

Abstract
To survive, fungal pathogens must acquire nutrient metals that are restricted by the host while also tolerating mechanisms of metal toxicity that are induced by the host. Given this dual vulnerability, we hypothesized that a pathogen’s access to and control of essential yet potentially dangerous metal ions would affect fungal tolerance to antifungal drug stress. Here, we show that Candida albicans becomes sensitized to both Cu limitation and Cu elevation during exposure in liquid culture to the antifungal drug fluconazole, a widely prescribed antifungal agent. Spectroscopic data confirm that while fluconazole forms a complex with Cu(II) in water, interactions of fluconazole with neither Cu(II) nor Cu(I) are observed in the cell culture media used for the cellular assays. This result is further supported by growth assays in deletion strains that lack Cu import machinery. Overall, we establish that increases in Cu levels by as little as 40 nM over basal levels in the growth medium reduce tolerance of C. albicans to fluconazole in a way that does not require formation of a Cu–fluconazole complex. Rather, our data point to a more complex relationship between drug stress and Cu availability that gives rise to metal-mediated outcomes of drug treatment.
Graphical Abstract
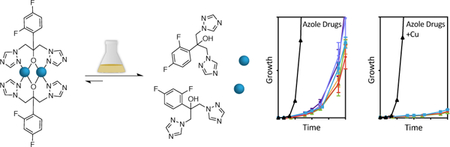
Modulating copper levels in the growth environment influences tolerance of Candida albicans to azole antifungal drugs.
Introduction
The opportunistic fungal pathogen Candida albicans typically exists as a commensal organism in its mammalian host but can invade the bloodstream of immunocompromised individuals and spread to multiple organs, giving rise to life-threatening infections.1 Like any successful pathogen, C. albicans has developed sophisticated adaptation mechanisms to propagate in hostile and varying host environments, including meticulous control of essential metals. For example, C. albicans has been shown to adjust its transcription circuitry to promote iron (Fe) acquisition during bloodstream infection, but protect from Fe toxicity while existing as a commensal in the gut.2 In another example of C. albicans’ adaptability to metal availability, it has been shown to fine-tune superoxide dismutase (SOD) expression as a function of copper (Cu) availability, utilizing Cu-requiring Sod1 under Cu-replete conditions and Mn-requiring Sod3 when intracellular Cu levels are low.3, 4 Similarly, in a mouse model of C. albicans infection, the pathogen senses elevated kidney Cu levels early in infection, and responds by upregulating Cu export and repressing import. As Cu levels decrease at later infection stages, export is repressed and import is induced.5
Given the importance of strategies to orchestrate the concerted movement, compartmentalization, and utilization of biometals to survive host-imposed stress, we posited that a pathogen’s access to and management of essential metals could also impact its ability to tolerate drug-induced stress. One of several azole antifungals, fluconazole is a mainstay in therapy to treat fungal infections, including those caused by C. albicans. Benefits of fluconazole include its low cost, limited toxicity, and ability to be administered orally.6 Its mechanism of action involves binding and inhibiting the heme center of lanosterol 14α-demethylase, Cyp51 (gene product of ERG11), thereby disrupting the biosynthesis of ergosterol, a key component of the fungal cell membrane.7 In the clinic for nearly three decades, fluconazole has been widely studied, including, to some extent, its relationship with metals and metal-dependent biological processes. For example, a metal complex of Cu and fluconazole was investigated for activity against several clinical isolates of C. albicans,8 and a variety of metal-azole complexes have been investigated for antimicrobial activity.9–14 Furthermore, Fe deprivation has been linked to increased fluconazole susceptibility.15, 16
In this work, we tested the consequences of modulating Cu levels in the growth medium on the efficacy of fluconazole against C. albicans. We find that small increases in Cu levels improve the efficacy of fluconazole in a concentration- and time-dependent manner. Investigations into speciation of growth media components lead us to conclude that this potentiation is not due to direct complex formation between fluconazole and Cu, but rather separate targets working in parallel.
Results
Fluconazole induces Cu-potentiated growth inhibition.
To determine the impact of Cu availability on fluconazole susceptibility, growth experiments were performed in the presence of fluconazole under conditions of Cu limitation and Cu supplementation. Cu-limited conditions were established by addition of membrane-impermeable extracellular Cu(I) chelator bathocuproinedisulfonic acid (BCS) to the broth, and Cu supplementation was achieved by adding CuCl2 or CuSO4. Because azole antifungals are fungistatic (inhibit cell division) not fungicidal (cause cell death),17 drug tolerance is often observed, which refers to the ability of a subpopulation of cells to grow at drug concentrations exceeding the minimal inhibitory concentration (MIC).18 For azoles specifically, the degree of tolerance is described by a phenomenon known as “trailing growth,” which is residual growth in the presence of an azole drug at a concentration above the MIC.19, 20 Importantly, tolerance is distinct from drug resistance in that it is epigenetic (reversible), in contrast to resistance, which results from stable genetic mutations.19
As shown in Fig 1a, trailing growth is observed for cells treated with fluconazole alone, with these cells recovering to 50% of the untreated control at 48 h in yeast peptone dextrose (YPD) medium, a complex and nutritionally rich medium derived from peptone and yeast extracts. Under these conditions, the 24-h MIC of fluconazole is 2.5 µM (ESI Fig. S1). Yet by 48 h, cells treated with even 50 µM fluconazole are able to grow (ESI Fig. S2). Strikingly, increasing the Cu content of the growth medium from basal levels (approx. 0.1—0.3 µM, depending on batch) to ~10 µM maintains suppressed growth up to 48 h. Even more surprising, the Cu effect on fluconazole activity appears at concentrations as low as 0.04 μM supplemental Cu added to a batch of media with background Cu measured to be 0.168 μM Cu. This amount of supplemental Cu is well below the 25-mM MIC of Cu in YPD (ESI Fig. S2). Analytical concentrations of other metals in this batch of YPD are reported in ESI Table S1. Given the high concentration of amino acids and other broth components capable of buffering Cu(II), it is notable that as little as 1.2-fold increase in total Cu has such an effect.
Figure 1 |. Impact of Cu levels on growth of fluconazole-treated cells.
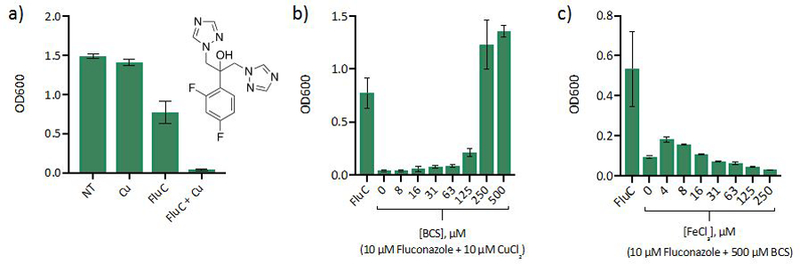
(a) Inhibition of C. albicans growth by fluconazole (FluC) improves when growth media is supplemented with Cu. Conditions: [CuCl2] = 10 µM, [FluC] = 10 µM, growth at 30 °C in YPD monitored at OD600 after 48 h. The structure of fluconazole is inset. (b) BCS treatment rescues growth of cells treated with fluconazole and Cu. Conditions: [CuCl2] = 10 µM, [FluC] = 10 µM, growth at 30 °C in YPD monitored at OD600 after 48 h. (c) Fe(III) supplementation does not rescue growth of cells co-treated with fluconazole and BCS. For controls, see ESI Fig. S3. Data are reported as means with error bars representing standard deviation of three replicate conditions.
Next, a growth assay was performed to determine whether Cu sequestration via BCS would rescue growth of cells co-treated with fluconazole and Cu. Indeed, BCS treatment reversed growth inhibition by fluconazole and Cu. In fact, the BCS-rescued cells grew more than cells treated with fluconazole alone (Fig. 1b). Interestingly, sequestration of basal Cu in the media via BCS also potentiated fluconazole activity (Fig. 1c). A similar response to BCS has been reported previously and was attributed to the requirement of Cu for high affinity Fe import.15 If this is the case, conditions with plentiful Fe would be expected to enable low affinity Fe import and overcome the Cu-deficient growth inhibition. Yet, supplementation of the media with Fe(III) does not rescue growth of cells co-treated with fluconazole and BCS (Fig. 1c). The observation that both Cu limitation and supplementation extend growth inhibition by fluconazole suggests a complex relationship between fluconazole tolerance and Cu availability.
To explore further the conflicting observations that BCS both potentiated fluconazole activity and rescued growth of cells treated with fluconazole and Cu, the kinetics of Cu sequestration via BCS were investigated by using UV-Vis spectroscopy. To simulate the conditions of our biological assays, the experiment was performed in YPD medium at 30 °C. Fluconazole, Cu, and BCS were added, and formation of [CuI(BCS)2]3– (λmax = 483 nm, ε = 13,000 M−1 cm−1)21 was monitored over time. As shown in Fig. 2, some complex formation is already evident at the first timepoint, which was taken just after BCS addition. However, full sequestration of Cu takes nearly 3 h under these conditions. Combined, these data suggest that BCS acts like a slow-turning knob that dials down Cu availability during the first couple of hours of the assay. During this time, some Cu is still available, and C. albicans acquires enough of it to support fluconazole tolerance. Once BCS has fully dialed down Cu access (complete sequestration), C. albicans does not acquire any more Cu. This halt in Cu acquisition appears to protect cells, allow for trailing growth, and gives rise to the growth recovery observed for BCS in the presence of fluconazole and Cu.
Figure 2 |. Formation of [CuI(BCS)2]3– in YPD over time.
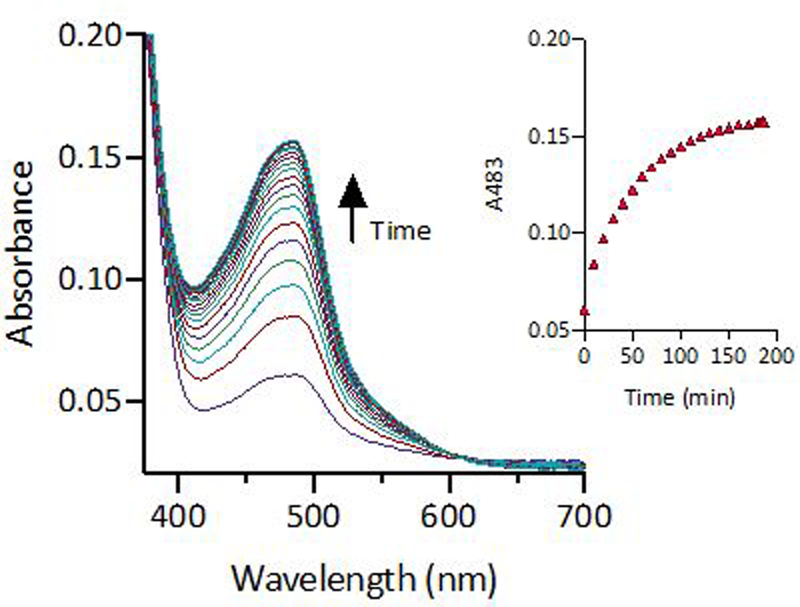
Increase in absorbance at 483 nm signals sequestration of Cu as the [Cu(BCS)2]3– complex. Conditions: [FluC]= 10 µM, [CuSO4] = 10 µM, [BCS] = 500 µM in YPD media at 30 °C.
Fluconazole does not form biologically relevant Cu complexes.
By design, azole antifungals have the ability to coordinate transition metal centers, notably the heme Fe in Cyp51. Fluconazole in particular seems well-suited to form complexes with transition metals due to the multiple donor atoms in its structure (Fig. 1a). Indeed, a crystal structure of a Cu(II)-fluconazole dimer is known,22 and characteristic absorption peaks at 327 nm and 670 nm signal its formation in HEPES buffer (Fig. 3). Interestingly, this fluconazole-Cu complex has been reported to be 10–40% more effective against C. albicans clinical isolates than the metal-free drug.8 Though determination of metal speciation in complex mixtures like cell culture media is a challenge, its consideration is important since speciation determines biological impact.23 To resolve whether fluconazole forms Cu complexes under the conditions of our biological assays, UV-Vis and EPR spectroscopy were used to probe Cu(II)-fluconazole complex formation in the presence of competing ligands in cell culture media. As shown in Fig. 3a, when Cu alone is added to YPD, an absorption band forms at 615 nm, signalling chelation of Cu(II) by media components. Yet when fluconazole is added to this solution, there is no shift in the absorbance band. A similar lack of spectroscopic change was observed by EPR spectroscopy: addition of CuSO4 to YPD produces a signal that is unchanged upon addition of fluconazole (ESI Fig. S4). Combined, these data indicate that fluconazole cannot compete with media components to chelate Cu.
Figure 3 |. Fluconazole does not form complexes with Cu(II) under conditions of biological assays.
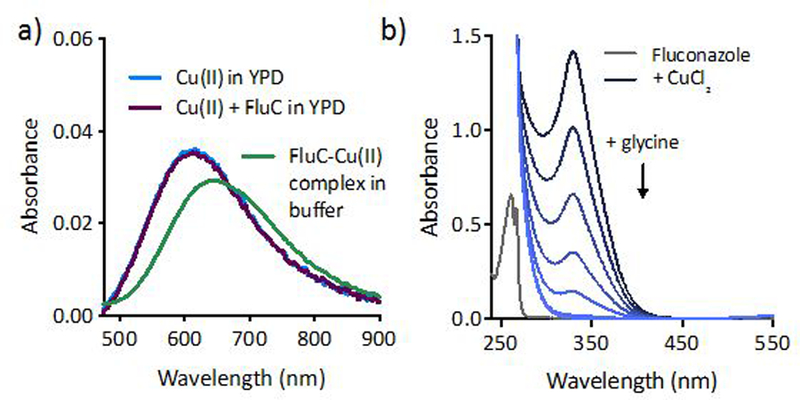
(a) The characteristic 670 nm absorbance feature of Cu(II)-fluconazole complex is not observed when fluconazole and Cu(II) are added to YPD, establishing that complexation does not occur in this medium. (b) Glycine readily competes with fluconazole for Cu(II), indicating that amino acids present in cell culture media prevent Cu(II)-FluC complexation. Conditions: [FluC] = 1 mM, [CuCl2] = 800 µM, [Glycine] = 0.25–2 mM in 50 mM HEPES, pH 7.4.
To further probe Cu-fluconazole complex formation, a competition experiment was performed with glycine, a weak Cu chelator (Kd ~10−9 for the bis-glycine complex)24, 25 that is present at millimolar concentrations in YPD. As shown in Fig. 3b, fluconazole binds Cu(II) in HEPES buffer in the absence of competing ligands. When glycine is titrated into the solution of pre-formed Cu(II)-fluconazole complex, it readily pulls Cu away from fluconazole, and the charge transfer band at 327 nm disappears, providing further evidence that fluconazole cannot compete with glycine and other YPD media components for Cu(II) binding.
Although Cu can cycle between Cu(I) and Cu(II) oxidation states in biological systems, evidence suggests that Cu(I) is the primary oxidation state of intracellular Cu,26, 27 levels of which are estimated between 5.1 × 10−21 and 8.9 × 10−17 M in yeast.28 In order to predict the ability of fluconazole to compete with cellular ligands for labile Cu(I), competitive binding assays were performed with colorimetric indicators bicinchoninate anion (BCA)21, 25, 29 and ferrozine (Fz).30 Fluconazole does not compete with BCA (Kd ~10−17)21 or weaker chelator Fz (Kd ~10−12–10−15)29, 30 for Cu (I) binding (Fig. 4). Given the high affinity (Kd ~10−11–10−20) of Cu chaperones and Cu binding proteins for Cu(I),28, 31–35 it is unlikely that Cu(I)-fluconazole forms intracellularly.
Figure 4 |. Fluconazole does not compete with chelators BCA or Fz for Cu(I) as determined by UV-Vis spectroscopy.
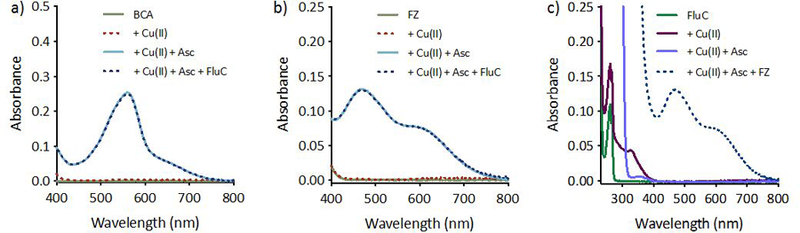
Fluconazole does not pull Cu(I) away from the pre-formed [CuI(BCA)2]3– complex (a), or from the weaker pre-formed [CuI(Fz)2]3- complex (b). Presence of fluconazole in solution does not prevent formation of the [CuI(Fz)2]3- complex (c). CuCl2 was used as the source of Cu(II), and ascorbate was added to generate Cu(I) in situ. Conditions: [BCA, Fz] = 100 µM, [CuCl2] = 40 µM, [Ascorbate] = 1 mM, [FluC] = 1 mM in 50 mM HEPES, pH 7.4.
Fluconazole is not an ionophore.
Our spectroscopic data do not support the presence of a Cu(I) or Cu(II) fluconazole complex in cell culture media, but they do not rule out possible ternary complex formation between fluconazole, Cu, and ligands in YPD. Therefore, in addition to testing Cu-fluconazole complex formation spectroscopically, biological assays were used to further probe whether a direct interaction between fluconazole and Cu could drive the phenotypic responses described above. Some small molecules that exhibit Cu-dependent biological activity do so by acting as ionophores, compounds capable of facilitating movement of the metal across cellular membranes.36 To determine whether fluconazole is capable of shuttling Cu into C. albicans cells, we tested its ability to recover growth of a C. albicans deletion strain lacking the high-affinity Cu(I) importer Ctr1, which the fungus needs to supply Cu for cytochrome c oxidase (COX) for respiration. Growth of C. albicans in YPEG medium, which contains ethanol and glycerol as the sole carbon sources, requires cells to respire. C. albicans utilizes two forms of respiration: Fe- and Cu-containing COX and an alternative Fe-only oxidase (AOX).37 Even when Cu availability is low, C. albicans utilizes COX for respiration.3 As shown in Fig. 5a, wild type cells can grow in YPEG, but ctr1Δ/Δ cells do not. When ctr1Δ/Δ cells were treated with fluconazole, no growth recovery was observed, suggesting fluconazole is unable to supply bioavailable Cu to these cells. This experiment was repeated in the fungal pathogen Cryptococcus neoformans (C. neoformans), which has two Cu importers, Ctr1 and Ctr4, but exclusively utilizes COX for respiration. Ionophores, such as pyrithione and 8-hydroxyquinoline, are able to recover growth of C. neoformans ctr1Δ ctr4Δ cells by delivering bioavailable Cu,38, 39 but as in C. albicans, fluconazole did not recover growth of this mutant (Fig. 5b). Collectively, these results demonstrate fluconazole is unable to deliver bioavailable Cu for use in COX and further support a lack of Cu-fluconazole complex formation under conditions of these assays.
Figure 5 |. Fluconazole does not recover growth of cells lacking Cu import machinery.
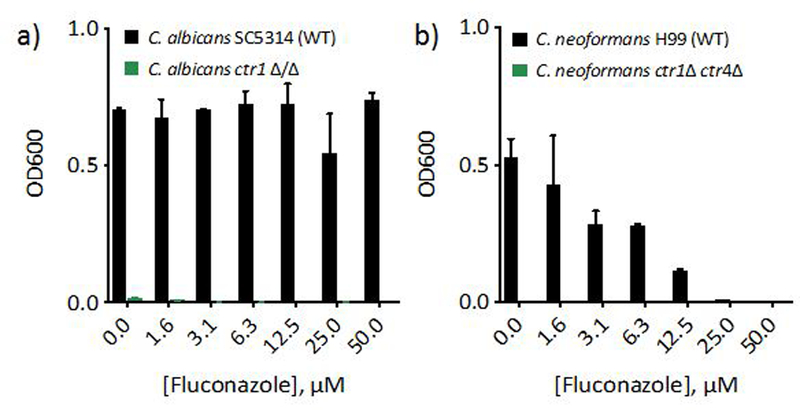
48 h-growth of C. albicans (a) or C. neoformans (b) WT (black bars) and Cu import mutants (green bars) treated with 0–50 µM fluconazole in YPEG media. No growth is observed for cells lacking Cu import genes under these conditions, even with fluconazole treatment. Data are reported as means with error bars representing standard deviation of three replicate conditions.
We reasoned that if the biological dependence on Cu is independent of Cu-fluconazole complex formation, other compounds in the azole class may also be impacted by Cu levels in the growth environment. Apart from their shared imidazole or triazole ring and halogenated aromatic ring, this class of antifungals is structurally diverse (Fig. 6a). With fewer donor atoms and no defined metal binding pockets, other molecules in the azole class are predicted to have a lower propensity for metal complex formation compared to fluconazole. To test whether the Cu effect applies to azoles broadly or only to fluconazole, we tested five other azole antifungals for Cu-dependent activity. As shown in Fig. 6b, all five azoles tested in addition to fluconazole exhibit trailing growth, and all show suppressed growth when the media is supplemented with Cu, relative to the azole alone. Furthermore, Cu-dependent activity is also apparent for the morpholine antifungal fenpropidin (Fig. 7), a drug used exclusively in agricultural applications. Fenpropidin is structurally distinct from the azoles and inhibits a different step in the ergosterol biosynthetic pathway.40 Interestingly, Cu supplementation of fenpropidin-treated cells gives rise to biphasic growth as a function of fenpropidin concentration.
Figure 6 |. Structurally diverse azole antifungals exhibit potentiation by Cu.
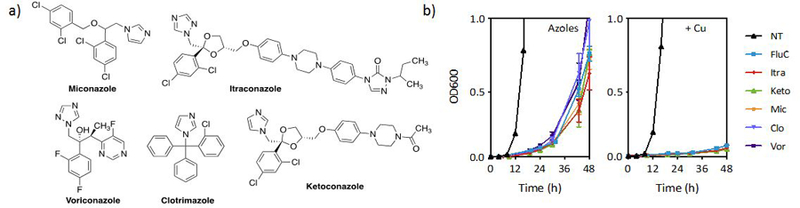
(a) Structures of azoles tested in addition to fluconazole. (b) Growth curves of azoles +/− 100 µM supplemental CuCl2 (colored lines) and control +/− CuCl2 (black lines). Cells were grown in YPD at 30 °C. Azole concentrations: Fluconazole (FluC) = 3.1 µM; Itraconazole (Itra), Ketoconazole (Keto), Miconazole (Mic) = 0.2 µM; Clotrimazole (Clo) = 0.06 µM; and Voriconazole (Vor) = 0.03 µM. Data are reported as means with error bars representing standard deviation of three replicate conditions.
Figure 7 |. Morpholine antifungal fenpropidin also exhibits Cu-dependent activity.
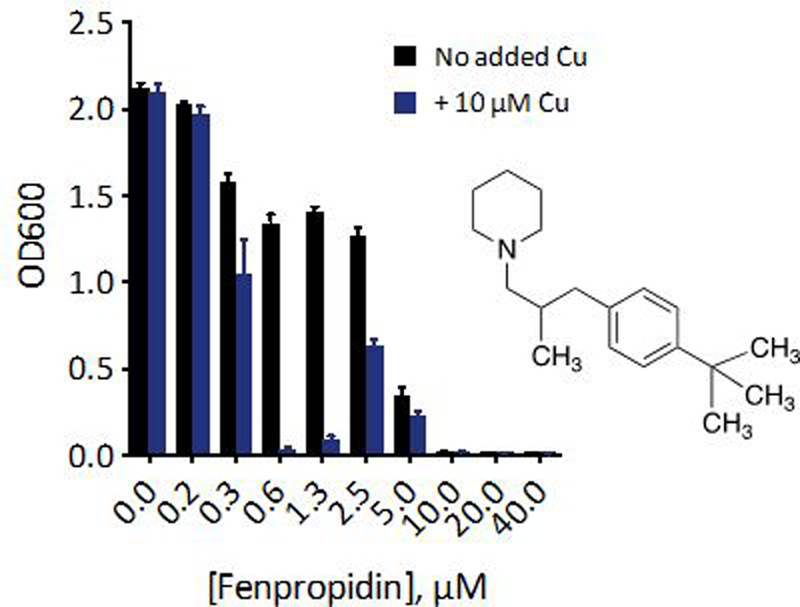
48-h growth of C. albicans treated with 0–40 µM fenpropidin alone (black bars) or with 10 µM supplemental CuSO4 (blue bars). The structure of fenpropidin is inset. Data are reported as means with error bars representing standard deviation of three replicate conditions.
The results of these structure-activity experiments further indicate that the impact of Cu on drug efficacy is a result of a more general response relating to drug stress and Cu availability as opposed to a phenomenon related directly to the structure of fluconazole. Taken together, our spectroscopic and biological data do not support biologically relevant Cu-fluconazole complex formation but suggest a more complex interplay between azole-induced stress and Cu homeostasis.
Fluconazole primes C. albicans for potentiation by Cu.
In our routine growth assays, cells incubated overnight in basal media are treated with fluconazole and Cu simultaneously. To ascertain the effect of intracellular Cu levels on fluconazole tolerance, cells were first grown overnight in Cu-supplemented media to increase cell-associated Cu.41 As shown by comparing the solid light blue and dark blue bars in Fig. 8a, cellular Cu enrichment does not impact fluconazole tolerance compared to normal cultures. However, both cultures, regardless of Cu pre-enrichment, are more susceptible to inhibition by fluconazole when Cu is added to the assay medium in conjunction with fluconazole (red and green dashed bars in Fig. 8a). In other words, pre-enrichment with Cu does not affect the outcome of the growth assay; the determinant is whether supplemental Cu is present during the assay.
Figure 8 |. Potentiation of fluconazole activity depends on timing of Cu supplementation.
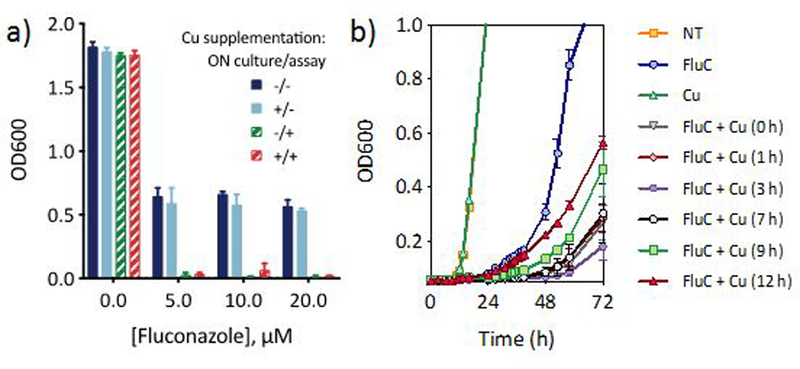
(a) Comparison of cell growth monitored 48 h after fluconazole addition for cells exposed to Cu supplementation during overnight (ON) culture and/or concurrently with fluconazole at time of assay, as designated in the figure legend. Overnight cultures were washed before fluconazole treatment. (b) Growth curve of fluconazole-treated cells without Cu addition (blue circles), or Cu added 0, 1, 3, 7, 9, or 12 h after fluconazole addition. Cu alone does not impact growth (green triangles) relative to untreated control (orange squares). Conditions: [FluC] = 10 µM, [CuSO4] = 10 µM. Data are reported as means with error bars representing standard deviation of three replicate conditions.
To further evaluate the importance of Cu availability as a function of timing, supplemental Cu was “spiked in” at timepoints between 0–12 h after fluconazole treatment, and growth was monitored over 72 h. As shown in Fig. 8b, the data indicate that delaying Cu addition correlates with higher trailing growth. Cu supplementation was most effective between 0 and 7 h after fluconazole treatment, while addition at 9 and 12 h showed a marked decrease in growth inhibition. Interestingly, Cu addition at 3 h gave rise to the least amount of trailing growth at 72 h. Notably though, any addition of Cu, even 12 h after fluconazole treatment, led to improved growth inhibition over the fluconazole-only control. Thus, fluconazole is most effective against C. albicans when Cu is elevated in the growth environment at the time of treatment.
Discussion
In this study, we characterized the effects of Cu restriction and addition on the activity of fluconazole against opportunistic pathogen C. albicans. We established that both Cu sequestration and Cu supplementation prolongs growth inhibition by fluconazole, and complex formation is not required for potentiation by Cu. Because Fe acquisition is dependent on Cu, restricting Cu availability is predicted to limit Fe uptake, thereby impeding restoration of the ergosterol biosynthesis pathway. The observation that sequestration of available Cu via BCS during fluconazole treatment increases growth inhibition is consistent with this hypothesis. Conversely, supplementing with low, non-toxic levels of Cu should enhance ergosterol recovery, thereby aiding cell recovery following fluconazole treatment. Instead, supplementing the growth media with Cu actually prolongs growth inhibition by fluconazole. In other words, Cu delays the onset of trailing growth normally observed during fluconazole treatment. The Cu effect observed here is therefore not a simple proxy of Fe availability and direct involvement in the ergosterol pathway but suggests more complex interactions involving metaltavailability and the cellular response to drug stress. Importantly, these supplemental amounts of Cu by no means exceed physiological ranges; human serum contains between 11–24 µM Cu.42
Some variability in the extent of trailing growth was evident between experiments performed on different days, with 48 h OD600 values ranging from ~0.4–0.8 for trailing growth of fluconazole-treated cells depending on the batch of media. In spite of these minor fluctuations, Cu supplementation always fully suppressed growth through 48 h.
Despite being described in the literature for more than 20 years, drug tolerance is poorly understood.18 Traditionally, tolerance has been considered a poor predictor of clinical susceptibility, and isolates exhibiting trailing growth in vitro are often classified as susceptible in vivo.43–45 However, recent work aiming to more thoroughly characterize azole tolerance in C. albicans provides proof-of-principle that tolerance, and adjuvants that impact tolerance, could in fact have clinical implications.18 Of note is the observation that tolerance correlated inversely with intracellular azole levels, though the ways in which more tolerant cells achieved lower drug concentrations was unclear.
Surprisingly little is known about how fluconazole gets into cells and where it goes once inside, but some light has been shed in the past decade. For a long time it was believed that fluconazole enters cells via passive diffusion, though recent evidence supports import through facilitated diffusion.46 This same study confirmed that CuSO4, even at 100x molar excess does not impact fluconazole accumulation in C. albicans, so it is unlikely that the Cu dependence we observe is the result of altered fluconazole accumulation, at least not due to competition between fluconazole and Cu. It has recently been proposed that fluconazole localizes, at least initially, to mitochondria.47 This suggestion is interesting for two reasons. First, the Cyp51 target of fluconazole resides in the endoplasmic reticulum, not mitochondria,48 but even more intriguing is the fact that mitochondria are sites of rich metallobiology with a number of metalloproteins being metallated there.49 However, it has more recently been reported that the identity of the fluorescent dye used in tracking the azoles seems to influence their subcellular location, limiting their use in providing information about localization of unmodified azoles.50, 51
Although biologically relevant Cu complexation by fluconazole has been posited,8, 52 our data suggest that fluconazole is unable to stabilize a Cu(II) or a Cu(I) complex in the presence of competing ligands, both in the growth medium and inside the fungal cell. More likely, the stress exerted by fluconazole, or C. albicans’ response to this stress, enables Cu in the growth environment to become problematic for C. albicans. This idea contrasts the thought that elevated Cu causes fluconazole to become more problematic for C. albicans. This subtle but important distinction is supported by the following facts: (1) C. albicans is extremely resilient to Cu, especially in rich growth medium like YPD.53 Under our conditions, the MIC of CuSO4 is 25 mM (ESI Fig. S2), confirming that Cu alone at concentrations employed in our assays poses no threat to C. albicans viability. (2) Preincubating cells with Cu does not change fluconazole susceptibility relative to cells that were not preincubated with Cu. If Cu primed cells for fluconazole damage, the expectation is that cellular Cu enrichment would increase susceptibility to fluconazole. Instead, Cu must be added at the same time, or within a certain timeframe after fluconazole treatment to extend growth inhibition. This observation supports the idea that fluconazole creates a window of vulnerability during which C. albicans struggles to manage normally innocuous levels of Cu. The timing of this response provides cues as to when cellular responses contributing to fluconazole tolerance occur.
It was surprising to observe that BCS both potentiated the activity of fluconazole and rescued growth of cells co-treated with fluconazole and Cu. At the highest BCS concentration tested, 50 times more BCS than Cu was present in the media. At such a large excess, BCS, which forms a 2:1 complex with Cu, should in principle chelate all available Cu. If this were the case, the expectation is that treatment with fluconazole, Cu, and BCS would give the same result as treatment with just fluconazole and BCS. However, BCS preferentially binds Cu(I), not Cu(II), and our data show that reductive transfer of Cu from media components to BCS is fairly slow (~3 h for full sequestration). Therefore, Cu would be initially accessible to C. albicans, with availability diminishing during the course of the assay. Thus, we posit that the rescue phenotype results from cells acquiring sufficient Cu early in the assay (due to slow binding between BCS and Cu), while avoiding deleterious effects from a surplus of Cu at later timepoints once BCS has effectively sequestered Cu.
Conversely, when no supplemental Cu was added during fluconazole and BCS treatment, cells were more readily Cu starved, and BCS potentiation of fluconazole activity was observed. Our data does not rule out the possibility that the combination of fluconazole and BCS has an additive effect on growth rate, since BCS alone slows cell growth (ESI Fig. S3). Regardless, the fact that Fe supplementation fails to rescue growth of cells co-treated with BCS and fluconazole supports the idea that BCS potentiates fluconazole activity in ways other than just indirect interference with Fe assimilation.
Conclusions
Our exploration of ergosterol biosynthesis inhibitors arose initially from the idea that fluconazole could impact metal-dependent processes through direct interaction with metal ions; however, our studies fail to identify direct evidence of metal-drug interaction, but rather uncover a broader relationship between Cu homeostasis and drug action. It is noteworthy that Cu potentiates the activity of at least seven drugs that target this pathway, including the six azoles and morpholine tested here. The observation that very low levels of Cu reduce tolerance of C. albicans to fluconazole points to a model in which small increases in Cu availability interfere with a tolerance mechanism employed by C. albicans when it senses assault on the ergosterol biosynthesis pathway. Notably, the timing of changes to Cu availability in relation to fluconazole treatment dramatically impacts the extent to which fluconazole suppresses growth. It is therefore possible the relative availability of Cu in C. albicans’ microenvironment during infection, which varies based on the site and progression of the disease, influences the outcomes of fluconazole therapy. The underlying molecular mechanisms of Cu’s effect on fluconazole tolerance are worthy of investigation.
Experimental
Materials and General Methods
Chemicals and solvents were obtained from commercial suppliers and used as received unless otherwise noted, and all solvents were reagent grade. Aqueous solutions were prepared using Milli-Q water. Buffers for metal-binding studies were prepared using biochemistry grade HEPES (Acros Organics). Stock solutions of metal salts were prepared in water and stock solutions of azoles were prepared in DMSO or water. Working solutions for each experiment were prepared by serial dilution into YPD growth media.
Yeast Strains and Culture Conditions
Fungal stocks were maintained in 25% glycerol in YPD at −80 °C. Unless otherwise noted, experiments were performed with C. albicans clinical isolate SC5314, which was obtained from the American Type Culture Collection (ATCC). The C. albicans ctr1Δ/Δ was generously provided by the Brown Lab of the University of Aberdeen. The C. neoformans H99 and ctr1Δ ctr4Δ strains were generously provided by the Thiele Lab of Duke University. C. albicans cells were cultured at 30 °C in yeast peptone dextrose (YPD, Gibco, catalog number A1374501) media, unless otherwise indicated.
Growth assays (General)
Prior to all experiments, C. albicans cells were streaked onto YPD agar plates from frozen glycerol stocks and incubated at 30 °C for 24 h. A single colony was used to inoculate 10 mL of YPD medium, which was then incubated overnight (~18 h) at 30 °C, 200 rpm. This overnight culture was diluted to an optical density (OD600) of 0.002 with fresh YPD medium and used as the working culture. Azole compounds to be tested were serially diluted 2-fold in YPD medium from DMSO stocks to final concentrations ranging from 0–50 µM, with <1% DMSO and plated in a clear, flat-bottomed 96-well plate. For experiments in which supplemental Cu was used, fresh working solutions of Cu(II) were prepared by diluting 100 mM aqueous stocks of CuSO4 or CuCl2 into YPD media, and aliquots of these working solutions were added to appropriate wells at final concentrations indicated in figure legends. Similarly, working solutions of BCS and FeCl3 were prepared by diluting 10 mM aqueous stocks into YPD media. These working solutions were serially diluted 2-fold to achieve final test concentrations indicated in figure legends. The working culture of C. albicans was then aliquoted to the 96-well plate to a final OD600 of 0.001 and a final volume of 200 µL per well. For each experiment, a compound-free positive growth control and a cell-free, negative control were included. Plates were incubated for 48 h at 30 °C, 200 rpm. Plates were covered with air-permeable AeraSeal film (Sigma) to minimize evaporation.
Fungal growth was evaluated by measuring OD600 using a PerkinElmer Victor3 V multilabel plate reader at 0, 24, and 48 h. For some experiments, additional timepoint data were collected. OD600 values were adjusted by subtracting the 0 h timepoint readings from other timepoint data to remove background signal from growth media. In cases where the OD was outside of the linear range (OD600 values approximately 0.0–0.8), cell suspensions were diluted 4-fold in fresh media and rescanned. To calculate actual OD values, the 0 h timepoint reading was subtracted from the diluted readings, and this value was multiplied by four. At least two biological replicates were performed with a minimum of three technical replicates per experiment. For a single experiment, each of the three replicate conditions were averaged and the error was calculated as standard deviation (SD), which is indicated by error bars in all figures. Final 48-h timepoint data is reported by plotting OD600 readings versus treatment conditions. Growth curves are reported by plotting OD600 readings versus time for each treatment condition.
ctr1Δ/Δ and ctr1Δ ctr4Δ Growth Recovery Assay
Overnight cultures of WT SC5314 and isogenic ctr1Δ/Δ (C. albicans) or WT H99 and isogenic ctr1Δ ctr4Δ (C. neoformans) in YPD were washed twice with PBS, pH 7.4 and resuspended in 10 mL YPEG medium with 2% EtOH and 3% glycerol. Cell suspensions were diluted to OD600 of 0.002 with fresh YPEG medium. WT was aliquoted to the top half of a 96 well plate and Cu importer deletion strain to the bottom half. Test compounds were added from DMSO stock solutions to final concentrations ranging from 0–50 µM. For each test run, a compound-free positive growth control and a cell-free, negative control were included. Plates were incubated at 30 °C and OD600 recorded at 0, 24, and 48 h. All tests were performed in triplicate for each condition in a single experiment, and two separate experiments were carried out. For a single experiment, each of the three replicate conditions were averaged and the error was calculated as standard deviation (SD).
UV-Vis and EPR Spectroscopy
UV-Visible absorption spectra were collected using a Varian Cary 50 UV-Visible spectrophotometer in quartz cuvettes with 1 cm pathlengths. Solutions were allowed to equilibrate and were scanned until no changes in the absorption spectra were observed. UV-Vis studies probing Cu(I) binding were conducted in air using CuCl2 with excess ascorbate added as a reducing agent, with the exception of Cu(I) binding by BCS, in which no ascorbate was added. Formation of [CuI(BCS)2]3- in YPD was monitored by taking scans every 10 min for a duration of 180 min. The sample was maintained at 30 °C to reproduce conditions of biological assays.
X-band continuous wave (CW) EPR spectroscopy was conducted on a Bruker ESP 300 spectrometer equipped with an Oxford Instruments ESR 910 continuous helium flow cryostat. Typical experimental
parameters were at 77 K, 9.37 GHz, 6.33 mW microwave power, and 5 G modulation amplitude. Solutions were prepared in YPD medium containing 20% glycerol (≥ 99.5%, Sigma Aldrich).
ICP-MS Analysis of YPD Metal Content
Samples of YPD media were submitted as received from the commercial supplier (Gibco, lot #2005064) for ICP-MS analysis. Kim Hutchison (Department of Soil Science, North Carolina State University) performed metal content analysis on a Varian 820 ICP-MS. Samples were run in triplicate, and the mean and standard deviation (SD) are reported.
Supplementary Material
Acknowledgements
We thank Prof. Dennis Thiele (Duke University, Durham, NC) for providing the two strains of C. neoformans (WT H99 and ctr1Δ ctr4Δ) and for valuable discussions, Prof. Alistair Brown (University of Aberdeen, Aberdeen, UK) for providing the two strains of C. albicans (WT SC5314 and ctr1Δ/Δ), and Kelley White and Tanvi Dange for performing initial spectroscopy experiments. This work was supported by the National Institutes of Health (Grant GM084176). E.W.H. acknowledges support from the United States Department of Education GAANN Fellowship (Award Number: P200A150114).
Footnotes
† Electronic Supplementary Information (ESI) available. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Seeliger HPR, Mycoses, 1976, 19, 87–97. [Google Scholar]
- 2.Chen C, Pande K, French Sarah D., Tuch Brian B. and Noble Suzanne M., Cell Host Microbe, 2011, 10, 118–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Broxton CN and Culotta VC, PLoS One, 2016, 11, e0168400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li CX, Gleason JE, Zhang SX, Bruno VM, Cormack BP and Culotta VC, Proc. Natl. Acad. Sci. USA, 2015, 112, E5336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mackie J, Szabo EK, Urgast DS, Ballou ER, Childers DS, MacCallum DM, Feldmann J and Brown AJP, PLoS One, 2016, 11, e0158683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whaley SG, Berkow EL, Rybak JM, Nishimoto AT, Barker KS and Rogers PD, Front. Microbiol, 2016, 7, 2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cowen LE, Nat. Rev. Microbiol, 2008, 6, 187–198. [DOI] [PubMed] [Google Scholar]
- 8.Ząbek A, Nagaj J, Grabowiecka A, Dworniczek E, Nawrot U, Młynarz P and Jeżowska-Bojczuk M, Med. Chem. Res, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gagini T, Colina-Vegas L, Villarreal W, Borba-Santos LP, de Souza Pereira C, Batista AA, Kneip Fleury M, de Souza W, Rozental S, Costa LAS and Navarro M, New J. Chem, 2018, 42, 13641–13650. [Google Scholar]
- 10.Kljun J, Scott AJ, Lanišnik Rižner T, Keiser J and Turel I, Organometallics, 2014, 33, 1594–1601. [Google Scholar]
- 11.Betanzos-Lara S, Chmel NP, Zimmerman MT, Barron-Sosa LR, Garino C, Salassa L, Rodger A, Brumaghim JL, Gracia-Mora I and Barba-Behrens N, Dalton Trans, 2015, 44, 3673–3685. [DOI] [PubMed] [Google Scholar]
- 12.Betanzos-Lara S, Gomez-Ruiz C, Barron-Sosa LR, Gracia-Mora I, Flores-Alamo M and Barba-Behrens N, J. Inorg. Biochem, 2012, 114, 82–93. [DOI] [PubMed] [Google Scholar]
- 13.Simpson PV, Nagel C, Bruhn H and Schatzschneider U, Organometallics, 2015, 34, 3809–3815. [Google Scholar]
- 14.Karaoun N and Renfrew AK, Chem. Commun, 2015, 51, 14038–14041. [DOI] [PubMed] [Google Scholar]
- 15.Prasad T, Chandra A, Mukhopadhyay CK and Prasad R, Antimicrob. Agents Chemother, 2006, 50, 3597–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fiori A and Van Dijck P, Antimicrob. Agents Chemother, 2012, 56, 3785–3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Venisse N, Grégoire N, Marliat M and Couet W, Antimicrob. Agents Chemother, 2008, 52, 937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenberg A, Ene IV, Bibi M, Zakin S, Segal ES, Ziv N, Dahan AM, Colombo AL, Bennett RJ and Berman J, Nat. Commun, 2018, 9, 2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delarze E and Sanglard D, Drug Resist. Updat, 2015, 23, 12–19. [DOI] [PubMed] [Google Scholar]
- 20.Marr KA, Rustad TR, Rex JH and White TC, Antimicrob. Agents Chemother, 1999, 43, 1383–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao Z, Brose J, Schimo S, Ackland SM, La Fontaine S and Wedd AG, J. Biol. Chem, 2011, 286, 11047–11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagaj J, Starosta R, Szczepanik W, Barys M, Mlynarz P and Jezowska-Bojczuk M, J. Inorg. Biochem, 2012, 106, 23–31. [DOI] [PubMed] [Google Scholar]
- 23.Levina A, Crans DC and Lay PA, Coord. Chem. Rev, 2017, 352, 473–498. [Google Scholar]
- 24.Thompsett AR, Abdelraheim SR, Daniels M and Brown DR, J. Biol. Chem, 2005, 280, 42750–42758. [DOI] [PubMed] [Google Scholar]
- 25.Xiao Z and Wedd AG, Nat. Prod. Rep, 2010, 27, 768–789. [DOI] [PubMed] [Google Scholar]
- 26.Yang L, McRae R, Henary MM, Patel R, Lai B, Vogt S and Fahrni CJ, Proc. Natl. Acad. Sci. USA, 2005, 102, 11179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ackerman CM, Lee S and Chang CJ, Anal. Chem, 2017, 89, 22–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wegner SV, Sun F, Hernandez N and He C, Chem. Commun, 2011, 47, 2571–2573. [DOI] [PubMed] [Google Scholar]
- 29.Xiao Z, Gottschlich L, van der Meulen R, Udagedara SR and Wedd AG, Metallomics, 2013, 5, 501–513. [DOI] [PubMed] [Google Scholar]
- 30.Alies B, Badei B, Faller P and Hureau C, Chem. Eur. J, 2012, 18, 1161–1167. [DOI] [PubMed] [Google Scholar]
- 31.Xiao Z, Loughlin F, George GN, Howlett GJ and Wedd AG, J. Am. Chem. Soc, 2004, 126, 3081–3090. [DOI] [PubMed] [Google Scholar]
- 32.Kaplan Jack H. and Maryon Edward B., Biophys. J, 2016, 110, 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scheller JS, Irvine GW, Wong DL, Hartwig A and Stillman MJ, Metallomics, 2017, 9, 447–462. [DOI] [PubMed] [Google Scholar]
- 34.Faller P, FEBS J, 2010, 277, 2921–2930. [DOI] [PubMed] [Google Scholar]
- 35.Morgan MT, Nguyen LAH, Hancock HL and Fahrni CJ, J. Biol. Chem, 2017, 292, 21558–21567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Helsel ME and Franz KJ, Dalton Trans, 2015, 44, 8760–8770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Helmerhorst EJ, Murphy MP, Troxler RF and Oppenheim FG, Biochim. Biophys. Acta, 2002, 1556, 73–80. [DOI] [PubMed] [Google Scholar]
- 38.Helsel ME, White EJ, Razvi SZ, Alies B and Franz KJ, Metallomics, 2017, 9, 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Festa RA, Helsel ME, Franz KJ and Thiele DJ, Chem. Biol, 2014, 21, 977–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mazu TK, Bricker BA, Flores-Rozas H and Ablordeppey SY, Mini-Rev. Med. Chem, 2016, 16, 555–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conklin SE, Bridgman EC, Su Q, Riggs-Gelasco P, Haas KL and Franz KJ, Biochemistry, 2017, 56, 4244–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Filippini T, Michalke B, Grill P, Malagoli C, Malavolti M, Vescovi L, Sieri S, Krogh V, Cherubini A, Maffeis G, Lucchini R, Ferrante M and Vinceti M, Mol. Med. Rep, 2017, 15, 3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zomorodian K, Bandegani A, Mirhendi H, Pakshir K, Alinejhad N and Poostforoush Fard A, Jundishapur J. Microbiol, 2016, 9, e28666–e28666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rueda C, Puig-Asensio M, Guinea J, Almirante B, Cuenca-Estrella M, Zaragoza O, Padilla B, Muñoz P, Guinea J, Paño Pardo JR, García-Rodríguez J, García Cerrada C, Fortún J, Martín P, Gómez E, Ryan P, Campelo C, de los Santos Gil I, Buendía V, Gorricho BP, Alonso M, Sanz FS, Aguado JM, Merino P, González Romo F, Gorgolas M, Gadea I, Losa JE, Delgado-Iribarren A, Ramos A, Romero Y, Sánchez Romero I, Zaragoza O, Cuenca-Estrella M, Rodriguez-Baño J, Isabel Suarez A, Loza A, Aller García AI, Martín-Mazuelos E, Pérez de Pipaón MR, Garnacho J, Ortiz C, Chávez M, Maroto FL, Salavert M, Pemán J, Blanquer J, Navarro D, Camarena JJ, Zaragoza R, Abril V, Gimeno C, Hernáez S, Ezpeleta G, Bereciartua E, Hernández Almaraz JL, Montejo M, Rivas RA, Ayarza R, Planes AM, Camps IR, Almirante B, Mensa J, Almela M, Gurgui M, Sánchez-Reus F, Martinez-Montauti J, Sierra M, Horcajada JP, Sorli L, Gómez J, Gené A, Urrea M, Valerio M, Díaz-Martín A, Puchades F and Mularoni A, Clin. Microbiol. Infect, 2017, 23, 49.e41–49.e48. [Google Scholar]
- 45.Arthington-Skaggs BA, Warnock DW and Morrison CJ, Antimicrob. Agents Chemother, 2000, 44, 2081–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mansfield BE, Oltean HN, Oliver BG, Hoot SJ, Leyde SE, Hedstrom L and White TC, PLoS Pathog, 2010, 6, e1001126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benhamou RI, Bibi M, Steinbuch KB, Engel H, Levin M, Roichman Y, Berman J and Fridman M, ACS Chem. Biol, 2017, 12, 1769–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bhattacharya S, Esquivel BD and White TC, mBio, 2018, 9, e01291–01218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Atkinson A and Winge DR, Chem. Rev, 2009, 109, 4708–4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benhamou RI, Jaber QZ, Herzog IM, Roichman Y and Fridman M, ACS Chem. Biol, 2018, 13, 3325–3332. [DOI] [PubMed] [Google Scholar]
- 51.Benhamou RI, Bibi M, Berman J and Fridman M, Angew. Chem. Int. Ed, 2018, 57, 6230–6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ali M, Ahmed M, Ahmed S, Imran Ali S, Perveen S, Mumtaz M, Moazzam Haider S and Nazim U, Pak. J. Pharm. Sci, 2017, 30, 187–194. [PubMed] [Google Scholar]
- 53.Weissman Z, Berdicevsky I, Cavari BZ and Kornitzer D, Proc. Natl. Acad. Sci. USA, 2000, 97, 3520–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.