

ABSTRACT
TP53INP2/DOR (tumor protein p53-inducible nuclear protein 2) contributes to mammalian macroautophagy/autophagy by carrying nuclear deacetylated MAP1LC3/LC3 to the cytoplasm. Here, we report that in the cytoplasm, TP53INP2 further functions in autophagosome biogenesis by promoting LC3B-ATG7 interaction. Cytoplasmic expression of the N-terminal region of TP53INP2, which includes the LC3-interacting region, effectively triggered LC3B–PE production and autophagosome formation. In the cytoplasm, TP53INP2 colocalized to early autophagic membrane structures containing ATG14, ZFYVE1/DFCP1 or WIPI2. While knockdown of TP53INP2 did not affect the formation of these autophagic structures, deletion of BECN1 or Atg5, or mutations preventing TP53INP2 from LC3 interaction, disrupted the membrane binding of TP53INP2. TP53INP2 interacted directly with ATG7 to form a LC3B-TP53INP2-ATG7 complex in the cytoplasm. Loss of TP53INP2-LC3 or TP53INP2-ATG7 interaction significantly reduced LC3B-ATG7 binding. Together, these results suggest that after shifting from the nucleus, cytoplasmic TP53INP2 is targeted to early autophagic membranes accompanied by LC3, where it contributes to autophagosome biogenesis by mediating LC3-ATG7 interaction.
Abbreviations: 3-MA, 3-methyladenine; 3NES, 3 repeated nuclear export signal; 3NLS, 3 repeated nuclear localization signal; ACTB, actin beta; ATG, autophagy related; BECN1, Beclin 1; mCherry, monomeric red fluorescent protein mCherry; GFP, green fluorescent protein; GST, glutathione S-transferase; KO, knockout; LC3B/MAP1LC3B, microtubule-associated protein 1 light chain 3 beta; LC3B[G120], LC3B mutant lacking amino acids after glycine 120; LDH, lactate dehydrogenase; LMNB1, lamin B1; LIR, LC3-interacting region; MTORC1, mechanistic target of rapamycin complex 1; PE, phosphatidylethanolamine; PtdIns3K, phosphatidylinositol 3-kinase; PtdIns3P, phosphatidylinositol 3-phosphate; rDNA, ribosomal DNA; RFP, red fluorescent protein; RNAi, RNA interference; SQSTM1, sequestosome 1; TP53INP2, tumor protein p53-inducible nuclear protein 2; TP53INP2[1-28], TP53INP2 mutant containing amino acids 1 to 28; TP53INP2[28-45], TP53INP2 mutant containing amino acids 28 to 45; TP53INP2[LIRΔ], TP53INP2 mutant lacking amino acids 1 to 144; TP53INP2[NLSΔ], TP53INP2 mutant lacking amino acids 145 to 221; TP53INP2W35,I38A, TP53INP2 mutant in which tryptophan 35 and isoleucine 38 are replaced with alanine; TP53INP2W35,I38A[NLSΔ], TP53INP2 mutant lacking amino acids 145 to 221, and tryptophan 35 and isoleucine 38 are replaced with alanine; TP53INP2W35,I38A[Δ1-28],[NLSΔ], TP53INP2 mutant lacking amino acids 1 to 28 and amino acids 145 to 221, and tryptophan 35 and isoleucine 38 are replaced with alanine; TP53INP2[Δ1-28],[NLSΔ], TP53INP2 mutant lacking amino acids 1 to 28 and amino acids 145 to 221; TP53INP2[Δ67-111],[NLSΔ], TP53INP2 mutant lacking amino acids 67 to 111 and amino acids 145 to 221; TP53INP2[Δ112-144],[NLSΔ], TP53INP2 mutant lacking amino acids 112 to 144 and amino acids 145 to 221; TUBB, tubulin beta class I; ULK1, unc-51 like autophagy activating kinase 1; VMP1, vacuole membrane protein 1; WIPI2, WD repeat domain phosphoinositide-interacting 2; WT, wild-type; ZFYVE1/DFCP1, zinc finger FYVE-type containing 1.
KEYWORDS: Autophagosome biogenesis, autophagy, cytoplasm, LC3-ATG7 binding, LC3-lipidation, TP53INP2
Introduction
Autophagy is an evolutionally conserved and lysosome-mediated intracellular degradation pathway. Eukaryotic cells depend on well-maintained basal autophagy and stimulus-responsive inducible autophagy to sustain homeostasis and to cope with environmental changes. The implementation and completion of autophagy depend on double-membrane autophagosomes, which are formed in cells by the expansion and elongation of phagophores derived from the endoplasmic reticulum membranes [1-4]. Accumulating evidence has demonstrated that the mammalian ortholog family of yeast Atg8, microtubule-associated protein 1 light chain 3 (MAP1LC3/LC3), is pivotal to the biogenesis of autophagosomes [5–9]. To carry out its function in autophagy, LC3 needs to be modified by a ubiquitination-like system, which conjugates it with phosphatidylethanolamine (PE) by interacting in turn with ATG7, ATG3, and ATG12–ATG5 [5,10,11]. This lipidation converts soluble LC3 to the membrane-bound form and enables the recruitment of LC3-interacting proteins for the growth of autophagic membranes [5,9,11,12]. However, the mechanism underlying the LC3-mediated protein recruitment and membrane growth remains elusive.
Recently, it has been found that the nuclear localization and the nucleus-to-cytoplasm translocation of LC3 are essential for autophagosome formation in mammalian cells [13]. In response to cell starvation, nuclear LC3 is deacetylated by the histone deacetylase SIRT1, which redistributes LC3 into the cytoplasm for lipidation [13]. The redistribution requires binding of deacetylated LC3 with TP53INP2, a nuclear protein which is enriched in cells with high metabolic levels and which was primarily identified as a coactivator for the thyroid hormone receptor [14] (Please note that the mouse acronym for TP53INP2 is TRP53INP2 but we will use TP53INP2 in both cases hereafter, for simplicity). Overexpression of TP53INP2 promotes basal autophagy and TP53INP2 knockdown inhibits autophagy induced by nutrient deprivation [13,15,16]. Intriguingly, besides interacting with the thyroid hormone receptor, nuclear TP53INP2 localizes to the nucleolus, where it facilitates ribosome biogenesis by promoting rDNA transcription [17]. A nucleolar localization signal (NoLS) located at the C-terminal domain of TP53INP2 has been identified, and a dual role of TP53INP2 in cell anabolism and catabolism has been suggested, depending on its subcellular localizations [17].
TP53INP2 can associate with autophagosomes after shifting to the cytoplasm [13,15,16], which suggests that TP53INP2 has a role in autophagy besides taking LC3 out of the nucleus. Biochemically, TP53INP2 interacts with vacuole membrane protein 1 (VMP1), a transmembrane protein which is detectable in autophagic membranes and whose expression is elevated in cells with nutrient deprivation or rapamycin treatment [15,18]. Based on this interaction, and on the observation that TP53INP2 knockdown blocks the recruitment of BECN1/Beclin 1 to autophagic membranes, it has been proposed that cytoplasmic TP53INP2 functions as a scaffold protein that recruits LC3 and/or LC3-related proteins to the autophagosome membranes [15]. However, the increase of BECN1 and LC3/LGG-1 on phagophore membranes when VMP1/epg-3 is silenced [19,20], the dependence of nuclear TP53INP2 export on class III PtdIns3K activity [16], and the failure of TP53INP2 in binding to autophagic membranes in atg5−/- and atg7−/- cells [13,15], suggest on the contrary that TP53INP2 may rely on LC3 for membrane association.
In this study, we have characterized the action of TP53INP2 in the cytoplasm. We present evidence that the recruitment of TP53INP2 to the phosphatidylinositol 3-phosphate (PtdIns3P)-positive early autophagic membranes relies on LC3. Through direct binding with ATG7, LC3-associated TP53INP2 promotes the interaction of LC3B and ATG7, thus enhancing autophagosome formation.
Results
Cytoplasmic TP53INP2 promotes autophagosome formation
To investigate the function of TP53INP2 in the cytoplasm, we first expressed in cells a TP53INP2 construct fused with 3 repeated nuclear export signals (3NES). This [3NES]-TP53INP2 construct exhibited a predominantly nuclear distribution, mimicking WT TP53INP2 (data not show), which suggests that TP53INP2 has a strong tendency to localize in the nucleus. We predicted a nuclear localization signal (NLS), which includes the NoLS in the C-terminal region of TP53INP2 [17], using NLStradamus: http://www.moseslab.csb.utoronto.ca/NLStradamus/. We then created a truncated TP53INP2 construct that retains the N-terminal LC3-interacting region (LIR) but lacks the C-terminal nuclear localization sequence (TP53INP2[NLSΔ]) (Figure 1(a)). In HeLa cells, transfected TP53INP2[NLSΔ] exhibited a nuclear and cytoplasmic distribution and formed puncta in the cells, while the WT protein and TP53INP2[LIRΔ] (which has the C-terminal nuclear localization sequence but not the N-terminal LIR) were only in the nucleus (Figure 1(a,b)). In stable GFP-LC3B-expressing HEK293 cells, TP53INP2[NLSΔ] expression triggered more GFP-LC3B puncta than TP53INP2 expression, while TP53INP2[LIRΔ] transfection failed to induce GFP-LC3B puncta (Figure 1(c,d)). Accordingly, transfection of TP53INP2[NLSΔ] produced more LC3B–PE and more cleavage of GFP-LC3B in the cells (Figure 1(e)). In addition, in stable mCherry-LC3B-expressing HEK293 cells, transfected GFP-[3NES]-TP53INP2[NLSΔ] localized selectively in the cytoplasm and strongly induced the formation of mCherry-LC3B puncta, while transfected GFP-[3NLS]-TP53INP2[NLSΔ] localized exclusively in the nucleus and resulted in neither cytoplasmic translocation of mCherry-LC3B nor the formation of mCherry-LC3B puncta (Figure 1(f)). Consistent with this, expression of the cytoplasm-located [3NES]-TP53INP2[NLSΔ], but not the nucleus-located [3NLS]-TP53INP2[NLSΔ], led to the production of LC3B–PE and autophagic sequestration of LDH (Figure 1(g,h)). Further, to exclude the possibility that the phenotypes induced by [3NES]-TP53INP2[NLSΔ] are due to an effect on the nucleus-to-cytoplasm translocation of LC3, we created 2 truncated mutants of the N-terminal region of TP53INP2, one of which (GFP-[3NES]-TP53INP2[28-45]) contains the LIR and is capable of binding to LC3B, while the other (GFP-[3NES]-TP53INP2[1-28]]) could not interact with LC3B (Fig. S1A). When these proteins were expressed via transfection into mCherry-LC3B-HEK293 cells, neither GFP-[3NES]-TP53INP2[1-28] nor GFP-[3NES]-TP53INP2[28–45] triggered autophagosome formation, even though GFP-[3NES]-TP53INP2[28–45] was competent to translocate mCherry-LC3B from the nucleus to the cytoplasm (Fig. S1B to D). This suggests that cytoplasmic TP53INP2 has a role in autophagosome formation other than altering the nucleo-cytoplasmic distribution of LC3. Taken together, these results suggest that cytoplasmic TP53INP2 effectively enhances basal autophagy.
Figure 1.
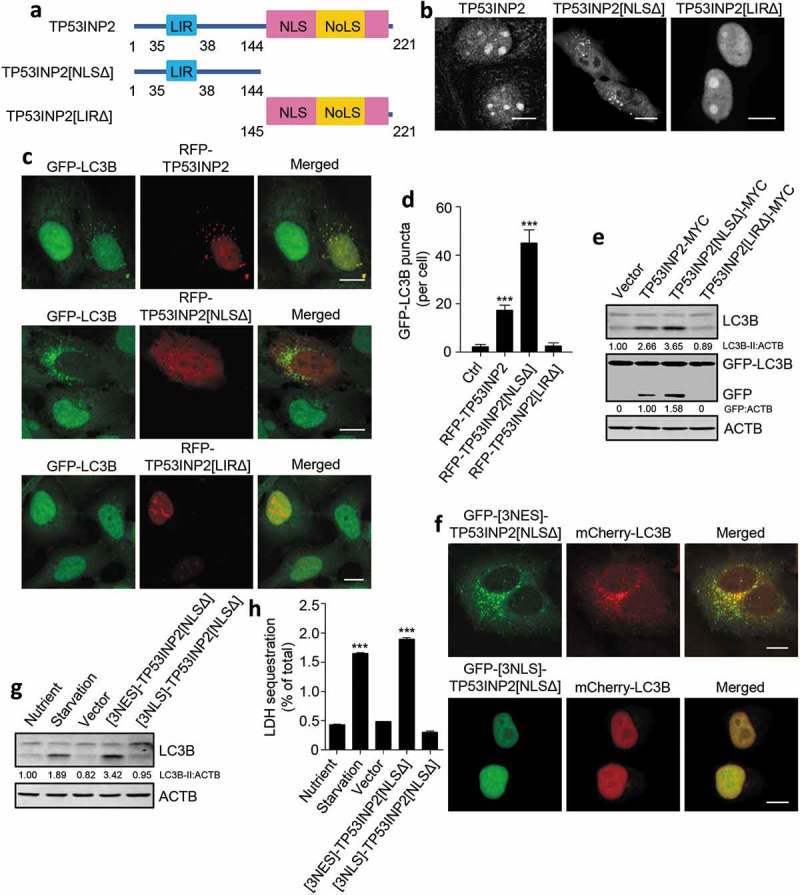
Cytoplasmic TP53INP2 stimulates autophagy. (a) Domain architecture of TP53INP2, TP53INP2[NLSΔ] and TP53INP2[LIRΔ]. LIR, LC3-interacting region; NLS, nuclear localization signal; NoLS, nucleolar localization signal. (b) The intracellular distribution of GFP-tagged TP53INP2, TP53INP2[NLSΔ] or TP53INP2[LIRΔ] in HeLa cells. (c) Confocal images of HEK293 cells stably expressing GFP-LC3B with RFP-tagged TP53INP2, TP53INP2[NLSΔ] or TP53INP2[LIRΔ] transfection. (d) Quantification of GFP-LC3B puncta in (c). The data are presented as mean ± SEM, n = 30 cells. (e) Western blot analysis of LC3B-II production and GFP-LC3B cleavage in HEK293 cells stably expressing GFP-LC3B and transfected with the indicated plasmids. (f) Confocal images of HEK293 cells stably expressing mCherry-LC3B and transfected with plasmids expressing GFP-[3NES]-TP53INP2[NLSΔ] or GFP-[3NLS]-TP53INP2[NLSΔ]. (g) Western blot analysis of intracellular LC3B in HEK293 cells that were starved or transfected with the GFP-tagged indicated plasmids. (h) LDH sequestration assay of HEK293 cells treated as in (g) in the presence of chloroquine. The data are presented as mean ± SEM of triplicates. ***, P < 0.001. Scale bars: 10 µm.
TP53INP2 associates with early autophagic membranes without affecting their formation
To understand why overexpression of TP53INP2 in the cytoplasm can stimulate autophagy, we first checked the location of TP53INP2 in the cytoplasm. We observed in starved MEFs that cytoplasmic RFP-TP53INP2 associated with the punctate structures containing GFP-tagged ULK1, ATG14, BECN1, ZFYVE1 or WIPI2 (Figure 2(a–e)). Intriguingly, in BECN1-deleted or 3-methyladenine (3-MA)-treated cells, no such associations were observed with RFP-TP53INP2 or RFP-TP53INP2[NLSΔ], even though ULK1 puncta formed normally when the cells were starved (Figure 2(f,g)). These results therefore suggest that cytoplasmic TP53INP2 can bind to early autophagic structures in a manner dependent on class III PtdIns3K activation. Because this phenomenon is inconsistent with a previous study showing that the membrane recruitment of BECN1 requires TP53INP2 [15], we then examined the formation of early autophagic membranes in cells with TP53INP2 RNAi. As expected, silencing TP53INP2 did not affect the formation of ULK1, BECN1 or ZFYVE1 puncta in fed cells and starved cells, whereas the production of LC3B-puncta and LC3B–PE in the cells was dramatically inhibited (Figure 2(h,i) and S2). Together, these results suggest that, in the cytoplasm, TP53INP2 associates with early autophagic membranes and is not essential to their formation.
Figure 2.
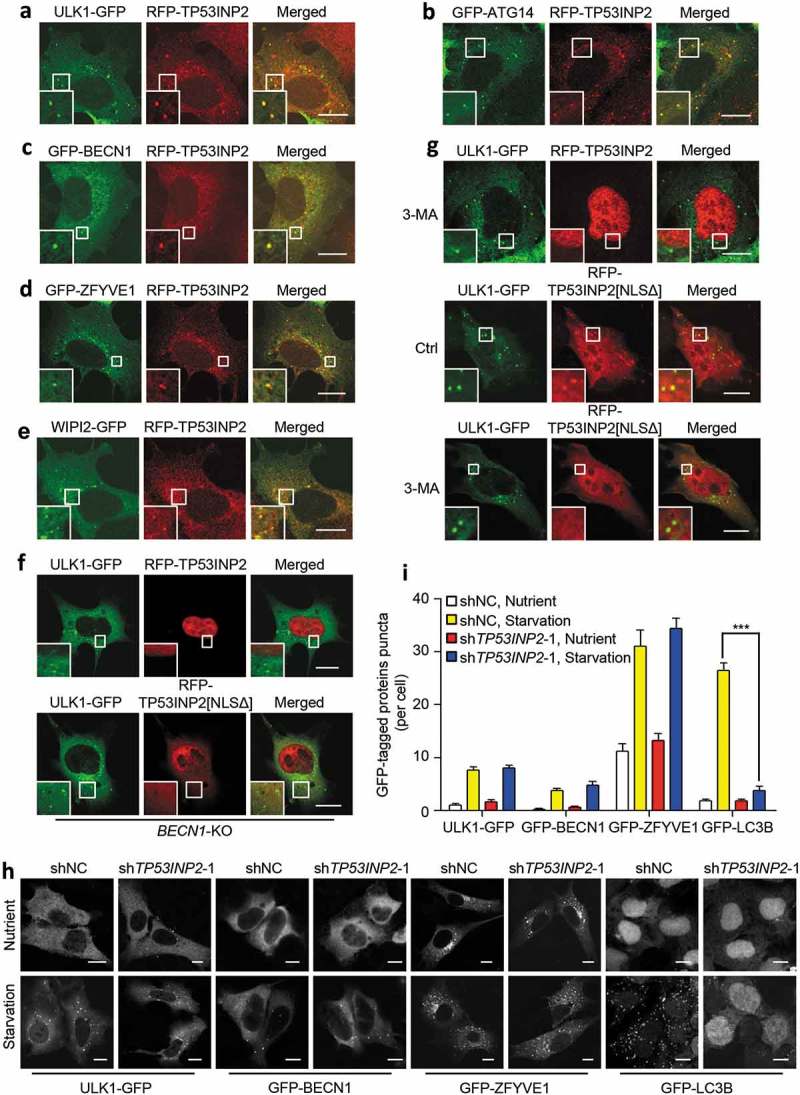
Association of TP53INP2 with early autophagic structures. (a-e) Colocalization of RFP-TP53INP2 with ULK1-GFP (a), GFP-ATG14 (b), GFP-BECN1 (c), GFP-ZFYVE1 (d) or WIPI2-GFP (e) in starved MEFs. (f and g) Localization of RFP-TP53INP2 or RFP-TP53INP2[NLSΔ] in starved BECN1-KO (f) or 3-MA-treated (g) HEK293 cells. The cells were imaged by confocal microscope 24 h after cotransfection of the plasmids. (h) Formation of puncta in HEK293 cells stably expressing ULK1-GFP, GFP-BECN1, GFP-ZFYVE1 or GFP-LC3B. The cells were infected with lentivirus expressing non-silencing control shRNA or TP53INP2 shRNA for 72 h, with or without cell starvation for 2 h. (I) Quantification of the puncta in (h). The data are presented as mean ± SEM, n = 30 cells. ***, P < 0.001. Scale bars: 10 µm.
Association of TP53INP2 with autophagic membranes depends on LC3
Under cell starvation, binding of nuclear deacetylated LC3 with TP53INP2 allows the 2 proteins to shift synchronously into the cytoplasm [13]. When LC3 is restricted to the nucleus, the nuclear export of TP53INP2 is unaffected, but TP53INP2 forms far fewer puncta in the cytoplasm [13]. Given that the membrane association of TP53INP2 depends on PtdIns3P production, and TP53INP2 lacks a typical PtdIns3P-binding motif, we speculated that the membrane association of TP53INP2 requires membrane-bound LC3. In starved atg5−/- and atg7−/- MEFs, RFP-TP53INP2 had a diffuse cytoplasmic distribution and failed to colocalize to GFP-ATG14 puncta (Figure 3(a)). The same phenomenon was observed in cells cooverexpressing RFP-TP53INP2 with nucleus-localized GFP-[3NLS]-LC3B or GFP-LC3BG120A, an LC3B mutant in which glycine 120 was replaced with alanine and is therefore incapable of membrane association [5,10] (Figure 3(b)). We also examined RFP-TP53INP2W35,I38A[NLSΔ], a W35,I38A LIR double mutant of TP53INP2[NLSΔ] with a RFP tag that is incapable of binding to LC3 [21]. RFP-TP53INP2W35,I38A[NLSΔ] showed no association with Flag-ATG14 puncta (Figure 3(c)). In these cells, the starvation-stimulated formation of GFP-LC3B puncta was dramatically reduced, suggesting that the autophagy-enhancing effect of TP53INP2[NLSΔ] was abolished by the W35,I38A LIR mutations. This was further confirmed by expressing only RFP-TP53INP2W35,I38A[NLSΔ] in GFP-LC3B-HEK293 cells. Overexpression of TP53INP2W35,I38A[NLSΔ] failed to induce the formation of GFP-LC3B puncta in normal cultured cells or enhance it in starved cells (Figure 3(d.e)). Together, these data strongly suggest that the binding of TP53INP2 to autophagic membranes is dependent on membrane-associated LC3, and interaction with LC3 is required for the autophagy-promoting function of cytoplasmic TP53INP2.
Figure 3.
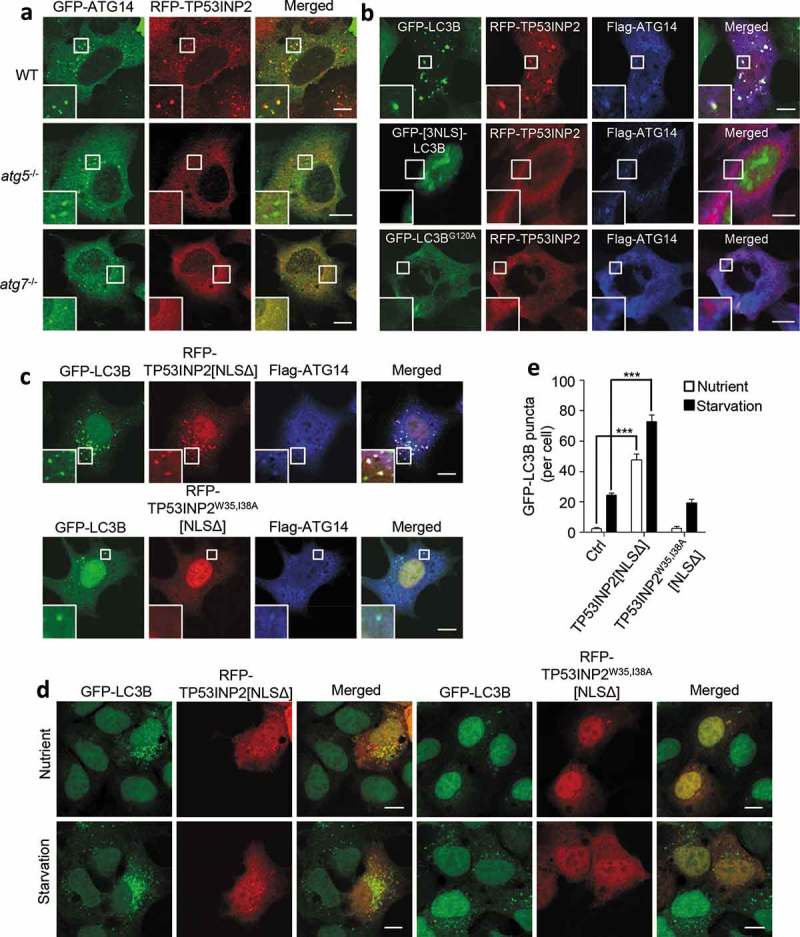
LC3 mediates the association of TP53INP2 with autophagic membranes. (a) Colocalization analysis of RFP-TP53INP2 and GFP-ATG14 in starved WT, atg5−/- or atg7−/- MEFs. (b) Colocalization analysis of RFP-TP53INP2 and Flag-ATG14 in starved HEK293 cells coexpressing GFP-LC3B, GFP-[3NLS]-LC3B or GFP-LC3BG120A. (c) HEK293 cells stably expressing GFP-LC3B were cotransfected with Flag-ATG14 and RFP-TP53INP2[NLSΔ], or with Flag-ATG14 and RFP-TP53INP2W35,I38A[NLSΔ]. Cells were then immunostained with anti-Flag and imaged with confocal microscopy. (d) GFP-LC3B punctum formation in HEK293 cells stably expressing GFP-LC3B with or without cell starvation. The cells were transiently transfected with RFP-TP53INP2[NLSΔ] or RFP-TP53INP2W35,I38A[NLSΔ]. (e) Quantification of GFP-LC3B puncta in (d). The data are presented as mean ± SEM, n = 30 cells. ***, P < 0.001. Scale bars: 10 µm.
TP53INP2 interacts with proteins involved in LC3–PE conjugation in cells
Our observations that interaction with LC3 is required for TP53INP2 membrane binding and for the autophagy-promoting function of cytoplasmic TP53INP2 suggest that TP53INP2 may be associated with LC3 in the cytoplasm and may contribute to LC3 lipidation. To test this, we first performed coprecipitation assays to check a potential interaction of TP53INP2 with ATG7, ATG3 or ATG12–ATG5. As expected, immunoprecipitation of cell-expressed GFP-TP53INP2 and GFP-TP53INP2[NLSΔ], but not GFP-TP53INP2[LIRΔ], coprecipitated endogenous ATG7, ATG3 and ATG12–ATG5, even in fed cells (Figure 4(a)). To deduce whether TP53INP2 interacts with ATG7, ATG3 or ATG12–ATG5 through LC3, we immunoprecipitated GFP-TP53INP2[NLSΔ] or GFP-TP53INP2W35,I38A[NLSΔ] from transfected cells. Intriguingly, ATG7, but not ATG3 or ATG12–ATG5, was coprecipitated by GFP-TP53INP2W35,I38A[NLSΔ] (Figure 4(b)), suggesting that TP53INP2 interacts directly with ATG7. We then performed an in vitro affinity-isolation assay using purified recombinant proteins. Purified LC3B[G120] (an LC3B mutant in which the residues after G120 were deleted and can conjugate with ATG7, ATG3 and PE) and ATG7 were incubated with recombinant GST-TP53INP2 or GST-TP53INP2W35,I38A. We found that much less LC3B[G120] was affinity-isolated by precipitated GST-TP53INP2W35,I38A than by precipitated GST-TP53INP2, but comparable amounts of ATG7 were affinity-isolated by both proteins (Figure 4(c)). This was verified by incubating purified ATG7 only with GST-TP53INP2 or GST-TP53INP2W35,I38A (Fig. S3A). Intriguingly, a similar in vitro affinity-isolation assay using purified proteins did not detect any direct interaction of ATG3 with GST-TP53INP2 or GST-TP53INP2W35,I38A (Fig. S3A).
Figure 4.
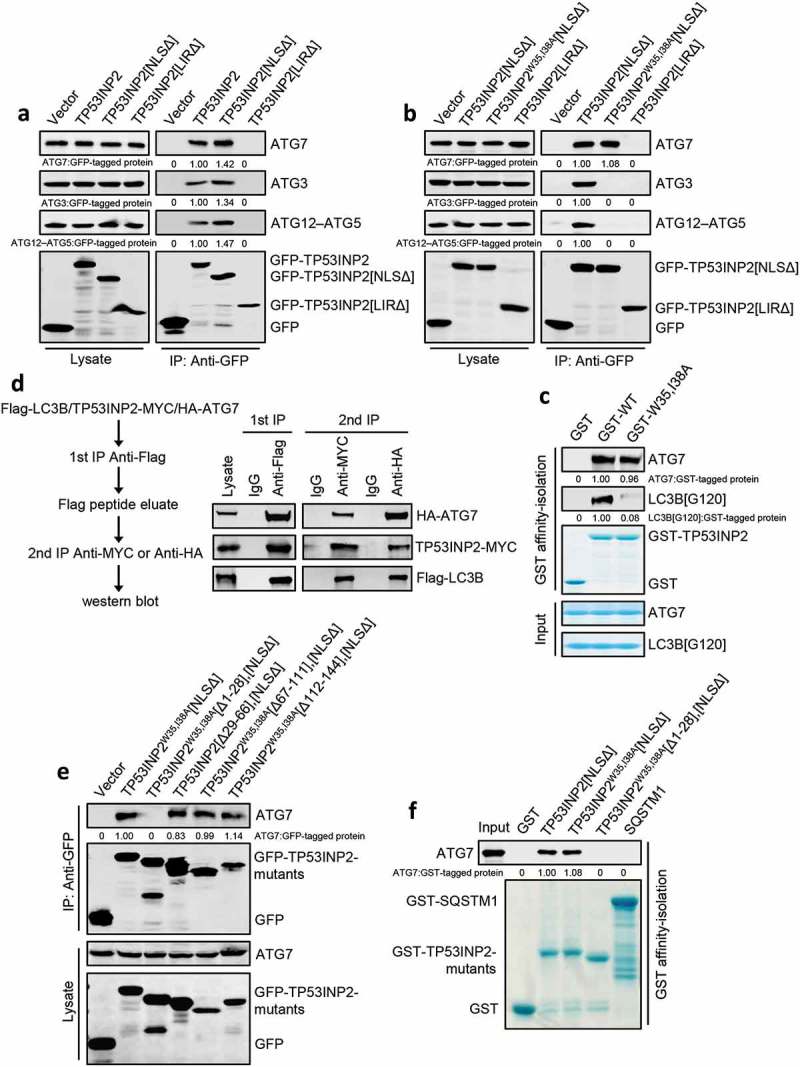
TP53INP2 forms a complex with LC3B and ATG7. (a) Coimmunoprecipitation of ATG7, ATG3 or ATG12–ATG5 with GFP-TP53INP2, GFP-TP53INP2[NLSΔ] or GFP-TP53INP2[LIRΔ] from HEK293 cells. TP53INP2 proteins were immunoprecipitated by anti-GFP. The coprecipitated ATG7, ATG3 or ATG12–ATG5 was detected by western blot using anti-ATG3, anti-ATG7 or anti-ATG5 respectively. (b) Coimmunoprecipitation of ATG7, ATG3 or ATG12–ATG5 with GFP-tagged TP53INP2[NLSΔ], TP53INP2W35,I38A[NLSΔ] or TP53INP2[LIRΔ]. GFP-tagged TP53INP2 mutants were immunoprecipitated using anti-GFP and the precipitates were analyzed using anti-ATG7, anti-ATG3 or anti-ATG5. (c) In vitro TP53INP2-ATG7 binding assay. Purified GST-TP53INP2 or GST-TP53INP2W35,I38A was incubated with purified LC3B[G120] and ATG7. After affinity-isolating GST-TP53INP2 or GST-TP53INP2W35,I38A with glutathione-sepharose 4B beads, the bound LC3B[G120] and ATG7 were analyzed by western blot. (d) HEK293T cells were cotransfected with Flag-LC3B, TP53INP2-MYC and HA-ATG7. The cells were lysed 48 h after transfection and Flag-LC3B was immunoprecipitated with anti-Flag. After incubation of the Flag-LC3B precipitates with Flag peptide, the eluate was used for immunoprecipitation with either anti-MYC or anti-HA. The immunoprecipitates were then analyzed by western blot by anti-Flag, anti-MYC and anti-HA respectively. (e) Coimmunoprecipitation of ATG7 with each of the indicated GFP-tagged truncated TP53INP2 mutants in HEK293 cells. TP53INP2 proteins were immunoprecipitated using anti-GFP and the precipitates were analyzed using anti-ATG7. (f) Purified GST-tagged TP53INP2[NLSΔ], TP53INP2W35,I38A[NLSΔ], TP53INP2W35,I38A[Δ1-28],[NLSΔ] or SQSTM1 was incubated with purified ATG7, then the GST-tagged proteins were affinity-isolated by glutathione-sepharose 4B beads and bound ATG7 was detected by western blot using anti-ATG7.
We then carried out a 2-step coimmunoprecipitation experiment to test whether LC3, TP53INP2 and ATG7 form a complex. In HEK293T cells transfected with Flag-LC3B, TP53INP2-MYC and HA-ATG7, immunoprecipitation of Flag-LC3B with anti-Flag coprecipitated TP53INP2-MYC and HA-ATG7 (Figure 4(d)). After washing the precipitates with Flag peptide to release Flag-LC3B from the beads, we immunoprecipitated TP53INP2-MYC by anti-MYC or HA-ATG7 by anti-HA from the Flag peptide eluate. This resulted in coprecipitation of Flag-LC3B and HA-ATG7 with TP53INP2-MYC, Flag-LC3B and TP53INP2-MYC with HA-ATG7 (Figure 4(d)). These results suggest that TP53INP2 can form a heterocomplex in the cytoplasm with LC3 and ATG7.
To find the region within TP53INP2 that is required for its interaction with ATG7, we created a number of GFP-tagged truncated forms of TP53INP2W35,I38A[NLSΔ] (Figure 4(e)), and performed coimmunoprecipitation experiments. We found that deletion of amino acids 1 to 28 in the N terminus prevented TP53INP2W35,I38A[NLSΔ] from coprecipitating ATG7 (Figure 4(e)). Further, in vitro affinity-isolation assays using purified ATG7 and recombinant GST-TP53INP2[NLSΔ], GST-TP53INP2W35,I38A[NLSΔ] or GST-TP53INP2W35,I38A[Δ1–28],[NLSΔ] confirmed that the region containing amino acids 1 to 28 in TP53INP2 is pivotal to TP53INP2-ATG7 interaction (Figure 4(f)). We also mapped the interacting region within ATG7 and found that amino acids 511 to 625 are responsible for TP53INP2 binding (Fig. S3B and S3C). Together, these results suggest that TP53INP2 can form a complex with LC3 and ATG7 in the cytoplasm by directly interacting with the 2 proteins.
TP53INP2 promotes autophagosome formation by enhancing LC3-ATG7 interaction
Given that LC3B-TP53INP2-ATG7 heterocomplex form in cells, we tested whether TP53INP2 can promote LC3-ATG7 interaction. Overexpression of TP53INP2-MYC increased the interaction between transfected Flag-LC3B and endogenous ATG7 both in fed and starved HEK293 cells (Figure 5(a)). In addition, in fed cells, overexpression of TP53INP2[NLSΔ], but not TP53INP2[LIRΔ], TP53INP2W35,I38A[NLSΔ] or TP53INP2[Δ1-28],[NLSΔ] enhanced the interaction (Figure 5(b)). It was previously shown that ATG7 only associates with cytoplasmic deacetylated LC3, so we assessed the role of TP53INP2 in the interaction between ATG7 and a deacetylation-mimetic LC3B mutant in which the lysines at positions 49 and 51 were replaced by arginines [13]. This LC3B with a double K49R K51R mutation (LC3BK49,51R), has a nuclear and cytoplasmic distribution [13]. Immunoprecipitation of cytosolic Flag-LC3BK49,51R coprecipitated much less endogenous ATG7 from TP53INP2-deleted cells (Figure 5(c)). These results suggest that cytoplasmic TP53INP2 has a mediating effect on LC3-ATG7 interaction, and that TP53INP2-LC3 and TP53INP2-ATG7 interactions are required for this mediation. To get more direct evidence, we carried out an in vitro affinity-isolation assay using purified recombinant GST-LC3B[G120]. We found that GST-LC3B[G120] affinity-isolated more ATG7 from the lysate of cells expressing TP53INP2[NLSΔ] than from the lysate of cells without TP53INP2[NLSΔ] expression, or with TP53INP2W35,I38A[NLSΔ] or TP53INP2[Δ1-28],[NLSΔ] expression (Figure 5(d)). In line with this, GST-LC3B[G120] also affinity-isolated more ATG3 from a TP53INP2[NLSΔ]-expressing cell lysate (Fig. S4A). Finally, we examined autophagosome formation in GFP-LC3B-HEK293 cells. Expression of TP53INP2W35,I38A[NLSΔ] or TP53INP2[Δ1-28],[NLSΔ] induced far fewer autophagosomes in these cells than TP53INP2[NLSΔ] or the other truncated mutants containing the LIR and amino acids 1 to 28 (Figure 5(e,f)). In addition, when an exclusively cytoplasm-localized TP53INP2[Δ1-28],[NLSΔ] (RFP-[3NES]-TP53INP2[Δ1-28],[NLSΔ]) was expressed in the cells, it failed to trigger autophagosome formation, even though it was competent to translocate nuclear GFP-LC3B to the cytoplasm (Fig. S4B and S4C). Taken together, these results suggest that TP53INP2 contributes to autophagosome formation in the cytoplasm by facilitating LC3-ATG7 interaction.
Figure 5.
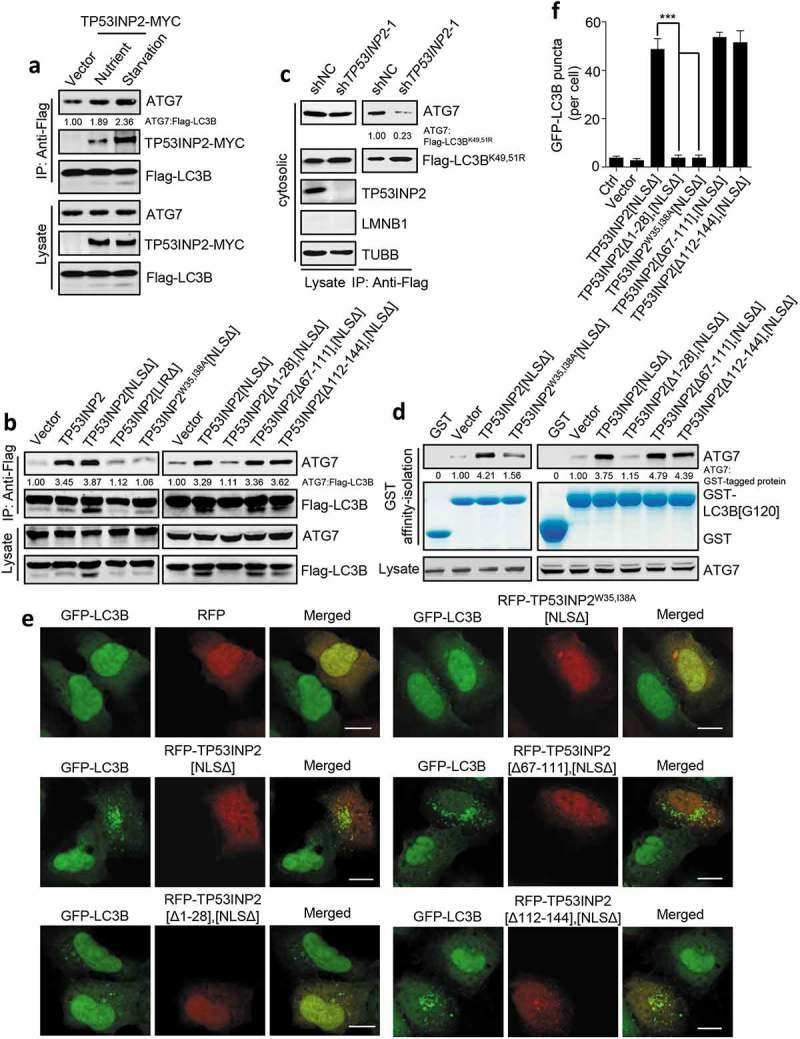
TP53INP2 facilitates LC3B-ATG7 interaction. (a) Coprecipitation of endogenous ATG7 with exogenous Flag-LC3B in TP53INP2-MYC cotransfected HEK293 cells with or without cell starvation. Flag-LC3B was immunoprecipitated using anti-Flag, then ATG7 and TP53INP2-MYC were detected by anti-ATG7 and anti-MYC respectively. (b) Coprecipitation of ATG7 with Flag-LC3B from HEK293 cells transiently expressing RFP-tagged TP53INP2 or each of the indicated TP53INP2 mutants. Flag-LC3B was immunoprecipitated using anti-Flag. (c) HEK293 cells stably expressing non-silencing shRNA or TP53INP2 shRNA were transfected with Flag-LC3BK49,51R and starved. The cells were then fractionated by differential centrifugation. Flag-LC3BK49,51R was immunoprecipitated from the cell cytosol using anti-Flag and the coprecipitated ATG7 was detected by western blot. (d) In vitro affinity-isolation assay of LC3B[G120]-ATG7 interaction. Purified GST-LC3B[G120] was incubated with cell lysate from HEK293 cells expressing the indicated RFP-tagged TP53INP2 mutants. After affinity-isolating GST-LC3B[G120] using glutathione-sepharose 4B beads, GST-LC3B[G120]-bound ATG7 was analyzed by western blot. (e) Confocal images of HEK293 cells stably expressing GFP-LC3B and transfected with plasmids expressing each of the indicated RFP-tagged TP53INP2 truncated mutants. (f) Quantification of GFP-LC3B puncta in (e). The data are presented as mean ± SEM, n = 30 cells. ***, P < 0.001. Scale bars: 10 µm.
Discussion
In this study, we have identified a novel function of TP53INP2 in autophagy by dissecting its action in the cytoplasm. Our results suggest that TP53INP2-LC3 binding not only targets TP53INP2 to autophagic membranes but also promotes autophagosome formation by facilitating LC3-ATG7 interaction.
Due to the predominantly nuclear distribution of TP53INP2, we used truncated nucleus-localized and cytoplasm-localized mutants to allow us to investigate the role of TP53INP2 in both basal autophagy and induced autophagy. Only an increase in the expression of LC3-interacting TP53INP2 in the cytoplasm triggers autophagy, which suggests that TP53INP2 has a specific function in the cytoplasm, in addition to serving as a driver for LC3 efflux from the nucleus [13,22]. This strategy also helps us to deduce that the association of TP53INP2 with autophagic membranes is mediated by LC3, even under basal conditions. This conclusion is supported not only by the use of an LC3B mutant that cannot associate with membranes and TP53INP2 mutants that cannot interact with LC3, but also by the observations in BECN1-KO cells suggesting that the membrane association of TP53INP2 depends on PtdIns3P production on the membranes. On the basis of a coimmunoprecipitation assay, it has previously been proposed that the membrane association of TP53INP2 depends on interaction between TP53INP2 and VMP1; however, this may reflect an indirect interaction of the proteins [15]. Besides, it has been suggested that VMP1 plays a role in autophagosome closure instead of the biogenesis of autophagosomal membranes [19,23].
The finding that TP53INP2 promotes LC3B-ATG7 interaction in cells suggests a potential involvement of TP53INP2 in the ubiquitination-like system for LC3 lipidation, although the main factors in the process have been identified and are well documented [11]. Consistent with the view, a recent study has defined an adaptor protein function of the molecular chaperone CLU, which enhances autophagy in response to cell stress by increasing the stability of the LC3-ATG3 heterocomplex [24,27]. Together, these observations suggest a more complex regulatory mechanism for the lipidation of LC3 in multicellular organisms than that of Atg8 in single-cell yeast. Most LC3-interacting proteins utilize the typical LIR in their structures for LC3-binding. However, the Atg7-Atg8 interaction involves the hydrophobic residues of the non-typical Atg8-interacting motif in the C-terminal of Atg7 [25]. Our results demonstrate a direct interaction between TP53INP2 and ATG7, which suggests that TP53INP2 promotes LC3 lipidation by stabilizing the LC3-ATG7 complex without competing with ATG7 for LC3 binding. In addition, we propose that ATG7 interacts with a TP53INP2-LC3 complex in vivo, because a previous study has demonstrated a TP53INP2-LC3 interaction in the nucleus, which drives the redistribution of the 2 proteins together to the cytoplasm in atg7−/- cells [13]. In addition to TP53INP2 and LC3, ATG7 has also been detected in the nucleus, and it undergoes deacetylation during cell starvation-induced autophagy [26-29]. However, only nuclear-exported LC3 interacts with ATG7 [13], and only cytoplasmic TP53INP2 shows a strong mediatory effect on LC3B-ATG7 interaction (Figure 5). These results indicate that the LC3-TP53INP2-ATG7 complex is formed in the cytoplasm. It is worth noting that although cytoplasmic TP53INP2 facilitates LC3B-ATG7 interaction and the subsequent LC3B–PE conjugation and autophagosome formation in cells, our in vitro ATG7-LC3B binding and LC3B–PE conjugation assays using purified TP53INP2 were unable to confirm that TP53INP2 promoted the formation of the ATG7-LC3B thioester bond and LC3B–PE (data not shown). A possible explanation for this is that TP53INP2 requires an unknown post-translational modification in vivo, because it has been shown that the efflux of TP53INP2 with LC3 from the nucleus is regulated by MTORC1 kinase and SIRT1 deacetylase [13,15–17]. However, identification of such a modification of TP53INP2 awaits future investigations.
Materials and methods
Cell cultures, reagents, and antibodies
HEK293, HEK293T, HeLa cells and MEFs were maintained in Dulbecco Modified Eagle Medium (Gibco, 11965092) supplemented with 10% fetal bovine serum (Gibco, 10091148) at 37°C in 5% CO2. HEK293 cells stably expressing ULK1-GFP, GFP-BECN1, GFP-ZFYVE1, GFP-LC3B or mCherry-LC3B were transfected with corresponding plasmids for 24 h and selected with G418. atg5−/- or atg7−/- MEFs were gifts from Han-Ming Shen (National University of Singapore, Singapore). BECN1-KO HEK293 cells have been described previously [30]. ATG7-KO HEK293 cells were generated by transient transfection of pEP-ATG7-KO plasmid followed by selection with puromycin. Insect Sf9 cells were maintained in SIM SF Medium (Sino Biological Inc., MSF1) with 10% fetal bovine serum at 27°C.
All chemicals were from Sigma. Chloroquine (C6628) was added to nutrient medium or starvation medium for 3 h at 200 µM. G418 (A1720) was added to medium for 7 days at 1mg/ml. Puromycin (P9620) was added to medium for 24 h at 2.5 µg/ml. 3-methyladenine (M9281) was used during starvation at 10 mM.
The following primary antibodies were used: rabbit polyclonal antibodies to LC3B (L7543), ATG7 (A2856) and ATG3 (A3606), mouse monoclonal antibodies to ACTB (A5316) and TUBB (T5293) were from Sigma; rabbit polyclonal antibodies to Flag (0912–1), MYC (R1208-1) and HA (0906–1) were from HuaBio; mouse monoclonal antibodies to Flag (SC-807), MYC (SC-40) and HA (SC-57592) were from Santa Cruz; rabbit polyclonal antibody to GFP (MBL, 598), mouse monoclonal antibody to GFP (MBL, M048-3); mouse monoclonal antibody to LMNB1 (Santa Cruz, SC-20682); rabbit polyclonal antibody to HIST1H3A (Cell Signaling Technology, 9715); rabbit polyclonal antibody to TP53INP2 (LifeSpan BioSciences, C119137); rabbit polyclonal antibody to ATG5 (Novus Biologicals, NB110-53818). The secondary donkey anti-mouse IRDye680 (926–32222) and anti-rabbit IRDye800CW (926–32213) antibodies for western blot were from LI-COR Biosciences.
Plasmid constructs and transfection
GFP-[3NLS]-LC3B, Flag-LC3B, RFP-TP53INP2, and GFP-TP53INP2, GST-TP53INP2, TP53INP2-MYC, and GFP-LC3B, mCherry-LC3B have been described previously [13,17,31]. GFP-TP53INP2[NLSΔ] and GFP-TP53INP2[LIRΔ] were made by cloning the corresponding Trp53inp2 DNA fragments into a pEGFP-C1 vector using EcoRI and KpnI restriction sites. RFP-TP53INP2[NLSΔ] and RFP-TP53INP2[LIRΔ] were made by cloning the corresponding Trp53inp2 DNA fragments into a pmRFP-C1 vector using EcoRI and KpnI restriction sites. TP53INP2[NLSΔ]-MYC and TP53INP2[LIRΔ]-MYC were made by cloning the corresponding Trp53inp2 DNA fragments into a pCDNA3.1-MYC vector using HindIII and KpnI restriction sites. The cDNA sequences coding the 3 repeated NES peptide (CTTGCACTCAAGGCGGGCTTGGATATCCTTGCACTCAAGGCGGGCTTGGATATC-CTTGCACTCAAGGCGGGCTTGGATATC) and the 3 repeated NLS peptide (GATCCAAAAAAGAAGAGAAAGGTAGATCCAAAAAAGAAGAGAAAGGTAGATC-CAAAAAAGAAGAGAAAGGTA) were synthesized, annealed and then inserted into GFP-TP53INP2[NLSΔ] to generate GFP-[3NES]-TP53INP2[NLSΔ] and GFP-[3NLS]-TP53INP2[NLSΔ] separately at XhoI and EcoRI sites. GFP-[3NES] vector and RFP-[3NES] vector were generated by inserting the 3 repeated NES peptide into a pEGFP-C1 vector and a pmRFP-C1 vector at XhoI and EcoRI sites; cloning the corresponding Trp53inp2 DNA fragments into a GFP-[3NES] vector at EcoRI and KpnI sites to get GFP-[3NES]-TP53INP2[1-28] and GFP-[3NES]-TP53INP2[28-45]; cloning the corresponding Trp53inp2 DNA fragments into a RFP-[3NES] vector at EcoRI and KpnI sites to get RFP-[3NES]-TP53INP2[NLSΔ] and RFP-[3NES]-TP53INP2[Δ1-28],[NLSΔ]. GFP-TP53INP2[Δ1-28],[NLSΔ], GFP-TP53INP2[Δ29-66],[NLSΔ], GFP-TP53INP2[Δ67-111],[NLSΔ] and GFP-TP53INP2[Δ112-144],[NLSΔ] were generated by cloning the corresponding Trp53inp2 DNA fragments into a pEGFP-C1 vector at EcoRI and KpnI sites. RFP-TP53INP2[Δ1-28],[NLSΔ], RFP-TP53INP2[Δ67-111],[NLSΔ] and RFP-TP53INP2[Δ112-144],[NLSΔ] were generated by cloning the corresponding Trp53inp2 DNA fragments to a pmRFP-C1 vector using EcoRI and KpnI restriction sites. GST-ATG3 was generated by cloning the human ATG3 protein ORF (GeneBank: NM_022488.4) into a pGEX-5X-1 vector (GE Healthcare, 27–4584-01) using EcoRI and BamHI restriction sites. GST-LC3B[G120] was generated by cloning the corresponding rat Map1lc3b DNA fragment into a pGEX-5X-1 vector at BamHI and EcoRI sites. GST-TP53INP2[NLSΔ] and GST-TP53INP2[Δ1-28],[NLSΔ] were generated by cloning the corresponding Trp53inp2 DNA fragments into a pGEX-4T-1 vector (GE Healthcare, 27–4580-01) at EcoRI and KpnI sites. Site-directed mutagenesis was performed using QuikChange II XL (Stratagene, 210522). GST-SQSTM1 was generated by cloning the human SQSTM1 protein ORF (GeneBank: NM_003900.4) into a pGEX-5X-1 vector using EcoRI and XhoI sites. The ULK1 cDNA was kindly provided by Tamotsu Yoshimori (Graduate School of Medicine, Osaka University, Osaka, Japan), cloned into a pEGFP-N1 vector at EcoRI and BamHI sites to generate ULK1-GFP. pEP-ATG7-KO plasmid was made by cloning the target DNA sequence of human ATG7 (GTAATAACCCTTAATGTCCT) into a pEP-KO-Z1779 vector using SapI. Insect expressing GST-ATG7 was a gift from Brenda A. Schulman (Department of Molecular Machines and Signaling, Max Planck Institute of Biochemistry, Martinsried, Germany). HA-ATG7 was generated by cloning the ATG7 cDNA into a pXF4H vector at NotI and SalI sites. ATG7-Flag, ATG7[1–325]-Flag, ATG7[326–676]-Flag, ATG7[Δ326-510]-Flag, ATG7[Δ511-625]-Flag and ATG7[Δ626-676]-Flag were generated by cloning the corresponding ATG7 DNA fragments into a pCMV-Tag4A vector at NotI and SalI sites. Flag-ATG14, GFP-ATG14 and the BECN1 cDNA were gifts from Qiming Sun (School of Medicine, Zhejiang University, Hangzhou, China). GFP-BECN1 was generated by cloning the BECN1 cDNA into a pEGFP-C1 vector at EcoRI and BamHI sites. WIPI2-GFP was a gift from Hong Zhang (Institute of Biophysics, Chinese Academy of Sciences, Beijing, China). GFP-ZFYVE1 was a gift from Nicholas T. Ktistakis (Babraham Institute, Cambridge, UK). For transient transfection, plasmids were transfected using Lipofectamine 2000 (Invitrogen, 11668019) according to the manufacturer’s instructions.
Autophagy induction
Cells were washed twice with PBS (Sangon Biotech, E607008), followed by addition of starvation medium (20 mM HEPES, 1% BSA [Sigma, A1933], 150 mM NaCl, 5 mM glucose, 1 mM CaCl2, 1 mM MgCl2, pH 7.4) and cultured for 2 h.
RNA interference
HEK293 cells expressing TP53INP2 shRNA were established by using lentivirus-based system. Briefly, scrambled sequence or TP53INP2 targeted sequence 5ʹ-GATCCGTGGACGGCTGGCTCATCATTCAAGAGATGATGAGCCAGCCGTCCACT-TTTTTG-3ʹ (shTP53INP2-1) or 5ʹ-GATCCGCGCCAGTTCAACTACTGATTCAAGAGAT-CAGTAGTTGAACTGGCGCTTTTTTG-3ʹ (shTP53INP2-2) was inserted into a lentivirus-based shuttle vector pGLV2 (GenePharma, C06002) and was cotransfected with pGag/Pol, pRev and pVSV-G into HEK293T cells for 72 h. Then the culture supernatant was collected and the virus particles were acquired. HEK293 cells were infected with the each virus for 72 h, the stable TP53INP2 shRNA-expressing HEK293 cells were selected with puromycin.
Immunostaining and confocal microscopy
HEK293 cells, HeLa cells or MEFs were fixed in 4% formaldehyde for 10 min at room temperature followed by permeabilization and blocking with PBS containing 0.1% saponin (Sigma, S7900) and 10% fetal calf serum (Sigma, F0685) for 20 min. Then the cells were incubated with appropriate primary and secondary antibodies in 0.1% saponin as indicated in the figure legends. The images were obtained in multi-tracking mode on a laser scanning confocal microscope (LSM510 Meta, Carl Zeiss, Oberkochen, Germany) with a 63× plan apochromat 1.4 NA objective.
A total of 30 cells were recorded and analyzed using the Axiovision Automatic measurement program on the Zeiss LSM510 Meta confocal microscope for counting the number of ULK1-GFP, GFP-BECN1, GFP-ZFYVE1, GFP-LC3B and mCherry-LC3B puncta.
Immunoprecipitation and Western Blot
For immunoprecipitation analysis, cells were lysed in NP-40 buffer (50 mM Tris-HCl, 150 mM NaCl, 1% NP-40 [Sangon Biotech, A100109], 10% glycerol, 2 mM EDTA, 1 mM DTT, pH 7.4) supplemented with protease inhibitor cocktail (Roche, 4693159001), then the cell lysate was mixed with antibodies at 4°C for overnight followed by the addition of protein A/G sepharose beads (Thermo Scientific, 20,421). Immunocomplexes were washed 5 times with NP-40 buffer and used to western blot.
Western blots were performed as described previously [32]. Briefly, immunoprecipitates, proteins from lysed cells or purified proteins were denatured and loaded on sodium dodecyl sulfate polyacrylamide gels. They were transferred to polyvinylidene difluoride membrane (PALL, bsp0161). The membrane was incubated in 5% (w/v) BSA and stained with the corresponding primary and secondary antibodies. The specific bands were analyzed by use of an Odyssey® infrared imaging system (Li-Cor Biosciences, Lincoln, NE, USA).
2-step coimmunoprecipitation
HEK293T cells cotransfected with Flag-LC3B, TP53INP2-MYC and HA-ATG7 plasmids were lysed with NP-40 buffer. Then the cell lysate was incubated with a Flag antibody at 4°C overnight followed by addition of protein A/G sepharose beads at 4°C for 4 h. Immunocomplexes were washed 5 times with NP-40 buffer and added Flag peptide (Sigma, F4799) to release the Flag-LC3B precipitates from the beads at room temperature for 30 min. The Flag peptide eluate was blotted for western blot or by addition of an anti-MYC antibody or an anti-HA antibody for immunopreciation of TP53INP2-MYC or HA-ATG7. The immunocomplexes were washed and blotted for western blot.
Cell fractionation
Cells were washed 3 times with cold PBS and scraped into hypotonic lysis buffer (10 mM HEPES, 10 mM KCl, 3 mM MgCl2, 0.5 mM DTT, pH 8.0) supplemented with protease inhibitor cocktail. The cell suspension in hypotonic lysis buffer was incubated on ice for 30 min and Dounce-homogenized (20 to 30 strokes) with a tight-fitting pestle. The homogenate was centrifuged at 500 × g for 5 min to pellet nuclei, the supernatant was used as the cytoplasmic fraction after centrifuged at 15,000 × g for 30 min.
Protein expression and purification
E. coli BL21 cells harboring pGEX-5X-1 encoding the GST-LC3B[G120], GST-ATG3 and GST-SQSTM1 constructs were grown in LB medium containing 50 µg/ml ampicillin (Sigma, A9518) at 37°C to an OD600 of ~ 0.5. The cells were then shaken at 37°C for 3 h in the presence of 0.5 mM isopropyl β-D-thiogalactopyranoside/IPTG (Sigma, PHG0010). To purify GST-TP53INP2, GST-TP53INP2W35,I38A and GST-TP53INP2W35,I38A[Δ1-28], the BL21 strains that carried pGEX-4T-1 encoding the proteins were used and induced at 28°C in the presence of 0.1 mM IPTG for 12 h. The proteins were purified by using glutathione-sepharose 4B beads (GE Healthcare Life Sciences, 17–0756-01), eluted with glutathione (Beyotime, S0073) or incubated with Factor Xa (New England Biolabs, P8010) at 4°C for 4 h to release the proteins from the GST. Then the protein eluate was concentrated with Amicon Ultra-4 filter (Millipore, UFC801024) and glycerol was added to a final concentration of 25% for storage at −80°C.
To purify ATG7, the baculovirus encoding GST-ATG7 was generated using the Bac-to-Bac Baculovirus Expression System (Thermo Scientific, 10359016). Protein expression was carried out in insect Sf9 cells at 27°C for 2.5 days. GST-ATG7 was purified using glutathione-sepharose 4B beads, and ATG7 was acquired by incubating GST-ATG7 with TEV protease (a gift from Qiming Sun, Zhejiang University, Hangzhou, China) at 4°C for 4 h to release the protein from the GST. The protein eluate was appropriately concentrated with Amicon Ultra-4 filter and glycerol was added to a final concentration of 25% for storage at −80°C.
LDH sequestration assay
Autophagic sequestration assay was measured by quantifying autophagosome lactate dehydrogenase activity [33]. Briefly, HEK293 cells were washed with 37°C PBS, addition of 200 µl Trypsin-EDTA to the cells until the cells have detached from the plate. Added 800 µl of 37°C PBS/5%/BSA then harvested the cells by centrifugation at 300 × g for 5 min. Cells were resuspended in 400 µl ice-cold 10% sucrose (Sangon Biotech, A100335) and then electrodisrupted by a single high-voltage pulse (800 V; 25 µF). Dilution of the cell lysate by addition of 400 µl cold phosphate buffer (2 mM DTT, 100 mM sodium monophosphate, 2 mM EDTA, 1.75% sucrose, pH 7.5). Transferred 600 µl of the sample to a new microcentrifuge tube and added 900 µl cold resuspension buffer (1 mM DTT, 1 mM EDTA and 50 mM sodium monophosphate) with 0.1% Tween-20 (Sangon Biotech, A100777) and 0.5% BSA then centrifuged for 50 min at 18,000 × g. The pellet and the remaining total-cell lysate (200 µl) were freeze-thawed for a cycle at −80°C. Then the pellet was dissolved in 400 µl resuspension buffer containing 1% Triton X-405 (Sangon Biotech, C604449), while the total-cell lysate was diluted with 300 µl resuspension buffer containing 1.5% Triton X-405, and incubated at 4°C for 30 min followed by a short centrifugation to remove cell debris (18,000 × g, 5 min). According to the manufacturer’s instructions, LDH Activity Kit (Sigma, MAK066) was used to measure the LDH activity. The amount of LDH sedimenting with the cell corpse pellet (sample value divided by 1.125) relative to the amount in the total cell lysate (sample value multiplied by 2.5) was expressed as ‘sequestered LDH (% of total)’ in the figure.
In vitro affinity-isolation assay
For the GST-TP53INP2 affinity-isolation assay, purified GST, GST-TP53INP2, GST-TP53INP2W35,I38A, GST-TP53INP2[NLSΔ], GST-TP53INP2W35,I38A[NLSΔ], GST-TP53INP2W35,I38A[Δ1-28],[NLSΔ] or GST-SQSTM1 proteins were incubated with purified ATG7, LC3B[G120] or ATG3 for 4 h at 4°C followed by addition of the glutathione-sepharose 4B beads to the mixture and further incubation for 2 h at 4°C. Immunocomplexes were washed and used for western blot. Protein bands were detected with Coomassie Brilliant Blue and quantified using the ImageJ software.
For the GST-LC3B[G120] affinity-isolation assay, HEK293 cells expressing TP53INP2[NLSΔ], TP53INP2[Δ1-28],[NLSΔ] TP53INP2W35,I38A[NLSΔ], TP53INP2[Δ67-111],[NLSΔ] or TP53INP2[Δ112-144],[NLSΔ] were lysed and the cell lysate was incubated with purified GST or GST-LC3B[G120] proteins at 4°C for 4 h. Then the glutathione-sepharose 4B beads were added to the mixture followed by incubation at 4°C for 2 h. Immunocomplexes were washed and used for western blot. Protein bands were detected with Coomassie Brilliant Blue and quantified using the ImageJ software.
Statistical analysis
All the statistical data are presented as mean ± SEM. Statistical significance of the differences was determined using the Student t test. P< 0.05 was considered statistically significant.
Funding Statement
This work was supported by the Ministry of Science and Technology of the People’s Republic of China [grant number 2017YFA0503402] and the National Natural Science Foundation of China [grant number 31790402, 31671434, and 31530040].
Acknowledgments
We are grateful to the Imaging Center of Zhejiang University School of Medicine for their assistance in confocal microscopy. We thank Brenda A. Schulman (Department of Molecular Machines and Signaling, Max Planck Institute of Biochemistry, Martinsried, Germany), Qiming Sun (School of Medicine, Zhejiang University, Hangzhou, China), Hong Zhang (Institute of Biophysics, Chinese Academy of Sciences, Beijing, China), Nicholas T. Ktistakis (Babraham Institute, Cambridge, UK) for providing GST-ATG7, and GFP-ATG14, Flag-ATG14, the BECN1 cDNA, and WIPI2-GFP, and GFP-ZFYVE1 respectively. We thank Tamotsu Yoshimori (Graduate School of Medicine, Osaka University, Osaka, Japan) for providing the ULK1 cDNA.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
The supplementary materials for this article can be accessed here.
References
- [1].Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007. November;8(11):931–937. [DOI] [PubMed] [Google Scholar]
- [2].Mizushima N. Autophagy: process and function. Genes Dev. 2007. November 15;21(22):2861–2873. [DOI] [PubMed] [Google Scholar]
- [3].He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ohsumi Y. Historical landmarks of autophagy research. Cell Res. 2014. January;24(1):9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000. November 1;19(21):5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Xie Z, Nair U, Klionsky DJ. Atg8 controls phagophore expansion during autophagosome formation. Mol Biol Cell. 2008. August;19(8):3290–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007. July 13;130(1):165–178. [DOI] [PubMed] [Google Scholar]
- [8].Weidberg H, Shvets E, Shpilka T, et al. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. Embo J. 2010. June 2;29(11):1792–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Weidberg H, Shpilka T, Shvets E, et al. LC3 and GATE-16 N termini mediate membrane fusion processes required for autophagosome biogenesis. Dev Cell. 2011. April 19;20(4):444–454. [DOI] [PubMed] [Google Scholar]
- [10].Kabeya Y, Mizushima N, Yamamoto A, et al. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004. June 1;117(Pt 13):2805–2812. [DOI] [PubMed] [Google Scholar]
- [11].Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008. 9 Sep(9):859–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wild P, McEwan DG, Dikic I. The LC3 interactome at a glance. J Cell Sci. 2014. January 1;127(Pt 1):3–9. [DOI] [PubMed] [Google Scholar]
- [13].Huang R, Xu Y, Wan W, et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell. 2015. February 5;57(3):456–466. [DOI] [PubMed] [Google Scholar]
- [14].Baumgartner BG, Orpinell M, Duran J, et al. Identification of a novel modulator of thyroid hormone receptor-mediated action. PLoS One. 2007. November 21;2(11):e1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nowak J, Archange C, Tardivel-Lacombe J, et al. The TP53INP2 protein is required for autophagy in mammalian cells. Mol Biol Cell. 2009. February;20(3):870–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mauvezin C, Orpinell M, Francis VA, et al. The nuclear cofactor DOR regulates autophagy in mammalian and Drosophila cells. EMBO Rep. 2010. January;11(1):37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Xu Y, Wan W, Shou X, et al. TP53INP2/DOR, a mediator of cell autophagy, promotes rDNA transcription via facilitating the assembly of the POLR1/RNA polymerase I preinitiation complex at rDNA promoters. Autophagy. 2016. July 2;12(7):1118–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ropolo A, Grasso D, Pardo R, et al. The pancreatitis-induced vacuole membrane protein 1 triggers autophagy in mammalian cells. J Biol Chem. 2007. December 21;282(51):37124–37133. [DOI] [PubMed] [Google Scholar]
- [19].Tian Y, Li Z, Hu W, et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell. 2010. June 11;141(6):1042–1055. [DOI] [PubMed] [Google Scholar]
- [20].Zhao YG, Chen Y, Miao G, et al. The ER-localized transmembrane protein EPG-3/VMP1 regulates SERCA activity to control ER-isolation membrane contacts for autophagosome formation. Mol Cell. 2017. September 21;67(6):974–989.e6. [DOI] [PubMed] [Google Scholar]
- [21].Sancho A, Duran J Garcia-Espana A, et al. Dor/tp53inp2 and tp53inp1 constitute a metazoan gene family encoding dual regulatorsof autophagy and transcription. Plos One. 2012;7(3):e34034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Liu X, Klionsky DJ. TP53INP2/DOR protein chaperones deacetylated nuclear LC3 to the cytoplasm to promote macroautophagy. Autophagy. 2015;11(9):1441–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian atg proteins. 770 Autophagy. 2010. August;6(6):764–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang F, Kumano M Beraldi E, et al. Clusterin facilitates stress-induced lipidation of lc3 and autophagosome biogenesis to enhance cancer cell survival. Nat Commun. 2014. December 12;5:5775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Noda NN, Satoo K Fujioka Y, et al. Structural basis of atg8 activation by a homodimeric e1, atg7. Mol Cell. 2011. November 4;44(3):462–475. [DOI] [PubMed] [Google Scholar]
- [26].Lee IH, Cao L Mostoslavsky R, et al. A role for the nad-dependent deacetylase sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S a. 2008. March 4;105(9):3374–3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lee IH, Kawai Y Fergusson MM, et al. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science. 2012. April 13;336(6078):225–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lee IH, Finkel T. Regulation of autophagy by the p300 acetyltransferase. J Biol Chem. 2009. March 6;284(10):6322–6328. doi: 10.1074/jbc.M807135200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sebti S, Prebois C Perez-Gracia E, et al. Bat3 modulates p300-dependent acetylation of p53 and autophagy-related protein 7 (atg7) during autophagy. Proc Natl Acad Sci U S a. 2014. March 18;111(11):4115–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Su H, Yang F Wang Q, et al. Vps34 acetylation controls its lipid kinase activity and the initiation of canonical and non-canonical autophagy. Mol Cell. 2017. September 21;67(6):907–921.e7. [DOI] [PubMed] [Google Scholar]
- [31].Guo Y, Chang C Huang R, et al. Ap1 is essential for generation of autophagosome from the trans-golgi network. J Cell Sci. 2012. April 1;125(Pt 7):1706–1715. [DOI] [PubMed] [Google Scholar]
- [32].Wan W, You Z, Xu Y, et al. mTORC1 phosphorylates acetyltransferase p300 to regulate autophagy and lipogenesis. Mol Cell. 2017. October 19;68(2):323–335.e6. [DOI] [PubMed] [Google Scholar]
- [33].Luhr M, Szalai P, Saetre F, et al. A simple cargo sequestration assay for quantitative measurement of nonselective autophagy in cultured cells. Methods Enzymol. 2017;587:351–364. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.