

ABSTRACT
Mutations in the γ-secretase complex are strongly associated with familial Alzheimer disease. Both proteolytic and non-proteolytic functions for the γ-secretase complex have been previously described in mammalian model organisms, but their relative contributions to disease pathology remain unclear. Here, we dissect the roles of orthologs of the γ-secretase components in the model system Dictyostelium, focusing on endocytosis, lysosomal activity and autophagy. In this model, we show that the orthologs of PSEN (psenA and psenB), Ncstn (nicastrin) and Aph-1 (gamma-secretase subunit Aph-1), are necessary for optimal fluid-phase uptake by macropinocytosis and in multicellular development under basic pH conditions. Disruption of either psenA/B or Aph-1 proteins also leads to disrupted phagosomal proteolysis as well as decreased autophagosomal acidification and autophagic flux. This indicates a general defect in lysosomal trafficking and degradation, which we show leads to the accumulation of ubiquitinated protein aggregates in cells lacking psenA/B and Aph-1 proteins. Importantly, we find that all the endocytic defects observed in Dictyostelium PSEN ortholog mutants can be fully rescued by proteolytically inactive Dictyostelium psenB and human PSEN1 proteins. Our data therefore demonstrates an evolutionarily conserved non-proteolytic role for presenilin, and γ-secretase component orthologs, in maintaining Dictyostelium lysosomal trafficking and autophagy.
Abbreviations: Atg8: autophagy protein 8a; Aph-1: gamma-secretase subunit Aph-1; crtA: calreticulin; ER: endoplasmic reticulum; GFP: green fluorescent protein; GSK3B: glycogen synthase kinase 3 beta; Ncstn: nicastrin; PSEN1: presenilin 1; psenA and psenB: Dictyostelium presenilin A and B; TRITC; tetramethylrhodamine isothiocyanate.
KEYWORDS: Alzheimer disease, autophagy, development, Dictyostelium, γ-secretase, lysosomal trafficking, presenilin
Introduction
Many studies have sought to explore a role for the γ-secretase complex (and PSEN [presenilin] proteins) in the pathology of Alzheimer disease [1–4]. One function of the mammalian complex (consisting of PSEN1 [presenilin 1], APH1 [aph-1 homolog, gamma-secretase subunit], NCSTN [nicastrin] and PSENEN/PEN2 [presenilin enhancer, gamma-secretase subunit]) or PSEN1 proteins alone is to regulate endocytosis, lysosomal acidification, and autophagy [5–7]. This role has been implicated in disease pathology, since mutations in PSEN1 proteins associated with familial Alzheimer disease result in elevated lysosomal pH, and aberrant autophagy in mouse models [7–9]. Dysfunctional endosomal-lysosomal and autophagic pathways have also been implicated in the pathogenesis of several other neurodegenerative disorders, including amyotrophic lateral sclerosis (ALS) and Parkinson disease [10–13]. These studies have given rise to a theory of neurodegenerative disease pathology relating to reduced protein clearance [13–15].
The role of the γ-secretase complex is primarily thought to be through its proteolytic activity and cleavage of target proteins [2–4,16]. Here, PSEN proteins contain two key aspartic acid residues necessary for the proteolytic activity of the complex in cleaving a range of substrates including the amyloid-β and NOTCH proteins [17]. However, the γ-secretase complex has also been proposed to act through non-proteolytic scaffolding functions in a variety of model organisms [18–24]. In mammalian models, these functions include stabilizing the binding of CTNNB1 and GSK3β, where Alzheimer disease causing mutations result in reduced stability of CTNNB1 [18,19,25,26], and in endoplasmic reticulum (ER) calcium regulation [27,28]. In D. melanogaster, presenilin proteins have been found to modulate the levels of CREBBP (cyclic-AMP Response Element Binding protein) independently of proteolytic activity [29]. A similar non-proteolytic function for presenilin proteins or the γ‐secretase complex has also been proposed in both C. elegans [30] and the moss P. patens [20]. However, although protease-independent functions of the γ-secretase complex have been observed in evolutionarily diverse species, our understanding of the mechanistic significance of these functions and relevance to Alzheimer pathology remains limited.
To better understand the role of the γ-secretase complex, several studies have employed the social amoeba Dictyostelium discoideum [22,31]. Dictyostelium contains orthologs of the core components of the complex including two PSEN (presenilin) proteins (psenA and B), Aph-1 and Ncstn [22,23,31]. In Dictyostelium, the complex components play roles in multicellular development, in cyclic AMP signalling and in intracellular calcium release [22] as well as in phagocytosis [31]. Although proteolytic targets(s) for the complex in Dictyostelium have not been defined, Dictyostelium psenA and psenB proteins show proteolytic activity in a Notch reporter assay [22]. In multicellular development, psenB plays a non-proteolytic (scaffold) function, since removal of the two key catalytic aspartic acids (348 and 394) does not block the formation of mature fruiting bodies. Furthermore, the roles of psenA and psenB proteins in Dictyostelium development are also complemented using the proteolytically inactive human PSEN1 protein, lacking the key catalytic aspartic acids 257 and 385 [22]. These studies have demonstrated the relevance of using Dictyostelium to examine the cellular and developmental roles of presenilin proteins and orthologs of other γ-secretase components.
Dictyostelium has been widely used as a model organism in a range of cell and molecular studies, often enabling translation to mammalian models [32,33]. It has been used in the study of endosomal- and autophagy-lysosomal systems and related pathways where WASH is required for lysosomal recycling and both autophagic and phagocytic digestion [34]; mucolipin is required for lysosomal exocytosis [35]; myosin I is involved in membrane recycling from early endosomes [36] as well as many other discoveries [37–41]. Here we advance our understanding of the role of the Dictyostelium orthologs of γ-secretase complex components in endocytosis and autophagy by demonstrating that these components are required for the efficient activity of these processes. We further show that regulation of endocytosis and autophagy activities occurs through a proteolysis-independent mechanism, where these activities are conserved in the human PSEN1 protein. We also show that loss of these proteolysis-independent functions leads to an increase in large poly-ubiquitinated autophagosome-like vesicles, demonstrating an ancient and conserved non-proteolytic role of presenilin and orthologs of other components of the γ-secretase complex in proteostasis.
Results
Dictyostelium mutants lacking γ-secretase component orthologs are unable to endocytose efficiently
Since the γ-secretase complex has been implicated in endocytosis in mammalian model organisms [5,6,42], we initially set out to investigate if orthologous components of this complex also play roles in Dictyostelium macropinocytosis – the dominant form of endocytosis and fluid-phase uptake in this organism. In wild-type cells, uptake of TRITC-Dextran was linear for 60 minutes and was therefore used to determine the rate of macropinocytosis (Figure 1(a,b), Figure S1). Macropinocytosis in the absence of the γ-secretase complex orthologs was analysed through the use of stable isogenic cells lacking Ncstn [22], Aph-1 or both presenilin proteins (psenA/B [22]). We observed that loss of all three γ-secretase component orthologs caused a 40–50% decrease in fluid-phase uptake (Figure 1(c)), and this effect was not due to altered exocytosis (Figure S2). These data suggest a role for the three γ-secretase component orthologs in regulating endocytosis in Dictyostelium.
Figure 1.
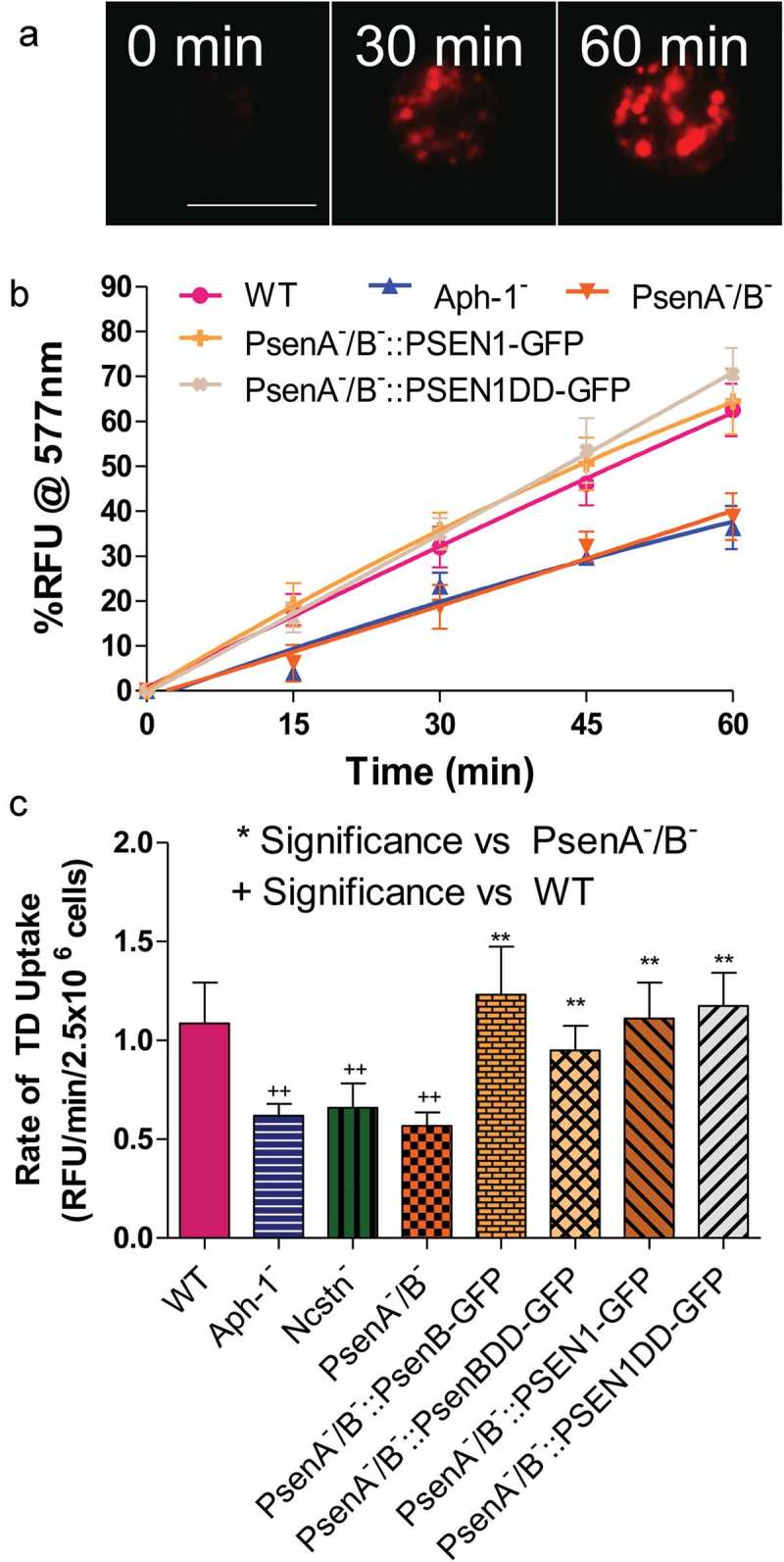
Dictyostelium mutants lacking γ-secretase component orthologs are unable to endocytose at wild type levels. Dictyostelium cells were shaken in media containing fluorescent TRITC-Dextran, and fluorescence uptake was used to monitor macropinocytosis over time, in wild-type cells, PsenA−/B−, Aph-1− and Ncstn− cells, and PsenA−/B− following rescue by the proteolytic and non-proteolytic human PSEN1 proteins (PsenA−/B−::PSEN1-GFP and PsenA−/B−::PSEN1DD). (a) Representative images showing uptake of TRITC-Dextran by wild-type cells over a 60-min period. Scale bar: 10μ m. (b) Quantification of macropinocytosis over 60 min (±SEM). (c) Rate of macropinocytosis over 60 min (±SD). Data are provided from at least three independent experiments with technical duplicates. ++p > 0.01 to wild type, **p > 0.01 to PsenA−/B−.
A previous study has suggested that the Dictyostelium γ-secretase complex shows both proteolytic and non-proteolytic functions of psenB [22], thus we sought to distinguish these roles in macropinocytosis. In these experiments, the Dictyostelium psenB-GFP protein was expressed in PsenA−/B− cells, in addition to a mutated version of the protein lacking the two key aspartic acid residues necessary for proteolytic activity (psenBDD-GFP) [22]. In both cases, wild type macropinocytosis levels were restored (Figure 1(b,c)), suggesting that this role of psenB is through a non-proteolytic function. Furthermore, expression of the wild type (PSEN1-GFP) and proteolytically inactive (PSEN1DD-GFP) human PSEN1 protein also rescued the macropinocytosis defect in PsenA−/B− cells (Figure 1(b,c), Figure S1) demonstrating the evolutionary conservation of this function, although expression of proteolytically active PSEN1-GFP did not restore the macropinocytosis defect in cells lacking Ncstn (Figure S3). These data suggest that, in Dictyostelium, macropinocytosis was dependent upon Aph-1, Ncstn and a non-proteolytic function of psenB.
Dictyostelium mutants lacking γ-secretase component orthologs show pH-dependent development
Since nutrients ingested in macropinosomes are degraded by fusion with lysosomes [43], and deficiencies in acidification decrease endocytic rate [44] we next investigated a role for orthologs of γ-secretase complex components in lysosomal acidification. For these experiments, we initially employed a qualitative development assay, where Dictyostelium mutants with severe acidification defects are unable to form mature fruiting bodies in neutral pH conditions [45]. We therefore tested the ability of mutants lacking orthologs of γ-secretase complex components to develop over 24 hours under neutral (pH 7) and basic conditions (pH 9). In both conditions, wild type cells were able to develop into mature fruiting bodies consisting of a basal disk, a stalk, and a spore head. However, while Ncstn− and Aph-1− cells were able to form fruiting bodies under neutral conditions, both failed to develop at pH 9 (Figure 2(b), Figure S4), consistent with defective lysosomal acidification. As reported previously [22], PsenA−/B− cells were unable to develop under neutral conditions showing more severe developmental defects than Ncstn− or Aph-1− cells, halting at the mound stage, but development was reduced further under basic conditions at pH 9 (Figure 2(a)).
Figure 2.
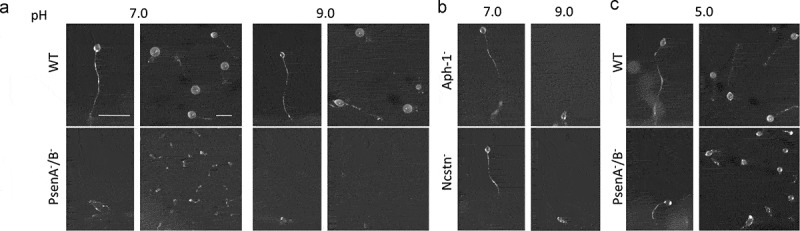
Dictyostelium mutants lacking γ-secretase component orthologs are unable to development under varying pH conditions. The development of Dictyostelium, through starvation over a 24-h period on nitrocellulose filters, leads to the formation of fruiting bodies consisting of round spore heads held aloft by a stalk, and provides a qualitative approach to monitor development in mutants. (a) At pH 7 and pH 9, wild type cells are capable of normal multi-cellular development forming fruiting bodies. In contrast, PsenA−/B− cells are unable to develop at pH 7, forming short variable structures, and development is further inhibited at pH 9 where cells are unable to aggregate. (b) Aph-1− and Ncstn− cells form wild-type fruiting bodies at pH 7 but development is blocked at pH 9 (see also Figure S4). (c) Under acidic conditions, pH 5, wild-type cells form morphologically normal fruiting bodies, and PsenA−/B− development is partially rescued, showing the formation of small fruiting bodies with round spore heads and stalks, similar to that shown for wild-type cells. Images are representative of triplicate experiments. Scale bar: 1 mm.
We then speculated that if the block in development of PsenA−/B− cells at neutral pH was related to defects in lysosomal acidification, development may be restored by simply reducing the extracellular pH. We therefore examined development of PsenA−/B− cells at pH 5 [22,45]. While acidic conditions had no observable effects on the development of wild type cells the development of PsenA−/B− cells was partially rescued, forming morphologically normal but smaller sized fruiting bodies (Figure 2(c)). This rescue suggested that the developmental defects observed are at least partly due to defects in pH homeostasis, consistent with a role for the Dictyostelium orthologs of γ-secretase complex components in lysosomal acidification. However, the differences in the developmental phenotype between PsenA−/B− cells and Ncstn− or Aph-1− mutants indicate that psenA and psenB proteins may also function through other independent roles in Dictyostelium development.
Importantly, the development of PsenA−/B− cells was fully rescued by expression of either proteolytically active or inactive Dictyostelium psenB or human PSEN1 proteins (Figure S5) [22] indicating a non-proteolytic role for these proteins in pH regulation.
Dictyostelium mutants lacking orthologs of γ-secretase components show defective lysosomal degradation
In order to directly test whether disruption of orthologs of the γ-secretase complex components leads to defects in lysosomal activity, we measured the ability of mutant cells lacking these components to degrade phagosomes. As a professional phagocyte, Dictyostelium cells readily engulf extracellular particles into endocytic vesicles which immediately fuse with lysosomes to be degraded. We therefore utilised a simple assay measuring the phagosomal proteolysis of beads coated with the self-quenching dye DQ-BSA, which becomes fluorescent upon proteolysis (Figure 3(a)) [46]. In this assay, we observed a significant decrease in proteolysis upon loss of multiple Dictyostelium orthologs of γ-secretase complex components. Ablation of Aph-1 or PsenA/B caused a reduction in proteolysis activity to 41.4%±4.1% (p = 0.05) and 42.1%+12.8% (p = 0.03) respectively relative to wild type cells (Figure 3(b)). Upon expression of proteolytically active, or inactive Dictyostelium psenB, relative proteolysis was restored to 78.9%±9.9% (p = 0.10) and 128.5%±25.1% (p = 0.36) and expression of active or inactive human PSEN1 resulted in a degradation rate of 98.8%±12.6% (p = 1.00) and 120.8%±7.9% (p = 0.35). The reduction in phagosomal proteolysis following loss of Aph-1 was not restored by expression of human PSEN1 (Figure S6). Although this assay does not distinguish between defective phagosome-lysosome fusion and decreased lysosomal activity, these data demonstrate that full degradative activity is dependent upon the presence of Aph-1, and psenB functioning through a non-proteolytic mechanism, and this activity is conserved in the human PSEN1 protein.
Figure 3.
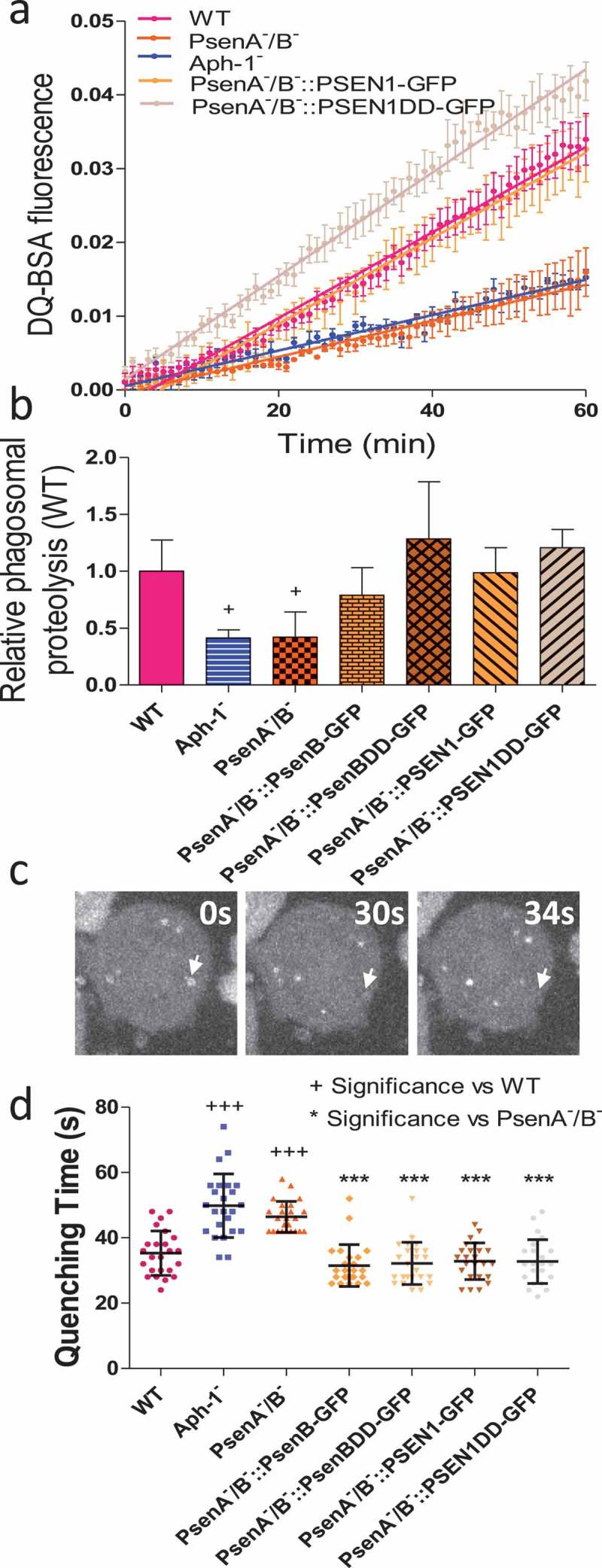
Dictyostelium mutants lacking γ-secretase component orthologs show abnormal lysosomal activity. Phagosome degradation is quantified by monitoring the increase in fluorescence of DQ-BSA-coated beads, taken up by phagocytosis, which becomes unquenched upon hydrolysis. This approach was used to assess in wild type cells, PsenA−/B−, and Aph-1− cells, and PsenA−/B− cells following rescue by the proteolytic and non-proteolytic Dictyostelium psenB or the equivalent human PSEN1 proteins (PsenB-GFP and PsenBDD-GFP or PSEN1-GFP and or PSEN1DD-GFP) respectively. (a) Quantification of proteolysis shows loss of a functional γ-secretase complex reduces phagolysosomal degradation, and this is restored by proteolytically active or inactive human PSEN1, from quadruplicate independent experiments (±SEM), which is reflected in (b) the rate of lysosomal acidification in (±SD). +P < 0.05. (c) Dictyostelium cells exhibit puncta of GFP-Atg8 that may be tracked (arrow) over time to determine the quenching time of GFP due to acidification. (d) Quantification of GFP-Atg8 quenching time shows an increase in the absence of a functional γ-secretase complex, and this is restored by both Dictyostelium and human presenilin proteins (both proteolytically active and inactive) (n = 25). +++p > 0.001 to wild type, ***p > 0.001 to PsenA−/B.
Loss of γ-secretase components orthologs causes defective autophagy
The degradation of intracellular components by autophagy is also dependent on lysosomal activity. As autophagy is required for normal development of Dictyostelium [37] and has been heavily implicated in supressing the accumulation of mis-folded proteins characteristic of Alzheimer and several other neurodegenerative diseases [11,13,15,47,48], we next investigated the effects of loss of orthologs of γ-secretase components on autophagy.
We first measured autophagosome acidification and degradation in mutant lacking Aph-1 and psenA/B activity. In these experiments, we employed a fluorescent marker, GFP-Atg8, which becomes lipidated and incorporated into the membrane of nascent autophagosomes as they expand. Following completion, autophagosomes rapidly fuse with lysosomes and acidify – quenching the luminal GFP fluorescence, while the exterior GFP-Atg8 is removed [38,41]. In Dictyostelium cells this can be clearly observed by microscopy and by quantifying the time required for GFP quenching after autophagosome formation, where a rate of acidification can be defined for individual vesicles (Figure 3(c); MovieS1) [38,41]. In wild type cells, GFP-Atg8 quenched in 35.3 ± 6.8 seconds (Figure 3(d)). In contrast, Aph-1− cells showed significantly increased acidification time to 49.8 ± 6.8 seconds (p < 0.001), and PsenA−/B− cells also significantly increased quenching time to 46.4 ± 9.7 seconds (p < 0.001). The acidification defect in PsenA−/B− cells was rescued upon expression of proteolytically active versions of Dictyostelium psenB (32.2 ± 4.7 seconds) as well as a proteolytically inactive version of the protein (31.5 ± 6.5 seconds) suggesting this effect was mediated by non-proteolytic functions of these proteins. The acidification defect in PsenA−/B− cells was also rescued by both proteolytically active and inactive versions of human PSEN1 protein (32.8 ± 6.4 and 28.8 ± 5.6 seconds respectively). Therefore, the defects in lysosomal activity we observed upon disruption of Aph-1 and psenA/B proteins also impact on autophagosome maturation, suggesting an explanation for both the observed defects in Dictyostelium development as well as cell pathology in the absence of these proteins.
Alterations in the process of vesicle acidification have been shown to regulate autophagy [15], in which cells recycle material for energy. In Dictyostelium, interruption in the autophagic pathway results in a decrease in the number and an increase in the size of GFP-Atg8 labelled structures [34,38,49]. As a result, we monitored their size and frequency in Aph-1− and PsenA−/B− cells for signs of autophagic dysfunction. Wild type cells expressing GFP-Atg8 show a majority of small (below 0.4μ m) diameter structures (79%), with very few structures above 0.8 μm (1.4%) representing normal functioning autophagosomes (Figure 4(a,b). In Aph-1− cells, a significant reduction was seen in the occurrence of small structures to 26% (p < 0.01) and a significant increase in the formation of large GFP-Atg8-positive structures, to 17% (p < 0.05) typical of cells with reduced autophagy [37,39]. Similarly, PsenA−/B− cells also showed single large GFP-Atg8 structures. These data suggest that loss of Aph-1 and psenA/B proteins caused autophagic dysfunction.
Figure 4.
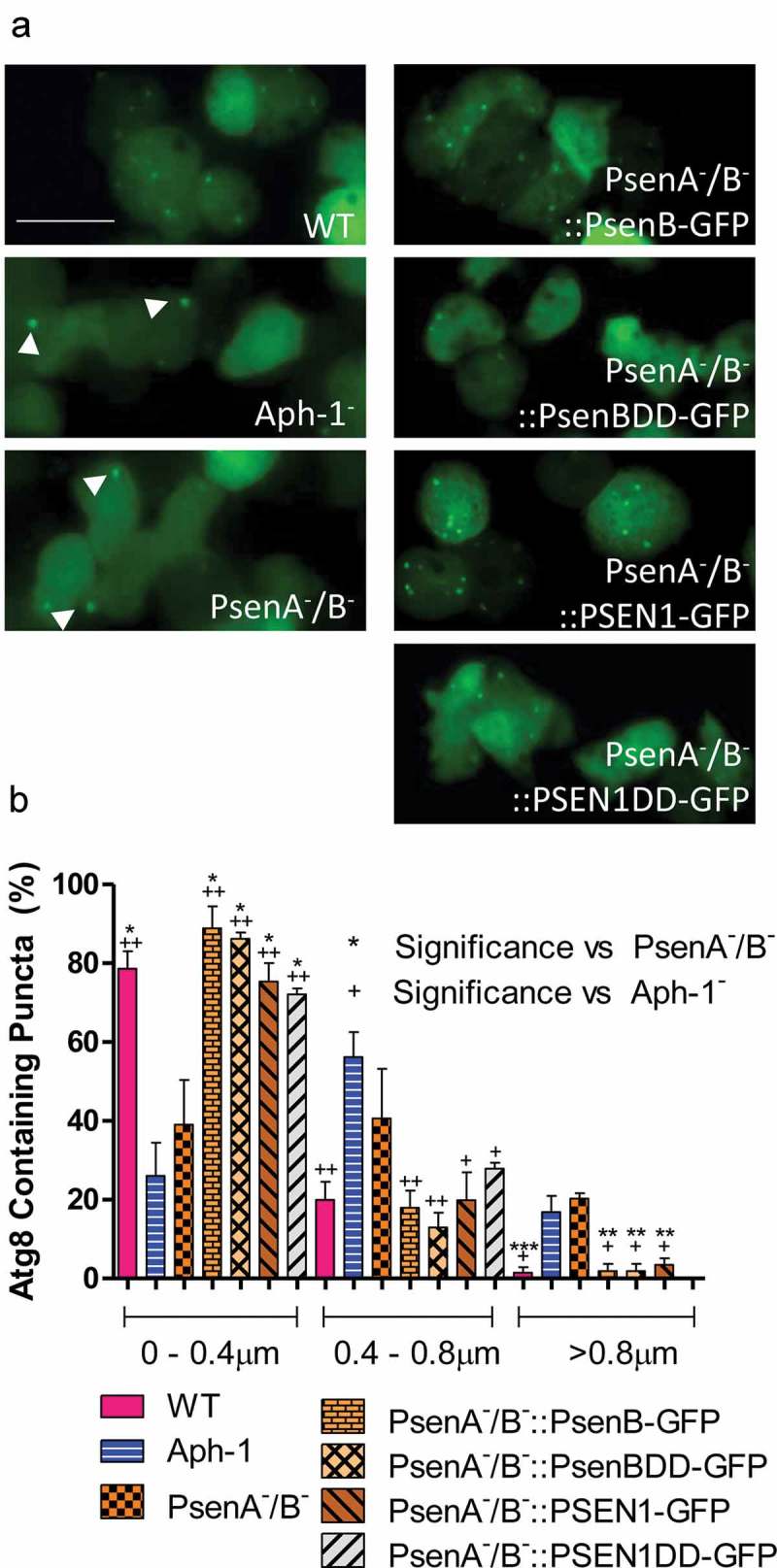
Dictyostelium mutants lacking γ-secretase component orthologs show aberrant size and localisation of GFP-Atg8. Visualization of GFP-Atg8 localisation in wild type cells, PsenA−/B− and Aph-1− and PsenA−/B− cells following rescue by the proteolytic and non-proteolytic Dictyostelium psenB or the equivalent human PSEN1 proteins (PsenB-GFP and PsenBDD-GFP or PSEN1-GFP and or PSEN1DD-GFP) respectively. (a) Wild-type cells show multiple small and distributed GFP-Atg8-containing autophagosomes, whereas a single large punctum is seen in a proportion of PsenA−/B− and Aph-1− cells, that is no longer observed when cells are rescued following expression of Dictyostelium psenB orhuman PSEN1 (proteolytically active or inactive). Scale bar: 10μ m. (b) Quantification of the size of GFP-Atg8 -containing autophagosomes shows an increase in the absence of a functional γ-secretase complex, and this is restored by both Dictyostelium and human presenilin proteins (both proteolytically active and inactive). Data are derived from triplicate experiments measuring approximately 50 cells per experiment. ‘+’ compares to Aph-1−, ‘*’compares to PsenA−/B−, where * or + is P < 0.05, ** or ++ is P < 0.01, *** or +++ is P < 0.05.
To test for the relevance of proteolytic activity in regulating autophagic function, we again expressed both active and inactive forms of both Dictyostelium psenB and human PSEN1 in PsenA−/B− cells expressing GFP-Atg8. In these live-cell experiments, fluorescence was primarily derived from GFP-Atg8 (Figure S7), rather than fluorescently labelled psenB, Aph-1 and Ncstn proteins that localize to the ER (Figure S8). Expression of each form of presenilin rescued the ability of cells to form numerous small GFP-Atg8 structures, confirming this effect was related to loss of presenilin, again independent of proteolytic activity, and with conserved function shown in the human protein (Figure 4(a,b)).
Finally, we investigated a role for the Dictyostelium γ-secretase component orthologs in autophagic degradation, by employing a western blot approach to examine autophagic flux as indicated by the cleavage of free GFP from GFP-Atg8 [50]. Comparing wild-type cells with PsenA−/B− and Aph-1− cells expressing GFP-Atg8, we measured the ratio of free GFP to total GFP-Atg8 in the absence and presence of lysosomal protease inhibitors as a measure of autophagic degradation. We showed that treatment of wild-type cells with protease inhibitor significantly inhibited the release of free GFP from GFP-Atg8 (p < 0.05) consistent with reduced autophagic degradation (Figure 5(a,b)). Ablation of psenA/B or Aph-1 resulted in a 39% and 49% decrease in the ratio of free GFP:GFP-Atg8, which was not significantly changed upon protease inhibitor treatment, suggesting a severe reduction in autophagic flux in PsenA−/B− and Aph-1− cells. Expression of the human PSEN1 in the PsenA−/B− cells restored wild-type autophagic levels. These experiments confirm a role for psenA/B and Aph-1 proteins in autophagic regulation and demonstrate that this cellular function is conserved with the human PSEN1 protein.
Figure 5.
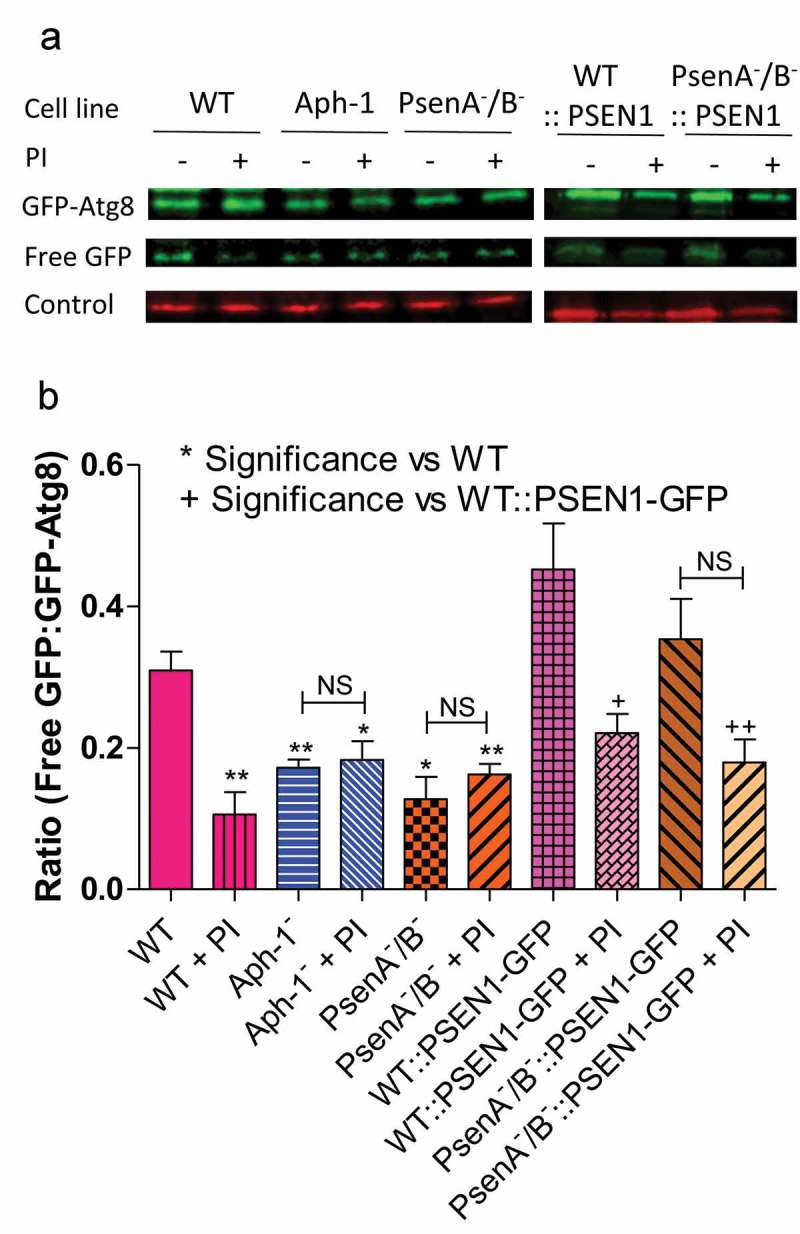
Dictyostelium mutants lacking γ-secretase component orthologs show decreased autophagic flux. Representative western blot analysis using an anti-GFP antibody enables comparison of levels of free GFP to GFP-Atg8 in the presence or absence of protease inhibitor (PI) treatment, allowing the comparison of wild type, Aph-1−, and PsenA−/B− cell autophagic flux, and following rescue by the proteolytically active human PSEN1 protein (PSEN1-GFP). (a) Levels of free GFP are reduced in wild-type cells following PI treatment. In both Aph-1− and PsenA−/B− cells, free GFP levels are reduced in the absence of PI, and remain low following PI treatment. Expression of PSEN1-GFP in the PsenA−/B− cells restores free GFP levels in untreated cells, and PI sensitivity. Endogenously biotinylated mitochondrial protein MCCC1 was used as a loading control. (b) Quantitation of free GFP to GFP-Atg8 ratios shows the absence of a functional γ-secretase complex reduces autophagic flux, and this is restored by the human PSEN1 protein. Data are derived from triplicate independent experiments (±SEM). ‘*’ compares to wild type, ‘+’ compares to PsenA−/B−, where * or + is P < 0.05, ** or ++ is P < 0.01; NS, not significant.
Loss of γ-secretase components orthologs results in large ubiquitin-positive structures
In Dictyostelium, large GFP-Atg8-positive structures typically consist of high molecular weight ubiquitinated protein that cannot be cleared when autophagy is impaired [39]. We therefore examined the colocalization of GFP-Atg8 with ubiquitin-positive structures in the mutants lacking psenA/B and Aph-1 proteins. In wild type cells, the multiple small GFP-Atg8 puncta that were observed did not contain ubiquitinated proteins (Figure 6(a)). In contrast, the large GFP-Atg8-positive structures observed following loss of the γ-secretase complex, in both Aph-1− and PsenA−/B− cells, colocalized with ubiquitinated protein. Rescue of this phenotype in the PsenA−/B− cells with both the proteolytically active and inactive version of the Dictyostelium PsenB and human PSEN1 proteins led to clearance of these ubiquitinated structures.
We then confirmed the effect of the loss of psenA/B and Aph-1 proteins on the accumulation of high molecular weight ubiquitinated proteins by western blotting (Figure 6(b,c)). Consistent with our results above, both the Aph-1− and PsenA−/B− cells showed a significant increase in high molecular weight ubiquitinated proteins (by 55% and 46% respectively; p = 0.0002 and p = 0.0001 respectively) again suggesting impaired protein degradation in these mutants. Accumulation of high molecular weight ubiquitinated protein was reversed by restoration of presenilin activity for both the Dictyostelium psenB (wild type and proteolytically inactive) and human PSEN1 (wild type and proteolytically inactive) rescue strains (Figure 6(b)). Importantly, accumulation of high molecular weight ubiquitinated protein indicated that these presenilin proteins and Aph-1 are physiologically important in maintaining basal autophagy levels and clearing misfolded proteins in Dictyostelium. Furthermore the function of these presenilin proteins are non-proteolytic and are conserved between Dictyostelium and humans.
Figure 6.
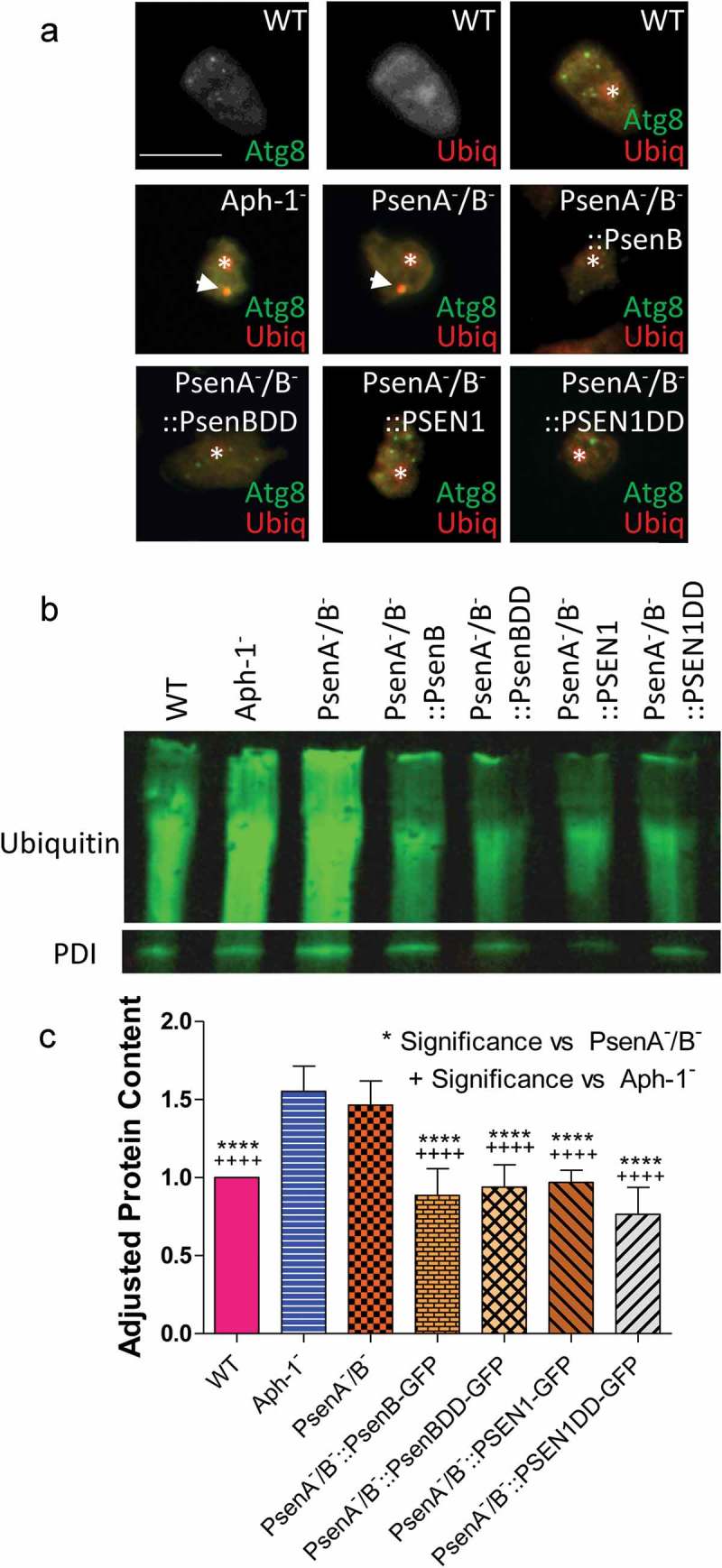
Dictyostelium mutants lacking γ-secretase component orthologs show ubiquitination defects. Analysis of ubiquitination levels in wild-type cells, PsenA−/B− and Aph-1− and PsenA−/B− cells following rescue by the proteolytic and non-proteolytic Dictyostelium PsenB or the equivalent human PSEN1 proteins (PsenB-GFP and PsenBDD-GFP or PSEN1-GFP and PSEN1DD-GFP) respectively. (a) In cells expressing GFP-Atg8, immunofluorescence analysis shows co-localization of GFP and ubiquitin in single large puncta in Aph-1− and PsenA−/B− cells that are absent in wild-type cells, and are absent following rescue with the presenilin proteins. Scale bar: 10μ m. (b) Representative western blot analysis using anti-ubiquitin in the wild type, mutants and following rescue as indicated. Anti-PDI was used as a loading control for normalisation. (c) Quantification of anti-ubiquitin western blot shows a ~ 50% increase in large molecular weight ubiquitinated protein in cells lacking a functional γ-secretase complex when compared to normalized wild type levels. This increase is restored to wild type levels in PsenA−/B− cells expressing Dictyostelium PsenB or human PSEN1 proteins regardless of proteolytic activity. Data are derived from 5 independent experiments.
Discussion
In this study, we have shown that in Dictyostelium, orthologous components of the γ-secretase complex have roles in macropinocytosis, phagocytosis and autophagy, most likely through mediating lysosomal acidification. Recent studies in mammalian models of Alzheimer disease have investigated the role of these components in vesicular trafficking [7,11,13,15,48,51–53], and suggest that dysfunction of these processes may contribute to the progression and pathology of Alzheimer disease. These studies typically describe aberrant intracellular degradation as a result of dysfunction of presenilin proteins, rather than the entirety of the complex [5,42,51,52]. Here, we used Dictyostelium discoideum as a model to investigate orthologous components of the γ-secretase complex, taking advantage of the ability to delete individual genes in stable isogenic cultures to monitor changes in cell and developmental function, and to restore protein activity using wild type and proteolytically inactive presenilin proteins to rescue mutant phenotypes [22,31]. Using these approaches we show that in Dictyostelium, ablation of Ncstn, Aph-1 or both psenA/B genes results in deficient endocytosis and pH-dependent development, and loss of Aph-1 and psenA/B proteins gives rise to defects in lysosomal acidification and autophagy. We further show that these functions are dependent on the non-proteolytic activity of the psenA/B proteins that are conserved in the human PSEN1 protein.
We show that loss of Ncstn, Aph-1 or both psenA/B proteins inhibits macropinocytosis, a process by which cells take up large quantities of extracellular fluid. However, the exact mechanism that drives and controls the process is not yet fully understood [54]. Dictyostelium has proven to be an important model organism in macropinocytosis research and has led to a number of related discoveries, including the role of RasS in maintaining normal actin function in this process [55], the roles of phosphatidylinositol 3-kinases in macropinocytosis control [56], and the role of WASH in macropinosome recycling [49,54]. Further, both axeB/neurofibromin [57] and ndkC/nucleoside diphosphate kinase C [58] function to negatively regulate macropinocytosis. In mammalian cells, a number of these key regulatory pathways are conserved across evolution, including the function of RAS proteins, phosphatidylinositol 3-kinases, F-actin and WASH [49,59–61]. Our data now adds to this list, suggesting that orthologous components of the γ-secretase complex are necessary for optimal fluid uptake in Dictyostelium [55,57,62], involving a non-proteolytic activity of psenB protein, and these defects can be rescued by expression of a human PSEN1 protein. These results are consistent with but extend those shown in mammalian Alzheimer disease models, where loss of γ-secretase function results in aberrant macropinocytosis [5,6], and dysfunctional protein aggregate clearance for affected neuronal cells [5,6,42,52].
After extracellular material has been internalized, vesicles move through the intracellular endocytic pathway before finally fusing with lysosomes for degradation of vesicular material [63–65]. We show here, that Dictyostelium cells lacking Ncstn, Aph-1 or both psenA/B proteins fail to develop under basic pH conditions, consistent with that seen in other mutants that are unable to appropriately acidify their vesicles [45]. We then confirmed this role directly, by measuring autophagosome acidification, phagosome proteolysis [34,41] and autophagic flux in cells lacking Aph-1 or both psenA/B proteins. In mammalian models, dysfunctional lysosomal pH regulation has been linked to Alzheimer disease [66], in addition to other disease such as Pompe [67] and Niemann-Pick disease [68], as well as cholesterol uptake, and atherosclerosis – all resulting in inefficient degradation of intra-endosome material. In regard to Alzheimer disease, presenilin proteins have been implicated in a non-proteolytic, complex-independent role in maintaining lysosomal pH [15,51,52]. These studies suggest that presenilin regulates lysosomal pH through vacuolar-ATPase-mediated lysosomal acidification, resulting in altered calcium levels [51]. In Dictyostelium, we have shown that Ncstn, Aph-1 and psenA/B proteins are localized in the endoplasmic reticulum and this localization is unaltered in the absence of other complex components (Figure S8), consistent with that shown for Vmp1 involved with lysosomal function and autophagy [39] and a role of the complex in interacting with v-ATPase proteins for vesicle acidification and lysosomal activity. It is important to note here that our data do not explicitly link macropinocytosis and acidification defects, and further studies will need to investigate this in detail.
Previous research in Dictyostelium suggested that the γ-secretase complex is important for phagocytosis, and that this process is dependent upon proteolytic activity [31]. Our study confirms the role of Aph-1 and psenA/B proteins in phagocytosis, but demonstrates that it is not reliant upon proteolytic activity [22]. These data quantify the loss of (phago-)lysosomal proteolysis in cells lacking the orthologs of multiple γ-secretase complex components, and together with data on macropinocytosis, suggest that material ingested through both processes are unlikely to be efficiently degraded, and these effects are commonly observed in cellular models for Alzheimer disease [5,6,66].
Another cellular process dependent upon lysosomal degradation is autophagy [69]. Dictyostelium has been widely used to investigate autophagy and the importance of autophagy, and its role in multicellular development [40]. Here we show that cells lacking psenA/B or Aph-1 proteins share a number of phenotypes with autophagy dysfunctional mutants, including defective GFP-Atg8 localization [37,39], co-localisation between GFP-Atg8 and ubiquitin aggregates [39], and an abundance of high molecular weight ubiquitinated protein [39,70], reduced autophagic flux, and aberrant development. These data demonstrate that orthologs of multiple γ-secretase complex components play a key role in the autophagic process in Dictyostelium. Consistent with this, neurons of patients diagnosed with Alzheimer disease also accumulate neurotoxic peptides due to inefficient lysosomal acidification and a resultant build-up of autophagosomes rich in amyloid precursor protein [7,8,52]. We further show that, in Dictyostelium, this deficiency in autophagy and accumulation of ubiquitinated proteins is not caused by loss of psenB proteolytic activity, and this activity can be fulfilled by the human PSEN1 protein [7,51].
In this study, we demonstrate that the Dictyostelium orthologs of γ-secretase complex components play a key role in regulating macropinocytosis, phagocytosis, acidification of lysosomal vesicles, and autophagy. The reduction of these activities following the loss of two (psenA/B and Aph-1) or three (psenA/B, Aph-1 and Ncstn) orthologs of γ-secretase complex components suggest that these phenotypes may be regulated by a Dictyostelium γ-secretase complex, but we cannot explicitly demonstrate that these roles are not independent. Similarly, the interrelatedness of these cellular function is also consistent with a common role for a Dictyostelium γ-secretase complex in this process, and that this function is not dependent on the proteolytic activity of the putative γ-secretase complex. Finally, we demonstrate that these roles of the psenA/B proteins can be rescued in Dictyostelium through expression of human PSEN1. Thus we propose that, in Dictyostelium, an orthologous γ-secretase complex plays a key role in maintaining endocytosis, lysosomal trafficking and autophagy through a non-proteolytic function, conserved between Dictyostelium and humans across the vast evolutionary gap that separates these two organisms.
Materials and methods
Cell culture and maintenance
Dictyostelium wild type strain Ax2 were grown in HL5 medium (Formedium, HLB0103) supplemented with 10% glucose (Sigma-Aldrich, G8270). PsenA−/B− cells and rescue strains, and Ncstn− cells have been previously described [22], and are maintained in HL5 medium supplemented with/without 10 μg/ml hygromycin (Formedium, HYG1000).
Plasmid construction and transformation
Aph-1− mutant cells were generated by homologous integration using the Cre-Lox system [71].
Briefly, a knockout vector was designed consisting of a 5ʹ and 3ʹ arm of homology (primers:
GTG GAT CCT ATA AGT ATT TTA AAG ATT/AAC TGC AGA GAT ATT TAA AAA TGT TTC TTA CC and TTA TTC CAT GGA GTT TAT AAC GTT TT/AAT GGT ACC TTG ATA ATG TTA AAA TGA) in order to ablate a central 762 base pair region of the gene. The linearized vector was electroporated into wild-type Dictyostelium cells, and transformants were screened by PCR in order to identify homologous recombinants [72].
Macropinocytosis and exocytosis assay
Rate of TRITC-Dextran uptake was measured as described previously [62]. Briefly, 2.5 × 107 cells were resuspended in 5 ml of HL5 medium supplemented with 100 μl or 100 mg/ml TRITC-Dextran (Sigma-Aldirch, T1162). At time points 0, 15, 30, 45, 60, 90, and 120 min, 500 μl of this suspension was removed, washed in phosphate buffer and measured on a Perkin Elmer LS50B spectrophotometer. Relative change in fluorescence was calculated and normalised against total protein content of each respective cell line. The rate of uptake was measured using the linear function in GraphPad Prism.
To determine whether efflux was also affected by γ-secretase ablation a simple assay was utilised [73]. Briefly, 2.5 × 107 cells were incubated in HL-5 with 100 mg/ml TRITC-Dextran for a period of 3 h. Following incubation, cells were washed and resuspended in 5 ml of phosphate buffer before shaking incubation. At time points 0, 15, 30, 45, 60, 90 and 120 min 500 μl of the suspension was removed and the fluorescence measured. Change in fluorescence was calculated relative to the measurement at time point 0 min of each respective cell line. The rate of efflux was calculated using GraphPad Prism.
Dictyostelium development at varying pH
In these assays, 1 × 107 cells were plated and allowed to develop on 47mm nitrocellulose filters (Millipore, HAWP04700) over 24 h [74]. Each filter was placed on an absorbent 3M paper pad (Millipore, AP1004700) soaked in phosphate buffer of varying pH. Phosphate buffer was prepared using varying amounts of KH2PO4 and K2HPO4 to modulate pH without altering ionic strength. Subsequent developmental phenotypes were imaged using a dissection microscope (Leica) and a QICAM FAST 1394 camera (QImaging).
Fluorescence microscopy and quantification
Cells were imaged on an Olympus IX71 wide-field fluorescence microscope. Images were captured using a QICAM FAST 1394 camera. For measurement of Atg8-positive structure size cells were analysed using ImageJ [75] and cell size was measured across the largest diameter. More than 100 GFP-Atg8 positive structures were measured, using around 50 cells per experiment including at least three independent experimental repeats. For live-cell imaging, cells were imaged under a layer of ~1.5-mm thick 1% phosphate buffered agarose [49].
Analysis of autophagosome maturation
The autophagy reporter GFP-Atg8 was expressed in cells using the extrachromosomal expression plasmid pDM430 [38]. To both reduce the movement of vesicles in the Z-plane and stimulate autophagosome formation cells were compressed under a thin layer of 1% agarose in HL-5 medium as previously described [38]. Cells were seeded in glass-bottomed microscopy dishes before removal of most of the medium, application of a ~ 2-mm thick agarose slab and compression by blotting with paper and capillary action. After 10 min, images were captured using a Perkin-Elmer Ultraview VoX inverted spinning disc microscope, using a 100 × 1.4NA objective and Hammamatsu C9100-50 EM-CCD camera. 4 Z planes at 1μM spacing were captured every 2 s. The point of autophagosome completion was determined by the characteristic enlargement and diming of GFP fluorescence [41]. The time until GFP-fluorescence was undetectable was subsequently measured from randomised, blinded movies captured from at least three independent experiments.
Phagosomal proteolysis assays
Phagosomal proteolytic activity was measured by feeding cells DQgreen/Alexa Fluor 594 (DQ-BSA; Invitrogen, D12050) co-labelled 3-μm silica beads (Kisker Biotech, PSI-3.0COOH) as previously described [76]. Briefly 3 × 105 cells/well were seeded in a 96-well plate before addition of beads, and fluorescence measured on a plate reader each minute in triplicate. Proteolysis was normalised to Alexa Fluor 594 fluorescence, over time to account for potential differences in bead uptake and rates normalized to wild-type cells to calculate relative activity.
Western blotting and immunofluorescence
Analysis of high molecular weight ubiquitinated protein was carried out by western blot as previously described [70]. Briefly, the proteins of 2 × 105 cells were separated by SDS gel electrophoresis and the membrane was probed with α-PDI [77] (an ER marker protein, disulfide isomerase a kind gift from Annette Muller-Taübenberger) as a loading control and α-ubiquitin (Cell Signalling Technology, P4D1) at 1:50, and 1:1000 dilutions, respectively. Relative amounts of high molecular weight ubiquitinated protein were calculated using Image Studio Lite (LI-COR Biosciences) and these were normalized against wild-type levels of ubiquitinated protein.
For colocalization of ubiquitin and GFP-Atg8, cells were fixed in −80°C methanol as previously described [78]. After fixation cells were probed with α-GFP (Chromotek, 3H9) and anti-ubiquitin (New England Biolabs, 3936S) antibodies at 1:500 and 1:200 dilution concentrations, secondary antibodies used were anti-rat Alexa Fluor 488 and anti-mouse Alexa Fluor 350 (Life Technologies, A-21,210 and A-31,552 respectively) at 1:1000 concentration dilutions. Cells were then imaged to visualise colocalization between ubiquitin and GFP-Atg8-positive structures.
To analyze autophagic flux a western blot based approach was utilised [50]. Briefly, 1.3 × 106 cells were seeded in a 6 well plate before treatment with protease inhibitors (Roche Life Science, 05892791001) for 1 h, when cells were harvested and proteins extracted. Western blots were probed with an anti-GFP antibody (1:1000 dilution) and levels of the endogenously biotinyated mitochondrial protein MCCC1 detected with a streptavidin-conjugated antibody used as a loading control [79] (Thermo Fisher Scientific, S21378). The fluorescent ratio of free GFP to GFP-Atg8 was calculated to determine a measure of autophagic flux.
To visualize Dictyostelium γ-secretase components localization, cells were fixed using 80°C methanol [78] and probed with antibodies. Briefly, cells overexpressing GFP-tagged components were seeded onto coverslips and fixed by submerging in −80°C methanol for 30 min. Following fixation, cells were washed in room temperature phosphate-buffered saline, stained with anti-GFP and anti-crtA (calreticulin) antibodies (a kind gift from Annette Muller-Taübenberger), subsequently stained with appropriate secondary antibodies to allow for ER visualization, and nuclei were stained with DAPI.
Statistical analysis
Statistical analysis depended upon the data analyzed. For normally distributed data a two-way ANOVA was utilised when comparing multiple data groups to each other. For data that is not normally distributed the Mann-Whitney test was used, and for data that was compared to a single normalised mean value a one sample T-test was used. Statistical analysis was carried out using GraphPad Prism Software.
Funding Statement
This work was supported by NC3Rs; Royal Society University Research Fellowship under Grant UF140624; MRC under Grant (G0700091); Wellcome Trust under Grant GR077544AIA, and a BBSRC studentship award.
Author contributions
DS, PB, JSK and RSBW designed the study. DS, GO, EW and JSK performed the research, data analysis and modelling. The paper was written by DS, PB, JSK and RSBW.
Data availability
All data related to this study are available in the paper or via supplementary material.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Scheuner D, Eckman C, Jensen M, et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimers disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimers disease. Nat Med. 1996. August;2(8):864–870. PubMed PMID: 8705854. [DOI] [PubMed] [Google Scholar]
- [2].De Strooper B, Annaert W, Cupers P, et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999. April 8;398(6727):518–522. PubMed PMID: 10206645. [DOI] [PubMed] [Google Scholar]
- [3].De Strooper B, Iwatsubo T, Wolfe MS.. Presenilins and gamma-secretase: structure, function, and role in Alzheimer Disease. Cold Spring Harb Perspect Med. 2012. January;2(1):a006304 PubMed PMID: 22315713; PubMed Central PMCID: PMC3253024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].De Strooper B, Saftig P, Craessaerts K, et al. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998. January 22;391(6665):387–390. PubMed PMID: 9450754. [DOI] [PubMed] [Google Scholar]
- [5].Zhang M, Haapasalo A, Kim DY, et al. Presenilin/gamma-secretase activity regulates protein clearance from the endocytic recycling compartment. FASEB J. 2006. June;20(8):1176–1178. PubMed PMID: 16645046. [DOI] [PubMed] [Google Scholar]
- [6].Tamboli IY, Prager K, Thal DR, et al. Loss of gamma-secretase function impairs endocytosis of lipoprotein particles and membrane cholesterol homeostasis. J Neurosci. 2008. November 12;28(46):12097–12106. PubMed PMID: 19005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lee JH, Yu WH, Kumar A, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010. June 25;141(7):1146–1158. PubMed PMID: 20541250; PubMed Central PMCID: PMCPMC3647462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pasternak SH, Bagshaw RD, Guiral M, et al. Presenilin-1, nicastrin, amyloid precursor protein, and gamma-secretase activity are co-localized in the lysosomal membrane. J Biol Chem. 2003. July 18;278(29):26687–26694. PubMed PMID: 12736250. [DOI] [PubMed] [Google Scholar]
- [9].Wolfe MS. Toward the structure of presenilin/gamma-secretase and presenilin homologs. Biochim Biophys Acta. 2013. December;1828(12):2886–2897. PubMed PMID: 24099007; PubMed Central PMCID: PMCPMC3801419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Frakes AE, Ferraiuolo L, Haidet-Phillips AM, et al. Microglia induce motor neuron death via the classical NF-kappaB pathway in amyotrophic lateral sclerosis. Neuron. 2014. March 05;81(5):1009–1023. PubMed PMID: 24607225; PubMed Central PMCID: PMCPMC3978641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ghavami S, Shojaei S, Yeganeh B, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014. January;112:24–49. PubMed PMID: 24211851. [DOI] [PubMed] [Google Scholar]
- [12].Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015. June;16(6):345–357. PubMed PMID: 25991442. [DOI] [PubMed] [Google Scholar]
- [13].Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013. August;19(8):983–997. PubMed PMID: 23921753. [DOI] [PubMed] [Google Scholar]
- [14].Ulamek-Koziol M, Furmaga-Jablonska W, Januszewski S, et al. Neuronal autophagy: self-eating or self-cannibalism in Alzheimers disease. Neurochem Res. 2013. September;38(9):1769–1773. PubMed PMID: 23737325; PubMed Central PMCID: PMCPMC3732752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wolfe DM, Lee JH, Kumar A, et al. Autophagy failure in Alzheimers disease and the role of defective lysosomal acidification. Eur J Neurosci. 2013. June;37(12):1949–1961. PubMed PMID: 23773064; PubMed Central PMCID: PMCPMC3694736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Capell A, Grunberg J, Pesold B, et al. The proteolytic fragments of the Alzheimers disease-associated presenilin-1 form heterodimers and occur as a 100-150-kDa molecular mass complex. J Biol Chem. 1998. February 6;273(6):3205–3211. PubMed PMID: 9452432. [DOI] [PubMed] [Google Scholar]
- [17].Parks AL, Curtis D. Presenilin diversifies its portfolio. Trends Genet. 2007. March;23(3):140–150. PubMed PMID: 17280736. [DOI] [PubMed] [Google Scholar]
- [18].Kang DE, Soriano S, Frosch MP, et al. Presenilin 1 facilitates the constitutive turnover of beta-catenin: differential activity of Alzheimers disease-linked PS1 mutants in the beta-catenin-signaling pathway. J Neurosci. 1999. June 1;19(11):4229–4237. PubMed PMID: 10341227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kang DE, Soriano S, Xia X, et al. Presenilin couples the paired phosphorylation of beta-catenin independent of axin: implications for beta-catenin activation in tumorigenesis. Cell. 2002. September 20;110(6):751–762. PubMed PMID: 12297048. [DOI] [PubMed] [Google Scholar]
- [20].Khandelwal A, Chandu D, Roe CM, et al. Moonlighting activity of presenilin in plants is independent of gamma-secretase and evolutionarily conserved. Proc Natl Acad Sci U S A. 2007. August 14;104(33):13337–13342. PubMed PMID: 17684101; PubMed Central PMCID: PMCPMC1948938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li D, Parks SB, Kushner JD, et al. Mutations of presenilin genes in dilated cardiomyopathy and heart failure. Am J Hum Genet. 2006. December;79(6):1030–1039. PubMed PMID: 17186461; PubMed Central PMCID: PMCPMC1698711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ludtmann MH, Otto GP, Schilde C, et al. An ancestral non-proteolytic role for presenilin proteins in multicellular development of the social amoeba Dictyostelium discoideum. J Cell Sci. 2014;127:1576–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Otto GP, Sharma D, Williams RS. Non-catalytic roles of presenilin throughout evolution. J Alzheimers Dis. 2016. April 12;52(4):1177–1187. PubMed PMID: 27079701; PubMed Central PMCID: PMCPMC4927835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Murayama M, Tanaka S, Palacino J, et al. Direct association of presenilin-1 with beta-catenin. FEBS Lett. 1998. August 14;433(1–2):73–77. PubMed PMID: 9738936. [DOI] [PubMed] [Google Scholar]
- [25].Zhang Z, Hartmann H, Do VM, et al. Destabilization of beta-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature. 1998. October 15;395(6703):698–702. PubMed PMID: 9790190. [DOI] [PubMed] [Google Scholar]
- [26].Xia X, Qian S, Soriano S, et al. Loss of presenilin 1 is associated with enhanced beta-catenin signaling and skin tumorigenesis. Proc Natl Acad Sci U S A. 2001. September 11;98(19):10863–10868. PubMed PMID: 11517342; PubMed Central PMCID: PMCPMC58565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shilling D, Muller M, Takano H, et al. Suppression of InsP3 receptor-mediated Ca2+ signaling alleviates mutant presenilin-linked familial Alzheimers disease pathogenesis. J Neurosci. 2014. May 14;34(20):6910–6923. PubMed PMID: 24828645; PubMed Central PMCID: PMCPMC4019804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tu H, Nelson O, Bezprozvanny A, et al. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimers disease-linked mutations. Cell. 2006. September 8;126(5):981–993. PubMed PMID: 16959576; PubMed Central PMCID: PMCPMC3241869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Boyles RS, Lantz KM, Poertner S, et al. Presenilin controls CBP levels in the adult Drosophila central nervous system. PLoS One. 2010. December 14;5(12):e14332 PubMed PMID: 21179466; PubMed Central PMCID: PMCPMC3001863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sarasija S, Norman KR. A gamma-secretase independent role for presenilin in calcium homeostasis impacts mitochondrial function and morphology in caenorhabditis elegans. Genetics. 2015. December;201(4):1453–1466. PubMed PMID: 26500256; PubMed Central PMCID: PMCPMC4676538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].McMains VC, Myre M, Kreppel L, et al. Dictyostelium possesses highly diverged presenilin/gamma-secretase that regulates growth and cell-fate specification and can accurately process human APP: a system for functional studies of the presenilin/gamma-secretase complex. Dis Model Mech. 2010. Sep-Oct;3(9–10):581–594. PubMed PMID: 20699477; PubMed Central PMCID: PMC2931536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chang P, Orabi B, Deranieh RM, et al. The antiepileptic drug valproic acid and other medium-chain fatty acids acutely reduce phosphoinositide levels independently of inositol in Dictyostelium. Dis Model Mech. 2012. January;5(1):115–124. PubMed PMID: 21876211; PubMed Central PMCID: PMCPMC3255550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Waheed A, Ludtmann MH, Pakes N, et al. Naringenin inhibits the growth of Dictyostelium and MDCK-derived cysts in a TRPP2 (polycystin-2)-dependent manner. Br J Pharmacol. 2014. May;171(10):2659–2670. PubMed PMID: 24116661; PubMed Central PMCID: PMCPMC4009007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].King JS, Gueho A, Hagedorn M, et al. WASH is required for lysosomal recycling and efficient autophagic and phagocytic digestion. Mol Biol Cell. 2013. September;24(17):2714–2726. PubMed PMID: 23885127; PubMed Central PMCID: PMCPMC3756923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lima WC, Leuba F, Soldati T, et al. Mucolipin controls lysosome exocytosis in Dictyostelium. J Cell Sci. 2012. May 01;125(Pt 9):2315–2322. PubMed PMID: 22357942. [DOI] [PubMed] [Google Scholar]
- [36].Neuhaus EM, Soldati T. A myosin I is involved in membrane recycling from early endosomes. J Cell Biol. 2000. September 04;150(5):1013–1026. PubMed PMID: 10973992; PubMed Central PMCID: PMCPMC2175260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Otto GP, Wu MY, Kazgan N, et al. Dictyostelium macroautophagy mutants vary in the severity of their developmental defects. J Biol Chem. 2004. April 09;279(15):15621–15629. PubMed PMID: 14736886. [DOI] [PubMed] [Google Scholar]
- [38].King JS, Veltman DM, Insall RH. The induction of autophagy by mechanical stress. Autophagy. 2011. December;7(12):1490–1499. PubMed PMID: 22024750; PubMed Central PMCID: PMCPMC3327616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Calvo-Garrido J, Escalante R. Autophagy dysfunction and ubiquitin-positive protein aggregates in Dictyostelium cells lacking Vmp1. Autophagy. 2010. January;6(1):100–109. PubMed PMID: 20009561. [DOI] [PubMed] [Google Scholar]
- [40].Otto GP, Wu MY, Kazgan N, et al. Macroautophagy is required for multicellular development of the social amoeba Dictyostelium discoideum. J Biol Chem. 2003. May 16;278(20):17636–17645. PubMed PMID: 12626495. [DOI] [PubMed] [Google Scholar]
- [41].Dominguez-Martin E, Cardenal-Munoz E, King JS, et al. Methods to monitor and quantify autophagy in the social amoeba dictyostelium discoideum. Cells. 2017. July 03;6(3). PubMed PMID: 28671610. doi: 10.3390/cells6030018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Fukumori A, Okochi M, Tagami S, et al. Presenilin-dependent gamma-secretase on plasma membrane and endosomes is functionally distinct. Biochemistry. 2006. April 18;45(15):4907–4914. PubMed PMID: 16605258. [DOI] [PubMed] [Google Scholar]
- [43].Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000. December 1;290(5497):1717–1721. PubMed PMID: 11099404; PubMed Central PMCID: PMCPMC2732363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Gekle M, Mildenberger S, Freudinger R, et al. Endosomal alkalinization reduces Jmax and Km of albumin receptor-mediated endocytosis in OK cells. Am J Physiol. 1995. May;268(5 Pt 2):F899–906. PubMed PMID: 7539587. [DOI] [PubMed] [Google Scholar]
- [45].Davies L, Farrar NA, Satre M, et al. Vacuolar H(+)-ATPase and weak base action in Dictyostelium. Mol Microbiol. 1996. October;22(1):119–126. PubMed PMID: 8899714. [DOI] [PubMed] [Google Scholar]
- [46].Gopaldass N, Patel D, Kratzke R, et al. Dynamin A, Myosin IB and Abp1 couple phagosome maturation to F-actin binding. Traffic. 2012. January;13(1):120–130. PubMed PMID: 22008230. [DOI] [PubMed] [Google Scholar]
- [47].Feng T, Tammineni P, Agrawal C, et al. Autophagy-mediated regulation of BACE1 protein trafficking and degradation. J Biol Chem. 2017. February 03;292(5):1679–1690. PubMed PMID: 28028177; PubMed Central PMCID: PMCPMC5290944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Li Q, Liu Y, Sun M. Autophagy and Alzheimers disease. Cell Mol Neurobiol. 2017. April;37(3):377–388. PubMed PMID: 27260250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Buckley CM, Gopaldass N, Bosmani C, et al. WASH drives early recycling from macropinosomes and phagosomes to maintain surface phagocytic receptors. Proc Natl Acad Sci U S A. 2016. October 04;113(40):E5906–E5915. PubMed PMID: 27647881; PubMed Central PMCID: PMCPMC5056073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Cardenal-Munoz E, Arafah S, Lopez-Jimenez AT, et al. Mycobacterium marinum antagonistically induces an autophagic response while repressing the autophagic flux in a TORC1- and ESX-1-dependent manner. PLoS Pathog. 2017. April;13(4):e1006344 PubMed PMID: 28414774; PubMed Central PMCID: PMCPMC5407849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lee JH, McBrayer MK, Wolfe DM, et al. Presenilin 1 maintains lysosomal Ca(2+) homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep. 2015. September 01;12(9):1430–1444. PubMed PMID: 26299959; PubMed Central PMCID: PMCPMC4558203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Neely KM, Green KN, LaFerla FM. Presenilin is necessary for efficient proteolysis through the autophagy-lysosome system in a gamma-secretase-independent manner. J Neurosci. 2011. February 23;31(8):2781–2791. PubMed PMID: 21414900; PubMed Central PMCID: PMCPMC3064964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Steele JW, Fan E, Kelahmetoglu Y, et al. Modulation of autophagy as a therapeutic target for Alzheimers disease. Postdoc J. 2013. February;1(2):21–34. PubMed PMID: 28286801; PubMed Central PMCID: PMCPMC5342246. [PMC free article] [PubMed] [Google Scholar]
- [54].Buckley CM, King JS. Drinking problems: mechanisms of macropinosome formation and maturation. FEBS J. 2017. November;284(22):3778–3790. PubMed PMID: 28544479. [DOI] [PubMed] [Google Scholar]
- [55].Chubb JR, Wilkins A, Thomas GM, et al. The Dictyostelium RasS protein is required for macropinocytosis, phagocytosis and the control of cell movement. J Cell Sci. 2000. February;113(Pt 4):709–719. PubMed PMID: 10652263. [DOI] [PubMed] [Google Scholar]
- [56].Hoeller O, Bolourani P, Clark J, et al. Two distinct functions for PI3-kinases in macropinocytosis. J Cell Sci. 2013. September 15;126(Pt 18):4296–4307. PubMed PMID: 23843627; PubMed Central PMCID: PMCPMC3772393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Bloomfield G, Traynor D, Sander SP, et al. Neurofibromin controls macropinocytosis and phagocytosis in Dictyostelium. Elife. 2015. March 27;4 PubMed PMID: 25815683; PubMed Central PMCID: PMCPMC4374526. doi: 10.7554/eLife.04940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Annesley SJ, Bago R, Bosnar MH, et al. Dictyostelium discoideum nucleoside diphosphate kinase C plays a negative regulatory role in phagocytosis, macropinocytosis and exocytosis. PLoS One. 2011;6(10):e26024 PubMed PMID: 21991393; PubMed Central PMCID: PMCPMC3186806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Commisso C, Davidson SM, Soydaner-Azeloglu RG, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013. May 30;497(7451):633–637. PubMed PMID: 23665962; PubMed Central PMCID: PMCPMC3810415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kozma R, Ahmed S, Best A, et al. The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol Cell Biol. 1995. April;15(4):1942–1952. PubMed PMID: 7891688; PubMed Central PMCID: PMCPMC230420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Sasaki AT, Janetopoulos C, Lee S, et al. G protein-independent Ras/PI3K/F-actin circuit regulates basic cell motility. J Cell Biol. 2007. July 16;178(2):185–191. PubMed PMID: 17635933; PubMed Central PMCID: PMCPMC2064438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hacker U, Albrecht R, Maniak M. Fluid-phase uptake by macropinocytosis in Dictyostelium. J Cell Sci. 1997. January;110(Pt 2):105–112. PubMed PMID: 9044041. [DOI] [PubMed] [Google Scholar]
- [63].Gruenberg J, Maxfield FR. Membrane transport in the endocytic pathway. Curr Opin Cell Biol. 1995. August;7(4):552–563. PubMed PMID: 7495576. [DOI] [PubMed] [Google Scholar]
- [64].Luzio JP, Rous BA, Bright NA, et al. Lysosome-endosome fusion and lysosome biogenesis. J Cell Sci. 2000. May;113(Pt 9):1515–1524. PubMed PMID: 10751143. [DOI] [PubMed] [Google Scholar]
- [65].Hu YB, Dammer EB, Ren RJ, et al. The endosomal-lysosomal system: from acidification and cargo sorting to neurodegeneration. Transl Neurodegener. 2015;4:18 PubMed PMID: 26448863; PubMed Central PMCID: PMCPMC4596472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Maxfield FR. Role of endosomes and lysosomes in human disease. Cold Spring Harb Perspect Biol. 2014. May 01;6(5):a016931 PubMed PMID: 24789821; PubMed Central PMCID: PMCPMC3996470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Fukuda T, Roberts A, Ahearn M, et al. Autophagy and lysosomes in Pompe disease. Autophagy. 2006. Oct-Dec;2(4):318–320. PubMed PMID: 16874053. [DOI] [PubMed] [Google Scholar]
- [68].Kirkegaard T, Roth AG, Petersen NH, et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature. 2010. January 28;463(7280):549–553. PubMed PMID: 20111001. [DOI] [PubMed] [Google Scholar]
- [69].Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct. 2002. December;27(6):421–429. PubMed PMID: 12576635. [DOI] [PubMed] [Google Scholar]
- [70].Xiong Q, Unal C, Matthias J, et al. The phenotypes of ATG9, ATG16 and ATG9/16 knock-out mutants imply autophagy-dependent and -independent functions. Open Biol. 2015. April;5(4):150008 PubMed PMID: 25878144; PubMed Central PMCID: PMCPMC4422124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Faix J, Kreppel L, Shaulsky G, et al. A rapid and efficient method to generate multiple gene disruptions in Dictyostelium discoideum using a single selectable marker and the Cre-loxP system. Nucleic Acids Res. 2004. October 26;32(19):e143 PubMed PMID: 15507682; PubMed Central PMCID: PMCPMC528815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Gaudet P, Pilcher KE, Fey P, et al. Transformation of Dictyostelium discoideum with plasmid DNA. Nat Protoc. 2007;2(6):1317–1324. PubMed PMID: 17545968. [DOI] [PubMed] [Google Scholar]
- [73].Rivero F, Maniak M. Quantitative and microscopic methods for studying the endocytic pathway. Methods Mol Biol. 2006;346:423–438. PubMed PMID: 16957305. [DOI] [PubMed] [Google Scholar]
- [74].Williams RS, Eames M, Ryves WJ, et al. Loss of a prolyl oligopeptidase confers resistance to lithium by elevation of inositol (1,4,5) trisphosphate. EMBO J. 1999. May 17;18(10):2734–2745. PubMed PMID: 10329620; PubMed Central PMCID: PMCPMC1171355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012. July;9(7):671–675. PubMed PMID: 22930834; PubMed Central PMCID: PMCPMC5554542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Sattler N, Monroy R, Soldati T. Quantitative analysis of phagocytosis and phagosome maturation. Methods Mol Biol. 2013;983:383–402. PubMed PMID: 23494319. [DOI] [PubMed] [Google Scholar]
- [77].Muller-Taubenberger A, Lupas AN, Li H, et al. Calreticulin and calnexin in the endoplasmic reticulum are important for phagocytosis. Embo J. 2001. December 03;20(23):6772–6782. PubMed PMID: 11726513; PubMed Central PMCID: PMCPMC125758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Hagedorn M, Neuhaus EM, Soldati T. Optimized fixation and immunofluorescence staining methods for Dictyostelium cells. Methods Mol Biol. 2006;346:327–338. PubMed PMID: 16957300. [DOI] [PubMed] [Google Scholar]
- [79].Davidson AJ, King JS, Insall RH. The use of streptavidin conjugates as immunoblot loading controls and mitochondrial markers for use with Dictyostelium discoideum. Biotechniques. 2013. July;55(1):39–41. PubMed PMID: 23834384. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data related to this study are available in the paper or via supplementary material.