

ABSTRACT
UVRAG (UV radiation resistance associated) is an important regulator of mammalian macroautophagy/autophagy by interacting with BECN1, PIK3C3, and RUBCN. Phosphorylation of UVRAG by MTORC1 negatively regulates autophagosome maturation under nutrient-enriched conditions. However, how UVRAG ubiquitination is regulated is still unknown. Here we report that UVRAG is ubiquitinated by SMURF1 at lysine residues 517 and 559, which decreases the association of UVRAG with RUBCN and promotes autophagosome maturation. However, the deubiquitinase ZRANB1 specifically cleaves SMURF1-induced K29 and K33-linked polyubiquitin chains from UVRAG, thereby increasing the binding of UVRAG to RUBCN and inhibiting autophagy flux. We also demonstrate that CSNK1A1-mediated UVRAG phosphorylation at Ser522 disrupts the binding of SMURF1 to UVRAG through PPxY motif and blocks UVRAG ubiquitination-mediated autophagosome maturation. Interestingly, ZRANB1 is phosphorylated at Thr35, and Ser209 residues by CSNK1A1, and this phosphorylation activates its deubiquitinating activity. Importantly, we provide in vitro and in vivo evidence that UVRAG ubiquitination at lysine residues 517 and 559 or prevention of Ser522 phosphorylation by D4476, a CSNK1A1 inhibitor, enhances the lysosomal degradation of EGFR, which significantly inhibits hepatocellular carcinoma (HCC) growth. Furthermore, UVRAG S522 phosphorylation levels correlate with ZRANB1 T35/S209 phosphorylation levels and poor prognosis in HCC patients. These findings identify a novel molecular mechanism by which ubiquitination and phosphorylation of UVRAG regulate its function in autophagosome maturation and HCC growth, encouraging further study of their potential therapeutic implications.
Abbreviations: ATG: autophagy related; BafA1: bafilomycin A1; BECN1: beclin 1; CHX: cycloheximide; CSNK1A1/CK1α: casein kinase 1 alpha 1; CQ: chloroquine; DUB: deubiquitinase; EBSS: Earle’s balanced salt solution; EGF: epidermal growth factor; GFP: green fluorescent protein; GST: glutathione S-transferase; HBSS: Hanks balanced salts solution; HCC: hepatocellular carcinoma; MAP1LC3B/LC3: microtubule associated protein 1 light chain 3 beta; MEFs: mouse embryo fibroblasts; mRFP: monomeric red fluorescent protein; PIK3C3/VPS34: phosphatidylinositol 3-kinase catalytic subunit type 3; PTMs: post-translational modifications; RUBCN: rubicon autophagy regulator; siRNA: small interfering RNA; SMURF1: SMAD specific E3 ubiquitin protein ligase 1; SQSTM1: sequestosome 1; Ub-AMC: ubiquitin-7-amido-4-methylcoumarin: a fluorogenic substrate; UVRAG: UV radiation resistance associated; ZRANB1/TRABID: zinc finger RANBP2-type containing 1
KEYWORDS: CSNK1A1/CK1α, phosphorylation, SMURF1, ubiquitination, UVRAG, ZRANB1/TRABID
Introduction
Autophagy, literally meaning ‘self-eating,’ is a conserved major cellular degradation pathway for damaged organelles and long-lived proteins [1]. It includes two key steps: 1) formation of double- membrane autophagosomes; 2) formation of a single-membrane autolysosome by the fusion of a mature autophagosome with a lysosome [2]. In response to changes in stress conditions and nutrient levels, autophagy is necessary to maintain the cellular homeostasis through degradation, recycling, and synthesis of cellular constituents [3]. Altered autophagy has been implicated in several major human diseases including neurodegenerative diseases, cancers, and immune disorders [1,4]. Since autophagy plays context-dependent roles in cancer, the clinical benefits of targeting autophagy may be unpredictable [4]. Hence, it is important to define the optimum cellular contexts or identify new biomarkers, which will be helpful in the therapeutic targeting of autophagy.
SMURF1 (SMAD specific E3 ubiquitin protein ligase 1) is a HECT-type ubiquitin ligase [5]. Previous studies indicate that SMURF1 regulates multiple biological networks including TGFB1 (transforming growth factor beta 1) and BMP signaling pathways, the Toll-like receptor pathway, and the mitogen-activated protein kinase pathway [6]. Therefore, SMURF1 is related to various diseases and disorders, such as embryonic development disorders and cancer [7]. Increasing evidence implies that SMURF1 could be a good candidate for further translational studies and a potential target for novel drug design. Recently, the role of SMURF1 in selective autophagy has been the subject of intensive study [8]. However, it is still mostly unclear about how SMURF1 affects the autophagy pathway. Thus, an in-depth studying of SMURF1 will help us to understand the etiopathological mechanisms of related disorders.
Recently, a kinome RNAi screen that identified CSNK (casein kinase) isoforms as constitutive autophagy-regulating kinases in human breast cancer cells [9]. The CSNK family is ubiquitously expressed and consists of 6 human isoforms (CSNK1A1/CK1α, CSNK1D/CK1δ, CSNK1E/CK1ϵ, CSNK1G1/CK1γ1, CSNK1G2/CK1γ2, and CSNK1G3/CK1γ3) [10]. All of them are evolutionarily conserved within eukaryotes. These isoforms regulate diverse cellular processes including RNA metabolism, DNA damage response, circadian rhythms, cytoskeleton maintenance, cell transformation, WNT signaling, and membrane trafficking [11]. Unlike its pro-oncogenic D/δ, E/ɛ, and G/γ isoforms, CSNK1A1/CK1α is a component of the CTNNB1/β-catenin destruction complex and thought to be mainly antiproliferative [12]. Depletion or pharmacologic inhibition of CSNK1A1 has been shown to increase autophagic flux in oncogenic RAS-driven human fibroblasts and multiple cancer cell lines [13]. CSNK1A1 also could increase phosphorylation of nuclear FOXO3A, thereby inhibiting transactivation of genes critical for RAS-induced autophagy [13]. However, so far the function of CSNK1A1 in the conversion of autophagosome to degradative autolysosome was unknown.
The deubiquitinase (DUB) ZRANB1/TRABID (zinc finger RANBP2-type containing 1), belongs to the A20 OTU family [14]. ZRANB1 has been reported to preferentially cleave K29-, K33-, and K63-linked ubiquitin chains [15]. Previous studies indicated that ZRANB1 functions as a positive regulator of WNT/CTNNB1 signal pathway, although this role of ZRANB1 remains controversial [15]. Recently, ZRANB1 has been shown to affect TLR4-mediated cellular immune and inflammation [14]. Given the tight relationship between the autophagy, inflammation, and tumor, it is possible that ZRANB1 plays a crucial role in autophagy system. However, so far, the involvement of ZRANB1 and underlying molecular mechanisms in autophagy pathway remains unknown.
For the first time, herein we show that UVRAG (UV radiation resistance associated) is ubiquitinated by SMURF1 and deubiquitinated by ZRANB1 at lysine residues 517 and 559. We also demonstrate that CSNK1A1 binds and phosphorylates UVRAG at Ser522, which has a positive effect on the interaction between UVRAG and RUBCN (rubicon autophagy regulator). Moreover, we provide in vitro and in vivo evidence that UVRAG ubiquitination at K517 and K559 or dephosphorylation at S522 significantly facilitates autophagosome maturation and lysosomal degradation of EGFR, reduces EGFR signaling, and suppresses hepatocellular carcinoma (HCC) cell proliferation and tumor growth in vivo. These results demonstrate that the CSNK1A1-ZRANB1-SMURF1 system is a previously unrecognized novel signaling network regulating the degree of autophagic flux via affecting the posttranslational modifications of UVRAG. Therefore, these findings offer insights into autophagy regulation and therapeutic combinations in HCC.
Results
UVRAG forms a complex with SMURF1 through the PPxY motif
In an attempt to elucidate the novel regulatory mechanism of UVRAG, we established a HEK293T derivative cell line with stably expressing Flag-HA double tagged-UVRAG. Tandem affinity purification (TAP) followed by mass spectrometry analysis allowed us to discover several UVRAG interacting proteins (Table S1). In addition to known UVRAG interactors, such as BECN1, RUBCN, and PIK3C3/VPS34 [16], we identified the E3 ubiquitin ligase SMURF1, as a potential new binding partner of UVRAG. We then set out to confirm whether the interaction exists between these two proteins. To this end, we co-expressed HA-UVRAG and MYC-SMURF1 in HEK293T cells and immunoprecipitated MYC-SMURF1. As shown in Figure 1(a), HA-UVRAG was co-immunoprecipitated with MYC-SMURF1. In a reciprocal coimmunoprecipitation experiment using an anti-HA antibody, similar results were obtained. We extended our analysis by investigating whether endogenous UVRAG and SMURF1 can interact with each other. When endogenous SMURF1 in Huh7 cells was immunoprecipitated by an anti-SMURF1 antibody, UVRAG was detectable in the immunoprecipitates by western blotting (Figure 1(b), left panel). Once endogenous UVRAG was immunoprecipitated, SMURF1 was also detectable in the immunoprecipitates (Figure 1(b), right panel). A GST affinity-isolation assay was performed to further confirm that SMURF1 interacts with GST-UVRAG, but not with GST alone (Figure 1(c)).
Figure 1.
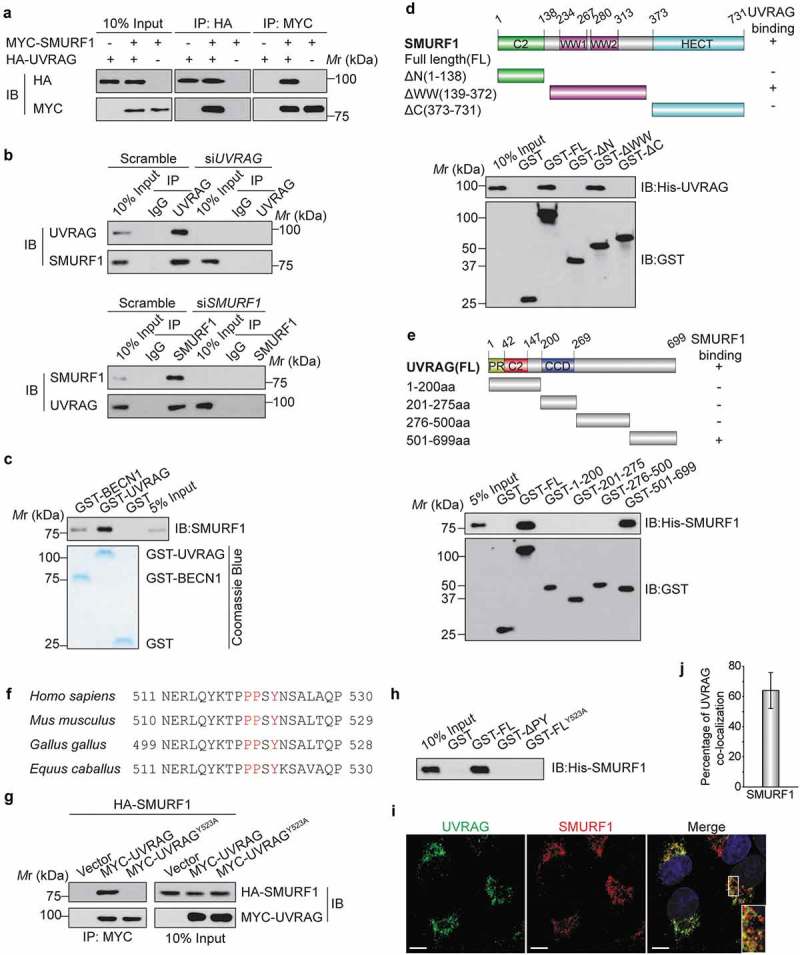
UVRAG forms a complex with SMURF1 through the PPxY motif. (a) HEK293T cells were co-transfected with HA-UVRAG and MYC-SMURF1 constructs. 24 h later, cell lysates were used for co-immunoprecipitation with anti-HA or anti- MYC antibodies and WB analyses. (b) Endogenous SMURF1 and UVRAG proteins interact with each other in Huh7 cells. Upper panel, Huh7 cell lysates were incubated with protein A/G Sepharose conjugated with either control IgG or UVRAG antibody. The immunoprecipitates were analyzed using western blotting with indicated antibodies. Lower panel, a similar assay was performed as the upper panel but immunoprecipitated using anti-SMURF1 antibody. (c) Recombinant SMURF1 protein was incubated with GST-tagged UVRAG, BECN1 (positive control), or GST protein. GST affinity-isolation assay was performed with glutathione-agarose and blotted as indicated. (d) Bacterially expressed GST fusion proteins of wild-type (WT), C2 domain (C2), WW domain (WW), HECT domain (HECT) mutants of SMURF1 were bound to glutathione-Sepharose beads and incubated with cell lysates of HEK293T cells transfected with the His-UVRAG. Numbers represent the amino acid (aa) residues in human SMURF1. The interaction between UVRAG and SMURF1 domains is indicated by the plus signs (+). Bound His-UVRAG was subjected to WB analyses with an anti-His antibody. (e) Various deletion mutation constructs of UVRAG are shown schematically (PR: proline-rich domain; CCD: coiled coil domain). GST-UVRAG FL or fragments were incubated with His-SMURF1, and western blotting was performed to detect the interaction with an anti-His antibody. (f) Protein sequence alignment of UVRAG orthologues from the different species and the conserved PPxY motif was highlighted in red color. (g) MYC-UVRAG, the WW-binding-defective mutant UVRAGY523A, or the vector was cotransfected with HA-tagged SMURF1 into HEK293T cells. UVRAG or UVRAGY523A was immunoprecipitated with anti-MYC antibody. Coprecipitated SMURF1 was detected by immunoblotting with an anti-HA antibody. The expression levels of indicated proteins in the cell lysates are shown. (h) Bacterially expressed GST fusion proteins of UVRAG deletion mutants were bound to glutathione-Sepharose beads as indicated and incubated with cell lysates of HEK293T cells transfected with His-SMURF1. The sample was subjected to WB analyses with indicated antibody. (i) The subcellular colocalization of overexpressed Flag-UVRAG and MYC-SMURF1 were detected by immunofluorescence in HEK293T cells subjected to glucose deprivation. Nuclei were stained with DAPI. The insets show a high magnification of the selected areas. Scale bars: 10 µm. (j) The quantitation analysis of the co-localization of UVRAG and SMURF1 in Figure 1(i). The quantitative values are expressed as the mean ± SD (n = 100 cells) obtained from three independent experiments).
To further map the domains of SMURF1 for UVRAG binding, we constructed and purified the full-length (FL) and three deletion fragments of GST-SMURF1 and then performed a GST affinity-isolation assay. As shown in Figure 1(d), SMURF1-WW domain, but not the C2 domain (C2), or HECT domain (HECT) mutant interacted with UVRAG. Thus, SMURF1 is capable of interacting with UVRAG through two WW domains. Next, the regions of UVRAG for SMURF1 binding were mapped by incubating the FL or fragments of GST-UVRAG with His-SMURF1. His-SMURF1 mainly interacted with the FL and the C-terminal fragment (501–699 aa) of GST-UVRAG, but not with the N-terminal fragment of UVRAG and GST alone (Figure 1(e)). All of the above data indicate that SMURF1 directly interacts with UVRAG.
SMURF1 belongs to the WW domain-containing family of E3 ubiquitin ligases that binds to the PPxY motif in its regulatory proteins or ubiquitination substrates [6,7]. UVRAG protein possesses a PPxY motif, which is highly conserved across a wide range of metazoans (Figure 1(f)). To determine whether the PPxY motif is the site of UVRAG interacting with SMURF1, we mutated Y523 to alanine, changing PPSY to PPSA, and then examined coimmunoprecipitation with SMURF1. As shown in Figure 1(g), while wild-type (WT) UVRAG coprecipitated SMURF1, the Y523A mutation eliminated the interaction of UVRAG with SMURF1, indicating that Y523 is required for UVRAG to bind to SMURF1. Moreover, an affinity-isolation assay showed that His-SMURF1 could pull down recombinant expressed GST-UVRAG, while deletion of the PPxY motif (ΔPY) or Y523 to alanine mutant entirely abolished the interaction between UVRAG and His-SMURF1 (Figure 1(h)). These data suggest the PPxY motif in UVRAG is indispensable for SMURF1 binding, and UVRAG can directly interact with SMURF1 in vitro in a PPxY motif-dependent manner.
To further verify the interaction between UVRAG and SMURF1, we examined whether these two proteins are localized to the same subcellular compartments. Because of difficulties in reliably detecting endogenous SMURF1 and UVRAG in HEK293T cells, we evaluated the colocalization of epitope-tagged proteins: MYC-SMURF1 and Flag-UVRAG. Using confocal microscopy, we observed extensive cytoplasmic colocalization between MYC-SMURF1 and Flag-UVRAG in transfected HEK293T cells subjected to glucose deprivation compared to the complete medium (Figures 1(i) and S1(a)). The quantitative analysis further confirmed a partial overlap between UVRAG and SMURF1 (Figure 1(j)). Together, these results indicated that UVRAG forms a complex with SMURF1 in vivo.
SMURF1 catalyzes K29- and k33-linked polyubiquitination of UVRAG
Given that SMURF1 is a HECT-type ubiquitin ligase that likely functions through ubiquitination of target proteins [5], we hypothesized that SMURF1 directly ubiquitinates UVRAG and that this ubiquitination regulates its function. To test this hypothesis, we assessed SMURF1-mediated ubiquitination of UVRAG. We found that coexpression of MYC-UVRAG and HA-ubiquitin with the wild-type SMURF1 significantly enhanced the polyubiquitination of UVRAG in HEK293T cells, while the ligase-dead SMURF1 mutant (SMURF1C699A) lost the ability to increase the polyubiquitination of UVRAG (Figure 2(a)). Silencing SMURF1 abolished the polyubiquitination of UVRAG (Figure 2(b)), indicating that UVRAG may be a substrate of SMURF1 and the E3 ligase activity of SMURF1 is required for the polyubiquitination of UVRAG.
Figure 2.
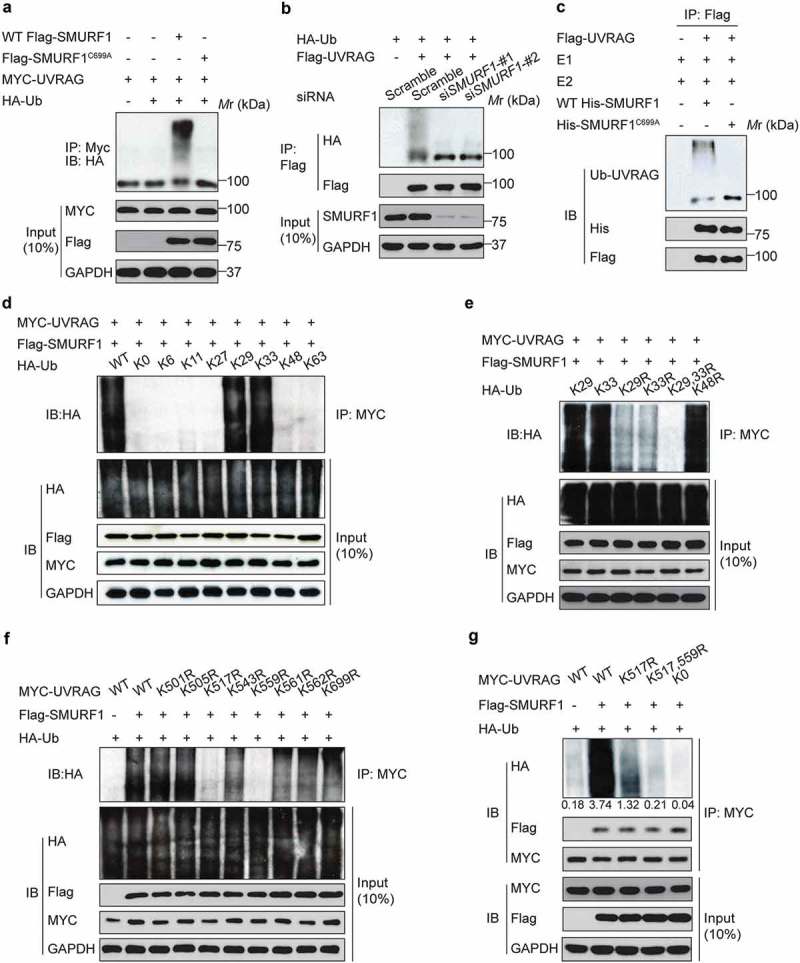
SMURF1 catalyzes the polyubiquitination of UVRAG via atypical chains K29 and K33. (a) Co-immunoprecipitation analysis of the UVRAG ubiquitination in HEK293T cells transfected with indicated plasmids. (b) UVRAG, SMURF1, or SMURF1 siRNA (#1 and #2) were introduced into HEK293 cells. Cells were treated with MG132. After 48 h, cell lysates were prepared for IP with an Ub antibody and IB against UVRAG. (c) In vitro ubiquitination assay of UVRAG by SMURF1 was carried out and analyzed using western blotting. (d) HA-ubiquitin mutants with only the indicated lysine residue were used, and an in vivo ubiquitin assay was performed. (e) According to the results from D, we mutated the lysine at K29, K33 or both K29 and K33 (K29R, K33R and K29, 33R) to arginine, and performed the in vivo ubiquitin assay. (f and g) Co-immunoprecipitation (co-IP) analysis of the polyubiquitination of WT-UVRAG and its mutants in HEK293T cells transfected with indicated plasmids. And the intensity of the western blot bands was quantified using NIH ImageJ software. In addition, co-IP assay indicates that K517 or K559 residue mutation to R does not block SMURF binding to UVRAG.
To further validate that SMURF1 can directly ubiquitinate UVRAG, we utilized an in vitro ubiquitination assay with HA-ubiquitin, His-SMURF1, Flag-UVRAG, and recombinant E1, E2 enzymes. Polyubiquitination of UVRAG was observed only in the presence of all defined proteins, while SMURF1C699A was unable to drive UVRAG ubiquitination under the same conditions (Figure 2(c)). Thus, these data confirm that SMURF1 directly ubiquitinates UVRAG. Also, it was found that the ubiquitination of UVARG by SMURF1 was significantly enhanced in HEK293T cells subjected to glucose depletion (Figure S1(b)).
We then sought to determine the type of SMURF1-mediated polyubiquitination of UVRAG. To this end, UVRAG and SMURF1 were cotransfected with individual ubiquitin constructs which could only form ubiquitin linkages at a single lysine. For example, K63 indicates every lysine except K63 changed to arginine (R), and K0 represented a construct that has no lysines available. It was observed that SMURF1 ubiquitinates UVRAG primarily through chains K29 and K33 (Figure 2(d)).
To further confirm these significant linkages, we mutated the lysines at K29 alone, K33 alone or both K29 and K33 to arginines and performed the assay described above. As shown in Figure 2(e), once K29 and K33 were both mutated to arginines (K29, 33R), SMURF1 no longer ubiquitinated UVRAG. Interestingly, K48R does not decrease ubiquitination, suggesting that SMURF1 does not target UVRAG to the proteasome-dependent degradation. Therefore, these data demonstrate that K29 and K33 are the predominant linkages.
To map the UVRAG ubiquitination sites, we generated a series of UVRAG mutants in which all lysine residues in a defined region are replaced with arginine. It was found that only mutation of all lysine residues in the 501–699 area (K501-699R) abolished UVRAG ubiquitination (data not shown), indicating that UVRAG ubiquitination sites are located in this region. The 501–699 region of UVRAG has 8 conserved lysine residues. To identify the lysine residues responsible for SMURF1-mediated polyubiquitination, we first generated the mutant UVRAG-K0, in which all of the lysine residues in UVRAG were replaced with arginine. Then, we reintroduced individual lysine residues into UVRAG-K0 to generate the single-lysine mutants. We observed that SMURF1 induced polyubiquitination of WT UVRAG and the UVRAGK517R and UVRAGK559R mutants (Figure 2(f)). Notably, compared with polyubiquitination of cells expressing WT UVRAG, SMURF1-induced that of the UVRAG mutants was decreased gradually with an increase in the number of lysine-to-arginine substitutions (Figure 2(g)). Interestingly, the K29- and K33-linked ubiquitination of UVRAGK517,559R dropped to a level similar to that of UVRAG-K0 (Figure 2(g)). Our data also indicate that K517 or K559 residue mutation to R does not block SMURF binding to UVRAG (Figure 2(g)). Together, these data demonstrated that SMURF1 catalyzes the K29- and K33-linked polyubiquitination of UVRAG on K517 and K559.
SMURF1-mediated UVRAG ubiquitination does not affect its stability
Given that SMURF1 has been reported to target many proteins for their degradation [6], we then focused our attention on exploring a possible role of SMURF1-mediated UVRAG ubiquitination. Surprisingly, when SMURF1 was overexpressed, no apparent decrease of UVRAG protein levels was detected (Figure 3(a)). Similarly, silencing SMURF1 did not affect UVRAG levels. We next used the proteasome inhibitor MG132. As shown in Figure 3(b), MG132 did not affect SMURF1-mediated UVRAG ubiquitination, indicating that the latter is not subject to proteasome-dependent degradation. We then measured whether the turnover rate of UVRAG is modulated by SMURF1 through cycloheximide (CHX) treatment, a standard method of blocking protein synthesis. As shown in Figure 3(c), the half-life of UVRAG was not reduced in SMURF1-overexpressed cells. Finally, SMURF1 overexpression also did not change the levels of UVRAG mRNA (Figure 3(d)). With these results combined, we conclude that SMURF1 does not target UVRAG for proteasome-dependent degradation.
Figure 3.
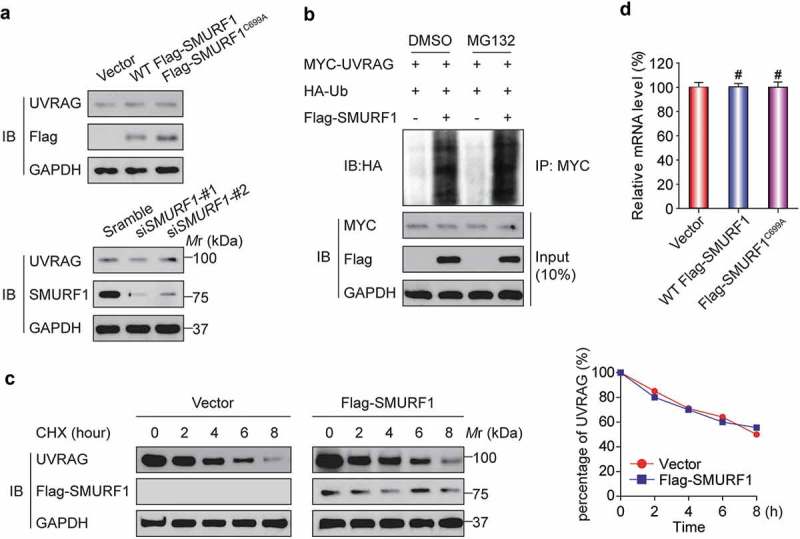
SMURF1-mediated UVRAG ubiquitination does not affect its stability. (a) No apparent change of UVRAG protein levels was detected in SMURF1-overexpressing or SMURF1-depleted cells. HEK293T cells were transfected with indicated siRNAs (lower) or plasmids (upper), and endogenous proteins were examined by western blotting. (b) MG132 did not change SMURF1-mediated UVRAG ubiquitination levels. In vivo ubiquitination assays were performed in HEK293T cells transfected with the indicated plasmids. Cells were treated with DMSO or MG132 (15 μM) for 4 h before harvest. (c) SMURF1 did not affect the turnover of UVRAG protein. HEK293T cells transfected with the indicated plasmids were treated with 15 μg/ml cycloheximide (CHX) for various times, and endogenous UVRAG was analyzed by western blotting. The graph in panel C represents the results from three independent experiments. (d) SMURF1 did not affect the levels of UVRAG mRNA. HEK293T cells were transfected with the indicated plasmids, and endogenous UVRAG was analyzed by qRT-PCR and shown.
CSNK1A1 phosphorylates UVRAG at Ser522
Because the PPxY motif in UVRAG is indispensable for SMURF1 binding and the conserved serine residue at 522 is exactly located within the PPxY motif (Figure 4(a)), we reasoned that S522 phosphorylation of UVRAG might influence the biological functions of SMURF1–mediated ubiquitination of UVRAG. To test this idea, we firstly aimed to identify the kinase responsible for the phosphorylation of serine residue at 522 on UVRAG. The residues surrounding S522 of UVRAG (PSYN/KSA), with acidic residues at +1 and +4, conform to a putative protein kinase CK1 phospho-motif (p-S/TXXS/T) [13], which makes S522 an optimal site for phosphorylation by CK1 (Figure 4(a)). Thus, we investigated whether CK1 mediated the phosphorylation of UVRAG at S522 and its potential functional relevance in various cell types including HCC cells.
Figure 4.
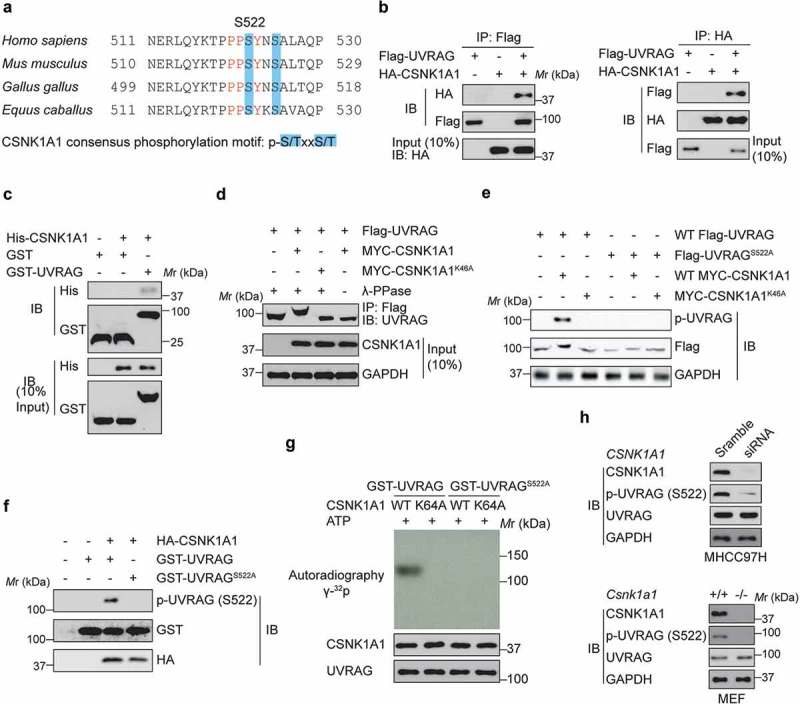
CSNK1A1 phosphorylates UVRAG at S522. (a) UVRAG consensus phosphorylation site (shown in blue) corresponding to the CSNK1A1 consensus motifs S/TXXS/T are presented. These consensus phosphorylation sites are exactly located within the consensus sequences of PPxY. (b) Flag-UVRAG was present in the HA-CSNK1A1 immunocomplex. Lysates were extracted from 293T cells transiently co-transfected with Flag-UVRAG and HA-Vector and HA-CSNK1A1, and immunoprecipitation was performed. (c) In vitro GST affinity-isolation assay as indicated. (d) Immunoprecipitation and immunoblot analysis of lysates from HEK293T cells expressing WT Flag-UVRAG and MYC-CSNK1A1 or MYC-CSNK1A1K46A with or without the phosphatase λ-PPase. GAPDH serves as a loading control throughout. (e) Immunoblot analysis of UVRAG and CSNK1A1 in lysates of HEK293T cells expressing WT Flag-UVRAG or Flag-UVRAGS522A and WT MYC-CSNK1A1 or MYC-CSNK1A1K46A. (f) Immunoblot analysis of phosphorylated p-UVRAG (S522) and total UVRAG and CSNK1A1 in HEK293T cell lysates. (g) In vivo kinase assay was carried out using UVRAG and its mutant proteins in the presence of labeled 32P-ATP and detected by SDS-PAGE and autoradiography. (h) The endogenous UVRAG phosphorylation in the Csnk1a1+/+ and csnk1a1−/- MEFs or MHCC97H cell lines with CSNK1A1 knockdown was examined using p-S522-UVRAG antibody.
The CSNK1 family includes six human isoforms [10]. Because siRNA-mediated knockdown of CSNK1E or CSNK1D or inhibition of their kinase activity by potent small molecules did not markedly change UVRAG phospho-serine abundance, it is possible that CSNK1A1 kinase activity is required for the regulation of UVRAG phosphorylation at S522. As expected, we confirmed that HA-CSNK1A1 physically associated with Flag-UVRAG (Figure 4(b)). We also found that endogenous UVRAG in HEK293T cells co-immunoprecipitated with endogenous CSNK1A1 (data not shown). Purified His-CSNK1A1 was able to pull down bacterially produced GST-UVRAG (Figure 4(c)), indicating that the interaction between UVRAG and CSNK1A1 is direct.
We then investigated the effect of CSNK1A1 on UVRAG. When co-expressed with WT CSNK1A1, but not CSNK1A1K46A, a kinase-inactive mutant of CSNK1A1 [17], UVRAG was found to migrate more slowly only in the presence of CSNK1A1 than did UVRAG on its own by SDS-PAGE (Figure 4(d)), suggesting that this differential migration was the result of phosphorylation. Given that S522 residue is conserved in UVRAG from various species and is homologous to the CSNK1A1-phosphorylated substrate motif (Figure 4(a)), we reasoned S522 is a CSNK1A1-dependent phosphorylation site. As expected, substitution of the serine at position 522 with alanine (S522A) abolished the CSNK1A1-induced slower migration of UVRAG by SDS-PAGE (Figure 4(e)). To further study the phosphorylation of UVRAG by CSNK1A1, we generated a rabbit polyclonal antibody to UVRAG phosphorylated at S522 (p-S522) by immunizing rabbits with the UVRAG peptide (data not shown). Co-expression of wild-type CSNK1A1, not CSNK1A1K46A, with UVRAG induced phosphorylation of UVRAG at S522, and the S522A substitution in UVRAG eliminated the phosphorylation of UVRAG by CSNK1A1 (Figure 4(e)). In an in vitro kinase assay, purified UVRAG was phosphorylated by CSNK1A1, but mutant UVRAGS522A did not (Figure 4(f)). In vitro kinase assay further indicated that the kinase-dead CSNK1A1 could not phosphorylate WT-UVRAG (Figure 4(g)). Moreover, by using the Csnk1a1+/+ and csnk1a1−/- MEFs or MHCC97H cell lines with CSNK1A1 knockdown, we provided the evidence that CSNK1A1 knockout or knockdown significantly decreased the endogenous UVRAG phosphorylation at S522 residue (Figure 4(h)). Collectively, our results suggest that CSNK1A1 directly phosphorylated UVRAG at S522.
Phosphorylation of UVRAG at S522 by CSNK1A1 inhibits SMURF1-mediated ubiquitination
To understand the functional consequence of the UVRAG phosphorylation at S522, we investigated whether the phosphorylation status could regulate the SMURF1-UVRAG interaction. Indeed, co-expression of CSNK1A1 with UVRAG and SMURF1 markedly inhibited the binding of UVRAG to SMURF1 in HEK293T cells, but co-expression of the kinase-inactive CSNK1A1K46A mutant did not (Figure 5(a)). Consistent with this finding, more UVRAGS522A than WT UVRAG or UVRAGS522E interacted with SMURF1 (Figure 5(bi)), which was further confirmed by proximity ligation (PLA) assay (Figure 5(bii)). Our data suggest that the binding of SMURF1 to UVRAG requires dephosphorylation of UVRAG at S522.
Figure 5.
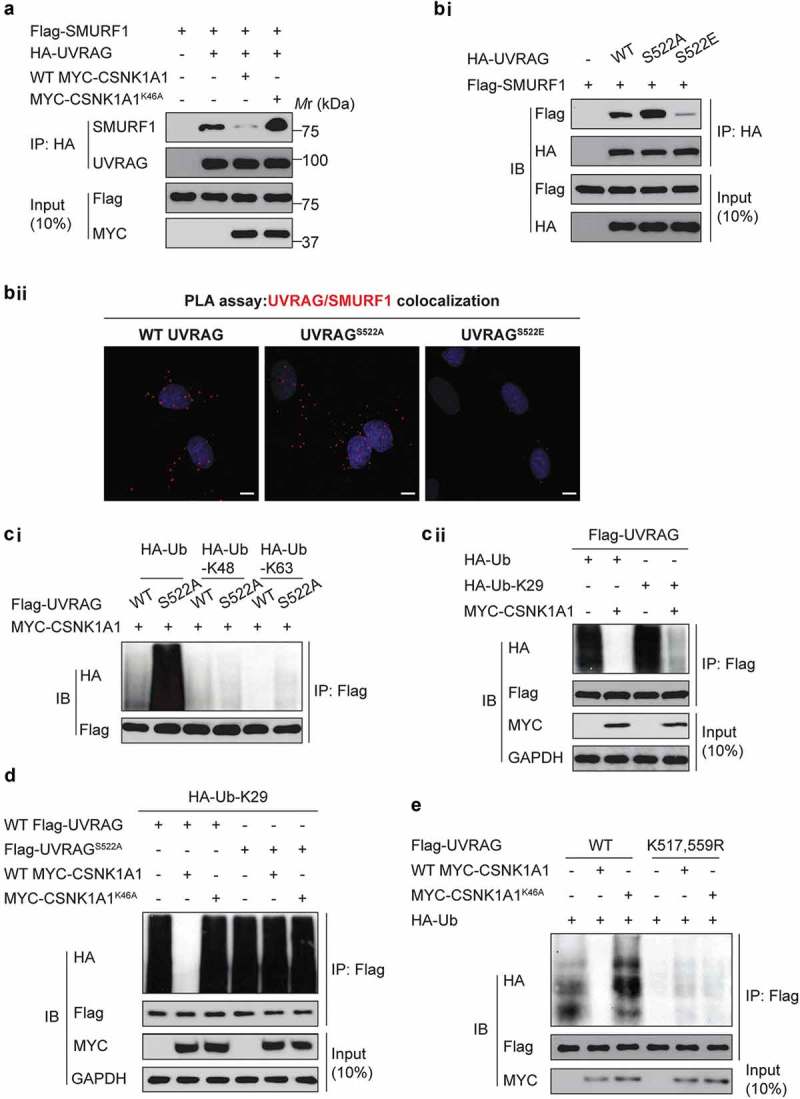
Phosphorylation of UVRAG at S522 inhibits SMURF1-mediated ubiquitination. (a) Immunoprecipitation and immunoblot analysis using HEK293T cells transfected with various combinations of plasmids as indicated. (bi) Immunoprecipitation and immunoblot analysis using HEK293T cells transfected with plasmid encoding empty vector, or WT HA-UVRAG, HA-UVRAGS522A, or HA-UVRAGS522E, plus Flag-SMURF1. (bii) Proximity ligation (PLA) assay was performed to confirm the interaction between SMURF1, UVRAG and their mutants. Scale bars: 10 µm. (ci) Immunoprecipitation and immunoblot analysis using HEK293T cells transfected with plasmid encoding HA-ubiquitin, or K63, or K48-linked ubiquitin with WT UVRAG or UVRAGS522A and MYC-CSNK1A1. (cii) Immunoprecipitation and immunoblot analysis using HEK293T cells transfected with plasmid encoding HA-K29-linked ubiquitin, with or without MYC-CSNK1A1. (d) Immunoprecipitation and immunoblot analysis using HEK293T cells transfected with indicated plasmids. (e) Overexpression of CSNK1A1 inhibits the ubiquitination of WT-UVRAG but not the K517R K559R mutant. Indicated plasmids were co-transfected with MYC-CSNK1A1 or vector into HEK293T cells.
Phosphorylation and ubiquitination are connected either negatively or positively [18]. As expected, although no substantial effect of CSNK1A1 on the K48- and K63-linked ubiquitination of UVRAG was observed (Figure 5(ci)), overexpression of CSNK1A1 markedly inhibited the K29- and K33-linked ubiquitination of UVRAG in HEK293T cells (Figure 5(cii)). Moreover, the S522A substitution of UVRAG markedly increased the K29- and K33-linked ubiquitination of UVRAG in HEK293T cells (Figure 5(d)), suggesting that phosphorylation of UVRAG at S522 by CSNK1A1 was critical for the K29- and K33-linked ubiquitination of UVRAG.
Moreover, the overexpression of CSNK1A1 decreased the ubiquitination of WT UVRAG but not K517R K559R mutant ubiquitination (Figure 5(e)). These results suggest that the CSNK1A1-mediated phosphorylation of UVRAG may reduce the subsequent interaction between UVRAG and SMURF1, thus inhibiting UVRAG ubiquitination. Since CSNK1A1-mediated phosphorylation of UVRAG may reduce the subsequent interaction between UVRAG and SMURF1, we reasoned that SMURF and CSNK1A1 molecules are mutually exclusive in binding with UVRAG. Indeed, as shown in Figure S2, SMURF and CSNK1A1 molecules are mutually exclusive in binding with UVRAG.
ZRANB1 antagonizes SMURF1-mediated K29- and K33-linked ubiquitination of UVRAG
Next, we wanted to identify the deubiquitinase (DUB) that targets K517 and K559 on UVRAG. Our mass spectrometric analysis of the UVRAG immunocomplex from HEK293T cells uncovered that ZRANB1 was present in the immunocomplex (Table S1). Given that ZRANB1 has been reported to preferentially cleaves K29-, K33-, and K63-linked ubiquitin chains [15], we hypothesized that ZRANB1 might reverse SMURF1-induced UVRAG ubiquitination. Next, we directly characterized the effect of ZRANB1 on ubiquitination of UVRAG. We found that inhibition of ZRANB1 by siRNA knockdown led to increases in the levels of K29- and K33-, but not K48-linked ubiquitination on endogenous UVRAG (Figure 6(a)). Conversely, overexpression of ZRANB1 significantly reduced the K29- and K33-, but not K48-, ubiquitination levels of UVRAG (Figure 6(b)). To demonstrate that ZRANB1 can directly deubiquitinate UVRAG K29- and K33-linked ubiquitination, we performed in vitro deubiquitination assays. Incubation of purified WT ZRANB1, but not an inactive ZRANB1C443A mutant, with K29- and K33-linked ubiquitinated UVRAG in vitro led to its deubiquitination (Figure 6(c)). These results suggest that ZRANB1 might play a role in regulating autophagy through inhibiting the UVRAG ubiquitination.
Figure 6.
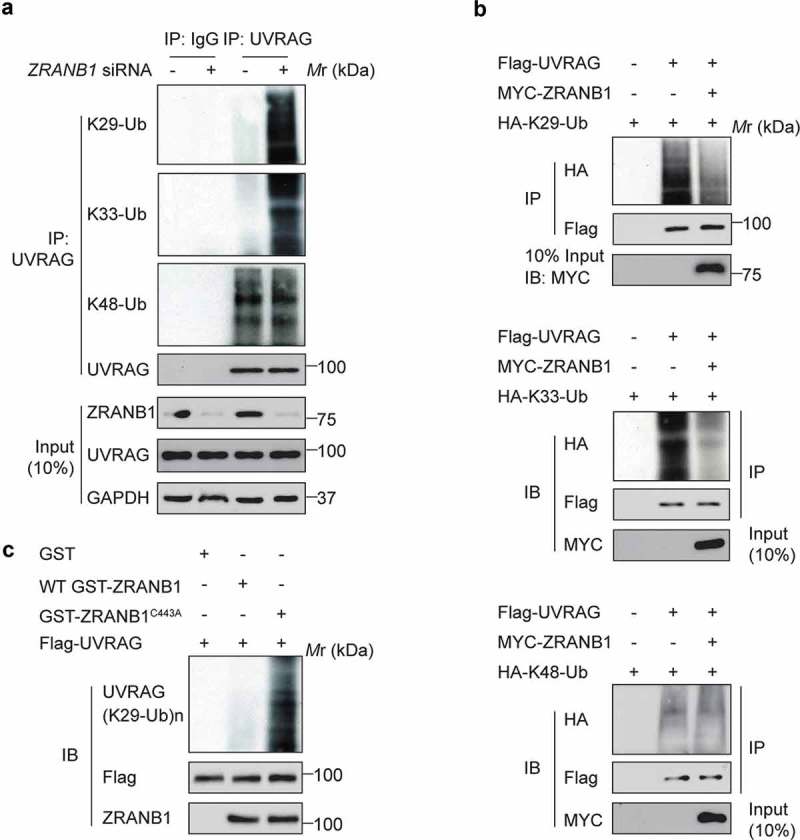
ZRANB1 removes UVRAG K29- and K33-linked ubiquitination. (a) Huh7 cells were transfected with siRNA pools against ZRANB1 for 72 h, and the lysates were incubated with anti-UVRAG antibody followed by western blot analysis to determine the K29-, K33-, and K48-linked ubiquitination on UVRAG. (b) ZRANB1 reduced the K29- and K33-, but not K48-linked ubiquitination levels of UVRAG. HEK293T cells were transfected with HA-K29-only ubiquitin, or K33-only ubiquitin, or K48-only ubiquitin and Flag-UVRAG as indicated with or without MYC-ZRANB1. 24 h later, cell lysates were used to perform an immunoprecipitation with anti-Flag antibody, and the samples were analyzed with an anti-HA antibody. (c) In vitro K29-linked deubiquitination of UVRAG by ZRANB1. K29-ubiquitinated UVRAG was incubated with purified recombinant ZRANB1 or ZRANB1C443A in vitro and then blotted with an anti-HA antibody.
CSNK1A1-induced phosphorylation regulates ZRANB1 DUB activity toward UVRAG
Since both CSNK1A1 and ZRANB1 inhibited SMURF1–mediated UVRAG ubiquitination, we next investigated the possible relationship between ZRANB1 and CSNK1A1. Among ZRANB1 orthologs, the amino acid sequences around Thr35 and Ser209 are highly evolutionarily conserved (Figure 7(a)), and both of them are predicted to be CSNK1A1 phosphorylation sites by Scansite. We, therefore, reasoned that ZRANB1 might be a substrate of CSNK1A1. In vitro kinase assay clearly demonstrated that co-incubation of recombinant ZRANB1 and CSNK1A1 led to ZRANB1 phosphorylation (Figure 7(bi)). As compared to WT ZRANB1, either T35A or S209A mutant showed partially decreased phosphorylation, while that of T35A, S209A mutant was completely abolished (Figure 7(bii)), suggesting both T35 and S209 as major phosphorylation sites of CSNK1A1. These results indicate that ZRANB1 is a CSNK1A1 substrate.
Figure 7.
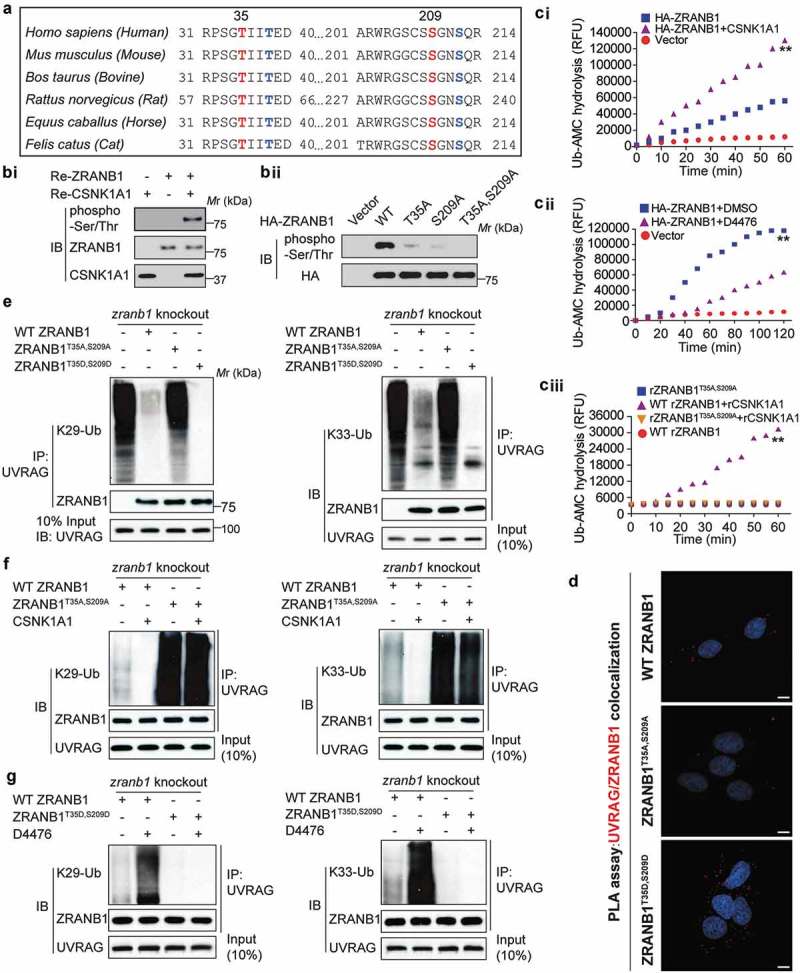
ZRANB1 is a CSNK1A1 substrate, and this phosphorylation activates the DUB activity of ZRANB1. (a) Putative CSNK1A1 phosphorylation sites predicted by Scansite within ZRANB1 orthologs. (bi) The phosphorylation of ZRANB1 was found using a phospho-Ser/Thr antibody. (bii) HEK293T cells were transfected with wild-type or mutant ZRANB1 constructs, then phosphorylation of ZRANB1 was detected using the phospho-Ser/Thr antibody. (ci and cii) CSNK1A1 activates ZRANB1 in cells. ZRANB1 was immunoprecipitated from HEK293T cells coexpressed with CSNK1A1 (ci) or treated with 10 μM D4476 (cii), and followed by Ub-AMC hydrolysis assay. (ciii) T35A S209A mutation blocks ZRANB1 activation by CSNK1A1. Recombinant ZRANB1 protein, T35A S209A mutation was mixed with or without CSNK1A1 in vitro, and Ub-AMC assay was performed. RFU represents relative fluorescence units. (d) Proximity ligation assay (PLA) was performed to confirm the interaction between UVRAG, ZRANB1 and their mutants. Scale bars: 10 µm. (e) zranb1−/− MEF cells were virally transfected with indicated plasmids, and the K29- and K33-ubiquitination of UVRAG was analyzed. (f) zranb1−/− MEF cells were virally transfected with indicated plasmids with or without CSNK1A1, and the K29- and K33-ubiquitination of UVRAG was analyzed. (g) zranb1−/− MEF cells were virally transfected with indicated plasmids with or without 10 μM D4476, and the K29- and K33-ubiquitination of UVRAG was analyzed.
To examine whether CSNK1A1-induced phosphorylation might affect the DUB activity of ZRANB1, we performed an Ub-AMC (ubiquitin-7-amido-4-methylcoumarin, a fluorogenic substrate) hydrolysis assay [19]. As shown in Figure S3, either purified ZRANB1 or CSNK1A1 alone showed trace hydrolyzing activity towards Ub-AMC, while ZRANB1 co-incubated with CSNK1A1 indicated a high activity (Figure S3). In vivo, ZRANB1 from HEK293T cells co-expressing CSNK1A1 showed higher activity than that expressed alone (Figure 7(ci)), whereas ZRANB1 activity in the Ub-AMC assay was reduced by D4476, a CSNK1A1 inhibitor (Figure 7(cii)). Notably, the stimulating effect of CSNK1A1 on the activity of ZRANB1 was substantially inhibited by T35A, S209A mutation (Figure 7(ciii)), suggesting that the phosphorylation of ZRANB1 at T35 and S209 is an important regulatory mechanism for its activity. Additionally, PLA assay indicated that CSNK1A1-induced phosphorylation of ZRANB1 promoted its binding to UVRAG (Figure 7(d)).
Then we studied whether phosphorylation of ZRANB1 by CSNK1A1 was required for its DUB activity toward K29- and K33-linked ubiquitination of UVRAG. As shown in Figure 7(e), either WT ZRANB1 or the ZRANB1T35D,S209D mutant, but not the ZRANB1T35A,S209A mutant, was able to inhibit the K29- and K33-linked ubiquitination of UVRAG. Even in the presence of CSNK1A1, ZRANB1T35A,S209A mutant failed to inhibit ubiquitination of UVRAG (Figure 7(f)). Upon inhibition of CSNK1A1 using D4476 compound [20], WT-ZRANB1 lost its deubiquitination activity toward UVRAG, while the ZRANB1T35D,S209D mutant was still able to reduce ubiquitination level of UVRAG (Figure 7(g)), indicating that the phosphorylation of ZRANB1 induced by CSNK1A1 promotes its K29- and K33-linked deubiquitinating activity toward UVRAG.
UVRAGK517,559R mutant or CSNK1A1-mediated phosphorylation at Ser522 inhibits autophagosome maturation by promoting the UVRAG-RUBCN interaction
Given that UVRAG plays a critical role in both autophagy and endocytosis by forming a set of interacting components [21], we hypothesized that the ubiquitination and phosphorylation of UVRAG might alter the UVRAG complex. To test this hypothesis, we expressed WT UVRAG, and UVRAGK517,559R, UVRAGS522A, or UVRAGS522D (a mutant with a replacement of S522 with aspartate) in HEK293T cells where endogenous UVRAG was silenced by shRNA. Then we examined the interaction between UVRAG and its binding partners in HEK293T cells. Compared to WT, UVRAGK517,559R did not affect the interaction of UVRAG with BECN1, while it increased the co-immunoprecipitation of UVRAG and RUBCN, which is a negative regulator of UVRAG activity (Figure 8(a)). Because RUBCN can inhibit the maturation of autophagosomes through binding to the UVRAG-PI3KC3 complex, and PI3KC3 consists of three core components, PIK3C3/VPS34, PIK3R4/p150 and BECN1 [22], we evaluated the effect of UVRAG mutants on the PIK3C3-RUBCN interaction. As shown in Figure 8(b), the UVRAGK517,559R mutant significantly increased the PIK3C3-RUBCN interaction. These findings were further confirmed when we used ZRANB1 MEF cells. We found that the interaction of UVRAG with RUBCN in zranb1−/- MEF cells was significantly decreased compared with that of Zranb1+/+ MEF cells (Figure S4), suggesting that ZRANB1 may have effect in inhibiting autophagosome maturation through regulating the UVRAG-containing PIK3C3 complex. Since UVRAG is one of the key components of the BECN1- PIK3C3 complex, we also examined whether UVRAG S522 phosphorylation by CSNK1A1 or ubiquitination by SMURF1 regulates BECN1- PIK3C3 activity. As shown in Figure S5(a,c), UVRAG-S522 phosphorylation by CSNK1A1 or ubiquitination by SMURF1 does not regulate BECN1- PIK3C3 interaction. However, UVRAG-S522 phosphorylation by CSNK1A1 decreases the PIK3C3 activity whereas ubiquitination by SMURF1 has the opposite effect. These findings suggest that UVRAGK517,559R deubiquitination or S522 phosphorylation might mainly promote binding of RUBCN to the UVRAG-PIK3C3 complex.
Figure 8.
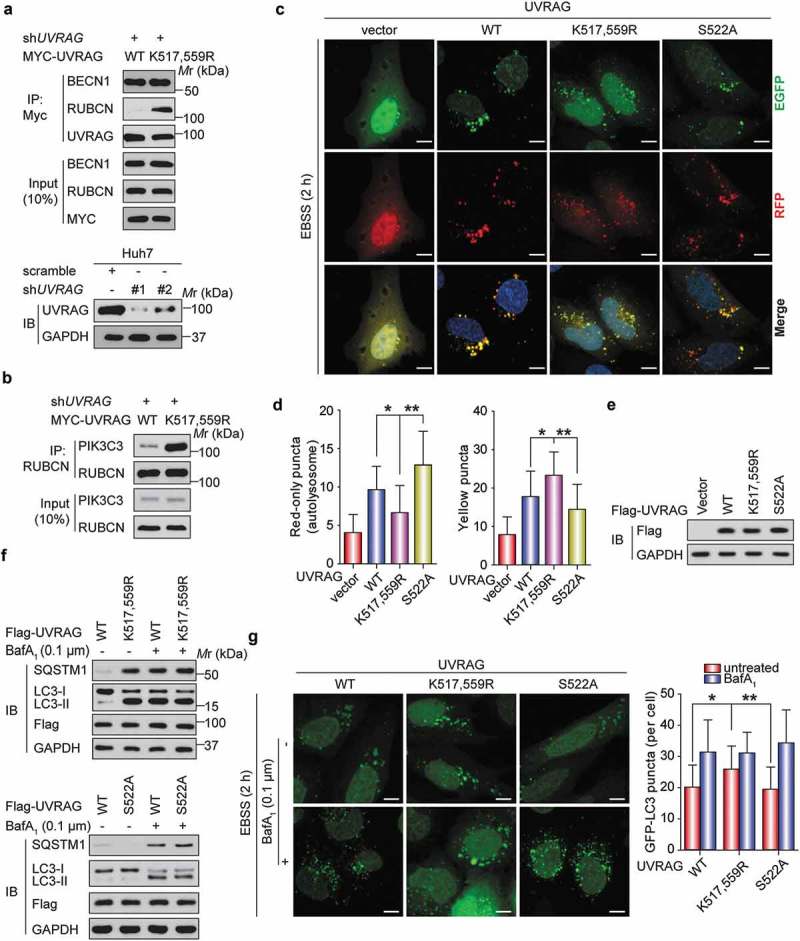
UVRAGK517,559R mutant or CSNK1A1-mediated phosphorylation at S522 inhibits autophagosome maturation by promoting the UVRAG-RUBCN interaction. (a) WT MYC-UVRAG or mutant was transiently expressed in UVRAG-silenced HEK293T cells. Immunoprecipitation and immunoblot were performed as indicated. (b) The indicated plasmids were expressed in UVRAG-silenced HEK293T cells. Immunoprecipitation and immunoblot were performed as indicated. (c) Huh7 cells stably expressing Vector or UVRAG constructs were transfected with mRFP-EGFP-LC3 with the treatment of EBSS. The confocal microscope was used to examine the effect of UVRAG mutant on the maturation of autophagosomes. Scale bars: 10 µm. (d) Quantification of mRFP-LC3-only puncta and the mRFP-EGFP overlay puncta in Huh7 cells treated as in (c). (e) Exogenous UVRAG expression was analyzed using immunoblot as in (c and d). (f) Huh7 cells stably expressing vector control or UVRAG constructs were treated with EBSS along with or without bafilomycin A1 (0.1 μM). 2 h later, the effect of UVRAG mutant on SQSTM1 and LC3B turnover was analyzed. (g) Huh7 cells stably expressing UVRAG constructs were transfected with GFP–LC3 and treated with bafilomycin A1 (0.1 μM). Then the effect of UVRAG mutants on the autophagosome formation was examined. Scale bars: 10 µm. Bars of quantification are mean ± SD of 40 cells.
Based on our novel findings and the previous reports that RUBCN is a negative regulator of autophagosome and endosome maturation [22], we reasoned that SMURF1 overexpression or CSNK1A1 inhibitor might mainly promote autophagosome maturation. To demonstrate this hypothesis, we stably transfected vector, WT UVRAG, UVRAGK517,559R, and UVRAGS522A into Huh7 cells respectively. We then assessed the effect of these UVRAG constructs on autophagosome maturation using the mRFP-EGFP (enhanced green fluorescent protein)-LC3 tripartite tandem vector, which was used to distinguish the autophagosomes (not acidic) and autolysosomes (acidic) [1]. Because the red fluorescence signal produced by mRFP is not sensitive to acid whereas the green fluorescence signal produced by GFP is, EGFP-positive puncta means the autophagosomes and mRFP-only represents autolysosomes upon the induction of autophagy [1]. As shown in Figure 8(c-e), Earle’s balanced salt solution (EBSS) treatment-induced the formation of yellow LC3 puncta in UVRAGK517,559R-expressing Huh7 cells was significantly higher than WT UVRAG and UVRAGS522A-expressing Huh7 cells. But EBSS produced higher induction of mRFP-LC3-only puncta in UVRAGS522A cells while the lower induction of mRFP-LC3-only puncta in UVRAGK517,559R cells compared to WT cells.
SQSTM1 is a protein removed by autophagy and often used as a marker for measuring the autophagic protein degradation rate [23]. To further evaluate autophagosome maturation, we performed western blot analyses of SQSTM1 and LC3B. In response to starvation, the level of SQSTM1 was decreased in the UVRAGS522A-expressing Huh7 cells while increased in the UVRAGK517,559R-expressing Huh7 cells (Figure 8(f)). However, treatment with bafilomycin A1 which disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and ATP2A/Ca-P60A/SERCA-dependent autophagosome-lysosome fusion [1], blocked SQSTM1 degradation in both the UVRAG mutant and WT cells (Figure 8(f)). Also, after BafA1 was added, the level of LC3B-II further accumulated in the WT UVRAG and UVRAGS522A-expressing Huh7 cells but not in the UVRAGK517,559R-expressing Huh7 cells (Figure 8(f)). Since SQSTM1 would be degraded after autophagosome maturation and fusion with lysosome [22], these data indicate that UVRAGS522A enhances but UVRAGK517,559R inhibits autophagosome maturation and autophagic degradation.
We also examined whether UVRAGK517,559R mutant or CSNK1A1-mediated phosphorylation at Ser522 could affect autophagosome formation using the GFP-LC3 vector and bafilomycin A1 in the Huh7 cells in response to starvation. It was observed that there was statistically significant increase in the number of GFP-LC3 puncta in UVRAGK517,559R-expressing Huh7 cells but not in the WT UVRAG and UVRAGS522A-expressing Huh7 cells without BafA1 treatment, but this difference was not visualized under BafA1 treatment (Figure 8(g)). All experiments above further demonstrate that UVRAGK517,559R mutant or CSNK1A1-mediated phosphorylation at Ser522 mainly inhibited the maturation of autophagosome but not the autophagosome formation.
UVRAG K29- and K33-linked ubiquitination or prevention of UVRAG (S522) phosphorylation facilitates autophagy-dependent degradation of EGFR and inhibits HCC growth
Due to the role of UVRAG in endocytic trafficking, UVRAG might promote the lysosomal degradation of EGFR [24]. Thus, we next examined whether the ubiquitination of UVRAG would affect the level of EGFR in HCC cells. When stimulated using epidermal growth factor (EGF), the degradation of EGFR was accelerated in Huh7 cells expressing WT UVRAG compared with Huh7 cells expressing UVRAGK517,559R, whereas EGFR was more stable in the vector cells compared with that in the UVRAG-overexpressing ones (Figure 9(a)). Also, the UVRAGS522A mutation also accelerated EGFR degradation compared with WT UVRAG in response to EGF (Figure 9(b)).
Figure 9.
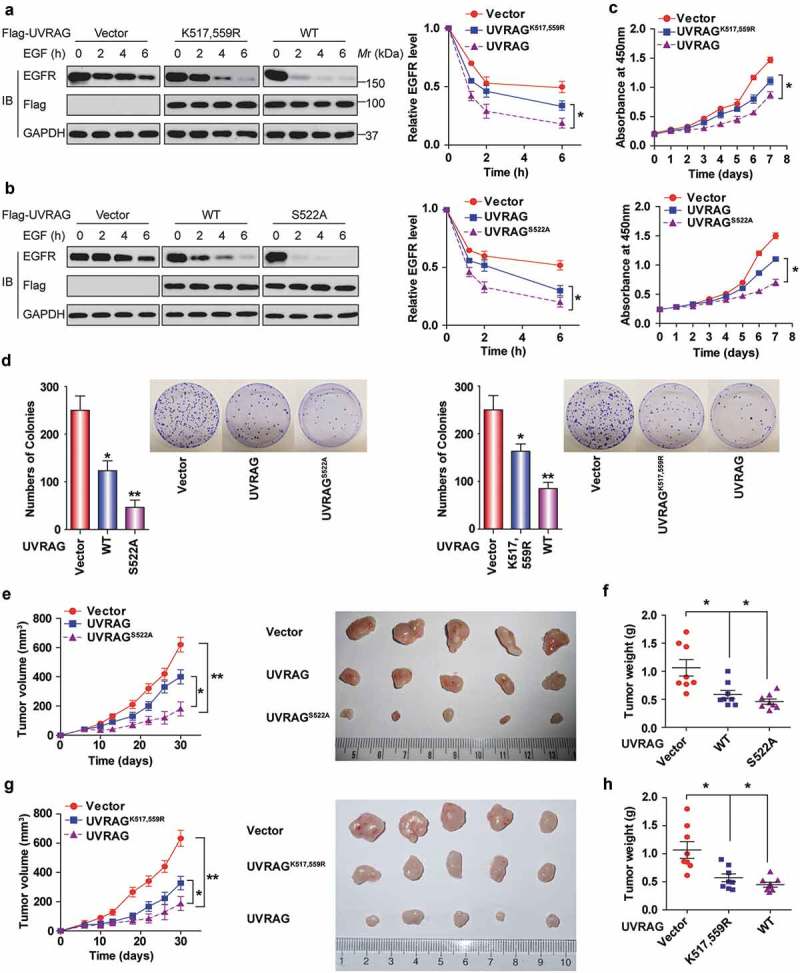
Deubiquitination and phosphorylation of UVRAG promotes HCC cell proliferation and tumorigenesis by decreasing EGFR degradation. UVRAGS522A mutation (b) enhances while UVRAGK517,559R (a) delays EGFR degradation. Huh7 cells stably transduced with empty vector or UVRAG constructs were starved of serum 24 h and then added with EGF (200 ng /ml) for the indicated time. EGFR level in cell lysate was measured by immunoblotting. The band intensity was measured and shown. (c) UVRAGS522A mutation inhibits whereas UVRAGK517,559R promotes Huh7 cell proliferation. Huh7 cells stably expressing vector control and UVRAG constructs were seeded at the same number per well. Then CCK-8 assay was performed every 24 h. (d) UVRAGS522A mutation significantly inhibits while UVRAGK517,559R promotes colony formation of Huh7 cells. Huh7 cells stably expressing vector control and UVRAG constructs were used for colony formation assays. UVRAGS522A mutation (e) significantly inhibits while UVRAGK517,559R (g) promotes tumor growth in mice. (n = 10/group). The effect of UVRAG-S522A mutation (f) and UVRAGK517,559R (h) on tumor weight relative to the whole body weight was analyzed.
Since EGFR promotes tumor growth, we then examined the effects of UVRAG ubiquitination and phosphorylation on HCC cell proliferation and tumor growth. As shown in Figure 9(c), it was observed that cells expressing WT UVRAG proliferated slower than the cells expressing UVRAGK517,559R mutant and vector. However, the suppressive effect of UVRAG became greater when S522A mutant was expressed instead of WT. The clonogenic assay indicated that WT UVRAG expression formed significantly fewer colonies than UVRAGK517,559R mutant and the vector group. In contrast, expression of S522A mutant in Huh7 cells dramatically suppressed colony formation compared to vector-transduced cells and WT cells (Figure 9(d)).
We also performed in vivo tumor xenograft studies. UVRAGS522A-expressing Huh7 xenografts grew at a slower rate than those derived from Huh7-expressing WT UVRAG and vector (Figure 9(e,f)). However, the sizes and weight of tumors formed by UVRAGK517,559R cells were significantly bigger than those formed by WT cells (Figure 9(g,h)). To exclude the artificial effect of UVRAG overexpression and interpret some of the data as being physiological, we knocked down endogenous UVRAG in Huh7 cell lines, and then put back shRNA-resistant WT UVRAG and UVRAG mutant. As shown in Figure S6, we made sure that the expression level of exogenous UVARG is similar to the endogenous UVRAG; these kinds of cells were used to examine the function of the UVRAG mutants in the animal study. Our data further confirmed our findings.
Taken together, these results demonstrate that deubiquitination and phosphorylation of UVRAG promote HCC cell proliferation and tumor growth.
CSNK1A1-dependent UVRAG (S522) and ZRANB1 (T35, S209) phosphorylation are associated with poor prognosis in HCC patients
We next analyzed 76 human primary HCC tissues with two specificity-validated antibodies: phospho-UVRAG (S522) and phopspho-ZRANB1 (T35, S209) antibodies. We showed that the phosphorylation levels of UVRAG (S522) were positively correlated with phosphorylation levels of ZRANB1 (T35, S209). And both of them were associated positively with SQSTM1 levels (Figure 10(a)). Quantification of the IHC staining indicated that these correlations were significant (Figure 10(b)). We compared the survival duration of the 76 HCC patients with tumor phosphorylation levels of UVRAG (S522) and phosphorylation levels of ZRANB1 (T35, S209). The median survival duration was 113.9 or 104.3 wk for HCC patients whose tumors had low UVRAG (S522) or ZRANB1 (T35, S209) phosphorylation levels, respectively, and 45.6 or 49.8 wk for those whose tumors had high phosphorylation levels of UVRAG (S522) or ZRANB1 (T35, S209), respectively (Figure 10(c)). These data support a model on the role of CSNK1A1-phosphorylated UVRAG and ZRANB1 and subsequent autophagy flux block in the clinical aggressiveness of human HCC (Figure 10(d)).
Figure 10.
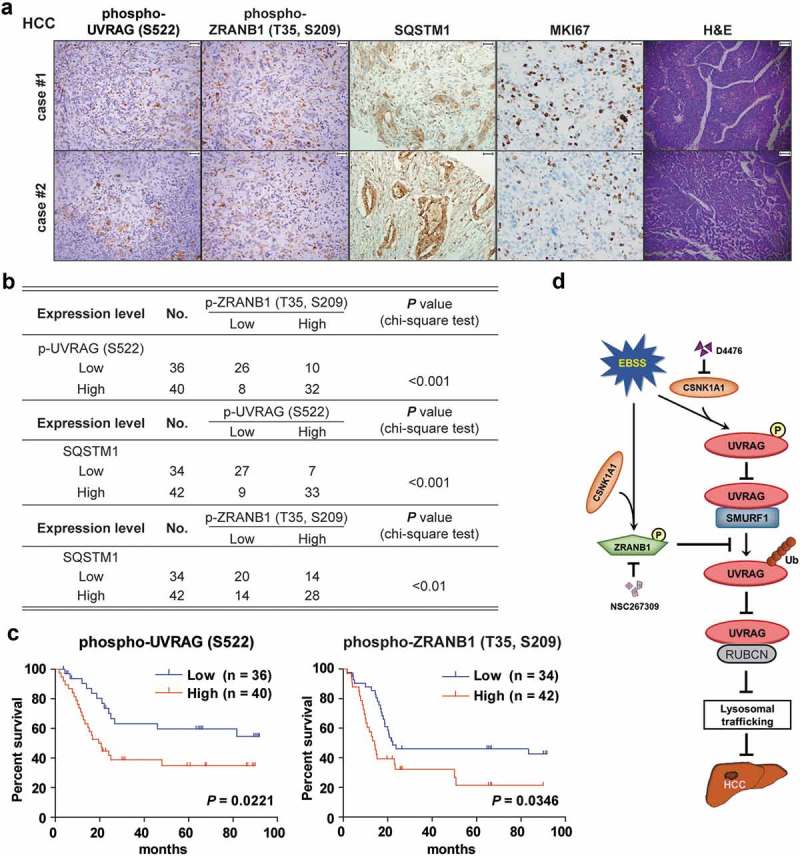
CSNK1A1-dependent UVRAG (S522) and ZRANB1 (T35, S209) phosphorylation are associated with poor prognosis in HCC patients. (a) Representative H&E (scale bar: 78.57 μm) and immunohistochemical staining (scale bar: 39.29 μm) with antibody against UVRAG (S522) or ZRANB1 (T35, S209) phosphorylation or SQSTM1, MKI67 of 76 human HCC tissues. Representative images are shown. (b) Chi-square analysis of data described in (a) was performed depending on the staining scores (low staining, 0–4; high staining, 5–8). (c) Kaplan-Meier analysis on the overall survival rates of 76 HCC patients in the groups of low or high expression of UVRAG (S522) and ZRANB1 (T35, S209) phosphorylation levels. (d) Schematic model of UVRAG ubiquitination and phosphorylation regulation by SMURF1-ZRANB1-CSNK1A1 system on autophagy maturation and HCC growth. Nutrition starvation or other autophagy-inducing signals activate CSNK1A1, which phosphorylates UVRAG at S522 and ZRANB1 at T35, S209 respectively. This phosphorylation disrupts SMURF1-mediated UVRAG K29-and K33-linked ubiquitination at K517 and K559 residues, which inhibits autophagosome maturation and EGFR degradation, thereby promoting HCC growth.
Discussion
Autophagy is well-known to be one of the most studied phenomena in cell biology and pathophysiology. Given its broad clinical implications, has become a major target for drug discovery. For the first time, the present study identifies the critical role of UVRAG ubiquitination in autophagy pathway and its clinical significance. We demonstrate the following: (1) SMURF1 induces K29- and K33-linked polyubiquitin of UVRAG at lysine residues 517 and 559; (2) UVRAG ubiquitination by SMURF1 decreases the association of UVRAG with RUBCN and enhances the activity of PIK3C3, which promotes autophagosome maturation; (3) ZRANB1 forms a complex with UVRAG and specifically cleaves SMURF1-induced K29- and K33-linked polyubiquitin chains from UVRAG, which enhances the association of UVRAG with RUBCN and inhibits autophagosome flux; (4) CSNK1A1-mediated phosphorylation at Ser522 disrupts the binding of SMURF1 to UVRAG through PPxY motif; (5) ZRANB1 is phosphorylated at Thr35, Ser209 residues by CSNK1A1, and this phosphorylation activates its deubiquitinating activity; (6) UVRAG ubiquitination by SMURF1 at lysine residues 517 and 559 or prevention of Ser522 phosphorylation by D4476 compound enhances the lysosomal degradation of EGFR, which significantly inhibits HCC initiation and development; and (7) Furthermore, UVRAG (S522) phosphorylation levels correlate with ZRANB1 (T35, S209) phosphorylation levels and poor prognosis in HCC patients. These findings uncover a novel molecular mechanism by which post-translational modifications (PTMs) regulate UVRAG function in autophagosome maturation and HCC growth, providing potential novel candidates for HCC therapy.
One interesting novel finding in this paper is that besides phosphorylation, ubiquitination also regulates the function of UVRAG. A recent study reported that UVRAG played a role in DNA damage repair and this might be affected by ubiquitination and could play a role in tumorigenesis [25]. Here, we identify SMURF1, not SMURF2, as an E3 ligase for UVRAG, suggesting that ubiquitination in turn can regulate the role of UVRAG in tumor progression. Previous studies indicated that SMURF1 could mediate K48, K63, and K29-linked polyubiquitination [6,26]. Recent studies also define SMURF1 as a newly recognized mediator of both mitophagy and viral autophagy [8]. In the current reports, we confirm and extend these findings by indicating that SMURF1 can mediate K29- and K33-linked polyubiquitination of UVRAG. Although there are many lysine residues in UVRAG protein, SMURF1 only mediates UVRAG ubiquitination at K517 and K559, suggesting that UVRAG ubiquitination can be mediated by other E3 ligases. Supporting this hypothesis, our MS/MS data reveals that there are several E3 ligases existing in the UVRAG IPs, including FBXL15, UBA1, and HECTD1. It remains unknown whether these E3 ligases also mediate UVRAG ubiquitination. These unsolved questions warrant further investigation. If UVRAG ubiquitination on individual lysine residues is probably determined by a specific E3 ligase, it will further demonstrate that the stability and function of UVRAG are strictly regulated by PTMs in which ubiquitination is a major modulating manner. Notably, although we thought that SMURF1 mediated the ubiquitination of UVRAG at K517 and K559 sites, loss of either K517 or K559 resulted in an almost complete loss of ubiquitination. If this is the case and not due to film exposure problems, this result may suggest that there is the potential for one site to influence the other, or ubiquitination of one site may cause a structural change upon mutation that abolishes ubiquitination at the other.
Many studies indicated that SMURF1 is probably an oncoprotein [7]. These studies found that SMURF1 was expressed at a high level in solid cancers and highly associated with oncogenesis and the advanced stage in various cancers including lung cancer and breast cancer [6,7]. However, we indicate that SMURF1 probably acts as a tumor suppressor in HCC, which is consistent with the previous reports [27]. Therefore, these findings suggest that SMURF1 functions in a context or tissue-dependent manner.
Another unexpected finding is that we identified ZRANB1 as a DUB responsible for cleaving SMURF1-mediated K29- and K33-linked polyubiquitin chains of UVRAG at K517 and K559, which has a positive effect on the interaction between UVRAG and RUBCN but negatively regulates the kinase activity of PIK3C3. Furthermore, deubiquitination of UVRAG by ZRANB1 inhibits the autophagy-dependent degradation of EGFR and thereby promotes HCC progression. Our report is consistent with the previous study in which ZRANB1 was found to cleave K63-linked ubiquitin chains of the APC tumor suppressor protein and facilitates TCF-mediated transcription [28]. Therefore, for the first time, these findings suggest that ZRANB1 can function as an oncogene in an autophagy-dependent and – independent manner.
Crosstalk between different types of PTMs are increasingly seen in eukaryotic biology. A major function of PTMs is to create binding sites for specific protein-protein interactions [18]. Particularly prominent is the crosstalk between phosphorylation and ubiquitination, which acts either positively or negatively in both directions [18]. Indeed, we reveal that CSNK1A1 is a novel kinase for UVRAG. The phosphorylation of UVRAG at S522 by CSNK1A1 enhances the UVRAG-RUBCN interaction and suppresses autophagosome maturation, which is consistent with the previous report that CSNK1A1 negatively regulates oncogenic RAS–induced autophagy [13]. Our finding is also reminiscent of the phosphorylation of UVRAG at S498, S550, and S571 by MTOR [24,29]. However, the phosphorylation of UVRAG at these residues does not affect the association of SMURF1 with UVRAG. Although our results support the role of S522 phosphorylation of UVRAG in regulating the ubiquitination, we do not rule out a possibility that other PTMs of UVRAG also have the similar function. The further elucidation of the crosstalk of UVRAG PTMs calls for more extensive information regarding which modifications coexist on UVRAG, which is worthy of further investigation.
To the best of our knowledge, we submitted the first report that identifies an ubiquitin ligase and a DUB for UVRAG ubiquitin modification. This study suggests that targeting crosstalk between CSNK1A1-mediated phosphorylation and ubiquitination of UVRAG may be a promising therapeutic strategy for HCC patients.
Materials and methods
GST affinity-isolation assay
Bacteria-expressed GST or GST–tagged UVRAG protein was immobilized on glutathione-Sepharose 4B beads (GE Healthcare, 17075601) and washed, then beads were incubated with His-SMURF1 or ZRANB1 expressed in Escherichia coli BL21 and purified with Ni-nitrilotriacetate-agarose beads (Qiagen, 30210) for 3 h at 4°C. Beads were washed with GST binding buffer (100 mM NaCl, 50 mM NaF, 2 mM EDTA, 1% NP40 [Thermo Fisher Scientific, 28324] and protease inhibitor cocktail and proteins were eluted, followed by western blotting.
In vivo ubiquitination assay
Indicated cells were transfected with combinations of plasmids including His-ubiquitin. Forty-eight h later, cells were treated with 10 μg/ ml MG132 (Selleckchem, S2619) for 6 h and lysed in buffer A (0.01 M Tris-HCl, pH 8.0, 5 mM imidazole, 6 M guanidium-HCl, 0.1 M Na2HPO4/NaH2PO4, 10 mM β-mercaptoethanol). Samples were mixed with Ni2+-NTA beads (Thermo Fisher Scientific, R90101) at room temperature for 4 h. The beads were sequentially rinsed using buffer A, buffer B (0.01 M Tris-Cl pH 8.0, 8 M urea [RPI Corp, 57-13-6], 10 mM β-mercaptoethanol, 0.1 M Na2PO4/NaH2PO4) and buffer C (0.1 M Na2PO4/NaH2PO4, 8 M urea, 0.01 M Tris-Cl, pH 6.3, 10 mM β-mercaptoethanol). Bound proteins were eluted with buffer D (30% glycerol [MilliporeSigma, G9012], 0.15 M Tris-Cl, pH 6.7, 0.72 M β-mercaptoethanol, 200 mM imidazole, 5% SDS [RPI Corp, 151-21-3]). Finally, all samples were subjected to immunoblotting analysis.
In vitro deubiquitination assay
Ubiquitinated UVRAG and recombinant MYC-ZRANB1 were purified from the cell extracts using an anti-FLAG affinity column (MilliporeSigma, A2220). The proteins were eluted with FLAG peptides (Sigma-Aldrich, F4799) after washing with lysis buffer. Ubiquitinated UVRAG protein was mixed with recombinant MYC-ZRANB1 in a 14.5-μL reaction mixture containing (10 mM DTT, 50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 1 mM EDTA, 5% glycerol) at 37°C for 1 h, and it was immunoprecipitated with anti-FLAG antibody (Abcam, ab205606) for immunoblot analysis. With the exclusion of the enzyme, a control reaction mixture was set up under identical conditions. The reaction mixture was stopped by addition of 5 × SDS-PAGE sample loading buffer, and then proteins were separated by SDS-PAGE.
Ub-AMC and ubiquitin cleavage assay
As described previously [30], Ub-AMC assays were conducted in a flat-bottom, low-flange 384-well plate in a 40-μl reaction with Ub AMC-conjugated proteins (Boston Biochem, U-550). Substrates and enzymes were incubated in Ub-AMC assay buffer (50 mM Tris-HCl, pH 7.5, 1 mM DTT, 1 mg/ml ovalbumin [RPI Corp, 9006-59-1], 1 mM EDTA, 5 mM MgCl2, 1 mM ATP [New England BioLabs, P0756L]). After adding of Ub-AMC, the reaction was initiated, and an Envision plate reader (PerkinElmer, MA, USA) was used to measure mixture at Ex345/Em445. For determination of KM, ZRANB1T35D,S209D was mixed with the indicated concentrations of Ub-AMC. Lineweaver–Burk analysis was carried out using a linear regression fit of the data with the equation to calculate KM: (1/ν) = KM/Vmax(1/[S]) + 1/Vmax.
In vitro kinase assay
Briefly, the assay was performed in a total volume of 50-μl reaction mixture. One μg recombinant ZRANB1 or ZRANB1 mutant protein or UVRAG or its mutant protein was incubated with 1 μg CSNK1A1. The kinase buffer contains 1 mM DTT, 10 μM cold ATP, 1 mM Na3VO4, 10 mM MgCl2, 50 mM HEPES, pH 7.4, and 5 μCi [γ-32P]-ATP for 1 h at 30°C. Then the reaction system was stopped by the addition of 10 μl 4× sample buffer and probed with phospho-specific antibodies (Abcam, ab17464).
EGFR degradation assay
Endocytic degradation of EGFR was assayed as described previously [31]. Briefly, cells starved of serum overnight were treated with EGF (100 ng/ml; R&D Systems, 236-EG) for the indicated periods of time and lysed in 0.3% CHAPS (Millipore Sigma, 10810118001)-containing lysis buffer. To analyze the activity of EGFR signaling, we stimulated serum-starved Huh7 or HEK293T cells with EGF for 10 min at 20 ng/ml or 10 ng/ml, respectively. The protein levels were analyzed by immunoblotting.
Antibodies and reagents
The antibodies used include the following: anti-ZRANB1 antibody (Abcam, ab103417), normal goat IgG (Invitrogen, 10200), and Alexa Fluor 488 goat anti-rabbit (Invitrogen, A-11008), anti-CSNK1A1/CK1α (C-19; Santa Cruz Biotechnology, sc-6477), anti-MYC (Santa Cruz Biotechnology, sc-90), anti-GAPDH (Bosterbio, 0411), K63-Ub (Selleckchem, UC-330), anti-FLAG (Sigma-Aldrich, F3165), anti-HA probe (F7; Santa Cruz Biotechnology, sc-7392), anti-SQSTM1 (2C11; Novus Biologicals, H00008878-M01), rabbit polyclonal anti-LC3B antibody (Sigma-Aldrich, L7543), anti-ubiquitin Lys63-specific antibody (Millipore Sigma, 2210353), glutathione S-transferase (GST) antibodies (Sigma-Aldrich, A7340), anti-His (Bethyl Laboratories, A190-112A), K48-Ub (Selleckchem, UC-230), rabbit polyclonal antibodies to phosphoserine (Abcam, ab9332), phosphothreonine (Abcam, ab9337), phosphoserine/threonine (Abcam, ab17464). UVRAG p-S522 antibody was generated by immunizing rabbits with a S522-phosphorylated peptide (ERLQYKTPPPSpYNSA, Rockland Immunochemicals Inc). The ZRANB1 p-T35, S209 antibody was generated by immunizing rabbits with a T35, S209-phosphorylated peptide (AQRPSGTpIITED, ARWRGSCSSpGN, Rockland Immunochemicals Inc). The tested sera were harvested after 4 more antigen boosters. Other antibodies used include anti-LC3B (Cell Signaling Technology, 2775). Anti-SMURF1 antibody (45-K) and secondary antibodies (Santa Cruz Biotechnology, sc-516102) were obtained from Santa Cruz Biotechnology (sc-100616). [γ-32P]ATP and ECL (enhanced chemiluminescence) reagent were purchased from Amersham Biosciences. Protein A/G PLUS Agarose (Santa Cruz Biotechnology, sc-2003), protease-inhibitor cocktail tablets (Roche, 4693116001), Tween 20 (Sigma-Aldrich, P9416), Nonidet P40 (Sigma-Aldrich, 74385) and Oligofectamine (Invitrogen, 12252-011) were from the indicated suppliers.
Proximity ligation assay
Proximity ligation assay was carried out using Duolink® In Situ Red Starter Kit Mouse/Rabbit (Sigma-Aldrich, DUO92101) according to the manufacturer’s instructions. Confocal microscopy image capture and analysis were performed on a Nikon A1 and the Nikon elements software suite.
Cycloheximide chase assay
After transfection with indicated plasmids for 24 h, HEK293 cells were treated with cycloheximide (CHX; Sigma-Aldrich, C7698; 100 μg/mL,) for 0 to 12 h. Cell lysates were then prepared by using a lysis buffer containing 2% SDS, followed SDS-PAGE and IB analyses with specific antibodies.
Confocal microscopy
HEK293T or HCC cells were transfected with indicated plasmids for 48 h. Cells were then cultured on sterile glass coverslips in 6-well plates to 80% confluence in Dulbecco’s modified Eagle medium and then stained with indicated antibody and visualized on a Nikon A1 and the Nikon elements software suite (Nikon, Japan).
Coimmunoprecipitation and western blot
Briefly, indicated cells were lysed using a low-stringency lysis buffer (50 mM Tris-HCl, pH 7.5, 120 mM NaCl, 0.5 mM EDTA, 0.5% Nonidet P-40, 10% glycerol) and approximately 0.5 mg of whole-cell extract was used for each immunoprecipitation. Indicated antibodies or control rabbit IgGs were used to carry out the immunoprecipitations overnight. Immunocomplexes were collected with a mixture of protein A and protein G-Sepharose and washed three times in washing buffer (50 mM Tris-HCl, pH 8, 150 mM NaCl, 0.1% Triton X-100, 5% glycerol, 0.5% dithiothreitol). Immunocomplexes were eluted in loading buffer and loaded onto a 7.5% SDS–PAGE gel and then transferred onto nitrocellulose membranes. The membranes were blocked overnight in 5% fat-free milk in TBST (0.1% Tween 20 plus TBS: 50 mM Tris-Cl, pH 7.6, 150 mM NaCl) buffer and were probed with the indicated primary antibodies overnight at 4°C.The following day, the primary antibodies were removed, the membranes were washed four times at room temperature and then incubated for 1 h at room temperature with the indicated secondary antibody. After applying electrochemiluminescence (MilliporeSigma, WBKL S0500), the protein bands were visualized using X-ray film (Labscientific, XARALF1318). And the intensity of the western blot bands was quantified using NIH ImageJ software.
Immunohistochemistry
Immunostaining was performed as described previously [12].
Glucose starvation assay
Glucose starvation performed in this study was carried out by switching the culture medium from complete medium to the same medium lacking glucose (Gibco, 11966-025).
Cell culture
HEK293T cells was purchased from the American Type Culture Collection (CRL-1573) and human HCC cells (MHCC97H and Huh7) were purchased from the Cell Bank of Chinese Academy of Sciences (SCSP-528, KCB 200970YJ). All cells were grown in Dulbecco’s modified Eagle medium (Invitrogen, 11965-084) a humidified atmosphere of 5% CO2 at 37°C. The medium was supplemented with 10% fetal bovine serum fetal calf serum (Sigma-Aldrich, F7524) and penicillin-streptomycin (100 μg/mL, Thermo Fisher Scientific, 15140163).
Plasmids and shRNA or siRNA
MYC-SMURF1 wild-type and MYC–SMURF1C699A were provided by Dr. Hui Wang (Rutgers University). Human UVRAG cDNA and its DNA fragments were cloned into the pRK5 vector (BD PharMingen, 556104) by PCR amplification. The pLKO.1 vector (Addgene, 8453, deposited by Bob Weinberg) was used to knock down UVRAG or RUBCN. HA-Ubs (WT, K29R, K33R, K48R, K63R, K29, K33, K48, and K63) were kindly provided by Dr. Jun Zhang (Rutgers University). Ub (including Ub-K6, Ub-K11, Ub-K27, Ub-K29, Ub-K33, Ub-K48, and Ub-K63) were from Addgene (17,605, 17,606, 17,607, and 17,608, deposited by Ted Dawson; 22,900, 22,901, 22,902, and 22,903 deposited by Sandra Weller). The mCherry-GFP-LC3 was kindly provided by Dr. Bing Wang (Rutgers University). The constructs pcDNA3.1-CSNK1A1, CSNK1D/CK1δ, and CSNK1E/CK1ϵ were purchased from GenScript ( OHu10853, OHu02012, OHu10985). CSNK1A1K46A, a kinase-dead mutant was created by using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies, 200521). Specific short interfering RNA (siRNA) oligonucleotides were used against human SMURF1 and synthesized by GenePharma. Their sequences were the following: SMURF1 siRNA-#1 (CCGACACUGUGAAAAACACTT), SMURF1 siRNA-#2 (GCGUUUGGAUCUAUGCAAATT), and the non-silencing siRNA from Qiagen (1022076) was used as the negative control. The target sequences of human CSNK1A1-, CSNK1D -, and CSNK1E-specific siRNAs (Dharmacon RNAi Technologies) were CSNK1A1 siRNA-#1 (GCGAUGUACUUAAACUAUU) and CSNK1A1 siRNA-#2 (GGAAUCAUUAGGAUAUGUU). Cells were transfected with 50 nM specific siRNAs using DharmaFECT Transfection Reagent (Dharmacon, T-2001–01) for 48–72 h according to the manufacturer’s instructions. The cDNAs for GST-tagged-, or HA-tagged-ZRANB1 constructs, and its point mutants have been described [28]. Based on the RNAi Codex algorithm, ZRANB1 shRNA targeting sequences were selected: shZRANB1-1, 5ʹ-TTGGAAAAGTCCGATTGCTCT-3′; shZRANB1-2, 5ʹ-CTGGCACATATTCTTAGACGA-3′; shZRANB1-3, 5ʹ-TGTCTCAACAAGCAGCAAAGT-3′; and shZRANB1-4, 5ʹ-TGATCATCCCAGACCTAATAA-3′. The shRNA-resistant MYC-ZRANB1, ZRANB1Δ, was made by performing site–directed mutagenesis to introduce 3 silent mutations against ZRANB1 shRNA. All other constructs have been previously described.
Cell proliferation assay
The cell proliferation assay was performed with the Cell Counting Kit-8 (Sigma-Aldrich, 96992) according to the manufacturer’s instructions.
Clonogenic proliferation analysis
Briefly, a single cell suspension including 300 cells per well was plated into a 10-mm dish. Two weeks later, every individual colony was fixed and stained with hematoxylin. Colonies were counted. All analysis was carried out in triplicate 3 times.
Mice xenograft assay
The project was conducted in strict accordance with the guidelines and protocols approved by the Institutional Animal Care and Use Committee of the university. Six-week-old male nude mice (BALB/c) were purchased from the Shanghai Experimental Animal Center, and randomly divided into the indicated groups (6–8 mice/group) before inoculation, and double-blinded evaluation was performed when measuring tumor weight and volume. The indicated HCC (5 × 106) cells in 0.2 ml PBS per mouse were subcutaneously injected into the right flank of each mouse. After the indicated time point, the mice were sacrificed, and tumor volume and weight were measured.
HCC patients’ samples collection
Human HCC tissues were collected from patients performing HCC resection from the First Hospital of Jilin and Anhui University. The diagnosis was established based on World Health Organization criteria. The clinical typing of tumors was determined according to the International Union against Cancer tumor-node-metastasis classification system. As described previously [12], the liver function was assessed by Child-Pugh score system and the staining index was calculated as follows: staining index = staining intensity × tumor cell staining grade. High expression was explained by a staining index score ≥4, while low expression was defined as a staining index <4. Ethical approval for using human subjects was obtained from the Research Ethics Committee of the First Hospital of University with the informed consent of patients.
Statistical analysis
All statistical analyses were performed using the SPSS 16.0 statistical software package (Abbott Laboratories, USA) and GraphPad Prism 5.0 software package for Windows. Quantitative values of all experiments are expressed as the mean ± SD. The significance of the correlation between the expression of indicated proteins and histopathological factors was determined using Pearson χ2 test. The Adobe Photoshop CS6 was used to compose images. Factorial design ANOVA was performed to test in vitro cell growth. Comparisons between groups were carried out with a 2-tailed paired Student’s t-test. The correlation between the expression levels of two proteins was determined using Spearman’s correlation analysis. A value of *P < 0.05 was considered to be statistically significant plus **P < 0.01 and ***P < 0.001.
Funding Statement
This work was supported by grants from Rutgers University Pilot Project, National Natural Science Foundation of China (81602149, 81702387, 31571241, and 31660266).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Song H, Pu J, Wang L, et al. ATG16L1 phosphorylation is oppositely regulated by CSNK2/casein kinase 2 and PPP1/protein phosphatase 1 which determines the fate of cardiomyocytes during hypoxia/reoxygenation. Autophagy. 2015;11(8):1308–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2016;12(1):1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Das G, Shravage BV, Baehrecke EH.. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb Perspect Biol. 2012;4(6):a008813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mokarram P, Albokashy M, Zarghooni M, et al. New frontiers in the treatment of colorectal cancer: autophagy and the unfolded protein response as promising targets. Autophagy. 2017;13(5):781–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Shimazu J, Wei J, Karsenty G. Smurf1 inhibits osteoblast differentiation, bone formation, and glucose homeostasis through serine 148. Cell Rep. 2016;15(1):27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Li X, Dai X, Wan L, et al. Smurf1 regulation of DAB2IP controls cell proliferation and migration. Oncotarget. 2016;7(18):26057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cao Y, Zhang L. Pharmaceutical perspectives of HECT-type ubiquitin ligase Smurf1. Curr Pharm Des. 2013;19(18):3226–3233. [DOI] [PubMed] [Google Scholar]
- [8].Orvedahl A, Sumpter R Jr, Xiao G, et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011;480(7375):113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lorenzi PL, Claerhout S, Mills GB, et al. A curated census of autophagy-modulating proteins and small molecules: candidate targets for cancer therapy. Autophagy. 2014;10(7):1316–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Knippschild U, Gocht A, Wolff S, et al. The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cell Signal. 2005;17(6):675–689. [DOI] [PubMed] [Google Scholar]
- [11].Shin S, Wolgamott L, Roux PP, et al. Casein kinase 1ϵ promotes cell proliferation by regulating mRNA translation. Cancer Res. 2014;74(1):201–211. [DOI] [PubMed] [Google Scholar]
- [12].Qu C, He D, Lu X, et al. Salt-inducible Kinase (SIK1) regulates HCC progression and WNT/β-catenin activation. J Hepatol. 2016;64(5):1076–1089. [DOI] [PubMed] [Google Scholar]
- [13].Cheong JK, Zhang F, Chua PJ, et al. Casein kinase 1α–dependent feedback loop controls autophagy in RAS-driven cancers. J Clin Invest. 2015;125(4):1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Afonina IS, Beyaert R. Trabid epigenetically drives expression of IL-12 and IL-23. Nat Immunol. 2016;17(3):227–228. [DOI] [PubMed] [Google Scholar]
- [15].Kristariyanto YA, Rehman SAA, Campbell DG, et al. K29-selective ubiquitin binding domain reveals structural basis of specificity and heterotypic nature of k29 polyubiquitin. Mol Cell. 2015;58(1):83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Nakajima S, Aikawa C, Nozawa T, et al. Bcl-xL affects group a streptococcus-induced autophagy directly, by inhibiting fusion between autophagosomes and lysosomes, and indirectly, by inhibiting bacterial internalization via interaction with beclin 1-UVRAG. PloS one. 2017;12(1):e0170138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Xiong W, Yang J, Wang M, et al. Vinexin β interacts with hepatitis C virus NS5A, modulating its hyperphosphorylation to regulate viral propagation. J Virol. 2015;89(14):7385–7400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Nguyen LK, Kolch W, Kholodenko BN. When ubiquitination meets phosphorylation: a systems biology perspective of EGFR/MAPK signalling. Cell Commun Signaling. 2013;11(1):52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lee JI, Woo SK, Kim KI, et al. A method for assaying deubiquitinating enzymes. Biol Proced Online. 1998;1(1):92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].He S, Liang C. Frameshift mutation of UVRAG: switching a tumor suppressor to an oncogene in colorectal cancer. Autophagy. 2015;11(10):1939–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Rena G, Bain J, Elliott M, et al. D4476, a cell‐permeant inhibitor of CK1, suppresses the site‐specific phosphorylation and nuclear exclusion of FOXO1a. EMBO Rep. 2004;5(1):60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang L, Tian Y, Ou J-H. HCV induces the expression of Rubicon and UVRAG to temporally regulate the maturation of autophagosomes and viral replication. PLoS Pathog. 2015;11(3):e1004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Moscat J, Karin M, Diaz-Meco MT. p62 in cancer: signaling adaptor beyond autophagy. Cell. 2016;167(3):606–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kim Y-M, Jung CH, Seo M, et al. mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol Cell. 2015;57(2):207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yang Y, He S, Wang Q, et al. Autophagic UVRAG promotes UV-induced photolesion repair by activation of the CRL4DDB2 E3 ligase. Mol Cell. 2016;62(4):507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fei C, Li Z, Li C, et al. Smurf1-mediated Lys29-linked nonproteolytic polyubiquitination of axin negatively regulates Wnt/β-catenin signaling. Mol Cell Biol. 2013;33(20):4095–4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tang X, Chen X, Xu Y, et al. CD166 positively regulates MCAM via inhibition to ubiquitin E3 ligases Smurf1 and βTrCP through PI3K/AKT and c-Raf/MEK/ERK signaling in Bel-7402 hepatocellular carcinoma cells. Cell Signal. 2015;27(9):1694–1702. [DOI] [PubMed] [Google Scholar]
- [28].Tran H, Hamada F, Schwarz-Romond T, et al. Trabid, a new positive regulator of Wnt-induced transcription with preference for binding and cleaving K63-linked ubiquitin chains. Genes Dev. 2008;22(4):528–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Munson MJ, Allen GF, Toth R, et al. mTOR activates the VPS34–UVRAG complex to regulate autolysosomal tubulation and cell survival. EMBO J. 2015;34(17):2272–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Xu D, Shan B, Lee B-H, et al. Phosphorylation and activation of ubiquitin-specific protease-14 by Akt regulates the ubiquitin-proteasome system. Elife. 2015;4:e10510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sun T, Li X, Zhang P, et al. Acetylation of Beclin 1 inhibits autophagosome maturation and promotes tumour growth. Nat Commun. 2015;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.