

ABSTRACT
Macroautophagy/autophagy plays important roles in health and disease, but mechanisms of its activation are unclear. Recently we established IPMK (inositol polyphosphate multikinase) as a physiological determinant of autophagy independent of its catalytic activity. Two signaling axes, IPMK-AMPK-SIRT1 and IPMK-AMPK-ULK1, appear to mediate the influence of IPMK on autophagy. IPMK enhances autophagy-related transcription by stimulating AMPK-dependent SIRT1 activation, which mediates the deacetylation of histone 4 lysine 16. Furthermore, direct binding of IPMK to ULK and AMPK forms a ternary complex that facilitates AMPK-dependent ULK phosphorylation. Deletion of Ipmk virtually abolishes lipophagy, promotes liver damage and impairs hepatocyte regeneration. Our study establishes the importance of IPMK in regulation of autophagy and as a drug target for autophagy-related diseases.
KEYWORDS: AMPK, autophagy, IPMK (Inositol polyphosphate multikinase), liver, ULK
IPMK (inositol polyphosphate multikinase) physiologically generates inositol 4-phosphate (IP4) and IP5. In mammalian cells IPMK also acts as a phosphoinositide 3-kinase. IPMK deleted mice die early in development, at approximately embryonic day 9.5 (E9.5), because of severe growth and morphological defects resembling the phenotype of mice with deletion of major autophagy-regulatory genes, such as Becn1/Beclin 1, FIP200, and Ambra1. In the present study, we assessed the impact of IPMK on autophagy [1]. We used several stimuli (glucose starvation and hydrogen peroxide) to induce autophagy in IPMK wild-type and knockout mouse embryonic fibroblasts (MEFs). Autophagic assays (LC3 puncta, LC3-II western blot, and transmission electron microscopy) established that deletion of IIPMK significantly diminishes autophagy. We also generated liver-specific IPMK knockout mice, which led to diminished hepatic autophagy.
Mechanistically, IPMK regulates autophagy in two different ways (Figure 1). (A) IPMK influences transcription of autophagy-related genes by regulating H4K16 deacetylation. (B) IPMK mediates AMPK-dependent ULK phosphorylation. (A) AMPK initiates autophagy by regulating the transcription of autophagic genes. Nutrient deprivation promotes AMPK-mediated SIRT1 activation and deacetylation of H4K16, followed by transcription of autophagy-related genes. Specifically, AMPK enhances dissociation of SIRT1 from its inhibitor DBC1. We showed that IPMK is essential for activation of AMPK and SIRT1. Thus, loss of IPMK hinders AMPK-mediated downstream effects on SIRT1 and transcription of Lc3b, Bnip3, Bnip3l, Sqstm1/p62, Gabarapl1, and Atg12. (B) Conversely, AMPK phosphorylates ULK and activates autophagy by recruiting the BECN1-containing complex and activating the class III phosphatidylinositol 3-kinase PIK3C3/VPS34. We showed that AMPK-dependent ULK phosphorylation is abolished with deletion of IPMK. IPMK might influence ULK phosphorylation by activating AMPK. However, with H2O2 treatment, IPMK deleted MEFs have increased levels of phospho-AMPK, comparable to the wild type, although ULK phosphorylation at the AMPK site is significantly diminished. Intriguingly, protein-protein interaction studies confirm that IPMK acts as a scaffold protein, linking AMPK with ULK, and promotes AMPK-mediated ULK phosphorylation.
Figure 1.
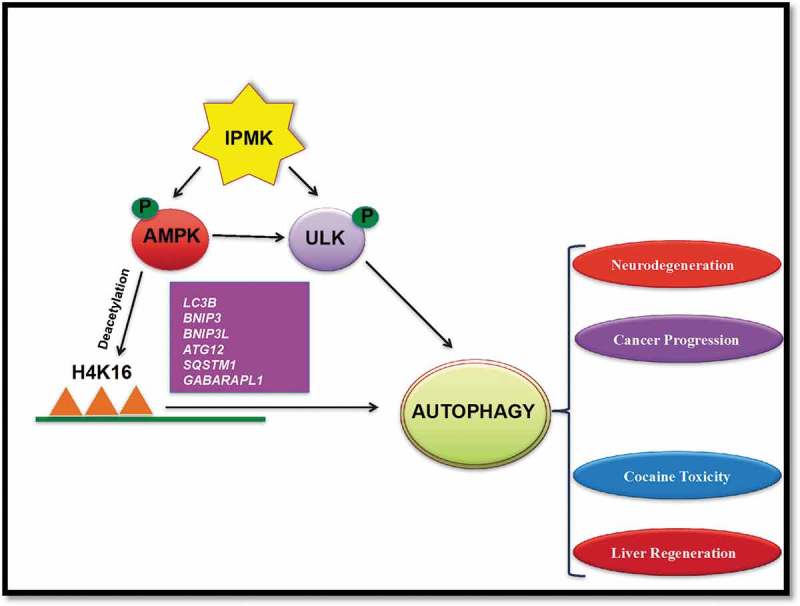
IPMK promotes AMPK activation and mediates deacetylation of histone K16 acetylation. It stimulates transcriptional activation of autophagic genes such as Lc3b, Bnip3, Bnip3l, Atg12, Sqstm1, and Gabarapl1. IPMK also forms a ternary complex of AMPK-IPMK-ULK. It facilitates AMPK-dependent ULK phosphorylation and activation of autophagy. Thus, IPMK could be a therapeutic target of autophagy-related diseases.
One form of macroautophagy, called lipophagy, contributes to hydrolysis of triacylglycerol stored in cytoplasmic lipid droplets. Accordingly, we evaluated a potential role for IPMK in regulating lipophagy. IPMK deleted MEFs display a doubling of lipid droplets both in regular medium and with oleate treatment, indicating substantial diminution of lipophagy. Starvation induces hepatic autophagy and increases delivery of free fatty acids from adipose tissue lipolysis to the liver. Deletion of IPMK in liver diminishes lipophagy and leads to accumulation of lipid droplets. We wondered whether IPMK deficiency affects overall liver function. In untreated preparations we find a mild increase of inflammatory cells in IPMK - deleted liver sections. Deleting IPMK increases the cytotoxic effects of liver toxicants such as carbon tetrachloride. A single dose of carbon tetrachloride promotes liver inflammation, but by 48 h hepatocyte regeneration stimulates wound healing. Strikingly, deletion of IPMK substantially inhibits mouse liver-regeneration. Thus IPMK-mediated autophagy appears to influence hepatic stem cell proliferation and liver regeneration.
Here we have reported IPMK as a prominent physiological regulator of autophagy. Accordingly, targeting IPMK may influence autophagy-driven diseases such as cancer progression, neurodegenerative disorders, cardiac diseases and neuroinflammation. We recently showed that autophagy promotes cocaine toxicity in the central nervous system. We hypothesize that inhibition of IPMK also regulates cocaine toxicity.
Funding Statement
This work was supported by NIH grant DA000266;National Institutes of Health [DA000266].
Disclosure statement
No potential conflict of interest was reported by the authors.
Reference
- [1].Guha P, Tyagi R, Chowdhury S, et al. IPMK mediates activation of ULK signaling and transcriptional regulation of autophagy linked to liver inflammation and regeneration. Cell Rep. 2019. March 5;26(10):2692–2703.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]