

Abstract
Fucosylated human milk oligosaccharides (HMOs) have important biological functions. Enzymatic synthesis of such compounds requires robust fucosyltransferases. A C-terminal 66-amino acid truncated version of Helicobacter pylori α1–3-fucosyltransferase (Hp3FT) is a good candidate. Hp3FT was biochemically characterized to identify optimal conditions for enzymatic synthesis of fucosides. The acceptor substrate specificity is N-acetyllactosamine (LacNAc) and lactose, with LacNAc as its preferred acceptor. At a low guanosine 5’-diphospho-β-L-fucose (GDP-Fuc) to acceptor ratio, Hp3FT selectively fucosylated LacNAc. Based on these enzymatic characteristics, diverse fucosylated HMOs, including 3-fucosyllactose (3-FL), lacto-N-fucopentaose (LNFP) III, lacto-N-neofucopentaose (LNnFP) V, lacto-N-neodifucohexaose (LNnDFH) II, difuco- and trifuco-para-lacto-N-neohexaose (DF-paraLNnH and TF-para-LNnH), were synthesized enzymatically by varying the ratio of the donor and acceptor as well as controlling the order of multiple glycosyltransferase-catalyzed reactions.
Keywords: fucosyltransferase, substrate specificity, human milk oligosaccharide, enzymatic synthesis
Graphical Abstract
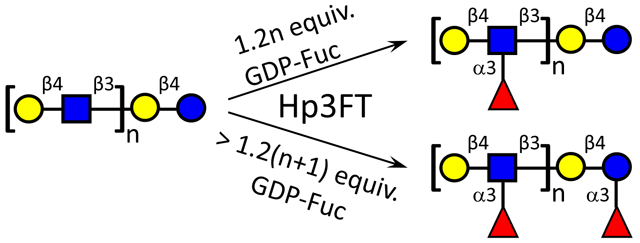
1. Introduction
Lactose (Lac), lipid, and human milk oligosaccharides (HMOs) are the three major components of human milk [1, 2]. HMOs are a group of oligosaccharides that can provide beneficial effects to the health of breast-fed infants [3, 4]. In a typical human milk sample, around 70 percent of HMOs are fucosylated [5], and they have crucial functions. For example, 3-fucosyllactose [3-FL, Galβ1–4(Fucα1–3)Glc] and lacto-N-fucopentaose III (LNFP III, Galβ1–4(Fucα1–3)GlcNAcβ1–3Galβ1–4Glc) can provide protection to infants as prebiotics to nourish specific gut bifidobacterial strains [6]. Fucosylated HMOs resemble intestinal cell surface glycans and act as soluble receptors of intestinal pathogens to prevent infection caused by their adhesion to intestinal epithelial surfaces [7]. Moreover, some fucosylated HMOs have potential pharmaceutical applications. LNFP III can improve glucose tolerance and insulin sensitivity [8] and reduce experimental allergic encephalomyelitis (EAE) severity and central nervous system (CNS) inflammation [9]. Therefore, synthesis of fucosylted HMOs is important for related functional studies. Compared with chemical synthesis, enzymatic fucosylation is an efficient approach to synthesize fucosylated HMOs since it does not require harsh chemicals.
A truncated version of Helicobacter pylori α1–3-fucosyltransferase (Hp3FT) lacking the C-terminal 66 amino acid residues was successfully cloned and expressed in E. coli and characterized [10, 11]. Its crystal structure was solved [12]. It can catalyze the transfer of L-fucose (Fuc) from sugar nucleotide donor guanosine 5’-diphospho-β-L-fucose (GDP-Fuc) to acceptors such as N-acetyllactosamine (LacNAc, Galβ1–4GlcNAc) to form Lewis X (Lex) antigen. The enzyme was first reported by the Lin group [10, 12] and has been applied for oligosaccharide synthesis by our labs [13-17] and others including the groups of Wu [18], Boons [19], Cao [20], Wang and Li [21, 22]. However, discrepancies on whether Lac moiety can be recognized as an acceptor by the enzyme were unclear. The Lin group reported that the enzyme was not able to recognize Lac as an acceptor [10]. In addition, the Cao group [20] and one of our groups [16] independently used it for the synthesis of LNFP III, a monofucosylated oligosaccharide, from Galβ1–4GlcNAcβ1–3Galβ1–4Glc successfully, without fucosylation of the Lac moiety in the acceptor. On the contrary, the Boons group reported that it catalyzed the transfer of two fucose residues to Siaα2–3Galβ1–4GlcNAcβ1–3Galβ1–4Glc to form Siaα2–3Galβ1–4(Fucα1–3)GlcNAcβ1–3Galβ1–4(Fucα1–3)Glc [19], indicating that Lac component was also suitable for fucosylation by the enzyme. Only the clarification of these conflicting results will enable a clear future application of Hp3FT in fucosides preparation. Therefore, in this study, its acceptor substrate specificity to Lac and LacNAc was elucidated by kinetic study. An HPLC-based assay that detects non-derivatized acceptor and product glycans for kinetic study was developed for the first time by combining hydrophilic interaction liquid chromatography (HILIC) separation and evaporative light scattering detector (ELSD). Meanwhile, systematic biochemical characterization of Hp3FT was carried out to obtain its optimal reaction condition. The information learned was applied in efficient synthesis of a series of fucosylated HMOs, including 3-FL, LNFP III, lacto-N-neofucopentaose (LNnFP) V [23], lacto-N-neodifucohexaose (LNnDFH) II [24], difuco- and trifuco-para-lacto-N-neohexaose (DF-paraLNnH and TF-para-LNnH) [25].
2. Results and discussion
2.1. Cloning, expression and purification of Hp3FT
A C-terminal 66 amino acids truncated Hp3FT was cloned from a codon-optimized synthetic gene of H. pylori α1–3-fucosyltransferase into pET15b vector and expressed in Escherichia coli BL21(DE3) as an N-terminal His6-tagged fusion protein. One-step affinity column purification using a nickel-nitrilotriacetic acid (Ni2+-NTA) agarose column was able to provide a purified sample containing more than 95% of the purified Hp3FT with molecular weight close to its theoretical value (44.2 kDa) (Fig. 1). An expression level of about 27 mg of purified protein per liter culture was typically achieved when Terrific Broth (TB) was used.
Fig. 1.

SDS-PAGE analysis of Hp3FT. The apparent molecular weight of the purified enzyme appears approximate 44 kDa (calculated molecular weight 44.2 kDa). Lane 1, whole cell extraction before induction; Lane 2, whole cell extraction after induction; Lane 3, lysate after induction; Lane 4, Ni2+-NTA agarose column purified protein; Lane M, protein markers (Blue Plus IV Protein Marker, 10–180 kDa, from TransGen Biotech, Beijing, China).
2.2. pH profile of Hp3FT and metal ion effects on Hp3FT
The pH profile study of Hp3FT showed that it was active in a broad pH range between pH 5.0 and pH 9.5 with an optimal activity at pH 7.5 in Tris-HCl buffer (Fig. 2).
Fig. 2.

pH profile of Hp3FT with LacNAcβPro2AA as the acceptor substrate. Buffers used were: sodium acetate, pH 4.0–4.5; MES, pH 5.0–6.5; HEPES, pH 7.0; Tris-HCl, pH 7.5–9.0; CAPSO, pH 9.5; and CAPS, pH 10.0–11.0.
Similar to E. coli α1–2-fucosyltransfearse EcWbgL [26] and H. pylori α1–3/4-fucosyltransferase (Hp3/4FT) [27], a metal cation is not required for the fucosyltransfer activity of Hp3FT, as shown in the metal effect studies using Mg2+, Mn2+, or ethylenediaminetetraacetic acid (EDTA) in Tris-HCl buffer at pH 7.5 (Fig. 3), despite the fact that it uses a nucleotide diphosphate sugar as the donor substrate. This can be explained by its GT-B structure fold [12] and the absence of a DXD domain in its sequence [28].
Fig. 3.

Metal cation effects on Hp3FT assayed using a reaction mixture containing Mn2+ (Black columns) or containing Mg2+ (grey columns), in the presence of EDTA (5 mM) (striped column) or in the absence of a metal (dotted column).
2.3. Kinetic study of Hp3FT
An HPLC-based assay using HILIC separation and ELSD was developed for kinetics studies of Hp3FT using non-derivatized LacNAc and Lac as acceptors. The obtained apparent kinetic parameters showed that, compared to Lac, LacNAc has a lower Km value and a higher kcat value, leading to a catalytic efficiency 161-fold higher than that of Lac. The results clearly indicate that LacNAc is a preferred acceptor for Hp3FT. A possible explanation for the preferential binding of Hp3FT to LacNAc is that, the hydrophobic interaction formed between the methyl group in the N-acetyl group on C2 of GlcNAc in LacNAc and the side chain of Leu-124 of Hp3FT [12] contributes to the better binding of LacNAc to the active site cleft of Hp3FT, whereas no such interaction is formed between the hydroxyl group on C2 of glucose residue in Lac and Leu-124.
2.4. Synthesis of fucosylated HMOs
With the kinetics toward the two acceptor substrates characterized, Hp3FT was subsequently employed to synthesize a series of fucosylated HMOs (Scheme 1). In our synthesis, oligosaccharides with only Lac moiety or both LacNAc and Lac moieties were used as acceptors, and effective fucosylation was achieved by the catalysis of Hp3FT. From Lac, 3-FL (1) can be efficiently synthesized by the enzyme. However, when synthesizing the monofucosylated LNFP III (4) from lacto-N-neotetraose (LNnT, 3), a tetrasaccharide containing two potential fucosylation sites (one Lac moiety and one LacNAc moiety), Fuc was preferentially transferred onto LacNAc rather than Lac at lower ratio of GDP-Fuc to acceptor (1.2:1), consistent with the kinetic study. The structure of the product was identified by tandem MS (Fig. 4a) and 2D NMR (the data is in match with the data in ref [27]). If the ratio of GDP-Fuc to LNnT was over 2:1 (in this case, 2.6:1 was used), two fucoses can be added and difucosylated LNnDFH II (5) was acquired and identified by tandem MS (Fig. 4b). According to tandem MS results, specific fragment of Galβ1–4(Fucα1–3)GlcNAc was found in LNFP III (4), whereas Galβ1–4(Fucα1–3)Glc was not (Fig. 4a). However, specific fragments of both Galβ1–4(Fucα1–3)GlcNAc and Galβ1–4(Fucα1–3)Glc can be found in LNnDFH II (5). These results are consistent with previously reported synthesis of fucosides, in which LNFP III was generated when GDP-Fuc to acceptor ratio was at most 1.5:1 [20], whereas LNnDFH II was formed with a GDP-Fuc to acceptor ratio at ~ 4:1 [19].
Scheme 1.

Synthesis of fucosylated HMOs by Hp3FT according to its acceptor specificity. (a) NmLgtA, UDP-GlcNAc, Mn2+; (b) NmLgtB, UDP-Gal, Mn2+; (c) Hp3FT, GDP-Fuc, Mn2+, GDP-Fuc to acceptor ratio = 1.2:1; (d) Hp3FT, GDP-Fuc, Mn2+, GDP-Fuc to acceptor ratio = 2.6:1; (e) Hp3FT, GDP-Fuc, Mn2+, GDP-Fuc to acceptor ratio = 2.4:1; (f) Hp3FT, GDP-Fuc, Mn2+, GDP-Fuc to acceptor ratio = 4:1.
Fig. 4.

Acceptor substrate specificity of Hp3FT was identified by tandem MS during the synthesis of (a) LNFP III (4), (b) LNnDFH II (5), (c) LNnFP V (7), (d) DF-para-LNnH (9), and (e) TF-para-LNnH (10).
For para-lacto-N-neohexaose (para-LNnH, 8) [25] with single Lac and double LacNAc moieties, the preferences of Hp3FT were similar to those for LNnT, with the first two Fuc residues transferred to the two LacNAc moieties to give DF-para-LNnH (9) at lower ratio (2.4:1) of GDP-Fuc to acceptor, and the third Fuc attached onto Lac moiety to generate TF-para-LNnH (10) only when the ratio of GDP-Fuc over acceptor was over 3:1 (in this case, 4:1 was used). The structures of both glycans (9 and 10) were identified by tandem MS (Fig. 4d and 4e).
Given its broad acceptor specificity and preferential fucosylation activity on LacNAc over Lac, an approach with altered sequence of glycosyltransferases was adopted to achieve site-specific fucosylation of Lac moiety at the reducing end with the existence of LacNAc moiety in the acceptor, similar to the strategy used for selective sialylation of galactose close to the reducing end of the acceptor catalyzed by a bacterial α2–6sialyltransferase with broad acceptor substrate specificity [29]. For example, by first using Hp3FT and then NmLgtB, LNnFP V (also named LNFP VI, 7), an isomer of LNFP III (4), can be obtained from lacto-N-triose II (LNT II, 2) (Scheme 1), as identified by tandem MS (Fig. 4c) and 2D NMR (Fig. S1). For the synthesis of all the 7 fucosylated compounds (compounds 1, 4, 5, 6, 7, 9, 10) shown in Scheme 1, no remaining acceptors were detected by MALDI-TOF MS, indicating the efficient fucosylation catalyzed by Hp3FT.
3. Conclusions
The commonly used fucosyltransferase Hp3FT has been previously reported to generate discrepant fucoside products from the same acceptor glycan [16, 19, 20]. According to the acceptor specificity study of recombinant Hp3FT performed in this study, the inconsistency can be explained by its recognition of both Lac and LacNAc as acceptors, but with a higher catalytic efficiency towards LacNAc. Therefore, lower donor (GDP-Fuc) to acceptor ratio used in synthetic reaction mixture in the previous reports resulted in fucosylation at LacNAc only [16, 20], whereas higher donor to acceptor ratio provided fully fucosylated glycans at both LacNAc and Lac sites [19]. In the case that Lac is present with multiple LacNAc moieties within the same glycan, Lac will be fucosylated only after LacNAc moieties are all fucosylated (as demonstrated by the synthesis of compounds 9 and 10 herein). If site-specific fucosylation of the reducing terminal Lac is desired with the coexistence of LacNAc in the target glycan (as illustrated by LNnFP V, compound 7 in this study), synthetic routes with appropriate sequences of glycosylation need to be designed. Based on this principle, fucosylated HMOs with fucosylation specifically on LacNAc, specifically on Lac, or both on LacNAc and Lac can be prepared, as exemplified in this study (including 3-FL, LNFP III, LNnFP V, LNnDFH II, DF-para-LNnH, and TF-para-LNnH). The products obtained are important probes for further biological studies on their nutraceutical and pharmaceutical potentials.
4. Experimental
4.1. Bacterial strains, vector, materials and general methods
Full-length H. pylori α1–3-fucosyltransferase synthetic gene with codons optimized for Escherichia coli expression system was custom-synthesized by Geneart, Inc. (Burlingame, CA, USA). E. coli DH5α electrocompetent cells and chemically competent BL21 (DE3) cells were from Invitrogen (Carlsbad, CA, USA). Vector pET15b was purchased from Novagen (EMD Biosciences Inc., Madison, WI, USA). Restriction enzymes NdeI and BamHI were purchased from New England BioLabs (Beverly, MA, USA). Chemicals were purchased and used without further purification. Fucose (Fuc), ethylenediaminetetraacetic acid (EDTA), adenosine 5'-triphosphate (ATP), guanosine 5'-triphosphate (GTP), magnesium chloride, manganese chloride, and nickel sulfate were purchased from Sigma Aldrich (Saint Louis, MO, USA). Gel filtration chromatography was performed with a column (100 cm × 2.5 cm) packed with BioGel P-2 Fine resins (Bio-Rad). BfFKP [30], PmPpA [31], NmLgtA [32], and NmLgtB [33] were overexpressed as reported. GDP-Fuc was prepared by one-pot multi-enzyme synthesis followed by gel filtration chromatography as previously reported [34].
4.2. Cloning of Hp3FT
N-His6-tagged and 66 amino acids at C-terminal truncated H. pylori α1–3-fucosyltransferase (Hp3FT) was cloned into pET15b vector. Primers used were: forward 5’-GATCCATATGTTTCAGCCGCTGCTG-3’ (NdeI restriction site is underlined) and reverse 5’-CGCGGATCCTTATTAGTAGTTAACACGCAGATC-3’ (BamHI restriction site is underlined). Plasmids were purified and positive clones were verified by restriction mapping and DNA sequencing performed by the Davis Sequencing Facility at the University of California-Davis.
4.3. Expression and purification of Hp3FT
The E. coli BL21 (DE3) cells containing recombinant plasmid with gene encoding Hp3FT were cultured in Terrific Broth (TB) containing 100 μg mL−1 of ampicillin at 37 °C with orbital shaking (250 rpm) until the OD600 nm reached 0.8–1.0. Overexpression of the target protein was achieved by adding 0.1 mM of IPTG to the E. coli culture followed by further incubation at 20 °C for 20 h. Then, the cells were harvested by centrifugation at 4,000 rpm at 4 °C for 30 min. The cell pellet was resuspended in lysis buffer (pH 8.0, 100 mM Tris-HCl containing 0.1% Triton X-100) (20 mL per liter cell culture) and the mixture went through two freeze-thaw cycles. Lysozyme (50 μg mL−1) and DNase I (3 μg mL−1) were then added and the mixture was incubated at 37 °C for 1 h with gentle shaking (125 rpm). Then, the supernatant was obtained by centrifugation at 11,000 rpm for 20 min at 4 °C, and was loaded onto a Ni-NTA column pre-equilibrated with binding buffer (50 mM Tris-HCl, pH 7.5, 0.5 M NaCl, 5 mM imidazole). The column was washed with 8 column volumes of binding buffer and 8 column volumes of washing buffer (50 mM Tris-HCl, pH 7.5, 0.5 M NaCl, 30 mM imidazole), and then the protein of interest was eluted with an elution buffer (50 mM Tris-HCl, pH 7.5, 0.5 M NaCl, 200 mM imidazole). The fractions containing purified recombinant protein were pooled and dialyzed against Tris-HCl buffer (pH 8.0, 20 mM) containing 10% glycerol and NaCl (100 mM). Dialyzed proteins were stored at 4 °C. The purified protein was analyzed by SDS-PAGE and was quantified by a BCA protein assay kit (Pierce Biotechnology, Rockford, IL).
4.4. pH profiles of Hp3FT
All assays were carried out in duplicate. Each reaction in a total volume of 10 μL contains a buffer (200 mM) with pH varying from 4.0 to 11.0, MnCl2 (20 mM), GDP-Fuc (1 mM), LacNAcβPro2AA (1 mM) which is labeled with 2-anthranilic acid (2AA), and 1.2 μg Hp3FT. The buffers used were sodium acetate, pH 4.0–4.5; MES, pH 5.0–6.5; HEPES, pH 7.0; Tris-HCl, pH 7.5–9.0; CAPSO, pH 9.5; and CAPS, pH 10.0–11.0. Reactions were carried out at 37 °C for 15 min before being quenched by adding pre-chilled 20% acetonitrile (20 μL). The samples were kept on ice until aliquots of 8 μL were injected and analyzed by a Shimadzu LC-2010A HPLC system equipped with a membrane online degasser, a temperature control unit, and a fluorescence detector. A reverse-phase Waters Spherisorb C18 column (250 × 4.6 mM i.d., 5 μm particle size) protected with a C18 guard column cartridge was used. The mobile phase was 18% acetonitrile. Fluorescently labeled compounds LacNAcβPro2AA and LexβPro2AA were detected by excitation at 315 nm and emission at 400 nm.
4.5. EDTA and metal effects on the α1–3-fucosyltransferase activity of Hp3FT
The reactions were carried out in duplicate in a total volume of 10 μL in Tris-HCl buffer (100 mM, pH 7.5) containing GDP-Fuc (1 mM), LacNAcβPro2AA (1 mM), and recombinant enzyme (1.2 μg) in the presence of EDTA (5 mM) or a divalent metal salt (MgCl2 or MnCl2) (5 mM, 10 mM, or 20 mM). Reactions without EDTA or a metal ion were used as controls. Reactions were in carried out in duplicate at 37 °C for 15 min, quenched and assayed using the same method as described above for pH profile.
4.6. Kinetics study for the α1–3-fucosyltransferase activity of Hp3FT
To obtain the apparent kinetic parameters of LacNAc and Lac, the kinetic assays were performed in triplicates in reaction mixtures of 20 μL containing Tris-HCl buffer (50 mM, pH 7.5), 10 mM of MgCl2, a fixed concentration of GDP-Fuc (15 mM), different concentrations of LacNAc (0.5, 0.8, 1, 1.5, 2, 3 and 5 mM) or Lac (1, 2, 3, 5, 6, 8 and 10 mM), and Hp3FT (1.6 μg for LacNAc, 100 μg for Lac). To obtain the apparent kinetic parameters of GDP-Fuc, the kinetic assays were performed in triplicates in reaction mixtures of 20 μL containing Tris-HCl buffer (50 mM, pH 7.5), a fixed concentration of LacNAc (3 mM) or Lac (10 mM), different concentrations of GDP-Fuc (2, 5, 8, 12, 18 and 20 mM), and Hp3FT (1.6 μg for LacNAc, 100 μg for Lac). All reactions were allowed to proceed at 37 °C for 20 min and the reactions were terminated by incubating the tubes containing the reaction mixtures in a boiling water bath for 5 minutes. After centrifugation at 13000 rpm in a table-top centrifuge for 20 min, the supernatant of each sample was obtained and 10 μL of each was injected and analyzed by a Waters e2695 Alliance system equipped with a 2424 evaporative light scattering detector (ELSD). An XBridge BEH Amide Column (130Å, 5 μm, 4.6 mm × 250 mm, Waters) was used. Mobile phase A: 100 mM ammonium formate, pH 3.4; Mobile phase B: acetonitrile; B% Gradient: 90% to 80% over 7 min, 80% to 70% over 10 min, 70% to 60% over 13 min, 10% for 10 min, 90% for 10 min; Flow rate: 1 mL min−1. LacNAc, Lac, and their fucosylated product were detected by ELSD. Apparent kinetic parameters were obtained by fitting the experimental data (the average values of triplicate assay results) into the Michaelis–Menten equation using Graph Pad 5.0.
4.7. HMOs synthesis
Reaction mixtures contained Tris-HCl (100 mM, pH 8.0), an acceptor glycan (as a pure compound or in the reaction mixture from the previous reaction, 10 mM), an appropriate sugar nucleotide (1.2 equiv. for UDP-GlcNAc or UDP-Gal, 1.2–4.0 equiv. for GDP-Fuc, depending on the number of fucose residues to be transferred), MnCl2 (10 mM), varying amounts of appropriate glycosyltransferases (NmLgtA, NmLgtB, or Hp3FT), and FastAP (1 U/200 μL, to digest the byproduct NDP and drive the reaction forward). Reactions were incubated at 37 °C for overnight and monitored by MALDI-TOF MS. When all acceptors were converted, the reactions were quenched by incubating the tubes containing the reaction mixtures in a boiling water bath for 5 min. If the product of a glycosylation step was not the final product, no purification was performed, and the reaction mixture was directly used for the next enzymatic extension. In the cases of the compounds being final compounds, purification was performed by HPLC with a semi-preparative amide column (130 Å, 5 μm, 10 mm × 250 mm). Mobile phase A: 100 mM ammonium formate, pH 3.4; Mobile phase B: acetonitrile; B% Gradient: 80% to 60% over 35 min; Flow rate: 4 mL min−1. HPLC eluent was monitored by absorption at 210 nm, and glycan-containing fractions were analyzed by MALDI-TOF MS. 1H NMR and 13C NMR spectra of the purified compounds were recorded on a Bruker Avance-III 600 MHz NMR spectrometer.
4.7.1. Galβ1–4(Fucα1–3)Glc (1).
5 mg, 88%; 1H NMR (600 MHz, D2O) δ 5.34 (d, J = 4.2, 0.5H), 5.28 (d, J = 4.2 Hz, 0.4H), 5.08 (d, J = 3.6 Hz, 0.4H), 4.80-4.72 (m, 1H), 4.55 (d, J = 7.8 Hz, 0.6H), 4.33 (d, J = 7.8 Hz, 1H), 3.84-3.35 (m, 16 H), 1.09-1.07 (m, 3H); 13C NMR (150 MHz, D2O) δ 101.77, 101.74, 98.48, 98.34, 95.79, 92.05, 76.97, 75.50, 75.33, 74.91, 74.68, 72.64, 72.58, 72.38, 71.93, 71.11, 70.88, 69.25, 69.20, 68.29, 68.09, 68.01, 66.47, 66.43, 61.51, 61.48, 59.74, 59.66, 15.22. MALDI-TOF m/z calculated for C18H32NaO15 (M+Na): 511.16, found 511.36.
4.7.2. Galβ1–4(Fucα1–3)GlcNAcβ1–3Galβ1–4Glc (4).
10 mg, 80%; NMR data see ref [27]. MALDI-TOF m/z calculated for C32H55NNaO25 (M+Na): 876.31, found 876.71.
4.7.3. Galβ1–4(Fucα1–3)GlcNAcβ1–3Galβ1–4(Fucα1–3)Glc (5).
8 mg, 68%; 1H NMR (600 MHz, D2O) δ 5.28 (s, 0.7H, 5.22 (s, 0.7 H), 5.03-4.98 (m, 2.6H), 4.56-4.49 (m, 5.4 H), 4.31 (d, J = 7.2 Hz, 1.4H), 4.26 (d, J = 7.8 Hz, 1.3H), 3.94 (s, 1H), 3.80-3.33 (m, 30H), 1.86 (s, 3H), 1.02-1.01 (m, 6H); 13C NMR (150 MHz, D2O) δ 174.72, 102.48, 101.73, 98.56, 98.42, 95.82, 92.11, 81.55, 81.49, 76.98, 75.53, 75.35, 75.06, 74.90, 74.73, 74.67, 74.47, 73.02, 72.66, 72.46, 72.35, 72.26, 71.90, 71.03, 70.93, 70.67, 70.63, 69.26, 69.18, 68.33, 68.25, 68.22, 68.00, 67.68, 66.66, 66.46, 61.49, 59.78, 59.62, 55.93, 22.22, 15.28, 15.19. MALDI-TOF m/z calculated for C38H68NNaO29 (M+Na): 1022.36, found 1022.46.
4.7.4. GlcNAcβ1–3Galβ1–4(Fucα1–3)Glc (6).
12 mg, 84%; 1H NMR (600 MHz, D2O) δ 5.33 (d, J = 4.2 Hz, 0.6H), 5.27 (d, J = 4.2 Hz, 0.5H), 5.08 (d, J = 4.2 Hz, 0.5H), 4.74-4.73 (m, 1H), 4.58 (dd, J = 2.4 and 8.4 Hz, 1H), 4.55 (d, J = 8.4 Hz, 0.5H), 4.32 (d, J = 7.8 Hz, 1H), 3.99 (d, J = 3.6 Hz, 1H), 3.87-3.33 (m, 22H), 1.93 (s, 3H), 1.07-1.06 (m, 3H); 13C NMR (150 MHz, D2O) δ 174.90, 102.76, 101.70, 98.51, 98.40, 95.81, 92.09, 81.44, 81.39, 76.97, 75.58, 75.32, 74.65, 74.46, 73.44, 72.65, 72.35, 72.27, 71.90, 70.90, 70.70, 70.65, 69.67, 69.23, 69.18, 68.23, 68.01, 67.98, 66.44, 61.43, 60.42, 59.76, 59.69, 55.60, 22.14, 15.20. MALDI-TOF m/z calculated for C26H45NNaO20 (M+Na): 714.25, found 714.67.
4.7.5. Galβ1–4GlcNAcβ1–3Galβ1–4(Fucα1–3)Glc (7).
6 mg, 80%; 1H NMR (600 MHz, D2O) δ 5.27 (d, J = 4.2 Hz, 0.5 H), 5.21 (d, J = 3.6 Hz, 0.5 H), 5.02 (d, J = 4.2 Hz, 0.5 H), 4.67-4.65 (m, 2H), 4.53 (d, J = 8.4 Hz, 1H), 4.49 (d, J = 7.8 Hz, 0.5 Hz), 4.32 (d, J = 7.8 Hz, 1H), 4.25 (d, J = 7.8 Hz, 1H), 3.94 (d, J = 3.0 Hz, 1H), 3.80-3.28 (m, 28H), 1.87 (s, 3H), 1.09-0.99 (m, 3H); 13C DEPT 135 NMR (150 MHz, D2O) δ 102.83, 102.67, 101.70, 98.52, 98.41, 95.81, 92.09, 81.54, 81.49, 78.12, 76.97, 75.52, 75.32, 74.66, 74.49, 74.46, 72.65, 72.47, 72.35, 72.26, 72.09, 71.90, 70.94, 70.64, 70.60, 69.24, 69.19, 68.22, 68.01, 67.98, 66.46, 66.41, 61.43, 59.81, 59.75, 59.68, 55.14, 22.14, 15.19. MALDI-TOF m/z calculated for C32H55NNaO25 (M+Na): 876.31, found 876.06.
4.7.6. Galβ1–4(Fucα1–3)GlcNAcβ1–3Galβ1–4(Fucα1–3)GlcNAcβ1–3Galβ1–4Glc (9).
4 mg, 80%; 1H NMR (600 MHz, D2O) δ 5.15 (d, J = 4.2 Hz, 0.4H), 5.06 (d, J = 3.6 Hz, 1H), 5.04 (d, J = 4.2 Hz, 1H), 4.65-4.63 (m, 2.6H), 4.59 (d, J = 8.4 Hz, 1H), 4.40-4.36 (m, 3H), 4.08 (d, J = 3.6 Hz, 1H), 4.02 (d, J = 3.0 Hz, 1H), 3.90-3.41 (m, 48H), 1.95 (s, 3H), 1.95 (s, 3H), 1.10 (d, J = 6.6 Hz, 3H), (d, J = 6.6 Hz, 3H). 13C DEPT 135 NMR (150 MHz, D2O) δ 102.84, 102.47, 101.67, 98.65, 98.54, 95.66, 91.76, 81.99, 81.52, 78.09, 74.98, 74.84, 74.67, 74.37, 73.70, 72.90, 72.64, 72.37, 71.83, 70.96, 70.45, 69.89, 69.09, 68.27, 67.60, 66.62, 61.46, 60.90, 59.96, 59.52, 59.27, 55.87, 22.16, 15.24. MALDI-TOF m/z calculated for C52H88N2NaO39 (M+Na) 1387.50, found 1387.45.
4.7.7. Galβ1–4(Fucα1–3)GlcNAcβ1–3Galβ1–4(Fucα1–3)GlcNAcβ1–3Galβ1–4(Fucα1–3)Glc (10).
4.8 mg, 94%; 1H NMR (600 MHz, D2O) δ 5.35 (d, J = 4.2 Hz, 1H), 5.29 (d, J = 3.6 Hz, 1H), 5.10 (d, J = 4.2 Hz, 1H), 5.05 (dd, J = 3.6, 8.4 Hz, 3H), 4.63 (d, J = 8.4 Hz, 3H), 4.57 (d, J = 7.8 Hz, 1H), 4.39-4.36 (m, 2H), 4.33 (d, J = 7.8 Hz, 1H), 4.02 (bs, 2H), 3.90-4.40 (m, 60H), 1.94 (s, 6H), 1.10-1.07 (m, 9H). 13C DEPT 135 NMR (150 MHz, D2O) δ 102.47, 101.72, 98.56, 95.82, 92.18, 81.60, 75.53, 75.35, 75.05, 74.90, 74.74, 74.43, 73.01, 72.75, 72.46, 71.90, 71.04, 70.52, 69.17, 68.34, 68.22, 68.00, 67.69, 66.67, 66.47, 61.45, 59.62, 55.94, 22.23, 15.29, 15.26, 15.20. MALDI-TOF m/z calculated for C58H98N2NaO43 (M+Na) 1533.55, found 1533.54.
4.8. MALDI-TOF/TOF MS analyses
One microliter of sample solution with the same volume of DHB (15 mg mL−1 in 50:50 ACN–water) was spotted on the MALDI plate (Bruker Daltonics, MTP 384 polished steel) for MS analysis. The MS and MS/MS spectra were acquired on an ultraflextreme MALDI TOF-TOF mass spectrometer (Bruker Daltonics, Bremen, Germany) equipped with 1 kHz Smartbeam-II laser in reflectron positive mode. The operation parameters for MS spectrum acquisition were ion source 1, 25.00 kV; ion source 2, 22.70 kV; lens, 8.30 kV; reflectron 1, 26.45 kV; and reflectron 2, 13.45 kV; detector gain, × 4.0. The operation parameters for MS/MS spectrum acquisition were ion source 1, 7.50 kV; ion source 2, 6.80 kV; lens 3.50 kV; reflectron 1, 29.50 kV; reflectron 2, 13.80 kV; LIFT 1, 19.00 kV; LIFT 2, 2.9 kV; detector gain, ×10.0. A total of 2000 shots were accumulated for each MS and MS/MS spectrum. The interpretation of the spectra was manually analyzed with GlycoWorkbench software (Euro-CarbDB) [35].
Supplementary Material
Table 1.
Apparent kinetic parameters of recombinant Hp3FT.
| Substrate | kcat (s−1) | Km (mM) | kcat/Km (s−1mM−1) |
|---|---|---|---|
| LacNAc | 0.3677±0.0080 | 0.4225±0.0393 | 8.7 × 10−1 |
| GDP-Fuca | 0.6168±0.0417 | 6.4480±1.1360 | 9.6 × 10−2 |
| Lac | 0.0201±0.0006 | 3.7290±0.2601 | 5.4 × 10−3 |
| GDP-Fucb | 0.0428±0.0007 | 5.3920±0.2868 | 7.9 × 10−3 |
Assayed with LacNAc as the acceptor for Hp3FT.
Assayed with Lac as the acceptor for Hp3FT.
Helicobacter pylori α1–3-fucosyltransferase (Hp3FT) is biochemically characterized
Hp3FT prefers LacNAc over Lac as an acceptor substrate
Hp3FT selectively fucosylates LacNAc at a low GDP-Fuc to acceptor ratio
Selective fucosylation of Lac can be achieved by controlling glycosylation sequence
Diverse fucosylated HMOs have been synthesized using Hp3FT
Acknowledgments
This work was supported by Hebei Science and Technology Plan Project grant 17226701D (to B.Z.), Hebei Provincial Department of Human Resources and Social Security grants C201812 and E2018100010 (to Z.W.), as well as United States National Institutes of Health grant numbers U01GM120419 and R01HD065122 (to X.C.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Jenness R, Semin Perinatol, 3 (1979) 225–239. [PubMed] [Google Scholar]
- [2].Newburg DS, J Mammary Gland Biol Neoplasia, 1 (1996) 271–283. [DOI] [PubMed] [Google Scholar]
- [3].Bode L, Glycobiology, 22 (2012) 1147–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gonia S, Tuepker M, Heisel T, Autran C, Bode L, Gale CA, J Nutr, 145 (2015) 1992–1998. [DOI] [PubMed] [Google Scholar]
- [5].Yu ZT, Chen C, Newburg DS, Glycobiology, 23 (2013) 1281–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ruiz-Moyano S, Totten SM, Garrido DA, Smilowitz JT, German JB, Lebrilla CB, Mills DA, Appl Environ Microbiol, 79 (2013) 6040–6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Newburg DS, Ruiz-Palacios GM, Morrow AL, Annu Rev Nutr, 25 (2005) 37–58. [DOI] [PubMed] [Google Scholar]
- [8].Bhargava P, Li C, Stanya KJ, Jacobi D, Dai L, Liu S, Gangl MR, Harn DA, Lee CH, Nat Med, 18 (2012) 1665–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhu B, Trikudanathan S, Zozulya AL, Sandoval-Garcia C, Kennedy JK, Atochina O, Norberg T, Castagner B, Seeberger P, Fabry Z, Harn D, Khoury SJ, Guleria I, Clin Immunol, 142 (2012) 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lin SW, Yuan TM, Li JR, Lin CH, Biochemistry, 45 (2006) 8108–8116. [DOI] [PubMed] [Google Scholar]
- [11].Sugiarto G, Lau K, Yu H, Vuong S, Thon V, Li Y, Huang S, Chen X, Glycobiology, 21 (2011) 387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sun HY, Lin SW, Ko TP, Pan JF, Liu CL, Lin CN, Wang AH, Lin CH, J Biol Chem, 282 (2007) 9973–9982. [DOI] [PubMed] [Google Scholar]
- [13].Yu H, Lau K, Li Y, Sugiarto G, Chen X, Curr Protoc Chem Biol, 4 (2012) 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sugiarto G, Lau K, Qu J, Li Y, Lim S, Mu S, Ames JB, Fisher AJ, Chen X, ACS Chem Biol, 7 (2012) 1232–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Santra A, Yu H, Tasnima N, Muthana MM, Li Y, Zeng J, Kenyond NJ, Louie AY, Chen X, Chem Sci, 7 (2016) 2827–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yu H, Li Y, Zeng J, Thon V, Nguyen DM, Ly T, Kuang HY, Ngo A, Chen X, J Org Chem, 81 (2016) 10809–10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wu Z, Liu Y, Ma C, Li L, Bai J, Byrd-Leotis L, Lasanajak Y, Guo Y, Wen L, Zhu H, Song J, Li Y, Steinhauer DA, Smith DF, Zhao B, Chen X, Guan W, Wang PG, Org Biomol Chem, 14 (2016) 11106–11116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wang W, Hu T, Frantom PA, Zheng T, Gerwe B, Del Amo DS, Garret S, Seidel RD 3rd, Wu P, Proc Natl Acad Sci U S A, 106 (2009) 16096–16101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Prudden AR, Chinoy ZS, Wolfert MA, Boons GJ, Chem Commun (Camb), 50 (2014) 7132–7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chen C, Zhang Y, Xue M, Liu XW, Li Y, Chen X, Wang PG, Wang F, Cao H, Chem Commun (Camb), 51 (2015) 7689–7692. [DOI] [PubMed] [Google Scholar]
- [21].Li L, Liu Y, Ma C, Qu J, Calderon AD, Wu B, Wei N, Wang X, Guo Y, Xiao Z, Song J, Sugiarto G, Li Y, Yu H, Chen X, Wang PG, Chem Sci, 6 (2015) 5652–5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang S, Zhang Q, Chen C, Guo Y, Gadi MR, Yu J, Westerlind U, Liu Y, Cao X, Wang PG, Li L, Angew Chem Int Ed Engl, 57 (2018) 9268–9273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Perret S, Sabin C, Dumon C, Pokorna M, Gautier C, Galanina O, Ilia S, Bovin N, Nicaise M, Desmadril M, Gilboa-Garber N, Wimmerova M, Mitchell EP, Imberty A, Biochem J, 389 (2005) 325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Donald AS, Feeney J, Carbohydr Res, 178 (1988) 79–91. [DOI] [PubMed] [Google Scholar]
- [25].Zibadi S, Watson RR, Preedy VR, Handbook of dietary and nutritional aspects of human breast milk, 1st ed., Wageningen Academic Publishers, Wageningen, 2013. [Google Scholar]
- [26].Engels L, Elling L, Glycobiology, 24 (2014) 170–178. [DOI] [PubMed] [Google Scholar]
- [27].Yu H, Li Y, Wu Z, Li L, Zeng J, Zhao C, Wu Y, Tasnima N, Wang J, Liu H, Gadi MR, Guan W, Wang PG, Chen X, Chem Commun (Camb), 53 (2017) 11012–11015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Albesa-Jove D, Giganti D, Jackson M, Alzari PM, Guerin ME, Glycobiology, 24 (2014) 108–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lu N, Ye J, Cheng J, Sasmal A, Liu CC, Yao W, Yan J, Khan N, Yi W, Varki A, Cao H, J Am Chem Soc, 141 (2019) 4547–4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yi W, Liu X, Li Y, Li J, Xia C, Zhou G, Zhang W, Zhao W, Chen X, Wang PG, Proc Natl Acad Sci U S A, 106 (2009) 4207–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chen Y, Thon V, Li Y, Yu H, Ding L, Lau K, Qu J, Hie L, Chen X, Chem Commun (Camb), 47 (2011)10815–10817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li Y, Xue M, Sheng X, Yu H, Zeng J, Thon V, Chen Y, Muthana MM, Wang PG, Chen X, Bioorg Med Chem, 24 (2016) 1696–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lau K, Thon V, Yu H, Ding L, Chen Y, Muthana MM, Wong D, Huang R, Chen X, Chem Commun (Camb), 46 (2010) 6066–6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhao G, Guan W, Cai L, Wang PG, Nat Protoc, 5 (2010) 636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ceroni A, Maass K, Geyer H, Geyer R, Dell A, Haslam SM, J Proteome Res, 7 (2008) 1650–1659. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.