

ABSTRACT
Mutations in the ER-associated VAPB/ALS8 protein cause amyotrophic lateral sclerosis and spinal muscular atrophy. Previous studies have argued that ER stress may underlie the demise of neurons. We find that loss of VAP proteins (VAPs) leads to an accumulation of aberrant lysosomes and impairs lysosomal degradation. VAPs mediate ER to Golgi tethering and their loss may affect phosphatidylinositol-4-phosphate (PtdIns4P) transfer between these organelles. We found that loss of VAPs elevates PtdIns4P levels in the Golgi, leading to an expansion of the endosomal pool derived from the Golgi. Fusion of these endosomes with lysosomes leads to an increase in lysosomes with aberrant acidity, contents, and shape. Importantly, reducing PtdIns4P levels with a PtdIns4-kinase (PtdIns4K) inhibitor, or removing a single copy of Rab7, suppress macroautophagic/autophagic degradation defects as well as behavioral defects observed in Drosophila Vap33 mutant larvae. We propose that a failure to tether the ER to the Golgi when VAPs are lost leads to an increase in Golgi PtdIns4P levels, and an expansion of endosomes resulting in an accumulation of dysfunctional lysosomes and a failure in proper autophagic lysosomal degradation.
Abbreviations: ALS: amyotrophic lateral sclerosis; CSF: cerebrospinal fluid; CERT: ceramide transfer protein; FFAT: two phenylalanines in an acidic tract; MSP: major sperm proteins; OSBP: oxysterol binding protein; PH: pleckstrin homology; PtdIns4P: phosphatidylinositol-4-phosphate; PtdIns4K: phosphatidylinositol 4-kinase; UPR: unfolded protein response; VAMP: vesicle-associated membrane protein; VAPA/B: mammalian VAPA and VAPB proteins; VAPs: VAMP-associated proteins (referring to Drosophila Vap33, and human VAPA and VAPB)
KEYWORDS: ALS8, autophagy, PI4KB, Vap33, VAPA/VAPB
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder that is characterized by progressive motor neuron degeneration and muscle weakness [1]. More than 20 ALS associated genes have been identified and these genes affect distinct cellular pathways including RNA processing, nuclear protein transport, and the unfolded protein response (UPR) [2]. One of the key pathological findings is the presence of TARDBP-positive protein aggregates in the cytoplasm of neurons in the brains and spinal cords of patients [1]. Accumulation of protein aggregates in the ER induces a UPR, which attenuates protein translation and promotes proteasome-mediated degradation as well as expression of numerous ER chaperones [3]. Several ALS-causing genes, including VAPB [4,5], VCP [6,7] and UBQLN2 [6], have been documented to play an important role in the ER, and the loss of these proteins promotes the UPR [8,9]. In addition, ER stress has also been documented in SOD1G93A heterozygous mice [4,10]. Whether ER stress is toxic or protective is still a matter of debate as some data argue that ER stress may be beneficial [11–14] whereas other data dispute this [15]. If the observed ER stress is protective, other defects may accelerate the demise of neurons given that defects in proteostasis are tightly linked to ALS [16].
Two major pathways regulate protein clearance: proteasome and autophagy-lysosome mediated degradation. Basal autophagy is required to maintain neuronal function, as loss of autophagy has been shown to induce neurodegeneration [17,18]. Emerging evidence indicates that 2 genes associated with ALS, including TARDBP and C9orf72, play a role in autophagy but how they achieve this is not well defined [19–23].
Various mutations (P56S, T46I, A145V, S160Δ, V234I) in the gene encoding the human VAPB protein cause ALS8 (OMIM: 608627), a form of ALS and spinal muscular atrophy [24–27]. Interestingly, mRNAs of VAPB are decreased in sporadic patients [28] and in neurons derived from ALS8 patients as well as in human SOD1G93A transgenic mice, suggesting that VAPB may play a role in many forms of ALS [29,30]. The VAPs belong to the VAMP-associated protein family and are highly conserved across species. There are 2 VAP homologs in mammals: VAPA and B (VAPA/B). However, Drosophila has a single VAP, Vap33 [31] which corresponds to VPR-1 in C. elegans [4]. Studies in Drosophila, C. elegans as well as mammalian cells have shown that VAPs (Vap33, VPR-1, VAPA/B) affect multiple cellular processes. These include the size and shape of neuromuscular junctions (NMJ) [31], the presence of a UPR [5], the transfer of lipids from the ER to the Golgi [32–35], mitochondrial calcium homeostasis [36,37] and muscle mitochondrial dynamics [38]. VAPA and B share an N-terminal major sperm protein (MSP) domain followed by a coiled-coil domain and a C-terminal transmembrane domain that targets the protein to the ER [4,39,40]. We previously documented that Drosophila Vap33 functions in a cell non-autonomous manner by releasing and secreting the MSP domain [4]. The MSP domain of the human VAPB is also detected in human blood and cerebrospinal fluid (CSF) [4,41] and the levels of MSP in the CSF is reduced in patients with sporadic ALS [41], indicating that loss of MSP secretion may be associated with different forms of ALS.
In addition to the cell non-autonomous function, VAPB also functions cell autonomously in non-vesicular lipid transfer. VAP proteins directly interact with lipid transport proteins, such as OSBP (oxysterol binding protein) and COL4A3BP/CERT through a FFAT motif (2 phenylalanines in an acidic tract) to facilitate lipid transfer [4,39,40]. Both the OSBP and COL4A3BP/CERT proteins contain a pleckstrin homology (PH) domain that interacts with PtdIns4P on the Golgi to promote membrane tethering and lipid transfer from the ER to the Golgi. The VAP-FFAT interaction is abolished in VAPBP56S, the most prevalent variant form of VAPB in ALS8 patients [5,29]. This P56S variant functions as a loss-of-function mutation in some phenotypic assays and as a dominant-negative mutation as it traps endogenous wild-type VAPA and VAPB proteins in aggregates [4,5,29], resulting in a partial loss of function of both VAPA and VAPB. The tethering of the ER to the Golgi facilitates the transfer of PtdIns4P from the Golgi to the ER for hydrolysis [42] and loss of VAPs affects PtdIns4P levels, including a general increase in the cytoplasm [43], a decrease in the Golgi [35], and an increase in endosomes [44]. However, little is known about the role of PtdIns4P in the autophagic-lysosomal degradation pathway.
Here, we provide both in vivo and in vitro evidence that loss of VAPs impairs endo-lysosomal degradation. We found that loss of VAPs leads to an obvious upregulation of the PtdIns4P levels in the Golgi, and a dramatic increase in the number of endosomes, lysosomes and autophagic vesicles. These compartments are defective because they do not acidify properly. Reducing the PtdIns4P levels significantly suppresses the autophagic and lysosomal defects, suggesting that the VAPs regulate autophagy-lysosomal degradation through a PtdIns4P-mediated endosomal trafficking pathway. Impairing this pathway causes a severe defect in lysosomal degradation that may play a critical role in ALS8 and other forms of ALS.
Results
PEK/eIF2α kinase activation may protect against neurodegeneration in ALS animal models
Three UPR sensors (ERN1/IRE1, EIF2AK3/PERK and ATF6) have been identified that activate distinct effectors to suppress protein translation and promote protein refolding [3]. We and others previously documented the upregulation of Xbp1 splicing in Vap33 null mutant flies as well as an increased level of EIF2S1 phosphorylation in VAPBP56S knockin mice [4,5,45], suggesting an upregulation of the ERN1/IRE1α and EIF2AK3/PERK/PEK pathways. The elevated ER stress is not tissue specific as we observed a general increase of phospho-eIF2α in whole pupae, 3rd instar larval brains and fat bodies of Vap33∆20 null mutants (Figure S1(a)). We also observed a very significant rough ER dilation in mutant salivary glands based on transmission electron microscopy (TEM), which is typically associated with ER stress [46] (Figure S1(b,c). Smooth ER dilations and ER expansions were also observed in the adult brains of Vap33 null mutant flies [5], showing that elevated ER stress in Vap mutant animals is present in many tissues.
An elevation of ER stress has also been described in heterozygous human SOD1G93A transgenic mice [47,48] and motor neurons of ALS patients [10,49]. Prolonged ER stress can induce apoptosis [50] suggesting that ER stress may contribute to the demise of neurons. However, removing one copy of EIF2AK3/PERK results in the formation of protein aggregates and premature lethality of heterozygous human SOD1G85R transgenic mice [11–14]. In addition, Jiang et al. [51] showed that Guanabenz, an activator of the EIF2AK3/PERK pathway, improves the motor performance and attenuates the motor neuron degeneration in heterozygous human SOD1G93A transgenic mice [51]. In contrast, others showed that the same drug accelerates ALS disease progression in a heterozygous human SOD1G93A transgenic mouse model [15]. Hence, the contribution of the EIF2AK3/PERK pathway in ALS is not well defined [52].
Recently, Sidrauski et al. [53] developed a drug named ISRIB (integrated stress response inhibitor) that reverses the phosphorylation effects of EIF2A and inhibits the EIF2AK3/PERK pathway. ISRIB treatments reverse cognitive deficits in mouse models of traumatic brain injury [54], arguing that suppressing ER stress is protective in some neurodegenerative diseases. We therefore treated heterozygous human SOD1G93A transgenic mice and Vap33P58S Drosophila mutants with ISRIB. Heterozygous human SOD1G93A transgenic mice displayed a short life span and an age-dependent decline in motor ability as monitored by rotarod assays [55] (Figure S2(a), blue line). ISRIB was introduced to SOD1G93A heterozygous and littermate control mice by daily intraperitoneal injection from day 40 to day 110 [56]. This treatment did not affect the performance of control mice on the rotarod assay (Figure S2(a), green line), suggesting that it is not toxic to wild-type animals. However, we did not observe any beneficial effect of the drug in motor activity of the SOD1G93A heterozygous mice (Figure S2(a), red line). Rather we observed a decrease in performance in these mice when compared to controls (Figure S2(a), red line vs blue line). Similar negative effects were also observed in Vap33P58S flies upon ISRIB treatment. Vap33 null flies are pupal lethal, but ~1% of the flies escape and survive for only a day [5]. Introduction of a genomic rescue construct carrying the P58S substitution (equivalent to P56S in ALS8 patients) (Vap33P58S flies) rescues the lethality, but the flies display an age-dependent decline in climbing ability (Figure S2(b)) and short life span (2 ~ 3 weeks) (Figure S2(b)) [5]. ISRIB treatment did not affect wild-type flies but also did not suppress the motor defect in aged Vap33P58S flies (Figure S2(b)). Consistent with mice, the life span of the ISRIB-treated Vap33P58S flies was reduced (Figure S2(c)). Hence, activation of the EIF2AK3/PERK/PEK pathway in ALS models may be protective [11–14] and beneficial by reducing the level of protein synthesis and protein aggregates.
Loss of VAP proteins impairs autophagy
ER stress induces autophagy [57,58]. Autophagy is required for cell survival in the presence of ER stress, and impairing autophagy promotes ER stress-induced cell death [57]. We therefore wondered if autophagy is induced in Vap33 mutants. To determine whether autophagy is altered, we characterize the autophagy flux in both Drosophila and mammalian cell lines. First, we stained the fly muscles for ref(2)P, the Drosophila homolog of SQSTM1/p62 [59]. Ref(2)P is a receptor protein for phagophores and it is typically elevated when the autophagic flux is impaired [60,61]. As shown in Figure 1(a), we observed a strong increase of ref(2)P puncta in late 3rd instar larval muscles of Vap33∆31 null flies. Similar increases in ref(2)P levels were observed in immunohistochemical stainings of mutant fat body cells and salivary gland cells (Figure 1(c)) as well as in brain and fat body tissue based on western blots (Figure 1(e)). The mRNA level of ref(2)P was not affected (Figure S3(a)), further confirming that ref(2)P accumulation is due to a degradation defect. In addition, the level of poly-ubiquitin was also significantly elevated upon loss of Vap33 (Figure 1(d)). Hence, loss of Vap33 impairs the autophagic flux in brain, muscle, fat body, and salivary glands.
Figure 1.
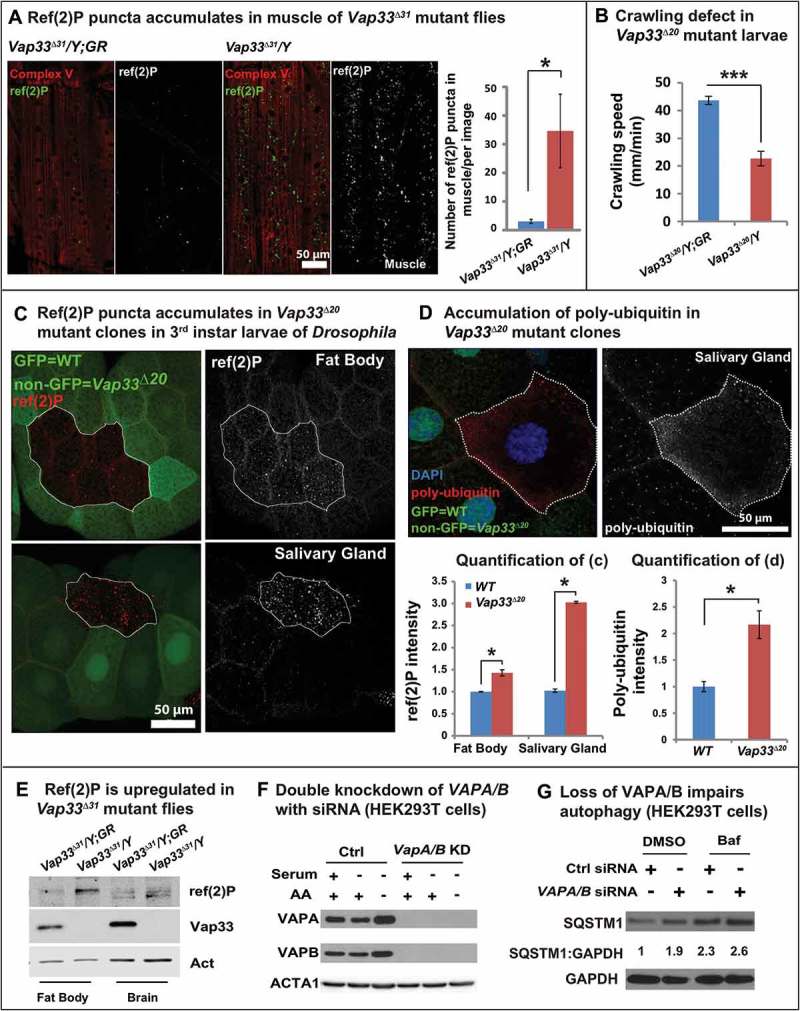
Loss of VAP proteins impairs autophagy. (a) Immunofluorescence staining and quantification of ref(2)P (green) in muscle tissue of the Vap33∆31 mutant. Images showing the muscle 6 and 7 of the abdominal segments 3. GR, 20-kb genomic rescue construct that contains the Vap33 locus and serves as a WT control [5] (b) Crawling assay showing the distance that individual larvae traveled within 1 min (n > 10). (c) Immunofluorescence staining and quantification of ref(2)P (red) in Vap33∆20 mutant clones (GFP negative) of Drosophila fat body and salivary gland. (d) Immunofluorescence staining and quantification of poly-ubiquitin (red) in Vap33∆20 mutant clones (GFP negative) of Drosophila salivary glands. (e) Western Blots (WBs) with ref(2)P antibody in flies. (f) Western blots of HEK293T cells transfected with control or VAPA/B siRNA for 4 days and starved with DMEM or HBSS for 4 h prior to blotting (g) Western blots with SQSTM1 antibody 4 days after VAPA/B siRNA transfection. HEK293T cells are treated with DMSO or bafilomycin A1 (0.1 mg/ml) for 4 h. Note the accumulation of SQSTM1. The quantification of SQSTM1 normalized to GAPDH is shown below each blot.
To assess muscle function, we performed a simple crawling assay of Vap33 null mutant larvae and controls and observed an obvious defect in crawling speed when compared to genomic rescue controls (Figure 1(b)). Hence, Vap33 is required for proper movement, and this behavioral assay can be used as a simple readout.
To assess if the function of VAPs in autophagy is conserved in mammals we simultaneously knocked down the 2 Vap33 homologs, VAPA and VAPB (VAPA/B) (Figure 1(f), Figure S3(b)). We tested 2 siRNA for each gene (Figure S3(b)) and knockdowns of VAPA or VAPB alone did not affect the expression levels of the other homolog (Figure S3(b)). Similar to flies, the level of SQSTM1/p62 was increased upon loss of VAPA/B (Figure 1(g), line 1 and 2). Moreover, treatment with bafilomycin A1, an autophagy inhibitor that blocks acidification of the lysosome, only marginally increased the level of SQSTM1 in VAPA/B-depleted cells compared to control cells (Figure 1(g), line 3 and 4), suggesting that increased accumulation of SQSTM1 is due to a defect in autophagic clearance. These data are not in agreement with a previous observation where loss of VAPB in HeLa cells was shown to promote autophagic flux and degradation [37]. Hence, we propose that the defect in autophagy that we observe is due to a lack of redundancy when both VAPA and VAPB are removed.
Loss of VAPs leads to severe autophagic vesicle accumulation
Autophagic flux can be separated into 3 main steps: formation of autophagosomes, fusion of autophagosomes with lysosomes, and degradation of autolysosomal contents. Defects in any step may lead to SQSTM1 accumulation. We therefore monitored autophagy initiation by assessing the levels of LC3 (mammalian homolog of Atg8a), an integral component of the autophagosome membrane. The Drosophila Atg8a antibody specifically recognizes both the non-lipidated (Atg8a-I) and lipidated (Atg8a-II) forms (Figure S3(c)). Loss of Vap33 led to an accumulation of Atg8a-II in brain, fat body and salivary gland. We also observe an increase of Atg8a-I in fat body and salivary gland (Figure 2(a)). A similar phenotype was also present in mammalian cell lines. We observed a 10-fold increase of steady state levels of LC3-II upon loss of both VAPs (Figure 2(b)). Treatment with bafilomycin A1 to block autophagic degradation led to a significant accumulation of LC3-II in control and VAPs-depleted cells. However, the level of LC3-II accumulation was almost 2-fold higher than that in control cells (Figure 2(b), line 3 and 4) showing that autophagy is initiated and even promoted when VAPs are lost.
Figure 2.
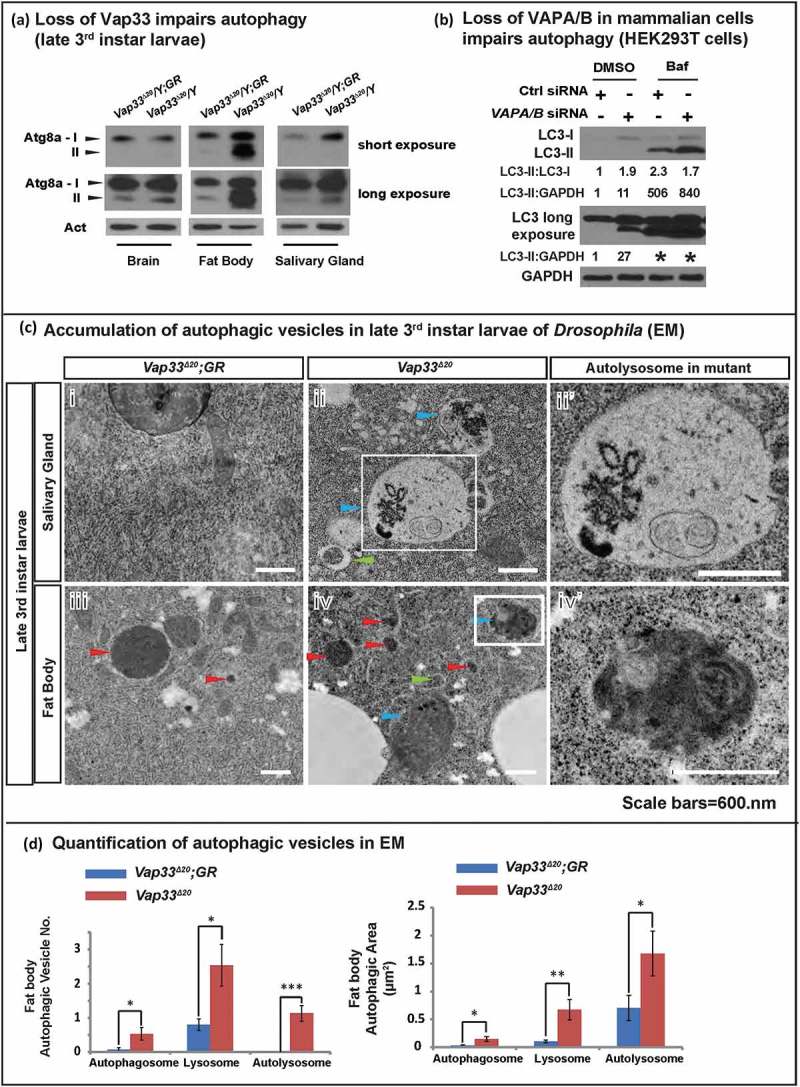
Loss of VAPs leads to autophagic vesicle accumulation. (a) Western blots with Atg8a antibody of late 3rd instar larvae. (b) HEK293T cells were transfected with control or VAPA/B siRNA for 4 days, then treated with DMSO or bafilomycin A1 (0.1 mg/ml) for 4 h. WBs show accumulation of LC3-I and LC3-II. Quantifications of LC3-II normalized to LC3-I and GAPDH are shown below each blot. LC3 long is a longer exposure of the LC3 blots. * indicates overexposure. (ci – iv) TEM images showing autophagic vesicles in salivary glands and fat body cells from Vap33∆20 and Vap33∆20;GR-Vap33WT 3rd instar larvae. Arrowheads indicate autophagosomes (green), lysosomes (red) and autolysosomes/amphisomes (blue). (cii’ and iv’) Higher magnification of autolysosomes/amphisomes in Vap33∆20 salivary gland and fat body. Scale bars: 600 nm. (d) Quantifications of the total area and number of autophagic vesicles in (c). Three animals were imaged from each genotype and 5 representative images were selected for each animal for quantification. Error bars represent s.e.m; *P < 0.05, **P < 0.01, ***P < 0.001).
To assess the population of autophagosomes, lysosomes, and autolysosomes we performed TEM of Vap33∆20 mutant flies. We observed a dramatic accumulation of all 3 compartments, especially autolysosomes in fat body and salivary glands of late 3rd instar larvae (Figure 2(c), quantified in Figure 2(d)). This defect is independent of the developmental stage of the larvae as similar phenotypes are observed in early 3rd instar larvae (Figure S3(d)), prior to the induction of developmental autophagy. Hence, in flies and mammalian cells, the abnormal accumulation of SQSTM1/ref(2)P and increased LC3-II/Atg8a-II argue that the autophagic phenotypes of VAP mutant cells are not due to failures in the initial steps of autophagy induction or autophagosome formation, but rather that fusion of autophagosomes with lysosomes or degradation of their content is affected.
In addition to an increase in autophagosomes, Vap33∆20 mutant flies also exhibited a more dramatic accumulation of autolysosomes (Figure 2(c,d). Because defective fusion of autophagosomes with lysosomes typically results in accumulation of autophagosomes and a reduction of autolysosomes, the accumulation of enlarged autolysosomes (Figure 2(c,d)) indicated that the fusion step was not impaired in the absence of Vap33. Indeed, immunostaining revealed that the colocalization of autophagosome and lysosome membrane proteins (LC3 and LAMP1) are similar in wild type and VAPs-depleted HEK293T cells (quantified in Figure 3(b), right panel). Hence, initiation of autophagy is normal or enhanced while the fusion of autophagosomes with lysosomes is not affected in VAP mutant cells.
Loss of VAP proteins impairs autophagic degradation
TEM analysis in Drosophila revealed that the size of autolysosomes was significantly enlarged in Vap33∆20 mutant salivary glands and fat body cells (Figure. 2(cii’,iv’)). These autophagosomes contained numerous structures that were not observed in controls (Figure. 2(ci and iii)). Indeed, mosaic Vap33 mutant clones in fat body labeled with mCherry-Atg8a, a marker for autophagosomes and autolysosomes, revealed expanded mCherry-Atg8a-positive domains with diminished signal intensity when compared to adjacent wild-type cells. This suggests that there was an accumulation of autolysosomes in these cells (Figure 3(a)).
Figure 3.
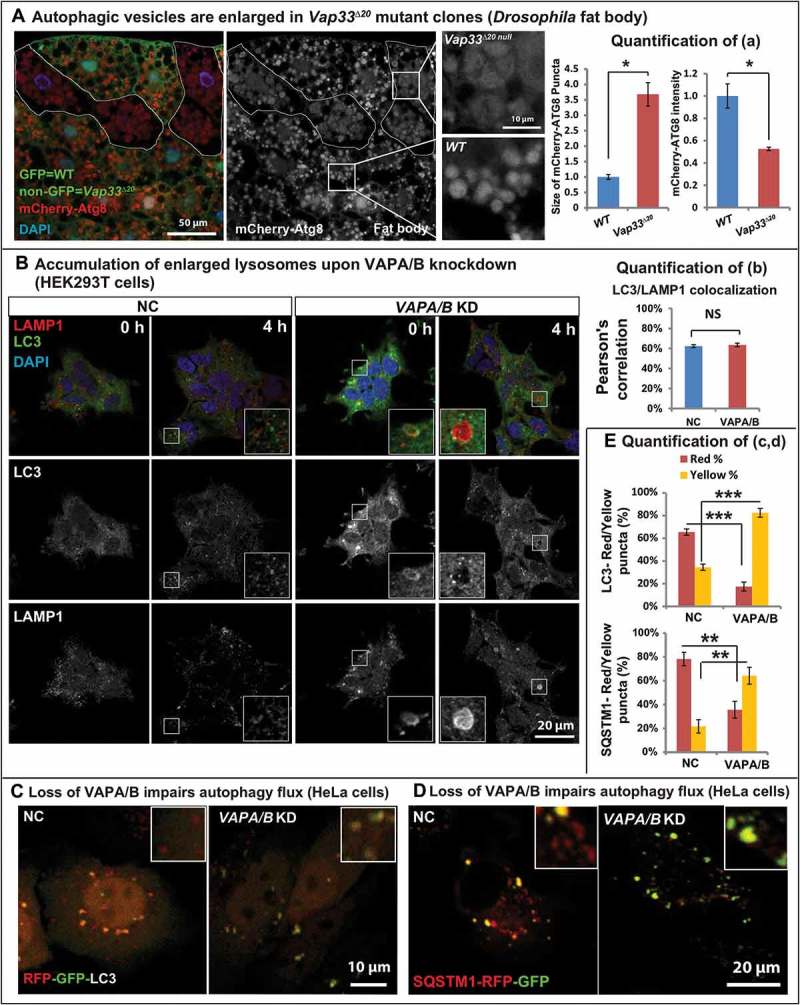
Loss of VAP proteins impairs autophagic degradation. (a) Left: mCherry-Atg8a (red) in Vap33∆20 mutant clones (GFP negative) of Drosophila fat body. Middle: Zooms of wild-type and mutant cells. Right: Quantification of the size and intensity of mCherry-Atg8a puncta. (b) Left: HEK293T cells were transfected with control or VAPA/B siRNA for 4 days and serum starved for 4 h prior to immunofluorescence staining of endogenous LC3 and LAMP1. Right: Quantification of the signal colocalization of LC3 and LAMP1 using Coloc 2 (ImageJ). (c,d) HeLa cells were transfected with control or VAPA/B siRNA for 3 days and then transfected with the RFP-GFP-LC3 or SQSTM1-RFP-GFP expression vector. Sixteen h after transfection, cells are starved with HBSS for 5 h and imaged live. (e) Quantification of the red and yellow puncta of RFP-GFP-LC3 or SQSTM1-RFP-GFP transfected cells.
In VAPs-depleted mammalian cells, we also observed an accumulation of autophagic vesicles. For example, in serum-starved cells, a treatment that promotes autophagosome formation and fusion with lysosomes, loss of VAPA/B led to an upregulation of LC3 levels (Figure 3B, Figure S4(a)) and the morphology of autophagosomes and lysosomes was also altered upon loss of VAPs. Indeed, we also observed an accumulation of enlarged LC3- and LAMP1-positive vesicles (Figure 3(b), embedded image) in mammalian cells. Autolysosomes are generally transient structures that are quickly degraded after fusion [62]. Enlarged autophagic vesicles typically accumulate in the presence of defective autophagosomal degradation [63–65], which was consistent with the observed ref(2)P accumulation in vivo upon loss of Vaps (Figure 1). In summary, these data suggest a defect in autolysosomal degradation in the absence of VAPs.
To explore the functional degradation capacity of autolysosomes in VAPA/B knockdown cells, we transfected cells with GFP-RFP-LC3. Neutral autophagosomes should be labeled yellow whereas acidic autolysosomes will quench GFP and should be labeled red [66]. Loss of VAPA/B led to a significant reduction of red-only LC3 puncta (Figure 3(c), quantified in Figure 3(e)), indicating an acidification defect in autolysosomes. We tested a second set of siRNAs against VAPA and VAPB (Figure S4(b), VAPA-2, VAPB-2) and observed a similar defect in the acidification of autolysosomes (Figure S4(b)). Similar quenching defects were also observed with SQSTM1-GFP-RFP transfected cells (Figure 3(d), quantified in Figure 3(e)). Loss of VAPA/B resulted in a significant increase of the colocalization of the GFP and RFP signal of SQSTM1-GFP-RFP protein (quantified in Figure S4(c)). Hence, the degradation capacity of autolysosomes is impaired in VAPA/B mutant cells.
Loss of VAP proteins results in lysosomal expansion
Typically, lysosomes are derived from late endosomes, which undergo acidification to activate the hydrolases that are targeted to lysosomes. The acidification process can be monitored with LysoTracker®, a chemical probe that stains acidified compartments. As shown in Figure 4(a), we observed a significant increase in LysoTracker Red staining in Vap33∆20 mutant clones of salivary gland cells. Similarly, the level of lysosomal protease, CtsB1 (Cathepsin B1), shown by Magic Red™ CtsB1 substrate staining, was significantly increased in Vap33∆20 mutant cells (Figure 4(b)). To further assess lysosome number and shape, we labeled lysosomes with the lysosomal membrane marker LAMP1-GFP [67]. The LAMP1-GFP construct expresses a fusion protein containing an N-terminal eGFP, a transmembrane domain in the middle and a C-terminal human LAMP1 cytoplasmic tail. The pH sensitive eGFP portion of this fusion protein is localized to the lysosomal lumen. Hence, the fluorescence signal of eGFP is quenched in the functionally active lysosomes, which exhibit a low pH (~ 4.5) [68] . As shown in Figure 4(c,d), we observed an accumulation of LAMP1-GFP in Vap33∆20 mutant clones in fat body and salivary gland cells, indicating a defect in lysosomal acidification and activation. These phenotypes were also present in early 3rd instar larvae with or without starvation, arguing that the expansion of lysosomes occurred independent of autophagy induction and the developmental stage (Figure S5(b)). Similarly, in mammalian cells, we also observed an accumulation of LAMP2 and LysoTracker Red when VAPA/B were knocked down (Figure 5(a,b), quantified in C), indicating an expansion of autolysosomal and lysosomal pools.
Figure 4.
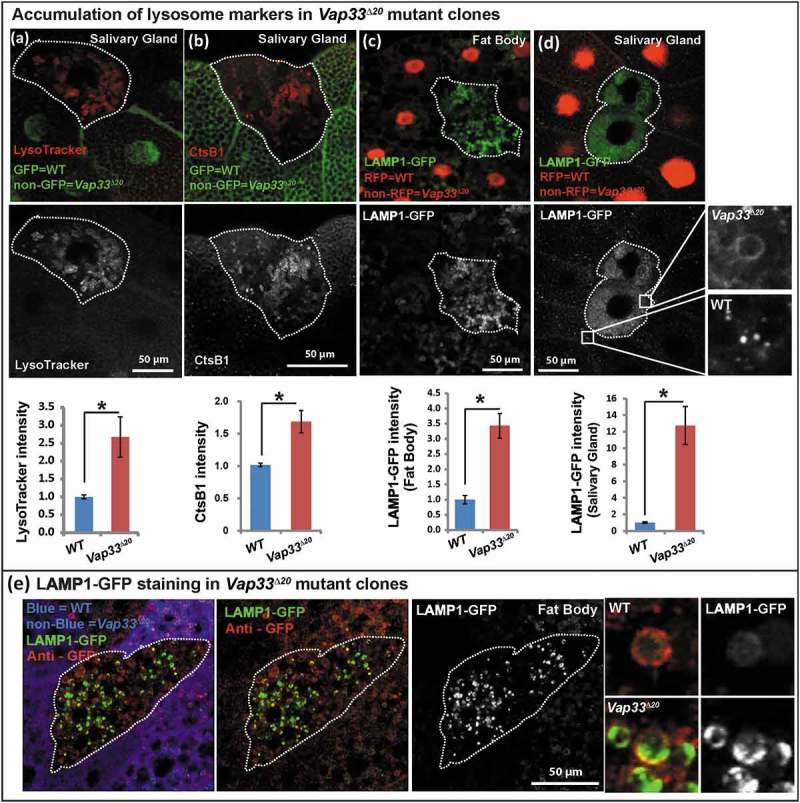
Loss of Vap33 in Drosophila leads to expansion of the lysosomal pool. (a) LysoTracker Red staining (red) and quantification in Vap33∆20 mutant clones (GFP negative) of Drosophila salivary glands. Wild-type cells are green. (b) Magic Red™ CtsB1 substrate staining and quantification in Vap33∆20 mutant clones of Drosophila salivary glands. Wild-type cells are green. (c,d) LAMP1-GFP (green) and quantification in Vap33∆20 mutant clones of Drosophila fat body and salivary gland. Wild-type nuclei are labeled with RFP. (e) Immunostaining of LAMP1-GFP (green) with GFP antibody (red) in Vap33∆20 mutant clones of Drosophila fat body. Acidic lysosomes should only be labeled in red. Wild-type cells are blue. Right panel, representative images showing lysosomes in wild-type and Vap33 mutant cells.
The increase in the LysoTracker Red-positive lysosomal pool is somewhat unexpected given the observed degradation defects. Based on the above data it is likely that these lysosomes are not functional. To further explore this possibility, we took advantage of the LAMP1-GFP construct and assessed GFP activity in Vap33∆20 mutant clones. As mentioned above, dysfunctional lysosomes will not quench the GFP signal and will show a bright GFP (green) signal. Conversely, the total LAMP1-GFP levels as assessed by anti-GFP staining (red) will not be quenched by low pH. Therefore, functional acidified lysosomes will be labeled as red-only puncta, whereas dysfunctional lysosomes will be stained in green. As shown in Figure 4(e), Vap33 mutant cells displayed an obvious accumulation of lysosomes with a green signal when compared to neighboring control cells. In contrast, we observed more red-only vesicles in wild-type tissue than in mutant cells showing that loss of Vap33 impairs the quenching of the LAMP1-GFP, and suggesting an acidification defect.
Because expansion of lysosomes may lead to accumulation of lysosomal proteases, we also assessed the levels of several lysosomal hydrolases, including CTSD, CTSB, PPT1 and TPP1, which are normally cleaved in an acidic environment. Although we did not observe dramatic differences between the different hydrolases and the total PPT1 levels were not affected, the levels of mature CTSD were reduced in cells that lacked VAPs. Conversely, there was an apparent small increase in both pro and mature forms of CTSB and TPP1 (Figure 5(d)). This accumulation of lysosomal enzymes was also observed in Drosophila (Figure 5(e)). However, given that there was a 2- to 3-fold increase in the number of lysosomes, we inferred that there was a significant disruption of the proper balance of various hydrolases per lysosome in cells that lacked VAPs versus wild-type cells (quantified in Figure S6(a,b)).
Figure 5.
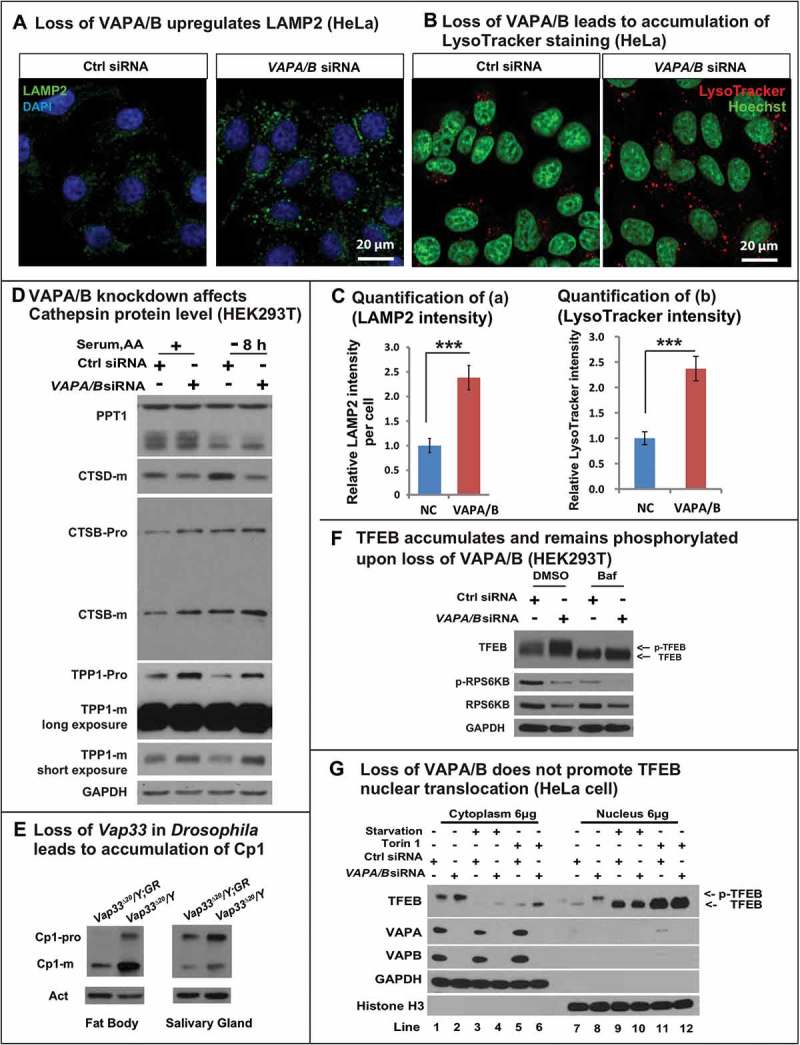
Loss of VAPs affects lysosomal degradation. (a) LAMP2 (green) staining in HeLa cells transfected with control or VAPA/B siRNA. (b) LysoTracker Red staining (red) in HeLa cells transfected with control or VAPA/B siRNA. (c) Quantification of LAMP2 and intensity from (a,b). (d) WBs of lysosomal hydrolases in control or VAPA/B siRNA-transfected HEK293T cells before or after HBSS starvation (8 h). (e) Western blots of Cp1/cathepsin L in Drosophila fat body and salivary glands. (f) HEK293T cells transfected with control or VAPA/B siRNA and treated with or without bafilomycin A1 for 6 h and probed for TFEB, p-RPS6KB/p70S6K and total RPS6KB/p70S6K. (g) HeLa cells transfected with control or VAPA/B siRNA and starved or treated with or without Torin for 2 h. The cytoplasm and nucleus are separated and then probed for TFEB. Starvation: Sixteen h serum starvation followed by 4 h serum and amino acid starvation.
Lysosomal biogenesis is not increased when VAPs are lost
An expansion of autolysosomes and lysosomes may be caused by 2 processes: an increase in biogenesis regulated by TFEB, which promotes transcription of many lysosomal and autophagic genes [69,70] or a defect in lysosomal degradation [71–73]. We therefore monitored the level of TFEB by western blot. As shown in Figure 5(f) and Figure S6(c), the level of TFEB protein was increased upon loss of VAPs in both HEK293T cells and HeLa cells. The activity of TFEB is regulated through phosphorylation, which inhibits its function, as p-TFEB is trapped in the cytoplasm and is inactive [74–76]. TFEB can be phosphorylated by MTORC1, and indeed treatment with bafilomycin A1 inactivates MTORC1, suppressing TFEB phosphorylation [77] and led to a mobility shift (Figure 5(f), line 1 and 3). Upon loss of VAPA/B, the observed mobility retardation of TFEB indicated that the protein was phosphorylated (Figure 5(f)), Figure S6(c)), and hence was not active. To further confirm the TFEB localization, we performed nuclear fractionation, shown in Figure 5(g). TFEB primarily localized to the cytoplasm in basal conditions (lane 1 and 7). MTOR inhibition dephosphorylated TFEB and significantly promoted TFEB nuclear translocation (lane 3–6 and 9–12). Loss of VAPA/B upregulated the total protein level of TFEB but the protein was still primarily localized to the cytoplasm (lane 2 and 8). Interestingly, we also observed a mobility shift of nuclear TFEB (lane 8) upon loss of VAPA/B. This argues that loss of VAPA/B leads to hyperphosphorylation of nuclear TFEB, and this process is upstream of MTOR. How VAPA/B regulate this process is beyond the scope of this study and requires further analysis.
Consistent with these observations, TFEB protein was still localized to the cytoplasm based on immunostaining (Figure S6(d)). In addition, the mRNA levels of numerous TFEB downstream targets were not altered or decreased upon loss of VAPs (Figure S6(e)). Hence, the observed lysosomal expansion in VAP mutant cells is not due to increased lysosomal biogenesis.
The above data raise the question as to how TFEB protein level is increased in VAP mutant cells. Given that Mitf/TFEB mRNA levels were not increased in either Drosophila or mammalian cells (Figure S6(e,f), the accumulation is likely due to a defect in its degradation, which requires chaperone-mediated autophagy [78]. This process requires the binding of chaperones to TFEB and the uptake of the complex by lysosomes. Given that we observed a lysosomal dysfunction, we assume that TFEB cannot be properly degraded and accumulates in cells that lack VAPs. We therefore treated control and mutant cells with bafilomycin A1 to block lysosomal degradation. This treatment led to an upregulation of TFEB in control cells, and loss of the VAPs did not further elevate the levels of TFEB (Figure 5(f), Figure S6(c)), indicating that TFEB accumulation in the absence of VAPs is primarily due to lysosomal dysfunction rather than activation of TFEB. These data further strengthen the model that loss of VAPs impairs lysosomal degradation, and that lysosomal expansion is due to a degradation defect, rather than increased biogenesis.
Loss of VAPs upregulates PtdIns4P and alters vesicle trafficking
We next examined how VAP proteins regulate lysosomal degradation. VAP proteins can interact with OSBPs to tether the ER to the Golgi membrane [33,35,42,79]. This tethering is required for ceramide or cholesterol transfer from the ER to the Golgi but it is also required for the transfer of PtdIns4P from the Golgi to the ER where SACM1L can hydrolyze PtdIns4P [42]. Hence, in the absence of VAPs, PtdIns4P may be upregulated in the Golgi. Although there is substantial evidence that loss of PtdIns4P affects endosome formation from the Golgi [80], if and how an increase in Golgi PtdIns4P levels affects endosome formation is not established. In addition, the published data related to VAPs and PtdIns4P are confusing: Peretti et al. [35] argue that there is a decrease of PtdIns4P in the Golgi based on the PH-OSBP marker; Forrest et al. [43] document an increase in PtdIns4P in cells that overexpress Vap33P58S using an antibody against PtdIns4P whereas Dong et al. [44] argue that PtdIns4P is elevated in endosomes upon loss of VAPA/B based on PH-OSBP and other markers. We therefore assessed if PtdIns4P is elevated upon loss of Vap33 and, if so, in which organelle. As shown in Figure 6(a), the level of PtdIns4P was obviously increased in Vap33 mutant clones of Drosophila fat body and salivary glands. As the Drosophila Golgi apparatus is distributed throughout the cytoplasm we stained mammalian cells where the Golgi is easily recognizable near the nucleus. Accumulation of PtdIns4P in the Golgi was obvious by co-staining of GOLGA2/GM130 in HEK293T cells (Figure 6(b), quantified in Figure S6(g)). Hence, loss of VAPs in mammalian cells leads to PtdIns4P accumulation in the Golgi.
Figure 6.
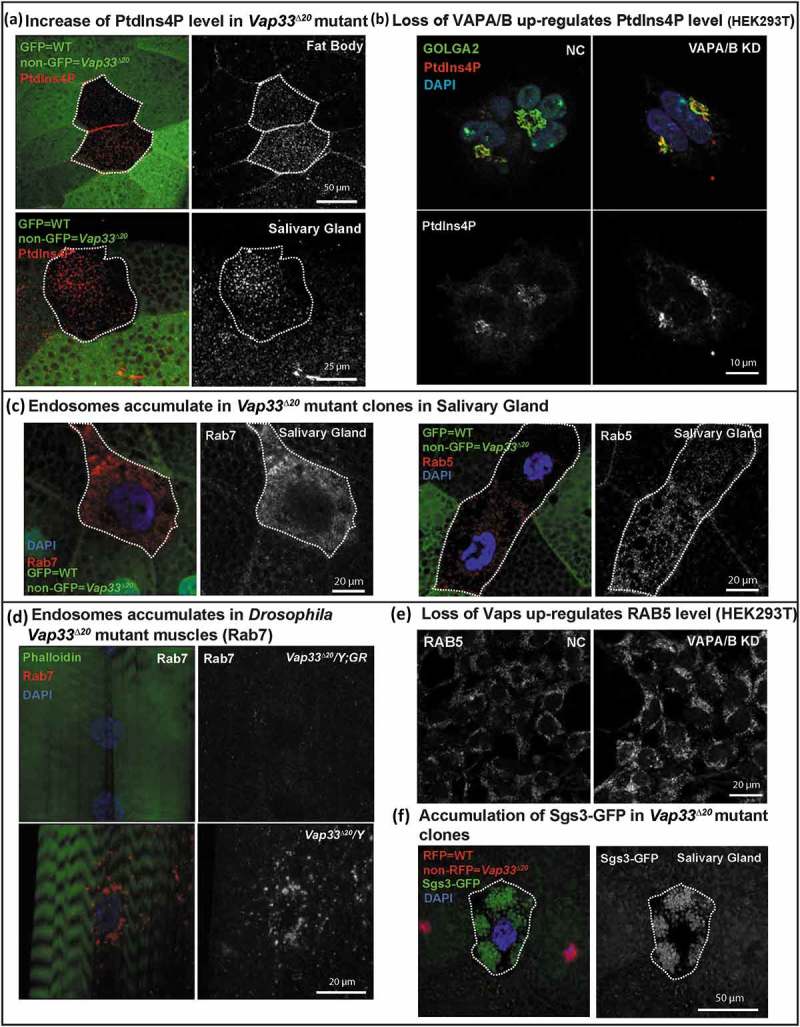
Loss of VAPs affects PtdIns4P and the endosome pathway. (a) Immunofluorescence staining of PtdIns4P (red) in Vap33 mutant clones (non-green) of Drosophila fat body and salivary gland cells. (b) Immunofluorescence staining of PtdIns4P in control or VAPA/B siRNA-transfected HEK293T cells. (c) Immunofluorescence staining of Rab5 and Rab7 (red) in Vap33 mutant clones (non-green) of Drosophila salivary glands (d) Immunofluorescence staining of Rab7 (red) in Drosophila muscles of control and Vap33 mutant animals (e) Immunofluorescence staining of RAB5 in control or VAPA/B siRNA-transfected HEK293T cells. (f) Sgs3-GFP (green, a marker for secretory granules) in Vap33 mutant clones of Drosophila salivary gland. Wild-type nuclei are labeled with RFP.
Interestingly, the increase in PtdIns4P was associated with an obvious accumulation of 2 endosomal markers, Rab5/RAB5 and Rab7/RAB7, in salivary gland and muscles of Vap33 mutant (Figure 6(c,d)) and HEK293T cells (Figure 6(e)). These data were also supported by other observations in salivary gland cells where we observed a severe secretory granule accumulation, a process that relies on proteins that affect or depend on the PtdIns4P pathway (Figure 6(f)). In addition, excess secretory granules are degraded via direct fusion with lysosomes, via a secretory granule-specific autophagic process known as crinophagy [81,82]. This suggests that secretory granule accumulation is due to an accumulation of PtdIns4P combined with a lysosomal dysfunction in salivary gland. Hence, PtdIns4P accumulates upon loss of VAPs promoting the formation of an endosomal population of vesicles. As some endosome mature into lysosomes [83,84], we propose that the PtdIns4P accumulation upregulates endosome levels and leads to the dilution of the lysosomal content mentioned in the previous section, resulting in the observed lysosomal degradation defect.
Modulating the PtdIns4P-mediated endosome pathway suppresses autophagic and lysosomal defects in VAPs mutants
If the elevated levels of PtdIns4P are at the root of the autophagic/lysosomal defects associated with the loss of VAPs, reducing the PtdIns4P levels should suppress the endosome accumulation and improve autophagic and lysosomal defects. PI4KIIIbeta-IN-10 is a potent PI4KB inhibitor with minor off-target inhibition of related lipid kinases [85]. The homolog of PI4KB in Drosophila is four-wheel drive (fwd). As shown in Figure 7(a), feeding Vap33 mutant larvae with PI4KIIIbeta-IN-10 significantly rescued endosome accumulation in muscles, arguing that the expansion of the endosome pool was due to PtdIns4P upregulation in the Vap33 mutant. Similarly, in mammalian cells, PI4KIIIbeta-IN-10 treatment in VAPA/B knockdown cells significantly reduced PtdIns4P to levels comparable to control cells (Figure 7(b)). Importantly, this PtdIns4P knockdown rescued the accumulation of enlarged lysosomal vesicles (Figure 7(b)). These data provide compelling evidence that the expansion of the endo-lysosomal pool is due to PtdIns4P accumulation.
Figure 7.
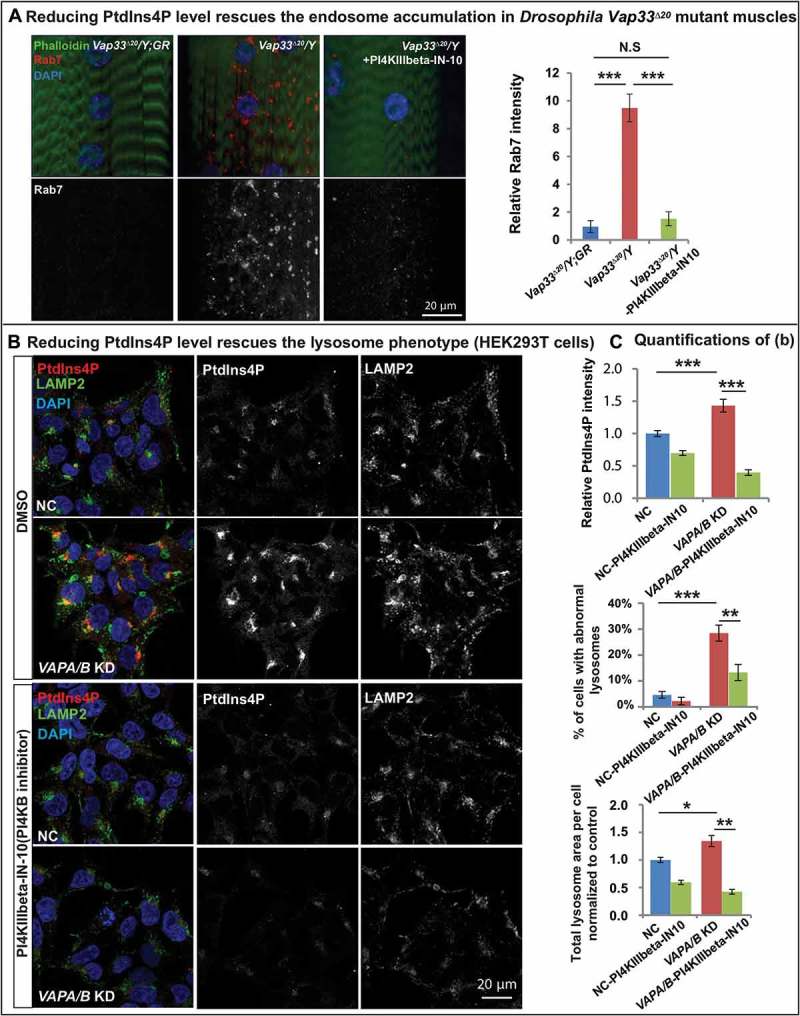
Reducing PtdIns4P levels rescues the endo-lysosomal defects. (a) Left: Immunofluorescence staining of Rab7 in muscles of control and Vap33 mutant larvae. Vap33 mutant larvae are treated with or without PI4KIIIbeta-IN-10 (5 nM, a PtdIns4K inhibitor). Right: Quantification of the Rab7 intensity. (b) HEK293T cells were transfected with control or VAPA/B siRNA and treated with DMSO or PI4KIIIbeta-IN-10 (25 nM) for 4 h prior to immunofluorescence staining of endogenous PtdIns4P and LAMP2. (c) Quantification of PtdIns4P intensity and the lysosomes in (b).
A rescue of the autophagic-lysosomal degradation defect (Figure 8(a)) was also observed in HEK293T cells as gauged by the suppression of the elevated levels of TFEB, CTSB and LC3-II (Figure 8(a,b). These data argue that increased PtdIns4P levels cause an endosome accumulation as well as autophagic-lysosomal degradation defect in the VAPs mutant. To further investigate if the autophagic degradation defect upon loss of VAPs is due to expansion of the endo-lysosomal pool, we removed one copy of Rab5 or Rab7 in Vap33 mutant animals, proteins required for endo-lysosomal maturation. As shown in Figure 8(c,d), loss of one copy of Rab5 or Rab7 significantly reduced the accumulation of ref(2)P-positive puncta observed in the muscle of Vap33 mutants. In addition, either PI4KB/FWD inhibition or removing one copy of Rab7 also partially rescued the crawling defect in Vap33 mutant larvae (Figure 8(e)). In summary, loss of VAP proteins dysregulates PtdIns4P-mediated endosome trafficking which in turn affects autophagic-lysosomal degradation.
Figure 8.
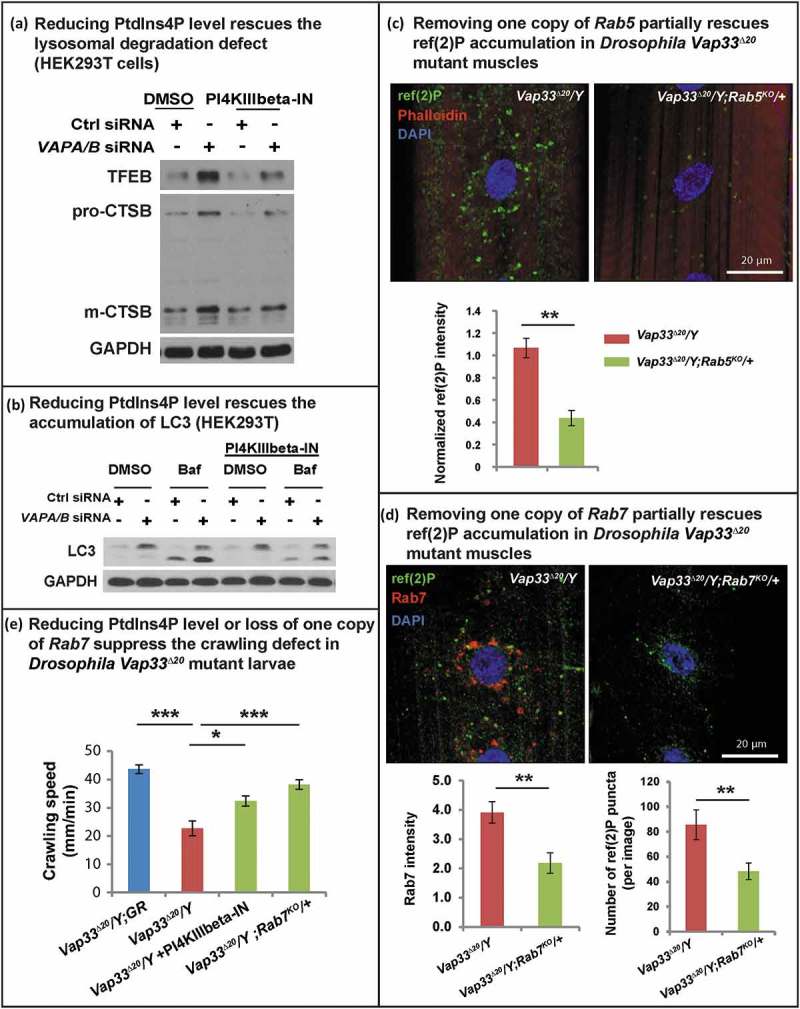
Reducing endosome levels rescues the lysosomal and autophagic defects. (a) Control or VAPA/B siRNA-transfected HEK293T cells were treated with DMSO or PI4KIIIbeta-IN-10 for 4 h. WB probed for TFEB and CTSB. (b) Control or VAPA/B siRNA-transfected HEK293T cells are treated with DMSO or PI4KIIIbeta-IN-10 for 18 h and treated with bafilomycin A1 for 4 h. WBs probed with LC3. (c) Immunofluorescence staining of ref(2)P/SQSTM1 in muscles of Vap33∆20 mutant and Vap33∆20; Rab5KO/+ larvae. Bottom: Quantification of the ref(2)P intensity. (d) Immunofluorescence staining of Rab7 and ref(2)P in muscles of Vap33∆20 mutant and Vap33∆20; Rab7KO/+ larvae. Bottom: Quantification of the Rab7 intensity and number of ref(2)P puncta. (e) Crawling assay of control, Vap33∆20 mutant, Vap33∆20 mutant treated with PI4KIIIbeta-IN-10 (5 nM) and Vap33∆20; Rab7KO/+ larvae. Ten to 20 animals are tested for each genotype/treatment.
Discussion
Based on our current studies and published data, we propose the following model (Figure 9(a)). VAP proteins localize to the ER and interact with lipid transfer proteins such as OSBP and COL4A3BP/CERT through their FFAT motif. The PH domains of OSBP and COL4A3BP/CERT interact with PtdIns4P anchored on the Golgi and tether the ER to the Golgi [5], facilitating PtdIns4P transfer from the Golgi to the ER for its hydrolysis by SACM1L [42]. We argue that this leads to an accumulation of PtdIns4P in the Golgi and increased production of RAB5- and RAB7-positive endosomes. These endosomes mature into lysosomes leading to an increase in the number of lysosomes with aberrant pH. These defective lysosomes affect protein degradation, and upon fusion with autophagosomes also impair autophagic degradation, resulting in an accumulation of autophagic vesicles.
Figure 9.
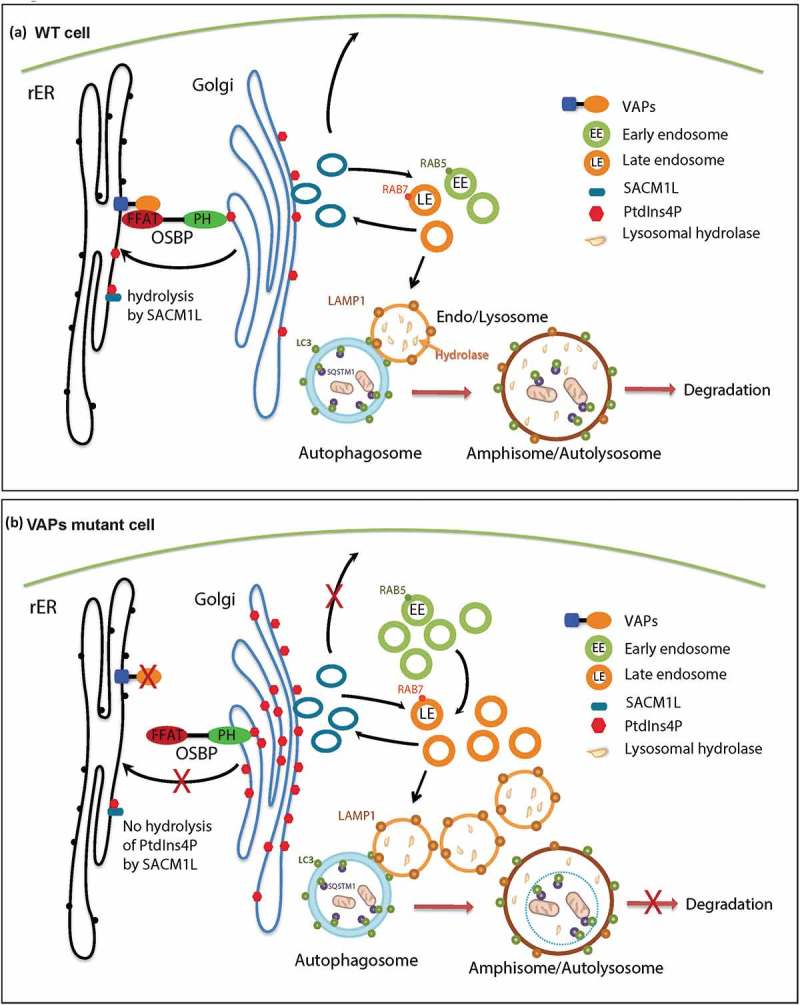
Model. (a) In WT cells, OSBP is localized to the Golgi through the interaction of its PH domain with PtdIns4P present on the Golgi. OSBP also interacts with the VAP proteins through a FFAT motif thereby tethering the ER to the Golgi. This membrane tethering facilitates the PtdIns4P transfer from the Golgi to the ER for hydrolysis by SACM1L. Golgi PtdIns4P mediates vesicle trafficking from the Golgi to various compartments, including the plasma membrane and endosomes. Endosomes mature to lysosomes, which play a key role in the degradation of autophagic compartments. (b) Upon loss of VAPs, OSBP is dissociated from the ER, and ER-Golgi tethering is disrupted. Therefore, hydrolysis of PtdIns4P in the ER is impaired and PtdIns4P accumulates on the Golgi. Elevated PtdIns4P levels promote endosome formation from the Golgi, resulting in an accumulation of endosomes. Expansion of the endosomal pool disrupts the degradation properties of lysosomes upon fusion by affecting lysosomal enzyme composition. This in turn impairs autophagy.
Our data argue that the defects in autophagic and lysosomal degradation in VAP mutant cells are due to PtdIns4P imbalance. Indeed, by reducing the PtdIns4P to more normal levels or removing one copy of the endosome proteins Rab5 or Rab7, we observed a significant suppression of endosome and autophagy-lysosomal defects in the Vap33 mutant (Figures. 7 and 8). Modulating the PtdIns4P and endosome pathway also rescues the locomotion deficit in Vap33 mutant animals, suggesting a strategy to modify the phenotype in patients. At the root of the elevated level of PtdIns4P is the loss of ER-Golgi tethering, as promoting ER-Golgi tethering by overexpression of an OSBP that does not require VAPs significantly suppresses the motor deficit and early lethality of mutant flies [5].
A recent study by Gomez-Suaga et al. [37] argues that the function of VAPB is to inhibit autophagy by promoting ER-mitochondria tethering. The authors argue that siRNA-mediated knockdown of VAPB in HeLa cells disrupts ER-mitochondria tethering through VAPB and its interaction with RMDN3/PTPIP51. Loss of this interaction promotes autophagy and does not impair degradation. Given that we observe dysfunctional autophagy in flies and mammalian cells upon loss of the VAPs we argue that VAPA and VAPB are redundant and that removing VAPB alone appears insufficient to impair lysosomal degradation, but seems sufficient to promote autophagy induction.
The combined loss of function of VAPA and B may be relevant to ALS8. Indeed, the most prevalent form of the VAPB mutation found in these patients is the P56S mutation, which functions as a dominant negative allele in some contexts and traps both VAPA and VAPB in aggregates [4,5,29]. Hence, reducing VAPA and B may better mimic the conditions of patients with the VAPBP56S mutation. This interpretation is also consistent with the observed accumulation of SQSTM1 in aged heterozygous VAPBP56S knockin mice [45].
The accumulation of lumenal tagged LAMP1-GFP argues that there is a defect in lysosomal acidification upon loss of Vap33 (Figure 4(c, d and e)). This phenotype needs to be reconciled with the increased LysoTracker Red staining and increased Magic Red CtsB1 staining observed in Vap33 mutant cells (Figure 4(a)). LysoTracker Red is activated at pH = 6.5 [86], a higher pH than what is required to quench GFP, which is 4.5 [68]. The lysosomal pH typically ranges from 4.5 to 5.0 [87]. Hence, LysoTracker Red should label lysosomes with a pH between 4.5 ~ 6.5, including many that may not be fully functional when VAPs are lost. Similarly, CTSB has been shown to have high proteolytic activity at pH>5 [88]. Hence, LAMP1-GFP reveals non-acidified lysosomes, whereas the increased LysoTracker Red and Magic Red CtsB1 staining indicate an expansion of lysosomes that may include acidified as well as poorly acidified lysosomes.” However, our data cannot exclude the possibility that loss of VAPs impairs the trafficking of some lysosomal proteins that are trapped in non-acidic endosomes. This trafficking defect would also result in dysfunctional lysosomes, consistent with our model.
Previously, Wijdeven et al. [89] have argued that OSBPL1A/ORP1L, an effector for cholesterol sensing, directly interacts with VAPA and that OSBPL1A/ORP1L inhibits autophagic maturation by regulating the fusion of autophagosomes with the endocytic pathway. They showed that loss of OSBPL1A/ORP1L results in a decrease of both LC3-II and SQSTM1 [89] but did not show that loss of VAPA and/or VAPB cause autophagic defects or promote autophagy. Given that we observe an accumulation of both LC3-II and SQSTM1 (Figures. 1 and 2) and that we do not observe a defect in autophagic fusion when the VAPs are lost, the degradation defects that we document are likely not related to the OSBPL1A/ORP1L pathway.
Our data show that there is an increase in the acidified lysosomal pool when VAPs are lost. Interestingly, lysosomal expansion is also observed in lysosomal storage diseases due to defects in lysosomal degradation [71–73]. Indeed, lysosomal degradation defects impair the processing of cargo as well as the renewal of the lysosomal compartment, leading to the accumulation of aberrant lysosomes [64,65,72]. Furthermore, loss of VAPs results in a significant disruption of the balance of the various hydrolases per lysosome (Figure 5(d,e), Figure S4(b)), and are consistent with the lysosomal phenotypes observed in lysosomal storage diseases.
The importance of autophagic and lysosomal function in ALS has only recently come into focus [90]. Loss of TARDBP was recently reported to elevate the levels of TFEB and impair the fusion of autophagosomes with lysosomes [19]. Conversely, C9orf72, the most prevalent ALS-causing gene [91,92], has been shown to decrease autophagic flux upon its loss [21,22,93], whereas others have argued that loss of C9orf72 promotes autophagy [23,94]. However, these studies were not performed in cells that carry the G4C2 hexanucleotide expanded repeat, and the role of autophagy in C9orf72 ALS patients therefore remains to be established. Our data in flies and human cells as well as the phenotypes associated with the mice carrying a single P56S mutation [5] argue that autophagic and lysosomal degradation may be impaired in ALS8 patients and that the primary defect is due to the upregulation of PtdIns4P upon loss of the VAP-mediated anchoring of ER to Golgi (Figure 9(b)).
Materials and methods
Fly stocks and genetics
Fly stocks are maintained at room temperature (~22°C) and all crosses for experiments are kept at 25°C. All Drosophila experiments are performed with either late 3rd instar larvae or adult flies. For clonal analysis in fat body and salivary gland, Drosophila eggs are heat shocked (37°C) for 1 h within 4 h after egg laying to induce clones. Collected eggs are kept at 25°C until the 3rd instar larva stage for further experiments. Null alleles of Vap33 (Vap33∆20 and Vap33∆31) were generated by P-element imprecise excision [4,31]. Transgenic flies carrying the genomic region of the Vap33 gene (WT or P58S) were generated by Moustaqim-Barrette et al. [5]. In short, the entire genomic region (approximately 20 kb) was amplified by PCR. The P58S mutant Vap33 construct was created by PCR with primers containing the mutation. These genomic fragments were subcloned into the pCasper 4 vector [95] and injected into VK31 attP docking sites [96]. The following fly lines were obtained from the Bloomington Drosophila Stock Center: Cg-Gal4 (7011),P[Ubi-mRFP.nls] (31418), P[Sgs3-GFP]3 (5884), Sb, w* P[hsFLP], P[neoFRT]19A; P[UASp-mCherry-ATG8a]2 [97] (37749), Rab52 P[neoFRT]40A/CyO (42702). UAS-LAMP1-GFP was a gift from Helmut Kramer (UT Southwestern) [67], and FRT82B rab7Gal4-knock-in/TM3, was a gift from Robin Hiesinger (Freie Universität Berlin) [98].
Experimental mouse lines
All animals used in this study were treated in compliance with US Department of Health and Human Services and Baylor College of Medicine IACUC guidelines. For the studies reported here, both male and female mice were considered for analyses and all animals were maintained on a normal 12-h light–dark cycle. B6SJL.SOD1-G93A mice were originally purchased and are available from Jackson Laboratories (002726). Mice used in this study were maintained as heterozygotes and bred to wild-type C57BL/6J female mice.
Chemicals
Chemicals were used at the following concentration: PI4KB inhibitor: PI4KIIIbeta-IN-10, 25 nM for mammalian cells and 5 nM for Drosophila treatments (MedChem Express, HY-100198), bafilomycin A1, 0.1mg/ml (Sigma, B1793), ISRIB: 5 mg/kg for intraperitoneal injection in mice, 40 µM mixed in food for Drosophila treatments (gift from Peter Walter, UCSF).
Drosophila larvae crawling assay
Late 3rd instar larvae were transfer to a 10-cm Petri dish containing 1.5% agar over graph paper with a 2-mm grid. The crawling assay is performed by counting the number of grid lines crossed in 1 min.
Mammalian tissue culture (siRNA and cell transfection)
Cells were grown in high glucose Dulbecco’s modified Eagle’s medium (ThermoFisher Scientific, 11960) supplemented with 10% fetal bovine serum (Sigma, F0926) 1% (v:v) GlutaMAX (ThermoFisher Scientific, 35050061), and 1% (v:v) penicillin-streptomycin (GenDEPOT, CA005-010) and grown in a humidified incubator at 37°C with 5% CO2. siRNAs used for transient interference of VAPA and VAPB: VAPA-1 (ThermoFisher Scientific, HSS113661): CACACAGUGUUUCACUUAAUGAUA; VAPA-2 (ThermoFisher Scientific, HSS113659): GGUAGCACAUUCGGAUAAACCUGGA; VAPB-1 (Sigma, SASI_Hs01_00190177): GUAAGAGGCUGCAAGGUGA. VAPB-2 (Sigma, SASI_Hs01_ 00190182): GCUUUCCGUGUCUUCAGUU. GFP-RFP-LC3 and SQSTM1-GFP-RFP tandem tagged constructs were provided by Marco Sardiello (Baylor College of Medicine).
Transmission electron microscopy
The fat body and salivary gland ultrastructure of Drosophila 3rd instar larvae were analyzed with standard electron microscopy procedures using a Ted Pella Bio Wave processing microwave with vacuum attachments. Briefly, dissected fat body and salivary glands were fixed in fixative (2% paraformaldehyde, 2.5% glutaraldehyde, 0.1 M sodium cacodylate, 0.005% CaCl2, pH 7.2) at 4°C overnight and then post-fixed in 1% OsO4. The fixed samples were dehydrated in ethanol and propylene oxide, and then embedded in Embed-812 resin (Electron Microscopy Sciences, 14900) using a standard protocol [99].
RNA-isolation and Q-PCR
For Drosophila: Total RNA was isolated from 10 third instar larvae per genotype, following standard procedures. Briefly, animals were flash frozen and subsequently homogenized in 1 ml TRIzol (ThermoFisher Scientific, 15596026). For mammalian cells: approximately 1 million cells were collected with 1 ml TRIzol. RNA-isolation and Q-PCR was then performed with a standard protocol [100]. In short, total mRNA was extracted with phenol-chloroform, precipitated with isopropanol, washed with 70% ethanol and resuspended in DEPC-treated water. Five micrograms of total RNA from each sample was reverse transcribed using Random Hexamer Primers and the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, 4368814). Real-time PCR was carried out in a thermal cycler (CFX96 Real Time System; Bio-Rad Laboratories), using iQ™ SYBR® Green Supermix (Bio-Rad Laboratories, 1725121). Data were collected and analyzed using the optical module (iQ5; Bio-Rad Laboratories). All primers used are listed below:
| Mammalian genes | Forward primer | Reverse primer |
|---|---|---|
| SQSTM1 | GACTACGACTTGTGTAGCGTC | AGTGTCCGTGTTTCACCTTCC |
| LC3B | CTGCGGGCTGAGGAGATACAAG | AGTCGGACATCTTCTACTCTTTGTT |
| TFEB | CCAGAAGCGAGAGCTCACAGAT | TGTGATTGTCTTTCTTCTGCCG |
| ATP6V0E1 | CATTGTGATGAGCGTGTTCTGG | AACTCCCCGGTTAGGACCCTTA |
| ATP6V1H | GGAAGTGTCAGATGATCCCCA | CCGTTTGCCTCGTGGATAAT |
| CLCN7 | TGATCTCCACGTTCACCCTGA | TCTCCGAGTCAAACCTTCCGA |
| CTSA | CAGGCTTTGGTCTTCTCTCCA | TCACGCATTCCAGGTCTTTG |
| CTSB | AGTGGAGAATGGCACACCCTA | AAGAAGCCATTGTCACCCCA |
| CTSD | AACTGCTGGACATCGCTTGCT | CATTCTTCACGTAGGTGCTGGA |
| GBA | TGGGTACCCGGATGATGTTA | AGATGCTGCTGCTCTCAACA |
| LAMP1 | ACGTTACAGCGTCCAGCTCAT | TCTTTGGAGCTCGCATTGG |
| PSAP | GCCAACAGTGAAATCCCTTCC | TCAGTGGCATTGTCCTTCAGC |
| ULK1 | ATGGAGTACTGCAACGGTGG | GGAAGAGCCTGATGGTGTCC |
| RPS6KB/p70S6K | GGGTACTTGGTAAAGGGGGC | CCTTTTTAAGCACCTTCATGGCA |
| RPS18 | TCGATGGGCGGCGGAAAATA | CGCCCTCTTGGTGAGGTCAA |
| GAPDH | TGCACCACCAACTGCTTAGC | GGCATGGACTGTGGTCATGAG |
| Drosophila genes | Forward primer | Reverse primer |
| ref(2)P | ATTGGGTTCCGCTACAAGTG | TCAAGTGCTCAGGATGCTTG |
| Atg8a | ATTCCACCAACATCGGCTAC | GCCATGCCGTAAACATTCTC |
| Mitf | GCGTTCTTCTTCAGGGATTG | ACTTACGCTCGGCGAAATAG |
| E(spl) | ATGGAATACACCACCAAGACCC | GGCGACAAGTGTTTTCAGGTT |
Live imaging of lysotracker red and magic redtm staining
Drosophila salivary glands were dissected in PBS (ThermoFisher Scientific, 10010023) and then stained with LysoTracker® Red DND-99 (ThermoFisher Scientific, L7528) or Magic Red cresyl violet-(RR)2 (Immunochemistry Technology, 937) for 1 min before washing and imaging with a Zeiss 880 confocal microscope.
Immunohistochemistry
For Drosophila:
Third instar wandering larvae were dissected and fixed for 20 min in, PBS with 4% formaldehyde and washed with PBT (PBS with 0.5% Triton X-100 (Sigma, X100). Then the tissues were incubated with primary antibodies diluted in PBT overnight at 4°C. Samples were washed in PBT prior to incubation with secondary antibodies diluted in PBT for 2 h at room temperature. Samples were then stained with DAPI for 10 min in PBT prior to washing and mounting. Images were taken with a Zeiss 880 confocal microscope. Antibodies were used at the following concentrations: Rabbit anti-ref(2)P, 1:2000 (gift from Sheng Zhang, University of Texas McGovern Medical School) [99]; guinea pig anti-Vap33, 1:1000 (Bellen Lab, Baylor College of Medicine) [31]; mouse anti-polyubiquitin, 1: 500 (Enzo, BML-PW8805-0500); rabbit anti-Atg8a, 1:1000 (gift from Linda Partridge, University College London) [101]; chicken anti-GFP, 1:1000 (Abcam, ab13970); mouse anti-Rab7, 1:200 (DSHB Hybridoma Product Rab7); mouse anti-Cp1/CTSL, 1:2000 (R&D systems, MAB22591).
For mammalian cells:
Cover slips were treated with poly-D-lysine overnight before plating the HEK293T cells. After different treatments, cells were fixed with 4% paraformaldehyde in PBS (Santa Cruz Biotechnology: 281692) for 10 min. Fixed cells were permeabilized with 0.25% saponin (Sigma, 47036) and blocked in PBS containing 10% fetal bovine serum. Samples were incubated with primary antibody at 4°C overnight followed by secondary antibody for 1 h at room temperature after washing. Cells were mounted in Fluoromount-G with DAPI (SouthernBiotech, 0100–20) before imaging with a Zeiss 880 confocal microscope. Antibodies were used at the following concentration: Mouse IgG2A anti-VAPA, 1/1000 (R&D Systems, MAB5820); rabbit anti-GAPDH, 1:10000 (Cell Signaling Technology, 2118); rabbit anti-LC3, 1:1000 (MBL, PM036; Cell Signaling Technology, 2775); rat anti-LAMP1, 1:500 (DSHB, 1D4B) [102]; mouse anti-LAMP2, 1:500 (Santa Cruz Biotechnology, sc-18822); mouse anti-TPP1, 1:500 (Santa Cruz Biotechnology, sc-393961); goat anti-CTSD, 1:100 (Santa Cruz Biotechnology, sc-6487); mouse anti-CTSB, 1:200 (Millipore, IM27L); mouse IgM anti-PtdIns4P, 1:200 (Echelon, Z-P004); rabbit anti-RAB5, 1:100 (Cell Signaling Technology, 3547); mouse anti-Flag, 1:1000 (Sigma, F1804), rabbit anti-Histone H3, 1:2000 (Cell Signaling Technology, 9717), mouse anti-ACTA1, 1:10000 (Millipore, MAB1501), mouse anti- TUBB, 1:5000 (DSHB Hybridoma Product E7). The anti-VAPB polyclonal antibody was generated by immunizing rabbit with MSP domain (amino acid 1–127) of the human VAPB protein. The antibody was used at concentration of 1:2000.
Western blotting
Images were acquired using a Bio-Rad ChemiDoc™ Imaging Systems and densitometric analyses of the bands were performed with ImageJ. Images were collected by the imaging system within the linear range unless specifically labeled (to better show the bands with lower intensity).
Funding Statement
This work was supported by the Howard Hughes Medical Institute; National Institute of Child Health and Human Development [U54HD083092]; National Institutes of Health [R01NS079618]; National Institutes of Health [R01NS069880]; Huffington Foundation; Robert A. and Renee E. Belfer Family Foundation; Target ALS.
Acknowledgments
We thank the BDSC (NIH P40OD018537) for Drosophila stocks. We thank Alberto Di Ronza and Rituraj Pal for providing valuable advice and technical support for the TFEB experiments in mammalian cells. We thank Robin Hiesinger for providing Drosophila Rab7 knockout flies. We thank Linda Partridge for providing Drosophila ATG8a antibody. We thank Hui Ye for providing antibodies and other reagent for Drosophila lysosome and endosome experiments. We thank Berrak Ugur, Karen Schulze and Kartik Venkatachalam for helpful discussions and comments. H.J.B. is an investigator of the Howard Hughes Medical Institute. Confocal microscopy was supported by the Neurovisualization Core of the BCM IDDRC, funded by NICHD U54HD083092. This work was supported by Target ALS, the Robert A. and Renee E. Belfer Family Foundation and the Huffington Foundation to H.J.B; NIH grant R01NS079618 to M. Sardiello and NIH grant R01NS069880 to S.Z.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary material for this article can be accessed here.
References
- [1].Peters OM, Ghasemi M, Brown RH Jr.. Emerging mechanisms of molecular pathology in ALS. J Clin Invest. 2015;125:1767–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Renton AE, Chio A, Traynor BJ.. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ron D, Walter P.. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. [DOI] [PubMed] [Google Scholar]
- [4].Tsuda H, Han SM, Yang Y, et al. The amyotrophic lateral sclerosis 8 protein VAPB is cleaved, secreted, and acts as a ligand for Eph receptors. Cell. 2008;133:963–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Moustaqim-Barrette A, Lin YQ, Pradhan S, et al. The amyotrophic lateral sclerosis 8 protein, VAP, is required for ER protein quality control. Hum Mol Genet. 2013;23:1975–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dai RM, Li CC. Valosin-containing protein is a multi-ubiquitin chain-targeting factor required in ubiquitin-proteasome degradation. Nat Cell Biol. 2001;3:740–744. [DOI] [PubMed] [Google Scholar]
- [7].Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414:652–656. [DOI] [PubMed] [Google Scholar]
- [8].Jarosch E, Geiss-Friedlander R, Meusser B, et al. Protein dislocation from the endoplasmic reticulum–pulling out the suspect. Traffic. 2002;3:530–536. [DOI] [PubMed] [Google Scholar]
- [9].Walters KJ, Kleijnen MF, Goh AM, et al. Structural studies of the interaction between ubiquitin family proteins and proteasome subunit S5a. Biochemistry. 2002;41:1767–1777. [DOI] [PubMed] [Google Scholar]
- [10].Atkin JD, Farg MA, Walker AK, et al. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol Dis. 2008;30:400–407. [DOI] [PubMed] [Google Scholar]
- [11].Wang L, Popko B, Roos RP. The unfolded protein response in familial amyotrophic lateral sclerosis. Hum Mol Genet. 2011;20:1008–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wang L, Popko B, Tixier E, et al. Guanabenz, which enhances the unfolded protein response, ameliorates mutant SOD1-induced amyotrophic lateral sclerosis. Neurobiol Dis. 2014;71:317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang L, Popko B, Roos RP. An enhanced integrated stress response ameliorates mutant SOD1-induced ALS. Hum Mol Genet. 2014;23:2629–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Das I, Krzyzosiak A, Schneider K, et al. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science. 2015;348:239–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Vieira FG, Ping Q, Moreno AJ, et al. Guanabenz treatment accelerates disease in a mutant SOD1 mouse model of ALS. PLoS One. 2015;10:e0135570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lin G, Mao D, Bellen HJ. Amyotrophic lateral sclerosis pathogenesis converges on defects in protein homeostasis associated with TDP-43 mislocalization and proteasome-mediated degradation overload. Curr Top Dev Biol. 2017;121:111–171. [DOI] [PubMed] [Google Scholar]
- [17].Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. [DOI] [PubMed] [Google Scholar]
- [18].Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. [DOI] [PubMed] [Google Scholar]
- [19].Xia Q, Wang H, Hao Z, et al. TDP-43 loss of function increases TFEB activity and blocks autophagosome-lysosome fusion. Embo J. 2016;35:121–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ying Z, Xia Q, Hao Z, et al. TARDBP/TDP-43 regulates autophagy in both MTORC1-dependent and MTORC1-independent manners. Autophagy. 2016;12:707–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sullivan PM, Zhou X, Robins AM, et al. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol Commun. 2016;4:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Webster CP, Smith EF, Bauer CS, et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. Embo J. 2016;35:1656–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yang M, Liang C, Swaminathan K, et al. A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci Adv. 2016;2:e1601167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chen HJ, Anagnostou G, Chai A, et al. Characterization of the properties of a novel mutation in VAPB in familial amyotrophic lateral sclerosis. J Biol Chem. 2010;285:40266–40281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kabashi E, El Oussini H, Bercier V, et al. Investigating the contribution of VAPB/ALS8 loss of function in amyotrophic lateral sclerosis. Hum Mol Genet. 2013;22:2350–2360. [DOI] [PubMed] [Google Scholar]
- [26].Nishimura AL, Mitne-Neto M, Silva HC, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75:822–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].van Blitterswijk M, van Es MA, Koppers M, et al. VAPB and C9orf72 mutations in 1 familial amyotrophic lateral sclerosis patient. Neurobiol Aging. 2012;33:2950 e1–4. [DOI] [PubMed] [Google Scholar]
- [28].Anagnostou G, Akbar MT, Paul P, et al. Vesicle associated membrane protein B (VAPB) is decreased in ALS spinal cord. Neurobiol Aging. 2010;31:969–985. [DOI] [PubMed] [Google Scholar]
- [29].Teuling E, Ahmed S, Haasdijk E, et al. Motor neuron disease-associated mutant vesicle-associated membrane protein-associated protein (VAP) B recruits wild-type VAPs into endoplasmic reticulum-derived tubular aggregates. J Neurosci. 2007;27:9801–9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mitne-Neto M, Machado-Costa M, Marchetto MC, et al. Downregulation of VAPB expression in motor neurons derived from induced pluripotent stem cells of ALS8 patients. Hum Mol Genet. 2011;20:3642–3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Pennetta G, Hiesinger PR, Fabian-Fine R, et al. Drosophila VAP-33A directs bouton formation at neuromuscular junctions in a dosage-dependent manner. Neuron. 2002;35:291–306. [DOI] [PubMed] [Google Scholar]
- [32].Kawano M, Kumagai K, Nishijima M, et al. Efficient trafficking of ceramide from the endoplasmic reticulum to the Golgi apparatus requires a VAMP-associated protein-interacting FFAT motif of CERT. J Biol Chem. 2006;281:30279–30288. [DOI] [PubMed] [Google Scholar]
- [33].Loewen CJ, Roy A, Levine TP. A conserved ER targeting motif in three families of lipid binding proteins and in Opi1p binds VAP. Embo J. 2003;22:2025–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sanhueza M, Chai A, Smith C, et al. Network analyses reveal novel aspects of ALS pathogenesis. PLoS Genet. 2015;11:e1005107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Peretti D, Dahan N, Shimoni E, et al. Coordinated lipid transfer between the endoplasmic reticulum and the Golgi complex requires the VAP proteins and is essential for Golgi-mediated transport. Mol Biol Cell. 2008;19:3871–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].De Vos KJ, Morotz GM, Stoica R, et al. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Genet. 2012;21:1299–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gomez-Suaga P, Paillusson S, Stoica R, et al. The ER-mitochondria tethering complex VAPB-PTPIP51 regulates autophagy. Curr Biol. 2017;27:371–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Han SM, Tsuda H, Yang Y, et al. Secreted VAPB/ALS8 major sperm protein domains modulate mitochondrial localization and morphology via growth cone guidance receptors. Dev Cell. 2012;22:348–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Nishimura Y, Hayashi M, Inada H, et al. Molecular cloning and characterization of mammalian homologues of vesicle-associated membrane protein-associated (VAMP-associated) proteins. Biochem Biophys Res Commun. 1999;254:21–26. [DOI] [PubMed] [Google Scholar]
- [40].Kagiwada S, Hosaka K, Murata M, et al. The saccharomyces cerevisiae SCS2 gene product, a homolog of a synaptobrevin-associated protein, is an integral membrane protein of the endoplasmic reticulum and is required for inositol metabolism. J Bacteriol. 1998;180:1700–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Deidda I, Galizzi G, Passantino R, et al. Expression of vesicle-associated membrane-protein-associated protein B cleavage products in peripheral blood leukocytes and cerebrospinal fluid of patients with sporadic amyotrophic lateral sclerosis. Eur J Neurol. 2014;21:478–485. [DOI] [PubMed] [Google Scholar]
- [42].Mesmin B, Bigay J, Moser von Filseck J, et al. A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell. 2013;155:830–843. [DOI] [PubMed] [Google Scholar]
- [43].Forrest S, Chai A, Sanhueza M, et al. Increased levels of phosphoinositides cause neurodegeneration in a Drosophila model of amyotrophic lateral sclerosis. Hum Mol Genet. 2013;22:2689–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dong R, Saheki Y, Swarup S, et al. Endosome-ER contacts control actin nucleation and retromer function through VAP-dependent regulation of PI4P. Cell. 2016;166:408–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Larroquette F, Seto L, Gaub PL, et al. Vapb/Amyotrophic lateral sclerosis 8 knock-in mice display slowly progressive motor behavior defects accompanying ER stress and autophagic response. Hum Mol Genet. 2015;24:6515–6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Schuck S, Prinz WA, Thorn KS, et al. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J Cell Biol. 2009;187:525–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Atkin JD, Farg MA, Turner BJ, et al. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J Biol Chem. 2006;281:30152–30165. [DOI] [PubMed] [Google Scholar]
- [48].Kikuchi H, Almer G, Yamashita S, et al. Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc Natl Acad Sci U S A. 2006;103:6025–6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ilieva EV, Ayala V, Jove M, et al. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain. 2007;130:3111–3123. [DOI] [PubMed] [Google Scholar]
- [50].Szegezdi E, Logue SE, Gorman AM, et al. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Jiang HQ, Ren M, Jiang HZ, et al. Guanabenz delays the onset of disease symptoms, extends lifespan, improves motor performance and attenuates motor neuron loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neuroscience. 2014;277:132–138. [DOI] [PubMed] [Google Scholar]
- [52].Hetz C, Saxena S. ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol. 2017;13:477–491. [DOI] [PubMed] [Google Scholar]
- [53].Sidrauski C, Acosta-Alvear D, Khoutorsky A, et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife. 2013;2:e00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chou A, Krukowski K, Jopson T, et al. Inhibition of the integrated stress response reverses cognitive deficits after traumatic brain injury. Proc Natl Acad Sci U S A. 2017;114:E6420–E6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Gurney ME, Pu H, Chiu AY, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. [DOI] [PubMed] [Google Scholar]
- [56].Placzek AN, Prisco GV, Khatiwada S, et al. eIF2alpha-mediated translational control regulates the persistence of cocaine-induced LTP in midbrain dopamine neurons. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ogata M, Hino S, Saito A, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Senft D, Ronai ZA. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci. 2015;40:141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Nezis IP, Simonsen A, Sagona AP, et al. Ref(2)P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. J Cell Biol. 2008;180:1065–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. [DOI] [PubMed] [Google Scholar]
- [61].Cheng X, Liu H, Jiang CC, et al. Connecting endoplasmic reticulum stress to autophagy through IRE1/JNK/beclin-1 in breast cancer cells. Int J Mol Med. 2014;34:772–781. [DOI] [PubMed] [Google Scholar]
- [62].Platt FM, Boland B, van der Spoel AC. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol. 2012;199:723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Mauvezin C, Nagy P, Juhasz G, et al. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat Commun. 2015;6:7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Yu L, McPhee CK, Zheng L, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Rong Y, Liu M, Ma L, et al. Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat Cell Biol. 2012;14:924–934. [DOI] [PubMed] [Google Scholar]
- [66].Yoshii SR, Mizushima N. Monitoring and measuring autophagy. Int J Mol Sci. 2017;18:1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Pulipparacharuvil S, Akbar MA, Ray S, et al. Drosophila Vps16A is required for trafficking to lysosomes and biogenesis of pigment granules. J Cell Sci. 2005;118:3663–3673. [DOI] [PubMed] [Google Scholar]
- [68].Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–544. [DOI] [PubMed] [Google Scholar]
- [69].Sardiello M, Palmieri M, Di Ronza A, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. [DOI] [PubMed] [Google Scholar]
- [70].Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kinghorn KJ, Gronke S, Castillo-Quan JI, et al. A drosophila model of neuronopathic gaucher disease demonstrates lysosomal-autophagic defects and altered mTOR signalling and is functionally rescued by rapamycin. J Neurosci. 2016;36:11654–11670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Onyenwoke RU, Brenman JE. Lysosomal storage diseases-regulating neurodegeneration. J Exp Neurosci. 2015;9:81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].de la Mata M, Cotan D, Oropesa-Avila M, et al. Pharmacological chaperones and coenzyme Q10 treatment improves mutant beta-glucocerebrosidase activity and mitochondrial function in neuronopathic forms of gaucher disease. Sci Rep. 2015;5:10903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Settembre C, Zoncu R, Medina DL, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. Embo J. 2012;31:1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Pena-Llopis S, Vega-Rubin-de-Celis S, Schwartz JC, et al. Regulation of TFEB and V-ATPases by mTORC1. Embo J. 2011;30:3242–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Martina JA, Chen Y, Gucek M, et al. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8:903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Zoncu R, Bar-Peled L, Efeyan A, et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334:678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Schneider JL, Villarroya J, Diaz-Carretero A, et al. Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging. Aging Cell. 2015;14:249–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Furuita K, Jee J, Fukada H, et al. Electrostatic interaction between oxysterol-binding protein and VAMP-associated protein A revealed by NMR and mutagenesis studies. J Biol Chem. 2010;285:12961–12970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Graham TR, Burd CG. Coordination of golgi functions by phosphatidylinositol 4-kinases. Trends Cell Biol. 2011;21:113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Csizmadia T, Lorincz P, Hegedus K, et al. Molecular mechanisms of developmentally programmed crinophagy in Drosophila. J Cell Biol. 2018;217:361–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Weckman A, Di Ieva A, Rotondo F, et al. Autophagy in the endocrine glands. J Mol Endocrinol. 2014;52:R151–63. [DOI] [PubMed] [Google Scholar]
- [83].Hu YB, Dammer EB, Ren RJ, et al. The endosomal-lysosomal system: from acidification and cargo sorting to neurodegeneration. Transl Neurodegener. 2015;4:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Huotari J, Helenius A. Endosome maturation. Embo J. 2011;30:3481–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Rutaganira FU, Fowler ML, McPhail JA, et al. Design and structural characterization of potent and selective inhibitors of phosphatidylinositol 4 kinase iiibeta. J Med Chem. 2016;59:1830–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Duvvuri M, Gong Y, Chatterji D, et al. Weak base permeability characteristics influence the intracellular sequestration site in the multidrug-resistant human leukemic cell line HL-60. J Biol Chem. 2004;279:32367–32372. [DOI] [PubMed] [Google Scholar]
- [87].Mindell JA. Lysosomal acidification mechanisms. Annu Rev Physiol. 2012;74:69–86. [DOI] [PubMed] [Google Scholar]
- [88].Werle B, Julke B, Lah T, et al. Cathepsin B fraction active at physiological pH of 7.5 is of prognostic significance in squamous cell carcinoma of human lung. Br J Cancer. 1997;75:1137–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Wijdeven RH, Janssen H, Nahidiazar L, et al. Cholesterol and ORP1L-mediated ER contact sites control autophagosome transport and fusion with the endocytic pathway. Nat Commun. 2016;7:11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Ramesh N, Pandey UB. Autophagy dysregulation in ALS: when protein aggregates get out of hand. Front Mol Neurosci. 2017;10:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Sellier C, Campanari ML, Julie Corbier C, et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. Embo J. 2016;35:1276–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Ugolino J, Ji YJ, Conchina K, et al. Loss of C9orf72 enhances autophagic activity via deregulated mTOR and TFEB signaling. PLoS Genet. 2016;12:e1006443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Pirrotta V. Vectors for P-mediated transformation in drosophila. Biotechnology. 1988;10:437–456. [DOI] [PubMed] [Google Scholar]
- [96].Venken KJ, He Y, Hoskins RA, et al. P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science. 2006;314:1747–1751. [DOI] [PubMed] [Google Scholar]
- [97].Nezis IP, Lamark T, Velentzas AD, et al. Cell death during drosophila melanogaster early oogenesis is mediated through autophagy. Autophagy. 2009;5:298–302. [DOI] [PubMed] [Google Scholar]
- [98].Cherry S, Jin EJ, Ozel MN, et al. Charcot-marie-tooth 2B mutations in rab7 cause dosage-dependent neurodegeneration due to partial loss of function. Elife. 2013;2:e01064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].David-Morrison G, Xu Z, Rui YN, et al. WAC regulates mTOR activity by acting as an adaptor for the TTT and pontin/reptin complexes. Dev Cell. 2016;36:139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Chen K, Lin G, Haelterman NA, et al. Loss of Frataxin induces iron toxicity, sphingolipid synthesis, and Pdk1/Mef2 activation, leading to neurodegeneration. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Castillo-Quan JI, Li L, Kinghorn KJ, et al. Lithium promotes longevity through GSK3/NRF2-dependent hormesis. Cell Rep. 2016;15:638–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Hughes EN, August JT. Characterization of plasma membrane proteins identified by monoclonal antibodies. J Biol Chem. 1981;256:664–671. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.