

Abstract
Mutation in the SHANK3 human gene leads to different neuropsychiatric diseases including Autism Spectrum Disorder (ASD), intellectual disabilities and Phelan-McDermid syndrome. Shank3 disruption in mice leads to dysfunction of synaptic transmission, behavior, and development. Protein S-nitrosylation, the nitric oxide (NO•)-mediated posttranslational modification (PTM) of cysteine thiols (SNO), modulates the activity of proteins that regulate key signaling pathways. We tested the hypothesis that Shank3 mutation would generate downstream effects on PTM of critical proteins that lead to modification of synaptic functions. SNO-proteins in two ASD-related brain regions, cortex and striatum of young and adult InsG3680(+/+) mice (a human mutation-based Shank3 mouse model), were identified by an innovative mass spectrometric method, SNOTRAP. We found changes of the SNO-proteome in the mutant compared to WT in both ages. Pathway analysis showed enrichment of processes affected in ASD. SNO-Calcineurin in mutant led to a significant increase of phosphorylated Synapsin1 and CREB, which affect synaptic vesicle mobilization and gene transcription, respectively. A significant increase of 3-nitrotyrosine was found in the cortical regions of the adult mutant, signaling both oxidative and nitrosative stress. Neuronal NO• Synthase (nNOS) was examined for levels and localization in neurons and no significant difference was found in WT vs. mutant. S-nitrosoglutathione concentrations were higher in mutant mice compared to WT. This is the first study on NO•-related molecular changes and SNO-signaling in the brain of an ASD mouse model that allows the characterization and identification of key proteins, cellular pathways, and neurobiological mechanisms that might be affected in ASD.
Introduction
Autism spectrum disorder (ASD) is a highly heritable neurodevelopmental syndrome, caused by genetic modifications [1–3] as well as non-genetic factors [4, 5]. These alterations may lead to improper development of synapses or brain circuits [6], resulting in impaired social behavior [6], along with stereotyped, repetitive behavior with narrowly restricted interests [7]. Human genetic studies have identified deletions/mutations of the SHANK3 gene as the cause of Phelan- McDermid Syndrome (PMS), a neurodevelopmental disorder with >50% of the patients having a diagnosis of ASD [8]. Recent genetic screens have identified many mutations/variants of the SHANK3 gene in ASD patients outside of a diagnosis of PMS suggesting that SHANK3 mutations also contribute to non-syndromic ASDs [9–12].
Shank3 is a member of the Shank family of proteins (Shank1–3) [13–16]. Shank interacts with many postsynaptic density (PSD) proteins. Most notably, Shank binds to SAPAP, which in turn binds to PSD95 to form the PSD95/SAPAP/Shank postsynaptic complex [17]. Altogether, these three groups of multi-domain proteins are proposed to form a key scaffold for assembling the macromolecular postsynaptic complex at glutamatergic synapses. This complex has been shown to play important roles in targeting, anchoring, and dynamically regulating synaptic localization of neurotransmitter receptors and signaling molecules [18, 19]. Shank is also connected to the metabotropic glutamate receptor (mGluR) pathway through its binding to Homer [20]. Together, these data strongly suggest that Shank proteins play key roles in synaptic development and function [21, 22]. Shank3 is highly enriched at corticostriatal glutamatergic synapses [16, 23], part of the neural circuits strongly implicated as dysfunctional in ASDs [24–27]. In the cortico-striatal-thalamo-cortical circuitry, information from various cortical areas converges on the striatum and the processed information returns to the cortex through the thalamic relay to guide motor and behavioral decisions. Thus, diverse synaptic defects in different parts of the circuitry will all end up affecting cortical activity that could be a converging mechanism for repetitive behaviors [28] with different genetic causes and in different disorders. Previous studies on mouse models showed that disruption of Shank3 resulted in both structural and functional changes in corticostriatal synapses [23, 27, 29–31], while oppositely, adult restoration of Shank3 expression rescued selectively the behavioral and physiological deficits in mice [32]. Thus, the Shank3 gene provides us with a unique opportunity to dissect cellular and molecular mechanisms of ASD-relevant behavioral abnormality [33]. It is not clear how such diverse genetic mutations and consequent synaptic dysfunctions converge to cause core ASD symptoms such as repetitive behaviors and social communication deficits. A plausible hypothesis is that ASD symptoms are the result of dysfunction of core behavior-relevant circuitry that could be caused by various forms of synaptic dysfunction [6, 34]. Such a dysfunction could be due to improper protein-protein interactions and altered molecular pathways. For instance, a mutation can lead to critical modifications in protein structure, which may affect its ability to interact with other proteins and its functionality. To study the proteomic consequences of Shank3 mutation in the cortico-striatal circuitry, we used the recently published human mutation-based mouse model for ASD (InsG3680(+/+)), in which a guanine nucleotide was inserted at cDNA position 3680 of Shank3 gene, leading to a frameshift that results in a premature stop codon [29]. These mice were shown to have striatal and cortico-striatal synaptic transmission and social behavioral defects, along with intense overgrooming repetitive behavior [29]. Understanding the nature of this circuitry imbalance and its consequences on circuitry output relevant to ASD behaviors is a critical step for defining converging neurobiological mechanisms and for developing new strategies for effective treatment.
Nitric oxide (NO•) is a signaling molecule in the central and peripheral nervous system and is one of the most important messenger molecules [35, 36]. NO• produced in the brain from L-arginine by three nitric oxide synthase isoforms (NOS1, NOS2, NOS3) [37]. NOS1 (also known as neuronal NOS - nNOS) is primarily expressed and regulated in neurons, NOS2 in microglia, astrocytes, and some neurons, and NOS3 in blood capillaries [38]. At low concentrations NO• acts as a signaling molecule, and at higher concentrations may cause modified phenotypes and cell death [39]. However, under some conditions NO• has neuroprotective properties and may have therapeutic value for brain injury [40]. NO• has a critical role in synaptic transmission in the brain [36] and plays a major role in signaling through activation of GMP cyclase [36], effects on ion transporters [36], and S-nitrosylation (SNO) of peptides and proteins [41, 42]. SNO, like phosphorylation, is a reversible posttranslational modification (PTM) in which a cysteine is converted to a nitrosothiol. SNO regulates the localization and activity of many key enzymes and receptors [39, 43, 44] leading to modulation of many canonical signaling pathways, synaptic plasticity, axonal elongation, movement of proteins to the cell membrane, and protein assembly [39, 43].
NO• also plays a major role in inflammation-induced oxidative/nitrosative stress, leading to damage to biological molecules, including protein tyrosine nitration (formation of 3-nitrotyrosine) [45–47]. S-nitrosoglutathione (GSNO) forms at diffusion-controlled rates in a radical recombination between NO• and GSH thiyl radicals (RS•), and can then transnitrosate other thiols on peptides and proteins [42, 48, 49].
In the brain, numerous examples of neuropathology exist that involve SNO [50–52] such as Alzheimer’s [39, 53, 54], Parkinson’s [55] and Huntington’s disease [39, 54]. In a mouse model of Alzheimer’s disease [53], NO• output greatly increases at the locus of the pathology, leading to changes of the SNO-proteome and consequently all of the functions mentioned above. However, previous literature has demonstrated that de-nitrosylation could lead to neuronal apoptosis [56] and dysregulated SNO may contribute to a range of human pathologies [57]. We now know that the status and composition of the SNO-proteome is regulated by a combination of transnitrosylating peptides and proteins [42, 58] balanced by de-nitrosylating peptides and proteins [57], but the details of this regulation are unknown. An example of a protein that can nitrosylate or de-nitrosylate, depending on its oxidative state is thioredoxin [57, 59].
In the case of ASD, there is currently no prior evidence for the involvement of NO• and S-nitrosylation in the pathology. The literature is dominated by genetic and epigenetic analysis of human brain, and the development of mouse models based upon the human database of autism-associated genes (SFARI). The mouse models validate the central role of specific mutations, but it is important to follow through and develop important biochemical information that may serve to identify potential therapeutic targets. Earlier works on PSD fraction of Shank3 mutated mice, and on post-mortem brain tissue from autism patients found altered levels of proteins that revealed changes in the systems biology affected by these alterations [60–62]. Recent research has identified hundreds of proteins and cysteine-sites (SNO-Cys) in the normal mouse that are changed in other neuro-pathological states [53, 63], and identification of hundreds of proteins permits analysis in Gene Ontology (GO) system and facilitates development of a systems biology model of normal and pathological function. In this study, our hypothesis is that multiple biochemical pathways are affected by aberrant S-nitrosylation of multiple proteins that contribute to the pathogenesis of neurological disorders. We further postulate that an initial pathological event, such as a mutation in a synaptic protein, could lead to an increase of calcium influx. Shank3 is part of the PSD at the glutamatergic synapse and is connected to receptors and channels that regulate calcium influx such as NMDAR, mGluRs and AMPAR [16]. Calcium influx could activate nNOS [64] that in turn leads to increased NO• formation [64] and controlled shift in the neuronal SNO-proteome [65]. Changes in protein modulation by SNO would affect cell signaling and consequential synaptic malfunction.
To test this hypothesis we have developed a mass spectrometric (MS) method, (SNO trapping by a TriAryl Phosphine (SNOTRAP)), that enables global, facile, and high-throughput identification of SNO-proteins [53, 66]. In this study we use this approach in analysis of the cortex and striatum of both 6 week-old (young) and 4 month-old (adult) of the InsG3680(+/+) mice. This process allows us to identify key proteins that are SNOed in the genetically modified animal but not in the control, or vice versa.
In order to minimize the dependence of our conclusions on the presence or absence of any individual protein in either mouse genotype, we took a systems biological approach to identifying biological pathways and processes significantly associated with the set of proteins differentially S-nitrosylated in this model of ASD. This approach is common in genomic and transcriptomic research, and has been applied to genes associated with ASD [67]. Similarly, systems biological approaches have been used in the analysis of post-translational modifications in proteomic approaches, most notably for phosphorylation [68]. Differentially S-nitrosylated proteins were analyzed using GO [69] to determine which biological processes are affected, KEGG [70] to identify biochemical pathways in which the affected proteins are enriched, and STRING [71] to identify protein-protein interactions between S-nitrosylated proteins.
Key proteins were verified by western blot (WB) and functionally analyzed biochemically. We have tested nNOS levels and nNOS localization in neurons, and GSNO concentrations in all WT and KO samples of cortex and striatum. It should be noted that due to diffusion and chemical reactions, NO• has a relatively short half-life in cells, and cannot be measured in intact brain. Therefore, we have measured GSNO, which is the proximate molecule for transnitrosylation. Finally, brain sections were analyzed by immunohistochemistry (IHC), in cortical and striatal regions that have functional relevance to the behavioral and physiological deficits in ASD, to detect protein damage by 3-nitrotyrosine (Ntyr), a product of the reaction of proteins with peroxynitrite.
Our results provide novel insights towards understanding the NO•-mediated molecular changes and their consequences on cell signaling in a human mutation-based Shank3 mouse model. To our knowledge, this is the first study on NO•- related molecular changes and SNO signaling in the brain of any ASD mouse model.
Materials and methods
Animal models
All animal-related work was performed under the guidelines of the Division of Comparative Medicine (DCM), approved by the Committee for Animal Care (CAC) of Massachusetts Institute of Technology and was consistent with the Guide for Care and Use of Laboratory Animals, National Research Council 1996 (institutional animal welfare assurance no. A-3125–01). The male InsG3680(+/+) mice (KO) harbor the ASD patient-linked single guanine nucleotide (G) insertion at cDNA position 3680, that leads to a frame shift and downstream premature stop codon, as was previously published [29].
MS data processing
Agilent Spectrum Mill MS proteomics Workbench B.05 was used for peak list generation, database searching, and FDR estimation, see supplementary information (SI) for detailed parameters.
GSNO analysis
For GSNO internal standard synthesis, sample preparation for GSNO quantification, and ESI + -QqQ-MS analyses, see reference [72] and SI.
Statistics and bioinformatics
To calculate the functional enrichment of Cellular Compartment (CC) and Biological Processes (BP) GO terms and KEGG pathways we uploaded the SNO-proteins into The Database for Annotation, Visualization and Integrated Discovery (DAVID) [73] Bioinformatics Resourses (version 6.8, https://david.ncifcrf.gov). STRING (version 10.0) was used to analyze the protein-protein interaction of SNO-proteins (http://string-db.org). For more information on DAVID, STRING, and the statistical analysis, see SI.
For materials and reagents, sample preparation of brain tissues for MS, MS analysis, WB and IHC methodology, and mathematical models, see SI.
Results
We studied two regions in the mouse brain, which are of special interest due to their role in ASD pathology, cortex and striatum. Because ASD is a neurodevelopmental disorder, we studied NO• involvement developmentally, using mice at two different ages: 6 week-old and 4 month-old. In total, 8 different groups were analyzed: 6 weeks-cortex-WT (6w-cor-WT), 6 weeks-cortex-KO (6w-cor-KO), 6 weeksstriatum-WT (6w-str-WT), 6 weeks-striatum-KO (6w-str-KO), 4 months-cortex-WT (4m-cor-WT), 4 months-cortex-KO (4m-cor-KO), 4 months-striatum-WT (4m-str-WT), and 4 months-striatum-KO (4m-str-KO). See Fig. 1 for a schematic of the study strategy.
Fig. 1.
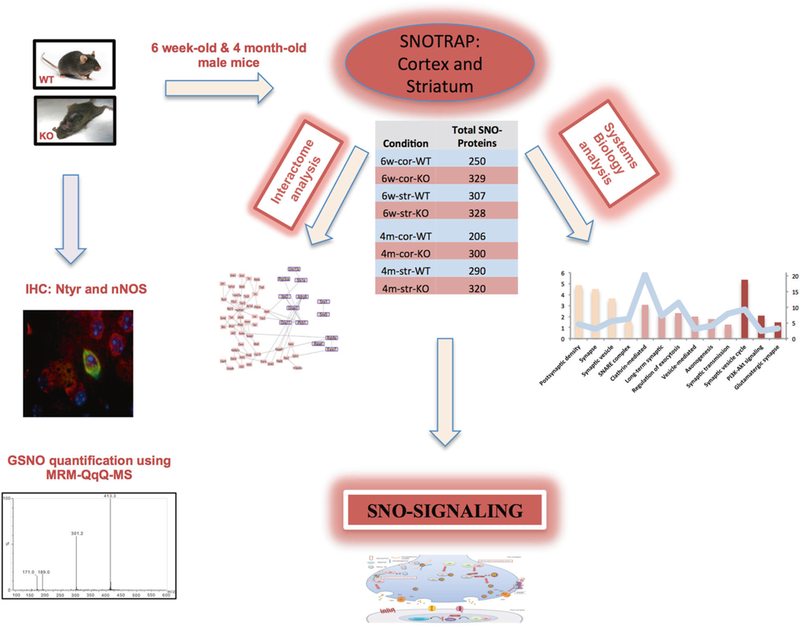
Schematic describes the steps done in this study. SNOTRAP sample preparation was conducted to identify SNO-proteins in the cortex and striatum of 6 week- and 4 month-old mice in both WT and InsG3680(+/+) (KO) groups. Each SNO-proteins set shown in the table was analyzed by GO, KEGG pathway and STRING protein–protein interactions. WB and IHC were conducted to validate SNO-signaling mechanisms. IHC was performed to identify differences in 3-Nitrotyrosine in 6 different regions of the cortex and striatum. Using an LC–MS quantitative method, we analyzed GSNO concentrations in all WT and KO samples. nNOS expression levels were measured by WB. nNOS co-localization with NeuN was examined
Shank3 mutation leads to changes in the SNO-proteome of 6w and 4 m InsG3680(+/+) mice
The SNO-proteomic analysis revealed differences in SNO-proteins between the KO and WT in both ages (6w and 4 m) and regions (cortex and striatum). For all proteins identified in the eight different groups, see Supplementary Table 1. Because tandem MS identifies a subset of sample proteins stochastically, we took three approaches to confirm that the proteins identified in WT and KO mice are not likely to be different subsets of the same overall pool. First, we built simple mathematical models describing the approach of our technical and biological replicates towards an asymptote under the assumption that each replicate within a condition stochastically sampled a fraction of an identical pool (Supplementary Figure 1). Second, for 10 proteins identified in the KO but not WT mice, we manually searched the WT MS data for evidence that the proteins were present at the correct chromatographic retention time and mass, but not fragmented for the second MS stage. For 7/10 proteins, we found no such evidence. For the remaining 3, an MS1 peak plausibly associated with the protein was found, but the protein was in insufficient quantity to be selected for fragmentation. Finally, WBs were performed in SNOTRAP eluates for two key proteins detected only in KO samples, calcineurin (CN) and syntaxin 1a (Stx1a). The proteins were seen only in KO eluates (Fig. 3a, b). Taken together, these results indicate that it is unlikely that the set of SNO-proteins is unaltered between the WT and KO conditions.
Fig. 3.
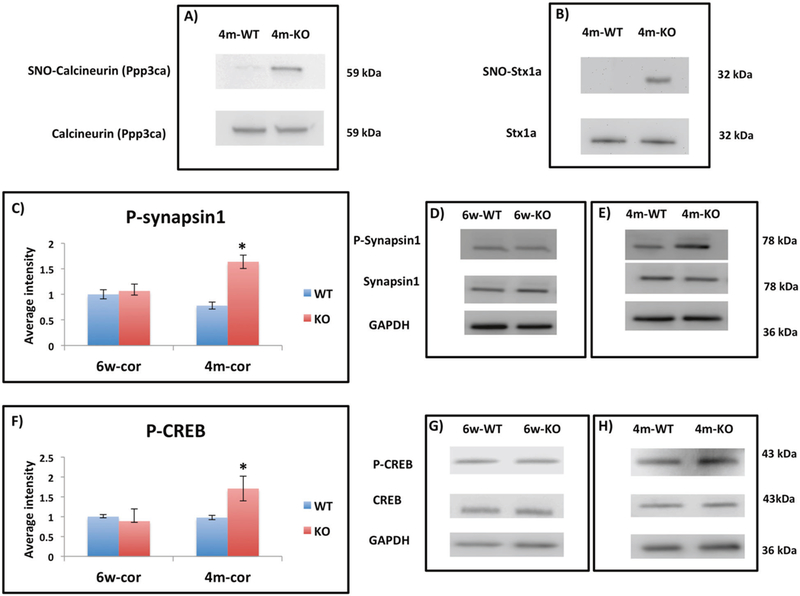
a Representative WB from eluted SNO-proteins prepared from 4m-WT and 4m-KO showing SNO-CN (Ppp3ca) in KO and not present in WT. CN shows similar levels in both groups. b Representative WB from eluted SNO-proteins prepared from 4m-WT and 4m-KO showing SNO-Stx1a in KO and not present in WT. Stx1a shows similar levels in both groups. c The relative average WB intensity of P-synapsin1 (Ser 62, Ser 67) comparing 6w-cor-WT to 6w-cor-KO and 4m-cor-WT to 4m-cor-KO. The data shows significant increase of P-synapsin1 in 4m-cor-KO compared to 4m-cor-WT. The data is normalized to synapsin1 and GAPDH and presented as mean ± SEM. One tailed t-test was conducted. *P < 0.05. WT mice (n = 4) and KO mice (n = 4). d Representative WB bands of P-synapin1, synapsin1, and GAPDH from cortex tissue prepared from 6w-WT and 6w-KO mice groups. e Representative WB bands of P-synapin1, synapsin1, and GAPDH from cortex tissue prepared from 4m-WT and 4m-KO. f The relative average WB intensity of P-CREB (Ser133) comparing 6w-cor-WT to 6w-cor-KO and 4m-cor-WT to 4m-cor-KO. The data shows significant increase of P-CREB in 4m-cor-KO compared to 4m-cor-WT. The data is normalized to CREB and GAPDH and presented as mean ± SEM. One tailed t-test was conducted. *P < 0.05. WT mice (n = 4) and KO mice (n = 4). g Representative WB bands P-synapin1, synapsin1, and GAPDH from cortex tissue prepared from 6w-cor and 6w-KO. h Representative WB from cortex tissue prepared from 4m-WT and 4m-KO
Proteins associated with processes and pathways known to be affected in ASD are enriched in KO SNOTRAP samples
To gain a systems-level insight into SNO-proteins’ functionalities, and to test whether enriched processes are related to ASD, we performed GO-based protein enrichment analysis of the cortex and striatum for both 6w-old and 4m-old WT and KO mice. Three categories were analyzed in DAVID: CC, BP, and KEGG pathways. We identified processes and pathways in each tissue type with statistically significant enrichment of S-nitrosylated proteins vs. a background of the total proteome, and then focused on the set of these processes and pathways that differentiated healthy from Shank3 mutant mice. For example, in adult KO cortex samples, 9/121 proteins identified by SNO-trapping were annotated with the KEGG pathway “synaptic vesicle cycle”, including Stx1a, synaptotagmin 1, N-ethylmaleimide sensitive fusion protein (Nsf), and others. This represents more than a 9-fold enrichment over the proteomic background, with a nominal p-value of 4.5 × 10−6 and a Benjamini-Hochberg corrected FDR of 2.7 × 10−4. By contrast, proteins associated with synaptic vesicle formation are not significantly enriched in the adult WT cortex sample.
6w-cor-KO showed a significant enrichment of different GO terms and KEGG pathways that in part are associated with ASD such as nervous system development (Fig. 2a). None of these was found in 6w-cor-WT, emphasizing the association between SNO and InsG3680(+/ + ) (see Supplementary Table 2). Testing the 4m-cor-KO group has also revealed enriched pathways such as synaptic vesicle cycle (detailed above) and oxidative phosphorylation (Fig. 2c, Supplementary Table 2). None of these processes was found in 4m-cor-WT (Supplementary Table 2).
Fig. 2.
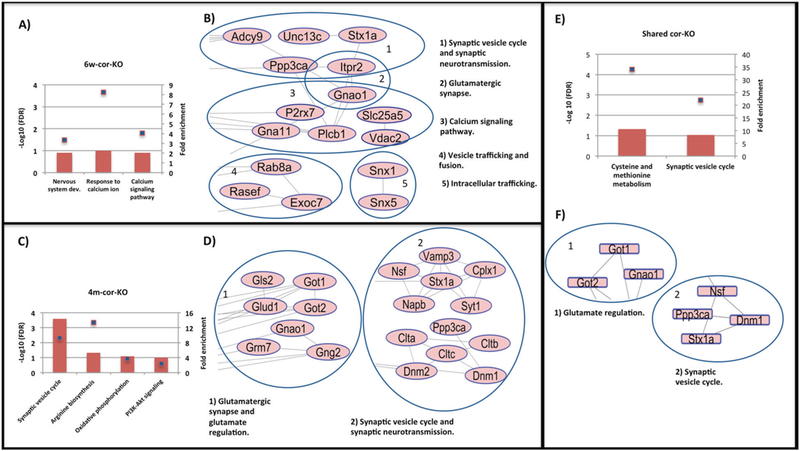
a BP and KEGG analysis of the SNO-proteins that were found exclusively in 6w-cor-KO. b Functional and physical protein interaction analysis of the SNO-proteins that were found exclusively in 6wcor-KO. c BP and KEGG analysis of the SNO-proteins that were found exclusively in 4m-cor-KO. d Functional and physical protein interaction analysis of the SNO-proteins that found exclusively in 4m-cor-KO. e BP and KEGG analysis of the shared SNO-proteins between 6w-cor-KO and 4m-cor-KO. f Functional and physical protein interaction analysis of the shared SNO-proteins between 6w-cor-KO and 4m-cor-KO. * Bars represent the −log10 of the Benjamini corrected false discovery rate (FDR) and the squares on bars represent the fold enrichment
For detailed lists of the CC, BP and KEGG annotations enriched in all 8 tested groups, see Supplementary Table 2. Further, Supplementary Table 3 lists 49 SNO-proteins (in 4m-cor-KO) in 5 important pathways and functions (from GO and KEGG) that are found in KO, not in WT, and may reasonably be linked to pathology. Also shown in Supplementary Table 3 are the results for 6w-cor-KO.
SNO-protein interactome analysis reveals synaptic proteins clusters in the cortex of InsG3680(+/+) mice
To test for functional and physical interactions among SNOed proteins in the InsG3680(+/+) mice, we used STRING to analyze protein-protein interactions. Clusters of SNO-proteins represent a group of proteins with a common function that are joined because of their proximity or physical interactions. In other words, it may be a concerted process. For all network clusters of the 8 groups, see Supplementary Figures 2 & 3.
Analysis of cor-KO mice at both ages showed networks of SNO-proteins related to processes involved in ASD. Proteins that were SNOed and are functionally related to synaptic vesicle-cycle and neurotransmission (Ppp3ca, Stx1a, VAMP3, and others) and to the glutamatergic synapse (mGluR7, Glud1, Gnao1, and others) were interconnected (See Fig. 2b, d). Proteins that were clustered in 6w-cor-KO are functionally associated with “synaptic vesicle cycle and neurotransmitters release regulation”, “vesicle trafficking and fusion” and also “glutamatergic synapse”, suggesting that S-nitrosylation may be involved in an early phase of InsG3680(+/+) mouse model pathology (Fig. 2b). 4m-cor-KO mice showed very similar results (Fig. 2d). Clusters related to “glutamatergic synapse and glutamate regulation” and “synaptic vesicle cycle and neurotransmission” were observed. Supplementary Figure 4 lists in details lists of proteins in cor-KO groups that were clustered and are functioning in processes known to be correlated with ASD. Regulatory proteins and processes were clustered in the cor-WT groups such as “ATPases” proteins in 6w-cor-WT and “cell cycle” and “transcript regulation” processes in 4m-cor-WT (Supplementary Figure 3).
Shared SNO-proteins of 6w-old and 4m-old mice in the cortex are enriched for processes and interactome clusters known to be affected in ASD
We studied the shared proteins that were SNOed and found in 6w-old and 4m-old mice (See Supplementary Figure 5 and Supplementary Table 1). GO analysis was conducted to decipher the possible impact of the shared proteins of both ages on the cellular systems. The shared proteins of the cor-KO groups showed an evidence of enriched process known to be affected in ASD such as “synaptic vesicle cycle” (Fig. 2e and Supplementary Table 2). None of these processes were enriched in the other comparisons (see Supplementary Figure 5).
Interactomic analysis of the shared proteins of cor-KO proteins showed clusters of SNO-proteins that function in “glutamate regulation” (Got1, Got2, and Gnao1) and “synaptic vesicle cycle” (Stx1a, Ppp3ca, Nsf, and Dnm1). (Fig. 2f).
Significant increase of 3-nitrotyrosine in the cortical regions of 4m-KO mice
Ntyr is a product of NO• biochemistry participants, particularly peroxynitrite, and proteins become nitrated under conditions of NO• overproduction [74]. Therefore, we examined brain regions with potential elevation of NO• levels. IHC staining of Ntyr was performed in cortical regions that have functional relevance to the behavioral and physiological deficits in ASD [6]: prefrontal cortex (PFC), motor cortex (MC) and somatosensory cortex (SSC), as well as striatal regions: central, medial, and lateral striatum. The 6 week-old mice showed no significant difference between the KO and the WT groups (Supplementary Figure 6). The 4m-KO mice had increased Ntyr levels in the cortex and the striatum (See Fig. 4a, b, and Supplementary Figures 7,8). Morphometric analysis of the Ntyr level in the cortical regions showed significant increase (p < 0.05) in 4m-KO compared to 4m-WT (Fig. 4c). Further, an increase of Ntyr was also found in the striatal regions of 4m-KO compared to 4m-WT mice (Fig. 4c). Supplementary Figures 6 and 7 show the intensity of Ntyr in each specific cortical and striatal region in both 6w-old and 4m-old mice, respectively. The presence of Ntyr indicates an increased level of nitrosative stress in the cortex and striatum of InsG3680(+/+) mice.
Fig. 4.
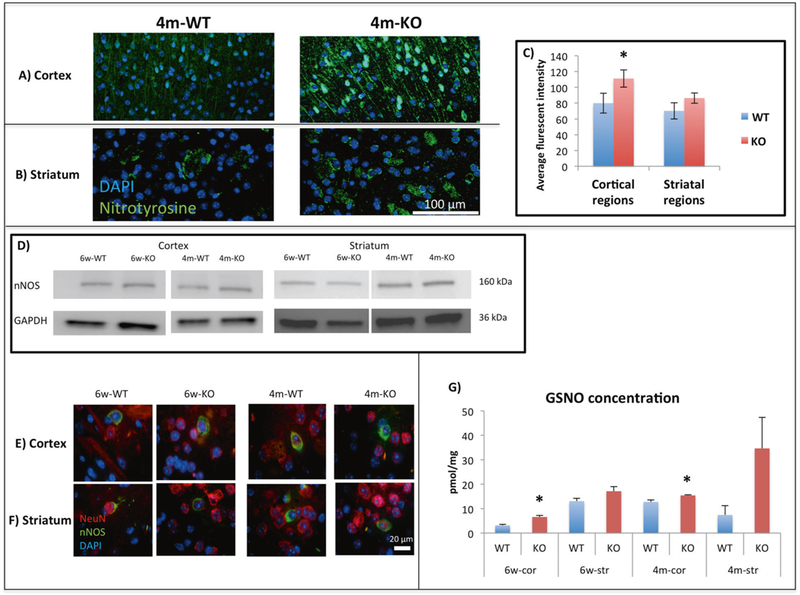
Representative staining of 3-nitrotyrosine (green) and DAPI (blue) for (a) Cortex and for (b) Striatum in 4m-WT and 4m-KO groups. c The average fluorescent intensity of 3-nitrotyrosine comparing 4m-WT to 4m-KO of the cortical regions (prefrontal cortex, motor cortex and somatosensory cortex) and striatal regions (central, medial, and lateral striatum). Significant increase was found in 4m-cor-KO compared to 4m-cor-WT. An increase was found in 4m-str-KO compared to 4m-str-WT. Presented as mean and ±SEM. One tailed t-test was conducted. *P < 0.05, WT mice (n = 9) and KO mice (n = 9). d Representative WB (for nNOS) from cortex and striatum tissues prepared from 6w-WT, 6w-KO, 4m-WT, and 4m-KO. See Supplementary Figure 9 for quantification. e Representative staining of the co-localization of nNOS (green), neurons (NeuN) (red), and DAPI (blue) in the cortex and f striatum of 6w-WT, 6w-KO, 4m-WT, and 4m-KO. g GSNO concentration of the 8 different tested groups. The multiple reaction monitoring (MRM) mode of a Triple Quadrupole mass spectrometer (QqQ MS) was used to establish a sensitive and selective quantification method. Y axis represents the concentration of GSNO in pmol/mg protein. Significant increase in the cor-KO compared to the cor-WT groups. Increase in the str-KO compared to the str-WT groups. Presented as mean and ±SEM. One tailed t-test was conducted. *P < 0.05, WT mice (n = 3) and KO mice (n = 3)
SNO-calcineurin leads to increase of phosphorylated (P) synapsin1 (ser62, ser67) and P-CREB (ser133) in 4m-cor-KO
The phosphatase calcineurin (CN, Ppp3ca-catalytic subunit) was SNOed in both ages of cor-KO mice. It was previously shown that nitrosylating compounds inhibit the activity of purified CN and of CN present in cell lysates [75]. To test the effect of S-nitrosylation on CN activity, we quantified two different known phosphorylated substrates: synapsin1, located in the presynapse [76] and cAMP-response element binding protein (CREB), located mainly in the postsynapse [77].
By WB, we found a significant increase of P-synapsin1 (Ser 62, Ser 67) in 4m-cor-KO compared to 4m-cor-WT (Fig. 3c, e). No significant changes were observed to P-synapsin in 6w KO vs. WT mice (Fig. 3c, d). We also found similar results with P-CREB (Ser 133), a significant increase of P-CREB in 4m-cor-KO but not in the 6w-cor-KO group (Fig. 3f–h). The no difference in the 6w groups may related to the fact that Ppp3cb (a different catalytic subunit in CN) was SNOed in 6w-cor-WT.
Examination of nNOS levels, nNOS localization, and GSNO levels in both WT and KO groups
In order to give a mechanistic explanation for the changes in SNO and Ntyr levels between the WT and KO groups, we have tested nNOS levels and its localization. We examined whether nNOS protein expression changed in the WT compared to KO groups in both cortex and striatum tissues of 6w and 4 m mice. We found no changes in protein expression between WT and KO groups see Fig. 4d and Supplementary Figure 9.
Further, We tested whether nNOS localization is different between WT and KO. The co-localization staining of nNOS and NeuN shown in Fig. 4e, f shows that nNOS is localized mainly in the cell body of the neuron cells in both the WT and KO groups.
To test whether the nNOS activity was altered, we measured S-nitrosoglutathione (GSNO) levels [72] in all WT and KO samples (Fig. 4g) and identified an increase of GSNO levels in the KO compared to the WT. The cortex of both ages showed significant differences in GSNO level, with an increase in both 6w-KO compared to 6w-WT and 4m-KO compared to 4m-WT. An increase was found in the str-KO compared to str-WT in both ages as well. The results point out the possibility of an increase of nNOS activity leading to increase of GSNO concentrations.
Discussion
Our study was designed to test the hypothesis that a mutated gene associated with human ASD would generate downstream effects on PTM of critical proteins that could lead to modification of synaptic functions. A significant body of published literature (reviewed in [39]) supports S-nitrosylation (SNO) as a protein PTM that leads to neuronal pathology.
The changes in the SNO-proteome found in the InsG3680(+/+) are a consequence of Shank3 mutation. That SNO-proteins were found exclusively in WT but not in KO, and vice versa shows that both nitrosylation and de-nitrosylation may be dysregulated in InsG3680(+/+) mice, leading to the small overlap between the groups.
Previous studies examined cellular, electrophysiological, and biochemical defects in the cortico-striatal circuits of the Shank3-KO model [16, 23, 29]. However only a few studies have looked at protein PTMs. One such study recently investigated changes in kinases and phosphatases that lead to alteration of the protein phosphorylation landscape [78].
In order to identify biology influencing or influenced by S-nitrosylation in the context of the ASD phenotype, we looked for processes and pathways significantly enriched among the proteins differentially nitrosylated in healthy and disease-model mice. GO and KEGG analysis revealed that deficiency of Shank3 leads to SNO-modulated proteins in the cortex that in part are functioning in ASD-related processes (Fig. 2). Earlier work has reported that Shank3-KO mice and other ASD models have a deficiency in glutamatergic synaptic neurotransmission, which include deficits in exocytosis, endocytosis and also imbalance of calcium regulation [16, 23, 29, 79, 80]. GO analysis of the shared synaptic proteins between 6w-cor-KO and 4m-cor-KO showed enrichment of the synaptic vesicle cycle and points out the possibility that these SNO-proteins may be involved in Shank3 pathology from young to adult age (Fig. 2e).
The network clusters in Supplementary Figures 2 and 3 analyzed by STRING demonstrate that age and regional locations lead to different changes in function for different classes of neurons in both cortex and striatum.
One of the shared SNO-proteins in the cortex of KO mice in both ages is CN, which is the only Protein Phosphatase (PP) in the brain activated by Ca2+ and a major regulator of key proteins essential for synaptic transmission and neuronal excitability [81, 82]. It has been shown that dysregulation of CN leads to different neurological disorders [83–85]. It was also previously shown that nitrosylating compounds inhibit the activity of purified CN and of CN present in cell lysates [75].
To confirm that SNO inhibits the phosphatase activity of CN, we have tested two CN substrates, synapsin1 and CREB. It is important to note that CN is the major Ser/Thr phosphatase that is a sensitive target for inhibition by NO. comparing to PP1 and PP2, meaning that the changes in the de-phosphorylation state of its substrates affected by nitrosative stress would be caused mainly by CN and not other PPs [75]. Further, synapsin1 dephosphorylation (P-Ser62, P-Ser 67) sites are highly specific to CN and not to PP1 or PP2 [86]. In the pre-synapse, CN is abundant and regulates key proteins involved in neurotransmission [82]. Synapsin1 is one of the key proteins controlling the vesicles for exocytosis by modulating its binding and trafficking as well. In the de-phosphorylated state, synapsin1 tethers synaptic vesicles and anchors them to actin-based cytoskeleton that maintains the vesicles in the reserve pool (RP) [87], but once phosphorylated, synapsin1 is detached from vesicles and leads to its mobilization from RP to a readily releasable pool (RRP) [76, 88] (See Fig. 5). The increased P-synapsin1 in 4m-cor-KO (Fig. 3c, e) supports the previous evidence that CN is inactivated by SNO and may lead to an increase of vesicle mobilization. To further validate CN inhibition by SNO, we have tested CREB, an important transcription factor that is also regulated by CN in neurons [89, 90]. CREB dephosphorylation is modulated by CN [91]. Our data showed consistency with the synapsin1 findings with an increase of P-CREB (Ser133) in 4m-cor-KO (Fig. 3f, h). P-CREB (Ser133) is involved in brain development and leads to an increase of gene expression by recruiting transcriptional co-activators [77] (Fig. 5). Our data are also consistent with previous studies that found an increase of P-CREB (Ser133) in an ASD murine model [92] and Fragile X mouse model [93]. Other work has also showed increase of different transcription factors in ASD [94, 95]. Here we provide a molecular interpretation of the elevated P-CREB levels.
Fig. 5.
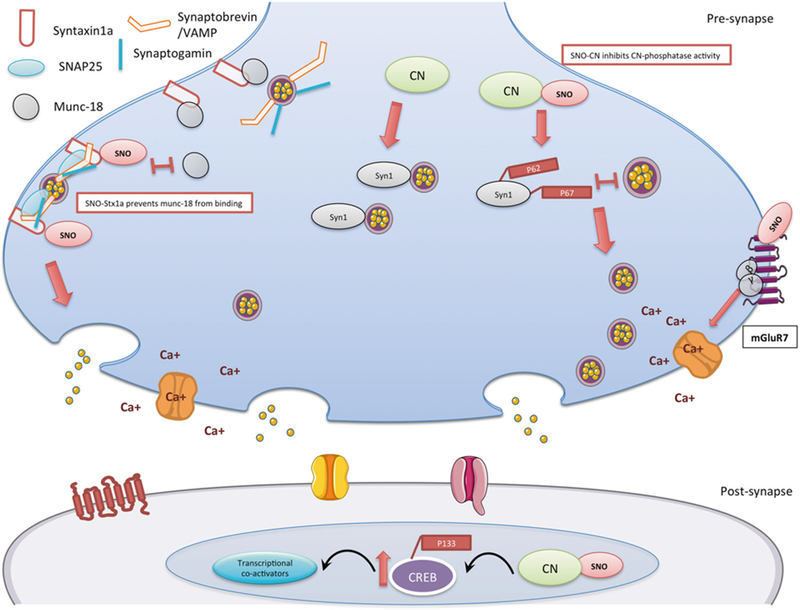
Schematic model summarizing the findings of the SNO-proteomics analysis and its consequences in 4m-cor-KO mice. In the pre-synapse, right side: S-nitrosylation of mGluR7 inhibits the receptor activity, once mGluR7 in a rest (or inhibited), β and γ subunits will not move to the calcium channel and inhibit the calcium channel activity. SNO-mGluR7 may lead to increase of calcium influx in the pre-synapse. In the pre-synapse, middle: S-nitrosylation of calcineurin (CN) leads to the inhibition of the phosphatase activity. This leads to an increase of its phosphorylated substrate P-synapsin1 (Syn1) (Ser62, Ser67). Phosphorylation of synapsin1 disconnects it from the vesicle and this leads to vesicle mobilization from the reserved pool to the readily released pool. In the pre-synapse, left side: SNO-Syntaxin1a prevents munc18 (also known as n-sec1) from binding to the closed conformation of syntaxin1a once SNOed. This allows syntaxin1a to unfold and bind to both VAMP on the vesicle and SNAP25 at the release site, which in turn enables the vesicle to dock to the membrane. In the post-synapse: The prolonged phosphorylation of CREB on Ser133 is a process mediated by Ca2+ dependent and -independent protein kinases. CREB phosphorylation is terminated by CN possibly through direct CREB dephosphorylation. SNO-CN inhibits CN activity and leads to increase of P-CREB (Ser 133) that leads to an increase of recruiting transcriptional co-activators
Another protein that was SNOed in the cortex of both ages is Stx1a which is a central coordinator for the vesicle exocytosis machine [96]. Stx1a interacts with synaptobrevin in the vesicle with SNAP25 and forms the SNARE complex [97] (Fig. 5). Munc18 also binds to Stx1a [96] in its closed conformation, inhibits Stx1a to bind to other SNAREs and execute vesicle fusion [98] (See Fig. 5). It was shown previously that incubation of proteins with nitrosative agents leads to increased formation of SNARE complex and inhibits binding of Munc18 to Stx1a [99]. Furthermore, S-nitrosylation of Stx1a was shown to act as a molecular switch inhibiting Munc18 binding to Stx1a [98]. This in turn leads to SNARE complex preclusion, facilitating Stx1a binding to the SNARE, and increasing the vesicle fusion machinery (Fig. 5) [98]. Towards this end, SNO-Stx1a in the cor-KO mice may have a critical role in facilitating vesicle fusion in Shank3-KO mice. Another mechanism that may lead to facilitating vesicle fusion is through mGluR7, which is highly localized in the pre-synapse [100] and SNOed in 4m-cor-KO. It is also proposed that in a resting state, mGluR7 binds Gβγ subunits; this binding inhibits the subunits from suppressing the voltage sensitive calcium channels leading to increase of Ca+2 influx in the pre-synapse [100] that could facilitate vesicle docking and fusion. We suggest that SNO of mGluR7 inhibits the receptor propagation by binding to the free cysteine [101] leading to the rest-state mode, which lead to increase of Ca+2 influx in the pre-synapse and facilitate vesicle fusion (see Fig. 5).
Our data, suggesting the potential increase of synaptic vesicle release and cortical activity, is consistent with previous data showing that deletion of Shank3 leads to an increase in cortical activity [102]. Further, the increase in P-CREB levels in the cortex supports our suggestion of increased cortical activity. Note that young or adult mice were not tested in the forementioned study, but only early postnatal mice [102].
Further, we have tested Ntyr levels (Fig. 4a–c) in both age groups and found a significant global increase of Ntyr in the 4m-KO mice in the cortical regions. The increased Ntyr formation could potentially be cytotoxic, leading to neuronal loss in certain neuron types [103, 104]. Generally, Ntyr forms from the radical recombination of NO• and O2•- to form peroxynitrite and is the fastest reaction of NO• in cells. Peroxynitrite reacts with tyrosine to form Ntyr and it is probable that the mutant cells develop a form of metabolic stress that increases the production of O •- by mitochondria, and with increased NO• leads to increased Ntyr.
To understand the mechanism behind the large changes in the SNO-proteome and Ntyr we measured nNOS levels and localization and found no change between the groups. It is now understood that one of the mechanisms for S-nitrosylation is transnitrosation by GSNO [42, 58]. Other nitrosylating agents are also responsible for S-nitrosylation such iNOS-S100A8/A9 [105], Thioredoxin, GAPDH, and others [58]. The elevated GSNO levels in the KO groups indicate the possibility that nNOS activity is increased, possibly through release of Ca++ from perinuclear proteins (based on unpublished experiments with embryonic neurons in vitro). This would lead to changes in NO•, SNO-proteins, and Ntyr levels in the KO group consistent with our observations.
In the case of protein phosphorylation there are kinases and phosphatases, and their balance determines cell function. We like to think of S-nitrosylation in a similar fashion with nitrosylases and de-nitrosylases. We now know about multiple SNO-peptides and proteins that drive transnitrosation and something about their specificity, but little about de-nitrosylases. We also know very little about the effect of SNO on protein function unless we have made that protein a specific target for further investigation.
In this study we did not account for de-nitrosylation that acts through different de-nitrosylases such GSNO redutase which regulates GSNO levels [106, 107], thioredoxin reductase, Xanthine oxidase, and others, although it is obvious that it is an important factor [58]. Genetic deletion of GSNO reductase in liver leads to a substantial increase in GSNO and subsequent liver cancer [108]. This study focused on the idea of looking at the entire SNO-proteome in WT and in a single genetic alteration and then looking at the biological effect using GO, pathway, and connectivity analysis to understand some of the downstream effects of the mutation.
Although our mouse model represents a mutation in a gene encoding a postsynaptic protein, substantial presynaptic modifications were characterized in our current study. These transsynaptic changes can be the result of the inability of Shank3 to signal properly to the presynaptic site [109].
A previous in vitro study found a robust changes in synaptic transmission, without alterations in presynaptic protein levels [109]. This suggests that posttranslational modifications, such S-nitrosylation may affect presynaptic proteins’ function, such as Stx1a, CN, VAMP and others that may alter neurotransmitter release.
To summarize, our findings show for the first time the involvement of NO. in an ASD mouse model (InsG3680 (+/+)). We have shown that S-nitrosylation is shifted in KO mice compared to WT. GO analysis demonstrated that SNO in KO mice affects processes and pathways known to be involved in ASD. SNO of critical proteins was identified in both ages of KO mice suggesting that NO. is involved in the pathology from a young age. We have shown that SNO-CN led to a significant increase in P-synapsin1 and P-CREB, which affect vesicle mobilization and gene transcription, respectively. We also suggest that SNO-Stx1a and SNO-mGluR7, which were identified in our study, regulate vesicle docking and calcium influx, respectively. Lastly, the nNOS and GSNO results point out the possibility of an increase of nNOS activity in the mutant mice.
Finally, while in this study we characterized a monogenic mouse model, there are hundreds of genes associated with ASD. The conceptual research approach we have taken here, of examining a specific modification of proteins allows a systems-level characterization and identification of key proteins, cellular pathways and neurobiological mechanisms that might be affected in ASD.
Supplementary Material
Acknowledgements
This work was supported by the MIT Center for Environmental Health Sciences Grant ES002109 and a grant from the Simons Foundation to the Simons Center for the Social Brain at MIT (SRT). Dr. Haitham Amal was supported by the Satell Technion-MIT Post-Doctoral Program. Research related to this project in the Feng lab was supported by NIMH (MH097104), Stanley Center for Psychiatric Research at Broad Institute of MIT and Harvard, Nancy Lurie Marks Family Foundation and Simons Center for the Social Brain at MIT. Dr. Boaz Barak was supported by postdoctoral fellowships from the Simons Center for the Social Brain at MIT and the Autism Science Foundation. Brian Joughin was supported by Army Research Office Institute for Collaborative Biotechnologies grant W911NF-09–0001.
Footnotes
Compliance with ethical standards
Conflict of interest The authors declare that they have no conflict of interest.
Contributor Information
Haitham Amal, Email: haitham.amal85@gmail.com.
Steven R. Tannenbaum, Email: srt@mit.edu.
References
- 1.Bourgeron T From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci 2015;16:551–63. [DOI] [PubMed] [Google Scholar]
- 2.Ronemus M, Iossifov I, Levy D, Wigler M. The role of de novo mutations in the genetics of autism spectrum disorders. Nat Rev Genet 2014;15:133–41. [DOI] [PubMed] [Google Scholar]
- 3.Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, Vitkup D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 2011;70:898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry 2011;68:1095–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Modabbernia A, Velthorst E, Reichenberg A. Environmental risk factors for autism: an evidence-based review of systematic reviews and meta-analyses. Mol Autism 2017;8:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barak B, Feng G. Neurobiology of social behavior abnormalities in autism and Williams syndrome. Nat Neurosci 2016;19: 647–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edition F, Association AP. Diagnostic and statistical manual of mental disorders Washington: American Psychological Association; 1994. [Google Scholar]
- 8.Phelan K, McDermid H. The 22q13. 3 deletion syndrome (Phelan-McDermid syndrome). Mol Syndromol 2011;2: 186–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet 2007;39:25–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moessner R, Marshall CR, Sutcliffe JS, Skaug J, Pinto D, Vincent J, et al. Contribution of SHANK3 mutations to autism spectrum disorder. Am J Human Genet 2007;81:1289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gauthier J, Spiegelman D, Piton A, Lafrenière RG, Laurent S, St‐Onge J, et al. Novel de novo SHANK3 mutation in autistic patients. Am J Med Genet Part B: Neuropsychiatr Genet 2009;150:421–4. [DOI] [PubMed] [Google Scholar]
- 12.Boccuto L, Lauri M, Sarasua SM, Skinner CD, Buccella D, Dwivedi A, et al. Prevalence of SHANK3 variants in patients with different subtypes of autism spectrum disorders. Eur J Hum Genet 2013;21:310–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du Y, Weed SA, Xiong W-C, Marshall TD, Parsons JT. Identification of a novel cortactin SH3 domain-binding protein and its localization to growth cones of cultured neurons. Mol Cell Biol 1998;18:5838–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, et al. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 1999;23:569–82. [DOI] [PubMed] [Google Scholar]
- 15.Boeckers TM, Winter C, Smalla K-H, Kreutz MR, Bockmann J, Seidenbecher C, et al. Proline-rich synapse-associated proteins ProSAP1 and ProSAP2 interact with synaptic proteins of the SAPAP/GKAP family. Biochem Biophys Res Commun 1999;264:247–52. [DOI] [PubMed] [Google Scholar]
- 16.Monteiro P, Feng G. SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat Rev Neurosci 2017;18:147–57. [DOI] [PubMed] [Google Scholar]
- 17.Sheng M, Kim E. The Shank family of scaffold proteins. J Cell Sci 2000;113:1851–6. [DOI] [PubMed] [Google Scholar]
- 18.Sheng M, Kim E. The postsynaptic organization of synapses. Cold Spring Harb Perspect Biol 2011;3:a005678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McAllister AK. Dynamic aspects of CNS synapse formation. Annu Rev Neurosci 2007;30:425–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, et al. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 1999;23: 583–92. [DOI] [PubMed] [Google Scholar]
- 21.Baron MK, Boeckers TM, Vaida B, Faham S, Gingery M, Sawaya MR, et al. An architectural framework that may lie at the core of the postsynaptic density. Science 2006;311:531–5. [DOI] [PubMed] [Google Scholar]
- 22.Kreienkamp H-J. Scaffolding proteins at the postsynaptic density: shank as the architectural framework. Handb Exp Pharmacol 2008;186:365–80. [DOI] [PubMed] [Google Scholar]
- 23.Peca J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, et al. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011;472:437–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graybiel AM. Habits, rituals, and the evaluative brain. Annu Rev Neurosci 2008;31:359–87. [DOI] [PubMed] [Google Scholar]
- 25.Ting JT, Feng G. Neurobiology of obsessive–compulsive disorder: insights into neural circuitry dysfunction through mouse genetics. Curr Opin Neurobiol 2011;21:842–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shepherd GM. Corticostriatal connectivity and its role in disease. Nat Rev Neurosci 2013;14:278–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muehlmann A, Lewis M. Abnormal repetitive behaviours: shared phenomenology and pathophysiology. J Intellect Disabil Res 2012;56:427–40. [DOI] [PubMed] [Google Scholar]
- 28.Welch JM, Lu J, Rodriguiz RM, Trotta NC, Peca J, Ding J-D, et al. Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature 2007;448:894–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou Y, Kaiser T, Monteiro P, Zhang X, Van der Goes MS, Wang D, et al. Mice with Shank3 mutations associated with ASD and schizophrenia display both shared and distinct defects. Neuron 2016;89:147–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bozdagi O, Sakurai T, Papapetrou D, Wang X, Dickstein DL, Takahashi N, et al. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol Autism 2010;1:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dichter GS. Functional magnetic resonance imaging of autism spectrum disorders. Dialog- Clin Neurosci 2012;14: 319–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mei Y, Monteiro P, Zhou Y, Kim J-A, Gao X, Fu Z, et al. Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 2016;530:481–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ting JT, Peça J, Feng G. Functional consequences of mutations in postsynaptic scaffolding proteins and relevance to psychiatric disorders. Annu Rev Neurosci 2012;35:49–71. [DOI] [PubMed] [Google Scholar]
- 34.Peça J, Feng G. Cellular and synaptic network defects in autism. Curr Opin Neurobiol 2012;22:866–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Snyder SH, Bredt DS. Biological roles of nitric oxide. Sci Am 1992;266:68–71. [DOI] [PubMed] [Google Scholar]
- 36.Bredt D, Snyder S. Nitric oxide: a physiologic messenger molecule. Annu Rev Biochem 1994;63:175–95. [DOI] [PubMed] [Google Scholar]
- 37.Bredt DS, Hwang PM. Cloned and expressed nitric oxide synthase structurally resembles cytochrome P-450 reductase. Nature 1991;351:714. [DOI] [PubMed] [Google Scholar]
- 38.Förstermann U, Schmidt HH, Pollock JS, Sheng H, Mitchell JA, Warner TD, et al. Isoforms of nitric oxide synthase characterization and purification from different cell types. Biochem Pharmacol 1991;42:1849–57. [DOI] [PubMed] [Google Scholar]
- 39.Nakamura T, Prikhodko OA, Pirie E, Nagar S, Akhtar MW, Oh C-K, et al. Aberrant protein S-nitrosylation contributes to the pathophysiology of neurodegenerative diseases. Neurobiol Dis 2015;84:99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garry P, Ezra M, Rowland M, Westbrook J, Pattinson K. The role of the nitric oxide pathway in brain injury and its treatment —from bench to bedside. Exp Neurol 2015;263:235–43. [DOI] [PubMed] [Google Scholar]
- 41.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol 2001;3:193–7. [DOI] [PubMed] [Google Scholar]
- 42.Smith BC, Marletta MA. Mechanisms of S-nitrosothiol formation and selectivity in nitric oxide signaling. Curr Opin Chem Biol 2012;16:498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakamura T, Tu S, Akhtar MW, Sunico CR, Okamoto Si, Lipton SA. Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron 2013;78:596–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seth D, Hess DT, Hausladen A, Wang L, Wang Y-j, Stamler JS. A multiplex enzymatic machinery for cellular protein S-nitrosylation. Mol Cell 2018;69:451–.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cakatay U, Telci A, Kayali R, Tekeli F, Akcay T, Sivas A. Relation of oxidative protein damage and nitrotyrosine levels in the aging rat brain. Exp Gerontol 2001;36:221–9. [DOI] [PubMed] [Google Scholar]
- 46.Darwish RS, Amiridze N, Aarabi B. Nitrotyrosine as an oxidative stress marker: evidence for involvement in neurologic outcome in human traumatic brain injury. J Trauma Acute Care Surg 2007;63:439–42. [DOI] [PubMed] [Google Scholar]
- 47.Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin JC, Smith CD, et al. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch Biochem Biophys 1992;298:431–7. [DOI] [PubMed] [Google Scholar]
- 48.Stamler JS, Lamas S, Fang FC. Nitrosylation: the prototypic redox-based signaling mechanism. Cell 2001;106:675–83. [DOI] [PubMed] [Google Scholar]
- 49.Stamler JS, Simon DI, Osborne JA, Mullins ME, Jaraki O, Michel T, et al. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci USA 1992;89:444–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qu J, Nakamura T, Cao G, Holland EA, McKercher SR, Lipton SA. S-Nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by β-amyloid peptide. Proc Natl Acad Sci USA 2011;108:14330–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Uehara T, Nakamura T, Yao D, Shi Z-Q, Gu Z, Ma Y, et al. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 2006;441:513–7. [DOI] [PubMed] [Google Scholar]
- 52.Shi Z-Q, Sunico CR, McKercher SR, Cui J, Feng G-S, Nakamura T, et al. S-nitrosylated SHP-2 contributes to NMDA receptor-mediated excitotoxicity in acute ischemic stroke. Proc Natl Acad Sci USA 2013;110:3137–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seneviratne U, Nott A, Bhat VB, Ravindra KC, Wishnok JS, Tsai L-H, et al. S-nitrosation of proteins relevant to Alzheimer’s disease during early stages of neurodegeneration. Proc Natl Acad Sci USA 2016;113:4152–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haun F, Nakamura T, Shiu AD, Cho D-H, Tsunemi T, Holland EA, et al. S-nitrosylation of dynamin-related protein 1 mediates mutant huntingtin-induced mitochondrial fragmentation and neuronal injury in Huntington’s disease. Antioxid Redox Signal 2013;19:1173–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chung KK, Dawson VL, Dawson TM. S‐Nitrosylation in Parkinson’s disease and related neurodegenerative disorders. Methods Enzymol 2005;396:139–50. [DOI] [PubMed] [Google Scholar]
- 56.Sun N, Hao J-R, Li X-Y, Yin X, Zong Y-Y, Zhang G, et al. GluR6-FasL-Trx2 mediates denitrosylation and activation of procaspase-3 in cerebral ischemia/reperfusion in rats. Cell Death Dis 2013;4:e771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Benhar M, Forrester MT, Stamler JS. Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat Rev Mol Cell Biol 2009;10:721–32. [DOI] [PubMed] [Google Scholar]
- 58.Anand P, Stamler JS. Enzymatic mechanisms regulating protein S-nitrosylation: implications in health and disease. J Mol Med 2012;90:233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barglow KT, Knutson CG, Wishnok JS, Tannenbaum SR, Marletta MA. Site-specific and redox-controlled S-nitrosation of thioredoxin. Proc Natl Acad Sci USA 2011;108:E600–E6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Broek JA, Guest PC, Rahmoune H, Bahn S. Proteomic analysis of post mortem brain tissue from autism patients: evidence for opposite changes in prefrontal cortex and cerebellum in synaptic connectivity-related proteins. Mol Autism 2014;5:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wesseling H, Xu B, Want E, Holmes E, Guest P, Karayiorgou M, et al. System-based proteomic and metabonomic analysis of the Df (16) A + /− mouse identifies potential miR-185 targets and molecular pathway alterations. Mol Psychiatry 2017;22: 384–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reim D, Distler U, Halbedl S, Verpelli C, Sala C, Bockmann J, et al. Proteomic analysis of post-synaptic density fractions from Shank3 mutant mice reveals brain region specific changes relevant to autism spectrum disorder. Front Mol Neurosci 2017;10:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Raju K, Doulias P-T, Evans P, Krizman EN, Jackson JG, Horyn O, et al. Regulation of brain glutamate metabolism by nitric oxide and S-nitrosylation. Sci Signal 2015;8:ra68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bredt DS, Snyder SH. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci USA 1990;87:682–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tannenbaum SR,White FM, Regulation and specificity of S-nitrosylation and denitrosylation. ACS Chem Biol 2006;1:615–8. [DOI] [PubMed] [Google Scholar]
- 66.Seneviratne U, Godoy LC, Wishnok JS, Wogan GN, Tannenbaum SR. Mechanism-based triarylphosphine-ester probes for capture of endogenous RSNOs. J Am Chem Soc 2013;135: 7693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wen Y, Alshikho MJ, Herbert MR. Pathway network analyses for autism reveal multisystem involvement, major overlaps with other diseases and convergence upon MAPK and calcium signaling. PLoS ONE 2016;11:e0153329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Miraldi ER, Sharfi H, Friedline RH, Johnson H, Zhang T, Lau KS, et al. Molecular network analysis of phosphotyrosine and lipid metabolism in hepatic PTP1b deletion mice. Integrative. Biology 2013;5:940–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Botstein D, Cherry JM, Ashburner M, Ball C, Blake J, Butler H, et al. Gene Ontology: tool for the unification of biology. Nat Genet 2000;25:25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 2016;45:D353–D61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRINGv10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2014;43:D447–D52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang X, Garcia TC, Gong G, Wishnok SJ, Tannenbaum RS. Automated online solid phase derivatization for sensitive quantification of endogenous S-nitrosoglutathione and rapid capture of other low-molecular-mass S-nitrosothiols. Anal Chem 2017;90:1967–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2008;4:44. [DOI] [PubMed] [Google Scholar]
- 74.Dedon PC, Tannenbaum SR. Reactive nitrogen species in the chemical biology of inflammation. Arch Biochem Biophys 2004;423:12–22. [DOI] [PubMed] [Google Scholar]
- 75.Sommer D, Coleman S, Swanson SA, Stemmer PM. Differential susceptibilities of serine/threonine phosphatases to oxidative and nitrosative stress. Arch Biochem Biophys 2002;404:271–8. [DOI] [PubMed] [Google Scholar]
- 76.Chi P, Greengard P, Ryan TA. Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron 2003;38:69–78. [DOI] [PubMed] [Google Scholar]
- 77.Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron 2002; 35:605–23. [DOI] [PubMed] [Google Scholar]
- 78.Bidinosti M, Botta P, Krüttner S, Proenca CC, Stoehr N, Bernhard M, et al. CLK2 inhibition ameliorates autistic features associated with SHANK3 deficiency. Science 2016;351:1199–203. [DOI] [PubMed] [Google Scholar]
- 79.Bourgeron T A synaptic trek to autism. Curr Opin Neurobiol 2009;19:231–4. [DOI] [PubMed] [Google Scholar]
- 80.Lee J, Chung C, Ha S, Lee D, Kim D-Y, Kim H, et al. Shank3-mutant mice lacking exon 9 show altered excitation/inhibition balance, enhanced rearing, and spatial memory deficit. Front Cell Neurosci 2015;9:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Baumgärtel K, Mansuy IM. Neural functions of calcineurin in synaptic plasticity and memory. Learn Mem 2012;19:375–84. [DOI] [PubMed] [Google Scholar]
- 82.Mansuy IM. Calcineurin in memory and bidirectional plasticity. Biochem Biophys Res Commun 2003;311:1195–208. [DOI] [PubMed] [Google Scholar]
- 83.Dineley KT, Kayed R, Neugebauer V, Fu Y, Zhang W, Reese LC, et al. Amyloid‐β oligomers impair fear conditioned memory in a calcineurin‐dependent fashion in mice. J Neurosci Res 2010;88:2923–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rachidi M, Lopes C. Molecular and cellular mechanisms elucidating neurocognitive basis of functional impairments associated with intellectual disability in Down syndrome. Am J Intellect Dev Disabil 2010;115:83–112. [DOI] [PubMed] [Google Scholar]
- 85.Miyakawa T, Leiter LM, Gerber DJ, Gainetdinov RR, Sotnikova TD, Zeng H, et al. Conditional calcineurin knockout mice exhibit multiple abnormal behaviors related to schizophrenia. Proc Natl Acad Sci USA 2003;100:8987–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jovanovic JN, Sihra TS, Nairn AC, Hemmings HC, Greengard P, Czernik AJ. Opposing changes in phosphorylation of specific sites in synapsin I during Ca2+-dependent glutamate release in isolated nerve terminals. J Neurosci 2001;21:7944–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ceccaldi P-E, Grohovaz F, Benfenati F, Chieregatti E, Greengard P, Valtorta F. Dephosphorylated synapsin I anchors synaptic vesicles to actin cytoskeleton: an analysis by videomicroscopy. J Cell Biol 1995;128:905–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Sci- New Y Then Wash 1993;259:780–5. [DOI] [PubMed] [Google Scholar]
- 89.Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca2+and stimulus duration–dependent switch for hippocampal gene expression. Cell 1996;87:1203–14. [DOI] [PubMed] [Google Scholar]
- 90.Siemann G, Blume R, Grapentin D, Oetjen E, Schwaninger M, Knepel W. Inhibition of cyclic AMP response element-binding protein/cyclic AMP response element-mediated transcription by the immunosuppressive drugs cyclosporin A and FK506 depends on the promoter context. Mol Pharmacol 1999;55:1094–100. [DOI] [PubMed] [Google Scholar]
- 91.Chang KT, Berg DK. Voltage-gated channels block nicotinic regulation of CREB phosphorylation and gene expression in neurons. Neuron 2001;32:855–65. [DOI] [PubMed] [Google Scholar]
- 92.He X, Thacker S, Romigh T, Yu Q, Frazier TW, Eng C. Cytoplasm-predominant Pten associates with increased region-specific brain tyrosine hydroxylase and dopamine D2 receptors in mouse model with autistic traits. Mol Autism 2015;6:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang H, Morishita Y, Miura D, Naranjo JR, Kida S, Zhuo M. Roles of CREB in the regulation of FMRP by group I metabotropic glutamate receptors in cingulate cortex. Mol Brain 2012;5:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Onore C, Yang H, Van de Water J, Ashwood P. Dynamic akt/ mTOr signaling in children with autism spectrum Disorder. Front Pediatr 2017;5:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gilbert J, Man H. Translational dysregulation in autism. Cell Dev Biol 2014;3:e124. [Google Scholar]
- 96.Wu MN, Fergestad T, Lloyd TE, He Y, Broadie K, Bellen HJ. Syntaxin 1A interacts with multiple exocytic proteins to regulate neurotransmitter release in vivo. Neuron 1999;23:593–605. [DOI] [PubMed] [Google Scholar]
- 97.Söllner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, et al. SNAP receptors implicated in vesicle targeting and fusion. Nature 1993;362:318–24. [DOI] [PubMed] [Google Scholar]
- 98.Palmer ZJ, Duncan RR, Johnson JR, Lian L-Y, Mello LV, Booth D, et al. S-nitrosylation of syntaxin 1 at Cys145 is a regulatory switch controlling Munc18–1 binding. Biochem J 2008;413:479–91. [DOI] [PubMed] [Google Scholar]
- 99.Meffert MK, Calakos NC, Scheller RH, Schulman H. Nitric oxide modulates synaptic vesicle docking/fusion reactions. Neuron 1996;16:1229–36. [DOI] [PubMed] [Google Scholar]
- 100.Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 2010;50:295–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Muto T, Tsuchiya D, Morikawa K, Jingami H. Structures of the extracellular regions of the group II/III metabotropic glutamate receptors. Proc Natl Acad Sci USA 2007;104:3759–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Peixoto RT, Wang W, Croney DM, Kozorovitskiy Y, Sabatini BL. Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B−/− mice. Nat Neurosci 2016; 19:716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Peluffo H, Shacka JJ, Ricart K, Bisig CG, Martìnez‐Palma L, Pritsch O, et al. Induction of motor neuron apoptosis by free 3‐ nitro‐l‐tyrosine. J Neurochem 2004;89:602–12. [DOI] [PubMed] [Google Scholar]
- 104.Kuhn DM, Sakowski SA, Sadidi M, Geddes TJ. Nitrotyrosine as a marker for peroxynitrite‐induced neurotoxicity: The beginning or the end of the end of dopamine neurons? J Neurochem 2004;89:529–36. [DOI] [PubMed] [Google Scholar]
- 105.Jia J, Arif A, Terenzi F, Willard B, Plow EF, Hazen SL, et al. Target-selective protein S-nitrosylation by sequence motif recognition. Cell 2014;159:623–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tang C-H, Wei W, Hanes MA, Liu L. Hepatocarcinogenesis driven by GSNOR deficiency is prevented by iNOS inhibition. Cancer Res 2013;73:2897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sips PY, Irie T, Zou L, Shinozaki S, Sakai M, Shimizu N, et al. Reduction of cardiomyocyte S-nitrosylation by S-nitrosoglutathione reductase protects against sepsis-induced myocardial depression. Am J Physiol-Heart Circ Physiol 2013;304:H1134–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wei W, Li B, Hanes MA, Kakar S, Chen X, Liu L. S-nitrosylation from GSNOR deficiency impairs DNA repair and promotes hepatocarcinogenesis. Sci Transl Med 2010;2: 19ra3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Arons MH, Thynne CJ, Grabrucker AM, Li D, Schoen M, Cheyne JE, et al. Autism-associated mutations in ProSAP2/ Shank3 impair synaptic transmission and neurexin–neuroliginmediated transsynaptic signaling. J Neurosci 2012;32: 14966–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.