

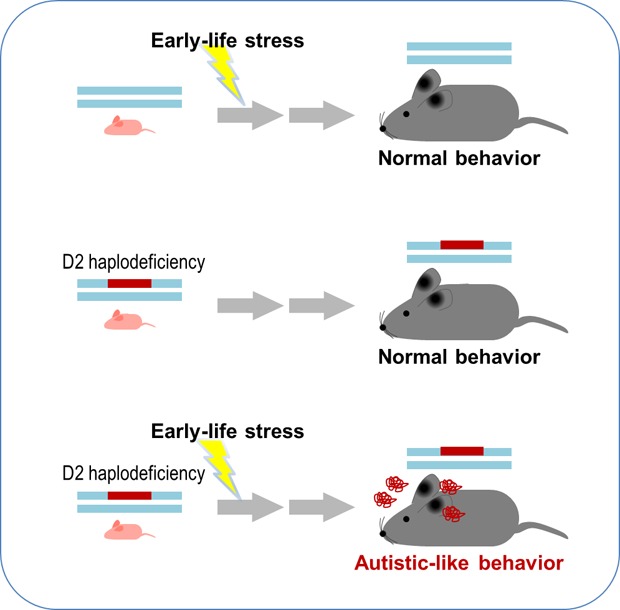
Abstract
A number of specific genetic variants including gene mutations and single nucleotide variations have been identified in genomewide association studies of autism spectrum disorder (ASD). ASD phenotypes in individuals carrying specific genetic variations are manifest mostly in a heterozygous state. Furthermore, individuals with most genetic variants show incomplete penetrance and phenotypic variability, suggesting that non-genetic factors are also involved in developing ASD. However, the mechanisms of how genetic and environmental factors interactively promote ASD are not clearly understood. In the present study, we investigated whether early-life stress (ELS) in D2 dopamine receptor heterozygous knockout (D2+/−) mice induces ASD-like symptoms. To address that, we exposed D2 heterozygous pups to maternal separation stress for 3 h daily for 13 days beginning on postnatal day 2. D2+/− adult mice that had experienced ELS exhibited impaired sociability in the three-chamber test and home-cage social interaction test and increased grooming behavior, whereas wildtype littermates exposed to ELS did not show those phenotypes. ELS-exposed D2+/− mice had decreased levels of BDNF, TrkB, phospho-ERK1/2 and phospho-CREB in the dorsal striatum. Administration of the TrkB agonist 7,8-dihydroxyflavone (7,8-DHF) to ELS-exposed D2+/− mice rescued the sociability deficits and repetitive behavior. In contrast, behavioral rescue by 7,8-DHF in ELS-exposed D2+/− mice was blocked when TrkB expression in the dorsal striatum was locally inhibited by the injection of TrkB-siRNA. Together, our results suggest that the interaction between ELS and defective D2 gene function promotes autistic-like behaviors by downregulating the BDNF-TrkB pathway in the dorsal striatum.
Keywords: Autism spectrum disorder, early-life stress, dorsal striatum, dopamine receptors, TrkB receptor
Graphical Abstract
INTRODUCTION
Autism spectrum disorder (ASD) is a group of neurodevelopmental disorders characterized by social communication defects and stereotyped repetitive behaviors [1]. ASD is usually diagnosed before the age of three [2]. Recent genome-wide studies have identified a number of specific gene mutations in individuals with ASD, including deletion, duplication and point mutations, and genetic variants such as single nucleotide variants and copy number variants. However, phenotypic penetrance of those genetic variants is incomplete [3]. For example, ASD symptoms are expressed in 84% of cases carrying 22q13.33 deletion or SHANK3 mutations [4], 30~60% of cases carrying NRXN1 deletion [5], and 24% of cases carrying 16p11.2 deletion and duplication [6,7]. Furthermore, the phenotypic spectrums of the individuals with those genetic variants are highly variable [8,9], partly because ASD phenotypes of individuals with those genetic variations are manifested mostly in a heterozygous state. Furthermore, ASD is produced by the interaction between environmental factors and genetic variants (gene×environment interaction) [10,11]. Therefore, understanding the interaction between environmental factors and genetic factors is important. However, the mechanisms by which gene×environment causes ASD are largely unknown.
Early-life stress (ELS) is believed to be one of that environmental factors affect ASD development [12,13]. The development of the brain network is sensitive to early-life adversity, depending on the intensity of the stress, timing of exposure, and genetic factors [14]. Specifically, a stressful migration after birth increased the risk of ASD along with stress-induced epigenetic changes [15]. Mothers with aberrant serotonin function (short allele variants of SLC6A4, known as an ASD risk gene) responded differently to stress, which increased the risk of ASD and ASD-like symptoms in their children [16]. Slc6a4+/− mice treated with prenatal stress exhibited impairment in social interaction and an increased embryonic methylation profile [17,18]. Those studies support the notion that ASD can be induced by the interaction between a genetic defect and environmental disturbance during development. However, that hypothesis has not been directly tested.
The dopamine system is closely related to ASD [19,20]. Polymorphisms in D2 dopamine receptors (A1 allele of Taq I; [21], rs1800498TT; [22]) and D1 dopamine receptors (rs265981-C and rs4532-A, rs686-T alleles) [23], and a de novo mutation (T356M) in the dopamine transporter [24] have been reported in ASD. Furthermore, mice whose D1 receptors were overactivated using a D1 receptor agonist (SKF38393) exhibited autistic-like behaviors. Mice lacking D2 dopamine receptors also showed autistic-like behaviors [20]. These results suggest that the dopamine system, in particular the D1 overactive state, is closely associated with ASD pathology. In contrast, D2 heterozygous mice, which have a hyperactive state of D1 dopamine receptors in comparison, did not exhibit autistic-like behaviors [20]. Considering that phenotypic penetrance of many specific genetic mutations is often incomplete [3,25,26], and ASD phenotypes of individuals carrying a known genetic mutation are highly variable [8,9], it is possible that the D2 heterozygous state might function as a genetic risk factor that promotes ASD under certain environmental condition. However, this possibility has not been carefully tested.
In this study, we investigated whether ELS can cause autistic-like behaviors in D2 heterozygous mice. We found that ELS in D2 heterozygous mice but not in wildtype mice produced autistic-like behaviors through the downregulation of BDNF-TrkB signaling in the dorsal striatum.
MATERIALS AND METHODS
Animals
C57BL/6 mice were purchased from Daehan BioLink (Eumsung, Chungbuk, Republic of Korea). D2 dopamine receptor knockout (KO) mice were described previously [20,27]. D2 KO mice were backcrossed to C57BL/6 mice for more than 10 generations. D2 KO mice and their wildtype (WT) littermates used in all experiments were from heterozygote crosses. For genotyping, the following primer sets were used: 5′-TGTGACTGCAACATCCCA CC-3′ and 5′-GCGGAACTCAATGTTGAAGG-3′ for D2 WT (105 bp); 5′-CTTG GGTGGAGAGGCTATTC-3′ and 5′-AGGTGAGATGACAGGA GATC-3′ for D2 KO (neo; 280 bp). The animal room was maintained at a temperature of 22~23℃ with 50~60% humidity and a normal light-dark cycle (light on at 7:00 a.m.) in a specific-pathogen-free environment. All animals were handled in accordance with the Guidelines of Animal Care at Ewha Womans University through the permission granted by EWU-IACUC (No. 16-020).
Early-life stress (ELS) treatment
ELS treatment was carried out as described previously [28]. Male D2 heterozygous KO mice were crossed with female C57BL/6 mice. Pregnant females were randomly assigned to one of maternal separation groups and control groups. Two to three days before giving birth, pregnant females were habituated to a signal cage filled with wood particles 30% less than before. The number of pups for each mother was adjusted to have six to eight. The pups were separated from their mother from 9:00 A.M. and placed together in a clean plastic cage maintained at 32~33℃ with 50~60% humidity, and then returned to their mother in the original cage after 3 hours of separation. Maternal separation began on postnatal day 2 and was repeated until postnatal day 14. For the control group, pregnant female were habituated to a single cage with normal wood particles two to three days before giving birth, and the control pups were reared without maternal separation in parallel with the ELS-exposed group. Control and ELS-exposed pups were weaned 4 weeks after birth. After genotyping, they were housed 2~3 mice with the same sex in a regular plastic cage. This housing condition was maintained until the behavioral tests, which was conducted between 8 and 10 weeks after birth.
Restraint stress treatment
Mice were restrained as described previously [29]. In brief, mice were individually placed in a 50-ml polypropylene conical tube that have many holes for ventilation and were restrained within this tube for 1 h.
Corticosterone measurement
Corticosterone measurements were carried out as described previously [29]. For these experiments, all animals were sacrificed between 12 p.m. and 1 p.m. ELS-exposed pups were sacrificed after the last treatment of maternal separation stress on postnatal day 14. Blood was collected from the hearts of the sacrificed mice. It was centrifuged at 1,500 g for 15 min to obtain serum, which was then stored at −80℃ until use. Plasma corticosterone levels were assessed using a commercial enzyme immunoassay kit (ADI-900-045, Enzo Life Science, NY, USA) and following the manufacturer's instructions. In brief, 1:40 diluted samples were loaded in each well in duplicate and probed with corticosterone ELISA antibody for 2 hours. Then, the wells were emptied, rinsed three times with wash buffer, and probed with pNPP-substrate for 1 hour at room temperature. Optical density was detected at 405nm using a spectrofluorometer (SpectraMax M5, Molecular Devices).
Stereotaxic injection
Stereotaxic injection of siRNA was carried out as described previously [20,30,31]. Mice were anesthetized with a mixture of ketamine hydrochloride (50 mg/mL) and xylazine hydrochloride (23.3 mg/ml) at a dose of 2.5 µl per body weight (g). While the mouse head was held in the stereotaxic device (Stoelting Company, Wood Dale, IL, USA), 1.5 µl (18 ng of siRNA) of siRNA mixture per each side of the dorsal striatum (AP, +1.0; ML, ±1.5; DV, −3.6 mm) was injected at the rate of 0.2 µl/min using a 30 G needle. The siRNA mixture was prepared 20 min before injection by mixing 1 µl of control-siRNA or TrkB-siRNA with siGLO Green (1/19 of target gene-siRNA), 0.5 µl of 50% sucrose and 2.5 µl of Neuro-FECT transfection reagent (T800075; Genlantis, San Diego, CA, USA).
Behavioral tests were conducted at the indicated time points. After the behavioral tests, the injection sites were confirmed by a histological analysis, and the mice with the wrong injection site were excluded from the final data as described [20,30,31]. The following siRNAs were used: control-siRNA (SN-1012) and Trk-BsiRNA (1393919, NM_001025074.2) were purchased from Bioneer Co. (Deajun, Korea), and FAM-labeled RISK-independent siRNA transfection control siGLO Green (D-001630-01-05) was purchased from Dharmacon Inc. (Chicago, IL, USA).
Drug administration
7,8-Dihydroxyflavone (7,8-DHF) was intraperitoneally administered once a day for three consecutive days at a dose of 10 mg/kg. The dose was chosen on the basis of a dose test in this study and a previous study [32]. 7,8-DHF was purchased from Sigma Aldrich (St. Louis, MO, USA).
Real-time PCR analysis
The real-time PCR analysis was performed as described previously [20,30]. Total RNA was extracted from the brain tissues of 3~4 animals from each group using TRI reagent (Sigma-Aldrich, St. Louis, MO, USA) with DNAase I to prevent genomic DNA contamination. Real-time PCR was conducted with iQTM SYBR Green Supermix (Bio-Rad Laboratories, Foster City, CA, USA) using a CFX 96 Real-Time PCR System Detector (Bio-Rad Laboratories; Foster City, CA, USA). The following primer sets were used: 5′-AAGGACTTTCATCGGGAAGCTG-3′ and 5′-TCGCCCTCCACACAGACAC-3′ for TrkB; 5′-TGTCGTATTCACCTTCAGTT-3′ and 5′-TGCTTTCAGTCATTTGGCTATA-3′ for Mecp2; 5′-CAGTGTGGCTCAGATTCCCT-3′ and 5′-GGGCAGCTCATTAGGGATCT-3′ for Hdac1; 5′-GGGACAGGCTTGGTTGTTTC-3′ and 5′-GAGCATCAGCAATGGCAAGT-3′ for Hdac2; 5′-AGAGAGGTCCCGAGGAGAAC-3′ and 5′-CTCTTGGGGACACAGCATC-3′ for Hdac3; 5′-CAATCCCACAGTCTCCGTGT-3′ and 5′-CAGCACCCCACTAAGGTTCA-3′ for Hdac4; 5′-TGTCACCGCCAGATGTTTTG-3′ and 5′-TGAGCAGAGCCGAGACACAG-3′ for Hdac5; 5′-TGGCTGACACTTTTGAGCAC-3′ and 5′-GTTTGCGGCATCCAGGTAAT-3′ for tBdnf; 5′-CCTGCATCTGTTGGGGAGAC-3′ and 5′-GCCTTGTCCGTGGACGTTTA-3′ for Bdnf1; 5′-CAGAGCAGCTGCCTTGATGTT-3′ and 5′-GCCTTGTCCGTGGACGTTTA-3′ for Bdnf4; 5′-GCTGCCATCTGTTTTACGG-3′ and 5′-TGACTGGTGCCTGATGAACT-3′ for Gapdh; and 5′-GCTGCCATCTGTTTTACGG-3′ and 5′-TGACTGGTGCCTGATGAACT-3′ for L32.
Western blot analysis
Western blot analyses were performed as described previously [20,30]. Brain tissues were homogenized in lysis buffer (50 mM Tris-HCl with pH 7.5, 150 mM NaCl, 0.5% Nonidet P-40, 0.5% Triton X-100, 0.25% sodium deoxycholate, 0.25% SDS, and 1 mM EDTA) with protease inhibitors (Complete Mini; Roche Applied Science). Protein concentrations were determined using the Bradford method. Proteins, 20 µg per lane, were loaded on SDS-PAGE, and transferred onto a PVDF membrane (Bio Rad., Hercules, CA, USA). Blots were blocked with 3% BSA in 0.1% TBS-T buffer and then probed with a primary antibody for p-ERK1/2 (4370, Cell Signaling Technology, Beverly, MA, USA), ERK1/2 (sc-135900, Santa Cruz Inc., Santa Cruz, CA, USA), p-CREB (06-519, Upstate Biotechnology Inc., Lake Placid, NY, USA), CREB (sc-186, Santa Cruz), MeCP2 (3456, Cell Signaling), TrkB (sc-136990, Santa Cruz), AKT (9272, Cell Signaling), S473-pAKT (9271, Cell Signaling), BDNF (ab108319, abcam), or β-actin (sc-47778, Santa Cruz). The following secondary antibodies were used; goat anti-rabbit IgG (sc-2004, Santa Cruz), goat anti-mouse IgG (sc-2005, Santa Cruz), and donkey anti-goat IgG (sc-2020, Santa Cruz). Specific signals were visualized using an enhanced chemiluminescence system (EBP-10073; Elpis Biotech. Inc., Daejon, Korea), and quantified using ImageJ (NIH, USA).
Behavioral assessments
Behavioral assessments were carried out using a computerized video tracking system (SMART, Panlab, Spain) as described previously [20,30]. The behavior testing room was lit by indirect illumination to 20 lux for the U-field assay, three-chamber test, reciprocal social interaction test, open field test, and repetitive behavior test. White noise (65 dB) was used in the behavior testing room. Behavior test equipment was cleaned frequently using 70% ethanol.
Three-chamber test
The three-chamber test was carried out as described previously [20,30]. The three-chamber apparatus consisted of an open-topped rectangular three-compartmented box (22×32 cm2 per compartment) made of clear polycarbonate, with two transparent dividing walls containing a retractable pass way in the middle of each wall (10-cm wide) to give the animal access to each chamber. A subject mouse was allowed to freely explore the three chambers, starting from the middle chamber, and the time spent and trajectory in the chambers were recorded for 10 min; this procedure was regarded as habituation. While a habituated subject mouse was in the middle chamber with the pass ways closed, a circular wire cage (12 cm in diameter) containing a social target (a naïve C57BL/6 mouse of the same sex and age as the subject) was placed in one side chamber, and an empty wire cage was placed in the other side chamber. By opening the pass ways, the subject mouse was allowed to freely explore both chambers, and the time spent and trajectory in the chambers, and the time spent sniffing the target mouse were recorded for 10 min.
Reciprocal social interaction test
The reciprocal social interaction test, also called home-cage social interaction test, was performed as described previously [20,30] with a minor modification. In brief, subject mice were individually placed in a home cage with new bedding for 20 min as habituation. After habituation, a target mouse (the same age and sex as the subject) that had not been seen before by the subject mouse was placed in the same cage for 10 min, during which the time spent in contact, such as facial and anogenital sniffing or mounting behavior, was recorded and regarded as social behavior.
Grooming behavior assessment
Grooming behavior assessment was carried out as described previously [20,30] with a minor modification. Mice were placed individually in an open field (45×45 cm) and allowed to move freely for 30 min. Grooming behavior was recorded for 15 min in the second half of the open field test. Grooming behavior was defined as rubbing the face, body, or head with the two forelimbs.
Open field test
The open field test was performed as described previously [30,33]. Mice were placed individually in the open field (45×45 cm) and allowed to move freely for 30 min. The distance and trajectory of locomotion in the open field was recorded.
Statistical analysis
Two-sample comparisons were performed using the Student's t-test, and multiple comparisons were performed using one-way and two-way ANOVA or two-way repeated measures ANOVA followed by a post hoc test. Statistical analyses were carried out using GraphPad Prism 6 (San Diego, CA, USA). All data are presented as mean±SEM or box plot diagrams, and statistical significance was accepted at the 5% level.
RESULTS
ELS in D2 heterozygous KO mice produced sociability deficits and stereotyped behavior
Maternal separation was chosen as the ELS with pups separated from their mothers for 3 hours per day from postnatal day 2 to postnatal day 14. (Fig. 1A and 1B). Wild-type (WT) mice exposed to ELS showed social interaction levels comparable to those of the normally raised WT mice in the three-chamber test (Fig. 1C and 1D). Normally raised D2+/− mice exhibited WT-like social interaction, whereas ELS-exposed D2+/− mice showed decreased social interaction in the three-chamber test (Fig. 1C and 1E). Sniffing time with a social target during the three-chamber test was also decreased in ELS-exposed D2+/− mice (Fig. 1F). In the home-cage social interaction test, ELS-exposed D2+/− mice, but not other groups, showed less social interaction time than WT control mice with a newly introduced social target (Fig. 1G). ELS-exposed D2+/− mice also exhibited increased grooming in the open field without changes in locomotion (Fig. 1H and 1I). These results suggest that ELS causes autistic-like behaviors in D2 heterozygous mice but not in WT mice.
Fig. 1. D2 heterozygous KO mice exposed to maternal separation stress exhibited autistic-like behaviors. (A, B) Experimental design for maternal separation stress, weaning, and subsequent behavioral tests (A). Diagram illustrating maternal separation of pups from their mothers for 3 hours a day and their return (B). Red arrow, time point for sacrifice. (C~E) Representative heatmaps showing exploratory activity in the three-chamber test of wildtype (WT) and D2+/− mice raised normally (CON) or after exposure to early-life stress (ELS) (C). The amount of time spent in the social target chamber (St) and empty chamber (E) for WT mice (D) and D2+/− mice (E). n= 10~16 animals (WT·CON, p<0.0001; WT·ELS, p=0.0014; D2+/−·CON, p=0.0009; D2+/− ·ELS, p=0.0543). (F) The amount of sniffing time with a social target for WT mice and D2+/− mice in the three-chamber test. n= 8~16 animals (ELS effect, F(1, 42)=9.518, p=0.0036; genotype effect, F(1, 42)=10.94, p=0.0019; ELS x genotype interaction, F(1, 42)=4.454, p=0.0408). (G) The amount of time spent in social interaction with facial and anogenital sniffing (G) of WT and D2+/− mice raised normally or after exposure to early-life stress. n= 8~16 animals (ELS effect, F(1, 43)=7.448, p=0.0092; genotype effect, F(1, 43)=7.947, p=0.0073; genotype x ELS interaction, F(1, 43)=7.301, p=0.0098). (H, I) Locomotion (H) and grooming behaviors (I) in the open field test of WT and D2+/−mice raised normally or after exposure to early-life stress. n= 6~16 animals. (Grooming: ELS effect, F(1, 35)=2.632, p=0.1137; genotype effect, F(1, 35)=3.995, p=0.0534; genotype x ELS interaction, F(1, 35) =6.035, p=0.0191). Data are presented as mean±SEM. * and ** denote differences between the indicated groups at p<0.05 and p<0.01, respectively (Student's t-test, two-way ANOVA and Holm-Sidak post hoc test).

D2 heterozygous pups had altered hypothalamus-pituitary-adrenal gland (HPA) axis responses to early-life stress
We examined whether the behavioral abnormalities of ELS-exposed D2+/− mice were associated with HPA axis dysfunction. At the end of their last 3-h maternal separation at postnatal day 14, WT pups exhibited corticosterone levels that were insignificantly increased compared with normally raised WT pups. In contrast, ELS-exposed D2 heterozygous pups exhibited significantly increased corticosterone levels at the end of their last 3-h maternal separation (Fig. 2A and 2B).
Fig. 2. D2 heterozygous pups exhibited increased HPA axis response to early-life stress. (A) Experimental design for treatment with early-life stress and time points for obtaining samples. Red arrows, time points for sacrifice. (B) Corticosterone levels of WT and D2+/− pups raised normally or after exposure to ELS. ELS-exposed groups and their control pups were sacrificed on postnatal day 14. n=4~5 animals for each group, each with duplicate. (ELS effect, F(1, 33)=7.421, p=0.0102; genotype effect, F(1, 33)=0.1043, p=0.7488; genotype x ELS interaction, F(1, 33)=2.466, p=0.1259). (C) Corticosterone levels of WT and D2+/− mice raised normally or after exposure to ELS, and sacrificed on postnatal day 70. To induce a stress response, the mice were exposed to 60-min restraint (rst-60) and then sacrificed. n=4~7 animals for each group, each with duplicate. Data are presented as mean±SEM. * and ** denote differences between the indicated groups at p<0.05 and p<0.01, respectively (One-way, two-way ANOVA and Holm-Sidak post hoc test).

Adult D2+/− mice raised normally had basal corticosterone levels similar to those of adult WT mice raised normally. ELS-exposed adult WT mice and D2+/− mice also showed similar levels of basal corticosterone. When exposed to 60-min restraint, ELS-exposed adult WT mice had markedly increased corticosterone levels, and ELS-exposed adult D2+/− mice also had increased corticosterone levels, but the increase caused by acute stress was slightly lower in the ELS-exposed adult D2+/− mice than in ELS-exposed adult WT mice (Fig. 2C). These results suggest that D2 heterozygous pups had higher activation of the HPA axis in response to maternal separation stress than wildtype littermate, and when ELS-exposed D2 heterozygous mice grew up, they had reduced HPA axis activation to restraint challenge compared to wildtype mice.
D2 heterozygous KO mice exposed to ELS had reduced expression of neurotrophic factors in the dorsal striatum
Brain-derived neurotrophic factor (BDNF) is associated with childhood maltreatment [34], autism [35,36,37] and ELS induced molecular changes [34]. Early-life adversity induces gene transcription alterations that last throughout the lifetime via epigenetic factors [38]. Therefore, we examined whether ELS exposure changed the expression levels of neurotrophic factors, TrkB, and key epigenetic factors known to regulate neurotrophic factors. Recent studies suggest that the dorsal striatum was the neural substrate whose dysfunction produced sociability deficits and grooming behaviors, the two core domains of ASD symptoms [19,20,30]. Furthermore, siRNA-mediated knockdown of D2 receptor in the dorsal striatum was sufficient to produce autistic-like behaviors [20]. Therefore, we focused on the molecular changes in the dorsal striatum in following experiments (Fig. 3A).
Fig. 3. Early-life stress in D2+/− mice induced altered expression of BDNF-TrkB signaling in the dorsal striatum. (A) Experimental design for treatment with early-life stress, weaning, and time points for sacrifice. A diagram showing the regions (circles) in the dorsal striatum used for tissue prep (right panel). (B, C) Real-time PCR data showing transcript levels of TrkB, tBdnf, Bdnf1, and Bdnf4 (B), and Mecp2, Hdac1, Hdac2, Hdac3, Hdac4, and Hdac5 (C) in the dorsal striatum of WT and D2 heterozygous KO pups raised normally or after exposure to early-life stress. Tissue samples were prepared on postnatal day 14. n=4~6 animals, 4~6 PCR repeats (TrkB: ELS effect, F(1, 12)=18.12, p=0.0011; genotype effect, F(1, 12)=0.1136, p=0.7419; ELS x genotype interaction, F(1, 12)=9.483, p=0.0095; tBdnf: ELS effect, F(1, 12)=4.962, p=0.0458; genotype effect, F(1, 12)=0.3740, p=0.5523; ELS x genotype interaction, F(1, 12)=0.2762, p=0.6088; Hdac1: ELS effect, F(1, 20)=0.5866, p=0.4527; genotype effect, F(1, 20)=23.25, p=0.0001; ELS x genotype effect; F(1, 20)=6.090, p=0.0227; Hdac3: ELS effect, F(1, 20)=0.0.8137, p=0.3778; genotype effect, F(1, 20)=11.24, p=0.0032; ELS x genotype effect, F(1, 20)=0.1040, p=0.7504). (D, E) Real-time PCR data showing transcript levels of TrkB, tBdnf, Bdnf4 (D), Mecp2, Hdac1, Hdac2, Hdac3, Hdac4, and Hdac5 (E) in the dorsal striatum of WT and D2 heterozygous KO mice raised normally or after exposure to ELS, and sacrificed on postnatal day 70. n=4–6 animals, 3~6 PCR repeats (TrkB: ELS effect, F(1, 14) =276.5, p<0.0001; genotype effect, F(1, 14)=53.77, p<00001; ELS x genotype interaction, F(1, 14)=179.5, p<0.0001; tBdnf: ELS effect, F(1, 12)=2.902, p=0.1142; genotype effect, F(1, 12)=10.83, p=0.0064; ELS x genotype interaction, F(1, 12)=4.888, p=0.0472; Bdn4f: ELS effect, F(1, 8)=2.734, p=0.1369; genotype effect, F(1, 8) =26.33, p=0.0009; ELS x genotype interaction, F(1, 8)=14.65, p=0.0050; Mecp2: ELS effect, F(1, 15)=17.80, p=0.0007; genotype effect, F(1, 15)=7.572, p=0.0148; ELS x genotype effect, F(1, 15)=9.961, p=0.0065; Hdac2; ELS effect, F(1, 16)=20.81, p=0.0003; genotype effect, F(1, 16)=6.247, p=0.0237; ELS x genotype effect, F(1, 16)=12.25, p=0.0030; Hdac3: ELS effect, F(1, 12)=7.365, p=0.0188; genotype effect, F(1, 12)=10.02, p=0.0081; ELS x genotype effect, F(1, 12)=0.08215, p=0.7739; Hdac5: ELS effect, F(1, 12)=0.06437, p=0.8040; genotype effect, F(1, 12)=1.741, p=0.2117; ELS x genotype effect, F(1, 12)=0.7854, p=0.3929). (F~I) Western blots showing TrkB, BDNF, MeCP2 (F), p-AKT, AKT, p-ERK1/2, ERK1/2, p-CREB, CREB and β-Actin (H) levels in the dorsal striatum of WT mice and D2 heterozygous KO mice raised normally or or after exposure to early-life stress, and sacrificed on postnatal day 70. Quantification of Western blots (G, I). n=6~8 animals, 3~5 repeats (TrkB: ELS effect, F(1, 8)=0.6597, p=0.4402; genotype effect, F(1, 8)=20.88, p=0.0018; genotype x ELS interaction, F(1, 8)=7.630, p=0.0246; BDNF: ELS effect, F(1, 8)= 3.428, p=0.1012; genotype effect, F(1, 8)=1.998, p=0.1953; genotype x ELS interaction, F(1, 8)=13.14, p=0.0067; MeCP2: ELS effect, F (1, 16)=5.085, p=0.0385; genotype effect, F(1, 16)=18.71, p=0.0005; genotype x ELS interaction, F(1, 16)=22.01, p=0.0002). Data are presented as mean±SEM. * and ** denote differences between the indicated groups at p<0.05 and p<0.01, respectively (Two-way ANOVA and Holm-Sidak post hoc test).

Real-time PCR analysis indicated that TrkB expression was decreased in the dorsal striatum of ELS-exposed D2+/− pups, but not ELS-exposed WT pups, at the end of ELS on postnatal day 14, and total Bdnf (tBdnf), Bdnf1, and Bdnf4 transcript levels also tended to decrease only in ELS-exposed D2+/− pups (Fig. 3B and 3C).
The transcript levels of TrkB, tBdnf, and Bdnf4 were reduced in the dorsal striatum of ELS-exposed adult D+/− mice but not in ELS-exposed adult WT mice (Fig. 3D). The transcript levels of Mecp2, Hdac2, and Hdac3 were also reduced in the dorsal striatum of ELS-exposed adult D2+/− mice but not in ELS-exposed adult WT mice (Fig. 3E).
Western blot analysis indicated that the protein levels of TrkB, BDNF, and MeCP2 were decreased in the dorsal striatum of ELS-exposed adult D2+/− mice but not in ELS-exposed adult WT mice (Fig. 3F and 3G). Furthermore, the protein levels of p-AKT, p-ERK1/2, and p-CREB, the downstream factors of TrkB receptors, tended to decrease in ELS-exposed D2+/− mice (Fig. 3H and 3I). These results suggest that early-life stress in D2 heterozygous pups resulted in the altered expression of factors in the BDNF-TrkB signaling system in the dorsal striatum of adult animals.
7,8-DHF treatment suppressed autistic-like behaviors of ELS-exposed D2 heterozygous KO mice
The therapeutic effects of TrkB receptor activation have been studied for the treatment of cognitive defects in a mouse model of Alzheimer's disease [39], Rett syndrome [40], and ASD [32]. 7,8-dihydroxyflavone (7,8-DHF) is a naturally occurring flavone that has a neurotrophic activity specific to TrkB [41]. ELS-exposed D2+/− mice and their WT controls were intraperitoneally injected with 7,8-DHF for 3 days, and their behavioral performance was examined 24 hours after the last drug injection (Fig. 4A). 7,8-DHF treatment in WT mice produced no significant change in social interaction in the three-chamber test. In contrast, 7,8-DHF treatment in ELS-exposed D2+/− mice increased social interaction in the three-chamber test (Fig. 4B and 4C) and tended to improve defective sociability in the home-cage social interaction test (Fig. 4D). 7,8-DHF treatment in ELS-exposed D2+/− mice also suppressed increased grooming to the level seen in WT controls (Fig. 4E).
Fig. 4. 7,8-DHF treatment rescued autistic-like behaviors in ELS-exposed D2+/− mice. (A) Experimental design for treatment with 7,8-DHF, behavioral tests, and time points for sacrifice. 7,8-DHF was intraperitoneally injected at the dose of 10 mg/kg/day for 3 days, and behavioral tests were carried out on day 4. (B, C) Representative heatmaps showing exploratory activity in the three-chamber test of normally raised WT mice and ELS-exposed D2+/− mice treated with 7,8-DHF or vehicle (B). The amounts of time spent in each chamber for the mouse groups are presented in (C). n= 5–7 animals (WT, CON+Veh: p<0.0001; WT, CON+DHF: p= 0.0042; D2+/−, ELS+Veh: p=0.5986, D2+/−; ELS+DHF: p=0.0002). (D, E) The social interaction time spent sniffing in the home-cage social interaction test (D) and grooming time (E) of normally raised WT mice and ELS-exposed D2+/− mice treated with 7,8-DHF or vehicle. n=5~7 animals (Home-cage social interaction: genotype effect, F(1, 19)=9.227, p=0.0068; DHF effect, F(1, 19)=0.5098, p=0.4839; genotype x DHF interaction, F(1,19)=2.703, p=0.1166; Grooming: genotype effect, F(1, 21)=10.04, p=0.0046; DHF effect, F(1, 21)=25.07, p<0.0001; genotype x DHF interaction, F(1, 21)=11.75, p=0.0025). (F) Real-time PCR data showing transcript levels of TrkB, tBdnf, and Bdnf4 in the dorsal striatum of normally raised WT mice and ELS-exposed D2+/− mice treated with 7,8-DHF or vehicle (F). n=3~4 animals, 3~5 PCR repeats (TrkB: genotype effect, F(1, 14)=5.693, p=0.0317; DHF effect, F(1, 14)=11.95, p=0.0039; genotype x DHF interaction, F(1, 14)=11.46, p=0.0044). (G~J) Western blots showing TrkB, BDNF (G), p-AKT, AKT, p-ERK1/2, ERK1/2, p-CREB, CREB and β-Actin (I) levels in the dorsal striatum of normally raised WT mice and ELS-exposed D2+/− mice treated with 7,8-DHF or vehicle. Quantification of Western blots (H, J). n=3~4 animals, 3~5 repeats (TrkB: genotype effect, F(1, 12)=9.596, p=0.0092; DHF effect, F(1, 12)=9.814, p=0.0086; genotype x DHF interaction, F(1, 12)=0.2431, p=0.6309; BDNF: genotype effect, F(1, 8)=1.936, p=0.2015; DHF effect, F(1, 8)=3.507, p=0.0980; genotype x DHF interaction, F(1, 8)=0.2157, p=0.6547). Data are presented as mean±SEM. * and ** denote differences between the indicated groups at p<0.05 and p<0.01, respectively (Student's t-test or two-way ANOVA followed by Holm-Sidak post hoc test).

Next, we examined whether the TrkB agonist also produced molecular restoration in the dorsal striatum. Real-time PCR analysis indicated that the decreased expression of TrkB, Bdnf, Bdnf4, Mecp2, and Hdac2 transcripts in the dorsal striatum of ELS-exposed D2+/− mice was reversed to WT levels by 7,8-DHF treatment. In particular, the total Bdnf transcript level was dramatically elevated in ELS-exposed D2+/− mice after 7,8-DHF treatment (Fig. 4F). Protein levels of TrkB and BDNF were increased by 7,8-DHF treatment in normally raised WT mice, although their expression levels in ELS-exposed D2+/− mice did not significantly increase (Fig. 4G and 4H). 7,8-DHF treatment in ELS-exposed D2+/− mice tended to restore the reduced level of p-AKT and p-ERK1/2 in the dorsal striatum, although its effect was not significant (Fig. 4I and 4J). These results suggest that 7,8-DHF treatment rescued the autistic-like behaviors of ELS-exposed D2+/− mice and weakly changed the TrkB and BDNF signaling system in the dorsal striatum.
Inhibition of TrkB in the dorsal striatum blocked the behavioral effects of 7,8-DHF treatment in ELS-exposed D2+/− mice
Next, we examined whether the behavioral effects of 7,8-DHF treatment were produced through the activation of TrkB in the dorsal striatum. To address that, we stereotaxically injected TrkB-siRNA or control-siRNA into the dorsal striatum on both sides, followed by 7,8-DHF injection for 3 days. The behavioral tests were performed on the day after the last 7,8-DHF injection (Fig. 5A). These experiments were verified that the siRNA-mediated knockdown of TrkB in the dorsal striatum reduced the expression levels of TrkB, p-AKT and p-ERK1/2 for more than 3 days (Fig. 5B and 5C), which is consistent with previous reports [42,43].
Fig. 5. Inhibition of TrkB in the dorsal striatum blocked the therapeutic effects of 7,8-DHF. (A) Experimental designs. In Exp.#1, CON-siRNA or TrkB-siRNA was injected in the dorsal striatum (AP, +1.0; ML, ±1.5; DV, −3.6 mm) of normally raised WT mice, and the mice were sacrificed on day 3. In Exp.#2, CON-siRNA or TrkB-siRNA was injected in the dorsal striatum of normally raised WT mice or ELS-exposed D2+/− mice, followed by 7,8-DHF injection (i.p., 10 mg/kg/day) and subsequent behavioral tests. (B, C) Western blots showing expression levels of TrkB, p-AKT, AKT, p-ERK1/2, ERK1/2, and β-Actin in the dorsal striatum injected with TrkB-siRNA or CON-siRNA (B). Samples were collected 2 days after siRNA injection. Quantification of Western blots (C). n= 4 animals, each, 3–4 repeats (TrkB: p=0.0002; p-AKT/AKT: p=0.0367; p-ERK/ERK: p=0.0014). (D~F) The amount of time spent in each chamber in the three-chamber test (D), social interaction time in the home-cage social interaction test (E), and grooming time (F) of normally raised WT mice and ELS-exposed D2+/− mice injected with TrkB-siRNA or CON-siRNA in the dorsal striatum and treated with vehicle or 7,8-DHF as indicated. n=5~6 animals (three-chamber test: WT, CON+siCON+Veh: p=0.0017; WT, CON+siTrkB+Veh; p=0.0015; D2+/−, ELS+siCON+Veh: p=0.5463, D2+/−, ELS+siCON+DHF: p=0.0025, D2+/−, ELS+siTrkB+DHF: p=0.9304; Grooming, WT, CON+siCON+Veh vs. WT, CON+siTrkB+Veh: p=0.9081; D2+/−, F(2, 14)=0.2332, p=0.7950). Data are presented as mean±SEM. * and ** denote differences between the indicated groups at p<0.05 and p<0.01, respectively (Student's t-test or one-way ANOVA and Holm-Sidak post hoc test).

Normally raised WT mice injected with TrkB-siRNA did not show behavioral changes compared with WT mice injected with control-siRNA. ELS-exposed D2+/− mice injected with control-siRNA and treated with 7,8-DHF showed increased sociability in the three-chamber test and the home-cage social interaction test. In contrast, ELS-exposed D2+/− mice injected with TrkB-siRNA and treated with 7,8-DHF exhibited social deficits in the three-chamber test (Fig. 5D). TrkB-siRNA injection in ELS-exposed D2+/− mice also partially blocked the effects of 7,8-DHF on sociability in the home-cage social interaction test (Fig. 5E). Regarding grooming behavior, TrkB-siRNA injection did not clearly block the effects of 7,8-DHF in ELS-exposed D2+/− mice (Fig. 5F). Together, these results suggest that TrkB-related changes in the dorsal striatum are critical to the behavioral effects of 7,8-DHF treatment in ELS-exposed D2+/− mice.
DISCUSSION
Early-life stress promoted autistic-like behaviors in D2 heterozygous KO mice but not WT mice
This study demonstrates that ELS produced sociability deficits and increased grooming behavior, the behavioral phenotypes relevant to the two core domains of ASD symptoms, in D2+/− mice but not WT mice (Fig. 1). D2 homozygous KO mice exhibited typical autistic-like behaviors [20], and when exposed to chronic stress, they showed severe depressive-like behaviors [44]. In contrast, D2 heterozygous KO mice did not show autistic-like behaviors (Fig. 1). Therefore, the autistic-like phenotypes displayed by ELS-exposed D2+/− adult mice likely resulted from the gene×stress interaction imposed during the early developing period. We have provided evidence that D2+/− pups had much higher activation of the HPA axis in response to ELS compared with WT pups exposed to ELS (Fig. 2). The increased stress response in D2+/− pups was correlated with the reduced expression of TrkB and BDNF in the dorsal striatum (Fig. 3B). Furthermore, expression levels of TrkB, Bdnf, Mecp2 and Hdac2 transcripts were reduced in the dorsal striatum of ELS-exposed D2+/− mice but not in the dorsal striatum of normally raised D2+/− mice (Fig. 3D and 3E). Although the detailed mechanism of how such changes cause ASD-like behaviors need to be studied in the future, our results suggest that ELS in animals with defective dopamine receptor function causes to the development of ASD-like phenotypes.
In developing pups, the HPA axis functionally reacts to stressors starting from the second postnatal week. During the first postnatal week, pups have low basal levels of corticosterone, and the levels of corticosterone and ACTH start to increase in response to stressors from postnatal day 12 but not at postnatal day 9 [45,46]. Therefore, it is possible that the ELS given to the pups from the middle of the second postnatal week might have induced corticosterone release. It will be necessary to investigate whether the promotion of ASD-like phenotypes in D2+/− mice exposed to ELS is caused primarily by increased corticosterone release or some other mechanism.
Striatal dysfunction induced by genetic and environmental factors promoted autistic-like behaviors
Recently, our lab and others' have reported that the dorsal striatum is the neuroanatomical correlate for ASD core symptoms [20,30,47]. Excessive activation of D1 receptors in the dorsal striatum promoted social deficits and repetitive behaviors [20]. AC5−/− and D2−/− mice show typical autistic-like behaviors, and siRNA-mediated knockdown of AC5 or D2 receptors within the dorsal striatum was sufficient to replicate autistic-like phenotypes [20,30]. Striatal dysfunction of SHANK3 [48], TSC1 [49], MeCP2 [50], FOXP1 [51], CNTNAP2 [52], and DGLα [53] also produced autistic-like behaviors. A polymorphism of D3 dopamine receptors (rs167771) was correlated with greater caudate nucleus volume and stereo-typed behavior [54]. Furthermore, specific knockout of MeCP2 or TSC1 in the dorsal striatum produced social deficits and repetitive behavior [31]. As demonstrated in the present study, repeated ELS in D2 heterozygous pups caused autistic-like behaviors through changes in the BDNF-TrkB signaling pathway in the dorsal striatum (Fig. 3, 4 and 5). Thus, specific mutations in the genes that function in the dorsal striatum or stress-induced TrkB-dependent changes in the dorsal stratum apparently promote autistic-like behaviors.
Stimulation of TrkB in the dorsal striatum was required for behavioral effects of 7,8-DHF
Recently, de novo mutations in TrkB were identified in individuals with ASD [55]. Abnormal BDNF levels in the cortex [37], hippocampus [37], striatum [35,36], and serum [56] were reported in ASD. The results of those studies suggest that the BDNF and TrkB receptor system correlates with ASD. ELS-exposed D2+/− mice exhibited autistic-like behaviors through reduced expression of BDNF and TrkB in the dorsal striatum, whereas 7,8-DHF treatment notably rescued the autistic-like phenotypes of ELS-exposed D2+/− mice (Fig. 3, 4 and 5). Our results suggest that the BDNF and TrkB system in the dorsal striatum is critical for the development of ASD core symptoms. Consistent with this interpretation, viral vector-mediated expression of BDNF in the dorsal striatum mitigated autistic-like behaviors in Mecp2 KO mice [57].
The increase of total Bdnf transcript level in ELS-exposed D2+/− mice following treatment with 7,8-DHF (Fig. 4F) was only partially manifested by BDNF protein level (Fig. 4G and 4H). The Bdnf transcription is regulated by various transcription factors that act at different promoter regions of the Bdnf gene in a different signaling context [58]. Furthermore, TrkB activation increases BDNF expression in a cell autonomous and non-autonomous manner [39,57,59,60]. TrkB activation by BDNF in rat primary cortical culture produced a biphasic expression of Bdnf exon IV~IX mRNA, with the primary induction 1~3 h after TrkB stimulation and the delayed and protracted induction 24~72 hours after the stimulation, in which the early induction was much higher than the delayed induction [61], thus suggesting that the expression of Bdnf exon IV~IX mRNA occurs in a complex time course. TrkB-dependent BDNF expression is regulated by cAMP response element binding protein (CREB) [62] and Methyl-CpG binding protein 2 (MeCP2) [63,64]. TrkB activation also increases TrkB expression via CREB activation [59,65]. When 7,8-DHF was treated for 3 days, the transcription mechanisms of the Bdnf transcription system might be presented into partially mismatched BDNF protein levels (Fig. 4G and 4H), although the underlying mechanisms need to be elaborated.
BDNF expression is regulated by multiple factors, including CREB, MeCP2, and HDACs [64,66]. Furthermore, genetic deficits of Mecp2, Hdac1, and Hdac2 in the dorsal striatum produced grooming behavior [67]. ELS-exposed D2+/− mice showed reduced expression of Mecp2, and Hdac2 transcripts in the dorsal striatum, and 7,8-DHF treatment partially reversed those changes (Fig. 3). Regarding these results, it might be worth investigating whether reduced expression of MeCP2 and HDAC2 is responsible for the downregulation of BDNF and TrkB in the dorsal striatum of ELS-exposed D2+/− mice. TrkB receptor activation promotes cell autocrine responses in the growth and differentiation of neurons [57,68]. It is also possible that 7,8-DHF treatment afforded behavioral rescue through the increased expression of MeCP2 and HDAC2, which enhanced BDNF and TrkB-dependent autocrine signaling in the dorsal striatum, although direct evidence for those links is unavailable.
ACKNOWLEDGEMENTS
This research was supported by a grant (HI15C1834) from the Ministry of Health and Welfare, and a grant (2018R1A2B2001535) from the Ministry of Science, ICT and Future Planning, Republic of Korea.
References
- 1.American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. American Psychiatric Publishing: Washington, D.C.; 2013. [Google Scholar]
- 2.DiCicco-Bloom E, Lord C, Zwaigenbaum L, Courchesne E, Dager SR, Schmitz C, Schultz RT, Crawley J, Young LJ. The developmental neurobiology of autism spectrum disorder. J Neurosci. 2006;26:6897–6906. doi: 10.1523/JNEUROSCI.1712-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9:341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Betancur C, Buxbaum JD. SHANK3 haploinsufficiency: a “common” but underdiagnosed highly penetrant monogenic cause of autism spectrum disorders. Mol Autism. 2013;4:17. doi: 10.1186/2040-2392-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al Shehhi M, Forman EB, Fitzgerald JE, McInerney V, Krawczyk J, Shen S, Betts DR, Ardle LM, Gorman KM, King MD, Green A, Gallagher L, Lynch SA. NRXN1 deletion syndrome; phenotypic and penetrance data from 34 families. Eur J Med Genet. 2019;62:204–209. doi: 10.1016/j.ejmg.2018.07.015. [DOI] [PubMed] [Google Scholar]
- 6.Shinawi M, Liu P, Kang SH, Shen J, Belmont JW, Scott DA, Probst FJ, Craigen WJ, Graham BH, Pursley A, Clark G, Lee J, Proud M, Stocco A, Rodriguez DL, Kozel BA, Sparagana S, Roeder ER, McGrew SG, Kurczynski TW, Allison LJ, Amato S, Savage S, Patel A, Stankiewicz P, Beaudet AL, Cheung SW, Lupski JR. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J Med Genet. 2010;47:332–341. doi: 10.1136/jmg.2009.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D'Angelo D, Lebon S, Chen Q, Martin-Brevet S, Snyder LG, Hippolyte L, Hanson E, Maillard AM, Faucett WA, Macé A, Pain A, Bernier R, Chawner SJ, David A, Andrieux J, Aylward E, Baujat G, Caldeira I, Conus P, Ferrari C, Forzano F, Gérard M, Goin-Kochel RP, Grant E, Hunter JV, Isidor B, Jacquette A, Jønch AE, Keren B, Lacombe D, Le Caignec C, Martin CL, Männik K, Metspalu A, Mignot C, Mukherjee P, Owen MJ, Passeggeri M, Rooryck-Thambo C, Rosenfeld JA, Spence SJ, Steinman KJ, Tjernagel J, Van Haelst M, Shen Y, Draganski B, Sherr EH, Ledbetter DH, van den Bree MB, Beckmann JS, Spiro JE, Reymond A, Jacquemont S, Chung WK Cardiff University Experiences of Children With Copy Number Variants (ECHO) Study; 16p11.2 European Consortium; Simons Variation in Individuals Project (VIP) Consortium. Defining the effect of the 16p11.2 duplication on cognition, behavior, and medical comorbidities. JAMA Psychiatry. 2016;73:20–30. doi: 10.1001/jamapsychiatry.2015.2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merikangas AK, Segurado R, Heron EA, Anney RJ, Paterson AD, Cook EH, Pinto D, Scherer SW, Szatmari P, Gill M, Corvin AP, Gallagher L. The phenotypic manifestations of rare genic CNVs in autism spectrum disorder. Mol Psychiatry. 2015;20:1366–1372. doi: 10.1038/mp.2014.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pizzo L, Jensen M, Polyak A, Rosenfeld JA, Mannik K, Krishnan A, McCready E, Pichon O, Le Caignec C, Van Dijck A, Pope K, Voorhoeve E, Yoon J, Stankiewicz P, Cheung SW, Pazuchanics D, Huber E, Kumar V, Kember RL, Mari F, Curró A, Castiglia L, Galesi O, Avola E, Mattina T, Fichera M, Mandarà L, Vincent M, Nizon M, Mercier S, Bénéteau C, Blesson S, Martin-Coignard D, Mosca-Boidron AL, Caberg JH, Bucan M, Zeesman S, Nowaczyk MJ, Lefebvre M, Faivre L, Callier P, Skinner C, Keren B, Perrine C, Prontera P, Marle N, Renieri A, Reymond A, Kooy RF, Isidor B, Schwartz C, Romano C, Sistermans E, Amor DJ, Andrieux J, Girirajan S. Rare variants in the genetic background modulate cognitive and developmental phenotypes in individuals carrying disease-associated variants. Genet Med. 2019;21:816–825. doi: 10.1038/s41436-018-0266-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rice D, Barone S., Jr Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect. 2000;(108 Suppl 3):511–533. doi: 10.1289/ehp.00108s3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, Miller J, Fedele A, Collins J, Smith K, Lotspeich L, Croen LA, Ozonoff S, Lajonchere C, Grether JK, Risch N. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry. 2011;68:1095–1102. doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grabrucker AM. Environmental factors in autism. Front Psychiatry. 2013;3:118. doi: 10.3389/fpsyt.2012.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singletary WM. An integrative model of autism spectrum disorder: ASD as a neurobiological disorder of experienced environmental deprivation, early life stress and allostatic overload. Neuropsychoanalysis. 2015;2:81–119. [Google Scholar]
- 14.Sandi C, Haller J. Stress and the social brain: behavioural effects and neurobiological mechanisms. Nat Rev Neurosci. 2015;16:290–304. doi: 10.1038/nrn3918. [DOI] [PubMed] [Google Scholar]
- 15.Crafa D, Warfa N. Maternal migration and autism risk: systematic analysis. Int Rev Psychiatry. 2015;27:64–71. doi: 10.3109/09540261.2014.995601. [DOI] [PubMed] [Google Scholar]
- 16.Hecht PM, Hudson M, Connors SL, Tilley MR, Liu X, Beversdorf DQ. Maternal serotonin transporter genotype affects risk for ASD with exposure to prenatal stress. Autism Res. 2016;9:1151–1160. doi: 10.1002/aur.1629. [DOI] [PubMed] [Google Scholar]
- 17.Jones KL, Smith RM, Edwards KS, Givens B, Tilley MR, Beversdorf DQ. Combined effect of maternal serotonin transporter genotype and prenatal stress in modulating offspring social interaction in mice. Int J Dev Neurosci. 2010;28:529–536. doi: 10.1016/j.ijdevneu.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sjaarda CP, Hecht P, McNaughton AJ, Zhou A, Hudson ML, Will MJ, Smith G, Ayub M, Liang P, Chen N, Beversdorf D, Liu X. Interplay between maternal Slc6a4 mutation and prenatal stress: a possible mechanism for autistic behavior development. Sci Rep. 2017;7:8735. doi: 10.1038/s41598-017-07405-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pavăl D. A dopamine hypothesis of autism spectrum disorder. Dev Neurosci. 2017;39:355–360. doi: 10.1159/000478725. [DOI] [PubMed] [Google Scholar]
- 20.Lee Y, Kim H, Kim JE, Park JY, Choi J, Lee JE, Lee EH, Han PL. Excessive D1 dopamine receptor activation in the dorsal striatum promotes autistic-like behaviors. Mol Neurobiol. 2018;55:5658–5671. doi: 10.1007/s12035-017-0770-5. [DOI] [PubMed] [Google Scholar]
- 21.Comings DE, Comings BG, Muhleman D, Dietz G, Shahbahrami B, Tast D, Knell E, Kocsis P, Baumgarten R, Kovacs BW, Levy DL, Smith M, Borison RL, Evans DD, Klein DN, MacMurray J, Tosk JM, Sverd J, Gysin R, Flanagan SD. The dopamine D2 receptor locus as a modifying gene in neuropsychiatric disorders. JAMA. 1991;266:1793–1800. [PubMed] [Google Scholar]
- 22.Hettinger JA, Liu X, Hudson ML, Lee A, Cohen IL, Michaelis RC, Schwartz CE, Lewis SM, Holden JJ. DRD2 and PPP1R1B (DARPP-32) polymorphisms independently confer increased risk for autism spectrum disorders and additively predict affected status in male-only affected sib-pair families. Behav Brain Funct. 2012;8:19. doi: 10.1186/1744-9081-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hettinger JA, Liu X, Schwartz CE, Michaelis RC, Holden JJ. A DRD1 haplotype is associated with risk for autism spectrum disorders in male-only affected sib-pair families. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:628–636. doi: 10.1002/ajmg.b.30655. [DOI] [PubMed] [Google Scholar]
- 24.Hamilton PJ, Campbell NG, Sharma S, Erreger K, Herborg Hansen F, Saunders C, Belovich AN, Sahai MA, Cook EH, Gether U, McHaourab HS, Matthies HJ, Sutcliffe JS, Galli A NIH ARRA Autism Sequencing Consortium. De novo mutation in the dopamine transporter gene associates dopamine dysfunction with autism spectrum disorder. Mol Psychiatry. 2013;18:1315–1323. doi: 10.1038/mp.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ijichi S, Ijichi N, Ijichi Y, Sameshima H, Morioka H. The genetic basis of phenotypic diversity: autism as an extreme tail of a complex dimensional trait. In: Deutsch S, editor. Autism spectrum disorders: the role of genetics in diagnosis and treatment. Rijeka: InTech; 2011. pp. 83–102. [Google Scholar]
- 26.Cooper DN, Krawczak M, Polychronakos C, Tyler-Smith C, Kehrer-Sawatzki H. Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum Genet. 2013;132:1077–1130. doi: 10.1007/s00439-013-1331-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelly MA, Rubinstein M, Asa SL, Zhang G, Saez C, Bunzow JR, Allen RG, Hnasko R, Ben-Jonathan N, Grandy DK, Low MJ. Pituitary lactotroph hyperplasia and chronic hyperprolactinemia in dopamine D2 receptor-deficient mice. Neuron. 1997;19:103–113. doi: 10.1016/s0896-6273(00)80351-7. [DOI] [PubMed] [Google Scholar]
- 28.Uchida S, Hara K, Kobayashi A, Funato H, Hobara T, Otsuki K, Yamagata H, McEwen BS, Watanabe Y. Early life stress enhances behavioral vulnerability to stress through the activation of REST4-mediated gene transcription in the medial prefrontal cortex of rodents. J Neurosci. 2010;30:15007–15018. doi: 10.1523/JNEUROSCI.1436-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim TK, Han PL. Physical exercise counteracts stress-induced upregulation of melanin-concentrating hormone in the brain and stress-induced persisting anxiety-like behaviors. Exp Neurobiol. 2016;25:163–173. doi: 10.5607/en.2016.25.4.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim H, Lee Y, Park JY, Kim JE, Kim TK, Choi J, Lee JE, Lee EH, Kim D, Kim KS, Han PL. Loss of adenylyl cyclase type-5 in the dorsal striatum produces autistic-like behaviors. Mol Neurobiol. 2017;54:7994–8008. doi: 10.1007/s12035-016-0256-x. [DOI] [PubMed] [Google Scholar]
- 31.Lee Y, Kim H, Han PL. Striatal inhibition of MeCP2 or TSC1 produces sociability deficits and repetitive behaviors. Exp Neurobiol. 2018;27:539–549. doi: 10.5607/en.2018.27.6.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kang MS, Choi TY, Ryu HG, Lee D, Lee SH, Choi SY, Kim KT. Autism-like behavior caused by deletion of vaccinia-related kinase 3 is improved by TrkB stimulation. J Exp Med. 2017;214:2947–2966. doi: 10.1084/jem.20160974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baek IS, Park JY, Han PL. Chronic antidepressant treatment in normal mice induces anxiety and impairs stress-coping ability. Exp Neurobiol. 2015;24:156–168. doi: 10.5607/en.2015.24.2.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roth TL, Lubin FD, Funk AJ, Sweatt JD. Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol Psychiatry. 2009;65:760–769. doi: 10.1016/j.biopsych.2008.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deogracias R, Yazdani M, Dekkers MP, Guy J, Ionescu MC, Vogt KE, Barde YA. Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2012;109:14230–14235. doi: 10.1073/pnas.1206093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hemmerle AM, Ahlbrand R, Bronson SL, Lundgren KH, Richtand NM, Seroogy KB. Modulation of schizophrenia-related genes in the forebrain of adolescent and adult rats exposed to maternal immune activation. Schizophr Res. 2015;168:411–420. doi: 10.1016/j.schres.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boschian C, Messina A, Bozza A, Castellini ME, Provenzano G, Bozzi Y, Casarosa S. Impaired neuronal differentiation of neural stem cells lacking the engrailed-2 gene. Neuroscience. 2018;386:137–149. doi: 10.1016/j.neuroscience.2018.06.032. [DOI] [PubMed] [Google Scholar]
- 38.Stroud H, Su SC, Hrvatin S, Greben AW, Renthal W, Boxer LD, Nagy MA, Hochbaum DR, Kinde B, Gabel HW, Greenberg ME. Early-life gene expression in neurons modulates lasting epigenetic states. Cell. 2017;171:1151–1164.e16. doi: 10.1016/j.cell.2017.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Devi L, Ohno M. 7,8-dihydroxyflavone, a small-molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of Alzheimer's disease. Neuropsychopharmacology. 2012;37:434–444. doi: 10.1038/npp.2011.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmid DA, Yang T, Ogier M, Adams I, Mirakhur Y, Wang Q, Massa SM, Longo FM, Katz DM. A TrkB small molecule partial agonist rescues TrkB phosphorylation deficits and improves respiratory function in a mouse model of Rett syndrome. J Neurosci. 2012;32:1803–1810. doi: 10.1523/JNEUROSCI.0865-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jang SW, Liu X, Yepes M, Shepherd KR, Miller GW, Liu Y, Wilson WD, Xiao G, Blanchi B, Sun YE, Ye K. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc Natl Acad Sci U S A. 2010;107:2687–2692. doi: 10.1073/pnas.0913572107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim H, Kim TK, Kim JE, Park JY, Lee Y, Kang M, Kim KS, Han PL. Adenylyl cyclase-5 in the dorsal striatum function as a molecular switch for the generation of behavioral preferences for cue-directed food choices. Mol Brain. 2014;7:77. doi: 10.1186/s13041-014-0077-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bortolozzi A, Castañé A, Semakova J, Santana N, Alvarado G, Cortés R, Ferrés-Coy A, Fernández G, Carmona MC, Toth M, Perales JC, Montefeltro A, Artigas F. Selective siRNA-mediated suppression of 5-HT1A autoreceptors evokes strong anti-depressant-like effects. Mol Psychiatry. 2012;17:612–623. doi: 10.1038/mp.2011.92. [DOI] [PubMed] [Google Scholar]
- 44.Sim HR, Choi TY, Lee HJ, Kang EY, Yoon S, Han PL, Choi SY, Baik JH. Role of dopamine D2 receptors in plasticity of stress-induced addictive behaviours. Nat Commun. 2013;4:1579. doi: 10.1038/ncomms2598. [DOI] [PubMed] [Google Scholar]
- 45.Schmidt MV, Enthoven L, van der Mark M, Levine S, de Kloet ER, Oitzl MS. The postnatal development of the hypothalamic-pituitary-adrenal axis in the mouse. Int J Dev Neurosci. 2003;21:125–132. doi: 10.1016/s0736-5748(03)00030-3. [DOI] [PubMed] [Google Scholar]
- 46.Dent GW, Okimoto DK, Smith MA, Levine S. Stress-induced alterations in corticotropin-releasing hormone and vasopressin gene expression in the paraventricular nucleus during ontogeny. Neuroendocrinology. 2000;71:333–342. doi: 10.1159/000054554. [DOI] [PubMed] [Google Scholar]
- 47.Fuccillo MV. Striatal circuits as a common node for autism pathophysiology. Front Neurosci. 2016;10:27. doi: 10.3389/fnins.2016.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, Feng G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011;472:437–442. doi: 10.1038/nature09965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Benthall KN, Ong SL, Bateup HS. Corticostriatal transmission is selectively enhanced in striatonigral neurons with postnatal loss of Tsc1. Cell Reports. 2018;23:3197–3208. doi: 10.1016/j.celrep.2018.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, Ekker M, Rubenstein JL, Noebels JL, Rosenmund C, Zoghbi HY. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bacon C, Schneider M, Le Magueresse C, Froehlich H, Sticht C, Gluch C, Monyer H, Rappold GA. Brain-specific Foxp1 deletion impairs neuronal development and causes autistic-like behaviour. Mol Psychiatry. 2015;20:632–639. doi: 10.1038/mp.2014.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peñagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, Sonnenblick LI, Gruver R, Almajano J, Bragin A, Golshani P, Trachtenberg JT, Peles E, Geschwind DH. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. 2011;147:235–246. doi: 10.1016/j.cell.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shonesy BC, Parrish WP, Haddad HK, Stephenson JR, Báldi R, Bluett RJ, Marks CR, Centanni SW, Folkes OM, Spiess K, Augustin SM, Mackie K, Lovinger DM, Winder DG, Patel S, Colbran RJ. Role of striatal direct pathway 2-arachidonoylglycerol signaling in sociability and repetitive behavior. Biol Psychiatry. 2018;84:304–315. doi: 10.1016/j.biopsych.2017.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Staal WG, Langen M, van Dijk S, Mensen VT, Durston S. DRD3 gene and striatum in autism spectrum disorder. Br J Psychiatry. 2015;206:431–432. doi: 10.1192/bjp.bp.114.148973. [DOI] [PubMed] [Google Scholar]
- 55.Hamdan FF, Myers CT, Cossette P, Lemay P, Spiegelman D, Laporte AD, Nassif C, Diallo O, Monlong J, Cadieux-Dion M, Dobrzeniecka S, Meloche C, Retterer K, Cho MT, Rosenfeld JA, Bi W, Massicotte C, Miguet M, Brunga L, Regan BM, Mo K, Tam C, Schneider A, Hollingsworth G, FitzPatrick DR, Donaldson A, Canham N, Blair E, Kerr B, Fry AE, Thomas RH, Shelagh J, Hurst JA, Brittain H, Blyth M, Lebel RR, Gerkes EH, Davis-Keppen L, Stein Q, Chung WK, Dorison SJ, Benke PJ, Fassi E, Corsten-Janssen N, Kamsteeg EJ, Mau-Them FT, Bruel AL, Verloes A, Õunap K, Wojcik MH, Albert DV, Venkateswaran S, Ware T, Jones D, Liu YC, Mohammad SS, Bizargity P, Bacino CA, Leuzzi V, Martinelli S, Dallapiccola B, Tartaglia M, Blumkin L, Wierenga KJ, Purcarin G, O'Byrne JJ, Stockler S, Lehman A, Keren B, Nougues MC, Mignot C, Auvin S, Nava C, Hiatt SM, Bebin M, Shao Y, Scaglia F, Lalani SR, Frye RE, Jarjour IT, Jacques S, Boucher RM, Riou E, Srour M, Carmant L, Lortie A, Major P, Diadori P, Dubeau F, D'Anjou G, Bourque G, Berkovic SF, Sadleir LG, Campeau PM, Kibar Z, Lafrenière RG, Girard SL, Mercimek-Mahmutoglu S, Boelman C, Rouleau GA, Scheffer IE, Mefford HC, Andrade DM, Rossignol E, Minassian BA, Michaud JL. High rate of recurrent de novo mutations in developmental and epileptic encephalopathies. Deciphering Developmental Disorders Study. Am J Hum Genet. 2017;101:664–685. doi: 10.1016/j.ajhg.2017.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Francis K, Dougali A, Sideri K, Kroupis C, Vasdekis V, Dima K, Douzenis A. Brain-derived neurotrophic factor (BDNF) in children with ASD and their parents: a 3-year follow-up. Acta Psychiatr Scand. 2018;137:433–441. doi: 10.1111/acps.12872. [DOI] [PubMed] [Google Scholar]
- 57.Sampathkumar C, Wu YJ, Vadhvani M, Trimbuch T, Eickholt B, Rosenmund C. Loss of MeCP2 disrupts cell autonomous and autocrine BDNF signaling in mouse glutamatergic neurons. eLife. 2016;5:e19374. doi: 10.7554/eLife.19374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bathina S, Das UN. Brain-derived neurotrophic factor and its clinical implications. Arch Med Sci. 2015;11:1164–1178. doi: 10.5114/aoms.2015.56342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lai TW, Zhang S, Wang YT. Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol. 2014;115:157–188. doi: 10.1016/j.pneurobio.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 60.English CN, Vigers AJ, Jones KR. Genetic evidence that brain-derived neurotrophic factor mediates competitive interactions between individual cortical neurons. Proc Natl Acad Sci U S A. 2012;109:19456–19461. doi: 10.1073/pnas.1206492109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yasuda M, Fukuchi M, Tabuchi A, Kawahara M, Tsuneki H, Azuma Y, Chiba Y, Tsuda M. Robust stimulation of TrkB induces delayed increases in BDNF and Arc mRNA expressions in cultured rat cortical neurons via distinct mechanisms. J Neurochem. 2007;103:626–636. doi: 10.1111/j.1471-4159.2007.04851.x. [DOI] [PubMed] [Google Scholar]
- 62.Koo JW, Mazei-Robison MS, LaPlant Q, Egervari G, Braunscheidel KM, Adank DN, Ferguson D, Feng J, Sun H, Scobie KN, Damez-Werno DM, Ribeiro E, Peña CJ, Walker D, Bagot RC, Cahill ME, Anderson SA, Labnté B, Hodes GE, Browne H, Chadwick B, Robison AJ, Vialou VF, Dias C, Lorsch Z, Mouzon E, Lobo MK, Dietz DM, Russo SJ, Neve RL, Hurd YL, Nestler EJ. Epigenetic basis of opiate suppression of BDNF gene expression in the ventral tegmental area. Nat Neurosci. 2015;18:415–422. doi: 10.1038/nn.3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 64.Choi J, Kwon HJ, Lee JE, Lee Y, Seoh JY, Han PL. Hyperoxygenation revitalizes Alzheimer's disease pathology through the upregulation of neurotrophic factors. Aging Cell. 2019;18:e12888. doi: 10.1111/acel.12888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gupta VK, You Y, Gupta VB, Klistorner A, Graham SL. TrkB receptor signalling: implications in neurodegenerative, psychiatric and proliferative disorders. Int J Mol Sci. 2013;14:10122–10142. doi: 10.3390/ijms140510122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 67.Mahgoub M, Adachi M, Suzuki K, Liu X, Kavalali ET, Chahrour MH, Monteggia LM. MeCP2 and histone deacetylases 1 and 2 in dorsal striatum collectively suppress repetitive behaviors. Nat Neurosci. 2016;19:1506–1512. doi: 10.1038/nn.4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cheng PL, Song AH, Wong YH, Wang S, Zhang X, Poo MM. Self-amplifying autocrine actions of BDNF in axon development. Proc Natl Acad Sci U S A. 2011;108:18430–18435. doi: 10.1073/pnas.1115907108. [DOI] [PMC free article] [PubMed] [Google Scholar]