57BL/6 mice. Analgesics including bupivacaine and buprenorphine were given to the mice, while meloxicam was not given as it may have an effect on the tumor immune microenvironment. Primary liver tumor formation was detected at day 4. Over 70% of the mice successfully develop lung metastasis at days 12–18. Mice were treated daily 7 d post-injection of tumor cells with 10 mg kg–1 of eFT508 or vehicle control through oral gavage.
Statistics.
Data were represented as mean ± s.d. Sample sizes were chosen on the basis of previous literature, or decided by using a power analysis (power/sample size calculator) on the basis of pilot experiments that provided an estimate of effect size. For in vivo experiments in mice, an n = 4 was the minimum amount used. Data were analyzed and statistics performed in Microsoft Excel and/or GraphPad Prism6. A two-tailed, unpaired Student’s t test was used for pair-wise comparison. For correlation analysis, a two-sided Pearson’s correlation test was used. For survival analysis, a log-rank test or Wilcoxon rank sum test was used. P < 0.05 was considered significant, and the exact P values are indicated in the figures.
Ethical compliance.
All experiments involving live vertebrates performed at UCSF were done in compliance with ethical regulations approved by the UCSF IACUC committee. Protocols for human sample collection and analysis were approved by UCSF, and all analyses of human data were carried out in compliance with the relevant ethical regulations.
Supplementary Material
Acknowledgements
We would like to thank members of the Ruggero laboratory for discussion and critical reading of the manuscript. We thank C. Her for his help with the mouse HCC cell line generation protocol, D. Wang for the help with hydrodynamic transfection, and K. Fujii and D. Simsek for advice and technique assistance. We thank B. Tiano for the contribution in developing and characterizing the MYC transgenic mice. This work was supported by the Damon Runyon Postdoctoral Fellowship (Y.X.), AACR-Incyte Corporation Fellowship In Basic Cancer Research (grant no. 17–40-46-JIN) (H.Y.J), Life Science Foundation Postdoctoral Fellowship (S.Z.), Department of Defense Physician training award (H.G.N), the Campini Foundation, The Leukemia and Lymphoma Foundation Career Development Grant and UCSF Department of Pediatrics K12 (grant no. 5K12HD072222–05) (C.M.F.), American Cancer Society Postdoctoral Fellowship and Conquer Cancer Foundation Young Investigator Award (J.D.G), Pew Scholars Award (M.B.), NIH grant no. 1R01HD086634 (M.B.), NIH grants (nos. R01CA140456, R01CA154916, and R01CA184624) (D.R.). M.B. is a New York Stem Cell Foundation Robertson Investigator. D.R. is a Leukemia and Lymphoma Society Scholar.
Footnotes
Competing interests
C.R.S., S.H.R. and K.R.W. are employees and shareholders of eFFECTOR Therapeutics, Inc. S.T.W. is the President and CEO of eFFECTOR Therapeutics, Inc. D.R. is a shareholder of eFFECTOR Therapeutics, Inc., and a member of its scientific advisory board.
Data availability
The sequencing data of the manuscript can be accessed using the following accession number: GSE105147.
Supplementary information is available for this paper at https://doi.org/10.1038/s41591-018-0321-2.
Reprints and permissions information is available at www.nature.com/reprints.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Chen DS & Mellman I Oncology meets immunology: the cancer-immunity cycle. Immunity 39, 1–10 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Casey SC et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 352, 227–231 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marzec M et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc. Natl Acad. Sci. USA 105, 20852–20857 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stewart BW & Wild C World Cancer Report 2014 (International Agency for Research on Cancer WHO Press, 2014). [Google Scholar]
- 5.Ally A et al. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 169, 1327 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schlaeger C et al. Etiology-dependent molecular mechanisms in human hepatocarcinogenesis. Hepatology 47, 511–520 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Marquardt JU et al. Sequential transcriptome analysis of human liver cancer indicates late stage acquisition of malignant traits. J. Hepatol. 60, 346–353 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaposi-Novak P et al. Central role of c-Myc during malignant conversion in human hepatocarcinogenesis. Cancer Res. 69, 2775–2782 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Dell MR et al. Kras(G12D) and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res. 72, 1557–1567 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saha SK et al. Mutant IDH inhibits HNF-4alpha to block hepatocyte differentiation and promote biliary cancer. Nature 513, 110–114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grivennikov SI, Greten FR & Karin M Immunity, inflammation, and cancer. Cell 140, 883–899 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sato E et al. Intraepithelial CD8+tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl Acad. Sci. USA 102, 18538–18543 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao Q et al. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J. Clin. Oncol. 25, 2586–2593 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Fu JL et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology 132, 2328–2339 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Fridman WH, Pages F, Sautes-Fridman C & Galon J The immune contexture in human tumours: impact on clinical outcome. Nat. Rev. Cancer 12, 298–306 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Tumeh PC et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mellman I, Coukos G & Dranoff G Cancer immunotherapy comes of age. Nature 480, 480–489 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joyce JA & Fearon DT T cell exclusion, immune privilege, and the tumor microenvironment. Science 348, 74–80 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Kortlever RM et al. Myc cooperates with Ras by programming inflammation and immune suppression. Cell 171, 1301–1315 e1314 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ingolia NT, Ghaemmaghami S, Newman JRS & Weissman JS Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Butte MJ, Keir ME, Phamduy TB, Sharpe AH & Freeman GJ Programmed death-1 ligand 1 interacts specifically with the B7–1 costimulatory molecule to inhibit T cell responses. Immunity 27, 111–122 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herbst RS et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gordon SR et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545, 495–499 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shalapour S et al. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature 521, 94–U235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khan AR et al. PD-L1hi B cells are critical regulators of humoral immunity. Nat. Commun. 6, 5997 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Lau J et al. Tumour and host cell PD-L1 is required to mediate suppression of anti-tumour immunity in mice. Nat. Commun. 8, 14572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gollwitzer ES et al. Lung microbiota promotes tolerance to allergens in neonates via PD-L1. Nat. Med. 20, 642–647 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Truitt ML & Ruggero D New frontiers in translational control of the cancer genome. Nat. Rev. Cancer 17, 332 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Sendoel A et al. Translation from unconventional 5′ start sites drives tumour initiation. Nature 541, 494–499 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stern-Ginossar N et al. Decoding human cytomegalovirus. Science 338, 1088–1093 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Calkhoven CF, Muller C & Leutz A Translational control of C/EBP alpha and C/EBP beta isoform expression. Gene Dev. 14, 1920–1932 (2000). [PMC free article] [PubMed] [Google Scholar]
- 32.Palam LR, Baird TD & Wek RC Phosphorylation of eIF2 facilitates ribosomal bypass of an inhibitory upstream ORF to enhance CHOP translation. J. Biol. Chem. 286, 10939–10949 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Starck SR et al. Translation from the 5′ untranslated region shapes the integrated stress response.Science 351, 3867 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sekine Y et al. Stress responses. Mutations in a translation initiation factor identify the target of a memory-enhancing compound. Science 348, 1027–1030 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sidrauski C, McGeachy AM & Ingolia NT & Walter P The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. Elife 4, 05033 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Furic L et al. eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc. Natl Acad. Sci. USA 107, 14134–14139 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Herdy B et al. Translational control of the activation of transcription factor NF-kappaB and production of type I interferon by phosphorylation of the translation factor eIF4E. Nat. Immunol. 13, 543–550 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ueda T, Watanabe-Fukunaga R, Fukuyama H, Nagata S & Fukunaga R Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Mol. Cell. Biol. 24, 6539–6549 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reich SH et al. Structure-based design of pyridone-aminal eFT508 targeting dysregulated translation by selective mitogen-activated protein kinase interacting kinases 1 and 2 (MNK1/2) inhibition. J. Med. Chem. 26, 3516–3540 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Pulko V et al. B7-H1 expressed by activated CD8 T cells is essential for their survival. J. Immunol. 187, 5606–5614 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu X et al. B7-H1 antibodies lose antitumor activity due to activation of p38 MAPK that leads to apoptosis of tumor-reactive CD8(+) T cells. Sci. Rep. 6, 36722 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Betts MR et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J. Immunol. Methods 281, 65–78 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Hart LS et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J. Clin. Invest. 122, 4621–4634 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kataoka K et al. Aberrant PD-L1 expression through 3′-UTR disruption in multiple cancers. Nature 534, 402 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Cerezo M et al. Translational control of tumor immune escape via the eIF4F-STAT1-PD-L1 axis in melanoma. Nat. Med. 24, 1877–1886 (2018). [DOI] [PubMed] [Google Scholar]
- 46.D’Cruz CM et al. c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nat. Med. 7, 235–239 (2001). [DOI] [PubMed] [Google Scholar]
- 47.Ying HQ et al. Oncogenic kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sharma P, Hu-Lieskovan S, Wargo JA & Ribas A Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matsuda T & Cepko CL Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc. Natl Acad. Sci. USA 101, 16–22 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miyazaki J et al. Expression vector system based on the chicken beta-actin promoter directs efficient production of interleukin-5. Gene 79, 269–277 (1989). [DOI] [PubMed] [Google Scholar]
- 51.Jackson EL et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Gene Dev. 15, 3243–3248 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rees S et al. Bicistronic vector for the creation of stable mammalian cell lines that predisposes all antibiotic-resistant cells to express recombinant protein. Biotechniques 20, 102–104,106, 108–110 (1996). [DOI] [PubMed] [Google Scholar]
- 53.Okada A, Lansford R, Weimann JM, Fraser SE & McConnell SE Imaging cells in the developing nervous system with retrovirus expressing modified green fluorescent protein. Exp. Neurol. 156, 394–406 (1999). [DOI] [PubMed] [Google Scholar]
- 54.Hsieh AC et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485, 55–61 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H & Durbin R Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li B & Dewey CN RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–U354 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen Y, Lun AT & Smyth GK From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. FI000Res. 5, 1438 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ritchie ME et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Law CW, Chen YS, Shi W & Smyth GK voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robinson MD & Oshlack A A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Robinson JT et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bindea G et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Szklarczyk D et al. STRINGv10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shannon P et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu F, Song Y & Liu D Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 6, 1258–1266 (1999). [DOI] [PubMed] [Google Scholar]
- 68.Kozak M Primer extension analysis of eukaryotic ribosome-mRNA complexes. Nucleic Acids Res. 26, 4853–4859 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ran FA et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.