

Abstract
BACKGROUND:
Erdheim Chester disease (ECD) is a rare form of non-Langerhans histiocytosis that still presents a diagnostic and clinical dilemma.
CASE PRESENTATION:
We present a rare case of ECD, young 31 male with atypical localisation and soft tissue presentation and no bone involvement. He started clinical investigations due to subcutaneous tumour mass in the lumbar spine that caused severe back pain. Skin biopsy revealed ECD with Immunohistochemistry CD68+, CD10+, CD11c+, vimentin+, S100A4+. Activating BRAFV600E mutation was positive from the tumour tissue. The patient was referred to the haematology department. PET CT was performed for initial disease staging. Treatment was started with corticosteroids (methylprednisolone 0.5 mg/kg per day), and after 7 days, a significant clinical improvement was noticed in terms of pain disappearance with no need for pain killers. After two weeks, treatment with interferon Alfa (IFN-α) was started in a dose of 3 million units 3 times per week. After 4 months of interim treatment PET, CT revealed a significant reduction of the tumour mass. Therapy with IFN-α was continued, and the patient is still clinically in good condition.
CONCLUSION:
It can be concluded that shortening the time of diagnosis of ECD is essential in treatment outcome of this disease. Still, large studies have to confirm the best treatment of this rare condition.
Keywords: Histiocytosis, Diagnosis, BRAF mutation, Interferon, Erdheim Chester Disease
Introduction
Erdheim Chester disease (ECD) is a rare form of Non – Langerhans histiocytosis with unclear pathogenesis and aetiology and has been considered to be a non-neoplastic inflammatory disorder as well as a clonal neoplastic disorder [1]. The recent discovery of BRAFV600E mutations in ECD described the oncogenic nature of the disease, as well ECD histiocytes have been found to express a pattern of proinflammatory cytokines and chemokines responsible for local activation and recruitment of histiocytes [2]. The revised 2008 WHO classification of malignant haematological tumours proposed to assign separate histiocytic proliferations and ECD is one of the three new entities that is supposed to origin from interstitial dendritic cells. The idea to create these provisional entities was to enable to collect new cases for further studies and to maintain the purity of well-defined categories. One disease site may dominate the clinical presentation and require focused treatment in addition to the treatment of underlying ECD. Clinically, some patients have indolent and asymptomatic disease. Symptomatic ECD can be further categorised as multi-organ affection (CNS cardiovascular, pulmonary, soft tissue, retroperitoneal, etc.). Severe involvement of essentially any organ system constitutes “high-risk” disease; therefore, the clinical phenotype of each patient is best characterised by the most pathophysiologically affected organ. ECD is a rare, multi-system disorder requiring multidisciplinary collaboration in its diagnosis and treatment. Guidelines for diagnosis and treatment of this disease are still to be refined in terms of generating trials with targeted molecular and immunologically based treatments which are essential to furthering therapeutic progress in ECD [3].
Case Presentation
A newly diagnosed case of ECD is presented in this article, a 31 years adult male patient with multiple nodular subcutaneous tumours and aches in his back. He has been admitted to the plastic surgery clinic for diagnostic surgical biopsy (Figure 1 and 3).
Figure 1.

Nodular subcutaneous tumours in the lower back of the spinal cord
Figure 3.

First skin biopsy No. 1130357: Oval skin excision measuring 1.9 x 0.8 cm with SFT measuring 1.1 cm has been received for histological analysis. Standard histological techniques have been used with H&E staining and immunohistochemical analysis for CD4, CD8, CD20, CD25, CD68, MCT
Histopathology revealed chronic lymphocytic vasculitis, and he was referred to a rheumatologist for further immunological investigations and diagnostic evaluation. Due to intensive pain in the lumbar spinal cord, he performed MRI of the spinal cord, and a tumour mass in the subcutaneous tissue was noticed all over the spinal cord and in the retroperitoneal soft tissue region (Figure 2).
Figure 2.

MRI of the lumbar spine; On MRI a tumour mass was found in the subcutaneous tissue all over the back of the patient and in the retroperitoneal soft tissue region
He was suggested to perform second skin biopsy that still didn’t clear the diagnosis and was in favour of inflammatory vasculitis (Figure 4).
Figure 4.
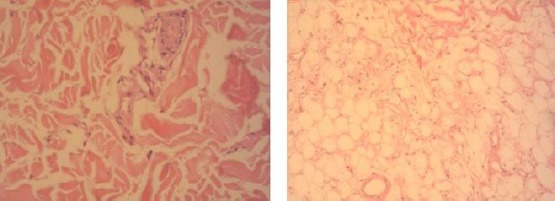
Second skin biopsy: Oval skin excision measuring 2.2x0.3cm with SFT measuring 1.1cm without peculiar features have been received for DIF analysis. IgG (-), IgM (-), IgA (-), C3c (-), C1q (-), Fibrin (-). Standard histological techniques have been used with H&E staining and PAS has been done
The patient clinically worsens with severe pain in the lower lumbar spine. He was treated with NSAIL and analgesics. Due to persistent pain and negative results from immunological tests he performed third needle biopsy in which lymphocytes, plasma cells, and a large number of histiocytic cells with pleomorphic features; some of them with foamy cytoplasm, or eosinophilic cytoplasm and multinuclear giant cells. Pseudocysts lined with cells with histiocytic features were found. Immunohistochemistry revealed CD68+, CD10+, CD11c+, vimentin +, S100A4 +, negative for CD21, CD23, MAC387, S-100. The histopathology was in favour of ECD (Figure 5).
Figure 5.

Third core needle biopsy No.1133255: hypocellular collagenous tissue with entrapped accumulations from polymorphous cells: lymphocytes, plasma cells, and a large number of histiocytic cells with pleomorphic features; some of them with foamy cytoplasm, or eosinophilic cytoplasm and multinuclear giant cells. Pseudocysts lined with cells with histiocytic features were found. The histology confirmed ECD (CD68+, CD10+, CD11c+, S100A4+, vimentin+)
One patient was evaluated with histopathology findings from three skin biopsies performed in the Institute of Pathology, Medical Faculty, University “Ss. Cyril and Methodius”, Skopje, Republic of Macedonia. With the diagnostic confirmation of ECD, he was referred to University Clinic for haematology. Oncogenic nature of ECD with BRAF mutation was performed with RT-PCR. Imaging was performed with MRI and PET-CT.
The patient was advised to continue evaluations and treatment at the haematology department. From haematological investigations activating BRAFV600E mutation was positive from the tumour tissue (RT PCR). Bone marrow biopsy was negative, with no infiltration for ECD. Laboratory blood tests were in the normal range. The patient performed initial PET CT for disease staging. PET CT revealed high metabolic activity in the gluteal region with deep subcutaneous infiltration and muscle affection (SUV 6.0).
Due to persistent lower back pain patient started treatment with corticosteroids (methylprednisolone) in a dose of 1mg/kg for one month. After one week he improved clinically with reduced pain, could perform the normal activity and he discontinued NSAIL and other analgesics. After diagnosis confirmation, his treatment followed with IFN alpha 3 million units 3 times per week for the next three months. After 3 months, a controlled PET CT was performed and revealed a significant reduction of tumour mass (SUV from 6 to 2.3). Therapy with IFN alpha was continued, and the patient is still clinically in good condition (Figure 6).
Figure 6.
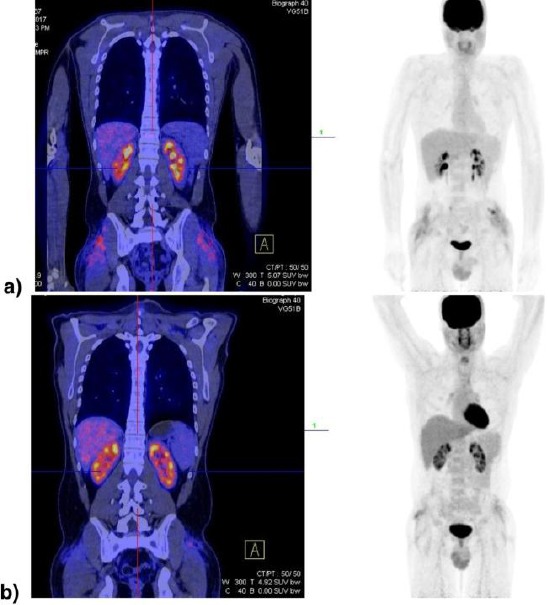
PET CT in a patient with ECD initially revealed high metabolic activity in the gluteal region with deep subcutaneous infiltration and muscle affection (SUV max 6.0) (a) and 4 months after treatment initiation with a significant reduction of the tumour mass and infiltration (SUV max 2.3) (b)
Discussion
ECD is a rare form of non-Langerhans’ cell histiocytosis, a disease with unclear pathophysiology, diagnostic dilemmas and still unknown treatment options. Individuals affected by this disease are typically adults between their 50 to 70 years, but patients between the ages of 7 to 84 years have been diagnosed [3]. Males and females are almost equally affected. The multisystemic form of ECD is associated with significant morbidity, which may arise due to histiocytic infiltration of critical organ systems. Among the more common sites of involvement are the skeleton, CNS, CVS, lungs, kidneys (retroperitoneum) and skin [4], [5]. The presented patient was 31 years of age male with no previous comorbidity and rare localisation of histiocytic infiltration, which caused diagnostic difficulties. The heterogeneous manifestations of ECD vary amongst different individuals. This results in a presentation that may vary from an indolent focal disease to life-threatening organ failure. The most common presenting symptom of ECD is bone pain. General symptoms are also described in most of the ECD cases like fever, fatigue, weight loss, loss of appetite and microcytic anaemia. The presented case was a rare soft tissue presentation of ECD with no bone lesions, which caused the diagnostic dilemma and became an ECD case with significance. The aetiology of ECD is unknown yet thought to be associated with an intense TH1 immune response. It may also be associated with the V600E BRAF mutation, as described in half of the patients in recent studies. Estimates of BRAFV600E mutation frequencies in ECD currently range between 38% and 68% in most reports, with one recent report suggesting that nearly 100% (18/18) of ECD patients have the mutation, which opened a new treatment perspective for novel targeted therapy [6].
The diagnostic criteria of ECD are based on radiographic and histologic findings. Two biopsies that were initiated in the presented case revealed features of inflammatory vasculitis that did not correlate with the clinical manifestations, laboratory tests and molecular analysis. This was the reason to advise the patient for the third needle biopsy that confirmed ECD. Skeletal imaging is a very important diagnostic tool because in most of the ECD cases osteosclerotic bilateral changes in large bones can be found. Also, gamma scan assessment in ECD reveals abnormal strong labelling of distal bone ends of the large bones. The presented case has performed bone scintigraphy that revealed no tracer accumulation in distal parts of the large bones. CT scans of the lumbar region on two occasions didn’t reveal the existence of soft tissue lesion. After an MRI of the spine and PET CT, the lesion was detected. However, a definite diagnosis of ECD is established only once CD68 (+), CD1a (−) histiocytes are identified within a biopsy specimen. (7)
There is no therapeutical consensus on ECD, but it is evident that initiation of therapy should be soon after diagnosis confirmation, there is no evidence for observational studies. It is still of limited alternatives. Currently, interferon-α is the most extensively studied agent in the treatment of ECD and serves as the first line of treatment [8]. The IFN-α optimal dose is 3MU/3 times per week in 3 years. There was no difference between IFN-α and pegylated formulations of IFN (Peg IFN-α). Dramatic efficacy was described in 3 cases of ECD treated with Vemurafenib, but enrollment in a prospective clinical trial is essential to document efficacy and potential toxicities as well as to determine the duration of therapy. Treatment with other agents anticytokine agents, cladribine (2CDA), tocilizumab, sirolimus, imatinib, infliximab and anakinra are currently advocated as promising second-line treatments for patients whose response to interferon-α is unsatisfactory [9], [10]. Overall, the 5 years survival of ECD is 68%. Disease surveillance is with organ-specific lesion imaging every 3 months after treatment initiation. After disease stabilisation, the intervals of the surveillance should be prolonged to 6 months. The presented case had PET CT imaging initially and 4 months after starting treatment with significant disease regression. There is no other cytokine marker that can be beneficial in disease surveillance period. C-reactive protein is usually elevated at the beginning of ECD and later decreased so that it can be used as a helpful biomarker in monitoring treatment. This case highlights the different clinical, radiological and pathological manifestations associated with ECD, the differential diagnoses and the various treatment options [11]. Due to a better characterisation of this entity, further studies that would shed new insights on the epigenetic and cytogenetic characteristics of ECD are needed, and further correlation with morphological and immunophenotypic features is also needed.
Footnotes
Funding: This research did not receive any financial support
Competing Interests: The authors have declared that no competing interests exist
References
- 1.Chester W. Lipoidgranulomatose. Virchows Arch Pathol Anat. 1930;279:561–602. https://doi.org/10.1007/BF01942684. [Google Scholar]
- 2.Haroche J, Charlotte F, Arnaud L, et al. High prevalence of BRAF V600E mutations in ErdheimChester disease but not in other non-Langerhans cell histiocytoses. Blood. 2012;120(13):2700–2703. doi: 10.1182/blood-2012-05-430140. https://doi.org/10.1182/blood-2012-05-430140 PMid: 22879539. [DOI] [PubMed] [Google Scholar]
- 3.Diamond EL, Dagna L, Hyman DM, Cavalli G, Janku F, Estrada-Veras J, Ferrarini M, Abdel-Wahab O, Heaney ML, Scheel PJ, Feeley NK, Ferrero E, McClain KL, Vaglio A, Colby T, Arnaud L, Haroche J. Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester disease. Blood. 2014;124(4):483–492. doi: 10.1182/blood-2014-03-561381. https://doi.org/10.1182/blood-2014-03-561381 PMid: 24850756 PMCid: PMC4110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dion E, Graef C, Miquel A, Haroche J, Wechsler B, Amoura Z, Zeitoun D, Grenier PA, Piette JC, Laredo JD. Bone involvement in Erdheim-Chester disease: imaging findings including periostitis and partial epiphyseal involvement. Radiology. 2006;238(2):632–9. doi: 10.1148/radiol.2382041525. https://doi.org/10.1148/radiol.2382041525 PMid: 16371583. [DOI] [PubMed] [Google Scholar]
- 5.Wittenberg KH, Swensen SJ, Myers JL. Pulmonary involvement with Erdheim-Chester disease: radiographic and CT findings. AJR Am J Roentgenol. 2000;174(5):1327–31. doi: 10.2214/ajr.174.5.1741327. https://doi.org/10.2214/ajr.174.5.1741327 PMid: 10789787. [DOI] [PubMed] [Google Scholar]
- 6.Diamond EL, Subbiah V, Lockhart AC, Blay JY, Puzanov I, Chau I, Raje NS, Wolf J, Erinjeri JP, Torrisi J, Lacouture M, Elez E, Martínez-Valle F, Durham B, Arcila ME, Ulaner G, Abdel-Wahab O, Pitcher B, Makrutzki M, Riehl T, Baselga J, Hyman DM. Vemurafenib for BRAF V600-Mutant Erdheim-Chester Disease and Langerhans Cell Histiocytosis: Analysis of Data From the Histology- Independent, Phase 2, Open-label VE-BASKET Study. JAMA Oncol. 2018;4(3):384–388. doi: 10.1001/jamaoncol.2017.5029. https://doi.org/10.1001/jamaoncol.2017.5029 PMid: 29188284 PMCid: PMC5844839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Young JR, Johnson GB, Murphy RC, Go RS, Broski SM. (18) F-FDG PET / CT in Erdheim - Chester Disease: Imaging Findings and Potential BRAF Mutation Biomarker. J Nucl Med. 2018;59(5):774–779. doi: 10.2967/jnumed.117.200741. https://doi.org/10.2967/jnumed.117.200741 PMid: 29097410. [DOI] [PubMed] [Google Scholar]
- 8.Fadi Braiteh, Cynthia Boxrud, Bita Esmaeli, Razelle Kurzrock. Successful treatment of Erdheim- Chester disease, a non - Langerhans - cell histiocytosis, with interferon - alfa. Blood. 2005;106(9):2992–2994. doi: 10.1182/blood-2005-06-2238. https://doi.org/10.1182/blood-2005-06-2238 PMid: 16020507. [DOI] [PubMed] [Google Scholar]
- 9.Adam Z, Petrášová H, Řehák Z, Koukalová R, Krejčí M, Pour L, Vetešníková E, Čermák A, Ševčíková S, Szturz P, Král Z, Mayer J. Evaluation of five years of treatment of Erdheim-Chester disease with anakinra: case report and overview of literature. Vnitr Lek Fall. 2016;62(10):820–832. [PubMed] [Google Scholar]
- 10.Cohen-Aubart F, Maksud P, Emile JF, Benameur N, Charlotte F, Cluzel P, Amoura Z, Haroche J. Efficacy of infliximab in the treatment of Erdheim-Chester disease. Ann Rheum Dis. 2018;77(9):1387–1390. doi: 10.1136/annrheumdis-2017-212678. https://doi.org/10.1136/annrheumdis-2017-212678 PMid: 29363511. [DOI] [PubMed] [Google Scholar]
- 11.Milne P, Bigley V, Bacon CM, Neel A, McGovern N, Bomken S, Haniffa M, Diamond EL, Durham BH, Visser J, Hunt D, Gunawardena H, Macheta M, McClain KL, Allen C, Abdel-Wahab O, Collin M. Hematopoietic origin of Langerhans cell histiocytosis and Erdheim-Chester disease in adults. Blood. 2017;130(2):167–175. doi: 10.1182/blood-2016-12-757823. https://doi.org/10.1182/blood-2016-12-757823 PMid: 28512190 PMCid: PMC5524529. [DOI] [PMC free article] [PubMed] [Google Scholar]