

Abstract
G protein-coupled receptors (GPCRs) are critical regulators of human physiology and make up the largest single class of therapeutic drug targets. Although GPCRs regulate highly diverse physiology, they share a common signaling mechanism whereby extracellular stimuli induce conformational changes in the receptor that enable activation of heterotrimeric G proteins and other intracellular effectors. Advances in GPCR structural biology have made it possible to examine ligand-induced GPCR activation at an unprecedented level of detail. Here, we review the structural basis for family A GPCR activation, with a focus on GPCRs for which structures are available in both active or active-like states and inactive states. Crystallographic and other biophysical data show how chemically diverse ligands stabilize highly conserved conformational changes on the intracellular side of the receptors, allowing many different extracellular stimuli to utilize shared downstream signaling molecules. Finally, we discuss the remaining challenges in understanding GPCR activation and signaling and highlight new technologies that may allow unanswered questions to be resolved.
Graphical Abstract
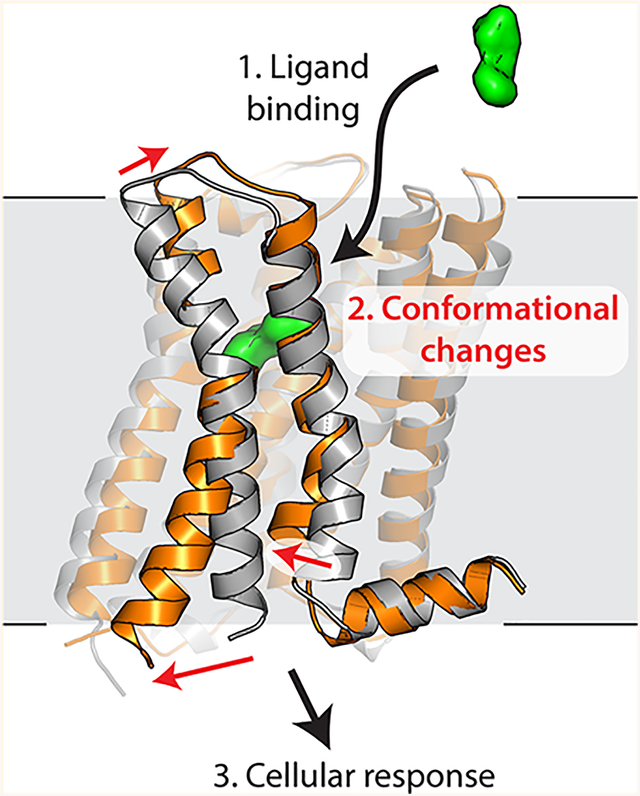
G protein-coupled receptors (GPCRs) constitute the largest family of transmembrane receptors in humans, and they have become the most successful class of therapeutic drug targets by virtually any metric. All GPCRs share a conserved seven-pass transmembrane fold, which connects an extracellular ligand binding site to an intracellular G protein binding surface. Until recently, the myriad challenges associated with biochemical manipulation and crystallization of GPCRs made high-resolution structural studies of ligand binding and receptor activation difficult or impossible. Over the past decade, a number of technical advances have changed this situation, including the use of fusion proteins such as T4 lysozyme,1 the development of new detergents,2 high-throughput lipidic mesophase crystallography,3 microfocus diffraction beamlines,4 and high-energy X-ray free electron lasers.5 Structural studies of GPCRs have become increasingly tractable, as evidenced by the 42 unique receptors that have been characterized by X-ray crystallography to date.
For a few GPCRs, high-quality structural data are available in multiple distinct structural states. While crystallography may never be able to fully capture the complete constellation of GPCR conformations, structures to date largely fall into two major classes: active or active-like states and inactive states. Active states are those in which the receptor adopts a conformation that is competent to interact with heterotrimeric G proteins or other effectors, exemplified by the β2 adrenergic receptor bound to the heterotrimeric G protein Gs.6 Inactive states are representative of conformations that are incapable of catalyzing G protein nucleotide exchange and typically show an occluded G protein binding surface. In addition, structures are available for several receptors bound to agonists but showing intracellular conformations resembling inactive states or with conformations intermediate between active and inactive states. These are often called “active intermediate” states and may represent one of several steps toward full receptor activation following agonist binding. These have been observed with particular frequency in studies of thermostabilized GPCRs such as the β1 adrenergic receptor7 and A2a adenosine receptor,8 as well as in studies of 5-hydroxytryptamine receptors bound to arrestin-biased agonists.9–11 Here, we focus on those receptors for which structural data are available for both fully active and inactive states (Table 1 and Figure 1). These structures offer insights into both shared and divergent aspects of GPCR activation. We restrict our discussion to receptors belonging to the rhodopsin-like GPCR family (family A), which is the largest and most well understood family.
Table 1.
Summary of Protein Data Bank (PDB) Entries for GPCRs Determined in Multiple Conformational States
| receptor | inactive-state PDB entries | active-state PDB entries |
|---|---|---|
| 5-HT2B | 4IB4,d 5TVN,d 4NC3d | 5TUD |
| rhodopsin | 1F88, 1U19, 2G87, 1L9H, 3C9L, 1HZX, 1GZM, 3OAX, 2HPY, 3C9M, 2PED, 2J4Y, 5TE5, 2I35, 2I36, 2I37 | 4X1H,c 5DYS, 4J4Q,c 4PXF,c 5EN0,c 3PQR,c 5TE3, 2X72,c 3PXO, 3CAP, 4BEY,c 3DQB,c 4A4M,c 4BEZ, 4ZWJ,b 5DGYb |
|
β2 adrenergic receptor |
2R4R,a 2R4S,a 2RH1, 3D4S, 3KJ6, 3NY9, 3NYA, 5D5B, 3NY8, 5D5A, 3PDS, 5JQHa | 3P0G,a 3SN6,a,b 4QKX,a 4LDO,a 4LDE,a 4LDLa |
| M2 muscarinic acetylcholine receptor | 3UON | 4MQS,a 4MQTa |
| μ opioid receptor | 4DKL | 5C1Ma |
| CB1 cannabinoid receptor | 5TGZ, 5U09 | 5XRA, 5XR8 |
| A2a adenosine receptor | 3PWH, 2YDO, 3QAK, 3REY, 3RFM, 4UG2, 4UHR, 3EML, 5UIG, 2YDV, 5MZJ, 5MZP, 5N2R, 3UZA, 3UZC, 3VG9,a 3VGA,a 5IU4, 5IU7, 5IU8, 5IUA, 5IUB, 5K2A, 5K2B, 5K2C, 5K2D, 5JTB, 5UVI, 4EIY | 5G53b |
| NTSR1 neurotensin receptor |
4BUO, 4BV0, 4BWB | 4XEE, 4XES, 4GRV,e 5T04e |
| GLP-1 receptor | 5VEX, 5VEW | 5VAI,b 5NX2 |
Antibody fragment-stabilized receptor.
G protein/arrestin-bound receptor.
Peptide-stabilized receptor.
Ambiguous state 1. Close to the inactive conformation, but with a slight opening of TM6. Bound to arrestin-biased agonists.
Ambiguous state 2. TM6 is in the fully active outward orientation, but TM7 occludes the G protein binding site.
Figure 1.

Family A GPCRs for which structural data are available. Most family A GPCRs that have been crystallized to date have been determined in only a single conformational state, usually an inactive conformation.
■ CHALLENGES IN STUDYING GPCR ACTIVATION
GPCRs are inherently challenging targets for structural study due in large part to the conformational plasticity that underlies their biological function.12,13 Though the range of this plasticity remains to be characterized in detail for the vast majority of GPCRs, biophysical studies using the prototypical β2 adrenergic receptor have begun to reveal the importance of this plasticity in GPCR function. Some of the earliest evidence supporting GPCR protein dynamics came from studies of the β2 adrenergic receptor that revealed agonist-dependent changes in signal from the fluorescently labeled receptor.14 Attempts to obtain an activated structure of the β2 adrenergic receptor bound to a covalent agonist captured the receptor in an inactive conformation,15 suggesting that the active conformation is not the lowest-energy state in such preparations. Indeed, this view has been supported by long time scale molecular dynamics simulations, which revealed both the complex dynamics of the inactive receptor16 and spontaneous relaxation of the agonist-bound, activated receptor to the inactive conformation.15,17 Subsequent studies with various spectroscopic techniques, including nuclear magnetic resonance (NMR) and electron paramagnetic resonance (EPR), have demonstrated that even picomolaraffinity full agonists do not completely stabilize the active conformation of the β2 adrenergic receptor.18,19 While such agonist-induced dynamics were initially observed for the β2 adrenergic receptor, a similar level of conformational heterogeneity has also been observed in the μ-opioid receptor and the A2a adenosine receptor, by both spectroscopic methods and simulation.20,21 Further supporting the generality of this model are the numerous agonist-bound GPCRs that have been crystallized in the inactive or active intermediate states.7–11,15,22–29
The conformational heterogeneity of agonist-bound receptors has made structural study of activated GPCRs challenging. Active-state GPCR structures have been determined primarily with the aid of proteins that stabilize the active conformation. These include G proteins or engineered fragments thereof,6,30–33 visual arrestin,34 and conformation-specific camelid antibody fragments called nanobodies.35 In one unusual case, antibody fragment-induced crystal packing was found to stabilize an active-state structure of the 5-HT2B receptor.36 Among the strategies employed to determine active-state structures, nanobodies have proven to be among the most useful tools in interrogating GPCR structure and conformational exchange. Nanobodies enabled determination of active-state structures for the β2 adrenergic receptor,6,37,38 M2 muscarinic receptor,39 μ-opioid receptor,40 and US28, a viral chemokine receptor.41 In each of these cases, nanobodies display pharmacological characteristics typical of effector proteins like the heterotrimeric G protein; i.e., they induce comparable enhancement of agonist affinity in radio-ligand binding assays. While it is possible in principle that nanobody-stabilized active states may differ from G protein-coupled states, the pharmacological features of nanobodies suggest they stabilize a conformation similar or identical to that which recognizes G proteins. Indeed, in the case of the β2 adrenergic receptor, structures of the active state stabilized by an active-state stabilizing nanobody and the heterotrimeric G protein revealed a nearly identical receptor conformation in the intracellular interface.6,37 It is important to note that while receptor conformations largely fall into fairly discrete classes of active and inactive states, structural data represent only a subset of receptor conformations, and it is clear from spectroscopic and simulation data that additional states exist but remain structurally uncharacterized.12,19,21
■CONSERVED STRUCTURAL FEATURES OF ACTIVATION
GPCRs possess a conserved structural fold, with seven transmembrane helices surrounding an extracellular-facing ligand binding site and a G protein binding site on the intracellular surface. Agonist binding stabilizes an active conformation on the intracellular side of the receptor, causing this conformation to be more frequently sampled among a number of conformational states. Importantly, the structural link between agonist binding and intracellular changes is not one to one, and thermal fluctuations between the agonist binding site and the intracellular domain result in a relatively weak allosteric coupling.12 Agonist binding enhances the propensity for intracellular conformational changes that are required for G protein binding, but agonist binding is not by itself sufficient to fully stabilize an active conformation of the entire receptor molecule.
Comparison of the intracellular changes upon GPCR activation reveals a striking degree of structural conservation and suggests a common evolutionary origin for the activation mechanism in most or all GPCRs. Without exception, GPCR activation involves a rotation and displacement of transmembrane (TM) helix 6 to create a cavity on the receptor intracellular face that can accommodate the G protein α subunit C-terminus (Figure 2). TM5 also rotates away from the receptor, further enlarging the G protein binding cavity. Recent structures of activated family B receptors in complex with heterotrimeric G proteins show similar overall structural features,30,31,42 including the outward rotation of TM6, suggesting that this activation mechanism is shared even among very distantly related receptors with few identifiable conserved features at the primary sequence level. Although there are no currently available active-state structures of family C and F receptors, it seems likely that these receptors will share similar structural features.
Figure 2.

Comparison of active and inactive states for a prototypical GPCR. (a) Inactive-state (gray, PDB entry 3UON) and active-state (orange, PDB entry 4MQS) structures of the human M2 muscarinic acetylcholine receptor are shown in a side view, parallel to the membrane plane. Red arrows indicate conformational changes upon activation. (b) Same structure, viewed from the intracellular side.
Conserved sequence motifs play important roles in GPCR activation. The highly conserved DRY/ERY motif found at the intracellular end of TM3 serves to stabilize the inactive conformation of the receptor through a conserved salt bridge to Glu6.40, a feature termed the ionic lock43 (all residue numbers are given using the Ballesteros−Weinstein system44). Mutagenic disruption of the ionic lock in rhodopsin enhances binding to a peptide derived from transducin, further attesting to its role as a barrier to receptor activation.45 Opening or closing of this salt bridge distinguishes two spectroscopically and structurally distinct inactive conformations for at least some receptors, and this has been experimentally observed for the β2 adrenergic receptor and the A2a adenosine receptor. In activated receptor conformations, a feature analogous to the ionic lock is formed through hydrogen bonds connecting the TM7 sequence motif NPxxY Tyr7.53 to the highly conserved Tyr5.58 via a bridging water (Figure 3a). In fact, this feature is observed in all family A GPCRs for which active- and inactive-state structural data are available, although the bridging water is not always detected in electron density maps because of the modest resolution of many active-state GPCR structures. In several cases, Tyr5.58 is additionally hydrogen bonded to Arg3.50 of the DRY/ERY motif, as seen in structures of the μ-opioid receptor and rhodopsin. This tyrosine “water lock” may play a role in the active state similar to that of the ionic lock in inactive conformations, stabilizing activated states to establish an energy minimum in a conformation capable of interacting with G proteins and other effectors. Although mutagenesis of Tyr5.58 has not been reported for most GPCRs, in the case of rhodopsin this residue has been investigated in depth and has been shown to increase the lifetime of the active metarhodopsin II state.46
Figure 3.

Intracellular motifs involved in GPCR activation. In each case, the inactive receptor is colored gray and the active state of the same receptor is colored orange. (a) Side view showing Tyr5.58 and Tyr7.53 engaged in a highly conserved hydrogen bond mediated by a bridging water molecule (red sphere), often also interacting with Arg3.50 as seen here for the μ-opioid receptor (PDB entries 4DKL for the inactive state and 5C1M for the active state). (b) Leucine ratchet of Leu6.37 past Tyr5.58 upon activation, exemplified by the β2 adrenergic receptor viewed from the extracellular direction (PDB entries 2RH1 for the inactive state and 4LDE for the active state). (c) Phe-Tyr switch in the β2 adrenergic receptor (same PDB entries as in panel b, also viewed from above/extracellular). (d) Structural sodium ion stabilization of the inactive-state receptor, shown here for the A2a adenosine receptor viewed from above (PDB entries 4EIY for the inactive state and 5G53 for the active state).
A related structural feature is a ratchetlike motion of a bulky hydrophobic residue in position 6.37 past Tyr5.58 coupled to the outward rotation of TM6 (Figure 3b). This exposes the tyrosine hydroxyl for hydrogen bonding through water to Tyr7.53, stabilizing the active state. The presence of a bulky residue at position 6.37 (most often Leu) may stabilize both inactive and active states by providing a kinetic barrier to opening and closing of TM6. Mutagenesis of this residue to alanine has thermo-stabilizing effects in some receptors,47 and in a thermostabilized neurotensin 1 receptor variant, a Leu6.37Ala mutation is one of three that are involved in preventing the fully thermostabilized receptor from undergoing activation in response to an agonist.27
Nearer to the ligand binding pocket, conformational changes upon receptor activation are more variable, although some conserved features are present. Structural and spectroscopic data for the β2 adrenergic receptor and other GPCRs have shown an activation-associated rearrangement of hydrophobic residues below (i.e., nearer the intracellular side) the ligand binding pocket. This rearrangement involves a ratchetlike motion of residue 6.44 (most often Phe) past residue 3.40 (most often Ile). Nearly identical rearrangements are seen in diverse receptors, including the β2 adrenergic receptor, rhodopsin, the A2a adenosine receptor, and the μ-opioid receptor (Figure 3c). Not all receptors share this feature, however, and those with smaller side chains in either the 3.40 or 6.44 position show smaller changes in this region, as seen in the M2 muscarinic receptor and the CB1 cannabinoid receptor.
Adjacent to this region, a structural sodium is found in most GPCRs, coordinated by side chains from TM2, −3, and −7 in the inactive conformation (Figure 3d). Structural rearrangements upon activation disrupt this site, requiring breakage of bonds with the sodium ion. Consequently, sodium serves as a negative allosteric modulator of receptor activation, stabilizing the inactive state of the receptor and decreasing agonist affinity. In fact, this effect of sodium was first identified in radioligand binding assays of opioid receptors,48 although it has only recently has been described in high-resolution structural detail.49
Within the ligand binding pocket itself, the structural differences between active and inactive states are quite diverse, reflecting the wide variety of ligands recognized by GPCRs (Figure 4). Most receptors seem to show relatively minor structural changes in the ligand binding pocket, as seen in the β2 adrenergic receptor, rhodopsin, and the A2a adenosine receptor, for instance. Other receptors show much larger changes, such as those seen in the M2 muscarinic receptor or the CB1 cannabinoid receptor. The ligand binding pockets have a low degree of sequence and structure conservation among different GPCRs, and the chemical details of ligand recognition are unique in each case with few discernible common themes. The only clear general trend is that in most receptors the ligand binding site undergoes a contraction when the receptor is in a fully active state. This is perhaps the most remarkable feature of GPCR activation: an array of molecules as diverse as lipids, nucleotides, and large proteins can trigger activation at highly divergent receptor binding sites. Despite this, the structural details of GPCR activation become increasingly similar close to the intracellular face of the receptors. In this way, a large family of receptors can serve as signaling adaptors to transduce binding of hundreds of different molecules into activation of only a few shared effector proteins.
Figure 4.

Agonist recognition. The molecular details of agonist recognition are highly diverse, although most agonist-bound activate-state GPCR structures show a modest contraction of the ligand binding site relative to their inactive-state counterparts. Here, the structures of the inactive and active β2 adrenergic receptor (PDB entries 2RH1 and 4LDL, respectively) and the CB1 cannabinoid receptor (PDB entries 5U09 and 5XRA for the inactive and active states, respectively) are shown as representative examples, showing contraction of the binding site upon activation, as well as the far more extensive nature of structural rearrangements upon activation of the CB1 receptor compared to the β2 receptor.
■FRONTIERS OF GPCR ACTIVATION
While recent years have seen increasingly rapid advances in our understanding of the structural basis for GPCR activation, many important questions remain to be answered in the years ahead. For instance, structural changes upon activation are now well understood for many GPCRs with small molecule and peptide agonists, but for receptors that respond to other types of stimuli, few structural data of any kind are available. This includes receptors that respond to large protein ligands such as the leucine-rich repeat (LRR)-containing receptors (LGRs), most of which bind to large protein agonists such as luteinizing hormone and thyroid-stimulating hormone. In these receptors, binding of agonist requires interactions with the large extracellular LRR domain, as well as with the seven-transmembrane bundle and extracellular loops. Other receptors like GPR4, GPR65, and GPR68 respond to changes in pH, relying on protonation of histidine side chains to trigger receptor activation under mildly acidic conditions such as inflamed tissue or under hypoxic conditions.50 Other GPCRs are activated by metal ions,51 odorants,52 or perhaps even mechanical stress,53 none of which is well understood in molecular detail.
A second major unresolved challenge is understanding the structural basis for biased signaling, in which ligands differentially activate GPCR signaling through downstream pathways. For instance, peptide TRV027 activates arrestin signaling through the angiotensin II type 1 receptor but has little effect on G protein signaling,54 while the endogenous agonist angiotensin II robustly activates both pathways. Currently, little is known about the structural or dynamic details that underlie biased signaling. Studies of 5-hydroxytryptamine receptors bound to arrestin-biased agonists ergotamine and LSD have provided some insight into this question. These structures show unusual conformations with features intermediate between those of fully active and inactive states and may be representative of an arrestin-preferring signaling conformation. Like every aspect of GPCR structural biology, however, a detailed understanding of signaling bias will require structural data for more than one receptor in multiple states, to gain insight into which features are shared, general mechanisms of biased agonism, and which details are receptor- or ligand-specific.
Finally, it is important to note that while family A is by far the largest GPCR family, many important receptors belong to other families, including family B receptors with critical roles in endocrinology and regulation of metabolism and family C receptors with pivotal importance in neurobiology. While the first structural data regarding activation mechanisms of family B receptors are beginning to become available, no active-state structure has yet been characterized for a family C GPCR. Despite negligible sequence similarity between GPCR families, activation of family B receptors resembles that for family A GPCRs, with an outward rotation of TM6 creating a G protein binding cavity. This and other shared structural features of receptor activation suggest that the GPCR activation mechanism, like the overall 7TM receptor fold, is an inherited ancient feature derived from a common ancestral receptor. While the lack of conserved sequence motifs between GPCR families implies that the structural basis for activation must necessarily differ in the details, it is likely that similar overall principles are important in activation of all GPCRs. In each case, sequence and structural diversity in the extracellular region allows GPCRs to recognize a wide range of agonists, which stabilize similar conformational changes on the intracellular side of the receptors, converging on activation of a small number of effector G proteins, arrestins, and kinases.
Funding
This work was supported in part by a grant from the Smith Family Foundation.
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.7b00747.
Summary of PDB entries for GPCRs determined in multiple conformational states (Table 1) (PDF)
REFERENCES
- (1).Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, and Kobilka BK (2007) GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science 318, 1266–1273. [DOI] [PubMed] [Google Scholar]
- (2).Chae PS, Rasmussen SG, Rana RR, Gotfryd K, Chandra R, Goren MA, Kruse AC, Nurva S, Loland CJ, Pierre Y, Drew D, Popot JL, Picot D, Fox BG, Guan L, Gether U, Byrne B, Kobilka B, and Gellman SH (2010) Maltose-neopentyl glycol (MNG) amphiphiles for solubilization, stabilization and crystallization of membrane proteins. Nat. Methods 7, 1003–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Caffrey M (2015) A comprehensive review of the lipid cubic phase or in meso method for crystallizing membrane and soluble proteins and complexes. Acta Crystallogr., Sect. F: Struct. Biol. Commun 71, 3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Smith JL, Fischetti RF, and Yamamoto M (2012) Micro-crystallography comes of age. Curr. Opin. Struct. Biol 22, 602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Johansson LC, Stauch B, Ishchenko A, and Cherezov V (2017) A Bright Future for Serial Femtosecond Crystallography with XFELs. Trends Biochem. Sci 42, 749–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, and Kobilka BK (2011) Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Warne T, Moukhametzianov R, Baker JG, Nehme R, Edwards PC, Leslie AG, Schertler GF, and Tate CG (2011) The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature 469, 241–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AG, and Tate CG (2011) Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 474, 521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wacker D, Wang C, Katritch V, Han GW, Huang XP, Vardy E, McCorvy JD, Jiang Y, Chu M, Siu FY, Liu W, Xu HE, Cherezov V, Roth BL, and Stevens RC (2013) Structural features for functional selectivity at serotonin receptors. Science 340, 615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wang C, Jiang Y, Ma J, Wu H, Wacker D, Katritch V, Han GW, Liu W, Huang XP, Vardy E, McCorvy JD, Gao X, Zhou XE, Melcher K, Zhang C, Bai F, Yang H, Yang L, Jiang H, Roth BL, Cherezov V, Stevens RC, and Xu HE (2013) Structural basis for molecular recognition at serotonin receptors. Science 340, 610–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wacker D, Wang S, McCorvy JD, Betz RM, Venkatakrishnan AJ, Levit A, Lansu K, Schools ZL, Che T, Nichols DE, Shoichet BK, Dror RO, and Roth BL (2017) Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell 168, 377–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Latorraca NR, Venkatakrishnan AJ, and Dror RO (2017) GPCR Dynamics: Structures in Motion. Chem. Rev 117, 139–155. [DOI] [PubMed] [Google Scholar]
- (13).Wacker D, Stevens RC, and Roth BL (2017) How Ligands Illuminate GPCR Molecular Pharmacology. Cell 170, 414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ghanouni P, Steenhuis JJ, Farrens DL, and Kobilka BK (2001) Agonist-induced conformational changes in the G-protein-coupling domain of the beta 2 adrenergic receptor. Proc. Natl. Acad. Sci.U. S. A 98, 5997–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, Rasmussen SG, Choi HJ, Devree BT, Sunahara RK, Chae PS, Gellman SH, Dror RO, Shaw DE, Weis WI, Caffrey M, Gmeiner P, and Kobilka BK (2011) Structure and function of an irreversible agonist-beta(2) adrenoceptor complex. Nature 469, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan Y, Xu H, and Shaw DE (2011) Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. U. S.A 108, 13118–13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Dror RO, Arlow DH, Maragakis P, Mildorf TJ, Pan AC, Xu H, Borhani DW, and Shaw DE (2011) Activation mechanism of the beta2-adrenergic receptor. Proc. Natl. Acad. Sci. U. S. A 108, 18684–18689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Nygaard R, Zou Y, Dror RO, Mildorf TJ, Arlow DH, Manglik A, Pan AC, Liu CW, Fung JJ, Bokoch MP, Thian FS, Kobilka TS, Shaw DE, Mueller L, Prosser RS, and Kobilka BK (2013) The dynamic process of beta(2)-adrenergic receptor activation. Cell 152, 532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Manglik A, Kim TH, Masureel M, Altenbach C, Yang Z, Hilger D, Lerch MT, Kobilka TS, Thian FS, Hubbell WL, Prosser RS, and Kobilka BK (2015) Structural Insights into the Dynamic Process of beta2-Adrenergic Receptor Signaling. Cell 161, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Okude J, Ueda T, Kofuku Y, Sato M, Nobuyama N, Kondo K, Shiraishi Y, Mizumura T, Onishi K, Natsume M, Maeda M, Tsujishita H, Kuranaga T, Inoue M, and Shimada I (2015) Identification of a Conformational Equilibrium That Determines the Efficacy and Functional Selectivity of the mu-Opioid Receptor. Angew. Chem., Int. Ed 54, 15771–15776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ye L, Van Eps N, Zimmer M, Ernst OP, and Prosser RS (2016) Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature 533, 265–268. [DOI] [PubMed] [Google Scholar]
- (22).Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, and Stevens RC (2011) Structure of an agonist-bound human A2A adenosine receptor. Science 332, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Liu W, Wacker D, Gati C, Han GW, James D, Wang D, Nelson G, Weierstall U, Katritch V, Barty A, Zatsepin NA, Li D, Messerschmidt M, Boutet S, Williams GJ, Koglin JE, Seibert MM, Wang C, Shah ST, Basu S, Fromme R, Kupitz C, Rendek KN, Grotjohann I, Fromme P, Kirian RA, Beyerlein KR, White TA, Chapman HN, Caffrey M, Spence JC, Stevens RC, and Cherezov V (2013) Serial femtosecond crystallography of G protein-coupled receptors. Science 342, 1521–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Srivastava A, Yano J, Hirozane Y, Kefala G, Gruswitz F, Snell G, Lane W, Ivetac A, Aertgeerts K, Nguyen J, Jennings A, and Okada K (2014) High-resolution structure of the human GPR40 receptor bound to allosteric agonist TAK-875. Nature 513, 124–127. [DOI] [PubMed] [Google Scholar]
- (25).Egloff P, Hillenbrand M, Klenk C, Batyuk A, Heine P, Balada S, Schlinkmann KM, Scott DJ, Schutz M, and Pluckthun A (2014) Structure of signaling-competent neurotensin receptor 1 obtained by directed evolution in Escherichia coli. Proc. Natl. Acad. Sci.U. S. A 111, E655–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhang J, Zhang K, Gao ZG, Paoletta S, Zhang D, Han GW, Li T, Ma L, Zhang W, Muller CE, Yang H, Jiang H, Cherezov V, Katritch V, Jacobson KA, Stevens RC, Wu B, and Zhao Q (2014) Agonist-bound structure of the human P2Y12 receptor. Nature 509, 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Krumm BE, White JF, Shah P, and Grisshammer R (2015) Structural prerequisites for G-protein activation by the neurotensin receptor. Nat. Commun 6, 7895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Shihoya W, Nishizawa T, Okuta A, Tani K, Dohmae N, Fujiyoshi Y, Nureki O, and Doi T (2016) Activation mechanism of endothelin ETB receptor by endothelin-1. Nature 537, 363–368. [DOI] [PubMed] [Google Scholar]
- (29).Ma Y, Yue Y, Ma Y, Zhang Q, Zhou Q, Song Y, Shen Y, Li X, Ma X, Li C, Hanson MA, Han GW, Sickmier EA, Swaminath G, Zhao S, Stevens RC, Hu LA, Zhong W, Zhang M, and Xu F (2017) Structural Basis for Apelin Control of the Human Apelin Receptor. Structure (Oxford, U. K.) 25, 858–866. [DOI] [PubMed] [Google Scholar]
- (30).Liang YL, Khoshouei M, Radjainia M, Zhang Y, Glukhova A, Tarrasch J, Thal DM, Furness SGB, Christopoulos G, Coudrat T, Danev R, Baumeister W, Miller LJ, Christopoulos A, Kobilka BK, Wootten D, Skiniotis G, and Sexton PM (2017) Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature 546, 118–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zhang Y, Sun B, Feng D, Hu H, Chu M, Qu Q, Tarrasch JT, Li S, Sun Kobilka T, Kobilka BK, and Skiniotis G (2017) Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature 546, 248–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Carpenter B, Nehme R, Warne T, Leslie AG, and Tate CG (2016) Structure of the adenosine A(2A) receptor bound to an engineered G protein. Nature 536, 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, and Ernst OP (2008) Crystal structure of opsin in its G-protein-interacting conformation. Nature 455, 497–502. [DOI] [PubMed] [Google Scholar]
- (34).Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, Barty A, White TA, Yefanov O, Han GW, Xu Q, de Waal PW, Ke J, Tan MH, Zhang C, Moeller A, West GM, Pascal BD, Van Eps N, Caro LN, Vishnivetskiy SA, Lee RJ, Suino-Powell KM, Gu X, Pal K, Ma J, Zhi X, Boutet S, Williams GJ, Messerschmidt M, Gati C, Zatsepin NA, Wang D, James D, Basu S, Roy-Chowdhury S, Conrad CE, Coe J, Liu H, Lisova S, Kupitz C, Grotjohann I, Fromme R, Jiang Y, Tan M, Yang H, Li J, Wang M, Zheng Z, Li D, Howe N, Zhao Y, Standfuss J, Diederichs K, Dong Y, Potter CS, Carragher B, Caffrey M, Jiang H, Chapman HN, Spence JC, Fromme P, Weierstall U, Ernst OP, Katritch V, Gurevich VV, Griffin PR, Hubbell WL, Stevens RC, Cherezov V, Melcher K, and Xu HE (2015) Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523, 561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Manglik A, Kobilka BK, and Steyaert J (2017) Nanobodies to Study G Protein-Coupled Receptor Structure and Function. Annu. Rev. Pharmacol. Toxicol 57, 19–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ishchenko A, Wacker D, Kapoor M, Zhang A, Han GW, Basu S, Patel N, Messerschmidt M, Weierstall U, Liu W, Katritch V, Roth BL, Stevens RC, and Cherezov V (2017) Structural insights into the extracellular recognition of the human serotonin 2B receptor by an antibody. Proc. Natl. Acad. Sci. U. S. A 114, 8223–8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, and Kobilka BK (2011) Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 469, 175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ring AM, Manglik A, Kruse AC, Enos MD, Weis WI, Garcia KC, and Kobilka BK (2013) Adrenaline-activated structure of beta2-adrenoceptor stabilized by an engineered nanobody. Nature 502, 575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hubner H, Pardon E, Valant C, Sexton PM, Christopoulos A, Felder CC, Gmeiner P, Steyaert J, Weis WI, Garcia KC, Wess J, and Kobilka BK (2013) Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL, Kato HE, Livingston KE, Thorsen TS, Kling RC, Granier S, Gmeiner P, Husbands SM, Traynor JR, Weis WI, Steyaert J, Dror RO, and Kobilka BK (2015) Structural insights into mu-opioid receptor activation. Nature 524, 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Burg JS, Ingram JR, Venkatakrishnan AJ, Jude KM, Dukkipati A, Feinberg EN, Angelini A, Waghray D, Dror RO, Ploegh HL, and Garcia KC (2015) Structural biology. Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science 347, 1113–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Jazayeri A, Rappas M, Brown AJH, Kean J, Errey JC, Robertson NJ, Fiez-Vandal C, Andrews SP, Congreve M, Bortolato A, Mason JS, Baig AH, Teobald I, Dore AS, Weir M, Cooke RM, and Marshall FH (2017) Crystal structure of the GLP-1 receptor bound to a peptide agonist. Nature 546, 254–258. [DOI] [PubMed] [Google Scholar]
- (43).Preininger AM, Meiler J, and Hamm HE (2013) Conformational flexibility and structural dynamics in GPCR-mediated G protein activation: a perspective. J. Mol. Biol 425, 2288–2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Ballesteros J, and Weinstein H (1995) Integrated methods for modeling G-protein coupled receptors. Methods Neurosci. 25, 366–428 [Google Scholar]
- (45).Janz JM, and Farrens DL (2004) Rhodopsin activation exposes a key hydrophobic binding site for the transducin alpha-subunit C terminus. J. Biol. Chem 279, 29767–29773. [DOI] [PubMed] [Google Scholar]
- (46).Goncalves JA, South K, Ahuja S, Zaitseva E, Opefi CA, Eilers M, Vogel R, Reeves PJ, and Smith SO (2010) Highly conserved tyrosine stabilizes the active state of rhodopsin. Proc. Natl. Acad. Sci. U. S. A 107, 19861–19866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Vaidehi N, Grisshammer R, and Tate CG (2016) How Can Mutations Thermostabilize G-Protein-Coupled Receptors? Trends Pharmacol. Sci 37, 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Pert CB, Pasternak G, and Snyder SH (1973) Opiate agonists and antagonists discriminated by receptor binding in brain. Science 182, 1359–1361. [DOI] [PubMed] [Google Scholar]
- (49).Katritch V, Fenalti G, Abola EE, Roth BL, Cherezov V, and Stevens RC (2014) Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci 39, 233–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Wang JQ, Kon J, Mogi C, Tobo M, Damirin A, Sato K, Komachi M, Malchinkhuu E, Murata N, Kimura T, Kuwabara A, Wakamatsu K, Koizumi H, Uede T, Tsujimoto G, Kurose H, Sato T, Harada A, Misawa N, Tomura H, and Okajima F (2004) TDAG8 is a proton-sensing and psychosine-sensitive G-protein-coupled receptor. J. Biol. Chem 279, 45626–45633. [DOI] [PubMed] [Google Scholar]
- (51).Alfadda TI, Saleh AMA, Houillier P, and Geibel JP (2014) Calcium-sensing receptor 20 years later. Am. J. Phys. - Cell Phys 307, C221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Antunes G, and Simoes de Souza FM (2016) Olfactory receptor signaling. Methods Cell Biol. 132, 127–145. [DOI] [PubMed] [Google Scholar]
- (53).Pires PW, Ko EA, Pritchard HAT, Rudokas M, Yamasaki E, and Earley S (2017) The angiotensin II receptor type 1b is the primary sensor of intraluminal pressure in cerebral artery smooth muscle cells. J. Physiol 595, 4735–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Violin JD, Crombie AL, Soergel DG, and Lark MW (2014) Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol. Sci 35, 308–316. [DOI] [PubMed] [Google Scholar]