

ABSTRACT
Notwithstanding the numerous drugs available for liver cancer, emerging evidence suggests that chemotherapeutic resistance is a significant issue. HGF and its receptor MET play critical roles in liver carcinogenesis and metastasis, mainly dependent on the activity of receptor tyrosine kinase. However, for unknown reasons, all HGF-MET kinase activity-targeted drugs have failed or have been suspended in clinical trials thus far. Macroautophagy/autophagy is a protective ‘self-eating’ process for resisting metabolic stress by recycling obsolete components, whereas the impact of autophagy-mediated reprogrammed metabolism on therapeutic resistance is largely unclear, especially in liver cancer. In the present study, we first observed that HGF stimulus facilitated the Warburg effect and glutaminolysis to promote biogenesis in multiple liver cancer cells. We then identified the pyruvate dehydrogenase complex (PDHC) and GLS/GLS1 as crucial substrates of HGF-activated MET kinase; MET-mediated phosphorylation inhibits PDHC activity but activates GLS to promote cancer cell metabolism and biogenesis. We further found that the key residues of kinase activity in MET (Y1234/1235) also constitute a conserved LC3-interacting region motif (Y1234-Y1235-x-V1237). Therefore, on inhibiting HGF-mediated MET kinase activation, Y1234/1235-dephosphorylated MET induced autophagy to maintain biogenesis for cancer cell survival. Moreover, we verified that Y1234/1235-dephosphorylated MET correlated with autophagy in clinical liver cancer. Finally, a combination of MET inhibitor and autophagy suppressor significantly improved the therapeutic efficiency of liver cancer in vitro and in mice. Together, our findings reveal an HGF-MET axis-coordinated functional interaction between tyrosine kinase signaling and autophagy, and establish a MET-autophagy double-targeted strategy to overcome chemotherapeutic resistance in liver cancer.
Abbreviations: ALDO: aldolase, fructose-bisphosphate; CQ: chloroquine; DLAT/PDCE2: dihydrolipoamide S-acetyltransferase; EMT: epithelial-mesenchymal transition; ENO: enolase; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GLS/GLS1: glutaminase; GLUL/GS: glutamine-ammonia ligase; GPI/PGI: glucose-6-phosphate isomerase; HCC: hepatocellular carcinoma; HGF: hepatocyte growth factor; HK: hexokinase; LDH: lactate dehydrogenase; LIHC: liver hepatocellular carcinoma; LIR: LC3-interacting region; PDH: pyruvate dehydrogenase; PDHA1: pyruvate dehydrogenase E1 alpha 1 subunit; PDHX: pyruvate dehydrogenase complex component X; PFK: phosphofructokinase; PK: pyruvate kinase; RTK: receptor tyrosine kinase; TCGA: The Cancer Genome Atlas
KEYWORDS: Biogenesis, combined treatment, glutaminolysis, targeted therapy, Warburg effect
Introduction
Liver cancer is currently the second leading cause of cancer-related mortality worldwide [1–3]. For instance, the 5-year survival rate among hepatocellular carcinoma (HCC) patients is <5% owing to poor prognosis [4]. Thus far, the most effective therapies for liver cancers are surgical excision, interventional radiological treatment, or liver transplantation [5,6]. Nevertheless, owing to delayed or indistinguishable appearance of clinical signs and symptoms, only few patients have the opportunity to receive treatment [7]. Regarding conventional chemotherapy, sorafenib, an approved small-molecule inhibitor targeting RAF1, BRAF, VEGFR2/KDR, FLT4/VEGFR3, PDGFRB, FLT3, KIT, and FGFR1 tyrosine kinases, has a limited survival benefit in unresectable advanced HCC [8,9]. Therefore, it is critical to investigate how liver cancers resist chemotherapy, and simultaneously develop new drugs or strategies to overcome chemotherapeutic resistance [10,11].
Because the liver is not only the largest metabolic organ in our body, but is also associated with almost all the central metabolic processes, tumorigenesis or tumor progression in the liver inevitably result in the reprogramming of metabolism [12]. In the case when nutrition supply is adequate, the Warburg effect and glutaminolysis are major characteristic metabolic modes in cancer [13]. Generally, in contrast with normal differentiated cells, most cancer cells or undifferentiated cells (such as stem cells) rely primarily on aerobic glycolysis rather than mitochondrial oxidative phosphorylation to metabolize glucose to generate energy for cellular processes [14,15]. This phenomenon was first described by Otto Warburg in 1924 [16], and is hence termed the ‘Warburg effect’ [17]. In addition, many cancer cells preferably metabolize glutamine, a nonessential amino acid; this phenomenon is called ‘glutaminolysis’ [18]. Glutamine is not only a nitrogen source for amino acid and nucleotide synthesis but is also a major carbon source for the tricarboxylic acid cycle and macromolecule biosynthesis; thus, cancer cells cannot survive without an exogenous supply of glutamine [19–21]. However, whether reprogrammed metabolism has a crucial impact on chemotherapeutic resistance in liver cancer is still unclear. More importantly, besides the Warburg effect and glutaminolysis, cancers also can depend on autophagy, a protective ‘self-eating’ metabolic process, to recycle obsolete components and supplement energy so as to support aberrant cell growth under metabolic dysfunction or when cells are suffering nutritional limitations [22], especially in the liver [23]. Of note, there is a longtime conjecture that the intimate connection and reciprocal conversion between the Warburg effect, glutaminolysis and autophagy determine cancer therapeutic efficacy. Even so, their significance and association with chemotherapeutic resistance in liver cancer are totally unknown.
HGF (hepatocyte growth factor) and its receptor tyrosine kinase MET/HGFR were first identified in the 1980s, owing to their hyperactivation in numerous liver cancers [24–27]. As the name indicates, the HGF-MET axis stimulates hepatocytes by sustaining proliferation, promotes epithelial-mesenchymal transition (EMT), and finally, causes invasion and metastasis during malignant transformation in liver cancer [28–31]. The importance of HGF-MET signaling renders them appropriate targets for at least 25 compounds currently being clinically developed [32–34]. However, almost all the HGF-MET-targeted drugs failed or were suspended in clinical trials, the underlying reason for which is unclear. Although the co-overexpression of human MET and mutant CTNNB1/beta-catenin can lead to HCC in mice, owing to RAS activation [35], the physio-pathological relevance of HGF-mediated growth signaling to MET-associated downstream pathways in chemotherapeutic resistance in liver cancer is largely unknown. In this study, we attempted to determine the role and detailed molecular mechanism of the HGF-MET axis in chemotherapeutic resistance in liver cancer. Using a CRISPR-Cas9-mediated knockout system, we found HGF-MET signaling facilitated cancer characteristic metabolic modes (Warburg effect and glutaminolysis) to support liver cancer biogenesis for tumor growth. Moreover, it is dependent on direct kinase effects of MET on the pyruvate dehydrogenase complex (PDHC) and GLS (glutaminase). However, we further found that the key residues for kinase activity in MET, also constituted a potential LC3-interacting region (LIR) motif. When treated with HGF-MET kinase activity-targeted drugs, tyrosine 1234/1235-dephosphorylated MET activated autophagy, which supports biogenesis for tumor survival under therapeutic stress. This switch-like relationship between the HGF-MET axis and autophagy was further confirmed in clinical tissue microarray and the TCGA database. Finally, we combined a MET-targeted inhibitor with an autophagy blocker, and found this simple strategy could overcome chemotherapeutic resistance in liver cancer.
Results
HGF-MET signaling stimulates the Warburg effect, glutaminolysis and biogenesis in liver cancer
Cancer cells are capable of adapting their metabolic processes to drive atypical synthesis of biological macromolecules to meet the requirements of rapid cell growth and proliferation, which is also referred to as ‘cancer biogenesis’. Initially and intriguingly, we found HGF stimulation visibly accelerated culture medium acidification of multiple liver cancer cell lines of human and murine origin cells, including HepG2, SMMC-7721, Huh-7, MHCC-97H, Hepa1-6, and H22 cell lines (Figure 1(a)), which usually indicated that cancer cell metabolism was increased. Since the liver plays a critical role in the transformation of metabolites, we further investigated the effects of HGF on cancer cell metabolism and biogenesis. We found HGF stimulation facilitated glucose consumption and lactate production across different liver cancer cells (Figure 1(b)), which are hallmarks of the Warburg effect; expedited glutamine consumption and glutamate production uniformly (Figure 1(c)), which are signs of glutaminolysis; and promoted the biosynthesis of DNA, triglyceride, and aspartate (Figure 1(d)), which indicate cancer biogenesis. Of note, the HepG2 cell line is universally recognized as a cell model in liver cancer research, and it showed strong, stable and significant phenotypes in all tests of metabolic marks and biosynthetic indexes under HGF stimulation. Therefore, we used it as the representative in most of the following analyses unless otherwise stated.
Figure 1.
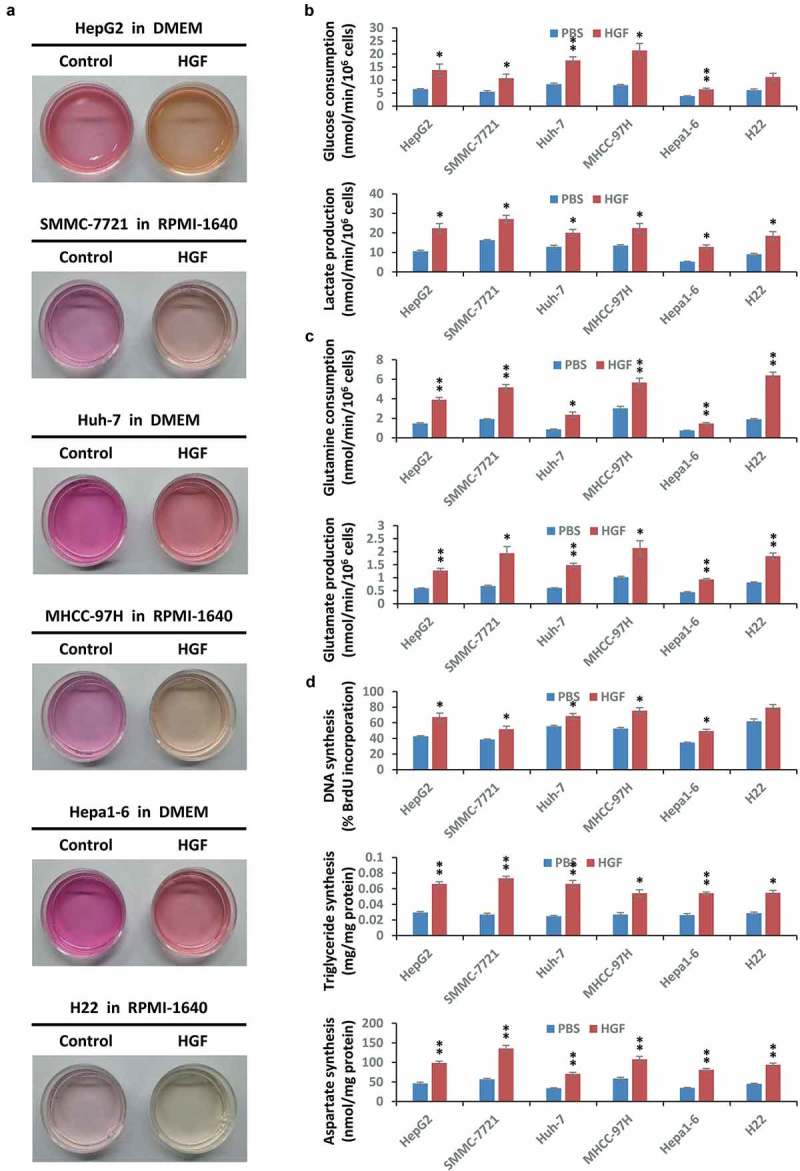
HGF facilitates liver cancer metabolism and biogenesis. (a) HGF accelerates acidification of liver cancer cell culture medium. After starvation overnight, HepG2, SMMC-7721, Huh-7, MHCC-97H, Hepa1-6 and H22 liver cancer cell lines (5 × 104) were individually treated with or without HGF (40 ng/ml) for 48 h, then used for photographs of the culture medium. The color of the medium indicated the degree of acidification. Abbreviations: DMEM, Dulbecco’s modified Eagle’s medium; RPMI-1640, Roswell Park Memorial Institute 1640 medium. (b) Impact of HGF on the Warburg effect in liver cancer cell lines. After starvation overnight, HepG2, SMMC-7721, Huh-7, MHCC-97H, Hepa1-6 and H22 cells (5 × 104) were individually stimulated with or without HGF (40 ng/ml) for 6 h, and subsequently subjected to analysis of glucose consumption and lactate production. (c) Effect of HGF on glutaminolysis in liver cancer cell lines. After starvation overnight, HepG2, SMMC-7721, Huh-7, MHCC-97H, Hepa1-6 and H22 cells (5 × 104) were individually stimulated with or without HGF (40 ng/ml) for 6 h, and subsequently subjected to analysis of glutamine consumption and glutamate production. (d) Impact of HGF on biogenesis in liver cancer cell lines. After starvation overnight, HepG2, SMMC-7721, Huh-7, MHCC-97H, Hepa1-6 and H22 cells (5 × 104) were individually stimulated with or without HGF (40 ng/ml) for 24 h, and subsequently subjected to analysis of nucleotide (DNA), lipid (triglyceride) and amino acid (aspartate) synthesis. Data are presented as the means ± SD from at least 3 independent experiments. Statistically significant differences with two-tailed Student’s t-test are marked as * (p < 0.05) or ** (p < 0.01). Abbreviations: HGF, hepatocyte growth factor.
Given that HGF stimulation may inevitably cause much indirect metabolism-related transformation, as well as the dominant role of MET in HGF signal transduction, we next investigated the metabolic alteration in MET-deficient conditions. We first generated a MET-knockout (KO) HepG2 cell line by the CRISPR-Cas9 system and mediated a double-strand break in a 5ʹ constitutive exon within the MET gene to disrupt its expression. We employed wild-type (WT) and MET KO HepG2 cells to perform an untargeted metabolomics analysis by a GC/LC-MS based assay, and the outcomes were basically consistent with the original conclusions under HGF stimulation. The landscape of MET deletion-caused metabolic alteration was presented in the heat-map, and the relative levels of all differential metabolites detected between WT and MET KO cells were quantified and clustered as indicated (Figure S1(a)). Moreover, statistically significant metabolite-metabolite connections in the case of MET deletion were presented to clarify the relationship between MET-controlled metabolites, such as the positive correlation between glucose and lactic acid, or L-glutamate and L-aspartic acid (Figure S1(b)). Subsequently, to figure out the potential influence of MET depletion on metabolic pathways, these differential metabolites were individually divided into main metabolic groups according to KEGG annotation (Figure S1(c) and Table S1). Detailed enrichment analysis then demonstrated that MET depletion indeed impaired the Warburg effect and glutaminolysis-associated metabolic pathways, including but not limited to carbohydrate metabolism, amino acid metabolism, lipid metabolism and energy metabolism (Figure S1(d) and Table S2). Together, the results of untargeted metabolomics analysis further confirmed the importance of MET signaling in cancer metabolism.
HGF-MET signaling facilitates the Warburg effect, glutaminolysis and biogenesis via inhibiting PDHC and activating GLS
It is well established that a few of the specific metabolic enzymes dominate the Warburg effect and glutaminolysis, mainly including HK (hexokinase), GPI/PGI (glucose-6-phosphate isomerase), PFK (phosphofructokinase), ALDO (aldolase, fructose-bisphosphate), GAPDH (glyceraldehyde-3-phosphate dehydrogenase), ENO (enolase), PK (pyruvate kinase), pyruvate dehydrogenase (PDH), LDH (lactate dehydrogenase), GLS (glutaminase), and GLUL/GS (glutamine-ammonia ligase). To determine how the HGF growth signal is transmitted and acts on liver cancer metabolism via the MET receptor, we conducted a small-scale activity-oriented screening for all these enzymes under conditions of HGF stimulation or/and MET deficiency to identify potential candidates which are probably regulated by HGF-MET signaling. Results clearly showed that HGF stimulation inhibited PDHC activity while it enhanced GLS activity; in contrast, MET deletion activated PDHC but restrained GLS (Figure 2(a)). Evidently, the HGF-MET axis presumably blocks PDHC and activates GLS, respectively. Meanwhile, by co-immunoprecipitation experiments, PDHC and GLS were also identified as direct interaction targets of MET for a few critical enzymes and transporters in cancer metabolism (Figure 2(b)). Furthermore, we designed MET-specific small interfering RNA to knock down MET in multiple other liver cancer cells (Figure S2(a)), and found that MET reduction generally and consistently activated PDHC and inhibited GLS (Figure 2(c,d)).
Figure 2.
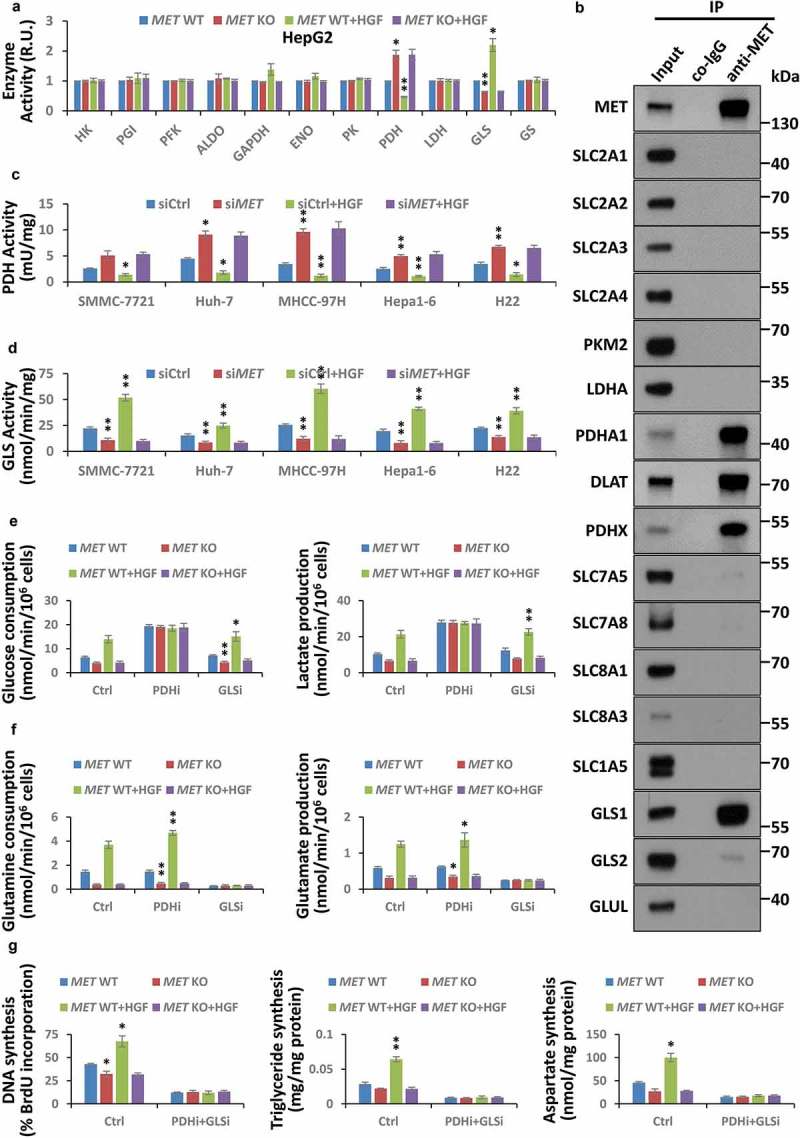
HGF-MET signaling promotes liver cancer metabolism and biogenesis via PDHC and GLS. (a) Screening for critical enzymes under HGF-MET regulation in cancer metabolism. After starvation overnight, HepG2-derived CRISPR-Cas9 system-mediated vehicle control (MET WT) or MET knockout (MET KO) cells (5 × 104) were treated with or without HGF (40 ng/ml) for 2 h, and subsequently subjected to activity analysis for the indicated enzymes. (b) Identification for interaction targets of MET from important enzymes and transporters in cancer metabolism. HepG2 cell lysates (5 × 105) were subjected to co-immunoprecipitation with anti-MET antibody, and then analyzed by western blot with the indicated antibodies. (c and d) Effect of MET on PDHC and GLS activity in liver cancer cell lines. SMMC-7721, Huh-7, MHCC-97H, Hepa1-6 and H22 cells (2 × 104) were individually transfected with siRNAs to knock down MET (siMET) or not (siCtrl). Seventy-two h after transfection, cells were starved overnight, then treated with or without HGF (40 ng/ml) for 2 h, and subsequently subjected to analyze activity of PDHC (c) and GLS (d). (e) Contribution of PDHC and GLS to the HGF-MET signaling-mediated Warburg effect. After pre-incubation with PDHC inhibitor (CPI-613, 100 μM) or GLS inhibitor (BPTES, 100 nM) overnight under starvation, HepG2 MET WT or KO cells (5 × 104) were treated with or without HGF (40 ng/ml) for 6 h, and subsequently subjected to analysis of glucose consumption and lactate production. (f) Contribution of PDHC and GLS to HGF-MET signaling-mediated glutaminolysis. HepG2 MET WT or KO cells (5 × 104) were treated as mentioned above, and subsequently subjected to analysis of glutamine consumption and glutamate production. (g) Contribution of PDHC and GLS to HGF-MET signaling-mediated biogenesis. After pre- and co-incubation with PDHC inhibitor (CPI-613, 100 μM) and GLS inhibitor (BPTES, 100 nM) overnight under starvation, HepG2 MET WT or KO cells (5 × 104) were treated with or without HGF (40 ng/ml) for 24 h, and subsequently subjected to analysis of DNA, triglyceride and aspartate contents. Data are presented as the means ± SD from at least 3 independent experiments. Statistically significant differences with two-tailed Student’s t-test are marked as * (p < 0.05) or ** (p < 0.01). Abbreviations: WT, wild type; KO, knockout; PDHC, pyruvate dehydrogenase complex; GLS, glutaminase; PDHi, PDHC inhibitor; GLSi, GLS inhibitor.
Given that PDHC and GLS are pivotal regulatory factors of the Warburg effect and glutaminolysis, and control carbon and nitrogen sources of cancer biogenesis, respectively, from glucose or glutamine, this observation suggested that the HGF-MET axis-mediated growth signal might expedite the metabolic processing of glucose-to-lactate and glutamine-to-glutamate transformation, and in consequence would lead to high-efficiency biogenesis in liver cancer cells. Indeed, on using the PDH inhibitor CPI-613 and the GLS inhibitor BPTES, we figured out that PDHC and GLS jointly contributed to HGF-MET axis-mediated cancer metabolism and biogenesis. In details, the HGF-MET axis lost its impact on the Warburg effect under PDHC inhibition (Figure 2(e)), was unable to control glutaminolysis under GLS inhibition (Figure 2(f)), and the HGF-MET axis-mediated cancer cell biogenesis was largely suppressed under combined inhibition of PDHC and GLS (Figure 2(g)). Moreover, using culture medium without glucose, glutamine, or both, we verified that HGF-MET signaling-mediated biogenesis was indeed dependent on glucose and glutamine (Figure S2(b)). Hence, the HGF-MET axis promoted liver cancer metabolism and biogenesis through PDHC and GLS.
In addition, metabolic functions of PDHC and GLS were largely attributed to enzyme catalytic activity, which were mainly regulated by reversible phosphorylation modification or phosphate [36–40]. To demonstrate whether the HGF-MET axis had the most direct effects on PDHC and GLS, we performed MET kinase activity-associated assays. First, in silico analysis revealed that PDHC and GLS contained conserved proline (P)-rich regions (PxPP motif) that were necessary for binding to MET-docking sites (Figure S3(a)). Next, an immunoprecipitation assay with anti-phospho-tyrosine also indicated that PDHC and GLS could be phosphorylated at tyrosine residues (Figure S3(b)). Furthermore, we found HGF-mediated MET activation stimulated phosphorylation of PDHC and GLS in vivo (Figure 3(a)). Finally, an in vitro kinase assay clearly shown that MET could directly phosphorylate PDHC (including PDHA1, DLAT/PDCE2 and PDHX) (Figure 3(b-d)) and GLS (Figure 3(e)). Obviously, all these lines of evidence supported the idea that both PDHC and GLS were direct substrates of MET kinase, which implied HGF-MET signaling mediated liver cancer metabolism and biogenesis through post-translational modification of PDHC and GLS.
Figure 3.
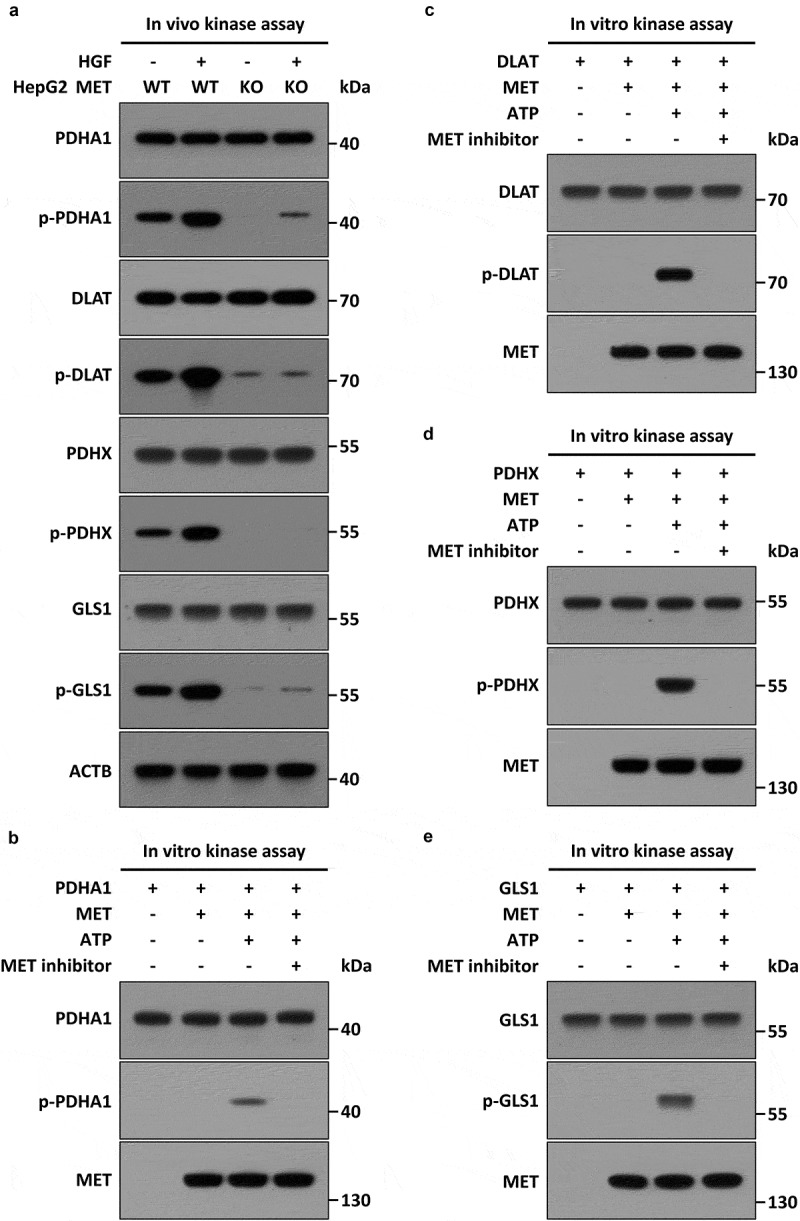
MET phosphorylates PDHC and GLS in vivo and in vitro. (a) Stimulation of HGF-MET signaling on phosphorylation of PDHC and GLS in vivo. WT and MET KO HepG2 cells (5 × 105) were individually treated with or without HGF (40 ng/ml) for 2 h, then subjected to immunoprecipitation with anti-PDHA1, anti-DLAT/PDCE2, anti-PDHX and anti-GLS antibodies, and subsequently analyzed by western blot using anti-phospho-tyrosine or the other indicated antobodies. (b–e) Direct phosphorylation effects of MET on PDHC and GLS in vitro. Recombinant HIS-PDHA1 (1 μg) (b), HIS-DLAT/PDCE2 (1 μg) (c), HIS-PDHX (1 μg) (d) or HIS-GLS (1 μg) (e) were individually incubated with FLAG-MET (400 ng) in kinase reaction buffer at 37°C for 45 min, with or without ATP (10 mM) and MET kinase inhibitor (JNJ-38877605, 50 nM). After the reaction, the mixtures were subjected to western blot analysis with anti-phospho-tyrosine or the other indicated antobodies. Abbreviations: PDHA1, pyruvate dehydrogenase E1 alpha 1 subunit; DLAT/PDCE2, dihydrolipoamide S-acetyltransferase; PDHX, pyruvate dehydrogenase complex component X; GLS, glutaminase.
HGF-MET kinase-targeted drugs only suppress the Warburg effect and glutaminolysis, but without disrupting biogenesis in resistant cells
Generally, the involvement of different receptor tyrosine kinases (RTKs) in diverse signaling pathways is primarily attributed to their kinase activities towards disparate substrates. Therefore, all the current HGF-MET-targeted drug candidates, such as Onartuzumab (Genentech) and Tivantinib (ArQule), mainly focus on inhibiting MET kinase activation or activity. Notwithstanding MET kinase inhibition, it is unclear why HGF-MET-targeted drugs have failed in clinical trials. To further investigate the reason underlying this discrepancy, we employed the MET kinase-specific small molecule inhibitor JNJ-38877605 and anti-MET monoclonal antibody 5D5 to establish MET-targeted inhibitor- or antibody-insensitive HepG2 cells. In contrast to MET KO HepG2 cells, MET-targeted inhibitor- or antibody-insensitive cells showed similar growth properties as WT HepG2 cells in cell proliferation, viability and colony formation capacity (Figure S4(a)). Moreover, these cells possessed a strong ability in resistance to MET-targeted drugs even after selection (Figure S4(b,c)). We further verified that phosphorylation in all kinase activity-associated residues of MET (including Y1003, Y1234/1235, Y1349, Y1356 and Y1365) was completely blocked in MET-targeted inhibitor- or antibody-insensitive cells, and the consequent inhibitory impacts on its downstream effector molecules (such as AKT, PDHC and GLS) were equally severe as well (Figure 4(a)). This indicated the kinase activity of MET was effectively repressed in drug-resistant cells.
Figure 4.
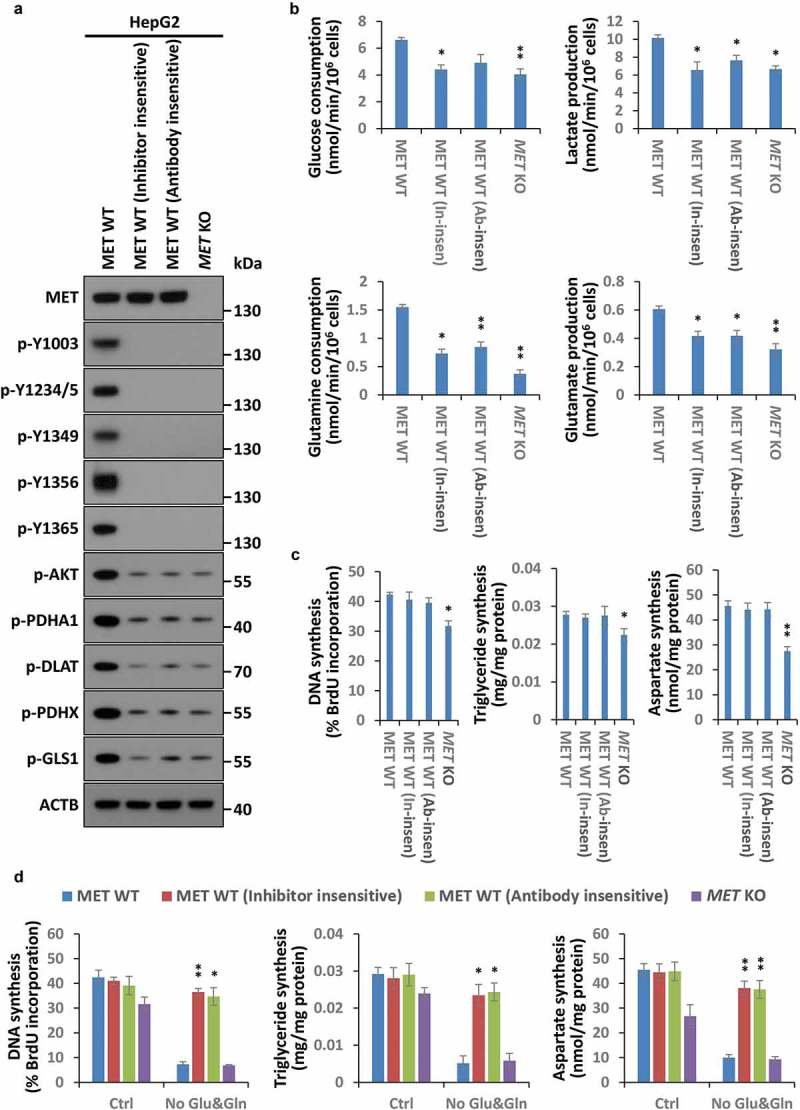
An atypical cancer biogenesis is actuated in HGF-MET-targeted drug-resistant cells. (a) Analysis of MET kinase activity in MET-targeted inhibitor- or antibody-insensitive cells. After 3 rounds of selection with the MET-specific small molecule inhibitor JNJ-38877605 (50 nM) and anti-MET monoclonal antibody 5D5 (40 μg/ml), MET-targeted inhibitor- or antibody-insensitive HepG2 cells (5 × 104) were harvested for western blot with the indicated antibodies. WT and MET KO HepG2 cells were used as controls. (b) Assessment of the Warburg effect and glutaminolysis in MET-targeted inhibitor- or antibody-insensitive cells. MET-targeted inhibitor- or antibody-insensitive HepG2 cells (5 × 104) were subjected to analysis of glucose consumption, lactate production, glutamine consumption and glutamate production. WT and MET KO HepG2 cells were used as controls. (c) Assessment of nucleotide, lipid and amino acid synthesis in MET-targeted inhibitor- or antibody-insensitive cells. MET-targeted inhibitor- or antibody-insensitive HepG2 cells (5 × 104) were subjected to analysis of DNA, triglyceride and aspartate contents. WT and MET KO HepG2 cells were used as controls. (d) Contribution of glucose and glutamine to biogenesis in HGF-MET-targeted drug-insensitive cells. After being pre-cultured in normal medium or medium without glucose and glutamine for 12 h, HepG2 MET WT or KO and MET-targeted inhibitor- or antibody-insensitive cells (5 × 104) were individually subjected to analysis of DNA, triglyceride and aspartate contents. Data are presented as the means ± SD from at least 3 independent experiments. Statistically significant differences with two-tailed Student’s t-test are marked as * (p < 0.05) or ** (p < 0.01). Abbreviations: p-Y, p-MET Y; Y1234/5, Y1234/1235; In-insen, inhibitor-insensative; Ab-insen, antibody-insensitive; Ctrl, normal medium; No Glu&Gln, both glucose- and glutamine-deleted medium.
However, despite the drastic metabolic suppression in the Warburg effect and glutaminolysis (Figure 4(b)), the biogenesis assay manifested that DNA, triglyceride and aspartate synthesis just displayed a little fluctuation in MET-targeted drug-resistant cells in comparison to MET KO cells (Figure 4(c)), that implied cancer biogenesis was not supported by the Warburg effect or glutaminolysis in these cells. On the contrary, these resistant cells still acted in response to HGF stimulation to accelerate the Warburg effect, glutaminolysis and biogenesis (Figure S4(d,e)). In addition, further analyses also revealed that biogenesis was indeed not dependent on glucose and glutamine in resistant cells (Figure 4(d)). Together, these findings indicated that HGF-MET kinase-targeted drugs could induce an atypical, glucose- and glutamine-independent cancer biogenesis in resistant cells.
HGF-MET kinase-targeted drugs reprogram liver cancer biogenesis via Y1234/1235 dephosphorylated MET-mediated autophagy
Besides the Warburg effect and glutaminolysis, emerging evidence suggests that autophagy could recycle obsolete components via ‘self-eating’ to maintain biogenesis under metabolic stress [41]. Indeed, autophagy was greatly activated in cells resistant to HGF-MET kinase-targeted drugs, whether or not JNJ-38877605 inhibitor or 5D5 antibody was present (Figure 5(a)). Moreover, we found that although the protein level of MET was quite stable under kinase-based inhibition (at least at 24 h when biogenesis was assessed in our system) (Figure S5(a)), the blockade of autophagy by ATG5 deletion greatly enhanced the inhibitory effects of MET-targeted drugs on biogenesis (Figure 5(b) and S5(b)). Furthermore, through a series of biochemical and morphological analyses, including western blot, GFP-LC3 reporter system and electron microscopy, we found that MET-targeted drugs significantly promoted LC3 turnover in multiple liver cancer cells (Figure 5(c)), induced LC3 punctate aggregation (Figure 5(d)), and increased autophagosome formation (Figure S5(c)). Furthermore, we demonstrated that kinase inhibition of MET obviously expedited autophagic flux in multiple liver cancer cells (Figure 5(e)), and a similar phenomenon was also observed in MET kinase-targeted drug-resistant cells (Figure 5(f)). Overall, HGF-MET kinase-targeted drugs promoted autophagy to sustain biogenesis in liver cancer.
Figure 5.
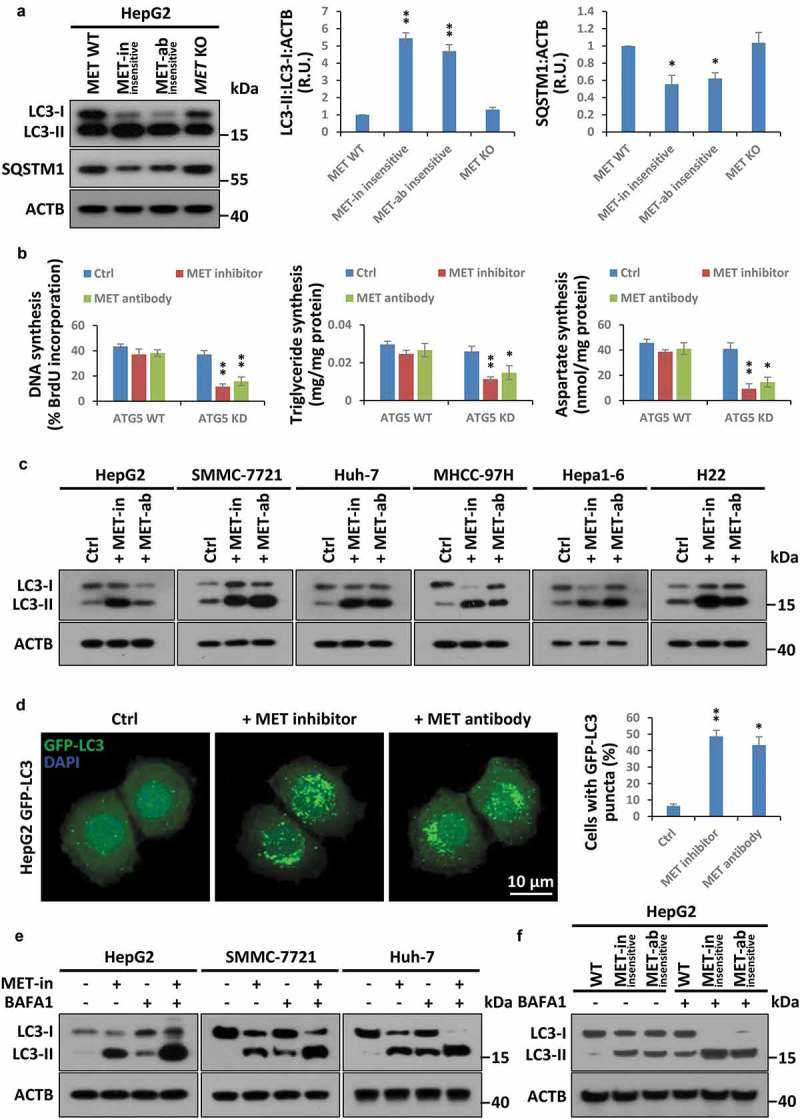
HGF-MET-targeted drugs induce autophagy to reset biogenesis in liver cancer. (a) Assessment of autophagy state in MET-targeted inhibitor- or antibody-insensitive cells. Cell lysates from MET-targeted inhibitor- or antibody-insensitive HepG2 cells (5 × 104) were analyzed by western blot with the indicated antibodies. WT and MET KO HepG2 cells were used as controls. Quantification of LC3 turnover and SQSTM1 degradation are shown on the right. (b) Contribution of autophagy to HGF-MET-targeted drugs-reprogrammed biogenesis. WT and ATG5 KD HepG2 cells (5 × 104) were individually treated with vehicle control, inhibitor of MET (JNJ-38877605, 50 nM) or anti-MET antibody (5D5, 40 μg/ml) for 24 h, and subsequently subjected to analysis of DNA, triglyceride and aspartate contents. (c) Impact of MET-targeted inhibitor or antibody on LC3 turnover in liver cancer cell lines. HepG2, SMMC-7721, Huh-7, MHCC-97H, Hepa1-6 and H22 cells (5 × 104) were individually treated with vehicle control, an inhibitor of MET (JNJ-38877605, 50 nM) or anti-MET antibody (5D5, 40 μg/ml) for 8 h, and subsequently subjected to western blot analysis with the indicated antibodies. (d) Effect of MET-targeted inhibitor or antibody on aggregation of GFP-LC3 puncta. HepG2 cells stably expressing GFP-LC3 (1 × 104) were individually treated with vehicle control, inhibitor of MET (JNJ-38877605, 50 nM) or anti-MET antibody (5D5, 40 μg/ml) for 6 h. Representative images and quantification of MET-targeted inhibitor or antibody-induced GFP-LC3 punctate cells were shown as indicated by confocal fluorescence microscopy. (e) Effect of MET inhibition on autophagic flux. After pre-incubation with or without BAFA1 (50 nM) for 2 h, HepG2, SMMC-7721 and Huh-7 cells (5 × 104) were individually treated with inhibitor of MET (JNJ-38877605, 50 nM) for 6 h or not treated, and subsequently subjected to western blot analysis with the indicated antibodies. (f) Analysis of autophagic flux in MET-targeted inhibitor- or antibody-insensitive cells. MET-targeted inhibitor- or antibody-insensitive cells (5 × 104) were individually treated with or without BAFA1 (50 nM) for 4 h, and subsequently subjected to western blot analysis with the indicated antibodies. Data are presented as the means ± SD from at least 3 independent experiments. Statistically significant differences with two-tailed Student’s t-test are marked as * (p < 0.05) or ** (p < 0.01). Abbreviations: MET-in, MET-targeted inhibitor; MET-ab, MET-targeted antibody; ATG5, autophagy related 5; KD, knockdown; BAFA1, bafilomycin A1; R.U., relative units.
Even so, it is still unknown how the HGF-MET axis coordinates conventional cancer cell metabolism and compensatory autophagy. To address this issue, we first employed bioinformatics analysis and found all essential tyrosine phosphorylation sites to be regulating MET kinase activity (including Y1003, Y1234/1235, Y1349, Y1356 and Y1365 as mentioned before), with all simultaneously being basic components of the potential LC3-interacting region (LIR) motifs (Figure 6(a)). LC3-I conversion to LC3-II initiates autophagosome formation, and thus acts as one of the leading participators in the autophagic process. To investigate whether MET really has the possibility to bind to LC3, we then performed an endogenous immunoprecipitation assay with anti-MET antibody, and found MET indeed interacted with LC3 in multiple liver cancer cell lines (Figure 6(b)). Consistent results were also obtained in anti-LC3 antibody-mediated reverse immunoprecipitation (Figure 6(c)). Given that dephosphorylation of the tyrosine residue in the LIR motif was supposed to enhance its interaction with LC3 [42], we next constructed tyrosine-dephosphorylated mutants of MET (Figure 6(d)). Subsequently, we conducted exogenous mapping analysis and the results suggested that the Y1234 and Y1235 residues were the most essential sites, because Y1234/1235 double dephosphorylated MET recruited more LC3 than did other mutants (Figure 6(e)). Moreover, through multiple sequence alignment, we found that Y1234/Y1235 x V1237 (YYxV), the functional LIR motif in MET, was highly conserved in evolution (Figure 6(f)). Further analyses demonstrated that Y1234/1235 dephosphorylated MET indeed promoted LC3 turnover and SQSTM1 degradation (Figure 6(g)), expedited autophagic flux (Figure S6(a)), and strengthened LC3 binding to SQSTM1 (Figure S6(b)). Finally, metabolic assays verified that Y1234/1235 dephosphorylated MET had no significant effects on the Warburg effect and glutaminolysis (Figure S6(c)), but could markedly enhance HGF-MET axis-controlled biogenesis (Figure S6(d)). Together, MET concealed a conserved LIR motif in Y1234/1235 to drive autophagy for cancer biogenesis under kinase inhibition.
Figure 6.
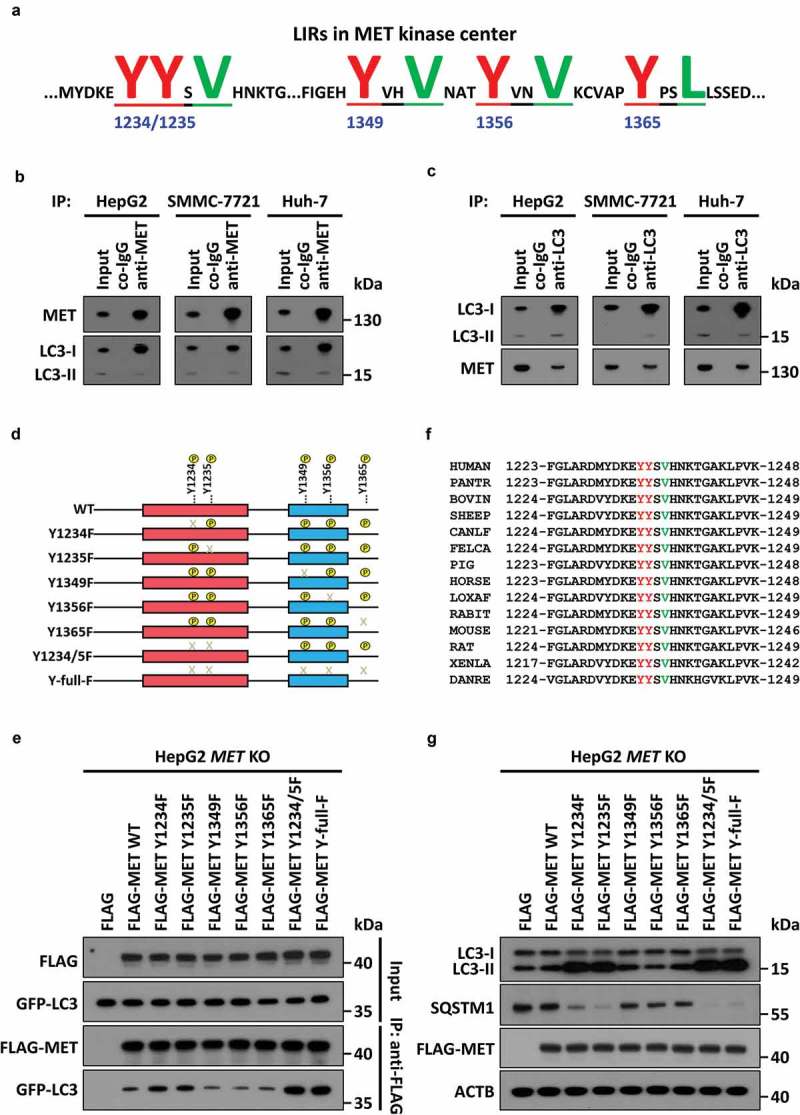
MET conceals a conserved LIR motif in Y1234/1235 to recruit LC3 and drive autophagy under kinase inhibition. (a) Schema of potential LIR motifs hidden in key tyrosine-phosphorylated modification sites of the MET kinase center. Underlined and capital highlighted letters indicate LIR constitution in MET. (b and c) Endogenous immunoprecipitation assay between MET and LC3. Cell lysates of HepG2, SMMC-7721 and Huh-7 cells (5 × 105) were individually immunoprecipitated with anti-MET antobody (b) or anti-LC3 antibody (c), and subsequently subjected to western blot analysis with the indicated antibodies. (d) Construction of MET dephosphorylated mutants. Schematic diagrams of MET and its dephosphorylated mutants are depicted as indicated. (e) Identification of Y1234/1235 as key points for MET-LC3 interaction. HepG2 MET KO cells (5 × 105) were individually co-transfected with vehicle control (Flag), or plasmids encoding WT or dephosphorylated mutants of MET and GFP-LC3. 24 h after transfection, cell lysates were subjected to immunoprecipitation and western blot analysis with the indicated antibodies. (f) Conservative analysis of Y1234, Y1235 and V1237-constituted LIR motif (YYxV) in MET. Alignment of amino acid sequences in evolutionarily different species is shown as indicated, and the YYxV motif is highlighted with red and green letters. (g) Effects of MET dephosphorylated mutants on autophagy. HepG2 MET KO cells (5 × 104) were individually transfected with vehicle control, or plasmids encoding WT or dephosphorylated mutants of MET. Twenty-four h after transfection, cell lysates were analyzed by western blot with the indicated antibodies. Abbreviations: LIR, LC3-interacting region; WT, wild-type; Y, tyrosine residue; Y1234/5F, both Y1234 and Y1235 mutated to F; Y-full-F, all Y sites mutated to F; V, valine residues.
The HGF-MET axis regulates MTOR activity, and controls serum starvation-mediated autophagy and biogenesis
To further figure out how the HGF-MET axis regulates or is regulated by autophagy, we tried to identify other potential players that were involved in HGF-MET signaling-mediated autophagic action. Interestingly, although none of the conventional autophagic machineries (including the ULK1 complex, the BECN1 complex and so on) existed in a MET-centric interactome (Figure S6(e)), we found that pharmacological inhibition of MTOR by rapamycin could suppress MET inhibitor-induced autophagy (Figure 7(a)). Correspondingly, overexpression of a constitutively active RRAGB mutant (Q99L, RRAGBGTP) to activate MTOR could largely rescue autophagy driven by MET inhibitor (Figure 7(b)). This indicated that MET inhibition-mediated autophagy was indeed MTOR dependent (at least largely if not completely). Moreover, phosphorylation analyses showed that HGF stimulated activation of MTOR signaling; in contrast, MET deficiency suppressed both basal level activity and HGF-mediated activation of MTOR (Figure 7(c)). In fact, there is evidence indicating the close connection between MTOR activity and states of oxidation-reduction reaction (redox), especially under metabolic stress. Coincidentally, PDHC and GLS are often linked to maintain redox homeostasis which is usually reflected by ratios of GSSG:GSH, NADP:NADPH and NAD:NADH. Therefore, to explore the regulatory mechanism of MTOR signaling through the HGF-MET axis in our system, we detected GSSG:GSH, NADP:NADPH and NAD:NADH under conditions of HGF stimulation or/and MET deletion, and found HGF-MET signaling indeed controlled redox states via PDHC and GLS (Figure 7(d)). In addition to the well-established RTK-PI3K-AKT cascade, this finding might also help explain in a logical way how MET inhibition blocked MTOR activity.
Figure 7.
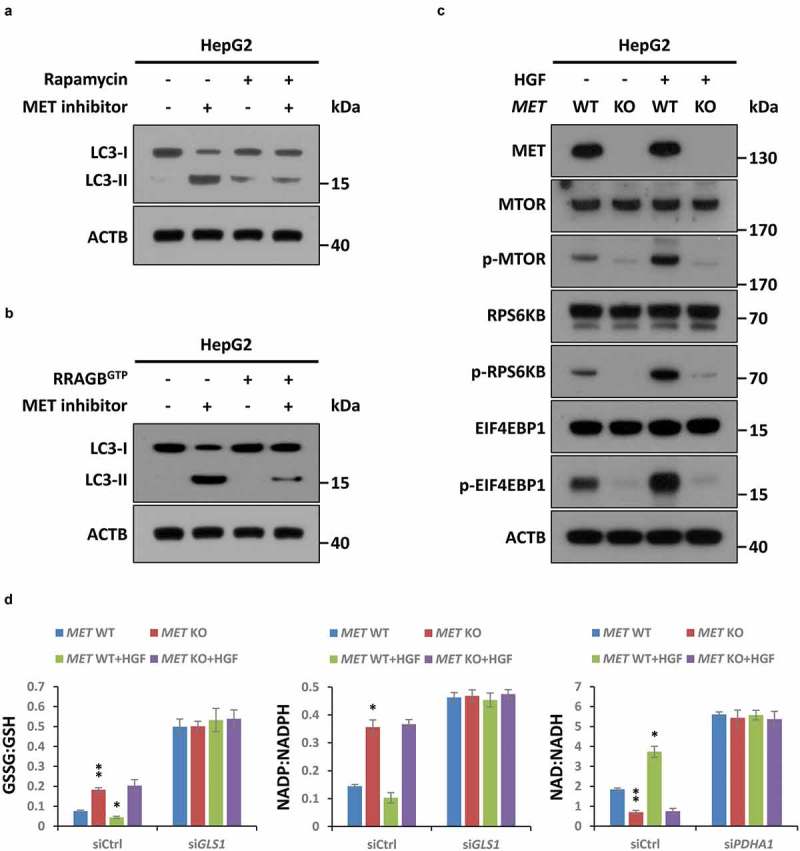
MTOR signaling is invovled in MET inhibition-induced autophagy. (a) Inhibitory effect of rapamycin on MET inhibition-induced autophagy. After pre-incubation with or without rapamycin (50 nM) for 4 h, HepG2 cells (5 × 104) were individually treated with an inhibitor of MET (JNJ-38877605, 50 nM) or vehicle control (DMSO) for 8 h, and subsequently subjected to western blot analysis with the indicated antibodies. (b) Rescue effect of MTOR activation on MET inhibition-induced autophagy. HepG2 cells (5 × 104) were individually transfected with or without a plasmid encoding the constitutively active RRAGBQ99L mutant (RRAGBGTP) for 48 h, and then treated with inhibitor of MET or vehicle control as above. Cell lysates were subjected to immunoblot with the indicated antibodies. (c) Impact of the HGF-MET axis on MTOR signaling. After stavation overnight, HepG2 MET WT or KO cells (5 × 104) were individually treated with or without HGF (40 ng/ml) for 2 h, and subsequently subjected to western blot analysis with the indicated antibodies. (d) Contribution of the HGF-MET axis to redox homeostasis through PDHC and GLS. HepG2 MET WT or KO cells (5 × 104) were individually transfected with GLS-specific siRNA (siGLS), PDHC-specific siRNA (siPDHA1) or non-targeting siRNA control (siCtrl). Seventy-two h after transfection, cells were starved overnight and treated with or without HGF (40 ng/ml) for 6 h, and then subjected to analysis of GSSG:GSH, NADP:NADPH and NAD:NADH ratios. Data are presented as the means ± SD from at least 3 independent experiments. Statistically significant differences with two-tailed Student’s t-test are marked as * (p < 0.05) or ** (p < 0.01). Abbreviations: GSSG:GSH, oxidized glutathione:reduced glutathione; NADP:NADPH, nicotinamide adenine dinucleotide phosphate:reduced NADP; NAD:NADH, nicotinamide adenine dinucleotide:reduced NAD.
Furthermore, we investigated the importance of the HGF-MET axis under serum starvation, in which condition the activity of autophagy rather than regular metabolism was supposed to be maximum in our system. The results turned out that serum starvation blocked the Y1234/1235 phosphorylation of MET (Figure S7(a)), and consequently enhanced the interaction between MET and LC3 (Figure S7(b)). Autophagic assessment also proved that in comparison to a parallel control, MET-deficient cells substantially lost the ability to accelerate autophagy flux under serum starvation (Figure S7(c)). Metabolic analyses further manifested that MET played critical roles in serum starvation-mediated biogenesis and that is primarily through autophagy, because disruption of autophagy flux by chloroquine (CQ) could block biogenesis under starvation, even in MET-targeted drug-resistant cells (Figure S7(d)). Thus, this also stated clearly that biogenesis in resistant cells were truly dependent on autophagy. On the contrary, instead of starvation, autophagy flux inhibition showed no significant impacts on biogenesis in the presence of HGF stimulation when normal metabolic activities rather than autophagy should be maximum in our system (Figure S7(e)). These results suggested that once highly activated by HGF, MET contributed to biogenesis in an autophagy-independent manner, or that there was no need for autophagy to support biogenesis in a growth factor-stimulated signaling pathway.
Y1234/1235 nonphosphorylated MET is associated with autophagy in clinical liver tissue microarray and the TCGA database
So far, we clarified the relationship between the phosphorylation status of MET in Y1234/1235, cancer biogenesis and autophagy in liver cancer cells. To further define the clinical significance of the aforementioned findings, we used liver tissue microarray to investigate the correlation between Y1234/1235 non-phosphorylated MET and autophagy in the clinic. Tissue sections of 208 patients, including 152 patients with liver hepatocellular carcinoma (LIHC), 12 with liver bile duct adenocarcinoma, 28 with liver metastatic adenocarcinoma, 8 with adjacent normal liver tissue, and 8 with normal liver tissue, were subjected to IHC analysis with H&E staining or antibodies against Y1234/1235 phosphorylated MET, the autophagy marker SQSTM1 or relevant isotype controls. Full-scale scanning of tissue chips was shown as indicated (Figure 8(a)), with representative images under high-power field microscopy (Figure 8(b)). Intriguingly, semi-quantified analyses clearly revealed that about 61% of cancer cases showed a positive correlation between p-MET (Y1234/1235) and SQSTM1; in other words, co-occurrence of p-MET (Y1234/1235) and SQSTM1 existed in a total of 61% of cancer samples; in contrast, only 36% of cancer cases showed a negative correlation between p-MET (Y1234/1235) and SQSTM1; more importantly, up to approximately 51% cancer cases shown neither p-MET (Y1234/1235) nor SQSTM1 staining signals; meanwhile, a similar expression tendency was observed in normal cases, just with the lower stained grade (Figure 8(c)). The results strongly suggested that the expression patterns of p-MET (Y1234/1235) and SQSTM1 were closely co-correlated in clinical liver cancer samples, which was actually consistent with our previous conclusion that Y1234/1235 dephosphorylated MET (presented as none or low staining of p-MET Y1234/1235) could promote autophagy, causing SQSTM1 degradation (presented as none or low staining of SQSTM1). Given the low level of phosphorylated MET, these findings also indicated that targeting MET kinase activity might be an unwise choice in liver cancer patients.
Figure 8.
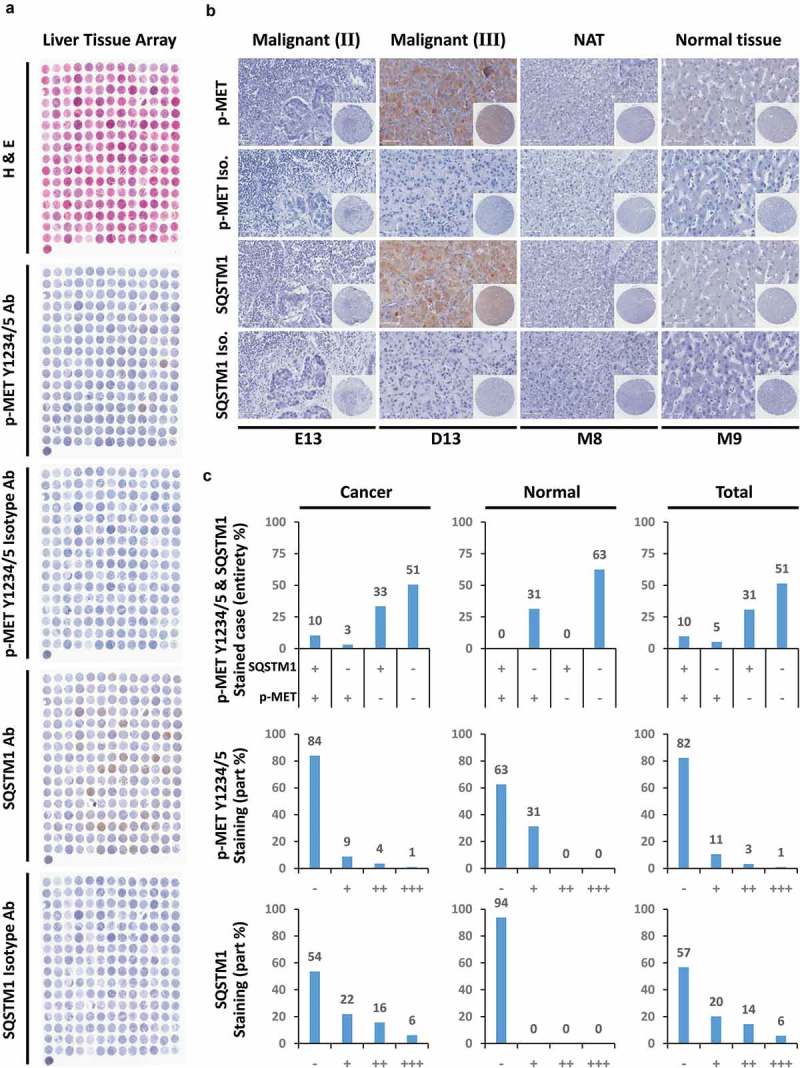
Y1234/1235 unphosphorylated MET correlates with autophagy in clinical liver cancer patients. (a) Clinical liver tissue microarray assay for correlation between Y1234/1235 unphosphorylated MET and the autophagic marker SQSTM1. A total of 208 cases of clinical liver tissue samples, including normal liver tissue (8 cases), cancer adjacent normal liver tissue (8 cases), hepatocellular carcinoma (152 cases), bile duct adenocarcinoma (12 cases) and metastatic adenocarcinoma (28 cases), were analyzed by immunohistochemistry (IHC) with the indicated antibodies and H&E staining. Full scans of tissue microarray are shown as indicated. (b) Representative images of Y1234/1235 unphosphorylated MET and SQSTM1 staining under high power field (HPF) from clinical liver tissue microarray. Scale bar: 100 µm. (c) Qualitative analysis for expression pattern and stained grade of Y1234/1235 unphosphorylated MET and SQSTM1 in clinical liver tissue microarray. Statistics in cancer, normal tissue or total samples were individually presented as indicated. Abbreviations: H&E staining, hematoxylin-eosin staining; ‘-’, negative; ‘+’, positive or weak; ‘++’, moderate; ‘+++’, strong.
In addition, because the sample size in clinical tissue microarray was not very large, we further employed the TCGA database and bioinformatics methods to conduct a series of supplementary analyses. First, we analyzed the differential expression of MET-HGF-SQSTM1 in paired tumor and normal tissues from multiple data sets. Of note, contrary to widely accepted viewpoints, we found compared with 2 other molecules, the expression level of HGF was much lower in liver tumor rather than normal liver tissue (Figure S8(a)). Given that MET is the principal receptor of HGF in physiological and pathological conditions, this indicated the phosphorylated level of MET might be downregulated in liver cancer, which was also in line with our latest findings in clinical liver tissue microarray (for instance, more than half of liver cancer samples in microarray without p-MET Y1234/1235). Furthermore, based on the clinical information available from the TCGA database (including copy number, mutation frequency, mRNA expression, and survival curve), we established a visual interactive network in liver hepatocellular carcinoma (LIHC), and found SQSTM1 was indeed involved in HGF-MET signaling pathways (Figure S8(b)). In fact, there were multiple connections between MET, HGF and SQSTM1. Overall, clinical data strongly suggested that in addition to HGF-MET kinase-targeted strategies, the importance of non-phosphorylated MET-mediated autophagy should be taken into simultaneous consideration in liver cancer therapeutics.
Combined treatment with MET-targeted inhibitor and an autophagy blocker overcomes chemotherapeutic resistance in liver cancer
Last, but perhaps most important, owing to the close association between the HGF-MET axis and autophagy, we speculated whether autophagy could be targeted to overcome HGF-MET-targeted chemotherapeutic resistance in liver cancer. Through combination therapy, we found that joint treatment with the autophagy flux blocker CQ remarkably heightened the repressive effects of the HGF-MET kinase-targeted inhibitor (JNJ-38877605) and antibody (5D5) on liver cancer cell proliferation (Figure 9(a)), viability (Figure 9(b)), and colony formation capacity (Figure 9(c)) in vitro. Representative images of clones under different treatments were shown as indicated (Figure 9(d)). Moreover, biogenesis analysis demonstrated that combined treatment with MET inhibitor and CQ considerably blocked synthesis of DNA, triglyceride and aspartate simultaneously in vitro (Figure 9(e)). Furthermore, to evaluate the synergistic effects of MET-targeted inhibitor and CQ on liver cancer in vivo, we used 2 conventional human liver cancer cell lines, SMMC-7721 and Huh-7, to constitute different xenograft models in mice. Athymic nu/nu mice bearing xenograft tumors were individually treated with vehicle control PBS, MET inhibitor only, CQ only, or MET inhibitor plus CQ, and were subsequently monitored as described. As expected, MET inhibitor and CQ in combination drastically suppressed tumor growth and reduced tumor surface area during growth (Figure 9(f)), as well as end-point tumor weight (Figure 9(g)). Therefore, autophagy blockage improved HGF-MET-targeted drug efficiency in liver cancer therapy.
Figure 9.
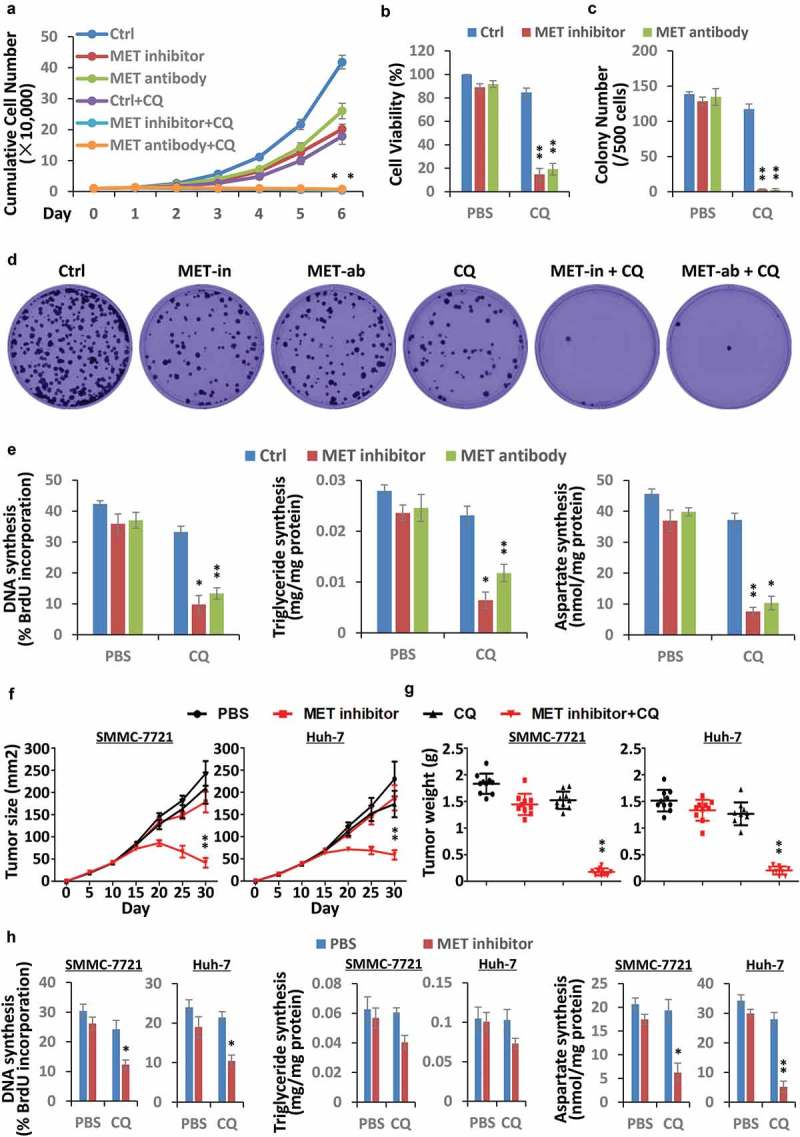
Autophagy blockage improves HGF-MET-targeted drug efficiency in liver cancer. (a) Synergistic effect of CQ and MET-targeted drugs on proliferation capacity of liver cancer cells. HepG2 cells (1 × 104) were individually treated with vehicle control, an inhibitor of MET (JNJ-38877605, 50 nM), anti-MET antibody (5D5, 40 μg/ml) or/and CQ (20 μM), and subsequently subjected to analysis of cell proliferation capacity at the indicated times. (b) Synergistic effect of CQ and MET-targeted drugs on viability of liver cancer cells. HepG2 cells (2.5 × 103) were individually treated with vehicle control, an inhibitor of MET (JNJ-38877605, 50 nM), anti-MET antibody (5D5, 40 μg/ml) or/and CQ (20 μM) for 8 h, and then subjected to analysis of cell viability. (c and d) Synergistic effect of CQ and MET-targeted drugs on colony-formation capacity of liver cancer cells. HepG2 cells (0.5 × 103) were individually treated with a single dose of vehicle control, an inhibitor of MET (JNJ-38877605, 50 nM), anti-MET antibody (5D5, 40 μg/ml) or/and CQ (20 μM) for 2 wk, and subsequently subjected to analysis of colony-formation capacity of cells (c). Representative images of clones are shown as indicated (d). (e) Synergistic impact of CQ and MET-targeted drugs on biogenesis of liver cancer cells. HepG2 cells (5 × 104) were individually treated with vehicle control, an inhibitor of MET (JNJ-38877605, 50 nM), anti-MET antibody (5D5, 40 μg/ml) or/and CQ (20 μM) for 24 h, and then subjected to analysis of DNA, triglyceride and aspartate contents. Data are presented as the means ± SD from at least 3 independent experiments. (f and g) Synergistic effect of autophagy blockage and HGF-MET inhibition on tumor growth in xenograft-bearing mice. SMMC-7721 and Huh-7 cells (1 × 106) were individually inoculated subcutaneously into the flanks of athymic nu/nu mice. Once palpable, the tumors were individually treated with PBS (vehicle control, 100 μl), MET inhibitor (20 mg/kg in 100 μl of PBS) or/and the autophagic blocker CQ (10 mg/kg in 100 μl of PBS) every 5 d. Tumor growth was recorded every 5 d during treatment, and reported as the mean tumor surface size ± SD with n = 10 animals per group (f). Tumor weight was detected after sacrifice and shown on the right (g). (h) Synergistic impact of autophagy blockage and HGF-MET inhibition on cancer biogenesis in xenograft tumors. Individual samples (n = 3) from each treated group in SMMC-7721 and Huh-7 xenograft tumors were randomly selected and subjected to analysis of DNA, triglyceride and aspartate contents. Statistically significant differences with two-tailed Student’s t-test are marked as * (p < 0.05) or ** (p < 0.01). Abbreviations: MET-in, MET inhibitor; MET-ab, MET antibody; CQ, chloroquine.
To further confirm that the beneficial effects of the combination of MET inhibitor and CQ really depend on simultaneous inhibition of MET kinase activity and autophagy flux, we investigated MET phosphorylation and autophagy state in randomly selected xenograft tumor samples. As shown, while MET inhibitor suppressed Y1234/1235 phosphorylation of MET that resulted in an increase of autophagy, combinatorial treatment of MET inhibitor and CQ drastically blocked autophagy in comparison to CQ only (Figure S9(a,b)). To determine whether combination therapy truly disrupted cancer biogenesis, excised xenografts were further used to analyze DNA, triglyceride, and aspartate contents. Consistent with the in vitro results, combinatorial treatment strongly decreased liver cancer biogenesis in xenograft mice (Figure 9(h)). Not to mention, the PDHC activity was enhanced in xenograft tumors, and meanwhile GLS activity was also blocked within expectation (Figure S9(c,d)). In addition, the body weight and liver weight of mice were not significantly affected during treatments (Figure S9(e,f)), which indicated the relative safety of combination treatment in mice. Together, autophagy-sustained biogenesis was indeed responsible for HGF-MET-targeted chemotherapeutic resistance in liver cancer.
In the end, a schematic diagram has been depicted to illustrate HGF-MET-coordinated metabolic modes involved in liver cancer therapeutic resistance, and a corresponding strategy for practicable optimization (Figure 10). In brief, HGF activates MET to facilitate the Warburg effect and glutaminolysis for biogenesis and growth in liver cancer. Upon treatment by HGF-MET kinase-targeted drugs, Y1234/1235 dephosphorylated MET recruits LC3 and promotes autophagy to maintain biogenesis homeostasis for survival under metabolic stress. Combined inhibition of HGF-MET signaling and autophagy flux improves liver cancer chemotherapeutic efficacy by disrupting the Warburg effect, glutaminolysis and autophagy, which co-contributed to cancer biogenesis.
Figure 10.
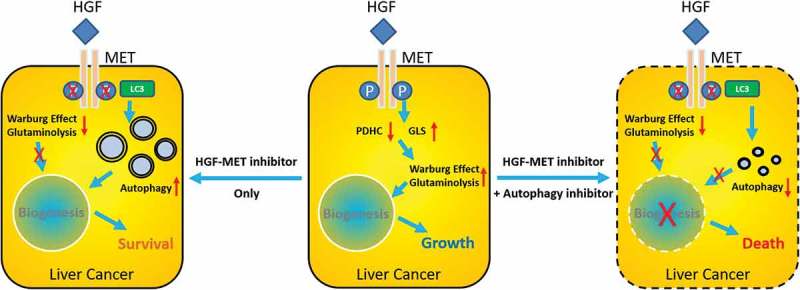
Schematic diagram for HGF-MET-coordinated metabolic modes in liver cancer therapeutic resistance. HGF activates MET to facilitate the Warburg effect and glutaminolysis for liver cancer biogenesis and growth. Upon treatment with HGF-MET-targeted drugs, dephosphorylated MET promotes autophagy to maintain biogenesis homeostasis for survival. Double inhibition of HGF-MET and autophagy overcomes liver cancer therapeutic resistance by disrupting biogenesis.
Discussion
Physiologically, various growth factors, such as EGF (epidermal growth factor) and FGF (fibroblast growth factor), serve as specific growth signals to stimulate the proliferation of different cells, primarily by activating metabolic enzymes and by upregulating metabolite transporters to expedite metabolic flux. Simultaneously, to avoid low effective production, growth factors also have inhibitory effects on essential autophagy machinery, including ULK1, BECN1, and the ATG12–ATG5-ATG16L1 complex [43,44]. However, in aberrant conditions, sustaining proliferative signaling and deregulating cellular energetics are 2 general hallmarks of cancer [45]. Although altered metabolism promotes cancer formation and growth, it also results in an increased demand for continuous nutrient supply. Thus, growth factor-mediated extensive metabolic reprogramming is deemed to be the Achilles’ heel of cancer, and it consequently offers a large number of ideal targets for cancer therapy [46,47]. Multiple enzyme-targeted agents have been used clinically, and some important candidates are under development [48]. For example, AGI-5198 (preclinical, Xcessbio) is designed to inhibit both wild-type and mutant IDH (isocitrate dehydrogenase), which demonstrates multipronged anticancer effects, presumably owing to reductions in 2-hydroxyglutarate and interference with glutamine metabolism.
In the present study, inspired by growth factor-driven cross-talk between metabolism and autophagy, we successfully addressed 2 questions. First, our findings explain why HGF-MET-targeted drugs always failed. Onartuzumab (Genentech), a single-arm MET-specific antibody that effectively prevents ligand-receptor interaction, dimerization, and activation, was terminated early in the phase III trial owing to its futility in patients or even slight worsening of symptoms. The dominant view is that its failure may have resulted from an aberrant design of the clinical trial [49]. Tivantinib (ArQule), a putative MET inhibitor, also failed in phase III trials, indicating that this staurosporine derivative is more like a cytoskeleton inhibitor rather than an inhibitor of MET [50]. In our opinion, this consideration is reasonable but not comprehensive. This is because it ignores the fact that MET itself, or in other words, HGF blockade-mediated MET dephosphorylation, still has critical roles in cancer. Considering our case as an example, we unmask the HGF-MET axis as a novel metabolic player in liver cancer and demonstrate that MET deficiency or HGF-MET-targeted drugs block an HGF-controlled Warburg effect and glutaminolysis. However, this action has no influence on cancer metabolism-mediated biogenesis. Although HGF-MET blockage indeed leads to MET Y1234/1235 dephosphorylation, kinase inactivation, and metabolic stress, Y1234/1235-dephosphorylated MET subsequently activates autophagy to replenish materials required for biogenesis and survival. We think that this is the reason for the failure of HGF-MET-targeted therapy. Furthermore, based on the experimental evidence and implications, we selected a novel strategy, wherein we combined a MET-targeted inhibitor with an autophagy blocker to investigate the synergistic effects on liver cancers. As described in the results, the combination of a MET inhibitor plus autophagy blocker drastically suppressed liver cancer growth in mice, which was unexpected yet comprehensible. Because our strategy is quite simple and practicable, we anticipate that medical practitioners or pharmaceutical companies might consider redesigning and reinitiating the suspended trials to evaluate the efficiency and feasibility of this treatment method in patients.
Of note, although MET was deemed as a receptor protein on the plasma membrane for transduction of extracellular signals for decades, recent studies have gradually disclosed the endocellular functions of MET. For instance, MET could enter into the nucleus and phosphorylate PARP1 to increase its enzymatic activity and reduce targeted drug binding [51]. Moreover, the p40 fragment of MET, generated by active caspase-mediated cleavage, could localize to mitochondria in a tyrosine kinase-independent manner, and cause its permeabilization so as to favor apoptosis [52]. Here, we report that through binding to LC3 and inhibiting MTOR activity, dephosphorylated MET drives autophagy to maintain cancer biogenesis, which is also an intracellular process. However, even MET has the chance to meet the autophagic machinery and MTOR regulatory complex when recycled through a conventional endo-lysosomal degradation pathway; however, the mechanistic significance of MET-LC3 interaction for autophagy induction is still poorly understood. Not to mention, we have identified several MET kinase activity-associated LIR motifs in this study, besides Y1234-Y1235-S1236-V1237. On account of the high frequency of genetic alterations in MET in liver cancer patients, it is necessary to further investigate the compensatory roles of clinic-derived MET mutation, especially at the other LIR motifs. Additionally, it is still unclear as to the potential functional impact of MET on MTOR signaling and how MET modulates MTOR activity under metabolic stress, particularly considering the complicated counteraction of MTORC1-independent autophagy to MET phosphorylation in an MTORC2-dependent manner [53]. Furthermore, in addition to AKT-dependent or redox-involved regulation, the direct connection between MET and MTOR could not be excluded so far. To figure out the interplay between MET, autophagy and MTOR, at least some of these important questions are worthy of further exploration for constructing a bigger interactive network of cancer biogenesis and chemoresistance.
Last but not least, our findings indicate HGF-MET together with autophagy as promising and valuable targets and highlight the scope for curing liver cancer; however we do not think an autophagy-based strategy would be sufficient enough to overcome chemotherapeutic resistance, in spite of the enormous potential in translation from bench to bed. Emerging evidence has suggested that autophagy itself has mutually contradictory effects on cancer progression [54]; e.g., autophagy might inhibit tumorigenesis but promote metastasis. Particularly in liver cancer, it has already been reported that genetic deletion or mutation in autophagy central machinery (e.g. Atg5, Atg7 and Becn1) or indirect regulatory factors (e.g. TSC)-mediated autophagy inhibition, to some extent, can promote liver tumorigenesis. Despite the fact that these studies did not completely exclude potential autophagy-independent effects of autophagy-associated genes or proteins, it still raises serious concerns about the feasibility and safety of autophagy-targeted therapeutic strategies in human, or at least that the possible drawbacks of any proposed therapy should be addressed in the near future. Conversely, therapeutic resistance in cancer was previously attributed to cellular immortality, e.g., avoiding apoptosis rather than autophagy; however, paradoxically, in the present study, autophagy seems to account for the majority of the effect. Considering the close association between autophagy and apoptosis [55–57], the role of each phenomenon in liver cancer should be further clarified. Therefore, our study is simply an initial attempt to elucidate the complicated roles of autophagy in chemotherapeutic resistance in liver cancer, exclusively in the context of HGF-MET signaling.
Materials and methods
Cell lines and cell culture
HepG2, SMMC-7721, Huh-7, MHCC-97H, Hepa1-6 and H22 liver cancer cell lines were individually obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) or KeyGEN BioTECH (Nanjing, China), where mycoplasma contamination detection and short tandem repeat (STR) profiling were performed for quality and identity guarantee. Mouse embryonic fibroblast Atg5 WT and atg5 KO cells were kindly provided by Quan Chen (Institute of Zoology, Chinese Academy of Sciences). All the cell lines and derived cells were maintained in a 37°C incubator with 5% CO2 under standard conditions as specified by the suppliers, in Dulbecco’s modified Eagle’s medium (Gibco, 11965092) or Roswell Park Memorial Institute 1640 medium (Gibco, 11875093), supplemented with 2 mM L-glutamine, 1% NEAA (Gibco, 11140050), 100 units/ml penicillin, 100 mg/ml streptomycin and 10% fetal bovine serum. No cell lines used in this work were commonly misidentified cell lines, according to the database from the International Cell Line Authentication Committee (ICLAC). All the cell lines were freshly thawed from the purchased seed cells, cultured for no more than 2 months, and regularly checked by virtue of their morphological features to avoid cross-contamination or misuse.
Generation of a CRISPR-Cas9-mediated MET knockout cell line
For generation of a MET knockout cell line via the CRISPR-Cas9 system, HepG2 cells were cotransfected with a MET CRISPR/Cas9 KO plasmid (Santa Cruz Biotechnology, sc-400101) and MET HDR plasmid (Santa Cruz Biotechnology, sc-400101-HDR) according to the manufacturer’s instructions. The MET CRISPR/Cas9 KO plasmid was designed to disrupt gene expression by causing a double-strand break in a 5ʹ constitutive exon within the MET gene. The MET CRISPR/Cas9 KO plasmid consists of a pool of 3 plasmids, each encoding the Cas9 nuclease and a target-specific 20 nucleotide guide RNA (gRNA) designed for maximum knockout efficiency. The MET HDR Plasmid consists of a pool of 2–3 plasmids, each containing a homology-directed DNA repair (HDR) template corresponding to the cut sites generated by the MET CRISPR/Cas9 KO plasmid. Each HDR template contains 2 800-base pair homology arms designed to specifically bind to the genomic DNA surrounding the corresponding Cas9-induced double-strand DNA break site. Simply, 1 × 105 HepG2 cells were seeded in 3 ml of antibiotic-free standard growth medium per well in a 6-well tissue culture plate, 12 h prior to transfection. When cell confluency was at 40%, 1 µg of CRISPR/Cas9 KO plasmids plus 1 µg of HDR plasmids DNA were mixed and cotransfected into HepG2 cells using UltraCruz® Transfection Reagent (Santa Cruz Biotechnology, sc-395739). Successful cotransfection of the CRISPR/Cas9 KO lasmid and HDR plasmid was visually confirmed by detection of the green and red fluorescent protein (GFP and RFP) via fluorescence microscopy. Seventy-two h post-transfection, 1 µg/ml puromycin (InvivoGen, ant-pr-1) was added into the growth medium for selection for at least 10 d. After selection, cells were suspended, diluted and re-seeded to ensure single clone formation. More than 20 clones were picked and the expression of MET in each single clone was evaluated by western blot with 2 different antibodies against different sites on MET. Further verification of positive candidate clones was done by sequencing the genomic DNA to make sure that the functional genomic editing occurred.
Establishment of a MET-targeted drug-insensitive cell line
To establish MET-targeted drug-insensitive cell lines, 1 × 106 HepG2 cells were individually treated with a single dose of 50 nM MET-specific small molecule inhibitor JNJ-38877605 (Selleck Chemicals, S1114) and 40 μg/ml anti-MET monoclonal antibody (Alphamab, 5D5) for 3 d. After treatment, the surviving population of HepG2 cells was cultured in normal growth medium for 3 d to avoid the therapeutic stress-mediated artificial effects. The recovered HepG2 cells were treated again with JNJ-38877605 and 5D5 as above for the other 2 rounds of selection. The concentration of JNJ-38877605 and 5D5 used to select out the insensitive cell line was based on clinical reports to apply a degree of clinical relevance to this in vitro study. To assess the resistance of MET-targeted drug-insensitive cell lines, cells were plated at a concentration of 1 × 103 cells per well in triplicate wells of 6-well tissue culture plates. Six h after plating, the triplicate wells were treated with JNJ-38877605 (0–100 nM) and 5D5 (0–80 μg/ml) for 3 d, and then subjected to cell proliferation, viability and colony formation assays as described. The resistant cells were incubated in very low concentrations of drugs to maintain their resistance in cell culture, but the drugs were withdrawn in formal experiments.
Generation of a stably expressing GFP-LC3 cell line
To generate a stably expressing GFP-LC3 reporter cell line, 1 × 104 HepG2 cells were seeded in 3 ml of antibiotic-free standard growth medium per well in a 6-well tissue culture plate overnight, and then transiently transfected with GFP-LC3 plasmids containing a Geneticin resistance marker using Lipofectamine 2000® (Invitrogen, 11668019). Following selection with the continuous presence of 200 μg/ml Geneticin antibiotic for 3 wk, individual clones were generated from the preliminary cell pool by single cell-sorting with a FACS VantageTM Flow Cytometer (BD Biosciences). After validation by western blot with antibody against LC3 and GFP, more than 10 stable clones with low expression of GFP-LC3 and few GFP puncta under normal conditions were picked up for further verification via GFP-LC3 puncta formation assays.
Antibodies, inhibitors and other reagents
Antibodies were obtained from the indicated sources: anti-MET (ThermoFisher, 700261, for WB; Proteintech Group, 25869-1-AP, for immunoprecipitation; Alphamab Co. Ltd, 5D5, for therapy), anti-p-MET Y1003 (Cell Signaling Technology, 3135), anti-p-MET Y1234/1235 (Cell Signaling Technology, 3077, for WB and immunohistochemistry [IHC]), anti-p-MET Y1349 (Cell Signaling Technology, 3133), anti-p-MET Y1356 (Abcam, ab73992), anti-p-MET Y1365 (Santa Cruz Biotechnology, sc-377548), anti-p-tyrosine (Cell Signaling Technology, 8954, for WB and IP), anti-PDHA1/PDH-E1α (Santa Cruz Biotechnology, sc-377092, for WB and IP), anti-DLAT/PDC-E2 (Santa Cruz Biotechnology, sc-271534, for WB and IP), anti-PDHX/E3BP (Santa Cruz Biotechnology, sc-377255, for WB and IP), anti-GLS/GLS1 (Abcam, ab93434, for WB and IP), anti-GLS2 (Abcam, ab150474), anti-GLUL/GS (Abcam, ab64613), anti-PKM/PKM2 (Santa Cruz Biotechnology, sc-365684), anti-LDHA (Santa Cruz Biotechnology, sc-137243), anti-SLC2A1/GLUT1 (Proteintech Group, 21829-1-AP), anti-SLC2A2/GLUT2 (Proteintech Group, 20436-1-AP), anti-SLC2A3/GLUT3 (Proteintech Group, 20403-1-AP), anti-SLC2A4/GLUT4 (Abgent, AP20792a), anti-SLC7A5/LAT1 (Proteintech Group, 13752–1-AP), anti-SLC7A8/LAT2 (Santa Cruz Biotechnology, sc-293242), anti-SLC8A1/SNAT1 (Proteintech Group, 12039-1-AP), anti-SLC8A3/SNAT3 (Proteintech Group, 14315-1-AP), anti-SLC1A5/ASCT2 (Abcam, ab187692), anti-MTOR (Cell Signaling Technology, 9862), Autophagy Antibody Sampler Kit (anti-BECN1, anti-ATG7, anti-ATG5, anti-ATG16L1) (Cell Signaling Technology, 4445), anti-LC3B (Novus, NB100-2220), anti-SQSTM1 (MBL, PM045, for WB and IP; Santa Cruz Biotechnology, sc-28359, for IHC), anti-ATG13 (ABclonal, A0690), anti-ULK1 (Abcam, ab128859), anti-PIK3C3/Vps34 (Abcam, ab124905), anti-UVRAG (Abcam, ab174550), anti-RB1CC1 (Proteintech Group, 17250-1-AP), anti-RUBCN/rubicon (Proteintech Group, 21444-1-AP), anti-RPTOR/raptor (Proteintech Group, 20984-1-AP), anti-Flag (Sigma, F1804), anti-GFP (Santa Cruz Biotechnology, sc-81045), anti-ACTB (Proteintech Group, HRP-60008), anti-rabbit IgG (HRP conjugated; GeneTex, GTX221666-01), and anti-mouse IgG (HRP conjugated; GeneTex, GTX221667-01). Unless explicitly stated, all the antibodies were used for western blot. Chemicals and growth factors were acquired from the designated suppliers: MET inhibitor JNJ-38877605 (Selleck Chemicals, S1114), PDH inhibitor CPI-613 (Selleck Chemicals, S2776), GLS inhibitor BPTES (Selleck Chemicals, S7753), rapamycin (Selleck Chemicals, S1039), bafilomycin A1 (Tocris/R&D, 1334), chloroquine (Sigma, C6628), and HGF (PeproTech, 100-39-2).
siRNAs and plasmids
For siRNA-mediated knockdown, 2 × 104 HepG2 cells were seeded in 3 ml of antibiotic-free standard growth medium per well in a 6-well tissue culture plate overnight, and then individually transfected twice (on day 1 and 2) with 25 nM indicated siRNAs (described below) by Lipofectamine RNAiMAX Transfection Reagent (Invitrogen, 13778150), according to the manufacturer’s instructions. Seventy-two h after transfection, cells were treated as indicated, and subsequently subjected to further experiments. Target sequences of each designed siRNA were as follows: siMET (5ʹ-GUGCCACUAACUACAUUUATT-3ʹ) and non-targeting control siCtrl (5ʹ-UUCUCCGAACGUGUCACGUTT-3ʹ). The knockdown efficiency was verified by western blot analysis with a specific antibody. All the siRNAs used in this study were widely validated, or obtained from a highly effective and specific siRNA online design site (http://sidirect2.rnai.jp/) as described previously. All siRNA oligonucleotides were synthesized by GenePharma. Full-length cDNA of MET was purchased from Sino Biological Inc. (HG10692-NF). Dephosphorylated point mutants of MET were synthesized by GeneScript, and subsequently subcloned into a modified pCDNA3.1(+)-3 × Flag vector (VT3106, MLCC, backbone from Invitrogen) by a standard PCR cloning strategy for transient expression. GFP-LC3 plasmid was kindly provided by Dr. Quan Chen (Institute of Zoology, Chinese Academy of Sciences) for construction of a reporter cell line. The pRK5-HA GST RagB Q99L (RRAGBGTP) plasmid from Addgene (19303; deposited by David Sabatini). All constructs were confirmed by DNA sequencing.
Co-immunoprecipitation and western blot
After rinsing 2 times with ice-cold PBS, 5 × 105 cells were resuspended in lysis buffer (25 mM HEPES, pH 7.5, 150 mM NaCl, 0.25% Triton X-100 [Sangon Biotech, A600198], 0.25% NP-40 [company, catalog number], 0.5% CHAPS [Sigma, 10810118001], 10% glycerol [Sigma, G6279], phosphatase inhibitor cocktail [Bimake, B15001] and protease inhibitor cocktail [Bimake, B14001]) on ice for 1.5 h, and then centrifuged at 14,000 g for 15 min. The supernatants were pre-cleared with protein A/G-coupled agarose (Santa Cruz Biotechnology, sc-2003), and subsequently incubated with 2 μg of the indicated antibodies or 20 μl Anti-DYKDDDDK (Flag) Affinity Gel (Bimake, B23101) overnight at 4°C, followed by addition of 20 μl protein A/G agarose (only for IP with unconjugated antibodies mentioned above) for 4 h. After washing 3 times with lysis buffer, immunoprecipitates were boiled in 1 × loading buffer for western blot analysis. Protein samples were resolved by SDS-PAGE with ExpressPlusTM PAGE Gel, 4–20%, 15 wells (GenScript, M42015C), and then transferred onto nitrocellulose membrane (PALL, 66485) following standard procedures. Membranes were blocked with 7.5% skim milk or BSA in TBST for 1 h, and subsequently incubated with the indicated primary antibodies overnight at 4°C according to the manufacturer’s recommendation. After washing with TBST 3 times, membranes were incubated with the appropriate HRP-labeled secondary antibodies. Immunolabeling was developed with SuperSignal™ West Femto Maximum Sensitivity Substrate (ThermoFisher, 34095). Visualized images were obtained using ImageQuantTM LAS-4000 (GE Fujifilm) or photographic film. Similar settings for exposure time, brightness, contrast and scanning condition were used for capture of the digital images.
Warburg effect and glutaminolysis assessment
For assessment of the Warburg effect and glutaminolysis, 5 × 104 cells were cultured and treated with HGF or other conditions as indicated, then subjected to metabolic analysis. Colorimetric assay kits were used according to the manufacturer’s instructions, and were as follows: glucose consumption assay kit (Sigma, MAK083-1KT), lactate production assay kit (Sigma, MAK065-1KT), glutamine consumption assay kit (BioVision, K556-100) and glutamate production assay kit (BioVision, K629-100). Consumption or production rate of glucose, lactate, glutamine and glutamate was measured using an iMarkTM Microplate Reader (Bio-Rad, 168–1130). The values were normalized to the cell number to exclude growth factor-stimulated proliferation-mediated side-effects.
Nucleotide, lipid and amino acid synthesis analysis
For analysis of nucleotide, lipid and amino acid synthesis, 5 × 104 cells were cultured and treated with HGF or other conditions as indicated, then subjected to analyze the biogenesis of metabolites with assay kits as follows: BrdU Cell Proliferation Assay Kit (BioVision, K306-200), Triglyceride Quantification Kit (BioVision, K622-100) and Aspartate Assay Kit (Sigma, MAK095-1KT). The percentages of BrdU-incorporated cells were counted and subjected to reflect DNA synthetic rate. The contents of intracellular triglyceride and aspartate were quantified using an iMarkTM Microplate Reader (Bio-Rad, 168–1130), and normalized to the protein concentration to exclude growth factor-stimulated proliferation-mediated side-effects.
Untargeted metabolomics analysis
GC-MS and LC-MS were conducted at BioNovoGene Co., Ltd. (Suzhou, China). In brief, 1 × 107 WT and MET KO HepG2 cells were individually collected, extracted, and subjected to a GC-MS assay (Agilent 7890 A/5975 C) using an HP-5MS capillary column (5% phenyl:95% methylpolysiloxane 30 m × 250 μm internal diameter, 0.25-μm film thickness, Agilent J & W Scientific, Folsom, CA, USA), and to LC-MS assay (Waters ACQUITY UPLC system, Waters, Milford, MA, USA) with an ACQUITY UPLC® HSS T3 column (1.8 μm, 2.1 × 150 mm, Ethylene Bridged Hybrid, Waters). The samples were randomized, the data acquisitions were conducted in one batch to eliminate system errors, and the metabolites were confirmed based on their exact molecular weight, mass spectra, and retention time index. The original GC-MS and LC-MS data were processed and analyzed using XCMS software (www.bioconductor.org) with optimized settings. The metabolite annotation of the GC-MS data was performed with an automatic processing and identification system (AMDIS), referenced to the databases of the National Institute of Standards and Technology (NIST) and the Wiley Registry of Mass Spectral Data (Wiley Online Library). The metabolite annotation of the LC-MS data was performed with the Compound Discoverer program and referenced to the mzCloud database (www.mzCloud.org), as well as the Human Metabolome Database (www.hmdb.ca), METLIN (metlin.scripps.edu), MassBank (www.massbank.jp), and LIPID MAPS (www.lipidmaps.org). Discriminating metabolites between 2 classes of samples were identified using a statistically significant threshold of Variable Importance in Projection (VIP) value (VIP ≥ 1), and further validated by Student’s t-test analysis (P ≤ 0.05). Principal components analysis (PCA) and partial least squares discriminant analysis (PLS-DA) were performed with SIMCA–P software (www.umetrics.com). Heat map was constructed using Euclidian distances and complete linkage grouping with the pheatmap package in R language (www.r-project.org), and the relative quantitative values of metabolites were normalized, transformed and clustered through Hierarchical Clustering. Metabolite correlation was assessed using Pearson Correlation Coefficient and constructed Cytoscape software (www.cytoscape.org). To further identify alterative metabolic pathways, differential metabolites were subjected to grouping and enrichment of metabolic pathway using MetaboAnalyst 4.0 software (www.metaboanalyst.ca) and KEGG database (www.kegg.jp). Univariate analysis of variance (ANOVA) was used to determine the significance of differences in relative contents between different groups.
Metabolic enzyme activity analysis
For analysis of metabolic enzyme activity, 5 × 104 cells were cultured and treated as indicated, then subjected to analyze activities of metabolic enzymes with specific assay kits, according to the manufacturer’s instructions. All used kits were listed as follows: Hexokinase (HK) Activity Assay Kit (Sigma, MAK091-1KT), Phosphoglucose Isomerase (GPI/PGI) Activity Assay Kit (Sigma, MAK103-1KT), Phosphofructokinase (PFK) Activity Assay Kit (Sigma, MAK093-1KT), Fructose-bisphosphate Aldolase (ALDO) Activity Assay Kit (Sigma, MAK223-1KT), Glyceraldehyde-3-phosphate Dehydrogenase (GAPDH) Activity Assay Kit (BioVision, K680-100), Enolase (ENO) Activity Assay Kit (Sigma, MAK178-1KT), Pyruvate Kinase (PK) Activity Assay Kit (Sigma, MAK072-1KT), Pyruvate Dehydrogenase (PDH) Activity Assay Kit (Sigma, MAK183-1KT), Lactate Dehydrogenase (LDH) Activity Assay Kit (Sigma, MAK066-1KT), Glutaminase (GLS) Activity Assay Kit (Nanjing Jiancheng Bioengineering Institute, A124) and Glutamine Synthetase (GLUL/GS) Activity Assay Kit (Nanjing Jiancheng Bioengineering Institute, A047). Enzyme activity was determined using an iMarkTM Microplate Reader (Bio-Rad, 168–1130). The values were normalized to the protein concentration.
MET kinase assay
FLAG-MET, HIS-PDHA1, HIS-DLAT/PDCE2, HIS-PDHX and HIS-GLS were individually expressed in HEK-293T cells and BL21 Escherichia coli, and the recombinant proteins were purified using anti-FLAG® M2 magnetic beads (Sigma, M8823) and HIS-Select® Nickel magnetic beads (Sigma, H9914), respectively. To perform assays of MET kinase-mediated phosphorylation, 1 μg of HIS-PDHA1, HIS-DLAT/PDCE2, HIS-PDHX or HIS-GLS were individually incubated with 400 ng of FLAG-MET in 50 µl of specific kinase buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 12.5 mM MgCl2, 1 mM DTT, 10 mM ATP [InvivoGen, tlrl-atpl] and protease inhibitors) for 45 min at 37°C. MET kinase inhibitor (JNJ-38877605, 50 nM) was used as a negative control to the reaction. The reaction was terminated by adding the same amount of 2 × SDS loading buffer and boiling at 100°C for 10 min. The mixture samples were subjected to western blot analysis as described above, and phosphorylated PDHA1, DLAT/PDCE2, PDHX and GLS were individually visualized with anti-phospho-tyrosine antibody.
Autophagy assessment
Autophagy was assessed in accordance with ‘Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition)’ by Daniel J. Klionsky, et al. in Autophagy [58], via western blot, confocal fluorescence microscopy and electron microscopy. Note: In the densitometry-based quantification of related protein level in the western-blot analyses, the ratio of ‘LC3-II:LC3-I:ACTB’ means LC3-II is first normalized to LC3-I and then normalized to ACTB.
Cell proliferation, viability and colony formation assay
For the cellular proliferation assay, 1 × 104 cells were seeded in 6-well tissue culture plates at day 0 in triplicates in 3 ml of normal growth medium per well, and subsequently treated as described. The medium was changed every day. Cell number at the indicated time points was counted after trypsin (Gibco, 25300054) digestion using a hemocytometer, and recorded for analysis. For cell viability assay, 2.5 × 103 cells were seeded in 96-well microplates at day 0 in triplicates in 100 μl of normal growth medium per well, and subsequently treated as described. After treatment, cells were further incubated with 0.25 mg/ml WST-8 solution (Bimake, B34302) at 37°C for 2 h, followed by addition with 10 μl of 3% SDS to end the reaction. The absorbance at 450 nm was measured, and the percentage of viable cells was calculated and averaged for each well. For clonogenic assay, the same amount of cells need to be seeded at a density to maintain control (untreated) cells in an exponential phase of growth during the entire experiment. After pre-tests for HepG2 cell line, 500 cells were seeded in 6-well plates at day 0 with triplicates in 3 ml of regular growth media supplemented with 20% fetal boine serum per well. Cells were then cultured and treated as indicated at 37°C under 5% CO2 for 2 wk. Growth medium with or without HGF or drugs was replaced every 2 d. Remaining cells were fixed with 4% cold paraformaldehyde for 45 min, and then stained with 0.1% (w:v) crystal violet (SunShineBio, SSI1047-1), dissolved in 10% methanol, for 2 h at room temperature. After extensive washing with distilled water until the background became clear, pictures of each replicate were photographed using a digital camera, and colony numbers were quantified under a microscope.
Bioinformatics analysis for the TCGA data set
According to data sets obtained from TCGA (https://tcga-data.nci.nih.gov/tcga/), differential expression of MET-HGF-SQSTM1 in paired tumor and normal tissues were individually investigated with FireBrowse (http://firebrowse.org) for clinical correlation. A visual interactive network between MET-HGF-SQSTM1 in liver hepatocellular carcinoma was set up via cBioPortal (http://www.cbioportal.org/).
Clinical liver tissue microarray
Liver tissue chips (LV2091) containing a total of 208 cases of clinical liver tissue samples were obtained from US Biomax Inc., including 152 cases of liver hepatocellular carcinoma, 12 cases of liver bile duct adenocarcinoma, 28 cases of liver metastatic adenocarcinoma, 8 cases of adjacent normal liver tissue and 8 cases of normal liver tissue. The slides were deparaffinized with xylene after baking at 60°C for 30 min, and subsequently rehydrated in a series of ethanol solutions. Antigen was retrieved using 0.01 M sodium citrate buffer (pH 6.0) at a sub-boiling temperature for 10 min after boiling in a microwave oven. After 1 h of pre-incubation in 5% normal goat serum (Cell Signaling Technology, 5425) to block nonspecific staining, the slides were individually incubated with antibodies against p-MET Y1234/1235 (Cell Signaling Technology, 3077, 1:50) or SQSTM1 (Santa Cruz Biotechnology, sc-28359, 1:100) at 4°C overnight. Negative control was obtained by replacing the primary antibody with normal IgG of the same isotype (rabbit mAb IgG [Cell Signaling Technology, 3900]; mouse mAb IgG1 [Cell Signaling Technology, 5415]). After washing with PBST (PBS [Sangon Biotech, B548117], 0.1% Tween-20 [Sangon Biotech, A600560]) for 3 × 5 min, the sections were incubated with secondary antibody (Fuzhou Maixin Biotech., KIT-9720), and subsequently developed using a DAB Detection Kit (Fuzhou Maixin Biotech., Kit-0017) according to the manufacturer’s protocol. A semi-quantitative scoring criterion for IHC of p-MET Y1234/1235 and SQSTM1 was used, in which both staining intensity and positive areas were integrated into account and recorded as a single staining index (‘-’, negative; ‘+’, positive or weak; ‘++’, moderate; ‘+++’, strong). Two independent pathologists, blinded to the clinic pathological information, performed the scorings. All the immunostained slides were scanned on the Aperio Digital Pathology Slide Scanner ScanScope® System (Aperio, SN: SS005396), and the captured pictures were subjected to quantification by ImageScope_v11.2.0.780 software (Aperio).
Tumor xenograft model
Athymic nu/nu mice (male, 4- to 6-weeks old) were obtained from the Comparative Medicine Center, Yangzhou University (Yangzhou, China), and maintained in specific pathogen-free conditions in the animal facility of the School of Medicine at Southeast University (SEU), with constant temperature and humidity, under 12-h light-dark cycles, and received food and water ad libitum. To evaluate synergistic therapeutic efficiency of autophagy blockage and HGF-MET inhibition, 1 × 106 equal numbers of SMMC-7721 or Huh-7 cells were individually prepared in 100 μl of PBS, and individually inoculated subcutaneously into the right flank of nu/nu mouse. After inoculation, mice were monitored daily and weighed twice weekly. When tumors became visible, tumor growth was routinely measured by caliper, and reported as tumor surface size (longest dimension × perpendicular dimension). Once a tumor reached 35–45 mm2, mice respectively received subcutaneous multi-point injection adjacent to the tumor every 5 d after caliper measurement, with PBS (vehicle control, 100 μl), MET inhibitor (20 mg/kg in 100 μl of PBS) or/and the autophagic blocker CQ (10 mg/kg in 100 μl of PBS). Mice were sacrificed when signs of ulceration in the tumor were evident or when tumors reached the maximum permitted condition. After sacrifice, tumors were dissected, weighed and processed for further analysis. All experiments contained 5 mice per group and were run at least twice. Each one of the total 10 mice under same treatment showed the same tendency, and were analyzed together. All animal research in this study was approved and supervised by the Institutional Animal Care and Use Committee (IACUC) of the Institute of Life Sciences (ILS) at SEU. All animals received humane care according to the criteria outlined in the ‘Guide for the Care and Use of Laboratory Animals’ prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication 86–23 revised 1985). Animals bearing neoplastic lesions that exceeded 20% of their body mass or 20 mm in the longest axis were euthanatized for ethical considerations.
Statistical analysis
Sample sizes were chosen by standard methods to ensure adequate power for proper statistical analysis. Statistical analysis was performed with Excel 2015 (Microsoft Corporation) and GraphPad Prism 5 (GraphPad Software, Inc.) software to assess the differences between experimental groups. Results are shown as means ± SD, which were calculated from at least triplicate technical or biological replicates. Statistical significance was analyzed by means of the Student’s t-test and expressed as a p-value. All p-values were obtained by means of two-tailed Student’s t-test. * p-value < 0.05, ** p-value < 0.01, as compared to the negative, untreated or scrambled control. p-value < 0.05 was considered statistically significant.
Funding Statement
This work was supported by grants from the National Natural Science Foundation of China (81502975 to X.H.), China Postdoctoral Science Foundation (2016T90413 and 2015M581693 to X.H.), and SEU-Alphamab Joint Center (SA2015001 to W.X.). The research was funded in part by Jiangsu Planned Projects for Postdoctoral Research Funds (1501002A to X.H.) and Fundamental Research Funds for the Central Universities (2242016R20027 and 2242016K41045 to X.H.).
Acknowledgments
We deeply thank Dr. David Sabatini (Whitehead Institute for Biomedical Research, Massachusetts, USA) and Dr. Quan Chen (Institute of Zoology, Chinese Academy of Sciences, Beijing, China) for the reagents, and Dr. Tao Xu (Bionovogene Co., Ltd., Suzhou, China) for metabolomics analysis. We deeply thank the TCGA project organizers for the open access database, and Editage for linguistic assistance. We deeply thank editors and reviewers for their efforts in improving the quality, clarity and impact of this study. Especially, the author Xing Huang would like to express deepest thanks to Dr. Guido Kroemer & Dr. Laurence Zitvogel for the technological training and ideological inspiration in their lab. Xing Huang conceived this study and wrote the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Chen QY, Jiao DM, Wang J, et al. miR-206 regulates cisplatin resistance and EMT in human lung adenocarcinoma cells partly by targeting MET. Oncotarget. 2016. April 26;7(17):24510–24526. PubMed PMID: 27014910; PubMed Central PMCID: PMCPMC5029718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015. March;65(2):87–108. PubMed PMID: 25651787. [DOI] [PubMed] [Google Scholar]
- [3].Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015. March 01;136(5):E359–E386. PubMed PMID: 25220842. [DOI] [PubMed] [Google Scholar]
- [4].Maluccio M, Covey A.. Recent progress in understanding, diagnosing, and treating hepatocellular carcinoma. CA Cancer J Clin. 2012 November-Dec;62(6):394–399. PubMed PMID: 23070690. [DOI] [PubMed] [Google Scholar]
- [5].Liu CY, Chen KF, Chen PJ. Treatment of liver cancer. Cold Spring Harb Perspect Med. 2015. July 17;5(9):a021535 PubMed PMID: 26187874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Villanueva A, Hernandez-Gea V, Llovet JM. Medical therapies for hepatocellular carcinoma: a critical view of the evidence. Nat Rev Gastroenterol Hepatol. 2013. January;10(1):34–42. PubMed PMID: 23147664. [DOI] [PubMed] [Google Scholar]
- [7].Bruix J, Sherman M. American association for the study of liver D. Management of hepatocellular carcinoma: an update. Hepatology. 2011. March;53(3):1020–1022. PubMed PMID: 21374666; PubMed Central PMCID: PMCPMC3084991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cheng AL, Kang YK, Lin DY, et al. Sunitinib versus sorafenib in advanced hepatocellular cancer: results of a randomized phase III trial. J Clin Oncol. 2013. November 10;31(32):4067–4075. PubMed PMID: 24081937. [DOI] [PubMed] [Google Scholar]
- [9].Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008. July 24;359(4):378–390. PubMed PMID: 18650514. [DOI] [PubMed] [Google Scholar]
- [10].Llovet JM, Villanueva A, Lachenmayer A, et al. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat Rev Clin Oncol. 2015. July;12(7):408–424. PubMed PMID: 26054909. [DOI] [PubMed] [Google Scholar]
- [11].Zucman-Rossi J, Villanueva A, Nault JC, et al. Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology. 2015. October;149(5):1226–1239 e4. PubMed PMID: 26099527. [DOI] [PubMed] [Google Scholar]
- [12].Takamura A, Komatsu M, Hara T, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011. April 15;25(8):795–800. PubMed PMID: 21498569; PubMed Central PMCID: PMCPMC3078705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011. May;11(5):325–337. PubMed PMID: 21508971. [DOI] [PubMed] [Google Scholar]
- [14].Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009. May 22;324(5930):1029–1033. PubMed PMID: 19460998; PubMed Central PMCID: PMCPMC2849637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Warburg O, Wind F, Negelein E. Über den Stoffwechsel der Tumoren in Körper. Klinische Wochenschrift. 1926;5:829–832. [Google Scholar]
- [16].Warburg O, Posener K, Negelein E. Über den Stoffwechsel der Carcinomzelle. Biochem Zeitschr. 1924;152:309–344. [Google Scholar]
- [17].Racker E. Bioenergetics and the problem of tumor growth. Am Sci. 1972 January-Feb;60(1):56–63. PubMed PMID: 4332766. [PubMed] [Google Scholar]
- [18].Wise DR, DeBerardinis RJ, Mancuso A, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008. December 02;105(48):18782–18787. PubMed PMID: 19033189; PubMed Central PMCID: PMCPMC2596212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yuneva M, Zamboni N, Oefner P, et al. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol. 2007. July 02;178(1):93–105. PubMed PMID: 17606868; PubMed Central PMCID: PMCPMC2064426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Son J, Lyssiotis CA, Ying H, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013. April 04;496(7443):101–105. PubMed PMID: 23535601; PubMed Central PMCID: PMCPMC3656466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. 2013. September;123(9):3678–3684. PubMed PMID: 23999442; PubMed Central PMCID: PMCPMC3754270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mathew R, White E. Autophagy, stress, and cancer metabolism: what doesn’t kill you makes you stronger. Cold Spring Harb Symp Quant Biol. 2011;76:389–396. PubMed PMID: 22442109. [DOI] [PubMed] [Google Scholar]
- [23].Madrigal-Matute J, Cuervo AM. Regulation of liver metabolism by autophagy. Gastroenterology. 2016. February;150(2):328–339. PubMed PMID: 26453774; PubMed Central PMCID: PMCPMC4728051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Xie Q, Su Y, Dykema K, et al. Overexpression of HGF promotes HBV-induced hepatocellular carcinoma progression and is an effective indicator for met-targeting therapy. Genes Cancer. 2013. July;4(7–8):247–260. PubMed PMID: 24167653; PubMed Central PMCID: PMCPMC3807646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Piguet AC, Medova M, Keogh A, et al. Impact of MET targeting on tumor-associated angiogenesis and growth of MET mutations-driven models of liver cancer. Genes Cancer. 2015. July;6(7–8):317–327. PubMed PMID: 26413215; PubMed Central PMCID: PMCPMC4575919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Takeda S, Liu H, Sasagawa S, et al. HGF-MET signals via the MLL-ETS2 complex in hepatocellular carcinoma. J Clin Invest. 2013. July;123(7):3154–3165. PubMed PMID: 23934123; PubMed Central PMCID: PMCPMC3696564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wu F, Wu L, Zheng S, et al. The clinical value of hepatocyte growth factor and its receptor–c-met for liver cancer patients with hepatectomy. Dig Liver Dis. 2006. July;38(7):490–497. PubMed PMID: 16627020. [DOI] [PubMed] [Google Scholar]
- [28].Ogunwobi OO, Puszyk W, Dong HJ, et al. Epigenetic upregulation of HGF and c-Met drives metastasis in hepatocellular carcinoma. PLoS One. 2013;8(5):e63765 PubMed PMID: 23723997; PubMed Central PMCID: PMCPMC3665785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jiang WG, Hallett MB, Puntis MC. Hepatocyte growth factor/scatter factor, liver regeneration and cancer metastasis. Br J Surg. 1993. November;80(11):1368–1373. PubMed PMID: 8252338. [DOI] [PubMed] [Google Scholar]
- [30].Junbo H, Li Q, Zaide W, et al. Increased level of serum hepatocyte growth factor/scatter factor in liver cancer is associated with tumor metastasis. In Vivo. 1999 March-Apr;13(2):177–180. PubMed PMID: 10363175. [PubMed] [Google Scholar]
- [31].Birchmeier C, Birchmeier W, Gherardi E, et al. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003. December;4(12):915–925. PubMed PMID: 14685170. [DOI] [PubMed] [Google Scholar]
- [32].Gherardi E, Birchmeier W, Birchmeier C, et al. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012. January 24;12(2):89–103. PubMed PMID: 22270953. [DOI] [PubMed] [Google Scholar]
- [33].Hu CT, Wu JR, Cheng CC, et al. The therapeutic targeting of HGF/c-Met signaling in hepatocellular carcinoma: alternative approaches. Cancers (Basel). 2017. May 26;9(6). PubMed PMID: 28587113; PubMed Central PMCID: PMCPMC5483877 DOI: 10.3390/cancers9060058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bladt F, Friese-Hamim M, Ihling C, et al. The c-Met inhibitor MSC2156119J effectively inhibits tumor growth in liver cancer models. Cancers (Basel). 2014. August 19;6(3):1736–1752. PubMed PMID: 25256830; PubMed Central PMCID: PMCPMC4190565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tao J, Zhang R, Singh S, et al. Targeting beta-catenin in hepatocellular cancers induced by coexpression of mutant beta-catenin and K-Ras in mice. Hepatology. 2017. May;65(5):1581–1599. PubMed PMID: 27981621; PubMed Central PMCID: PMCPMC5397318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kolobova E, Tuganova A, Boulatnikov I, et al. Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem J. 2001. August 15;358(Pt 1):69–77. PubMed PMID: 11485553; PubMed Central PMCID: PMCPMC1222033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Roche TE, Baker JC, Yan X, et al. Distinct regulatory properties of pyruvate dehydrogenase kinase and phosphatase isoforms. Prog Nucleic Acid Res Mol Biol. 2001;70:33–75. PubMed PMID: 11642366. [DOI] [PubMed] [Google Scholar]
- [38].O’Donovan DJ, Lotspeich WD. Activation of kidney mitochondrial glutaminase by inorganic phosphate and organic acids. Nature. 1966. November 26;212(5065):930–932. PubMed PMID: 6005667. [DOI] [PubMed] [Google Scholar]
- [39].Curthoys NP, Watford M. Regulation of glutaminase activity and glutamine metabolism. Annu Rev Nutr. 1995;15:133–159. PubMed PMID: 8527215. [DOI] [PubMed] [Google Scholar]
- [40].Cassago A, Ferreira AP, Ferreira IM, et al. Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc Natl Acad Sci U S A. 2012. January 24;109(4):1092–1097. PubMed PMID: 22228304; PubMed Central PMCID: PMCPMC3268272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Galluzzi L, Pietrocola F, Levine B, et al. Metabolic control of autophagy. Cell. 2014. December 04;159(6):1263–1276. PubMed PMID: 25480292; PubMed Central PMCID: PMCPMC4500936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Liu L, Feng D, Chen G, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012. January 22;14(2):177–185. PubMed PMID: 22267086. [DOI] [PubMed] [Google Scholar]
- [43].van der Vos KE, Coffer PJ. Glutamine metabolism links growth factor signaling to the regulation of autophagy. Autophagy. 2012. December;8(12):1862–1864. PubMed PMID: 22996802; PubMed Central PMCID: PMCPMC3541306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wei Y, Zou Z, Becker N, et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell. 2013. September 12;154(6):1269–1284. PubMed PMID: 24034250; PubMed Central PMCID: PMCPMC3917713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011. March 04;144(5):646–674. PubMed PMID: 21376230. [DOI] [PubMed] [Google Scholar]
- [46].Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. 2011. August 31;10(9):671–684. PubMed PMID: 21878982. [DOI] [PubMed] [Google Scholar]
- [47].Cheong H, Lu C, Lindsten T, et al. Therapeutic targets in cancer cell metabolism and autophagy. Nat Biotechnol. 2012. July 10;30(7):671–678. PubMed PMID: 22781696; PubMed Central PMCID: PMCPMC3876738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Galluzzi L, Kepp O, Vander Heiden MG, et al. Metabolic targets for cancer therapy. Nat Rev Drug Discov. 2013. November;12(11):829–846. PubMed PMID: 24113830. [DOI] [PubMed] [Google Scholar]
- [49].Garber K. MET inhibitors start on road to recovery. Nat Rev Drug Discov. 2014. August;13(8):563–565. PubMed PMID: 25082276. [DOI] [PubMed] [Google Scholar]
- [50].Katayama R, Aoyama A, Yamori T, et al. Cytotoxic activity of tivantinib (ARQ 197) is not due solely to c-MET inhibition. Cancer Res. 2013. May 15;73(10):3087–3096. PubMed PMID: 23598276; PubMed Central PMCID: PMCPMC3759033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Du Y, Yamaguchi H, Wei Y, et al. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat Med. 2016. February;22(2):194–201. PubMed PMID: 26779812; PubMed Central PMCID: PMCPMC4754671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lefebvre J, Muharram G, Leroy C, et al. Caspase-generated fragment of the Met receptor favors apoptosis via the intrinsic pathway independently of its tyrosine kinase activity. Cell Death Dis. 2013. October 17;4:e871 PubMed PMID: 24136235; PubMed Central PMCID: PMCPMC3824686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lampada A, O’Prey J, Szabadkai G, et al. mTORC1-independent autophagy regulates receptor tyrosine kinase phosphorylation in colorectal cancer cells via an mTORC2-mediated mechanism. Cell Death Differ. 2017. June;24(6):1045–1062. PubMed PMID: 28475179; PubMed Central PMCID: PMCPMC5442471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Galluzzi L, Pietrocola F, Bravo-San Pedro JM, et al. Autophagy in malignant transformation and cancer progression. Embo J. 2015. April 01;34(7):856–880. PubMed PMID: 25712477; PubMed Central PMCID: PMCPMC4388596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Huang X, Wu Z, Mei Y, et al. XIAP inhibits autophagy via XIAP-Mdm2-p53 signalling. Embo J. 2013. August 14;32(16):2204–2216. PubMed PMID: 23749209; PubMed Central PMCID: PMCPMC3746193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Marino G, Niso-Santano M, Baehrecke EH, et al. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014. February;15(2):81–94. PubMed PMID: 24401948; PubMed Central PMCID: PMCPMC3970201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005. September 23;122(6):927–939. PubMed PMID: 16179260. [DOI] [PubMed] [Google Scholar]
- [58].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. PubMed PMID: 26799652; PubMed Central PMCID: PMCPMC4835977. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.