

Abstract
Toxin-antitoxin (TA) systems were initially discovered as plasmid addiction systems on low-copy-number plasmids. Thousands of TA loci have since been identified on chromosomes, plasmids and mobile elements in bacteria and archaea with diverse roles in bacterial physiology and in maintenance of genetic elements. Here, we identified and characterised a plasmid mediated type II TA system in Enterobacteriaceae as a member of the ParDE super family. This system (hereafter, ParDEI) is distributed among IncI and IncF-type antibiotic resistance and virulence plasmids found in avian and human-source Escherichia coli and Salmonella. It is found that ParDEI is a plasmid stability and stress response module that increases tolerance of aminoglycoside, quinolone and β-lactam antibiotics in E. coli by ~100–1,000-fold, and thus to levels beyond those achievable in the course of antibiotic therapy for human infections. ParDEI also confers a clear survival advantage at 42 °C and expression of the ParEI toxin in trans induces the SOS response, inhibits cell division and promotes biofilm formation. This transmissible high-level antibiotic tolerance is likely to be an important factor in the success of the IncI and IncF plasmids which carry it and the important pathogens in which these are resident.
Subject terms: Bacteriology, Bacterial infection
Introduction
Self-transferable (conjugative) antibiotic resistance plasmids promote their own retention in bacterial populations via “addiction systems”, typically toxin-antitoxin (TA) modules that inhibit or kill plasmid-free daughter cells and serve to maintain plasmids in bacterial populations. The antibiotic resistance conferred on major pathogens such as Escherichia coli and Klebsiella pneumoniae in this way is a major problem worldwide1,2.
The archetypal TA system is a two-gene operon, producing a protein toxin and an antitoxin that opposes it in the bacterial cell3. TA systems were first recognised by their role in plasmid maintenance by post segregational killing of plasmid-free cells4. In a similar way, TA systems found on other mobile genetic elements like transposons, prophages and genomic islands also contribute to their maintenance5–7. Hundreds of TA systems have been identified in bacterial and archaeal chromosomes but the functions of many remain elusive. It is reported that numerous chromosomal TA systems contribute to bacterial stress responses, biofilm formation, intestinal colonisation, virulence, antibiotic tolerance and persister cell formation8–12. The roles of chromosomal TA systems in antibiotic tolerance and persister cell formation have received a great deal of attention8,9,13,14 although the molecular mechanism behind persister formation is not fully understood14.
Persister cells are a phenotypic subpopulation of bacteria that form under different environmental cues and remain viable after exposure to usually lethal concentrations of antibiotics15. Antibiotic tolerance is important in the survival of bacterial biofilms. Several chromosomal TA systems are induced in biofilms11, where persister cells are implicated in antimicrobial tolerance and treatment failure16,17.
TA system toxins typically exert their function by impairing bacterial replication or RNA translation3,18,19 and are commonly divided into several types, the most common and best understood of which are the type II TA systems, with protein toxin-antitoxin pairs. In classic type II TA systems, the antitoxin is encoded upstream of the toxin and the antitoxin protein usually forms a dimer, each monomer generally being composed of an N-terminal dimerisation and DNA-binding domain and a C-terminal domain for toxin binding and neutralisation20 (Fig. S1). Chromosomal type II TA systems are thought to be heavily involved in antibiotic tolerance and formation of persister cells8,9,15 but there is ongoing debate arising from published data13,14,21–25. Recent studies24,25 have shown no effect on antibiotic tolerance or persister cell formation from the deletion of as many as ten E. coli chromosomal type II TA systems, but other E. coli type II TA modules such as vapBC and pasTI have been shown to increase persister cell formation upon exposure to different antibiotics26,27 and mutations in some TA modules also enhance the formation of antibiotic-tolerant persisters28,29. Expression of (type II) chromosomal RelE, MazF and E. coli F plasmid-borne CcdB toxins in the absence of their cognate antitoxins (RelB, MazE and CcdA respectively) have been shown to increase antibiotic survival30–32 and other types of TA systems including TisB-istR-1 and GhoST have also been shown to be involved in persistence in E. coli33,34 but their exact molecular mechanisms are largely unknown.
One of the best-known systems is the chromosomal Type II TA system RelBE, homologues of which are widely distributed in Gram-positive and Gram-negative bacterial chromosomes and in the archaea, where they appear to be important in environmental and nutrient stress adaptation35. ParDE, another type II TA system, was first identified in the broad-host range plasmid RK2 as a plasmid maintenance/stability (“addiction”) system36. The ParE toxins are in the RelE toxin superfamily and have significant primary DNA sequence homologies with RelE but distinct cellular targets and mechanisms of action. ParE is a gyrase inhibitor that blocks DNA replication37 while RelE is an endoribonuclease that inhibits translation by promoting cleavage of mRNA in the ribosomal A-site38–40. Here we describe a RelE/ParE superfamily toxin-containing TA system in IncI and IncF conjugative plasmids, these plasmids being major vectors of β-lactam antibiotic resistance in the Enterobacteriaceae. Our results indicate that this ParDEI TA system is a plasmid maintenance system with an additional role in bacterial stress management particularly in antibiotic tolerance and heat tolerance. This TA system is therefore expected to contribute to the success of IncI and IncF plasmids in the global spread of antibiotic resistance and may play a vital role in treatment failure of major classes of antibiotics.
Results
A ParDE-type TA system in IncI1 plasmid pJIE512b
A putative type II TA system with a RelE-like toxin was predicted in our sequenced IncI1 plasmid pJIE512b (Accession No. HG970648.1) by TA-finder (http://202.120.12.133/TAfinder/index.php). A BLASTp search revealed identical toxin proteins in GenBank, variably annotated as ‘putative hypothetical protein’, YacB, RelE toxin, RelE/ParE family protein, RelE/StbE family protein, plasmid stabilisation system protein, mRNA interferase toxin RelE or RelE/StbE. Although the amino acid sequence is only 18%, 15% and 15% identical with E. coli chromosomal RelE, plasmid RK2 ParE and Caulobacter crescentus chromosomal ParE1 (ParE1-CC, hereafter), respectively (Fig. S2a), the predicted secondary structure of the putative toxin (α1α2β1β2β3) is closer to the secondary structure of ParE1-CC (α1α2β1β2) than RK2 ParE (α1β1β2α2) and RelE (β1α1α2α3β2β3β4α4) (Fig. S2b). Conserved domain search for the toxin protein identifies it as a member of ParE toxin superfamily, and three highly conserved amino acids in RelE (61R, 81R and 87Y) that are thought to be important for mRNA cleavage39 are conspicuously absent from the putative toxin and from other ParE toxins (Fig. S2a).
The putative antitoxin protein is predicted as RHH-like by TA-finder. BLASTp search of GenBank found a ‘hypothetical protein’, ATPase component of ABC transporter, ‘prevent host death’ protein, addiction module antitoxin RelB/DinJ family protein, RHH domain protein, Phd antitoxin, TA system protein, etc. The amino acid sequence has 15%, 11% and 21% identity with E. coli chromosomal RelB, plasmid RK2 ParD and Caulobacter crescentus chromosomal ParD1 (ParD1-CC, hereafter), respectively (Fig. S3a), and the predicted secondary structure (β1α1α2α3) is the same as ParD1-CC41, but different from E. coli RelB (β1α1α2α3β2β3β4α4) and RK2-ParD which has an additional α-helix (β1α1α2α3α4) at the C-terminus (Fig. S3b). A conserved domain database search identifies the putative antitoxin as a COG3905 superfamily protein, although it has the ribbon-helix-helix (RHH) motif of the ParD and RelB antitoxins. Amino acid sequence and predicted secondary structure places the putative antitoxin closest to the ParD antitoxin and we will therefore use the term ParDEI (the superscriptI denoting initial recognition in an IncI plasmid).
Distribution of ParDEI
A BLASTn search using the complete coding region for ParDEI identified 153 plasmid sequences (as on October 2018) in GenBank that included at least 80% nucleotide sequence identity and 90% of the length of ParDEI coding region. Searching for rep genes and information available in published sequences and papers indicated that 48% (74) and 47% (72) of plasmids were of IncI and IncF type, respectively, and most from E. coli (72%) and Salmonella species (24%) (Fig. 1 and Table S1). The sources of those isolates were identified as birds (especially poultry) and animals (39%), vegetables and foods (5%) and human samples (53%). 72% of plasmids carrying ParDEI have at least one and many have multiple antibiotic resistance genes. 19% of plasmids in which the ParDEI system was identified (irrespective of antibiotic resistance) carried important virulence genes including shiga toxin and colicins (Fig. 1 and Table S1).
Figure 1.
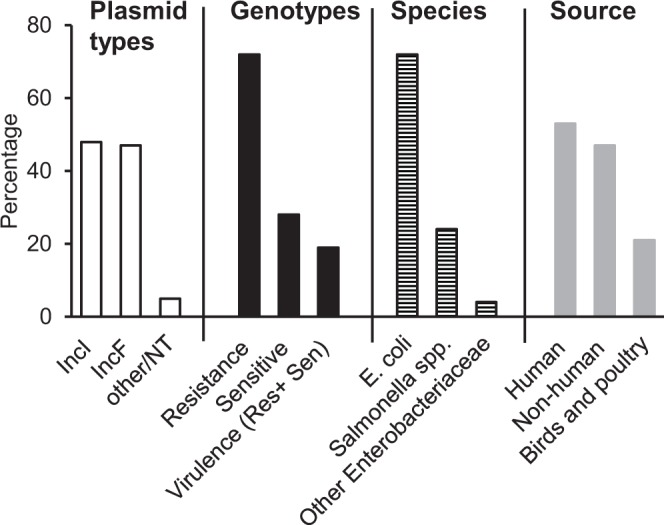
Distribution of the ParDEI TA system in plasmid incompatibility types (□), in antibiotic-resistance (‘resistance’), plasmids without identified antibiotic resistance genes (‘sensitive’) and plasmids identified as “virulence plasmids” (■); and the bacterial hosts of those plasmids (⊟) and their sources ( ).
).
ParDEI TA is a plasmid maintenance system
The BPROM42 software was used to predict the −35, −10 promoter sequences and the ribosome binding site (RBS) of parDEI (Fig. S4). parDEI was cloned with its putative promoter and RBS into low (pACYC184) and high copy (pBCSK+) vectors and plasmid stability assayed in two E. coli K12 strains, DH5α (recA mutant) and BW25113 (recA proficient wild type). ParDEI ensured complete retention of both plasmids in antibiotic free media in both strains, while control plasmids lacking ParDEI were lost from 50–60% of cells within 96 h (Fig. 2). This indicates that ParDEI is an effective plasmid maintenance (addiction) system.
Figure 2.

Plasmid stability assay. Plasmid stability was assessed for cloned ParDEI TA system in low copy (a,b) and high copy number plasmids (c,d) in E. coli BW25113 (a,c) and DH5α (b,d), expressed as relative plasmid retention over time. Plasmids carrying cloned ParDEI TA were very stable (■), irrespective of plasmid type and host strain, while ~60% of control plasmids (vector only) were lost (▲) within 96 h without antibiotic selection. Each experiment was repeated three times, and the mean values with standard deviations (error bars) are presented.
Effects of the ParEI putative toxin on bacterial growth and cell morphology
To examine the effects of ParEI toxin on bacterial growth, parEI and parDI coding regions were cloned under the control of the arabinose-inducible promoter of pBAD24 and pBAD33-Gm vectors to construct pJIMK78 and pJIMK99 respectively. A C-terminal truncated version of parEI toxin gene was cloned into pBAD24 under the control of the arabinose-inducible promoter to construct pJIMK92. Plasmids pBAD2443 (with pBR322 ori and an ampicillin resistance marker) and pBAD33-Gm44 (p15A ori and a gentamicin resistance marker) are expression vectors with arabinose-inducible promoters but different replication systems and resistance markers and thus allow controlled simultaneous expression of both toxin and antitoxin gene in the same bacteria. Induction of parEI expression from J53(pJIMK78) extended lag phase and inhibited growth (Fig. 3a). The effect of ParEI toxin on growth was neutralised when both toxin and antitoxin were simultaneously expressed in trans from J53(pJIMK78 + pJIMK99) after induction with arabinose. A truncated or non-toxic version of parEI was generated by removing amino acid residues from the C-terminus region of ParEI known to be essential for stability and toxicity of ParE protein45. Expression of truncated ParEI toxin from J53(pJIMK92) with 0.2% arabinose did not result in growth inhibition. OD600 and viable cell counts were both decreased after parEI was induced in exponentially growing J53 cells (Fig. 3b,c) compared to simultaneously expressed toxin and antitoxin and compared to expression of truncated toxin. Induction of ParEI toxin produced elongated E. coli cell morphology (Fig. 3d) which is very similar to that of ciprofloxacin-treated J53 cells and to that ascribed to DNA gyrase inhibition by other ParE toxins45,46 but normal cell morphology was observed from cells in which both toxin and antitoxin genes were co-expressed in trans or those in which only truncated toxin was expressed (Fig. 3d).
Figure 3.

Effects of ParEI toxin on E. coli growth, survival and cell morphology. J53(pJIMK78) growth is significantly inhibited compared to J53(pBAD24) empty vector, J53(pJIMK92) expressing the truncated ParEI toxin and J53(pJIMK78 + pJIMK99) expressing both toxin and antitoxin, respectively, with ParEI continually expressed from beginning of culture (a). Optical density (b) and viable bacterial count (c) were also reduced after toxin induction in mid-logarithmic phase. (d) Elongated cell morphology in J53(pJIMK78) with ParEI toxin expressed (top panel) is similar to CIP-treated (0.25 µg/mL) J53 cells (bottom panel), while normal cell morphology is observed in the presence of the truncated toxin (2nd panel from bottom) or empty vector (2nd panel from top) or with the co-expression of toxin and antitoxin (3rd panel from top). Each experiment was repeated three times, and the mean values with standard deviations (error bars) are presented.
ParEI triggers the SOS response
Unopposed ParE expression in V. cholerae is associated with elongated cell morphology, DNA damage and induction of the SOS response46. In addition to cell elongation, parEI expression from pJIMK78 in trans resulted in significantly increased recA and lexA gene expression in E. coli, consistent with induction of the SOS response (Fig. 4b,c). We found no significant increase in rpoS mRNA (Fig. 4a) to indicate general stress response (GSR) pathway induction.
Figure 4.

Relative expression of stress response genes rpoS (a), recA (b) and lexA (c), in the presence of ParEI toxin induction or repression in E. coli J53(pJIMK78). Toxin-induced state (with 0.2% arabinose) (■), toxin-repressed state (with 0.2% glucose) (□) is compared with CIP-treated (0.25 µg/mL) cells ( ) and J53(pBAD24) vector-only control cells (
) and J53(pBAD24) vector-only control cells ( ). Each experiment was repeated three times, and the mean values with standard deviations (error bars) are presented.
). Each experiment was repeated three times, and the mean values with standard deviations (error bars) are presented.
ParDEI provides antibiotic tolerance and promotes persister cell formation
Bacterial tolerance of ciprofloxacin (CIP, a fluoroquinolone), gentamicin (GEN, an aminocyclitol aminoglycoside) and cefotaxime (CTX, a third-generation cephalosporin) were assessed in E. coli J53 with or without parDEI and after specific activation of full-length or truncated ParEI toxin. Experiments were performed five times independently in supplemented M9 media (described in the methods section) and tolerance was assessed at 3 h and 5 h after exposure to high doses of antibiotics. The baseline MIC of E. coli J53 was low (very susceptible) to all 3 antibiotics tested (MIC = 0.25 µg/mL, 0.25 µg/mL and 0.064 µg/mL for CTX, GEN and CIP antibiotics, respectively). There was no change in MIC between J53 alone and with recombinant constructs used in antibiotic tolerance assay for above mentioned antibiotics. We compared (i) pJIMK86 (with cloned parDEI) with pACYC184 (low-copy vector-only control), (ii) a naturally-occurring antibiotic-susceptible IncI1 plasmid with (pJIMK56) or without (specific deletion of) parDEI (pJIMK82) and (iii) pJIMK78 (parEI under control of an arabinose-inducible promotor), pJIMK92 (C-terminus truncated parEI toxin, under control of arabinose-induced promoter) and with pBAD24 (vector-only control), all in a J53 E. coli background. All bacteria expressing the ParDEI TA system had a clear survival advantage in the presence of high concentrations of all 3 antibiotics at both time points examined (Figs 5a, S5a), with at least ~1,000-fold better survival of supra-MIC CIP (5 µg/mL) and GEN (16 µg/mL) exposure and at least ~10-fold better survival of supra-MIC CTX (16 µg/mL) exposure (Figs 5b, S5b). Specific activation of toxin alone (without the putative antitoxin ParDI) provided the same or slightly superior survival advantage than ParDEI together (~1,000 fold) in the presence of high concentrations of CIP and GEN and at least ~100-fold better survival in the presence of high concentration of CTX (Figs 5a,b, S5a,b). Expression of truncated toxin from pJIMK92 did not show any additional survival advantage in E. coli than are those carrying empty vector (Figs 5a,b, S5a,b). Note that these experiments were performed with pJIMK56 and pJIMK82, these being derivatives of pJIE512b from which we have deleted the powerful antibiotic resistance gene blaCMY-247,48.
Figure 5.

Role of ParDEI TA system in antibiotic tolerance/persister cell formation. Survival after 5 h exposure to high doses of antibiotics CIP (□), GEN (■) and CTX ( ). E. coli J53 cells carrying ParDEI (cloned in pJIMK86 or from the native plasmid pJIMK56) or ParEI toxin alone (pJIMK78) induced with 0.2% arabinose increased survival by ~100 to 1,000-fold for CIP and GEN and ~10 to 100-fold for CTX; expressed as (a) relative survival and (b) as fold-change. (c) Expression of ParDEI TA genes after exposure to sub-MIC concentration of different antibiotics (CIP, GEN and CTX antibiotic) is increased 3–4 folds within 60 min of CIP and GEN exposure but only slightly after CTX exposure. Antibiotic tolerance experiments (a,b) were repeated five times and gene expression experiment (c) was repeated 3 times. Mean values and standard deviations (error bars) are shown.
). E. coli J53 cells carrying ParDEI (cloned in pJIMK86 or from the native plasmid pJIMK56) or ParEI toxin alone (pJIMK78) induced with 0.2% arabinose increased survival by ~100 to 1,000-fold for CIP and GEN and ~10 to 100-fold for CTX; expressed as (a) relative survival and (b) as fold-change. (c) Expression of ParDEI TA genes after exposure to sub-MIC concentration of different antibiotics (CIP, GEN and CTX antibiotic) is increased 3–4 folds within 60 min of CIP and GEN exposure but only slightly after CTX exposure. Antibiotic tolerance experiments (a,b) were repeated five times and gene expression experiment (c) was repeated 3 times. Mean values and standard deviations (error bars) are shown.
Expression of parDEI mRNA gradually increased with time and was 3- and 4-fold higher after 60 min exposure to sub-MIC concentrations of CIP (0.05 µg/mL) and GEN (0.20 µg/mL), respectively (Fig. 5c). Expression of parDEI was initially downregulated at 15 min and only slightly increased after 30 or 60 min exposure to sub-MIC concentrations of CTX (0.20 µg/mL).
Role of ParDEI in other stress conditions
The presence of ParDEI appeared to make no significant difference to bacterial growth and survival in nutrient limiting (supplemented M9 containing casaminoacids but no glucose) or starvation conditions (supplemented M9, no glucose or casaminoacids) (Fig. 6a,b), but there was a significant benefit in bacterial growth was observed at 42 °C in the presence of ParEI toxin alone and ParDEI together. In the absence of ParDEI or ParEI, growth in supplemented M9 media is reduced by ~50% or more within 90 min at 42 °C (Fig. 6c). Normalised expression of parDEI was also relatively increased at 42 °C, consistent with a role in the observed increased survival (Fig. 6d).
Figure 6.

The effect of ParDEI on E. coli (J53) growth in stress conditions. E. coli with cloned ParDEI (pJIMK86) is not significantly different from the vector-only control in nutrient limitation (a) and nutrient starvation (b). A significant benefit is evident in E. coli J53(pJIMK86) and after activation of toxin from pJIMK78 (induction with 0.2% arabinose) for growth at 42 °C (c). Expression of ParDEI is also increased at 42 °C (d). Each experiment was repeated 3 times and mean values and standard deviations (error bars) are presented.
The putative ParEI toxin promotes biofilm formation in E. coli
Arabinose-induced ParEI toxin expression in E. coli J53 (from pJIMK78) was associated with significantly increased biofilm formation, as determined by a simple crystal violet elution assay (Fig. S6a,b) and by direct microscopic observation (Fig. S6c). No significant biofilm formation was observed by J53 E. coli carrying pBAD24 vector only, or after induction of C-terminus truncated ParEI toxin from plasmid pJIMK92 (Fig. S6a–c).
Discussion
The ParDEI type II TA system was mostly found in conjugative IncI and IncF plasmid types and predominantly in E. coli and Salmonella species (Fig. 1, Table S1). IncI and IncF type plasmids are major vectors of β-lactamase (AmpC, Extended-spectrum β-lactamase, Carbapenemase), aminoglycoside and quinolone resistance genes among the Enterobacteriaceae49. ParDEI is evidently important for the stable maintenance of mobile genetic elements (here, plasmids) in E. coli, like other plasmid and chromosome borne ParDE-homologues6,36. Deletion of ParDE from chromosome II of Vibrio cholerae, for example, results in loss of the chromosome and death of the cell from unopposed residual toxicity of the ParE toxin46.
Persister cells are metabolically inactive or dormant cells which can be produced by TA toxins that inhibit vital cellular processes34,50–52 and are thought to be less vulnerable to (more tolerant of) antibiotics largely because of the reduced activity in the cellular processes they target30. Biofilm formation is often depicted in terms of a stress response53 and chromosomal TA systems are well known to participate in the stress response, antibiotic tolerance and persister cell formation8,9,11,12 but such functions have only been reported for the plasmid-borne ccdAB type II TA system32.
Studies of antibiotic tolerance and perister cell formation mediated by chromosomal type II TA systems51 led to some discrepancies21,54 and debate in recent years. Recent studies24,25 have confirmed no role for E. coli chromosomal 10 type II TA systems in antibiotic tolerance or persister cell formation and identified several factors influenced previously published persister data, including contamination of experiments by cryptic prophage, concentration of antibiotics and growth media in different studies. Taking in to account the observations from both papers24,25, we confirmed that our experimental strains are free from prophage ɸ80 contamination and used concentrations of antibiotics >50-fold higher than the MIC value for the CIP, GEN and CTX and performed antibiotic killing assays at two time points, 3 h and 5 h after exposure to antibiotics. The tolerance assay was performed in supplemented M9 media25 (in which all strains behaved consistently), but E. coli K12 strains (DH5α, UB5201 and J53) exhibited different and variable growth kinetics in MOPS-based media, a recommended alternative24.
We show here that ParDEI contributes substantially to tolerance of three of the most important major classes of antibiotics for E. coli (Figs 5a,b, S5a,b), with >1,000-fold increased survival in the presence of supra-MIC concentrations of CIP (fluoroquinolone, targeting DNA gyrase) and GEN (aminoglycoside, targeting the 30 S ribosomal subunit) in the presence of the conjugative plasmid in which ParDEI naturally occurs, and we show that this is specifically mediated by RecA-dependent SOS response by activation of the ParEI toxin, similar to that found in activation of the CcdB toxin32. Expression of the ParEI toxin alone increased bacterial survival >100-fold in the presence of CTX (cephalosporin, targeting cell wall remodelling) but while the ~10-fold increased survival in the presence of ParDEI satisfies traditional definitions of ‘tolerance’ (survival at more than 4-fold MIC;55). ParDEI expression from its natural promoter is not induced in the presence of sub-MIC CTX concentrations, in contrast to the effects of GEN and CIP (Fig. 5c). Unlike the β-lactams (e.g. CTX), the aminoglycoside and quinolone antibiotics typically result in RecA-dependent SOS stress responses, with promotion of error-prone polymerase V expression, increased biofilm formation, and increased production of metabolically inactive small cells that are relatively unaffected by antibiotics56,57.
ParEI (the putative toxin) is therefore predicted to target DNA gyrase and participate in a RecA-dependent SOS response (Fig. 4b,c), much like the ParE1 and ParE2 toxins of V. cholerae chromosome II and the CcdB toxin of E. coli F-plasmids32. The elongated cell morphology observed after ParEI expression in E. coli is similar to that of CIP treated cells and to that described for the ParE-toxin of ParDE-RK2 or V. cholerae ParE (Fig. 3d). Also consistent with a response to DNA damage, ParDEI expression is induced at 42 °C (Fig. 6d), and ParDEI (or the ParEI toxin) markedly improves E. coli growth at 42 °C (Fig. 6c). ParDEI does not appear to protect bacteria from nutrient stress, which ordinarily results in an RpoS-dependent general stress response (Fig. 6a,b), and we found no evidence of significantly increased rpoS expression in the presence of the ParEI toxin (Fig. 4a). Close examination of Fig. 3a shows growth to be delayed and reduced, with an upward trend only evident 180 minutes after ParEI induction in J53(pJIMK78). Similar growth trend is also seen in the same bacteria transferred to 42 °C, 180 minutes after induction of ParEI toxin expression (Fig. 6c).
Detailed mechanistic studies are needed to understand the exact mechanism of ParDEI mediated tolerance but this is likely to be similar to that of CcdB toxin mediated antibiotic tolerance32. We hypothesise that (e.g.) antibiotic stress elevates the level of Lon and/or other proteases that degrades the ParDI antitoxin and releases ParEI from ParDI-ParEI toxin-antitoxin complex. The free toxin interacts with DNA gyrase to induce DNA damage and the SOS response, which in turn activates other cellular systems participating in persister cell formation.
Plasmid-borne ParDEI thus increases tolerance of temperatures that might occur in febrile humans and in healthy birds. The relative frequency of E. coli and Salmonella species in poultry, foods and other non-human samples (Fig. 1) is noteworthy. Salmonella species are commonly associated with leafy greens, poultry meat, and eggs58 and are important agents of food-borne disease. Heat treatment is one of the more effective ways to eliminate Salmonella and other pathogens from food products59; pasteurisation and other heat treatments are often used in processing milk, vegetables, juices, meat, and poultry carcasses59,60. Plasmid-encoded temperature tolerance is thus an important acquired virulence advantage in foodborne pathogens such as Salmonella and E. coli, which must adapt to core body temperatures of ~42 °C in many avian hosts (e.g. chickens).
Finally, homologous chromosomal TA systems including RelBE, MazEF and MqsRA are known to be involved in bacterial biofilm production12,61,62 and the RecA-dependent SOS response associated with DNA damage is well known to promote biofilm formation63. We show here that ParEI expression induces biofilm formation in E. coli (Fig. S6) and characteristic RNA signatures of the SOS response. Biofilms have increased resistance to antimicrobial agents64,65 and stress responses that stimulate biofilm formation may be expected to enhance resistance to the host immune system.
In summary, ParDEI appears necessary and sufficient to stabilise important (mostly IncI and IncF-type) conjugative antibiotic resistance plasmids in bacterial cells. Importantly, this type II TAS is also induced by and strongly protects bacteria from insults normally associated with a RecA-dependent SOS response (heat, quinolone and aminoglycoside antibiotics). ParDEI contributes to biofilm formation and allows survival in vitro at antibiotic concentrations that exceed those that can be achieved in vivo by several orders of magnitude. This high-level antibiotic tolerance is spread among the top-tier human pathogens, E. coli and Salmonella, on common self-transmissible antibiotic resistance plasmids and can be expected to promote the survival of these bacterial populations and of the resistance plasmids that protect them.
Materials and Methods
Bacterial strains and growth conditions
E. coli K12 strains DH5α (F−, 80lacZ∆M15, recA1, endA1, hsdR17, phoA, supE44, λ-thi-1, gyrA96, relA1), UB5201 (F−, pro, met, recA56, gyrA)66, J53 (F−, lac+, pro, met)67 and BW25113 (lacIq, rrnBT14, ∆lacZWJ16, hsdR514, ∆araBADAH33, ∆rhaBADLD78) strains were grown on LB broth or LB agar at 37 °C except where stated otherwise.
Preparation of supplemented M9 media
We suspended 10.5 g of M9 Medium Broth Powder (VWR, AMRESCO, Fountain Parkway, OH) (which contains 6 gm of Na2HPO4.7H2O, 3 gm of KH2PO4, 0.5 gm of NaCl and 1.0 gm of NH4Cl) in 1 L of distilled, deionized water and dissolved with gentle stirring and adjusted the pH to 7.4 with sodium hydroxide. The media was then autoclaved for 15 min at 15 psi at 121 °C. Supplemental components and antibiotics were added to the solution after cooled it to 50 °C. For supplementation, we added 2 mL of 1 M MgSO4, 0.1 mL of 1 M CaCl2, 50.0 µL of 10 mg/mL FeSO4.7H2O, 0.1 mL of 10 mg/mL thiamine, 20 mL of 20% glucose and 20 mL of 20% casaminoacids. All supplemental solutions were sterilised by passing through 0.22 µ filter (Millipore). Media were then stored at 4 °C.
Sequence and structural analysis
BLASTn was used to determine the distribution of TA systems in GenBank data. Amino acid identity and comparison of toxin and antitoxin proteins were performed by ClustalW alignment of MEGA7 software68. The secondary structures of toxin and antitoxin were predicted using PSIPRED69 software, with default settings.
Construction of plasmids
Construction of plasmid pJIMK82 are described below and all other plasmids used in this study are described in Table 1. Primers used for plasmids construction, PCR and sequencing are described in Table S2. All the constructs were verified by PCR and sanger sequencing.
Table 1.
Plasmids used in this study.
| Plasmids | Characteristics | Source/Reference |
|---|---|---|
| pBCSK+ | High copy number cloning vector, CmR | Catalog # 212215, Stratagene |
| pACYC184 | Low copy number cloning vector, CmR, TetR | New England Biolabs, Ipswich, MA; GenBank Accession # X06403 |
| pBAD24 | Expression vector with arabinose inducible promoter, AmpR, pBR322 replicon | 43 |
| pBAD33-Gm | Expression vector with arabinose inducible promoter, GenR marker and p15A replicon | 44, Addgene ID: 65098 |
| pJIE512b | A naturally occurring antibiotic resistance IncI1 plasmid carrying the ParDEI TA system | 47 |
| pJIMK77 | The ParDEI TA nucleotide sequence with its own putative promoter and RBS was cloned into the XbaI and BamHI site of pBCSK+ | This study |
| pJIMK86 | The ParDEI TA nucleotide sequence with its own putative promoter and RBS was cloned into the XbaI and BamHI site of pACYC184 | This study |
| pJIMK78 | The parEI toxin coding sequence was amplified from pJIE512b and cloned into EcoRI and XbaI sites of pBAD24 plasmid under the control of an arabinose inducible promoter | This study |
| pJIMK92 | The C-terminus truncated parEI toxin gene (201/282-bp) was cloned into EcoRI and XbaI sites of pBAD24 under an arabinose inducible promoter | This study |
| pJIMK56 | Derivative of pJIE512b which has an intact copy of ParDEI TA but the large antibiotic resistance region (~28.8 kb) and pndC toxin of the PndBCA TA system have been deleted. | 48 |
| pJIMK82 | Derivative of pJIE512b where parEI toxin, antibiotic resistance region and pndC toxin were deleted. | This study |
| pJIMK99 | The parDI toxin coding sequence was amplified from pJIE512b and cloned into EcoRI and XbaI sites of pBAD33-Gm plasmid under the control of an arabinose inducible promoter | This study |
pJIMK82
Derivative of pJIE512b where parEI toxin, antibiotic resistance region and pndC toxin were deleted. The deletion of pndC region was performed as described previously48. ParDEI and the antibiotic resistance region are closely located in the plasmid pJIE512b (Fig. S7) and were deleted in a single deletion event by allelic exchange70 using fosA3-specific primers, Long-F and Long-R (Table S2), targeting 50-bp upstream and downstream of the target region for deletion.
Plasmid stability assay
Plasmid stability was assessed as previously described71, with minor modifications. A single colony of E. coli bacteria carrying plasmid was grown in LB broth at 37 °C with shaking at 225 rpm without antibiotic. Bacterial cultures were transferred into fresh LB medium at 1:1,000 dilution twice daily for 4 days. Samples were taken before every transfer, diluted in saline and plated on to LB agar without antibiotic and incubated at 37 °C for 18 h. From each plate, 120 colonies were replica plated onto LB agar plates with and without relevant antibiotic to estimate plasmid retention.
Growth assays and bacterial cell morphology
Single colonies of E. coli J53 carrying recombinant plasmids pJIMK78 (pBAD24 with parEI toxin) or pJIMK92 (pBAD24 with truncated parEI) or pBAD24 (vector control) were inoculated into 10 mL LB broth in 100 mL conical flasks, with ampicillin 50 µg/mL and J53 with pJIMK78 and pJIMK99 (parEI toxin in pJIMK78 and parDI antitoxin in pJIMK99) was inoculated in to the LB broth with ampicillin 50 µg/mL and gentamicin 8 µg/mL and grown overnight (O/N) at 37 °C with shaking at 225 rpm. Cultures were adjusted to optical density (OD600) of 1.00 and diluted at 1:1,000 ratio into fresh LB broth and then inoculated (200 µl) into 96 well plate with 0.2% arabinose, ampicillin and gentamycin as needed. An automated multi-mode microplate reader SpectraMax iD5 (Molecular Devices, LLC; CA) was used to assay results. Bacteria were grown in microplates at 37 °C with medium shaking. Absorbance of the culture was measured at a wavelength of 600 nm (OD600) every 10 min over 10 h. Experiments were performed in triplicate and mean and standard deviations of nine data points for each sample were calculated and plotted.
To evaluate the effects of ParEI toxin on exponential growing cells, J53(pJIMK78) or J53(pJIMK92) were inoculated from O/N culture into 10 mL LB broth with ampicillin at 1:1,000 ratio and grown for 4 h at 37 °C with shaking and then 0.2% arabinose (for induction of the ara promotor) was added to the culture. To evaluate the neutralisation of toxin by cognate antitoxin, J53(pJIMK78 + pJIMK99) was inoculated from O/N culture into 10 mL LB broth at 1:1,000 ratio with ampicillin and gentamycin and grown for 4 h at 37 °C with shaking and then 0.2% arabinose (for induction of the ara promotor) was added. The bacterial cultures were sampled at 0, 30, 60 and 90 min after addition of arabinose, OD600 was measured and cell viability calculated by serial dilution onto solid growth media. Bacterial cell morphology was observed under phase contrast using an Olympus BX43 light microscope (Olympus corporation, Tokyo, Japan).
Determination of minimum inhibitory concentration (MIC)
MICs of ciprofloxacin, gentamicin and cefotaxime antibiotics for E. coli strain J53 with and without pBAD24 vector control and derived constructs pJIMK78, pJIMK92, pACYC184 vector control and pJIMK86, pBCSK+ vector control and pJIMK77, pJIMK56, pJIMK82 and pJIMK99, as well as J53 carrying both pJIMK78 and pJIMK99 together, were determined by E-test (BioMérieux, France).
Antibiotic treated bacterial persister assay
Antibiotic tolerance and bacterial persister formation was assayed in supplemented M9 media described earlier in this section and previously25. Single colonies of E. coli K12 bacteria J53 carrying plasmids pACYC184 (vector only) or pJIMK86 (pACYC184 with ParDEI TA) or pJIMK82 (derivative of pJIE512b without ParDEI) or pJIMK56 (derivative of pJIE512b with ParDEI) were grown O/N in 10 mL of supplemented M9 media in 100 mL conical flasks at 37 °C with shaking at 225 rpm with appropriate antibiotics. After O/N growth the cultures were adjusted to OD600 = 1.0 and sub-cultured at 1:100 ratio in 10 mL supplemented M9 media and grown at 37 °C with shaking at 225 rpm for about 4 to 4.5 h to reach the OD600 at 0.5. Supra-MIC concentrations of ciprofloxacin (5 µg/mL), gentamicin (16 µg/mL) or cefotaxime (16 µg/mL) were added as indicated and continued growth for further 5 h. Bacterial cultures were collected before and after 3 h and 5 h of antibiotic exposure and serial dilutions were plated onto LB agar plates and incubated at 37 °C incubator for 18 h. Colonies were then counted and percent survival and log survival rate calculated.
Direct effects of ParEI in antibiotic tolerance were measured as follows. Single colonies of E. coli J53 carrying plasmid pBAD24 (expression vector), or pJIMK78 (parEI in pBAD24) or pJIMK92 (truncated parEI in pBAD24) were grown O/N in 10 mL of supplemented M9 media in 100 mL conical flasks at 37 °C with shaking at 225 rpm with appropriate antibiotics. After O/N growth the cultures were adjusted to OD600 = 1.0 and sub-cultured at 1:100 ratio in 10 mL supplemented M9 media (without glucose, because glucose is a repressor of ara promoter; casaminoacids in the media as a carbon source in absence of glucose) and grown at 37 °C with shaking at 225 rpm for about 4 to 4.5 h to reach the OD600 at 0.5 and then 0.2% arabinose was added to each culture flask to induce toxin expression. Bacteria were then grown for another 3 h and then supra-MIC concentrations of antibiotic were added and continued growth for further 5 h. Bacterial cultures were collected before and after 3 h and 5 h of antibiotic exposure and survival calculated as above.
Real time reverse transcriptase assay
mRNA from recA, lexA, rpoS and parDEI was measured by quantitative reverse-transcriptase (qRT) PCR. For recA, lexA and rpoS gene expression, ParEI toxin was expressed or repressed from J53(pJIMK78) by adding 0.2% arabinose or 0.2% glucose respectively and cells were collected at t = 0, 30, 60, and 90 minutes. Bacteria with the empty vector, J53(pBAD24), and ciprofloxacin (0.25 µg/mL)-treated J53 bacteria were used as negative and positive controls respectively and assayed at the same time points. mRNA from parDEI in exponentially growing bacterial cultures exposed to sub-MIC concentrations of CIP (0.05 µg/mL), or GEN (0.2 µg/mL), or CTX (0.2 µg/mL) and cells were collected at t = 0, 30, 60 and 90 minutes.
RNA extraction, cDNA preparation and qRT-PCR were performed as previously described72. Briefly, total RNA was isolated using NucleoSpin RNA kit (Macherey Nagel, Germany). RNA was treated with DNase (TURBO DNA-free Kit, Ambion) and freedom from DNA contamination was further confirmed by PCR. cDNA was synthesized by high-capacity cDNA reverse transcriptase kit (Applied Biosystems). One microgram of the initially isolated RNA was used in each reverse transcription reaction. cDNA was diluted 1:10 and 2 μl were used for PCR (Table S2 for primers). Expression of E. coli rpoB was used as a reference. The stability of rpoB expression was assessed using the variation of threshold cycle (CT) values across samples and experimental conditions. Efficiency comparisons of target gene primers were performed against rpoB primers. The 2−ΔΔCT (Livak) method73 was used to determine the relative difference in expression using the geometric mean of rpoB ΔCT values for normalisation. For normalisation of expression, we first normalised CT of the target gene to that of rpoB for test samples (samples at different test conditions) and calibrator samples (samples just before exposing in test conditions, here, at 0 min time point). The ΔCT of the test samples were then normalised to the ΔCT of the calibrators and the normalised expression ratio calculated (2−ΔΔCT). Mean 2−ΔΔCT values and standard deviations for each target gene were derived in three biological replicates, each with three technical replicates, and presented in the graph.
Other stress tolerance assays
For heat stress experiments, J53 bacteria with plasmid pACYC184 (vector) or pJIMK86 (cloned parDEI) were grown in supplemented M9 media at 37 °C O/N with appropriate antibiotic and then subcultured in fresh supplemented M9 media at 1:100 ratio and incubated for a further 4 hours. Bacteria were then transferred to 42 °C. Bacterial cultures were sampled at different time intervals (0, 30, 60, 90 min) and dilutions were spread onto LB agar plates and grown at 37 °C for a further 18 h. To examine the direct effect of ParEI toxin on heat stress tolerance, overnight cultures of E. coli J53(pJIMK78) and the vector-only control J53(pBAD24) were subcultured in fresh supplemented M9 media without glucose at 1:100 ratio and grown for 4 h, then 0.2% arabinose was added to induce ParEI toxin expression from J53(pJIMK78) and grown for another 3 h. Bacterial cultures were then transferred to 42 °C and sampled at different time intervals (0, 30, 60, 90 min) for viable cell count and for qRT PCR analysis as described above.
For nutrient stress, J53(pACYC184), J53(pJIMK86), J53(pBCSK+) and J53(pJIMK77) bacteria were grown overnight in supplemented M9 media (with/out 0.4% glucose and 0.4% casamino acid supplementation, as specified). Overnight cultures were adjusted to an OD600 of 1.0 and subcultured at 1:100 ratio in fresh supplemented M9 media and OD600 was subsequently measured hourly for 8 h (no glucose supplementation) and cell viability was calculated for 48 h (no glucose and casamino acid supplementation).
Biofilm assay
E. coli J53(pBAD24), J53(pJIMK78) and J53(pJIMK92) were grown in LB medium with ampicillin at 37 °C overnight. Bacterial cultures were adjusted to OD600 = 1.0 and subcultured at 1:100 ratio in LB broth with ampicillin. After 3 h growth, 0.2% arabinose was added to induce toxin expression. After 3 h incubation with shaking at 37 °C, 500 µL of cultures were transferred to polystyrene tubes and incubated at 37 °C without shaking. Biofilm formation was measured by staining with crystal violet as described previously74 after 24 h and 48 h and cells were also observed under microscope.
Supplementary information
Acknowledgements
This work was funded by grant numbers G1084672 and G1145914, National Health and Medical Research Council (NHMRC), Australia. Thanks to Dr. Juan M. Tomas (Universidad de Barcelona) and Addgene for expression vector pBAD33-Gm.
Author Contributions
M.K. and J.I. designed and conceived the study. M.K. performed the experiments. M.K. and J.I. analysed data and wrote manuscript.
Data Availability
All data generated or analysed during this study are included in this published article and its Supplementary Information File.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-46318-1.
References
- 1.Nordmann P, Dortet L, Poirel L. Carbapenem resistance in Enterobacteriaceae: here is the storm! Trends Mol. Med. 2012;18:263–272. doi: 10.1016/j.molmed.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 2.Schultsz C, Geerlings S. Plasmid-mediated resistance in Enterobacteriaceae: changing landscape and implications for therapy. Drugs. 2012;72:1–16. doi: 10.2165/11597960-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 3.Yamaguchi Y, Park JH, Inouye M. Toxin-antitoxin systems in bacteria and archaea. Annu. Rev. Genet. 2011;45:61–79. doi: 10.1146/annurev-genet-110410-132412. [DOI] [PubMed] [Google Scholar]
- 4.Yarmolinsky MB. Programmed cell death in bacterial populations. Science. 1995;267:836–837. doi: 10.1126/science.7846528. [DOI] [PubMed] [Google Scholar]
- 5.Huguet KT, Gonnet M, Doublet B, Cloeckaert A. A toxin antitoxin system promotes the maintenance of the IncA/C-mobilizable Salmonella Genomic Island 1. Sci. Rep. 2016;6:32285. doi: 10.1038/srep32285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szekeres S, Dauti M, Wilde C, Mazel D, Rowe-Magnus DA. Chromosomal toxin-antitoxin loci can diminish large-scale genome reductions in the absence of selection. Mol. Microbiol. 2007;63:1588–1605. doi: 10.1111/j.1365-2958.2007.05613.x. [DOI] [PubMed] [Google Scholar]
- 7.Wozniak RA, Waldor MK. A toxin-antitoxin system promotes the maintenance of an integrative conjugative element. PLoS Genet. 2009;5:e1000439. doi: 10.1371/journal.pgen.1000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerdes K, Christensen SK, Lobner-Olesen A. Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol. 2005;3:371–382. doi: 10.1038/nrmicro1147. [DOI] [PubMed] [Google Scholar]
- 9.Gerdes K, Maisonneuve E. Bacterial persistence and toxin-antitoxin loci. Annu. Rev. Microbiol. 2012;66:103–123. doi: 10.1146/annurev-micro-092611-150159. [DOI] [PubMed] [Google Scholar]
- 10.Lobato-Marquez D, Diaz-Orejas R, Garcia-Del Portillo F. Toxin-antitoxins and bacterial virulence. FEMS Microbiol. Rev. 2016;40:592–609. doi: 10.1093/femsre/fuw022. [DOI] [PubMed] [Google Scholar]
- 11.Wang X, Wood TK. Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl. Environ. Microbiol. 2011;77:5577–5583. doi: 10.1128/AEM.05068-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, et al. Functional RelBE-family toxin-antitoxin pairs affect biofilm maturation and intestine colonization in Vibrio cholerae. PLoS One. 2015;10:e0135696. doi: 10.1371/journal.pone.0135696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balaban NQ, Gerdes K, Lewis K, McKinney JD. A problem of persistence: still more questions than answers? Nat. Rev. Microbiol. 2013;11:587–591. doi: 10.1038/nrmicro3076. [DOI] [PubMed] [Google Scholar]
- 14.Kaldalu N, Hauryliuk V, Tenson T. Persisters-as elusive as ever. Appl. Microbiol. Biotechnol. 2016;100:6545–6553. doi: 10.1007/s00253-016-7648-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harms Alexander, Maisonneuve Etienne, Gerdes Kenn. Mechanisms of bacterial persistence during stress and antibiotic exposure. Science. 2016;354(6318):aaf4268. doi: 10.1126/science.aaf4268. [DOI] [PubMed] [Google Scholar]
- 16.Fauvart M, De Groote VN, Michiels J. Role of persister cells in chronic infections: clinical relevance and perspectives on anti-persister therapies. J. Med. Microbiol. 2011;60:699–709. doi: 10.1099/jmm.0.030932-0. [DOI] [PubMed] [Google Scholar]
- 17.Levin BR, Rozen DE. Non-inherited antibiotic resistance. Nat. Rev. Microbiol. 2006;4:556–562. doi: 10.1038/nrmicro1445. [DOI] [PubMed] [Google Scholar]
- 18.Hayes F. Toxins-antitoxins: plasmid maintenance, programmed cell death, and cell cycle arrest. Science. 2003;301:1496–1499. doi: 10.1126/science.1088157. [DOI] [PubMed] [Google Scholar]
- 19.Unterholzner SJ, Poppenberger B, Rozhon W. Toxin-antitoxin systems: Biology, identification, and application. Mobile genetic elements. 2013;3:e26219. doi: 10.4161/mge.26219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loris R, Garcia-Pino A. Disorder- and dynamics-based regulatory mechanisms in toxin-antitoxin modules. Chem. Rev. 2014;114:6933–6947. doi: 10.1021/cr400656f. [DOI] [PubMed] [Google Scholar]
- 21.Kim JS, Wood TK. Persistent persister misperceptions. Front. Microbiol. 2016;7:2134. doi: 10.3389/fmicb.2016.02134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim, J. S. & Wood, T. K. Tolerant, growing cells from nutrient shifts are not persister cells. mBio8, 10.1128/mBio.00354-17 (2017). [DOI] [PMC free article] [PubMed]
- 23.Van Melderen L, Wood TK. Commentary: what is the link between stringent response, endoribonuclease encoding type II toxin-antitoxin systems and persistence? Front. Microbiol. 2017;8:191. doi: 10.3389/fmicb.2017.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goormaghtigh, F. et al. Reassessing the role of type II toxin-antitoxin systems in formation of Escherichia coli type II persister cells. mBio9, 10.1128/mBio.00640-18 (2018). [DOI] [PMC free article] [PubMed]
- 25.Harms, A., Fino, C., Sorensen, M. A., Semsey, S. & Gerdes, K. Prophages and growth dynamics confound experimental results with antibiotic-tolerant persister cells. mBio8, 10.1128/mBio.01964-17 (2017). [DOI] [PMC free article] [PubMed]
- 26.Norton JP, Mulvey MA. Toxin-antitoxin systems are important for niche-specific colonization and stress resistance of uropathogenic Escherichia coli. PLoS Pathog. 2012;8:e1002954. doi: 10.1371/journal.ppat.1002954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ronneau, S. & Helaine, S. Clarifying the link between toxin-antitoxin modules and bacterial persistence. J. Mol. Biol., 10.1016/j.jmb.2019.03.019 (2019). [DOI] [PubMed]
- 28.Fridman O, Goldberg A, Ronin I, Shoresh N, Balaban NQ. Optimization of lag time underlies antibiotic tolerance in evolved bacterial populations. Nature. 2014;513:418–421. doi: 10.1038/nature13469. [DOI] [PubMed] [Google Scholar]
- 29.Moyed HS, Bertrand KP. hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 1983;155:768–775. doi: 10.1128/jb.155.2.768-775.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keren I, Shah D, Spoering A, Kaldalu N, Lewis K. Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J. Bacteriol. 2004;186:8172–8180. doi: 10.1128/JB.186.24.8172-8180.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tripathi A, Dewan PC, Siddique SA, Varadarajan R. MazF-induced growth inhibition and persister generation in Escherichia coli. J. Biol. Chem. 2014;289:4191–4205. doi: 10.1074/jbc.M113.510511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tripathi A, Dewan PC, Barua B, Varadarajan R. Additional role for the ccd operon of F-plasmid as a transmissible persistence factor. Proc. Natl. Acad. Sci. USA. 2012;109:12497–12502. doi: 10.1073/pnas.1121217109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng HY, et al. Toxin GhoT of the GhoT/GhoS toxin/antitoxin system damages the cell membrane to reduce adenosine triphosphate and to reduce growth under stress. Environ. Microbiol. 2014;16:1741–1754. doi: 10.1111/1462-2920.12373. [DOI] [PubMed] [Google Scholar]
- 34.Dorr T, Vulic M, Lewis K. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 2010;8:e1000317. doi: 10.1371/journal.pbio.1000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Christensen SK, Mikkelsen M, Pedersen K, Gerdes K. RelE, a global inhibitor of translation, is activated during nutritional stress. Proc. Natl. Acad. Sci. USA. 2001;98:14328–14333. doi: 10.1073/pnas.251327898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roberts RC, Strom AR, Helinski DR. The parDE operon of the broad-host-range plasmid RK2 specifies growth inhibition associated with plasmid loss. J. Mol. Biol. 1994;237:35–51. doi: 10.1006/jmbi.1994.1207. [DOI] [PubMed] [Google Scholar]
- 37.Jiang Y, Pogliano J, Helinski DR, Konieczny I. ParE toxin encoded by the broad-host-range plasmid RK2 is an inhibitor of Escherichia coli gyrase. Mol. Microbiol. 2002;44:971–979. doi: 10.1046/j.1365-2958.2002.02921.x. [DOI] [PubMed] [Google Scholar]
- 38.Christensen SK, Gerdes K. RelE toxins from bacteria and Archaea cleave mRNAs on translating ribosomes, which are rescued by tmRNA. Mol. Microbiol. 2003;48:1389–1400. doi: 10.1046/j.1365-2958.2003.03512.x. [DOI] [PubMed] [Google Scholar]
- 39.Neubauer C, et al. The structural basis for mRNA recognition and cleavage by the ribosome-dependent endonuclease RelE. Cell. 2009;139:1084–1095. doi: 10.1016/j.cell.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pedersen K, et al. The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell. 2003;112:131–140. doi: 10.1016/S0092-8674(02)01248-5. [DOI] [PubMed] [Google Scholar]
- 41.Dalton KM, Crosson S. A conserved mode of protein recognition and binding in a ParD-ParE toxin-antitoxin complex. Biochemistry. 2010;49:2205–2215. doi: 10.1021/bi902133s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Solovyev, V. & Salamov, A. In Metagenomics and its Applications in Agriculture, Biomedicine and Environmental Studies (ed Li, R. W.) 61–78 (Nova Science Publishers, 2011).
- 43.Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jimenez N, et al. Genetics and proteomics of Aeromonas salmonicida lipopolysaccharide core biosynthesis. J. Bacteriol. 2009;191:2228–2236. doi: 10.1128/JB.01395-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fiebig A, Castro Rojas CM, Siegal-Gaskins D, Crosson S. Interaction specificity, toxicity and regulation of a paralogous set of ParE/RelE-family toxin-antitoxin systems. Mol. Microbiol. 2010;77:236–251. doi: 10.1111/j.1365-2958.2010.07207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yuan J, Yamaichi Y, Waldor MK. The three Vibrio cholerae chromosome II-encoded ParE toxins degrade chromosome I following loss of chromosome II. J. Bacteriol. 2011;193:611–619. doi: 10.1128/JB.01185-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tagg KA, Iredell JR, Partridge SR. Complete sequencing of IncI1 sequence type 2 plasmid pJIE512b indicates mobilization of blaCMY-2 from an IncA/C plasmid. Antimicrob. Agents Chemother. 2014;58:4949–4952. doi: 10.1128/AAC.02773-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kamruzzaman M, Shoma S, Thomas CM, Partridge SR, Iredell JR. Plasmid interference for curing antibiotic resistance plasmids in vivo. PLoS One. 2017;12:e0172913. doi: 10.1371/journal.pone.0172913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carattoli A. Resistance plasmid families in. Enterobacteriaceae. Antimicrob. Agents Chemother. 2009;53:2227–2238. doi: 10.1128/AAC.01707-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keren I, Kaldalu N, Spoering A, Wang Y, Lewis K. Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 2004;230:13–18. doi: 10.1016/S0378-1097(03)00856-5. [DOI] [PubMed] [Google Scholar]
- 51.Maisonneuve E, Shakespeare LJ, Jorgensen MG, Gerdes K. Bacterial persistence by RNA endonucleases. Proc. Natl. Acad. Sci. USA. 2011;108:13206–13211. doi: 10.1073/pnas.1100186108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Verstraeten N, et al. Obg and membrane depolarization are part of a microbial bet-hedging strategy that leads to antibiotic tolerance. Mol. Cell. 2015;59:9–21. doi: 10.1016/j.molcel.2015.05.011. [DOI] [PubMed] [Google Scholar]
- 53.Landini P. Cross-talk mechanisms in biofilm formation and responses to environmental and physiological stress in Escherichia coli. Res. Microbiol. 2009;160:259–266. doi: 10.1016/j.resmic.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 54.Ramisetty BC, Ghosh D, Roy Chowdhury M, Santhosh RS. What is the link between stringent response, endoribonuclease encoding type II toxin-antitoxin systems and persistence? Front. Microbiol. 2016;7:1882. doi: 10.3389/fmicb.2016.01882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.French GL. Bactericidal agents in the treatment of MRSA infections–the potential role of daptomycin. J. Antimicrob. Chemother. 2006;58:1107–1117. doi: 10.1093/jac/dkl393. [DOI] [PubMed] [Google Scholar]
- 56.Fadlallah SM, Rahal EA, Sabra A, Kissoyan KA, Matar GM. Effect of rifampicin and gentamicin on Shiga toxin 2 expression level and the SOS response in Escherichia coli O104:H4. Foodborne Pathog. Dis. 2015;12:47–55. doi: 10.1089/fpd.2014.1824. [DOI] [PubMed] [Google Scholar]
- 57.Recacha, E. et al. Quinolone resistance reversion by targeting the SOS response. mBio8, 10.1128/mBio.00971-17 (2017). [DOI] [PMC free article] [PubMed]
- 58.Foley SL, et al. Population dynamics of Salmonella enterica serotypes in commercial egg and poultry production. Appl. Environ. Microbiol. 2011;77:4273–4279. doi: 10.1128/AEM.00598-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jarvis N, et al. An overview of Salmonella thermal destruction during food processing and preparation. Food Control. 2016;68:280–290. doi: 10.1016/j.foodcont.2016.04.006. [DOI] [Google Scholar]
- 60.Zhang L, Singh P, Lee HC, Kang I. Effect of hot water spray on broiler carcasses for reduction of loosely attached, intermediately attached, and tightly attached pathogenic (Salmonella and Campylobacter) and mesophilic aerobic bacteria. Poult. Sci. 2013;92:804–810. doi: 10.3382/ps.2012-02504. [DOI] [PubMed] [Google Scholar]
- 61.Kim Y, Wang X, Ma Q, Zhang XS, Wood TK. Toxin-antitoxin systems in Escherichia coli influence biofilm formation through YjgK (TabA) and fimbriae. J. Bacteriol. 2009;191:1258–1267. doi: 10.1128/JB.01465-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun C, et al. MqsR/MqsA Toxin/Antitoxin system regulates persistence and biofilm formation in Pseudomonas putida KT2440. Front Microbiol. 2017;8:840. doi: 10.3389/fmicb.2017.00840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bernier SP, et al. Starvation, together with the SOS response, mediates high biofilm-specific tolerance to the fluoroquinolone ofloxacin. PLoS Genet. 2013;9:e1003144. doi: 10.1371/journal.pgen.1003144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mah TF, O’Toole GA. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 2001;9:34–39. doi: 10.1016/S0966-842X(00)01913-2. [DOI] [PubMed] [Google Scholar]
- 65.Muranaka LS, Takita MA, Olivato JC, Kishi LT, de Souza AA. Global expression profile of biofilm resistance to antimicrobial compounds in the plant-pathogenic bacterium Xylella fastidiosa reveals evidence of persister cells. J. Bacteriol. 2012;194:4561–4569. doi: 10.1128/JB.00436-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.de la Cruz F, Grinsted J. Genetic and molecular characterization of Tn21, a multiple resistance transposon from R100.1. J. Bacteriol. 1982;151:222–228. doi: 10.1128/jb.151.1.222-228.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Coetzee JN, Datta N, Hedges RW. R factors from Proteus rettgeri. J. Gen. Microbiol. 1972;72:543–552. doi: 10.1099/00221287-72-3-543. [DOI] [PubMed] [Google Scholar]
- 68.Kumar S, Stecher G, Tamura K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 70.Murphy KC, Campellone KG. Lambda Red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol. Biol. 2003;4:11. doi: 10.1186/1471-2199-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Deane SM, Rawlings DE. Plasmid evolution and interaction between the plasmid addiction stability systems of two related broad-host-range IncQ-like plasmids. J. Bacteriol. 2004;186:2123–2133. doi: 10.1128/JB.186.7.2123-2133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kamruzzaman M, et al. Relative strengths of promoters provided by common mobile genetic elements associated with resistance gene expression in Gram-negative bacteria. Antimicrob. Agents Chemother. 2015;59:5088–5091. doi: 10.1128/AAC.00420-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔCT) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 74.Zhu J, et al. Quorum-sensing regulators control virulence gene expression in Vibrio cholerae. Proc. Natl. Acad. Sci. USA. 2002;99:3129–3134. doi: 10.1073/pnas.052694299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analysed during this study are included in this published article and its Supplementary Information File.