

Summary
Many neurodegenerative diseases are caused by unstable trinucleotide repeat (TNR) expansions located in disease-associated genes. siRNAs based on CAG repeat expansions effectively kill cancer cell lines in vitro through RNA interference (RNAi). They also cause significant reduction in tumor growth in a human ovarian cancer mouse model with no toxicity to the treated mice. This suggests that cancer cells are particularly sensitive to CAG TNR-derived siRNAs and explains a reported inverse correlation between the length of CAG TNRs and reduced global cancer incidences in some CAG TNR diseases. This review discusses both mutant proteins and mutant RNAs as a cause of TNR diseases with a focus on RNAi and its role in contributing to disease pathology and in suppressing cancer.
Keywords: TNR, cancer, RNAi, Huntington’s disease, SBMA, AR
The TNR Diseases
Trinucleotide repeat (TNR) expansions in a number of genes are the cause of many degenerative diseases characterized by amplification of DNA triplet motifs [1]. The following triplets (when present on the coding strand) have been associated with neurodegenerative syndromes: CAG, CTG, GAA, CCG, and CGG. Most of them exhibit a negative correlation between the repeat length and the onset of the diseases and/or the severity of disease progression [2]. However, the mechanism of how the TNRs cause disease is quite diverse and is linked to the location of the TNRs within the affected genes. Traditionally, the diseases have been viewed as either caused by extended toxic protein stretches, toxic repeat RNAs, or by deregulated gene expression (Table 1). The most frequently amplified triplet is CAG. It has been implicated in Dentatorubral-pallidoluysian atrophy (DRPLA) [3]. Spinal and Bulbar Muscular Atrophy (SBMA) [4], and in Huntington’s disease (HD) [5]. In addition, a number of Spinocerebellar Ataxias (SCAs) exhibit elongated CAG repeats. They include SCA1 [6], SCA2 [7], SCA3 [8], SCA6 [9], SCA7 [10], SCA12 [11], and SCA17 [12]. Diseases caused by CTG-based TNRs overall are less frequent and include spinocerebellar ataxias type 8 (SCA8) [13]. Huntington disease-like 2 (HDL2) [14], and myotonic dystrophy type 1 (DM1). Even fewer diseases are caused by the expansion of the repeats GAA, CGG, and CCG. GAA-repeat expansion is associated with Friedreich’s ataxia (FRDA) [15]. CGG repeats underlie fragile X syndrome (FXS), fragile X-associated primary ovarian insufficiency (FXPOI) [16], and fragile X-associated tremor (FXTAS) [17], and CCG repeats cause fragile XE syndrome [18]. While this review focuses on TNRs, there are additional repeat diseases that have been identified with other repeat sizes: tetra-, penta-, or hexamers and even longer repeats [19]. They are present in myotonic dystrophy type 2 (DM2) (CCTG) [20, 21], SCA31 (TGGAA) [22], amyotrophic laterals sclerosis (ALS) and/or frontotemporal dementia (GGGGCC) [23, 24]. Detailed descriptions of disease etiology and neurological symptoms have been extensively reviewed elsewhere [2, 25]. In this review we will discuss the molecular causes of disease pathology as well as the connections between TNR diseases and cancer with a focus on RNAi.
Table 1:
Trinucleotide Repeat Expansion Diseases
| Disease | Full disease name | Gene name | TNR | AA coded for | Functional disease group | Alternative mechanism | Chr band | Chr location (Homo sapiens Annotation Release 108, GRCh38.p7) | Location within gene | Normal repeat length | Intermediate repeat length | Mutant repeat length | Inheritance | Estimated frequency / Prevalence | Ref |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SCA1 | Spinocerebellar ataxia type 1; olivopontocerebellar atrophy I | ATXN1/ATX1/SCA1 | CAG | Glu | Toxic protein | 6p22.3 | chr6:16,299,112 to 16,761,490 | Exon-7 | 4 to 39 | 36 to 38 | 39 to 44 | Autosomal dominant | 1 to 2:100,000 | 6 | |
| SCA2 | Spinocerebellar ataxia type 2 | ATXN2 | CAG | Glu | Toxic protein | 12q24.12 | chr12:111,452,214 to 111,599,676 | Exon-1 | ≤ 31 | 32 | ≥ 33-200 | Autosomal dominant | 40:100,000 | 7 | |
| SCA3/MJD/MJD1 | Spinocerebellar ataxia type 3; Machado-Joseph disease; Azorean disease | ATXN3/AT3 | CAG | Glu | Toxic protein | Toxic RNA | 14q32.12 | chr14:92,058,552 to 92,106,621 | Exon-10 | 12 to 43 | 45 to 60 | 60 to 87 | Autosomal dominant | unknown | 8 |
| SCA6 | Spinocerebellar ataxia type 6 | CACNA1A/APCA | CAG | Glu | Toxic protein | 19p13.13 | chr19:13,206,442 to 13,506,460 | Exon-47 | 4 to 18 | 19 | 20 to 33 | Autosomal dominant | 1:100,000 | 9 | |
| SCA7/ADCAII/OPCA3 | Spinocerebellar ataxia type 7 | ATXN7 | CAG | Glu | Toxic protein | 3p13.1 | chr3:63,864,557 to 64,003,462 | Exon-1 | 4 to 19 | 28-36 | > 36-460 | Autosomal dominant | <1:100,000 | 10 | |
| SCA17 | Spinocerebellar ataxia type 17 | TBP | CAG | Glu | Toxic protein | 6q27 | chr6:170,554,333-170,572,869 | Exon-3 | 25 to 42 | 41 to 48 | 43 to 66 | Autosomal dominant | 0.47:1,000,000. to 0.16:100,000 | 12 | |
| DRPLA | Dentatorubral-pallidoluysian atrophy; Naito Oyanagi disease | ATN1/DRLPA | CAG | Glu | Toxic protein | 12p13.31 | chr12:6,924,463 to 6,942,321 | Exon-5 | 6 to 35 | unknown | ≥ 48-93 | Autosomal dominant | 0.48:100,000 (in Japan) | 147 | |
| SBMA | X-linked Spinal and bulbar muscular atrophy; Kennedy’s disease | AR | CAG | Glu | Toxic protein | Toxic RNA | Xq12 | chrX:67,544,032 to 67,730,619 | Exon-1 | 7 to 34 | 36 to 37 | 38 to 68 | X-Linked Recessive | 1:150,000 | 4 |
| HD | Huntington’s disease | HTT/IT15 | CAG | Glu | Toxic protein | Toxic RNA; siRNAs, RAN translation/poly Ala (S), poly Ser (S), poly Leu (AS), poly Cys (AS) | 4p16.3 | chr4:3,074,681-3,243,959 | Exon-1 | ≤ 26 | 27 to 35 | >36 | Autosomal dominant | 3 to 7:100,000 | 5 |
| SCA8 | Spinocerebellar ataxia type 8 | ATXN8/ATXN8OS/(overlapping genes) | CTG (CAG repeat in ATX8) | n.a. | Toxic RNA | RAN translation/poly Ala (AS) | 13q21/13q21.33 | chr13:70,107,213-70,139,429 / ? | 3’UTR | 15 to 50 | 50 to 70 maybe | 80 to 1300 | Autosomal dominant | <1:100,000 | 13 |
| SCA12 | Spinocerebellar ataxia type 12 | PPP2R2B | CAG | n.a. | Toxic RNA suspected | In vitro data: Increased gene expression | 5q32 | chr5:146,589,504 to 147,081,520 | 5’UTR | 4 to 32 | unknown | 55 to 78 | Autosomal dominant | rare, mostly found in Northern India | 11 |
| DM1/MD1 | Myotonic dystrophy type 1; Steinert’s Disease | DMPK | CTG | n.a. | Toxic RNA | siRNAs; RAN translation/poly Glu (AS) | 19q13.32 | chr19:45,769,709 to 45,782,557 | 3’UTR | 5 to 34 | 35 to 49 | > 50-1000 | Autosomal dominant | 1:10,000 - 1:100,000 | 21 |
| HDL2 | Huntington disease-like 2 | JPH3 | CTG | n.a. | Toxic RNA | 16q24.2 | chr16:87,602,905-87,698,156 | 3’UTR; alternatively spliced exon-2A | 6 to 28 | 29 to 39 | 41-58 | Autosomal dominant | rare | 14 | |
| FXTAS | Fragile X-associated tremor/ataxia syndrome | FMR1 | CGG | n.a. | Toxic RNA | RAN translation/poly Gly (S) | Xq27.3 | chrX:147,912,052-147,951,125 | 5’UTR | 5 to 55 | NA | 55 to 200 | X-Linked | 1:4000 (males); 1:7800 (females) | 84 |
| FRAXA/FXS | Fragile X syndrome | FMR1 | CGG | n.a. | Loss of function/gene silencing | Xq27.3 | chrX:147,912,052-147,951,125 | 5’UTR | 5 to 55 | NA | > 200 | X-Linked | 1:3600-4000 (males); 1:40006000 (females) | 85 | |
| FRDA | Friedreich’s ataxia | FXN/X25 | GAA | n.a. | Loss of function | 9p | chr9:69,035,259-69,074,213 | Intron-1 | 5 to 33 | 34 to 65 | 66 to ~1300 | Autosomal recessive | 1:50,000 | 15 | |
| FRAXE | Fragile XE syndrome | FMR2/AFF2 | CCG | n.a. | Loss of function/gene silencing | Xq28 | chrX:148,500,619-149,000,663 | 5’UTR | <50 | 5-200 | >200 | X-Linked | 1:50,000 to 1:100,000 | 81, 148 | |
The Mechanisms of how TNRs Cause Disease
TNR diseases have been classified according to the mechanism causing pathology, which is directly connected to the location of the TNRs in the affected gene [25]. Originally, most TNRs located in open reading frames (ORFs) were believed to cause disease through translation of stretches of poly amino acids to produce toxic proteins. In contrast, TNRs located in UTRs may cause pathology through toxic RNAs. In addition to these gain-of-function mechanisms, loss-of-function TNR diseases are believed to result from insertion of TNRs in either the 5’UTRs or introns of genes. We present the TNRs sorted according to these three groups and discuss, in each case, examples of multiple mechanisms that contribute to individual diseases as well as highlight alternative mechanisms.
The Contribution of Toxic Proteins to Disease Pathology
Two of the first diseases identified associated with a TNR expansion had CAG expansions: SBMA [4] and HD [5].
SBMA is caused by an extended CAG repeat present in exon 1b of the androgen receptor (AR). It is an X-linked sex-limited recessive adult-onset neurodegenerative disorder that involves the death of the spinal and bulbar motor neurons and dorsal root ganglia [26, 27]. The disease is likely caused by the fact that the normal AR gene is expressed in the brain, as shown for mouse embryonic development [28]. Patients’ age at the time of disease onset correlates negatively with the length of the CAG repeat [29]. The mean length of the CAG repeat in the AR gene in different SBMA patient populations around the world is between 44 and 48 with repeat lengths of up to 68, whereas the normal length is between 9 and 34 [27, 30].
HD is marked by progressive degeneration of neurons [1, 31]. It is caused by expansion of a CAG repeat in the huntingtin (HTT) gene. HTT is expressed in all mammalian cells, is highest in brain and testes, and deletion of HTT is embryonically lethal [32–34]. Anyone who inherits the expanded CAG TNR in the mutant (m)HTT gene in the full penetrance range (>39 repeats) will develop the disease. Similar to SBMA the length of the CAG amplifications determines the severity and age of onset of the disease [31]. Symptoms most often start between 30 to 50 years, and worsen over a 10 to 25-year period. Patients develop symptoms when their HTT gene contains more than 27 CAG repeats. Gene silencing experiments in mouse models have shown that when the expression of mutant (m)HTT is reduced, symptoms improve [35].
It was shown that the polyQ expansions in AR and HTT are toxic to cells [2, 4, 36]. The discovery of additional genes with expanded CAG repeats also supported the assumption that repeats present in exons are eventually translated into polyQ stretches, which subsequently cause toxicity and neuronal tissue degradation. This was shown for SCA1 [6]; SCA2 [7], SCA3 [8], and DRPLA [3].
A connection between mutant protein and disease progression may be best supported by data on mutant ATXN1—the CAG containing gene that causes SCA1. SCA1 was described as a polyQ disorder with a gain-of-function mechanism that involves nuclear aggregation of ATXN1 [2]. Resultant phenotypes in different transgenic mice suggested that the nuclear localization of the ATXN1 protein was required to initiate pathogenesis, whereas aggregation of ATXN1 was not [37].
Different mechanisms were proposed to explain the toxicity of mutant proteins containing extended polyQ stretches. It was suggested that mHTT contributes to HD pathogenesis by promoting the fibrillogenesis of wild-type (wt) HTT, which causes its aggregation [38]. In HD, considerable toxic activity seems to come from a proteolytic polyQ fragment of HTT [39]. Recently, disruption of the nuclear pore complex inhibiting nucleocytoplasmic transport was identified as a central component of HD [40, 41]. Most recently it was shown for SCA1 that an ATXN1-cognate partner capicua (CIC) complex is a primary driver of disease pathology through a gain-of-function mechanism [42]. In addition, in SCA1, phosphorylation of ATXN1 at Ser776 plays a role in the pathogenesis. Replacing Ser776 with a phospho-mimicking Asp converted ATXN1 with a wt glutamine tract into a pathogenic protein when expressed as a transgene in mice [43]. Subsequently, however, it was determined that while the Ser776 phosphorylation is important for neuronal dysfunction, it is not involved in neuronal death [44]. This suggests that while the protein context matters in the case of SCA1, it appears that different mechanistic aspects of the disease can have different causes. In summary, while a large number of data suggest that disease pathology in CAG TNR diseases might be caused by aggregation of mutant protein [45], there are conflicting data with this hypothesis [37, 46–49]. In most studies, involvement of mutant RNA could not completely be ruled out unless CAG TNRs were replaced with glutamine-coding CAA repeats and this has rarely been done.
The Contribution of Toxic RNA to Disease Pathology
While there is clear evidence for protein toxicity in TNR diseases, a number of observations suggest a role for RNA in the disease process [1, 50]. A shift from studying protein to RNA began with the recognition that many of the diseases-causing TNRs were located, not in the coding region of a gene, but in regions not expected to be translated [2].
The most widely studied disease caused by toxic repeat RNAs is DM1 [21], the most common muscular dystrophy presenting in adults. Distal skeletal muscles are typically involved and muscle atrophy occurs preferentially in type 1 fibers. In DM1, CTG TNRs are located in the 3’UTR of the myotonic dystrophy protein kinase (DMPK). Unaffected individuals carry less than 50 CTG TNRs in this gene and expansions of up to more than 1000 CTGs were reported in DM1 patients. A major breakthrough on how CUG repeats located in the 3’UTR of a gene could be toxic came in 1995 with the observation of nuclear foci containing the DMPK transcript in mutant cells [51]. The subsequent identification that RNA-binding proteins are present in nuclear foci [52, 53] and interact with the expanded repeat spurred a new hypothesis that repeat expansion leads to a toxic gain-of-function mediated through RNA. Such toxic RNA was found to sequester muscleblind like splicing regulator 1 (MBNL1) [54–56], which regulates alternative splicing, antagonizing the CUG binding protein 1 (CUG-BP1/CELF1) [57, 58]. The mechanism of sequestering RNA binding proteins has been shown for CUG in DM1 [56]. CAG in HD [59], and SCA3 [60]. A new mechanistic twist to RNA-induced toxicity in TNR diseases, including HD, was demonstrated in a recent report that such RNAs can undergo a sol-gel phase transition that contributes to nuclear RNA aggregate formation [61]. This suggests that TNR diseases with toxic polyQ proteins can also have a RNA component.
For SBMA, the contribution of the mutant AR protein to disease progression is well established [62]. Increasing the CAG repeat length in AR results in reduced transactivation activity of AR and promotes its degradation [63, 64]. However, disease progression seems to be regulated by androgens. Removal of testosterone from young mutant transgenic male mice blocked development of disease [65], while in older mutant males castration restored normal function [66]. Correspondingly, female mice carrying a mutant AR gene developed disease upon administration of testosterone [65]. Recently, expression of polyglutamine expanded AR in a mouse model of SBMA was shown to upregulate the gene encoding calcitonin gene-related peptide (CGRP1) [67]. CGRP1 expression was found to be high in motor neurons of patients with SBMA providing an explanation for how AR protein contributes to this disease. However, even though these data provide strong support for the contribution of the AR protein to the disease pathology, there is also room for a RNA contribution to the disease. The first evidence arose during generation of a mouse model for SBMA. After a number of unsuccessful attempts to model the disease in mice [68, 69] and the generation of transgenic mice that showed the expected pathology [70–72], a mouse model mimicking the pathology seen in SBMA patients was created by introducing a transgene of 239 CAG repeats driven by the AR promoter with no other AR sequences [73]. The mice showed disease pathology mimicking aspects of SBMA suggesting that androgen-regulated AR protein may not be the only factor involved in the etiology of SBMA. It needs to be noted that for unknown reasons in this and many other mouse models of TNR diseases disease causing repeat lengths are much larger than the alleles typically observed in affected humans. There is an alternative explanation for how AR stimulation could be driving disease without necessarily requiring AR protein. Androgen has been shown to induce upregulation of AR in a number of normal tissues such as bone and neuronal cells [74, 75]. While in many situations androgen represses its own expression, in certain disease relevant tissues androgen may drive upregulation of AR mRNA thus promoting the disease both at the protein and the RNA level. While there is strong evidence that mutant AR protein contributes to the neurodegenerative disease, in particular the last finding suggests that RNA-based toxicity can contribute to disease pathology.
Evidence to support a role for RNA in TNR expansion toxicity also comes from other model organisms. It was shown in Drosophila that the toxicity of the CAG repeat disease gene for SCA3, ATXN3, is in large part due to the TNR RNA and not the polyQ protein [76]. Replacing some of the glutamine coding CAG repeats with the other codon coding for glutamine, CAA, mitigated the toxicity despite similar polyQ protein expression levels. A direct toxicity of mRNAs with extended CAG repeats was also demonstrated in mice [77].
While the location of TNR expansions in untranslated gene regions points at the RNA being toxic, one has to recognize that repeat-associated, non-ATG translation (RAN translation) was discovered as another translation-level pathogenic mechanism of CAG repeat-containing mRNAs in a number of TNR diseases giving rise to multiple poly amino acid stretches [78] (Table 1). These peptides are the result of bidirectional transcription and translation from all three reading frames of a repeat without the need of an AUG start codon.
Loss-of-function mechanisms
In addition to localizing to the ORF or the 3’UTR of mutant genes, TNRs are also found in the 5’UTRs or in introns. In 1996, a disease-associated gene was identified containing an intronic GAA repeat expansion in Friedreich’s ataxia patients [15]. This TNR results in loss-of-function as the affected gene FXN is no longer expressed due to formation of a stable triple helix at the gene locus that blocks transcription [79]. FRAXA (CGG TNR), and FRAXE (CCG TNR) are both caused by TNRs present in their 5’UTRs. In both cases this insertion results in epigenetic silencing of the promoter reducing protein expression [80, 81]. TNR pathology can also be caused by increasing gene expression. This is found in SCA12 [11]. In this disease a CAG TNR present in the 5’UTR of the PPP2RE2B phosphatase resulting in increased gene expression, at least in vitro [82].
One of the most interesting cases, mechanistically, is the situation found with the CGG TNR inserted into the 5’UTR of the FMR1 gene. Depending on the repeat length within this gene, three different diseases can arise [83]. The normal repeat length is <55 CGGs. At a repeat length between 55 and 200 (the premutation) elderly carriers can develop the progressive neurodegenerative disorder fragile X-associated tremor/ataxia syndrome (FXTAS) caused by toxic RNA. [84]. Females who carry the premutation are also at risk for FXPOI [16]. However, a repeat size above 200 CGGs results in fragile X syndrome (FXS/FRAXA) [85], and this is caused by silencing of the FMR1 gene through methylation of the repeat and the promoter region. Taken together, the data suggests that just because a TNR is located in specific sites in a gene does not allow one to predict whether pathology arises from protein or RNA components.
Involvement of RNAi in TNR Diseases
RNA toxicity in TNR diseases is thought to be mostly mediated through the formation of hairpin structures [86]. While such hairpins have been posited to be toxic through the recruitment of RNA-binding proteins to nuclear RNA foci [54], they can also be processed into small interfering RNAs [87]. The RNA interference (RNAi) machinery converts double-stranded RNAs into small RNAs that negatively regulate gene expression [88, 89]. Endogenous small RNAi-active RNAs are micro(mi)RNAs, which are small 19–22 nucleotide long noncoding RNAs generated in the nucleus from longer precursor primary miRNAs. These primary miRNAs are processed by the microprocessor complex Drosha/DGCR8 and exported into the cytosol where they are further trimmed by Dicer/TRBP. Mature miRNAs are then loaded into the RNA-induced silencing complex (RISC) [90, 91]. Once in the RISC, the single-stranded miRNA guide strand then regulates expression of certain genes by targeting in most cases the 3’UTR, resulting in either degradation of the mRNA or translational silencing through various well-studied mechanisms [92, 93]. The extent of reverse complementarity determines the level of interaction between a miRNA and its mRNA targets. Small interfering RNAs (siRNAs) act through a similar mechanism as miRNAs. However, they are usually designed by investigators to be fully complementary to the targeted gene and result in degradation of the targeted mRNA.
There is convincing evidence that CAG/CUG TNRs can give rise to RNAi-active small RNAs. In human neuronal cells, expression of the CAG expanded exon 1 of HTT (above the threshold for complete penetrance which is >40) [77] caused an increase in small CAG repeat-derived RNAs (sCAG) of about 21 nt in length. Above a certain length, CAG/CUG repeats were found to be cleaved by Dicer and to have RNAi activity. For a TNR sequence to be cleaved by Dicer it must form a hairpin stem structure that resembles a miRNA. TNRs of the CNG type (CAG, CUG, CCG and CGG) have been shown to be able to form stable hairpins that can be cleaved by Dicer [87]. CNGs of 17 repeats in length were shown not to be cleaved. However, CNG repeats as short as 25 repeats were Dicer substrates [87]. Interestingly, both CAG and CUG repeat structures were found to be much more susceptible to Dicer cleavage than those composed of CCG or CGG repeats. In the context of entire mRNAs, 21 to 26 nt long Dicer cleavage products were produced from SCA1 transcripts with 53 CAG repeats, or HTT transcripts with 21 to 44 CAG repeats. In contrast, DMPK transcripts with 15 CUG repeats (normal) were not processed by Dicer, yet transcripts with 80 CUG repeats (disease) were [87]. Hence, Dicer activity controls the levels of the mutant transcripts, and at the same time produces small RNAi-active CAG/CUG-derived double stranded RNAs. One study identified sCAGs as a disease-causing agent. sCAGs, isolated from HD human brains, when transfected into neurons reduced viability [50], suggesting that these sequences affect cell viability through RNAi by targeting genes that regulate cell survival. RNAi-active sCAG RNAs were also isolated from a R6/2 HD mouse model that expresses human HTT with 90 CAG repeats and were shown to kill neuronal cell lines after transfection [94].
Compelling evidence of sCAG RNAs contributing substantially to disease pathology came from studies that used locked nucleic acid–modified antisense oligonucleotides complementary to the CAG repeat (LNA-CTG), an inhibitor that unlike siRNAs or conventional antisense oligonucleotides, blocks the activity of sCAGs without reducing the level of either HTT mRNA or protein [94]. In the R6/2 HD mouse model, treatment with LNA-CTG produced a rapid and sustained improvement of motor deficits.
Direct evidence for involvement of RNAi in the toxicity of TNR-based small RNAs was provided in a study that demonstrated that pathogenic ATXN3 with amplified CAG repeats showed strongly enhanced toxicity in HeLa cells after knockdown of Dicer [95]. In addition, it was previously shown in Drosophila that impairing miRNA processing dramatically enhanced neurodegeneration caused by the CAG repeat gene ATXN3 [96]. This data can be interpreted as sCAGs getting better access to the RISC in the absence of most miRNAs. Indeed, these authors concluded that miRNA pathways normally play a protective role on TNR induced pathology.
We recently screened the toxicity of all possible TNR-derived siRNAs on two cancer cell lines (one human and one mouse) [97]. We found that siRNA based on the members of one family of TNRs (three frames: CAG, AGC, GCA and their reverse complements, CUG, UGC, and GCU) effectively killed all transfected cancer cells in quantities as low as 10 pM [97]. A hybrid duplex between the CAG and CUG based disease molecules was also previously tested in a well-established Drosophila model of DM1 [98]. Expression of the two transcripts led to the generation of Dicer-2 (Dcr-2) and Argonaute-2-dependent 21-nt TNR-derived siRNAs resulting in high toxicity to the cells. In a separate study, it was shown that expression of these complementary repeat RNAs leads to Dcr-2-dependent neurodegeneration [99]. These results suggest that co-expression of CAG and CUG repeat-derived sequences may dramatically enhance toxicity in human repeat expansion diseases through RNAi in which anti-sense transcription occurs. Antisense transcription that has the potential to create CAG/CUG repeat duplexes occurs in SCA8 in two genes that overlap on these TNRs: ATXN8 (CAG repeat), on the sense strand and ATXN8OS (CTG repeat) on the antisense strand [100].
TNR Diseases and Cancer
The majority of the TNR diseases belong to the CAG/CUG family [101] (Table 1). For most of the diseases, a connection to cancer has not been established, likely due to the fact that patient numbers in many of these diseases are too small to correlate disease genotype with cancer incidences. However, for a number of the CAG/CUG TNR diseases such clinical analyses could be performed.
Patient populations in Sweden with CAG-based TNR diseases had an overall reduced risk of cancer (1510 patients with HD, 471 with SBMA, and 3425 with HA; standardized incidence ratios (SIR) were 0.47 (95% confidence interval (CI) 0.38–0.58), 0.65 (0.45–0.91), and 0.77 (0.70–0.85), respectively). This affected cancer of the digestive tract (HD, SBMA, hereditary ataxia [HA] - encompassing spinocerebellar ataxia types 1, 2, 3, 6, 7, and 17), the lung (HD, HA), the breast (HD), female genital organs (HD, HA), prostate (HD, HA), hematopoietic, lymphoid tissues (HD), and other sites (HD) [102]. Patients in France had a generally reduced risk of cancer (372 patients with HD and 134 with SCA; SIRs were 0.21, 95% CI 0.13–0.31) in HD and 0.23, 95% CI 0.08–0.42 in SCA patients) [103], and patients in Denmark (all HD) were reported to have reduced cancer (694 HD patients; SIR 0.6 with a 95% CI 0.5– 0.8). This affected cancers of the digestive organs, lung, breast, female genital organs, male genital organs, urinary system, skin, lymphatic and hematopoietic tissue, and other sites [104]. An HD patient cohort in England was reported to have reduced cancer (4865 HD patients; SIR 0.71, 95% CI 0.61–0.83) of the breast, colon, kidney, lung, esophagus, pancreas, rectum, stomach, and the upper gastrointestinal tract. Malignant melanoma, non-melanoma skin cancer, and Non-Hodgkin’s lymphoma was also reduced. In addition, secondary cancers were also less frequent [105]. Finally, a strong inverted relationship between CAG repeat length and cancer incidence was also reported for SCA3 (154 patients; SIR 0.27, 95% CI 0.13 to 0.58) [106].
There are a few studies that did not find an inverse correlation between CAG length and cancer incidence. One study reported that mHTT accelerates breast tumor development and metastasis through ErbB2/HER2 signaling, both in patients and in a HD mouse model [107]. Another study found an increased risk of melanoma in HD patients [103]. The reason for these discrepancies is unclear at this moment but it could be that certain cancers benefit from having either the mutant RNA or protein expressed. In addition, the tumor suppressive effect of TNR sequences seems to be restricted to CAG TNRs and is not found with CUG TNRs. Patients with DM1 were reported to have increased risk of skin cancer (1061 DM1 patients; hazard ratio 55.44, 95% CI 3.33–8.89) [108] or cancer of various anatomic sites (1658 myotonic muscular dystrophy patients; SIR 2.0; 95% CI 1.6-2.4) [109]. More recently, it was suggested that this increased risk may be caused by downregulation of miR-200c/141, through an unknown mechanism particularly in women and for gynecologic, brain, and thyroid cancer [110]. In contrast the miR-200 target BMI1, an oncogenic polycomb protein, was upregulated in these patients possibly outweighing the effects of a CUG derived toxic siRNA.
Perhaps the most convincing inverse relationship between CAG repeat length and the risk/severity of a human cancer was reported between the CAG extension in AR and prostate cancer (PCa). Of all TNR containing genes, AR is the gene with the longest continuous CAG repeat in unaffected individuals. It is located in the amino-terminal domain. The number of CAG repeats in AR is polymorphic; the average number is 22 in Caucasian males [111]. The CAG repeat length in the AR has been inversely linked to cancer. Carriers of short polymorphic CAG repeats have increased susceptibility to prostate cancer [112]. A shorter CAG repeat has been shown to result in a two-fold increased risk of PCa [113], a more aggressive disease, a high risk of distant metastases [114–116], and a younger age of disease onset [111]. The odds of having <17 CAG TNRs is 8-times greater in men with lymph-node positive disease [117]. In contrast, a longer repeat (>20 CAGs) confers a protective effect among the PCa patients 45 years or older [118]. Most significantly, shortening of CAG repeat length was found in in situ lesions of PCa and its possible precursors [119], suggesting that PCa selects for shorter repeats. Interestingly, not only prostate cancer is affected by the TNR expansion in the AR gene. Shorter AR CAG repeats length and a resulting increased AR activity are associated with increased ovarian cancer risk [120].
These last two observations are consistent with our recent report on siRNAs based on CAG/CUG TNRs (siCAG/CUG) that are super toxic to all cancer cells [97]. We did not see any toxicity to normal cells in mice treated with siCAG/CUG coupled to nanoparticles [97]. This suggests that cancer cells are more sensitive to this form of cell death than normal cells, possibly because normal cells are protected from RNAi-mediated toxicity by miRNAs which are more highly expressed in normal cells than in cancer cells [121]. This interpretation is also supported by our data demonstrating that toxic siRNA derived from the gene FASLG when delivered in vivo affected cancer cells without toxicity to normal cells [122]. Mechanistically we showed that titering in a non toxic siRNA ameliorated toxicity of such a toxic siRNA to cancer cells [123] and as a reverse we demonstrated that when most miRNAs were eliminated by knocking out Drosha or Dicer, cells became hypersensitive to any toxic siRNA [97, 124].
Even though the toxicity in the TNR diseases is ultimately detrimental to the affected patients, genes carrying the TNR amplifications (i.e. AR or HTT) are often abundantly expressed in multiple tissues (Figure 1). However, the disease only affects specific tissues and only after years of exposure suggesting that most tissues are relatively resistant to the toxic effects of either the mutant proteins or the extended RNAs, assuming that most tissues have the machinery to process long repeat containing transcripts and utilize them in the RNAi pathway. An evolutionary trade-off between reduced cancer and HD has been discussed before [105]. In fact, HD has been recognized as one of a number of diseases that may persist in the human population because the disease-causing mutation may provide an evolutionary advantage, a phenomenon called antagonistic pleiotropy [125]. Based on these data we recently proposed to use siRNAs based on the CAG TNR to treat cancer (Figure 1).
Figure 1: CAG-based TNR diseases point at a new form of cancer treatment.

Left: CAG TNR disease patients have reduced rates of multiple cancers. Symptoms in neurodegenerative diseases usually do not manifest in the first few decades of life and the disease often affects selected tissues, usually the brain. This suggests that the disease-causing agent is not generally highly toxic to most cells. Right: Recently, it was found that CAG-derived siRNAs are highly toxic to all cancers and can be administered to mice to reduce tumor growth without signs of toxicity [97]. This suggests an evolutionary trade-off (disease outbreak but reduced cancer rate) and a possible use of TNR-based siRNAs as a novel form of cancer therapy.
A Model to Explain Reduced Cancer Incidences in Certain CAG-based TNR Diseases
In our recent analysis the toxicity of all 60 TNR-based siRNAs to cancer cells correlated well with the number of reverse complementary TNR repeats in the ORFs of genes [97]. Interestingly, siRNAs based on either CAG or CUG triplets were the most toxic TNRs out of all TNRs tested. In addition, both CAG and GUG repeats (>18 nt in length) are highly abundant in both human and mouse coding genes - with CUG being the most abundant of all 60 TNRs in both humans and mice [97]. Based on these findings we are now proposing a model in which TNR expansions are part of an anti-cancer mechanism (Figure 2). In order to understand the rationale behind the model, one needs to understand how TNR expansions occur.
Figure 2: A model to explain the reduced cancer incidences in certain CAG-based TNR diseases.
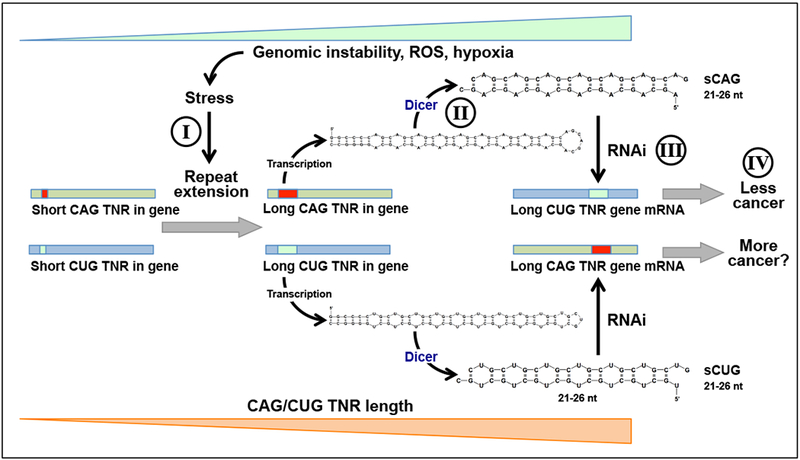
Genotoxic or oxidative stress as well as hypoxia all often found in neoplastically transformed cells, cause an increase in the length of both CUG and CAG repeats in certain genes through replication slippage (I). Both TNRs are transcribed and these transcripts are subject to Dicer processing resulting in short RNAi-active CAG (sCAG) and CUG (sCUG) RNAs (II). sCAGs attack multiple CUG TNR containing genes through RNAi (III) that in combination are critical for cell survival, particularly of cancer cells (IV). sCUGs may also be RNAi active but the consequences of this activity are not clear (?). In addition, there are more genes in the human and mouse genomes that contain long CUG TNRs (>18nt) than genes containing long CAG TNRs that could be targeted [97]. Because both CAG and CUG repeats are unstable and may increase in size with progressing genomic instability, this system may function as a mutational/cancer sensor eliminating cancer cells, resulting in an apparent reduction of cancer incidences in affected patients.
Repetitive regions such as TNR expansions are prone to slippage during DNA replication and this can result in a small loop in either DNA strand. With impaired mismatch repair function, these loops may become permanent, and alleles of different sizes can be formed during the next round of replication [126, 127]. Inactivation of mismatch repair genes (i.e. hMLH1, hMSH2, hPMS1, hPMS2, GTBP/hMSH6) is found in cancer. It is responsible for the microsatellite instability seen in more than 90% of hereditary non-polyposis colorectal cancers (HNPCC), also known as Lynch syndrome [126]. Two components of mismatch repair machinery (MutSβ, a heterodimer of MSH2 and MSH3 and MutL, a heterodimer of MLH1 and PMS2) have been directly linked to CAG/CTG repeat expansions [128–130]. While MutSβ plays a major role in the repair of larger insertion/deletion mispairs [131], it was also shown to be essential for CAG/CTG repeat expansions in mice [132, 133]. Mechanistically, MutSβ recognizes (CAG)n or (CTG)n hairpins formed in the nascent DNA strand, and recruits Pô to the complex, which then utilizes the hairpin as a primer for extension, fixing the hairpin structure and leading to CAG/CTG repeat expansion. All these mechanisms make CAG/CUG repeats highly unstable. Consequently, at least 10% of HD cases are due to a new mutation [134, 135].
We are proposing that MutSβ is tumor suppressive in two ways: 1) Through its activities in p53−/− mice by being involved in double strand break repair [136] through mediating ATR activation [137]. 2) By driving expansion of CAG/CTG repeats by forming a complex with Pô binding to (CAG)n or (GTG)n hairpins.
In general, repeat expansions can be triggered by a number stresses such as cold, heat, hypoxic, and oxidative stress [14], some of which arise in cancerous cells. H2O2 exposure was shown to cause a significant increase in repeat length [138]. Direct evidence that oxidative stress causes TNR expansions in vivo was obtained for the R6/1 HD mouse model [138]. In addition, repeated exposure to genotoxic agents inducing oxidative DNA damage also gave a significant and dose dependent increase in somatic triplet expansion. Increased oxidative stress is also found in HD individuals, and oxidative DNA damage preferentially accumulates at CAG repeats in a length dependent manner in HD mouse models [139–141].
The pathway for stress-induced TNR mutagenesis shows intriguing parallels with pathways that lead to cancer. Cancer cells survive in hostile environments by increasing the expression of key stress response factors (reviewed in [142]). One of the consequences of stress response factor expression in hypoxic cancer cells is increased mutagenesis [143], similar to the stress induced mutagenesis of TNRs discussed here. Pathways for tumorigenesis and TNR mutagenesis may therefore intersect at TNR repeats. Interestingly, TNR repeats are five times more frequent in cancer-related genes, and may make those genes more prone to mutagenesis [144]. With increasing genomic instability and increasing stress, CAG/CTG expansions increase in size (Figure 2, I). We are proposing that the initial event that results in repeat expansion is random and affects precancerous cells. This may involve any gene that already has CAG/CUG repeats of a certain nonpathologic length, explaining the lack of species conservation. When CAG/CUG repeats are expanded, they give rise to hairpins that are cleaved by Dicer into double stranded RNAi-active RNAs 21 to 26 nt in length (Figure 2, II). The small CAG/CUG RNAs then target mRNAs of survival genes that contain fully complementary CUG/CAG repeat regions in genes present in the genome (Figure 2, III). It was shown that the silencing effect of short CAG/CUGs depends on the length of the targeted complementary CUG/CAG repeat. It was efficient in cells expressing an EGFP reporter attached to a 70 or 200 nt long targeted repeat but not a 30 nt long repeat [87]. We found that of all TNRs present in human and mouse genes the CUG TNRs are the longest and most abundant in both genomes [97] making them the best target for CAG TNR-derived siRNAs. As a result cancer cells and normal cells undergoing neoplastic transformation will die, reducing cancer load (Figure 2, IV).
While longer CAG expansions are correlated with reduced cancer risk there is little evidence that this is the case for CUG expansions. On the contrary, it appears that longer CUG repeats correlate with an increase in cancer risk (Figure 2), even though both repeat siRNAs were highly toxic to cancer cells in our screen [97]. There are a number of possible explanations for this observation. Just like CAG TNRs, CUG TNRs may also give rise to small RNAi-active siRNAs, but since there are fewer long CAG TNR containing potential target genes [97], in vivo they may not be as toxic as CAG TNR-derived siRNAs. Alternatively, targeted CAG and CUG TNRs could be located in genes with different functions in tumorigenesis. Finally, we recently found that the 7mer seed sequence of the CAG-based siRNA is identical to one of the most tumor suppressive miRNAs, miR-15a-5p/16-1-5p (AGCAGCA). In contrast, a major oncogenic miRNA, miR-93-3p, shares a 6mer seed sequence with the CUG-containing siRNA (CUGCUG). It is therefore possible that in addition to their toxicity the CAG/CUG siRNAs may also target a set of oncogenic and tumor suppressive targets, respectively, providing a possible explanation for the apparent opposite correlations between CAG and CUG repeat expansions on cancer risk in affected patients. This can now experimentally be tested.
Our model provides an explanation for the observed reduced cancer incidence in multiple CAG TNR diseases and points at a path to develop a novel form of cancer treatment based on CAG/CUG repeats. We have recently shown that a siRNA based on CAG/CUG TNRs can be used to treat ovarian cancer in mice with no detectable toxicity to the animals [97].
Concluding Remarks
TNRs can give rise to toxic gain-of-function proteins, however, there is growing evidence that toxic RNAs are also involved in the disease etiology, irrespective of where the TNR is located in the mutated gene. Ever since the discovery of the genetic basis of TNR-induced degenerative diseases it has been a fundamental goal to target the repeat derived poly amino acids or the coding RNA as a means to treat these diseases. Promising strategies are being developed involving targeting the proteins by using translation blockers, antisense oligonucleotides, or siRNAs (reviewed in [145, 146]). It is now becoming apparent that RNAi may also be a disease-causing mechanism. Data on the connection between TNR diseases and reduced global cancer incidence and the discovery that TNR-derived siRNAs have the potential to kill all cancer cells, now points at a new field of study located at the interface between neurodegenerative diseases and cancer. This could be beneficial to the understanding of disease progression and the development of new treatments for TNR-based diseases, and for the development of a new form of cancer treatment. What needs to be considered is the fact that the mechanisms that cause neurodegenerative diseases and affect cancer could be separate. One could involve toxic proteins/RNAs and the other RNAi. Future studies (see outstanding questions) will have to address the precise contribution of protein, RNA and other mechanisms to the disease progression and cancer.
Box -. Outstanding Questions.
Why are all CAG/CUG derived siRNAs toxic to cancer cells? There are 64 different TNRs but most of them are not toxic when delivered as siRNAs.
What could protect normal cells from this toxicity? Cells lacking most miRNAs are hypersensitive to toxic siRNAs.
Why do patients with certain TNR diseases have reduced while others have increased cancer incidence? Do TNR-derived small RNAs differentially target tumor suppressors and oncogenes?
Are nonconserved CAG/CUG TNRs found throughout the genome part of an anti-cancer mechanism? The location of TNRs in the genome is poorly conserved - including in exons - suggesting that they may not be required for protein function. Are small CAG or CUG derived RNAs ever released in cells to kill cancerous cells?
Can si- or shRNAs targeting TNR repeats be used to treat cancer patients? Is there a therapeutic window?
Can the TNR-cancer connection be modeled in a disease mouse model? Will Huntington’s disease or SBMA mouse models have reduced cancer incidences?
Box 1: TNRs in the Genome of Higher Organisms.
While the CAG repeat amplifications in the human HTT and AR genes can cause neuropathology, their location and length are not well conserved between human and mouse (Figure I, Text Box 1). Despite this fact, overall both genes show a high degree of sequence similarity between these two species. The sequence of HTT outside of the CAG repeat region has more than 86% sequence identity at the nucleotide level, that of the AR gene is 83 to >90% identical outside three TNR regions in this gene (data not shown). Even in monkeys and great apes, closely related to humans, the CAG repeat length is only poorly conserved (Figure 2, bottom). This argues against polyQ stretches playing an important role in these otherwise highly conserved proteins. The observation of low conservation can be extended to other TNRs and genes. We recently analyzed the open reading frames (ORFs) and 3’UTRs of all human and mouse genes for the presence of CTG repeats (the targets of CAG repeat-based siRNAs) of >18 nucleotides in length [97]. We found between human and mouse, a total of 92 genes carrying such longer repeats in their ORF. Of these genes only 7.6% were shared between the two species. In addition, among the 31 mouse and human genes carrying CTG repeats longer than 18 nucleotides in their 3’UTRs, none of them were conserved between the two species. These data suggest that while different mammalian species all have TNRs, they may not play an important role in either normal protein function or gene regulation. This raises the question of why these sequences exist and what their role is in the genome. To begin to address this question one may have to ask how these sequences are being generated. One observation in mice expressing TNRs as transgenes and consistent with patient data of multiple diseases is that they are notoriously unstable, and the length can vary from one generation to the next [149, 150]. There are a few theories that explain how TNRs are created. According to one theory they are generated by replication slippage [150], and according to another by defects in the single nucleotide repair machinery [151].
Figure I, Text Box 1: The location and length of the CAG TNRs in the HTT and AR genes are poorly conserved among mammalian species.
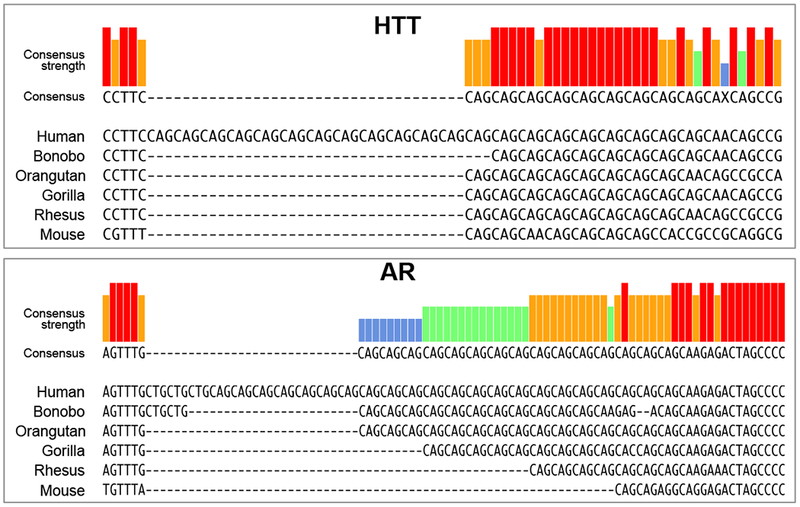
DNA alignment of the regions in human HTT (top) and human AR (bottom), containing the CAG TNRs in exon-1 with other primates and murine orthologs. The degree of conservation is indicated. X = nucleotide position without consensus.
Acknowledgements
This work was funded by R35CA197450 (to M.E.P.).
References
- 1.Nalavade R et al. (2013) Mechanisms of RNA-induced toxicity in CAG repeat disorders. Cell Death Dis 4, e752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Orr HT and Zoghbi HY (2007) Trinucleotide repeat disorders. Annu Rev Neurosci 30, 575–621. [DOI] [PubMed] [Google Scholar]
- 3.Komure O et al. (1995) DNA analysis in hereditary dentatorubral-pallidoluysian atrophy: correlation between CAG repeat length and phenotypic variation and the molecular basis of anticipation. Neurology 45 (1), 143–9. [DOI] [PubMed] [Google Scholar]
- 4.La Spada AR et al. (1991) Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352 (6330), 77–9. [DOI] [PubMed] [Google Scholar]
- 5.The_Huntington’s_Disease_Collaborative_Research_Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 72 (6), 971–83. [DOI] [PubMed] [Google Scholar]
- 6.Orr HT et al. (1993) Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet 4 (3), 221–6. [DOI] [PubMed] [Google Scholar]
- 7.Sanpei K et al. (1996) Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet 14 (3), 277–84. [DOI] [PubMed] [Google Scholar]
- 8.Kawaguchi Y et al. (1994) CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet 8 (3), 221–8. [DOI] [PubMed] [Google Scholar]
- 9.Zhuchenko O et al. (1997) Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet 15 (1), 62–9. [DOI] [PubMed] [Google Scholar]
- 10.David G et al. (1997) Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet 17 (1), 65–70. [DOI] [PubMed] [Google Scholar]
- 11.Holmes SE et al. (1999) Expansion of a novel CAG trinucleotide repeat in the 5’ region of PPP2R2B is associated with SCA12. Nat Genet 23 (4), 391–2. [DOI] [PubMed] [Google Scholar]
- 12.Fujigasaki H et al. (2001) CAG repeat expansion in the TATA box-binding protein gene causes autosomal dominant cerebellar ataxia. Brain 124 (Pt 10), 1939–47. [DOI] [PubMed] [Google Scholar]
- 13.Koob MD et al. (1999) An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). Nat Genet 21 (4), 379–84. [DOI] [PubMed] [Google Scholar]
- 14.Holmes SE et al. (2001) A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet 29 (4), 377–8. [DOI] [PubMed] [Google Scholar]
- 15.Campuzano V et al. (1996) Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271 (5254), 1423–7. [DOI] [PubMed] [Google Scholar]
- 16.Brouwer JR et al. (2009) The FMR1 gene and fragile X-associated tremor/ataxia syndrome. Am J Med Genet B Neuropsychiatr Genet 150B (6), 782–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sellier C et al. (2013) Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep 3 (3), 869–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gu Y and Nelson DL (2003) FMR2 function: insight from a mouse knockout model. Cytogenet Genome Res 100 (1-4), 129–39. [DOI] [PubMed] [Google Scholar]
- 19.Mirkin SM (2007) Expandable DNA repeats and human disease. Nature 447 (7147), 932–40. [DOI] [PubMed] [Google Scholar]
- 20.Liquori CL et al. (2001) Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293 (5531), 864–7. [DOI] [PubMed] [Google Scholar]
- 21.Brook JD et al. (1992) Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein kinase family member. Cell 68 (4), 799–808. [DOI] [PubMed] [Google Scholar]
- 22.Niimi Y et al. (2013) Abnormal RNA structures (RNA foci) containing a penta-nucleotide repeat (UGGAA)n in the Purkinje cell nucleus is associated with spinocerebellar ataxia type 31 pathogenesis. Neuropathology 33 (6), 600–11. [DOI] [PubMed] [Google Scholar]
- 23.DeJesus-Hernandez M et al. (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72 (2), 245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Renton AE et al. (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72 (2), 257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nelson DL et al. (2013) The unstable repeats--three evolving faces of neurological disease. Neuron 77 (5), 825–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kennedy WR et al. (1968) Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-linked recessive trait. Neurology 18 (7), 671–80. [DOI] [PubMed] [Google Scholar]
- 27.Lund A et al. (2001) Multiple founder effects in spinal and bulbar muscular atrophy (SBMA, Kennedy disease) around the world. Eur J Hum Genet 9 (6), 431–6. [DOI] [PubMed] [Google Scholar]
- 28.Crocoll A et al. (1998) Expression of androgen receptor mRNA during mouse embryogenesis. Mech Dev 72 (1-2), 175–8. [DOI] [PubMed] [Google Scholar]
- 29.Atsuta N et al. (2006) Natural history of spinal and bulbar muscular atrophy (SBMA): a study of 223 Japanese patients. Brain 129 (Pt 6), 1446–55. [DOI] [PubMed] [Google Scholar]
- 30.Katsuno M et al. (2012) Pathogenesis and therapy of spinal and bulbar muscular atrophy (SBMA). Prog Neurobiol 99 (3), 246–56. [DOI] [PubMed] [Google Scholar]
- 31.Gatchel JR and Zoghbi HY (2005) Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet 6 (10), 743–55. [DOI] [PubMed] [Google Scholar]
- 32.Duyao MP et al. (1995) Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science 269 (5222), 407–10. [DOI] [PubMed] [Google Scholar]
- 33.Lee JM et al. (2013) Dominant effects of the Huntington’s disease HTT CAG repeat length are captured in gene-expression data sets by a continuous analysis mathematical modeling strategy. Hum Mol Genet 22 (16), 3227–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nasir J et al. (1995) Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 81 (5), 811–23. [DOI] [PubMed] [Google Scholar]
- 35.Boudreau RL et al. (2009) Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol Ther 17 (6), 1053–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ross CA (2002) Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington’s disease and related disorders. Neuron 35 (5), 819–22. [DOI] [PubMed] [Google Scholar]
- 37.Klement IA et al. (1998) Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell 95 (1), 41–53. [DOI] [PubMed] [Google Scholar]
- 38.Busch A et al. (2003) Mutant huntingtin promotes the fibrillogenesis of wild-type huntingtin: a potential mechanism for loss of huntingtin function in Huntington’s disease. J Biol Chem 278 (42), 41452–61. [DOI] [PubMed] [Google Scholar]
- 39.Ross CA and Tabrizi SJ (2011) Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 10 (1), 83–98. [DOI] [PubMed] [Google Scholar]
- 40.Gasset-Rosa F et al. (2017) Polyglutamine-Expanded Huntingtin Exacerbates Age-Related Disruption of Nuclear Integrity and Nucleocytoplasmic Transport. Neuron 94 (1), 48–57 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grima JC et al. (2017) Mutant Huntingtin Disrupts the Nuclear Pore Complex. Neuron 94 (1), 93–107 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rousseaux MWC et al. (2018) ATXN1-CIC Complex Is the Primary Driver of Cerebellar Pathology in Spinocerebellar Ataxia Type 1 through a Gain-of-Function Mechanism. Neuron. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Emamian ES et al. (2003) Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron 38 (3), 375–87. [DOI] [PubMed] [Google Scholar]
- 44.Duvick L et al. (2010) SCA1-like disease in mice expressing wild-type ataxin-1 with a serine to aspartic acid replacement at residue 776. Neuron 67 (6), 929–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Powers ET et al. (2009) Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem 78, 959–91. [DOI] [PubMed] [Google Scholar]
- 46.Saudou F et al. (1998) Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95 (1), 55–66. [DOI] [PubMed] [Google Scholar]
- 47.Cummings CJ et al. (1999) Mutation of the E6-AP ubiquitin ligase reduces nuclear inclusion frequency while accelerating polyglutamine-induced pathology in SCA1 mice. Neuron 24 (4), 879–92. [DOI] [PubMed] [Google Scholar]
- 48.Arrasate M et al. (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431 (7010), 805–10. [DOI] [PubMed] [Google Scholar]
- 49.Watase K et al. (2002) A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron 34 (6), 905–19. [DOI] [PubMed] [Google Scholar]
- 50.Banez-Coronel M et al. (2012) A pathogenic mechanism in Huntington’s disease involves small CAG-repeated RNAs with neurotoxic activity. PLoS Genet 8 (2), e1002481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Taneja KL et al. (1995) Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J Cell Biol 128 (6), 995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mankodi A et al. (2001) Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum Mol Genet 10 (19), 2165–70. [DOI] [PubMed] [Google Scholar]
- 53.Miller JW et al. (2000) Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J 19 (17), 4439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wojciechowska M and Krzyzosiak WJ (2011) Cellular toxicity of expanded RNA repeats: focus on RNA foci. Hum Mol Genet 20 (19), 3811–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goodwin M et al. (2015) MBNL Sequestration by Toxic RNAs and RNA Misprocessing in the Myotonic Dystrophy Brain. Cell Rep 12 (7), 1159–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ho TH et al. (2005) Colocalization of muscleblind with RNA foci is separable from mis-regulation of alternative splicing in myotonic dystrophy. J Cell Sci 118 (Pt 13), 2923–33. [DOI] [PubMed] [Google Scholar]
- 57.Ho TH et al. (2005) Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy. Hum Mol Genet 14 (11), 1539–47. [DOI] [PubMed] [Google Scholar]
- 58.Echeverria GV and Cooper TA (2012) RNA-binding proteins in microsatellite expansion disorders: mediators of RNA toxicity. Brain Res 1462, 100–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lin L et al. (2016) Transcriptome sequencing reveals aberrant alternative splicing in Huntington’s disease. Hum Mol Genet 25 (16), 3454–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mykowska A et al. (2011) CAG repeats mimic CUG repeats in the misregulation of alternative splicing. Nucleic Acids Res 39 (20), 8938–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jain A and Vale RD (2017) RNA phase transitions in repeat expansion disorders. Nature 546 (7657), 243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kratter IH and Finkbeiner S (2010) PolyQ disease: too many Qs, too much function? Neuron 67 (6), 897–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chamberlain NL et al. (1994) The length and location of CAG trinucleotide repeats in the androgen receptor N-terminal domain affect transactivation function. Nucleic Acids Res 22 (15), 3181–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lieberman AP et al. (2002) Altered transcriptional regulation in cells expressing the expanded polyglutamine androgen receptor. Hum Mol Genet 11 (17), 1967–76. [DOI] [PubMed] [Google Scholar]
- 65.Katsuno M et al. (2002) Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 35 (5), 843–54. [DOI] [PubMed] [Google Scholar]
- 66.Chevalier-Larsen ES et al. (2004) Castration restores function and neurofilament alterations of aged symptomatic males in a transgenic mouse model of spinal and bulbar muscular atrophy. J Neurosci 24 (20), 4778–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Minamiyama M et al. (2012) Naratriptan mitigates CGRP1-associated motor neuron degeneration caused by an expanded polyglutamine repeat tract. Nat Med 18 (10), 1531–8. [DOI] [PubMed] [Google Scholar]
- 68.Bingham PM et al. (1995) Stability of an expanded trinucleotide repeat in the androgen receptor gene in transgenic mice. Nat Genet 9 (2), 191–6. [DOI] [PubMed] [Google Scholar]
- 69.La Spada AR et al. (1998) Androgen receptor YAC transgenic mice carrying CAG 45 alleles show trinucleotide repeat instability. Hum Mol Genet 7 (6), 959–67. [DOI] [PubMed] [Google Scholar]
- 70.McManamny P et al. (2002) A mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet 11 (18), 2103–11. [DOI] [PubMed] [Google Scholar]
- 71.Merry DE et al. (1998) Cleavage, aggregation and toxicity of the expanded androgen receptor in spinal and bulbar muscular atrophy. Hum Mol Genet 7 (4), 693–701. [DOI] [PubMed] [Google Scholar]
- 72.Kobayashi Y et al. (2000) Chaperones Hsp70 and Hsp40 suppress aggregate formation and apoptosis in cultured neuronal cells expressing truncated androgen receptor protein with expanded polyglutamine tract. J Biol Chem 275 (12), 8772–8. [DOI] [PubMed] [Google Scholar]
- 73.Adachi H et al. (2001) Transgenic mice with an expanded CAG repeat controlled by the human AR promoter show polyglutamine nuclear inclusions and neuronal dysfunction without neuronal cell death. Hum Mol Genet 10 (10), 1039–48. [DOI] [PubMed] [Google Scholar]
- 74.Lu S et al. (1999) Neural androgen receptor regulation: effects of androgen and antiandrogen. J Neurobiol 41 (4), 505–12. [DOI] [PubMed] [Google Scholar]
- 75.Wiren KM et al. (1997) Transcriptional up-regulation of the human androgen receptor by androgen in bone cells. Endocrinology 138 (6), 2291–300. [DOI] [PubMed] [Google Scholar]
- 76.Li LB et al. (2008) RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature 453 (7198), 1107–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hsu RJ et al. (2011) Long tract of untranslated CAG repeats is deleterious in transgenic mice. PLoS One 6 (1), e16417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cleary JD and Ranum LP (2017) New developments in RAN translation: insights from multiple diseases. Curr Opin Genet Dev 44, 125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gacy AM et al. (1998) GAA instability in Friedreich’s Ataxia shares a common, DNA-directed and intraallelic mechanism with other trinucleotide diseases. Mol Cell 1 (4), 583–93. [DOI] [PubMed] [Google Scholar]
- 80.Kunst CB et al. (1996) FMR1 in global populations. Am J Hum Genet 58 (3), 513–22. [PMC free article] [PubMed] [Google Scholar]
- 81.Hillman MA and Gecz J (2001) Fragile XE-associated familial mental retardation protein 2 (FMR2) acts as a potent transcription activator. J Hum Genet 46 (5), 251–9. [DOI] [PubMed] [Google Scholar]
- 82.Lin CH et al. (2010) The CAG repeat in SCA12 functions as a cis element to up-regulate PPP2R2B expression. Hum Genet 128 (2), 205–12. [DOI] [PubMed] [Google Scholar]
- 83.Willemsen R et al. (2011) CGG repeat in the FMR1 gene: size matters. Clin Genet 80 (3), 214–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hagerman RJ et al. (2001) Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 57 (1), 127–30. [DOI] [PubMed] [Google Scholar]
- 85.Verkerk AJ et al. (1991) Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65 (5), 905–14. [DOI] [PubMed] [Google Scholar]
- 86.Napierala M and Krzyzosiak WJ (1997) CUG repeats present in myotonin kinase RNA form metastable “slippery” hairpins. J Biol Chem 272 (49), 31079–85. [DOI] [PubMed] [Google Scholar]
- 87.Krol J et al. (2007) Ribonuclease dicer cleaves triplet repeat hairpins into shorter repeats that silence specific targets. Mol Cell 25 (4), 575–86. [DOI] [PubMed] [Google Scholar]
- 88.Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116 (2), 281–97. [DOI] [PubMed] [Google Scholar]
- 89.Hannon GJ (2002) RNA interference. Nature 418 (6894), 244–51. [DOI] [PubMed] [Google Scholar]
- 90.Krol J et al. (2010) The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet 11 (9), 597–610. [DOI] [PubMed] [Google Scholar]
- 91.Ha M and Kim VN (2014) Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15 (8), 509–24. [DOI] [PubMed] [Google Scholar]
- 92.Meister G (2013) Argonaute proteins: functional insights and emerging roles. Nat Rev Genet 14 (7), 447–59. [DOI] [PubMed] [Google Scholar]
- 93.Ghildiyal M and Zamore PD (2009) Small silencing RNAs: an expanding universe. Nat Rev Genet 10 (2), 94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rue L et al. (2016) Targeting CAG repeat RNAs reduces Huntington’s disease phenotype independently of huntingtin levels. J Clin Invest 126 (11), 4319–4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bilen J et al. (2006) MicroRNA pathways modulate polyglutamine-induced neurodegeneration. Mol Cell 24 (1), 157–63. [DOI] [PubMed] [Google Scholar]
- 96.Jiang F et al. (2005) Dicer-1 and R3D1-L catalyze microRNA maturation in Drosophila. Genes Dev 19 (14), 1674–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Murmann AE et al. (2018) Small interfering RNAs based on huntingtin trinucleotide repeats are highly toxic to cancer cells. EMBO Rep 19, e45336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yu Z et al. (2011) Triplet repeat-derived siRNAs enhance RNA-mediated toxicity in a Drosophila model for myotonic dystrophy. PLoS Genet 7 (3), e1001340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lawlor KT et al. (2011) Double-stranded RNA is pathogenic in Drosophila models of expanded repeat neurodegenerative diseases. Hum Mol Genet 20 (19), 3757–68. [DOI] [PubMed] [Google Scholar]
- 100.Moseley ML et al. (2006) Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet 38 (7), 758–69. [DOI] [PubMed] [Google Scholar]
- 101.Jasinska A et al. (2003) Structures of trinucleotide repeats in human transcripts and their functional implications. Nucleic Acids Res 31 (19), 5463–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ji J et al. (2012) Cancer incidence in patients with polyglutamine diseases: a population-based study in Sweden. Lancet Oncol 13 (6), 642–8. [DOI] [PubMed] [Google Scholar]
- 103.Coarelli G et al. (2017) Low cancer prevalence in polyglutamine expansion diseases. Neurology 88 (12), 1114–1119. [DOI] [PubMed] [Google Scholar]
- 104.Sorensen SA et al. (1999) Significantly lower incidence of cancer among patients with Huntington disease: An apoptotic effect of an expanded polyglutamine tract? Cancer 86 (7), 1342–6. [PubMed] [Google Scholar]
- 105.Turner MR et al. (2013) Reduced cancer incidence in Huntington’s disease: record linkage study clue to an evolutionary trade-off? Clin Genet 83 (6), 588–90. [DOI] [PubMed] [Google Scholar]
- 106.Souza GN et al. (2017) Cancer in Machado-Joseph disease patients-low frequency as a cause of death. Cancer Genet 212-213, 19–23. [DOI] [PubMed] [Google Scholar]
- 107.Moreira Sousa C et al. (2013) The Huntington disease protein accelerates breast tumour development and metastasis through ErbB2/HER2 signalling. EMBO Mol Med 5 (2), 309–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang Y et al. (2018) Risk of skin cancer among patients with myotonic dystrophy type 1 based on primary care physician data from the U.K. Clinical Practice Research Datalink. Int J Cancer 142 (6), 1174–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gadalla SM et al. (2011) Cancer risk among patients with myotonic muscular dystrophy. JAMA 306 (22), 2480–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fernandez-Torron R et al. (2016) Cancer risk in DM1 is sex-related and linked to miRNA-200/141 downregulation. Neurology 87 (12), 1250–7. [DOI] [PubMed] [Google Scholar]
- 111.Hardy DO et al. (1996) Androgen receptor CAG repeat lengths in prostate cancer: correlation with age of onset. J Clin Endocrinol Metab 81 (12), 4400–5. [DOI] [PubMed] [Google Scholar]
- 112.Qin Z et al. (2017) Association between polymorphic CAG repeat lengths in the androgen receptor gene and susceptibility to prostate cancer: A systematic review and meta-analysis. Medicine (Baltimore) 96 (25), e7258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ingles SA et al. (1997) Association of prostate cancer risk with genetic polymorphisms in vitamin D receptor and androgen receptor. J Natl Cancer Inst 89 (2), 166–70. [DOI] [PubMed] [Google Scholar]
- 114.Giovannucci E et al. (1997) The CAG repeat within the androgen receptor gene and its relationship to prostate cancer. Proc Natl Acad Sci U S A 94 (7), 3320–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Platz EA et al. (1998) The androgen receptor gene GGN microsatellite and prostate cancer risk. Cancer Epidemiol Biomarkers Prev 7 (5), 379–84. [PubMed] [Google Scholar]
- 116.Irvine RA et al. (1995) The CAG and GGC microsatellites of the androgen receptor gene are in linkage disequilibrium in men with prostate cancer. Cancer Res 55 (9), 1937–40. [PubMed] [Google Scholar]
- 117.Hakimi JM et al. (1997) Androgen receptor variants with short glutamine or glycine repeats may identify unique subpopulations of men with prostate cancer. Clin Cancer Res 3 (9), 1599–608. [PubMed] [Google Scholar]
- 118.Gu M et al. (2012) The CAG repeat polymorphism of androgen receptor gene and prostate cancer: a meta-analysis. Mol Biol Rep 39 (3), 2615–24. [DOI] [PubMed] [Google Scholar]
- 119.Tsujimoto Y et al. (2004) In situ shortening of CAG repeat length within the androgen receptor gene in prostatic cancer and its possible precursors. Prostate 58 (3), 283–90. [DOI] [PubMed] [Google Scholar]
- 120.Zhu H et al. (2017) The role of the androgen receptor in ovarian cancer carcinogenesis and its clinical implications. Oncotarget 8 (17), 29395–29405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lu J et al. (2005) MicroRNA expression profiles classify human cancers. Nature 435 (7043), 834–838. [DOI] [PubMed] [Google Scholar]
- 122.Murmann AE et al. (2017) Induction of DISE in ovarian cancer cells in vivo. Oncotarget 8, 84643–84658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Putzbach W et al. (2017) Many si/shRNAs can kill cancer cells by targeting multiple survival genes through an off-target mechanism. eLife 6, e29702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gao QQ et al. (2018) 6mer Seed Toxicity Determines Strand Selection in miRNAs. Nature Commun, accepted with minor revisions. Preprint at BiorXiv, 10.1101/284406. [DOI] [Google Scholar]
- 125.Carter AJ and Nguyen AQ (2011) Antagonistic pleiotropy as a widespread mechanism for the maintenance of polymorphic disease alleles. BMC Med Genet 12, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wheeler JM et al. (2000) DNA mismatch repair genes and colorectal cancer. Gut 47 (1), 148–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shelbourne P and Johnson K (1992) Myotonic dystrophy: another case of too many repeats? Hum Mutat 1 (3), 183–9. [DOI] [PubMed] [Google Scholar]
- 128.Guo J et al. (2016) MutSbeta promotes trinucleotide repeat expansion by recruiting DNA polymerase beta to nascent (CAG)n or (CTG)n hairpins for error-prone DNA synthesis. Cell Res 26 (7), 775–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Iyer RR et al. (2015) DNA triplet repeat expansion and mismatch repair. Annu Rev Biochem 84, 199–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Schmidt MH and Pearson CE (2016) Disease-associated repeat instability and mismatch repair. DNA Repair (Amst) 38, 117–26. [DOI] [PubMed] [Google Scholar]
- 131.Kolodner R (1996) Biochemistry and genetics of eukaryotic mismatch repair. Genes Dev 10 (12), 1433–42. [DOI] [PubMed] [Google Scholar]
- 132.Manley K et al. (1999) Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat Genet 23 (4), 471–3. [DOI] [PubMed] [Google Scholar]
- 133.Foiry L et al. (2006) Msh3 is a limiting factor in the formation of intergenerational CTG expansions in DM1 transgenic mice. Hum Genet 119 (5), 520–6. [DOI] [PubMed] [Google Scholar]
- 134.Falush D et al. (2001) Measurement of mutational flow implies both a high new-mutation rate for Huntington disease and substantial underascertainment of late-onset cases. Am J Hum Genet 68 (2), 373–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Almqvist EW et al. (2001) High incidence rate and absent family histories in one quarter of patients newly diagnosed with Huntington disease in British Columbia. Clin Genet 60 (3), 198–205. [DOI] [PubMed] [Google Scholar]
- 136.van Oers JM et al. (2014) The MutSbeta complex is a modulator of p53-driven tumorigenesis through its functions in both DNA double-strand break repair and mismatch repair. Oncogene 33 (30), 3939–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Burdova K et al. (2015) The Mismatch-Binding Factor MutSbeta Can Mediate ATR Activation in Response to DNA Double-Strand Breaks. Mol Cell 59 (4), 603–14. [DOI] [PubMed] [Google Scholar]
- 138.Jonson I et al. (2013) Oxidative stress causes DNA triplet expansion in Huntington’s disease mouse embryonic stem cells. Stem Cell Res 11 (3), 1264–71. [DOI] [PubMed] [Google Scholar]
- 139.Bogdanov MB et al. (2001) Increased oxidative damage to DNA in a transgenic mouse model of Huntington’s disease. J Neurochem 79 (6), 1246–9. [DOI] [PubMed] [Google Scholar]
- 140.Goula AV et al. (2009) Stoichiometry of base excision repair proteins correlates with increased somatic CAG instability in striatum over cerebellum in Huntington’s disease transgenic mice. PLoS Genet 5 (12), e1000749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Browne SE et al. (1997) Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol 41 (5), 646–53. [DOI] [PubMed] [Google Scholar]
- 142.Chatterjee N et al. (2015) Environmental stress induces trinucleotide repeat mutagenesis in human cells. Proc Natl Acad Sci U S A 112 (12), 3764–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Fitzgerald DM et al. (2017) Stress-Induced Mutagenesis: Implications in Cancer and Drug Resistance. Annu Rev Cancer Biol 1, 119–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Haberman Y et al. (2008) Trinucleotide repeats are prevalent among cancer-related genes. Trends Genet 24 (1), 14–8. [DOI] [PubMed] [Google Scholar]
- 145.Urbanek MO et al. (2017) Reduction of Huntington’s Disease RNA Foci by CAG Repeat-Targeting Reagents. Front Cell Neurosci 11,82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wild EJ and Tabrizi SJ (2017) Therapies targeting DNA and RNA in Huntington’s disease. Lancet Neurol 16 (10), 837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Koide R et al. (1994) Unstable expansion of CAG repeat in hereditary dentatorubral-pallidoluysian atrophy (DRPLA). Nat Genet 6 (1), 9–13. [DOI] [PubMed] [Google Scholar]
- 148.Hamel BC et al. (1994) Segregation of FRAXE in a large family: clinical, psychometric, cytogenetic, and molecular data. Am J Hum Genet 55 (5), 923–31. [PMC free article] [PubMed] [Google Scholar]
- 149.Wheeler VC et al. (1999) Length-dependent gametic CAG repeat instability in the Huntington’s disease knock-in mouse. Hum Mol Genet 8 (1), 115–22. [DOI] [PubMed] [Google Scholar]
- 150.McMurray CT (2010) Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet 11 (11), 786–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Liu Y and Wilson SH (2012) DNA base excision repair: a mechanism of trinucleotide repeat expansion. Trends Biochem Sci 37 (4), 162–72. [DOI] [PMC free article] [PubMed] [Google Scholar]