

Abstract
A technology to record membrane potential from multiple neurons, simultaneously, in behaving animals will have a transformative impact on neuroscience research1, 2. Genetically encoded voltage indicators are a promising tool for these purposes, but were so far limited to single-cell recordings with marginal signal to noise ratio (SNR) in vivo3-5. We developed improved near infrared voltage indicators, high speed microscopes and targeted gene expression schemes which enabled recordings of supra- and subthreshold voltage dynamics from multiple neurons simultaneously in mouse hippocampus, in vivo. The reporters revealed sub-cellular details of back-propagating action potentials and correlations in sub-threshold voltage between multiple cells. In combination with optogenetic stimulation, the reporters revealed brain state-dependent changes in neuronal excitability, reflecting the interplay of excitatory and inhibitory synaptic inputs. These tools open the possibility for detailed explorations of network dynamics in the context of behavior.
Archaerhodopsin-derived genetically encoded voltage indicators (GEVIs) are excited by red light and emit in the near infrared part of the spectrum, have sub-millisecond response times and report neuronal action potentials with a ~40% increase in fluorescence6. A transgenic mouse expressing the Arch-based GEVI QuasAr2 and the channelrhodopsin CheRiff (together called ‘Optopatch2’) enabled optical stimulation and recording in sparsely expressing acute brain slices3, but did not attain adequate signal-to-noise ratio (SNR) for voltage imaging in brain in vivo due to poor GEVI trafficking and high background fluorescence.
We rationally designed multiple constructs and tested them for the SNR of optogenetically induced action potentials in culture and in brain slices. This screen led to a construct, QuasAr3, that showed high expression and excellent trafficking in vivo and reported action potentials in acute slices with fast kinetics and high SNR (21 ± 12 in a 1 kHz bandwidth, n = 10 neurons, mean ± s.d., Extended Data Fig. 1). We made a transgenic mouse with Cre-dependent QuasAr3 and CheRiff in the highly expressing TIGRE locus 7. Acute slices from these animals yielded high SNR genetically targeted all-optical electrophysiology recordings and clearly showed cell type-specific differences in firing patterns (Extended Data Fig. 2).
We then tested a point mutation in QuasAr3, V59A, whose homolog in bacteriorhodopsin (V49A) causes photoswitching behavior8 and enhances the population of the fluorescent Q state9. Under continuous red excitation (λexc = 640 nm, 10 W/mm2), illumination with moderate intensity blue light (λact = 488 nm, 100 mW/mm2) reversibly increased near infrared fluorescence (λem = 660 – 740 nm) of QuasAr3(V59A) by a factor of 2.9 ± 0.7, n = 8 cells (Extended Data Fig. 3). Blue light enhanced voltage-dependent fluorescence in HEK cells (Extended Data Fig. 3) and cultured neurons (Fig. 1b) by 2–3-fold. We called QuasAr3(V59A) ‘photoactivated QuasAr3’, or paQuasAr3. A detailed characterization of the kinetic, spectroscopic, and voltage-dependent properties of paQuasAr3 (Extended Data Fig. 3) suggested the photocycle model shown in Fig. 1c. Blue light transferred population from a dark state, D1, to a voltage-sensitive equilibrium between a second dark state, D2, and a fluorescent state, F.
Figure 1. Photo-activated QuasAr3 (paQuasAr3) reports neuronal activity in vivo.

(a) PaQuasAr3 construct. (b) Blue light photoactivation enhanced red light-excited voltage signals in cultured neurons expressing paQuasAr3 (representative example of 4 repeats). (c) Model of the photocycle of paQuasAr3. (d) Confocal images of sparsely expressed paQuasAr3 in brain slices. Scale bar 50 μm. Representative images, repeated in N = 3 mice. (e) Simultaneous fluorescence and patch clamp recordings from a neuron expressing paQuasAr3 in acute brain slice. Inset shows boxed regions. (f) Simultaneous fluorescence and patch clamp recordings of IPSPs in an L2/3 neuron induced via electrical stimulation of L5–6 in acute slice. (g) ΔF/F and SNR of optically recorded PSPs as a function of the PSP amplitude. The voltage sensitivity was ΔF/F = 40 ± 1.7%/100 mV. The SNR was 0.93 ± 0.07/mV in a 1 kHz bandwidth (n = 42 PSPs from 5 cells, mean ± s.d.). (h) Optical measurements of paQuasAr3 fluorescence in CA1 region of the hippocampus (top) and glomerular layer of the olfactory bulb (bottom) of anesthetized mice (representative traces from n = 7 CA1 cells and n = 13 OB cells, N = 3 mice). (i) Spike-triggered average fluorescence from 88 spikes in a CA1 Oriens neuron. (j) Frames from the spike-triggered average movie showing the delay in the back-propagating action potential in the dendrites relative to the soma. (k) Sub-Nyquist fitting of the action potential delay and width show electrical compartmentalization in the dendrites. Experiment in k–m repeated in n = 2 cells from N = 2 mice.
PaQuasAr3 expressed in vivo by viral transduction trafficked well in soma and dendrites (Fig. 1d). Recordings in acute cortical slices showed that photoactivation by blue light significantly improved the SNR for spike detection (Fig. 1e, Extended Data Fig. 3, QuasAr3: SNR = 20 ± 12, paQuasAr3 red only: SNR = 27 ± 10, paQuasAr3 red and blue: SNR = 37 ± 14, n = 10 cells in each group). PaQuasAr3 resolved 1–2 mV fluctuations, including sub-threshold post-synaptic potentials (PSPs, Fig. 1 f,g).
The enhanced SNR from paQuasAr3 enabled single-neuron optical voltage measurements in CA1 region of the hippocampus and in the glomerular layer of the olfactory bulb of anesthetized mice (Fig. 1h). In a CA1 Oriens interneuron expressing paQuasAr3 we observed back-propagating action potentials in dendrites (Fig. 1i-k). A spike-triggered average movie showed a conduction delay between the cell body and the nearby dendrites. Using a sub-Nyquist interpolation algorithm6 we observed electrical compartmentalization of the distal dendrites (Figs. 1i-k; Methods), as anticipated 10.
To record from multiple neurons in densely expressing samples, we fused paQuasAr3 with a trafficking motif from the soma-localized KV2.1 potassium channel11, 12, which led to largely soma-localized expression (Figs. 2a,b). We called this construct paQuasAr3-s. Patch clamp recording in acute brain slices confirmed that expression of paQuasAr3-s and the other QuasAr3 constructs used in this study did not affect membrane electrical properties (Extended Data Fig. 3).
Figure 2. Optical recording of neuronal activity in hippocampus of walking mice.
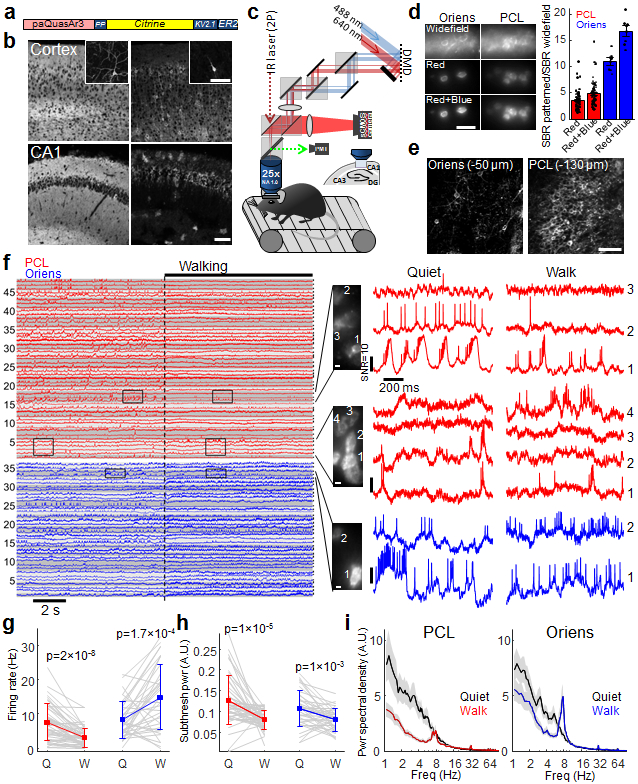
(a) Soma-localized paQuasAr3 (paQuasAr3-s). (b) Confocal images of brain slices. Broadly expressed paQuasAr3 filled the neuropil, preventing optical resolution of individual cells (left), while paQuasAr3-s resolved cell bodies (right). Insets: sparsely expressed constructs showing the difference in dendritic expression between paQuasAr3 and paQuasAr3-s. Scale bars 100 μm. Representative images, repeated in N = 2 mice per condition. (c) Optical system for simultaneous 2-photon imaging and patterned illumination with red and blue light. (d) Left, top: epifluorescence images with wide-field red illumination of paQuasAr3-s in the CA1 region of the hippocampus, in vivo. Middle: same fields of view with patterned red illumination. Bottom: Addition of patterned blue illumination increased the image contrast. Scale bar 50 μm. Right: Effect of patterned red and blue light on signal-to-background ratio (SBR) in the Oriens and PCL (PCL, n = 64 cells from N = 2 mice, Oriens, n = 7 cells from N = 2 mice, mean ± s.e.m). (e) Two-photon images of paQuasAr3-s expression in the Oriens (left) and the PCL (right). Scale bar 100 μm. (f) Fluorescence recordings from PCL (red) and Oriens (blue, N = 5 mice). Traces with similarly shaded backgrounds were acquired simultaneously. Right: Magnified views showing complex spikes, bursts, correlated activity between cells, and modulation of the spiking by subthreshold dynamics. (g–i) Effect of brain state (Quiet or Walking) on (g) firing rate (h) total power in the subthreshold oscillations, and (i) population-average power spectra in the PCL (red) and the Oriens (blue). (f–i) n = 48 cells in PCL, 36 cells in Oriens, N = 5 mice. g,h, mean ± s.d, paired t-test. i, Shading shows mean ± s.e.m.
We performed optical recordings from the hippocampus, in the Oriens layer (20–60 μm below the hippocampal surface), and in the pyramidal cell layer (PCL, up to 130 μm depth). To enhance the signal-to-background ratio in vivo, we developed a custom dual-wavelength micromirror-based illumination system to deliver both the red excitation and the blue sensitization light (Fig. 2c). The red illumination was patterned to impinge just on the cell bodies, avoiding interstitial regions which contributed to background. Patterned red illumination (5–15 mW per cell in the PCL, 5–30 mW per cell in the Oriens) enhanced the signal-to-background ratio (SBR) by a factor of 3.5 ± 0.2 in the PCL and 11 ± 1 in the Oriens (Fig. 2d, Extended Data Fig. 4). Adding patterned blue illumination, (10–60 μW per cell), enhanced the SBR by an additional 40–50%, leading to a net improvement over wide-field illumination of 4.9 ± 0.3 in the PCL and 16.7 ± 1.0 in the Oriens (Fig. 2d, Extended Data Fig. 4). Patterned red and blue illumination also substantially improved the SNR for spike detection (Extended Data Fig. 4).
To test for laser-induced heating or photodamage, we quantified the effect of the red illumination on neuronal spontaneous firing rate. We observed no change in spontaneous firing rate at up to 80 mW into the tissue (spread over 10 pyramidal cells). Numerical simulations of laser-induced heating showed that for 100 mW into the tissue, brain temperature remained < 37 °C (Extended Data Fig. 4). Optical recordings showed ~50% photobleaching over a 10 min recording (Extended Data Fig. 5), but reporter expression recovered within 1 week (Extended Data Fig. 8). High quality recordings were obtained throughout times tested (3 weeks to 3 months after viral gene delivery).
To probe brain state-dependent changes in activity, we recorded from awake, head-fixed mice walking on a motorized treadmill, during 10–15 s of rest, followed by 15 s of walking (Methods). Recordings were made from n = 5 mice and probed 48 spiking neurons in the PCL from 19 fields of view and 36 spiking neurons in the Oriens from 20 fields of view (Fig. 2f). We adapted an activity-based segmentation method13 to resolve single-neuron signals from densely expressing fields of view and validated the method via extensive simulations, composite movies, and patch clamp recordings in acute slices (Extended Data Fig. 6, Methods).
Typical recordings resolved 2–4 spiking cells simultaneously in the PCL and up to 3 spiking cells simultaneously in the Oriens (Fig. 2f). In an anesthetized animal we recorded from up to 7 spiking cells simultaneously (Extended Data Fig. 7). All fluorescence traces are displayed at the native 1 kHz recording bandwidth, without smoothing. Fluorescence traces clearly resolved hallmarks of CA1 intracellular activity, including simple and complex spikes, bursts, sub-threshold depolarizations, and θ-frequency oscillations (Fig. 2f)14-17.
Forced walking decreased the mean spike rate of PCL neurons (7.6 ± 5.3 Hz to 3.1 ± 2.8 Hz) but, consistent with prior reports 18, increased the mean spike rate of Oriens neurons (8.3 ± 5.4 Hz to 14.9 ± 9.5 Hz, Fig. 2g). Walking decreased the overall power of the subthreshold oscillations in both the PCL (1.9 ± 1.9-fold reduction) and Oriens (1.5 ± 0.8-fold reduction, Fig. 2h). In the Oriens, walking significantly increased θ-band power (6.7–8.3 Hz, 146 ± 24% increase, p = 2×10−5, n = 36 cells, paired t-test) and decreased low frequency power (2–6 Hz, 37 ± 5.4% decrease, p = 4×10−7), consistent with prior LFP recordings in freely walking animals 19. In the PCL, walking did not significantly affect θ-band power (23 ± 18% increase, p = 0.46, n = 48 cells, paired t-test) but significantly decreased low frequency power (52 ± 5.9% decrease, p = 8×10−7). Walking significantly decreased the rate of complex spikes (CS) in the PCL (0.38 ± 0.41 Hz to 0.09 ± 0.25 Hz, mean ± s.d., p = 4×10−4 paired t-test).
Intercellular correlations in subthreshold voltage reflect shared synaptic inputs. In paired patch recordings in cortex in vivo, subthreshold oscillations have been decomposed into a shared correlated component, and an uncorrelated residuum 1, 20-23. When more than two cells are measured simultaneously, one can test for multiple subthreshold signals, shared among different subsets of cells24, 25. Figures 3a, e and Extended Data Fig. 9 show that in simultaneously recorded trios, the three pairwise cross-correlations can be distinctly different in shape and amplitude. In 10 FOVs in the PCL containing more than 2 cells, the cell-to-cell cross-correlation functions were only correlated between different pairs by a mean of 0.44 ± 0.5 during rest and 0.32 ± 0.48 during walking (mean ± s.d., n = 54 pairs of pairs), establishing that there exist multiple sub-threshold signals in distinct sub-ensembles.
Figure 3. Behavior-dependent intercellular correlations in the hippocampus.
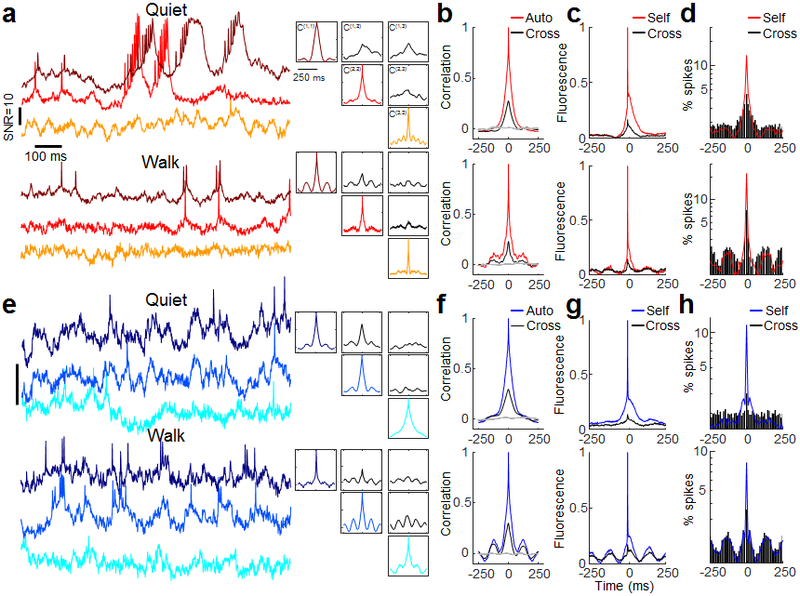
(a,e) Left: Samples of recordings from a trio of cells in (a) the PCL and (e) Oriens. Top: during quiet. Bottom: during walking. Representative traces from 19 FOVs in the PCL and 20 FOVs in the Oriens. Right: auto- and cross-correlations of the fluorescence traces, calculated from 9 s recordings in each brain region and behavioral state. The auto- and cross-correlations clearly show enhanced θ-rhythm in both brain regions during walking and differing cross-correlations between simultaneously recorded pairs of cells. (b,f) Grand average auto- and cross-correlations during quiet (top) and walking (bottom) in the PCL (n = 46 cells for autocorrelations, 43 pairs for cross correlations) and Oriens (n = 29 cells for autocorrelations, 19 pairs for cross correlations). Grey: Mean cross-correlation between randomly selected cells from different fields of view. (c,g) Spike-triggered grand average fluorescence during quiet (top) and walking (bottom) in the PCL (n = 86 pairs) and Oriens (n = 38 pairs). During walking, spikes in both layers occurred on average on the rising edge of the STA θ-rhythm, leading to a 22-degree phase shift in the PCL and 42-degree shift in the Oriens between the mean spike and the peak of the mean intracellular θ -rhythm. (d,h) Spike-triggered grand average spiking probability during quiet (top) and walking (bottom) in the PCL (d) (quiet: n = 4796 spikes, walking: n = 1983 spikes) and Oriens (h) (quiet: n = 2719 spikes, walking: n = 4258 spikes). Y axis is on a log scale.
Spike-triggered averages (STA) of the fluorescence (Fig. 3g) reflect the relation between the sub-threshold inputs and the spiking output. STA fluorescence of neighboring cells (cross-STA)20, 21 were highly heterogeneous in both layers. Some pairs of cells showed nearly identical subthreshold components of self- and cross-STA, while others showed anti-correlated self- and cross-STA (Extended Data Fig. 9). The grand average cross-STA waveforms showed a strong θ-rhythm component during walking, confirming that on average θ-rhythm inputs were strongly shared between cells (Fig. 3c,g).STA plots of the spike probability (Fig. 3d,h) revealed that in the PCL, when a cell spiked, its neighbors also had a slightly elevated spike probability, likely a consequence of the shared subthreshold inputs. In the 150 ms surrounding a spike, however, spiking cells had a period of suppressed spike probability, possibly due to a cell-autonomous refractory period26. In the Oriens, cells spiked at a relatively constant rate during the quiet period and at a rate modulated by the θ-rhythm during walking.
Passive voltage imaging alone cannot distinguish the relative contributions of excitatory (E) and inhibitory (I) inputs to the membrane potential. Optogenetic depolarization of a cell can, in principle, resolve this ambiguity by amplifying the impact of I inputs and diminishing the impact of E inputs. We used a transgenic somatostatin-Cre (SST-Cre) mouse to target expression of paQuasAr3-s and soma-localized CheRiff, and imaged in the Oriens (Fig. 4a) 27. Cells were stimulated with a variety of illumination intensities during rest and locomotion (Fig. 4b). Both red and blue illumination were patterned to illuminate only the cell bodies. For display purposes only, we subtracted blue light photo-artifact from the traces (Methods).
Figure 4. Simultaneous optogenetic stimulation and voltage imaging in hippocampal SST cells in walking mice.

(a) Optopatch construct for viral Cre-dependent co-expression of CheRiff-s and paQuasAr3-s. (b) Top: Protocol for optogenetic stimulation during quiet and walking. Middle: fluorescence of a single SST Oriens cell showing optogenetically and behaviorally modulated firing. Bottom: Spike raster from n = 25 SST interneurons from 17 FOVs in N = 2 mice. (c) Optically recorded activity of a single Oriens interneuron during 500 ms steps of optogenetic stimulation from 0 to 10 mW/mm2. Left: Quiet. Right: Walking. (d) Spike rate as a function of optogenetic stimulus strength (F-I curve) during quiet and walking (mean ± s.e.m., n = 25 SST neurons in Oriens). (e) Spontanous spike rate (no optogenetic stimulation) during quiet and walking. (f) Cell-by-cell excitability during quiet vs. walking, measured as the slope of the F-I curve between 0 to 8 mW/mm2. (g) Subthreshold dynamics during quiet and walking, with and without optogenetic stimulation (from the dataset in b). Gray: raw fluorescence. Blue, black: subthreshold dynamics (spikes digitally removed, bandpass filtered 3–20 Hz). (h) Effect of brain state and optogenetic stimulation on population-average power spectra. (i) Grand-average spike waveforms showing increased afterhyperpolarization during optogenetic stimulation (arrow). (j) Two SST cells, recorded before and after a 3-week interval. The full dataset is presented in Extended Data Fig. 10. (k) Correlation of the firing rate and excitability (F-I slope) of n = 19 SST cells recorded before and after a 3-week interval. Population-average firing rates slightly increased after 3 weeks (Quiet: 3.2 ± 2.2 to 4.2 ± 2.5 Hz, p = 0.04. Walking 11.8 ± 5.9 to 16.6 ± 11.6 Hz, p = 0.03, mean ± s.d, paired t-test). The F-I slope was stable between sessions (quiet p = 0.82, walking p = 0.67, paired t-test).
We collected a dataset of n = 25 SST cells from N = 2 mice. Single cells showed enhanced firing during walking (with no optogenetic stimulation) (Fig. 4b, Extended Data Fig. 10). We then measured the relation of mean spike rate, F, to optogenetic stimulus strength, I (F-I curve), during quiet and walking (Fig. 4c,d). The slope of the F-I curve during walking was only 75 ± 32% of the slope during quiet (n = 25 cells, mean ± s.d., p = 3×10−5, paired t-test, Fig. 4f), reflecting increased inhibition in the walking state.
To dissect further the change in E-I balance during walking, we studied the impact of targeted optogenetic stimulation on the subthreshold dynamics. As in the broader Oriens population (Fig. 2i), SST cells showed an increase in the θ-band (6.7–8.3 Hz) power during walking (125 ± 21% increase, n = 25 cells, mean ± s.d., p = 3×10−5, paired t-test, Fig. 4h). Strikingly, in the presence of cell-targeted tonic optogenetic stimulation, these same sells showed a significantly larger increase in θ-band power during walking (261 ± 43% increase, p = 4×10−4, paired t-test; θ power +/− ChR stimulation: p = 0.005, paired t-test, Fig. 4h, Extended Data Fig. 10), suggesting that the θ-rhythm in CA1 Oriens INs is predominantly driven by oscillatory inhibition, possibly from the medial septum 19,28.
Voltage-gated ion channels within each cell also contribute to both excitatory and inhibitory drives. We compared STA spike waveforms with and without optogenetic stimulation. In animals at rest, optogenetic depolarization dramatically enhanced the spike after-hyperpolarization (AHP) (Fig. 4i), revealing the role of voltage-gated K+ channels in governing the spike waveform.
To test the capability for chronic recordings, we imaged and then re-imaged SST cells at a 3 week interval (Fig. 4j,k, Extended Data Figs. 5,10). Pair-wise comparisons of n = 19 single cells at t = 0 and 3 weeks showed persistent correlations in firing rate during quiet (R = 0.74, p = 3×10−4, Pearson’s correlation), during walking (R = 0.67, p = 2×10−3), and in the effect of optogenetic stimulation on firing rate (F-I slope) during quiet (R = 0.5, p = 0.03) and during walking (R = 0.67, p = 0.001).
Genetically encoded voltage indicators promise to be a powerful tool for studying neuronal dynamics in vivo in the context of behavior, but so far have been used mostly for technical demonstrations. To create a robust tool required improvements in signal-to-noise ratio in scattering brain tissue, which came from (a) improved membrane localization, (b) photo-activation, (c) soma-localized expression, (d) soma-targeted illumination, and (e) algorithms for robust separation of in-focus signal from sources of crosstalk and background. A unique advantage of NIR GEVIs is their spectral compatibility with optogenetic actuators and GFP-based fluorescent reporters. The ability to probe subthreshold dynamics and excitability over multiple days opens the possibility to study changes in circuit dynamics associated with learning and memory.
Methods
Protein engineering
Screening pipeline.
Performance of a genetically encoded voltage indicator (GEVI) depends on several independent parameters: brightness, sensitivity, speed of response, expression level and membrane trafficking. QuasAr2 showed excellent sensitivity and speed6, but dim fluorescence and poor membrane trafficking in vivo limited application in vivo (Extended Data Fig. 1). Trafficking efficiency and expression level can differ dramatically between cell types and growth conditions, so it is important to test candidate GEVIs in neurons, ultimately in vivo.
We used the signal to noise ratio (SNR) for single spikes in cultured neurons as the screening parameter. The SNR integrates all relevant molecular features and is the parameter most relevant to applications in vivo. To facilitate rapid evaluation of constructs we used the Optopatch configuration for simultaneous optical stimulation and optical measurement of action potentials, comprising bicistronic expression of the candidate GEVI with the blue shifted channelrhodopsin, CheRiff, using a P2A peptide6.
The starting material comprised QuasAr2 fused to dark mOrange2 (QuasAr2-dmOrange2-TS-ER2), driven by the CamKII promoter. We previously found that the presence of an appended fluorescent fusion was beneficial for trafficking6, but we wished to reserve the spectrum for other uses so we introduced the mutation Y71A into the mOrange2 backbone to destroy the chromophore3, 29. TS represents the trafficking signal from Kir2.130 and ER2 represents the endoplasmic reticulum export signal FCYENEV.30
QuasAr variants were then transfected into primary neurons using the calcium phosphate transfection method and tested for spike SNR using blue light stimulation. Membrane trafficking in vivo was often worse than in vitro so constructs that showed SNR improvements in cultured primary neurons were then expressed in vivo using in utero electroporation (IUE) and tested for spike SNR in acute brain slices (Extended Data Fig. 1).
QuasAr3.
The primary goal in developing QuasAr3 was to solve the membrane trafficking problem. We tested multiple modifications. Some other microbial rhodopsins (e.g. CheRiff, ChR86 and ChR131) showed good membrane trafficking in rodent brain in vivo, so we explored chimeras comprising N-terminal fusions derived from these microbial rhodopsins. The N-terminus was added either appended to the QuasAr N-terminus or added after truncation of the first 16 residues from the QuasAr N-terminus. We also tried the N-terminal LUCY and LUCY-Rho tags which were shown to improve the surface expression of olfactory receptor32 and multiple repeats of the TS sequence30. Most modifications had neutral or negative effects on the SNR. Adding a second TS sequence in the linker between QuasAr2 and mOrange improved the spike SNR in vitro by 28%. Concatenating 3 TS sites instead of one at the C-terminus further improved SNR in vitro by 17% (Extended Data Fig. 1d-f). Mutating a putative ubiquitination site in an intracellular loop (K171R) resulted in higher expression levels, quantified by dividing the QuasAr fluorescence with the fluorescence of the GFP fused to the co-expressed CheRiff (Extended Data Fig. 1g).
In parallel to rational designs, we used a hierarchical screening approach for rapid evaluation of membrane trafficking and brightness of QuasAr point mutant and linker libraries (Extended Data Fig. 1c). We found that lentiviral gene delivery was superior to conventional lipid-based transfection in primary cultured neurons as measured by homogeneity of expression, neuronal morphology and homogeneity of membrane trafficking. Therefore, developed a screening strategy in which constructs that showed good brightness in E. coli could immediately be packaged in lentiviral vectors and tested in neurons, without laborious re-cloning. We inserted a putative prokaryotic promoter sequence BBaJ23101: TTTACAGCTAGCTCAGTCCTAGGTATTATGC and an E. coli ribosome binding site (AAAGAAGAGAAA) between the CAMKII promoter and the Kozak sequence of plasmid FCK-Arch-GFP (Addgene #22217).33 The resulting vector is termed FCK_DuEx1.0 for gene expression in both prokaryotic and eukaryotic cells as well as lentivirus production.
We created libraries of QuasAr2 fused to either eGFP, Citrine, mKate, or mRuby with a linker containing 2 randomized amino acids between QuasAr2 and the FP fusion. For library screening, E. coli colonies were transformed with linker libraries in FCK_DuEx1.0. The colonies with the brightest fluorescence were picked for lentivirus production in HEK cells and secondary screening in primary neuronal culture. For lentivirus production, HEK cells were co-transfected with the QuasAr variants in FCK_DuEx1.0, along with packaging plasmids pCMV-dR8.2 dvpr (Addgene #8455) and pCMV-VSV-G (Addgene #8454). The media from the HEK cell culture was collected 48 hours after transfection and 10% of the lentivirus-containing media was added into the primary hippocampal neuronal cultures for transduction. After 2–5 days, the membrane trafficking of QuasAr mutants was evaluated using fluorescence microscopy. The best constructs in this screen included Citrine as the fluorescent protein fusion with 4 different linkers – GE, GP, PP or PP. These constructs were then cloned in the Optopatch configuration and tested for spike SNR as described above. Two of these linkers, PP and PT, showed better SNR compared with QuasAr2-mOrange (Extended Data Fig. 1e).
We next combined the best modifications into a new construct: QuasAr2(K171R)-TS-Citrine-TS-TS-TS-ER2 which we call QuasAr3. We packaged QuasAr3 into AAV and validated its performance in acute slices (Extended Data Fig. 1h-j).
paQuasAr3.
We explored the effects of previously described Arch point mutations T27A, V59A, T99C, P196S, I129V, I129T, and A225M34 on membrane trafficking, brightness and sensitivity of QuasArs 1 and 2 using the hierarchical screening pipeline described above. We introduced these mutations separately into QuasAr2-Citrine, and estimated brightness by expressing these constructs in primary neurons and dividing the QuasAr fluorescence by the Citrine fluorescence. Initially it seemed that none of the mutations improved the QuasAr brightness. However, when we tested mutation V59A for spike SNR using blue light optogenetic stimulation, we found that fluorescence of the V59A mutant was potentiated by blue light. The photoactivation property of the V59A mutant may be related to the photoswitching property observed in the homologous bacteriorhodopsin mutant (V49A).9
To our surprise, we found that the V59A mutation led to near perfect membrane trafficking. This improved trafficking may be related to the enhanced thermal stability observed in the homologous bacteriorhodopsin mutant.9 The photoactivation property, combined with the excellent trafficking, motivated us to further characterize QuasAr3(V59A) (Fig. 1, Extended Data Fig. 3).
paQuasAr3-s and CheRiff-s.
The trafficking sequence of the KV2.1 channel was previously shown to restrict the expression channelrhodopsin 2 to the soma and proximal dendrites 11, 12. To produce soma-localized paQuasAr3 and CheRiff, we removed all the Kir2.1 trafficking sequence (TS) and introduced the KV2.1 sequence: QSQPILNTKE MAPQSKPPEE LEMSSMPSPV APLPARTEGV IDMRSMSSID SFISCATDFP EATRF either in the linker, at the C-terminus, or in both positions. In the QuasAr construct with a single KV2.1 sequence in the C-terminus, we used PP for the linker (see above). CheRiff was directly fused with GFP. These 6 constructs were then expressed in primary hippocampal neurons and visually inspected for localization. For both CheRiff and paQuasAr3, expression was most soma-restricted when the KV2.1 motif was at the C-terminus. These constructs were further characterized both in slices and in vivo (Fig. 2-4, Extended Data Fig. 3).
Spectroscopic studies.
To probe the spectroscopic properties of the QuasAr3 and paQuasAr3 proteins, we expressed them in E. coli as described before 6. Briefly, E. coli cultures were grown overnight in liquid LB medium with 200 μg/mL ampicillin. The next day the cultures were supplemented with 50 μM all-trans-retinal and 0.1% arabinose and incubated for 4 hours at 37 °C. Cell pellets were collected by centrifugation and kept in −80 °C until use. We measured the excitation and photoactivation spectra using a previously described microscope equipped with a wavelength-tunable supercontinuum light source and an EMCCD detector 35.
Optical systems
Imaging HEK cells and primary neurons.
Experiments were conducted on a home-built inverted fluorescence microscope equipped with 405 nm, 488 nm, 532 nm, and 640 nm laser lines and a scientific CMOS camera (Hamamatsu ORCA-Flash 4.0). Laser beams were combined using dichroic mirrors and sent through an acousto-optic tunable filter (AOTF; Gooch and Housego TF525-250-6-3-GH18A) for temporal modulation of intensity of each wavelength. The beams were then expanded and focused onto the back-focal plane of a 60× water immersion objective, numerical aperture 1.2 (Olympus UIS2 UPlanSApo 60×/1.20 W). Imaging of fluorescent proteins was performed at illumination intensities of 20–40 mW/mm2. Imaging of QuasAr fluorescence was performed at an illumination intensity of 4 W/mm2. Stimulation of CheRiff was performed at an illumination intensity of 0.4–1.2 mW/mm2. For fast data acquisition, a small field of view around the cell of interest was chosen at the center of the camera to achieve a frame rate of 1 kHz.
Imaging acute slices and live mice.
Experiments were conducted on a home-built upright fluorescence microscope equipped with 488 nm and 640 nm laser lines and a scientific CMOS camera (Hamamatsu ORCA-Flash 4.0). The 488 nm line was sent through an acousto-optic tunable filter (Gooch and Housego 48058-2.5-.55) for intensity modulation, and then expanded and focused onto the back-focal plane of the objective.
In the first generation of the microscope, we used 140 mW 640 nm laser expanded to a single ~50 μm circular spot, and a 20x NA1.0 objective (Olympus XLUMPLFLN 20×/1.0 W). This configuration was used for the olfactory bulb in vivo experiments (Fig. 1h). Data were acquired on a small field of view around the cell of interest at a frame rate of 1 kHz.
In the second generation of the microscope, we used an 8 W, 640 nm diode bar laser (DILAS MB-638.3-8C-T25-SS4.3) and patterned the illumination using a digital micromirror device (DMD, Vialux, V-7000 UV, #9515) to illuminate specific sub-cellular structures and to avoid illuminating interstitial regions of the brain slice. We used a 16× 0.8 N.A. objective with 3 mm working distance (Nikon CFI75 LWD 16xW). Sampling rate was 1 kHz. This configuration was used for the single cell in vivo measurements in the hippocampus (Fig. 1i-k).
In the third generation of the microscope, we patterned both the 488 nm and 640 nm lasers to achieve overlapping red and blue spots in the sample. The beams were then combined on a dichroic beamsplitter and aligned to be side-by-side and propagating parallel. The combined beam was then sent to a DMD, so half of the DMD patterned one wavelength and half of the DMD patterned the other wavelength. The two wavelengths were then split and recombined with a pair of dichroic mirrors to form a single beam with independently patterned blue and red excitation (Fig. 2c). Cylindrical lenses were used to correct for aberrations introduced by slight warping of some dichroic mirrors.
The patterned epi-illumination was combined with a home-built scanning two photon system. The visible and near-infrared beams were combined using an 875 nm long-pass dichroic mirror (Semrock). A 532 nm notch dichroic mirror (Semrock) directed green emission (from Citrine or GFP) to a photomultiplier, while near infrared fluorescence (from QuasAr) was sent to a Hamamatsu sCMOS camera. This configuration was used for all multicellular recordings in the hippocampus in vivo (Figures 2-4) using a 25x NA1.0 objective with 4 mm working distance (Olympus XLPLN25XSVMP2). Red laser intensity was 12 W/mm2 and sampling rate was 1 kHz.
This configuration was also used for the patch clamp experiments in slices (Fig. 1e-j, Extended Data Figs. 1j, 3i-k) with a 20x objective (Olympus XLUMPLFLN 20×/1.0 W), red laser intensity 12 W/mm2 and sampling rate 1 kHz, and for measurements from brain slices of transgenic mice (Extended Data Fig. 2) at 500 Hz sampling rate.
Primary neuronal culture and gene delivery
All procedures involving animals were in accordance with the National Institutes of Health Guide for the care and use of laboratory animals and were approved by the Harvard University Institutional Animal Care and Use Committee (IACUC).
Hippocampal neurons from P0 rat pups were dissected and cultured on rat glial monolayers as described before6. Briefly, P0 neurons were plated in neurobasal-based medium (NBActiv4, BrainBits LLC.) at a density of 30,000–40,000 cm−2 on the pre-established glial monolayers. At one day in vitro (DIV), cytarabine was added to the neuronal culture medium at a final concentration of 2 μM to inhibit further glial growth.
Neurons were transfected between DIV 7–10 via the calcium phosphate transfection method. Measurements on neurons were taken between DIV 14–18.
Imaging and electrophysiology in HEK cells and primary neurons
HEK293T cells (ATCC; CRL-11268) were cultured and transfected as described before6. Briefly, HEK-293 cells were grown at 37 °C, 5% CO2, in DMEM supplemented with 10% FBS and penicillin-streptomycin. Cells were tested negative for mycoplasma. Cells were transfected with GEVI constructs under the upstream CMV promoter of the FCK plasmid. 200–400 ng of plasmid DNA was transfected using Transit 293T (Mirus) following the manufacturer’s instructions and assayed 48 hours later. The day before recording, cells were re-plated onto Matrigel coated glass-bottom dishes (In Vitro Scientific) at a density of ∼10,000 cells/cm2.
All imaging and electrophysiology were performed in extracellular buffer containing (in mM): 125 NaCl, 2.5 KCl, 3 CaCl2, 1 MgCl2, 15 HEPES, 30 glucose (pH 7.3) and adjusted to 305–310 mOsm with sucrose. A gap junction blocker, 2-aminoethoxydiphenyl borate (50 μM, Sigma), was added to HEK cells to eliminate electrical coupling between cells. Primary neurons were supplemented with excitatory synaptic blockers (either 20 μM NBQX or 20 μM CNQX, both from Tocris).
Optopatch measurements and the simultaneous whole-cell patch clamp and fluorescence recordings were acquired on the home-built, inverted epifluorescence microscope described above. For simultaneous electrophysiology and imaging, filamented glass micropipettes (WPI) were pulled to a tip resistance of 5–10 MΩ, and filled with internal solution containing (in mM):125 potassium gluconate, 8 NaCl, 0.6 MgCl2, 0.1 CaCl2, 1 EGTA, 10 HEPES, 4 Mg-ATP, 0.4 Na-GTP (pH 7.3); adjusted to 295 mOsm with sucrose. Pipettes were positioned with a Sutter MP285 manipulator. Whole-cell, voltage and current clamp recordings were acquired using an Multiclamp 700B amplifier (Molecular Devices), filtered at 2 kHz with the internal Bessel filter and digitized with a National Instruments PCIE-6323 acquisition board at 10 kHz.
Gene targeting in ES cells and generation of knock-in Cre-dependent reporter mice
To generate the Ai155 targeting vector, Optopatch3 comprising QuasAr2(K171R)-TS-Citrine-TSX3-ER2-P2A-CheRiff-TS-HA was inserted into the cre-dependent TIGRE 2.0 construct36. Gene targeting was performed at the Allen Institute for Brain Science. Targeting of the transgene cassettes into the TIGRE locus was accomplished via Flp-recombinase mediated cassette exchange (RMCE) using circularized targeting vector, a CAG-FlpE vector (Open Biosystems), and a Flp recombinase landing pad ES cell line derived from G4 cells7. Correctly targeted ES cells were identified via PCR, qPCR, and Southern blots. ES clones were karyotyped and verified to be chromosomally normal. ES clone injection was performed at the Harvard University Genetic Modification Facility. Optopatch-positive ES clones were injected into C57BL/6J blastocysts to obtain chimeric mice. Chimeric mice were bred with C57BL/6J mice to obtain F1 Floxopatch+/− mice. The Ai155 mouse line is on a mixed C57BL/6J;C57BL/6N genetic background. The Ai155 mouse line has been deposited in Jackson lab with Stock No: 029679.
Ai155 mice were crossed with the following Cre driver lines: CKII-Cre (gift from Venkatesh Murthy), Somatostatin (SST)-IRES-Cre (JAX #013044) and Rbp4-Cre (MMRRC-031125-UCD, gift from Bernardo L. Sabatini,)
Genotyping.
The presence of QuasAr3 was determined with the primer pair: 5’-GCTGGTCTCCAACTCCTAATC −3’ 5’-CTGTATCTGGCTATGGCCG −3’. A 1.07 kb amplicon indicated presence of the gene. Homozygous mutant insertion was determined with the primer triplets: 5’- GTG TAG CCC TGG CTT TTC TG-3’, 5’-GAA CTC ACA GTG GCC AGT CA-3’, 5’- TCC CCT GGC ACA ACG TAA G-3’. These yielded a 295 bp amplicon for mutant band and a 468 bp amplicon for wild type band.
Imaging and electrophysiology in acute slices
In utero electroporation (IUE).
In utero electroporation was performed in timed pregnant CD-1 mice as previously described.37 The day of vaginal plug was designated as embryonic day 0.5 (E0.5), while the day of birth was designated as postnatal day 0 (P0). Briefly, 1 μL of endonuclease-free purified DNA (3.75 μg/μL comprising 2.75 μg/μL of the candidate GEVI construct and 1 μg/μL of tdTomato control) in sterile PBS mixed with 0.005% Fast Green was injected into the lateral ventricle of embryonic day 14.5 mice under ultrasound guidance (Vevo 770, VisualSonics). Five 35-volt pulses of 50 ms duration at 1 s intervals were delivered outside the uterus in appropriate orientation using 1 cm diameter platinum electrodes and a CUY21EDIT square-wave electroporator (Nepa Gene). Tested constructs were co-electroporated with either CBIG-tdTomato plasmid (gift from Jeffery Macklis) or pCAG-GFP (gift from Connie Cepko, also available as Addgene 11150) for visual identification of electroporated embryos using a fluorescence stereoscope.
AAV virus preparation.
Constructs were cloned into an AAV transfer plasmid either with hSyn promoter (Addgene #51697) or for Cre-dependent expression with the CAG promoter (Addgene #22222)33. To pack the Cre-dependent Optopatch constructs into the 4.5 kb size limitation of AAV, we had to modify the both the construct and the transfer plasmid. First, we replaced the GFP fusion of CheRiff with a short HA tag. Second, we used a Cre-dependent transfer plasmid with an hSyn promoter instead of CAG (Addgene #44362) 38. Third, we swapped the 480 bp bGH polyA sequence with 120bp SV-40 polyA. Viruses were produced by the Gene Transfer Vector Core at Massachusetts Eye and Ear Infirmary & Schepens Eye Research Institute (MEEI), Harvard Medical School. All experiments used AAV serotype 2/9. All AAV plasmids were deposited with Addgene as follows:
hSyn-QuasAr2-mOrange (Extended Data Fig. 1h, Addgene #107705)
hSyn-QuasAr3-Citrine-P2A-CheRiff (Extended Data Fig. 1h, Addgene #107700)
CAG-FLEX-QuasAr3-Citrine (Extended Data Figs. 1i-j, 3i, Addgene #107701)
CAG-FLEX-paQuasAr3-Citrine (Fig. 1d-k, Extended Data Fig. 3i, Addgene #107702)
CAG-FLEX-paQuasAr3s-Citrine (Figs. 2-3, Extended Data Figs. 3k, 4, 6-9, Addgene #107703)
hSyn-Dio-paQuasAr3s-Citrine-P2A-CheRiff-s (Fig. 4, Extended Data Figs. 5, 10, Addgene #107704)
Virus injection for acute slices measurement.
For acute brain slice experiments, we injected AAV virus in C57BL/6 P0–P1 mice of either sex. Virus was diluted and injected at final titers of 1×1013 GC/mL. To achieve sparse expression, Cre dependent constructs were mixed with hSyn-Cre virus at final titers of 1 to 5×1010 GC/mL.
P0–P1 pups were cryo-anesthetized and immobilized dorsal side up under a stereotaxic setup. Injections were made using home-pulled micropipettes (Sutter P1000 pipette puller), mounted in a microinjection pump (World Precision Instruments Nanoliter 2010) controlled by a microsyringe pump controller (World Precision Instruments Micro4). The micropipette was positioned using a stereotaxic instrument (Stoelting). Pups were injected in the left hemisphere, 0.9 mm lateral and 0.9 mm anterior to lambda. Starting at a depth of 1.0 mm beneath the surface of the skull, virus injections (40 nL, 5 nL/s) were performed at 0.1 mm increments as the pipette was withdrawn. Pups were placed back in their home cage once they were awake.
Acute slice preparation.
Acute brain slices were prepared from P14–P28 mice as described before.3 Briefly, mice were anesthetized with isoflurane, and then subjected to intracardiac perfusion with ice-cold slicing solution containing (in mM): 110 choline chloride, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, 0.5 CaCl2, 7 MgCl2, 11.6 Na-ascorbate, and 3.1 Na-pyruvate and saturated with carbogen (95% O2, 5% CO2). Mice were then decapitated and the brains were rapidly dissected and sliced into 300 μm coronal sections using a vibratome (Leica VT 1200S). Mice < P21 were directly decapitated without intracardiac perfusion. Slices were incubated for 45 min at 34 °C in a carbogenated artificial CSF (ACSF) containing (in mM): 127 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, 2 CaCl2, and 1 MgCl2. The osmolarity of all solutions was adjusted to 300–310 mOsm and the pH was maintained at 7.3 under constant bubbling with carbogen.
Imaging and electrophysiology in acute slices.
Acute slices from either in the visual cortex or in CA1 were imaged on a home built upright microscope described above. Measurements were conducted in carbogen-saturated ACSF at room temperature in slices from transgenic mice experiments or at ~31 °C in all the patch clamp experiments. ACSF was perfused at a rate of 2 mL/minute. For simultaneous imaging and whole cell patch clamp measurements, filamented glass micropipettes (WPI) were pulled to a tip resistance of 4–6 MΩ, and filled with internal solution containing (in mM): 125 potassium gluconate, 8 NaCl, 0.6 MgCl2, 0.1 CaCl2, 1 EGTA, 10 HEPES, 4 Mg-ATP, 0.4 Na-GTP (pH 7.3); adjusted to 295 mOsm with sucrose. Pipettes were positioned with a Sutter MP285 manipulator. Whole-cell, current clamp recordings were acquired using an MultiClamp 700B amplifier (Molecular Devices), filtered at 2 kHz with the internal Bessel filter and digitized with a National Instruments PCIE-6323 acquisition board at 10 kHz. Action potentials were evoked by 10 s ramps of current injection, from 0 to 200 pA. To evoke subthreshold PSPs we positioned a concentric bipolar electrode (FHC) in cortical layer 5–6 and delivered trains of stimuli (10 × 100 μs at 2 Hz). Stimulus strength was ramped during the trains and was adjusted to evoke subthreshold PSPs.
Confocal imaging.
Acute slices were fixed in 4% paraformaldehyde (PFA) and confocal fluorescence imaging was performed on an Olympus FV1000 confocal microscope at the Harvard Center for Brain Sciences microscope facility.
Cranial windows, virus injections, training, and in vivo imaging
Virus injection and cranial window surgery.
10–18-week-old C57BL/6 or heterozygous Somatostatin-Cre mice (male and female) were deeply anesthetized with 2% isoflurane and maintained with ~1% isoflurane throughout the surgery. The skull was exposed and thoroughly dried and a 3 mm round craniotomy was opened using a biopsy punch (Miltex). For Olfactory bulb (OB) imaging, the craniotomy covered both OBs as previously described39. For CA1 imaging, the craniotomy center was 1.8 mm lateral, 2.0 mm caudal of bregma. Virus was then injected in 1–3 locations in the center of the craniotomy.
For single-cell measurements with paQuasAr3 (Fig. 1), sparse expression was achieved by mixing CAG-FLEX-paQuasAr3 AAV virus (final titer 2×1013 GC/mL) with hSyn-Cre virus (final titer 2×1010 to 1×1011 GC/mL). For dense expression of paQuasAr3-s, we used low titer CAG-FLEX-paQuasAr-s virus (final titer 6×1011 GC/mL) or hSyn-DiO-paQuasAr3-s-P2A-CheRiff-s (final titer 1.6×1012 GC/mL), mixed with higher titer CKII(0.4)-Cre virus (UPenn vector core, final titer 7×1011 GC/mL). Injections were made using home-pulled micropipettes (Sutter P1000 pipette puller), mounted in a microinjection pump (World Precision Instruments Nanoliter 2010) controlled by a microsyringe pump controller (World Precision Instruments Micro4). The micropipette was positioned using a stereotaxic instrument (Stoelting Digital Mouse Stereotaxic Instrument). For CA1 expression, injections were made −1.5 mm to −1 mm from Dura, at 0.1 mm increments (40 nL per depth, 5 nL/s). For OB expression, injections were made from −0.4 to 0 mm from dura at 0.1 mm increments (40 nL per depth, 5 nL/s). Brain surface was kept moist with saline throughout the injection.
In the OB, a 3 mm round #1 cover glass (Harvard apparatus) was then placed on the OB surface, and sealed with cyanoacrylate and dental cement (C&B Metabond). A titanium bar was glued behind the window, skin was sutured and animal was sent to recovery.
The procedure for imaging in CA1 followed Dombeck and coworkers.40. Briefly, a cannula was prepared prior to the surgery and comprised a 1.5 mm segment of a 3 mm outer diameter thin walled stainless steel tube (MicroGroup). A 3 mm diameter #1 round coverglass (Harvard apparatus) was cemented to one end of the tube using UV curable adhesive (Norland Products) and cured for at least 5 minutes on a standard lab UV table. Following hippocampal virus injection, we removed the dura, and then slowly aspirated the cortex while continuously irrigating with saline until bleeding stopped. After exposure of the external capsule, a small region of the capsule in the center was gently removed, exposing the CA1 surface. To reduce brain motion during locomotion, in some of the animals a small amount of Kwiksil (WPI) was applied to the surface of the brain. The cannula was then inserted and cemented to the skull with dental cement (C&B metabond). After the cannula cured, a titanium headplate (similar to Ref. 41) was glued around the cannula and any exposed skull was covered with dental cement. Animals were returned to their home cage for recovery and treated for 3 days with Carprofen (5 mg/kg) and Buprenorphine (0.1mg/kg) twice a day. To avoid damage to the implant, mice were housed in separate cages.
Imaging anesthetized animals.
Imaging typically started 3 weeks post-surgery. Mice were lightly anesthetized (0.7–1% isoflurane) and head-fixed under the upright microscope (see above) using the titanium head plate. Eyes were kept moist using ophthalmic eye ointment. Body temperature was continuously monitored and maintained at 37 °C using a heating pad (WPI). A typical imaging session lasted 1–2 hours, and then animals quickly recovered and returned to their home cage.
Motorized treadmill.
A home-built treadmill was composed of two 3D printed plastic wheels (5 cm wide, 10 cm in diameter). We used a 5 cm wide, 180 cm long velvet belt (McMaster Carr #88015K1). Treadmill speed was regulated using a computer controlled small electric motor (Pololu #3042). Linear speed was 5 to 10 cm/s.
Imaging awake, walking animals.
Head-fixed animals were imaged while walking on a home-built motorized treadmill. Before imaging sessions started, mice were habituated to head restraint and walking by training them at least 3 times, every 24 hours, for 15–30 minutes. During each training session mice were first habituated to head restraint until completely relaxed, and then started walking on the treadmill. Each walking period lasted 1 minute followed by at least 2 minutes of rest. Walking speed was 5 to 10 cm/s. To adjust mice to imaging conditions, at least one training session took place under the microscope with the objective on top of the cranial window. Awake animals were imaged 3 weeks to 3 months from virus injection without any detectable effects on cell health. >95% of SST cells could be reidentified and re-recorded after a 3-weeks interval (N = 2 mice).
For experimental runs, we first used a protocol of 10 s rest following by 3× 15 s periods of increased speed (5, 7.5 and 10 cm/s) followed by additional 10 s rest (65 s in total, Extended Data Fig. 8). We observed that the difference in firing patterns was more starkly different between rest and walking than between different walking speeds, so we switched to a protocol of 15 s rest followed by 15 s walking at 10 cm/sec repeated twice (60 s in total). In each field of view (FOV), we repeated the protocol 2 to 5 times. Imaging session lasted up to 1 h and then animals were returned to their home cages.
For combined voltage imaging and optogenetic stimulation (Fig. 4, Extended Data Fig. 10), we performed a 75 s protocol, composed of 500 ms steps of blue light at successively greater intensities (nominally 0.2–10 mW/mm2, not accounting for light scatter), 5 s ramps of blue light (0–10 mW/mm2) and 8 s periods of constant illumination (5 mW/mm2), repeated during quiet and walking epochs.
Data analysis
Statistics.
Statistical tests were performed using standard Matlab functions (Mathworks). For two-sample comparisons of a single variable, a two-sided Student’s t-test (paired or unpaired) was used unless otherwise noted. The experiments were not randomized, and the investigators were not blinded to the experimental condition. Sample size was based on reports in related literature and was not predetermined by calculation.
Extracting fluorescence from single cell movies.
First, an estimate of the photobleaching baseline was constructed from the mean intensity of the whole FOV by applying sliding minimum filter, followed by a sliding mean filter. Each frame of the movie was then divided by this baseline. Fluorescence values were extracted from corrected movies in one of two ways. When single cells were well isolated from other fluorescent sources, we used the maximum likelihood pixel weighting algorithm described in Kralj et al.42 Briefly, the fluorescence at each pixel was correlated with the whole-field average fluorescence. Pixels that showed stronger correlation to the mean were preferentially weighted. This algorithm automatically found the pixels carrying the most information, and de-emphasized background pixels. Alternatively, we used a user-defined region of interest and calculated fluorescence from the unweighted mean of pixel values within this region. For calculations of ΔF/F, background fluorescence from a cell-free region was subtracted from the baseline fluorescence of the cell.
Sub-frame interpolation of AP timing (SNAPT).
Dendritic propagation spike width and delay were calculated using the SNAPT algorithm as described previously.6 Briefly, spikes were identified from the whole-cell fluorescence trace. Spike timing was used to construct a spike-triggered average movie. The mean spike waveform was then used as a template and fit to the spike waveform at each pixel, using as fitting parameters: vertical offset, amplitude, time-shift, and width (uniform dilation in time). The time-shift and width parameters are displayed in Fig. 1k.
Extracting fluorescence and subcellular voltages.
Inference of subthreshold voltages required explicit steps to account for (1) possible contamination from motion artifacts, (2) possible crosstalk between nearby cells whose true subthreshold fluctuations were correlated, and (3) possible crosstalk from out-of-focus background created by one or more cells.
Extracting fluorescence from sparse multicellular movies in behaving animals (Oriens data).
Movies were first corrected for motion using the NoRMCorre algorithm.43 Movies were then corrected for photobleaching by dividing the movie by an exponential fit of the mean fluorescence. Activity-based image segmentation was performed on the spiking component of the signal. To remove subthreshold signals for segmentation purposes, movies were high-pass filtered in time with a 50 Hz high-pass filter. Movies were then segmented semi-automatically using principal components analysis followed by time-domain independent components analysis (PCA/ICA).44 ICA produces an arbitrary number of candidate cells. We set the maximum number of sources up to 15 and then further eliminated traces that did not correspond to cells by manually inspecting the traces and corresponding spatial maps. In some cases we divided the movie into sub-movies based on patterns of illumination from the DMD masks (Fig. 2d), and performed the PCA/ICA analysis separately in each sub-movie. The spatial masks from PCA/ICA were then applied to the movies without high-pass filtering to extract fluorescence traces that included subthreshold dynamics.
Extracting fluorescence from dense multicellular movies in behaving animals (PCL data).
The image segmentation pipeline was based on Ref. 13, with modifications as follows.
Main steps:
NoRMCorre correction of x-y motion 43
Trimming edges of movie to remove pixels that entered or exited the field of view
Photobleach correction with b-spline fit
PMD denoiser with parameters optimized on simulated data
From each pixel, regress out residual motion artifacts using NoRMCorre-estimated motion trajectories
Manually crop blood vessels
Select areas of background to initialize background estimator
Superpixel-based NMF demixing to calculate spatial footprints of cells and background
Regress the spatial footprint of cells out of the background (using a constant intercept term) to update spatial footprint of cells and background
Apply the updated spatial and background footprints to calculate the temporal traces from the full denoised movie
Movies were first corrected for x-y translational motion using the NoRMCorre algorithm 43, and trimmed to the largest frame size that was in the field of view at all times. Each pixel of the movie was then corrected for photobleaching by subtracting a b-spline fit of the pixel’s fluorescence with knots 5000 frames (5 s) apart. A penalized matrix decomposition (PMD) denoiser was next used to remove temporally uncorrelated noise from the movie13. This method is a generalization of principal components analysis: it divides the movie into blocks and within each block decomposes the movie into the sum of low-rank “signal” and residual ‘noise’ that is uncorrelated pixel-to-pixel; this residual is then discarded.
Brain motion caused shifts of cells relative to the patterned illumination masks targeted to individual cells. Frame-by-frame translations corrected for brain motion, but introduced spurious apparent motion into the patterned illumination masks. This relative motion of cells and masks introduced spurious pixel-level motion artifacts, even in the motion-corrected movies. To reduce the effect of these artifacts, motion-correlated signals were projected out at the level of each pixel in the denoised movie, using a general linear model. The regressors were the motion signals x, y, x2, and y2 and xy, as computed by NoRMCorre. This procedure was performed independently in successive epochs of 5000 frames (5 s). Regions of the movie with visible blood flow were then manually masked to avoid high-frequency noise from passing blood cells.
Following Ref. 13, we defined “superpixels” as collections of nearby pixels with correlated dynamics. We defined “pure” superpixels via an iterative procedure that chose the sparsest temporal signals from the remaining superpixels and then projected out this signal from the other superpixels. These “pure superpixels” served as seeds to extract single-cell activity from the movie. Regions without pure superpixels, were manually selected as “background” to seed the estimator of the background dynamics. A singular value decomposition on the background pixels yielded temporal and spatial components that initialized the background in the demixing analysis described below.
The movies were decomposed into temporal and spatial components through a local non-negative matrix factorization (NMF) on the denoised data; this NMF minimized the summed squared residual of the movie after modelling both cell activity and a rank r background13. To enforce non-negativity, a pixel-wise minimum subtraction was first performed on the movie. For the in vivo movies analyzed, a rank-2 background model was used to capture different time varying components of the background, while preserving cell signal. Higher rank background models did not improve performance and sometimes spuriously assigned cell signals to background.
The spatial components, B, of the background extracted from the NMF demixer typically contained contamination from cell footprints, A. With a background of rank r, n total cells in the FOV, and d pixels in a frame, B represents the d×r matrix of images of the background components, and A is a d×n matrix of cell spatial footprints. To remedy this crosstalk, the spatial footprints of the cells were removed from the images of the background by a least-squares fit to create updated background spatial components B*. The use of B* facilitated the recovery of correlations between the cells and background lost in the demixer.
Finally, a least-squares estimate was used to regress the cell spatial footprints and updated background images on the full denoised and motion-corrected movie to find the corresponding fluorescence traces for signal and background. No non-negativity constraints were imposed on these traces.
Validation of image segmentation algorithm with simulated data.
We used numerical simulations to test and optimize the image segmentation code. First, we produced synthetic movies containing n partially overlapping ‘cells’ (disks) and a spatially heterogeneous time-varying background. We assigned a unique time-trace to each cell and to the background, as follows. We simulated n + 1 independent waveforms using an Ornstein-Uhlenbeck process, a numerically simple model that captures the approximate statistics of neuronal sub-threshold voltage dynamics 45. We then used a Gram-Schmidt procedure to ensure that these waveforms were precisely orthonormal. To challenge the image segmentation algorithms with realistic correlated subthreshold signals, we then constructed correlated source waveforms, as follows. We selected a desired degree of sub-threshold cross-correlation between the cells, and between each cell and the background. These correlations ranged from 0 (easy to segment) to 1 (impossible to segment). We used a Cholesky decomposition of the correlation matrix to compute the required linear combinations of orthonormal input functions. These n + 1 linear combinations were assigned as the sub-threshold fluctuations to the n cells and to the background.
We then assigned spikes to the cells with a mean spike rate of 8 Hz and Poisson-distributed interspike intervals. Finally, we added Poisson-distributed shot noise to each pixel at a level that approximately matched the noise level in our recordings. These simulations were designed to match the spatial and temporal correlation structure, and noise statistics, of real data as closely as possible—but with known ground-truth.
We then ran the movies through the image segmentation algorithm, and compared the extracted waveforms to the input waveforms. We quantified performance by calculating the cross-correlation of the extracted signals and the input signals. We sought algorithms that accurately extracted the input signals and that did not introduce spurious cell-cell or cell-background correlations. Extended Data Fig. 6 shows that under realistic conditions the image segmentation algorithm reliably captured the waveforms and correlational structure of the input.
Validation of image segmentation algorithm with composite data.
To test the image segmentation algorithm further, we challenged it with composite movies comprised of real data. We took a series of movies in vivo of individual well-isolated cells either in-focus or at 20 μm defocus. We ran the image segmentation algorithm on each movie to extract the underlying single-cell waveforms. We then created composite movies by adding together pairs of independently recorded movies, translated such that ~50% of each cell overlapped with the other. In this way, mixed signals contributed substantially to the movie, and the signal and noise properties were the same as in real data. These composite movies represented a ‘worst case’ scenario: in real data, we did not observe cells with such large overlap and such small focal offsets.
We ran the composite movies through the image segmentation algorithm, and compared the extracted waveforms to the waveforms extracted from the individual single-cell movies. Extended Data Fig. 6 shows that the image segmentation algorithm faithfully reproduced the input signals and their correlational structure.
Selection and scaling of fluorescence recordings.
Some morphologically distinct cells did not spike during the recording session, consistent with previous whole cell patch clamp recordings in the CA116, 17, and were excluded from the analysis. Due to cell-to-cell variations in DC background level, the offset of fluorescence traces was not judged to be meaningful. For presentation purposes, traces were scaled by their baseline noise, defined as the standard deviation after removal of the spikes and high-pass filtering at 100 Hz (to remove slow oscillations). For presentation purposes, in the ‘all-optical’ data (Fig. 4, Extended Data Fig. 10), we removed the blue light photoactivation artifacts, as follows. We averaged together the fluorescence traces from all cells to create a template function characterizing the photo-artifact. We used a linear regression to scale this crosstalk function for each single cell, and subtracted the estimated crosstalk. We also divided by the crosstalk function to correct for blue light-dependent changes in voltage sensitivity. All analyses of spiking and subthreshold dynamics were performed on the raw data, without crosstalk correction. To ensure that the subthreshold power spectrum analysis (Fig. 4g-h, Extended Data Fig. 10b was not biased by the blue light photoactivation of paQuasAr3, we only used data from the 8 s steady illumination epochs and we normalized traces by the spike amplitude. This normalization enabled comparisons of subthreshold amplitudes between conditions (+/− walking, +/− optogenetic stimulation) within a population of cells. Due to systematic low-pass filtering of the spike amplitudes by the reporter kinetics and camera exposure, we did not attempt to convert this normalization into an absolute millivolt scale.
Spike detection, correlations and spike-triggered averaging.
A simple threshold-and-maximum procedure was applied for spike detection. Fluorescence traces were first high-pass filtered, and initial threshold was set at 3 times the noise level. This threshold was then manually adjusted if needed. All analyses of brain state-dependent activity (Fig. 3) were performed on segments of the recordings comprising the last 9 s of the first rest period (quiet) and the first 9 s of the first walk period.
Fluorescence signals were first converted to ΔF/F0 where F0 was the mean fluorescence in the epoch. The data were mean-subtracted in 1 s windows to correct for slow drifts in baseline fluorescence. To estimate the statistical errors in the cross correlations, we calculated the cross-correlations between randomly selected cells from distinct fields of view. Spikes were not removed from the traces for calculations of auto- and cross-correlations.
Spike triggered averages of the fluorescence (Figs. 3c,g, Extended Data Fig. 9) were calculated for each pair of simultaneously recorded cells, normalized to the range 0–1, and then averaged across all cell pairs. Spike triggered histograms of spike timing (Figs. 3d,h) were constructed for each spike with 10 ms bins. These histograms were then summed over all spikes and all cells and divided by the total number of spikes to yield a percentage of spikes in each time bin.
Subthreshold power and power spectra.
To compare subthreshold powers (Fig. 2h) and power spectra (Figs. 2i, 4h) across conditions, signals were normalized by the optically recorded spike height. To determine whether changes in the power spectra were due to changes in spike-rate or changes in subthreshold dynamics, we compared power spectra calculated from ‘raw’ traces with power spectra calculated from traces where the spikes were digitally removed and replaced with smooth interpolations of the surrounding data. These two approaches yielded qualitatively similar power spectra, establishing that the bulk of the power in the traces was in the sub-threshold components.
Heating simulation.
To simulate optical tissue heating, we adapted a finite differencing model from Ref. 46, further optimized in Ref. 47. This model accommodates heat conduction, metabolic heating and cooling by blood prefusion and by heat sinking from the objective, glass window and immersion water. We further modified the model for our imaging conditions: Illumination of an r = 60 μm area, glass window attached to a metal cannula, and 4 mm working distance objective. We used tabulated optical properties of brain tissue at 640 and 920 nm to compare heating effects from 1-photon QuasAr imaging vs. conventional 2-photon microscopy.
Extended Data
Extended Data Fig 1. Development and characterization of QuasAr3.

(a) Screening pipeline. Rationally designed constructs were cloned in the Optopatch configuration, expressed in primary neurons and tested for spike SNR using light-induced spikes. Constructs with improved SNR were then expressed in vivo using in utero electroporation (IUE) and tested for spike SNR in acute slices. The process was repeated iteratively. (b) Examples of SNR measurements in cultured neurons. Left: wide-field epifluorescence images of GFP fused to CheRiff, an opsin with excellent membrane trafficking. Middle: Fluorescence of QuasAr mutants. Scale bar 10 μm. Right: QuasAr fluorescence transients in response to optogenetically induced spikes (10 ms blue light stimulation at 1 mW/mm2). Each construct was tested on at least 5 cultured neurons. (c) Hierarchical screen for improved membrane trafficking of QuasAr variants (see Methods for details). Diagram: schematic of the FCK_DuEx1.0 construct and overview of the screening pipeline. E. coli colonies were transformed with libraries in FCK_DuEx1.0. The colonies with the brightest fluorescence were picked for lentivirus production and secondary screening in primary neuronal culture. Images: example images of the FP channel of QuasAr2-FP fusions: i. mOrange; ii. mRuby2; iii. mKate2; iv. Citrine. Scale bar 10 μm. (d) SNR of N-terminal modifications compared with QuasAr2. All constructs showed reduced SNR (see Methods for details). (e) Replacing mOrange2 with Citrine as a fusion protein improved the trafficking only with two specific linkers. (f) Adding additional TS sequences at the linker and C terminal improved the spike SNR. (g) The mutation K171R increased the QuasAr expression level, quantified by normalizing QuasAr fluorescence by the fluorescence of the co-expressed CheRiff-GFP. (d-g) all error bars are mean ± s.e.m., 1-tail t-test. (h) Top: diagram of the QuasAr2 and QuasAr3 constructs. Bottom: Confocal images of brain slices expressing QuasAr2 and QuasAr3. Scale bar 500 μm. Insets: single cell bodies, scale bar 10 μm. Representative images from N = 2 mice (QuasAr2) and N = 3 mice (QuasAr3). (i) Confocal images of brain slices expressing Cre-dependent QuasAr3 with sparsity controlled by co-expression of hSyn-Cre. Scale bar 50 μm. (j) Simultaneous fluorescence and patch clamp recordings from two neurons expressing QuasAr3 using AAV virus in acute brain slice. Left: image of QuasAr3 fluorescence in the soma. Scale bar 10 μm. Middle: spiking during ramp current injection. Right: mean spike, overlay of fluorescence and voltage. Inset: boxed regions showing correspondence of optical and electrical recordings of sub-threshold voltage overlaid. See Extended Data Fig. 3 for statistics. Scale bar 10 μm.
Extended Data Fig. 2. Mapping neuronal excitability in Cre-on Optopatch3 transgenic mice (line Ai155).

(a) Construct design for a Cre-dependent Optopatch3 transgenic mouse. (b) Representative traces for all-optical electrophysiology recordings in acute brain slices from Optopatch3 transgenic mice crossed with different Cre driver lines. Scale bar 10 μm. (c) Confocal images showing Citrine fluorescence from QuasAr3-Citrine, in offspring of crosses between Optopatch3 mice and different Cre driver mice. Acute brain slices were prepared from mice ages 14 to 17 days and imaged in the cortex. Scale bar 50 μm. (d) Composite bright-field image of a coronal brain slice from an Rbp4-Cre+/−;Optopatch3+/− transgenic mouse, with locations of optical recordings marked with white spots. (e) Spike raster showing 94 cells recorded sequentially from a single Rbp4-Cre+/−;Optopatch3+/− acute brain slice. (f) Optogenetic stimulus intensity-dependent firing rates in acute slices with different Cre drivers. Left: slices homozygous for Optopatch3. Right: slices heterozygous for Optopatch3. (g) Mean firing rate, during a 500 ms stimulus as a function of stimulus intensity, I, calculated from the data in (f). Error bars show mean ± s.e.m. In the mice with Optopatch3 expression driven by CamKII-Cre, the F vs I curve for the Optopatch3+/+ mice is compressed along the x-axis relative to the Optopatch3+/− mice, indicating a stronger optogenetic drive for a given optical stimulation strength in the mice homozygous for CheRiff. The decrease in firing rate at strong stimulus in these mice is a signature of depolarization block. Data are from 128 cells from 5 slices and 2 mice for SST-Cre+/−; Optopatch3+/+; 25 cells from 1 slice and 1 mouse for CamKII-Cre+/−; Optopatch3+/+; 152 cells from 6 slices and 4 mice for Rbp4-Cre+/−; Optopatch3+/−; 89 cells from 2 slices and 2 mice for CamKII-Cre+/−; Optopatch3+/−.
Extended Data Fig. 3. Photophysical properties, kinetics and electrical properties of photoactivated QuasAr3 (paQuasAr3).

(a) Photoactivation by blue light. Red lines: fluorescence of HEK cells expressing paQuasAr3 during voltage steps under constant red illumination (10 W/mm2) and variable blue illumination (average of n = 8 cells). Grey lines: same experiment in HEK cells expressing QuasAr3 (average of n = 7 cells). (b) PaQuasAr3 fluorescence vs. blue light intensity at two membrane voltages (n = 8 cells, mean ± s.e.m.). Photoactivation showed saturation behavior, with 50% maximum enhancement at I488 = 27 mW/mm2. (c) Voltage-dependent near infrared fluorescence of paQuasAr3 and QuasAr3 with and without blue light (150 mW/mm2). All fluorescence values are normalized to fluorescence with red only illumination at Vm = −75 mV. (d) Same data as (c) but each fluorescence trace was normalized to its value F0 at Vm = −75 mV. Blue illumination enhanced the absolute fluorescence and the absolute voltage sensitivity, but the fractional voltage sensitivity (ΔF/F0) was the same between QuasAr3 and paQuasAr3 and was not affected by blue illumination (n = 7 cells in each condition, p = 0.91, one-way ANOVA). (e) Kinetics of QuasAr3 and paQuasAr3 measured in HEK293T cells. Cells were subjected to a square wave from −60 mV to +40 mV at 5 Hz (see panel a). Response transients were fit to a double exponential. QuasAr3, n = 5 cells, paQuasAr3, n = 9 cells. All values are mean ± s.e.m. Red intensity, 10 W/mm2, blue intensity, 150 mW/mm2. (f) Response of paQuasAr3 and QuasAr3 to steps of blue illumination. The blue light enhancement arose with a 50 ± 14 ms time-constant and subsided with a 167 ± 26 ms time-constant (mean ± s.d.). Blue light activated paQuasAr3 was ~2-fold brighter than QuasAr3 (mean of n = 10 cells). (g) Action spectrum for photosensitization, measured in E. coli expressing paQuasAr3. Fluorescence was excited at λexc = 640 nm and emission was collected from λem = 660 nm – 740 nm. The activation wavelength was scanned from λact = 450 – 650 nm. Peak activation was at λact = 470 nm. (h) Fluorescence excitation spectra ± blue sensitization (40 mW/mm2). The sensitized state of paQuasAr3 had a fluorescence excitation spectrum similar to QuasAr3, with peak excitation at λexc = 580 nm. (i) SNR of single spikes in acute slices for QuasAr3 (n = 10 cells) and paQuasAr3 (n=10 cells) with either red only or red and blue illumination (mean ± s.d. paQuasAr3 ± blue, paired t-test, paQuasAr3 red and blue vs. QuasAr3 red only, t-test). (j) Voltage clamp recordings in CA1 pyramidal cell expressing paQuasAr3-s showed no photocurrents in response to illumination with red light (640 nm, 12 W/mm2), blue light (488 nm, 90 mW/mm2) or combination of the two, both when the cell was held at −70 mV and at 0 mV (repeated in n = 2 cells). (k) Electrical properties measured by patch clamp in acute slices. QuasAr3 (n = 11 cells, 6 slices, 2 mice) and paQuasAr3 (n = 9 cells, 6 slices, 3 mice) were expressed in the visual cortex and compared with non-expressing cortical cells (n = 9 cells, 7 slices, 7 mice). paQuasAr3-s (n = 7 cells, 7 slices, 5 mice) was expressed in CA1 pyramidal cell layer and compared with non-expressing cells in that layer (n = 8 cells, 6 slices, 5 mice). Error bars are mean ± s.d.
Extended Data Figure 4. Patterned illumination improves brain imaging and minimizes brain heating.

(a) A single Oriens IN was illuminated with red and blue light projected precisely onto the cell (1x) or with an oval mask whose area was either twice (2x) or 10 times bigger (10x) than the cell (scale bar 20 μm). Illumination intensity was held constant across measurements. (b) Spontaneous spiking activity of the Oriens IN (a) in an awake resting mouse. (c) Mean spike SNR was similar with 2x mask compared with 1x mask (p = 0.2, two-sided paired t-test) and significantly reduced in the 10x mask (p = 0.001, two-sided paired t-test, n = 10 cells from N = 2 mice, error bars ± s.e.m.). (d) Cross section of NIR fluorescence of Oriens or PCL cells imaged in anesthetized mice and illuminated with either wide-field red illumination, patterned red illumination or patterned red and patterned blue illuminations. The lower SBR in the PCL is attributed to the greater density of expressing cells. All cells visible in the focal plane were targeted with illumination, leading to higher background in the PCL. In the Oriens, blue illumination increased signal but not background. In the PCL, blue illumination modestly increased background, a consequence of light scattering between neighboring cells. SBR values were: PCL: wide field: 0.27 ± 0.02, Red only: 0.95 ± 0.063, Red + Blue: 1.31 ± 0.08, n = 64 cells. Oriens: wide field: 0.27 ± 0.05, Red only: 2.9 ± 0.19, Red + Blue: 4.5 ± 0.27, n = 7 cells, mean ± s.e.m. Shading represents s.e.m. These data were used to calculate the improvement in SBR in Fig. 2d. (e) Left, spontaneous activity of a representative Oriens neuron in an awake resting mouse illuminated with the indicated laser intensity. Right, population average shows no change in the firing rate in the illumination range tested (corresponding to 12.5 to 34 mW into the tissue, n = 10 cells from 2 mice, error bars show mean ± s.d., p = 0.22, two-sided paired t-test). (f) FOVs with single Oriens INs were imaged while illuminating with increasing number of masks around the cell to simulate the multicell imaging conditions used in the PCL. Left, typical illumination pattern with the indicated number of masks. Middle, representative traces from an Oriens FOV imaged in an awake, resting mouse with the indicated number of masks. Right, mean spontaneous firing rate as a function of the total laser power. Firing rates were stable with illumination of up to 10 masks (80 mW). Projection of 15 masks (120 mW) caused a modest increase in the spontaneous firing rates (n = 10 cells from 2 mice, error bars show mean ± s.d., p = 0.05, two-sided paired t-test). Subsequent experiments were restricted to 10 or fewer masks. (g) Simulated spatial temperature profile in brain tissue with an imaging cannula and immersion water. Profiles are shown for no illumination, 100 mW 640 nm illumination and 100 mW 920 nm illumination, corresponding to a two-photon imaging experiment.
Extended Data Fig 5. Photostability of paQuasAr3-s in vivo during ‘all-optical’ excitability measurements.

(a) 10-minute recording of fluorescence in mouse hippocampus. Oriens SST cells expressed paQuasAr3-s and CheRiff-s and were illuminated with red light at 12 W/mm2 and stimulated with blue light (up to 10 mW/mm2) using the stimulation protocol presented at the top, during quiet and walking periods. The trace shows the fluorescence from two user-defined ROIs after subtraction of the background from a cell-free region. The baseline signal photobleached in both cells by ~50% during this interval. Top: total acquisition. Dashed lines denote separate movies. Bottom: Close-up views of the indicated regions from the top graph. Similar recordings were performed in 5 FOVs. (b) The same two cells were imaged 3 weeks later. While at the end of the initial 10-minute recording the SNR was low due to photobleaching, 3 weeks later the signal had recovered. Images show wide-field epi-fluorescence images of the cells in the two imaging sessions. Scale bar 20 μm. (c) Spike SNR was on average stable in repeated recordings over 3 weeks (p = 0.34, two-sided paired t-test).
Extended data Fig. 6. Validation of the pipeline for signal extraction from dense PCL movies using penalized matrix decomposition denoising followed by non-negative matrix factorization demixing (PMD-NMF).

(a) Illustration of the segmentation pipeline: raw movies were first corrected for motion, followed by photobleaching correction, penalized matrix decomposition denoising, manual removal of blood vessels, and demixing using non-negative matrix factorization. The pipeline produced waveforms corresponding to individual cell traces and to the background (see Methods for details). (b) Testing the pipeline using simulated data composed of two cells partially overlapping in space and with varying levels of correlation in their sub-threshold voltages and with the background. Poisson-distributed shot noise was added to each pixel to mimic experimental noise. Left: image of the input movie pixel-wise s.d. and of the output cell footprints. Middle: input waveforms and the pipeline output waveforms. Right top: correlation matrix of input signals (cells C1, C2, and background B). Right bottom: cross-correlation of output waveforms with input waveforms, mean ± s.d. for n = 5 simulations. (c) Performance of the pipeline as a function of input parameters (n = 5 simulations per condition). Top: output to input correlation and output C1 to C2 cross correlation as a function of the pixel noise level. Noise is scaled to the spike amplitude, input C1 to C2 cross correlation is 0.5 and the input correlation with the background is also 0.5. Middle: output to input correlation and output C1 to C2 cross correlation as a function of the correlation between the input and the background, at two noise levels. Input C1 to C2 correlation is 0.5. Bottom: output to input correlation and output C1 to C2 cross correlation as a function of the cross correlation between input C1 and C2, at two noise levels. Input correlation with background is 0.5. (mean ± s.e.m.). (d) Testing the pipeline with composite movies composed from real data. We imaged FOVs with single Oriens neurons spontaneously spiking in awake resting mice. Each cell was imaged in the focal plane and then at 20 μm defocus. The two movies were first processed with the pipeline to extract the ground-truth input signals and then the movies were summed such that the focused and defocused cells were ~50% overlapping. We then ran the blended movies through the pipeline and compared outputs to the input traces using cross-correlation analysis. (e) Mean cross-correlograms of 5 FOVs processed as described in (d) showing that the segmentation pipeline accurately reproduced the correlational structure of the inputs even under these challenging conditions. Shading represents s.e.m. (f) Validation of the image segmentation pipeline using patch clamp recording as the ground-truth. Top left: FOV with dense expression of paQuasAr3-s in CA1 PCL in acute brain slice. The FOV was imaged while the voltage in the blue outlined cell was recorded by manual patch clamp. Bottom left: two of the spatial footprints identified by the PMD-NMF pipeline. Middle: Ground-truth voltage recording (black), flat average ROI around the cell (blue), and two PMD-NMF units (magenta and red). The flat average ROI trace showed fluctuations not present in the patch clamp recording, presumably from an out-of-focus cell. These events were absent in the magenta PMD-NMF demixed trace. Right: zoom-in on the indicated inset. This experiment was performed once.
Extended Data Fig. 7. Simultaneous optical recording from 7 spiking cells in the PCL of an anesthetized mouse.

The mouse expressed paQuasAr3-s. (a) Left: wide-field epifluorescence image of Citrine fluorescence. Middle: same field of view with patterned blue illumination. Right: same field of view with patterned red illumination. (b) Simultaneous fluorescence recordings from 7 cells. Close-ups show synchronized complex spikes and action potentials riding atop sub-threshold oscillations. This experiment was performed once.
Extended Data Fig. 8. Chronic recordings of Oriens neurons in hippocampus.

(a) Top left: wide-field epifluorescence and two-photon (2P) images showing two Oriens interneurons. Scale bar 50 μm. Middle: fluorescence recordings from the two cells during three consecutive repeats of a 65 s protocol. The protocol comprised of 10 s of rest, followed by 15 s epochs of walking at speeds of 5, 7.5, and 10 cm/s, successively, followed by 10 s of rest. Inset shows the optical traces with clearly resolved spikes and sub-threshold events. Bottom: average spike rate across three trials of the two cells (mean ± s.d.). (b) Same two neurons and same protocol as in (a), recorded 7 days later. Trial 1 was only spontaneous activity without locomotion.
Extended Data Fig. 9. Examples of brain state-dependent intercellular correlations and spike triggered averages for single pairs of cells (same analysis as in Fig. 3).

(a,b,c) Left: Magnified sections of recordings from trios of cells in (a,b) the PCL and (c) Oriens. Top: recordings during quiet. Bottom: recordings from the same cells during walking. Right: auto- and cross-correlations of the fluorescence traces, calculated from the complete 9 s recording in each brain region and behavioral state. The auto- and cross-correlations clearly show enhanced θ-rhythm in both brain regions during walking and differing cross-correlations between simultaneously recorded pairs of cells. (d,e) Distribution of equal-time correlation coefficients between pairs of simultaneously recorded cells in the PCL (d) and Oriens (e). (f) Three examples of single-pair spike-triggered average fluorescence during quiet (left) and walking (right) in the PCL (top) and Oriens (bottom).
Extended Data Fig. 10. Optopatch measurements of hippocampal SST neurons.

(a) Fluorescence waveforms of all 25 SST cells shown in Fig. 4. (b) Cell-by-cell comparison of the change in θ-band (6.7 to 8.3 Hz) power between walking and quiet brain states. Left: without optogenetic stimulation. Middle: same cells with tonic optogenetic stimulation. Right: ratio of θ-band enhancement (walking vs. quiet) with optogenetic stimulation vs. without (cell-by-cell comparison for the data in Fig. 4h). (c) Spike raster of 19 SST cells imaged at week 0 and week 3 and stimulated with the blue light protocol at the top (full dataset for the data in Fig. 4j-k).
Supplementary Material
Acknowledgments
We thank M.S. Lee and V. Joshi for help with tissue culture, A.H. Gheorghe for help with spectroscopy experiments, N. Rollins and S. Brownsberger for technical help, L. Yapp for advice on hippocampal surgeries, A. Ruangkittisakul and K. Ballanyi for neuronal cultures, K. Podgorski for heating simulation code, G. Buzsaki and members of the Cohen lab for helpful discussions, and D. Hochbaum, C. Straub, J.L. Saulnier, B.L. Sabatini, V. Kapoor and V. Murthy for help at early stages of the project. Y.A. was supported by fellowships from the Human Frontiers Science Program and the Edmund and Lili Safra Center for Brain Sciences. This work was supported by the Howard Hughes Medical Institute.
Footnotes
Competing interests
AEC is a founder of Q-State Biosciences.
Data availability
The datasets generated during the current study and custom data analysis codes are available from the corresponding author on reasonable request.
References
- 1.Petersen CC Whole-Cell Recording of Neuronal Membrane Potential during Behavior. Neuron 95, 1266–1281 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Lee AK & Brecht M Elucidating Neuronal Mechanisms Using Intracellular Recordings during Behavior. Trends Neurosci. (2018). [DOI] [PubMed] [Google Scholar]
- 3.Lou S et al. Genetically targeted all-optical electrophysiology with a transgenic Cre-dependent Optopatch mouse. J. Neurosci. 36, 11059–11073 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gong Y et al. High-speed recording of neural spikes in awake mice and flies with a fluorescent voltage sensor. Science 350, 1361–1366 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang HH et al. Subcellular imaging of voltage and calcium signals reveals neural processing in vivo. Cell 166, 245–257 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hochbaum DR et al. All-optical electrophysiology in mammalian neurons using engineered microbial rhodopsins. Nat. Methods 11, 825–833 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Madisen L et al. Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron 85, 942–958 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Millerd JE et al. Improved sensitivity in blue-membrane bacteriorhodopsin films. Opt. Lett. 24, 1355–1357 (1999). [DOI] [PubMed] [Google Scholar]
- 9.Wagner NL, Greco JA, Ranaghan MJ & Birge RR Directed evolution of bacteriorhodopsin for applications in bioelectronics. J. R. Soc. Interface 10, 20130197 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hausser M, Spruston N & Stuart GJ Diversity and dynamics of dendritic signaling. Science 290, 739–744 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Wu C, Ivanova E, Zhang Y & Pan Z rAAV-mediated subcellular targeting of optogenetic tools in retinal ganglion cells in vivo. PLoS One 8, e66332 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baker CA, Elyada YM, Parra A & Bolton MM Cellular resolution circuit mapping with temporal-focused excitation of soma-targeted channelrhodopsin. Elife 5, 10.7554/eLife.14193 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buchanan EK et al. Penalized matrix decomposition for denoising, compression, and improved demixing of functional imaging data. bioRxiv, 334706 (2018). [Google Scholar]
- 14.Klausberger T, Magill PJ, Márton LF & Roberts JDB Brain-state-and cell-type-specific firing of hippocampal interneurons in vivo. Nature 421, 844 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Harvey CD, Collman F, Dombeck DA & Tank DW Intracellular dynamics of hippocampal place cells during virtual navigation. Nature 461, 941–946 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bittner KC et al. Conjunctive input processing drives feature selectivity in hippocampal CA1 neurons. Nat. Neurosci. 18, 1133–1142 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Epsztein J, Brecht M & Lee AK Intracellular determinants of hippocampal CA1 place and silent cell activity in a novel environment. Neuron 70, 109–120 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varga C, Golshani P & Soltesz I Frequency-invariant temporal ordering of interneuronal discharges during hippocampal oscillations in awake mice. Proc. Natl. Acad. Sci. U. S. A. 109, E2726–34 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buzsaki G Theta oscillations in the hippocampus. Neuron 33, 325–340 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Poulet JF & Petersen CC Internal brain state regulates membrane potential synchrony in barrel cortex of behaving mice. Nature 454, 881–885 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Gentet LJ, Avermann M, Matyas F, Staiger JF & Petersen CC Membrane potential dynamics of GABAergic neurons in the barrel cortex of behaving mice. Neuron 65, 422–435 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Lampl I, Reichova I & Ferster D Synchronous membrane potential fluctuations in neurons of the cat visual cortex. Neuron 22, 361–374 (1999). [DOI] [PubMed] [Google Scholar]
- 23.Okun M & Lampl I Instantaneous correlation of excitation and inhibition during ongoing and sensory-evoked activities. Nat. Neurosci. 11, 535–537 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Briggman KL, Abarbanel HD & Kristan WB Jr From crawling to cognition: analyzing the dynamical interactions among populations of neurons. Curr. Opin. Neurobiol. 16, 135–144 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Cunningham JP & Byron MY Dimensionality reduction for large-scale neural recordings. Nat. Neurosci. (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harris KD, Hirase H, Leinekugel X, Henze DA & Buzsáki G Temporal interaction between single spikes and complex spike bursts in hippocampal pyramidal cells. Neuron 32, 141–149 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Bezaire MJ & Soltesz I Quantitative assessment of CA1 local circuits: knowledge base for interneuron‐pyramidal cell connectivity. Hippocampus 23, 751–785 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Unal G, Joshi A, Viney TJ, Kis V & Somogyi P Synaptic Targets of Medial Septal Projections in the Hippocampus and Extrahippocampal Cortices of the Mouse. J. Neurosci. 35, 15812–15826 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shaner NC et al. Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat. Methods 5, 545–551 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gradinaru V et al. Molecular and Cellular Approaches for Diversifying and Extending Optogenetics. Cell 141, 154–165 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klapoetke NC et al. Independent optical excitation of distinct neural populations. Nat. Meth. 11, 338–346 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shepard BD, Natarajan N, Protzko RJ, Acres OW & Pluznick JL A cleavable N-terminal signal peptide promotes widespread olfactory receptor surface expression in HEK293T cells. PLoS One 8, e68758 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chow BY et al. High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature 463, 98–102 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McIsaac RS et al. Directed evolution of a far-red fluorescent rhodopsin. Proc. Natl. Acad. Sci. U. S. A. 111, 13034–13039 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maclaurin D, Venkatachalam V, Lee H & Cohen AE Mechanism of voltage-sensitive fluorescence in a microbial rhodopsin. Proc. Natl. Acad. Sci. USA 110, 5939–5944 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daigle TL et al. A suite of transgenic driver and reporter mouse lines with enhanced brain cell type targeting and functionality. bioRxiv, 224881 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Molyneaux BJ, Arlotta P, Hirata T, Hibi M & Macklis JD Fezl is required for the birth and specification of corticospinal motor neurons. Neuron 47, 817–831 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Krashes MJ et al. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J. Clin. Invest. 121, 1424–1428 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adam Y & Mizrahi A Long-term imaging reveals dynamic changes in the neuronal composition of the glomerular layer. J. Neurosci. 31, 7967–7973 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dombeck DA, Harvey CD, Tian L, Looger LL & Tank DW Functional imaging of hippocampal place cells at cellular resolution during virtual navigation. Nat. Neurosci. 13, 1433–1440 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goldey GJ et al. Removable cranial windows for long-term imaging in awake mice. Nat. Protoc. 9, 2515–2538 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kralj JM, Douglass AD, Hochbaum DR, Maclaurin D & Cohen AE Optical recording of action potentials in mammalian neurons using a microbial rhodopsin. Nat. Meth. 9, 90–95 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pnevmatikakis EA & Giovannucci A NoRMCorre: An online algorithm for piecewise rigid motion correction of calcium imaging data. bioRxiv, 108514 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Mukamel EA, Nimmerjahn A & Schnitzer MJ Automated analysis of cellular signals from large-scale calcium imaging data. Neuron 63, 747–760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ricciardi LM & Sacerdote L The Ornstein-Uhlenbeck process as a model for neuronal activity. Biol. Cybern 35, 1–9 (1979). [DOI] [PubMed] [Google Scholar]
- 46.Stujenske JM, Spellman T & Gordon JA Modeling the spatiotemporal dynamics of light and heat propagation for in vivo optogenetics. Cell reports 12, 525–534 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Podgorski K & Ranganathan G Brain heating induced by near-infrared lasers during multiphoton microscopy. J. Neurophysiol 116, 1012–1023 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.