

Abstract
Gasotransmitters are endogenous small gaseous messengers exemplified by nitric oxide (NO), carbon monoxide (CO), and hydrogen sulfide (H2S or sulfide). Gasotransmitters are implicated in myriad physiologic functions including many aspects of reproduction. Our objective was to comprehensively review basic mechanisms and functions of gasotransmitters during pregnancy from conception to uterine involution and highlight future research opportunities. We searched PubMed and Web of Science databases using combinations of keywords nitric oxide, carbon monoxide, sulfide, placenta, uterus, labor, and pregnancy. We included English language publications on human and animal studies from any date through August 2018 and retained basic and translational articles with relevant original findings. All gasotransmitters activate cGMP signaling. NO and sulfide also covalently modify target protein cysteines. Protein kinases and ion channels transduce gasotransmitter signals, and co-expressed gasotransmitters can be synergistic or antagonistic depending on cell type. Gasotransmitters influence tubal transit, placentation, cervical remodeling, and myometrial contractility. NO, CO, and sulfide dilate resistance vessels, suppress inflammation, and relax myometrium to promote uterine quiescence and normal placentation. Cervical remodeling and rupture of fetal membranes coincide with enhanced oxidation and altered gasotransmitter metabolism. Mechanisms mediating cellular and organismal changes in pregnancy due to gasotransmitters are largely unknown. Altered gasotransmitter signaling has been reported for preeclampsia, intrauterine growth restriction, premature rupture of membranes, and preterm labor. However, in most cases specific molecular changes are not yet characterized. Nonclassical signaling pathways and the crosstalk among gasotransmitters are emerging investigation topics.
Keywords: gasotransmitter, nitric oxide (NO), nitric oxide synthase (NOS), carbon monoxide (CO), heme oxygenase (HO), hydrogen sulfide (H2S), cystathionine-β-synthase (CBS), cystathionine-γ-lyase (CSE), 3-mercaptosulfurtransferase (3-MST), pregnancy, decidua, maternal–fetal interface, extravillous trophoblast (EVT), placenta, paraventricular nucleus (PVN), myometrium, calcium-gated potassium channel (BKCa), ATP-gated potassium channel (KATP), parturition, uterus
Gasotransmitters modulate mammalian pregnancy via conserved ion channels, receptors, and second messenger-mediated signal transduction pathways.
Nitric oxide (NO), carbon monoxide (CO), and hydrogen sulfide (H2S; hereafter referred to as “sulfide”) are volatile autocrine and paracrine gaseous signaling compounds known as “gaso-transmitters” [1–3]. Gasotransmitters participate in myriad aspects of pregnancy, and signaling can vary by gestational age. Genetic or pharmacologic impairment of gasotransmitter biosynthesis adversely impacts placentation [4, 5], maternal- and feto-placental hemodynamic adaptations of pregnancy [6, 7], the timing of parturition [8–10], and long-term neonatal outcomes [11].
The purpose of this review is threefold:
First, to survey gasotransmitter molecular and cellular biochemistry.
Second, to evaluate reports of NO, CO, and sulfide in pregnancy from embryo fallopian tube transit though postpartum uterine involution.
Third, to discuss opportunities for future gasotransmitter research in pregnancy.
Defogger: an overview of gasotransmitter biochemistry and signaling
The consensus among biochemists a century ago was that NO, CO, and sulfide were environmental toxins with no physiologic functions, although they were useful as reagents and indicators. For example, the odor of sulfide indicated bacterial contamination in the early days of food quality control [12]; the reaction of NO and methemoglobin established early pH testing [13], and Archibald Hill exploited CO’s hemoglobin affinity to establish enzymatic cooperativity [14]. The first discussion of endogenous biosynthesis of NO, CO, and sulfide emerged in the 1940s when the Fromageot and Smythe labs discovered a cysteine-dependent sulfide synthase [15, 16]. Sjöstrand [17] later observed heme degradation-dependent CO production in human erythrocytes, which inspired Tenhunen and Schmid's characterization of heme oxygenase (HO) in 1969 [18]. Research establishing the second messenger paradigm in the 1950s and 1960s, exemplified by 3′,5′-cyclic adenosine monophosphate (cAMP) [19, 20], then facilitated the seminal discoveries of gasotransmitter signal transduction.
In the 1970s, Furchgott and Zawadzki first postulated the existence of endothelium-derived relaxing factor (EDRF) mediating acetylcholine vasodilation [21]. By 1977, Murad's lab reported that NO stimulates soluble guanylate cyclase (sGC) to produce 3′,5′-cyclic guanosine monophosphate (cGMP) [22]. Ignarro and Chaudhuri confirmed that EDRF is NO [23], and Moncada's group published the biosynthetic route from L-arginine to nitric oxide via NO synthase (NOS) in 1988 [24]. These pivotal discoveries defined the so-called classical NO pathway by which NO activates sGC to make cGMP, causing vasodilation. Subsequently, the Ulrich and McGrath groups reported CO-dependent vasodilation via classical sGC-cGMP signaling [25, 26], and Snyder's lab showed that brain cGMP correlates with HO-2 transcription [27]. Sulfide research resumed in the 1990s with Abe and Kimura's observation of sulfide synthesis in rat brain [28], and Wang's group demonstrated that sulfide is also a vasodilator [29, 30]. These discoveries established the signaling family of gasotransmitters and their general transduction pathways.
Nitric oxide
NO is a nonpolar, selectively reactive gas with an unpaired electron. It reacts with ferrous (Fe2+) hemoglobin, molecular oxygen (O2), and superoxide (O2−), respectively, to promote the three established NO signal transduction mechanisms: classical sGC-cGMP signaling, S-nitrosation, and the peroxynitrite (ONOO−) cytotoxic pathway (Figure 1A) [31].
Figure 1.

Nitric oxide metabolism and regulation. (A) Intermediates, enzymes (bold, italics), and biochemical effects (shaded boxes) of classical, S-nitrosothiol, and cytotoxic NO signaling. Gray text and arrows indicate excretion pathways. Arg: arginine. Cit: citrulline. GR: GSH reductase. SM: smooth muscle. (B–D) Transcriptional and post-translational regulation of neuronal (B), inducible (C), and endothelial (D) nitric oxide synthase (NOS) isoforms. Kinases, phosphoregulatory sites, and inhibitory ubiquitin ligases are shown. Green +’s and red X’s indicate positive and negative regulation, respectively.
In the classical pathway, NO’s unpaired electron attacks the Fe2+ heme of sGC, inducing increased cGMP synthesis. cGMP activates protein kinase G (PKG), which phosphorylates targets including myosin light chain phosphatase (MLCP; activating phosphorylation) and voltage-gated calcium (Ca2+) channels (Cav; inactivating phosphorylation). Cav closure reduces cytosolic Ca2+ entry, thereby curtailing Ca2+ binding to calmodulin (CaM) and reducing CaM-dependent myosin light chain kinase (MLCK) activity to promote smooth muscle relaxation [32].
S-nitrosation is the addition of a nitroso group (—NO) to a cysteine thiol (—SH) resulting in an S-nitrosothiol (SNO). Models suggest that cysteine S-nitrosation is indirect [33]. NO and O2 undergo radical–radical coupling to produce nitroso-oxide intermediates that rearrange to nitrous anhydride (N2O3). Subsequently, glutathione's (GSH) thiol group nucleophilically attacks N2O3 to produce nitrite (NO2−) and S-nitrosoglutathione (GSNO), which is the primary agent of S-nitrosation [34]. GSH-independent S-nitrosation has been detected in bacteria [35], but a role in mammals is uncertain. S-nitrosation protects thiols from oxidation and can thereby alter cysteine-dependent enzyme activity, although nitrosation is vulnerable to reducing agents [33, 36, 37]. Mass spectrometry has identified thousands of nitrosated proteins [38, 39], but the biophysical basis for selective cysteine modification is unknown [40]. Sulfide and SNO react to form nitrosopersulfide (ONSS−), which enhances NO-dependent cGMP production by an unknown mechanism [41, 42].
In the cytotoxic pathway, NO and O2− radicals form ONOO−, a strong oxidant that damages DNA and promotes inflammation [43]. All nucleated cells can produce ONOO− in response to environmental oxidants, infection, or inflammatory mediators [44], but macrophages specifically use cytotoxic ONOO− to attack microorganisms [45]. NO and O2− must accumulate to produce ONOO− [46]. Therefore, both downregulation of the O2−-degrading enzyme superoxide dismutase (SOD) and upregulation of NO synthesis favor the cytotoxic pathway [47].
Living systems produce NO via arginine oxidation and nitrate (NO3−) reduction [48]. Arginine oxidation to NO and citrulline requires NOS enzymes, which are homodimers containing N-terminal oxidase and C-terminal reductase domains. A salvage pathway also exists through which NO3− is reduced to NO2− and then NO [48]. The purine catabolic enzyme xanthine oxidoreductase (XOR) can reduce NO3− to NO [49]; however, the importance of XOR for endogenous NO synthesis is not known.
There are three NOS isoforms: neuronal (nNOS or NOS1), inducible (iNOS or NOS2), and endothelial (eNOS or NOS3) (Figure 1B–D). All NOSs require the same cofactors (O2, Fe2+-heme, flavins, NADPH, HSP90, tetrahydrobiopterin, and Ca2+-bound CaM [50]), but each NOS has distinct tissue expression, regulation, and function. Interaction with the membrane protein PSD95 and N-terminal acylation enable membrane localization of nNOS and iNOS/eNOS, respectively [51]. Neuronal NOS and eNOS are constitutively expressed (nNOS in neurons and skeletal muscle; eNOS in endothelium, myocardium, and syncytiotrophoblast [STB]). In contrast, iNOS expression requires inflammatory signaling [52–56]. The cellular redox state is important for all three NOSs, as oxidative stress can “uncouple” the flow of electrons to produce O2− and promote the cytotoxic pathway [57]. Although nNOS, iNOS, and eNOS each require CaM, the EC50 for CaM activation of nNOS/eNOS is 100–300 nM Ca2+, whereas iNOS is essentially Ca2+independent [58, 59]. Because resting intracellular [Ca2+] is 10–100 nM [60], full nNOS/eNOS activation requires external Ca2+ entry via membrane channels (CaV) or internal release from endoplasmic reticulum stores. Phosphorylation and ubiquitination also regulate NOS activity. Phosphorylation at Akt/PKA consensus sites (S1417 of nNOS and S1177 of eNOS) reduces nNOS/eNOS Ca2+ dependence [61, 62]. Conversely, phosphorylation by CaMKII (S852 of nNOS) and PKC/AMPK (T495 of eNOS) diminishes NOS activity even at maximal Ca2+ concentrations [63–65]. The tyrosine kinase Src decreases iNOS activity via Y1055 phosphorylation, and the ubiquitin ligases ECS-SPSB and SCFFBXO45 facilitate iNOS and eNOS/nNOS degradation [66–69]. Compared with synthesis and signaling, NO diffusion and catabolism have received less attention. NO binds reversibly to circulating hemoglobin [70], so NO carried by red blood cells may exert endocrine actions in addition to autocrine and paracrine effects. Inhibition of the cGMP-degrading enzyme phosphodiesterase-5 (PDE5) by drugs like sildenafil (Viagra®) enhances penile erection [32], underscoring the importance of the classical pathway in vascular biology. NO spontaneously oxidizes to NO3−, which is eliminated in urine. GSNO reductase (GSNOR) couples GSH oxidation to SNO reduction [71], and S-nitrosation decreases GSNOR activity [72].
Carbon monoxide
Carbon monoxide (CO) is a relatively nonpolar, chemically stable gas composed of carbon triple bonded to oxygen. The two known CO signaling mechanisms are the classical (cGMP) pathway and a cGMP-independent pathway (Figure 2A). Classical CO signaling is mechanistically identical to classical NO signaling: CO activates sGC to increase cGMP stimulation of PKG. CO and NO bind sGC with similar affinity, and both elicit smooth muscle relaxation. However, NO-sGC is 25–50 times more active than CO-sGC [73]. Hence, in some circumstances CO competes with NO and can attenuate NO-mediated cGMP production [74]. In the cGMP-independent CO pathway, CO directly modulates protein function. CO binds heme groups of Ca2+-gated large conductance K+ channels (BKCa), which increases BKCa channel opening [75, 76]. CO also inhibits K+ inward rectifier channels (Kir) by an unidentified cGMP-independent mechanism [77]. Organometallic compounds (e.g. heme) probably mediate nonclassical CO signaling; CO is not very reactive in the absence of transition metals except at extreme temperature and pressure [78]. CO cannot covalently modify proteins. Thus, cGMP-independent NO and CO pathways do not directly interact or compete, and it is not known if the cGMP-independent NO and CO signals are physiologically additive or antagonistic.
Figure 2.
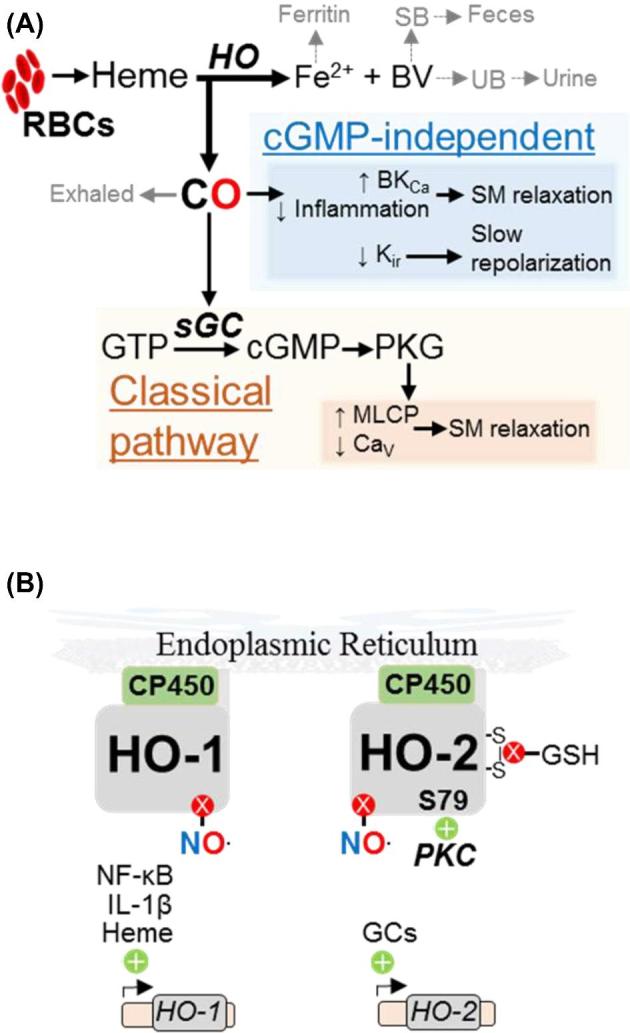
Carbon monoxide metabolism and regulation. (A) Intermediates, enzymes (bold, italics), and biochemical effects (shaded boxes) of classical and cGMP-independent CO signaling. RBCs: red blood cells, (B) Transcriptional and post-translational regulation of HO-1 and HO-2.
Two homodimeric heme oxygenases, HO-1 and HO-2 (Figure 2B), synthesize CO. HO-1 and HO-2 localize to the endoplasmic reticulum and require O2 and NADPH to oxidize Fe2+-heme to CO, biliverdin IX-α (BV), and free Fe2+. HO-1 and HO-2 associate with cytochrome P450 reductase (CP450), which reduces ferric (Fe3+) heme from red blood cells to the HO substrate Fe2+ heme [79]. The HO reaction producing CO is the first step of porphyrin degradation [18]. NO inhibits both HO enzymes [80, 81], suggesting complex interactions between NO and CO.
Due to differences in protein structure and tissue expression, HO-1 and HO-2 are not redundant. Like iNOS, HO-1 is expressed during inflammation and oxidative stress. HSF1, NF-kB, HIF-1α, and heme upregulate HO-1 transcription and translation [82], and HO-1 in turn upregulates anti-inflammatory cytokines such as IL-10 [83]. HO-2 is constitutively expressed by neurons, glia, vascular endothelium, and endometrium [84]. Glucocorticoids stimulate HO-2 transcription, and post-translational modification activates (e.g. PKC serine-79 phosphorylation) or inhibits (e.g. GSH reduction of intramolecular disulfides) HO-2 activity [85, 86].
Excretion of HO products involves multiple organ systems. Endogenous CO, chemically stable and too dilute to influence O2 transport, is exhaled during respiration [87]. Intracellular ferritin sequesters Fe2+ for use in iron-containing proteins. Porphyrin degradation enzymes process BV to urobilin and stercobilin, which are eliminated in the urine and feces, respectively [88].
Sulfide
In living systems, sulfide is a mixture of polar hydrogen sulfide gas (H2S), hydrogen sulfide anion (HS−), and nonpolar polysulfide (H2Sn). The [H2S]:[HS-] ratio approaches unity in vivo, and H2S spontaneously oxidizes to H2Sn. Because these are all bioactive and difficult to distinguish [89], they are collectively referred to as “sulfide.” Sulfide signals through cysteine persulfidation (Cys-SH + sulfide → Cys-SSH, also called sulfhydration) and by transactivation of classical cGMP signaling via 8-HS-cGMP (Figure 3A). Persulfidation and S-nitrosation sometimes compete at target cysteines that alter enzyme activity [90, 91]. NFκB persulfidation reduces TNFα-stimulated apoptosis [90], while ATP-gated K+ (KATP) and BKCa channel persulfidation hyperpolarizes cell membranes [92]. 8-HS-cGMP forms by persulfidation of 8-nitro-cGMP, a cGMP derivative that promotes autophagy and oncogenesis [93]. Compared with cGMP, 8-HS-cGMP resists degradation by PDE5. As such, 8-HS-cGMP augments cGMP signaling [3]. Recent reports suggest PDE5 inhibition contributes to sulfide-dependent smooth muscle relaxation [94, 95].
Figure 3.
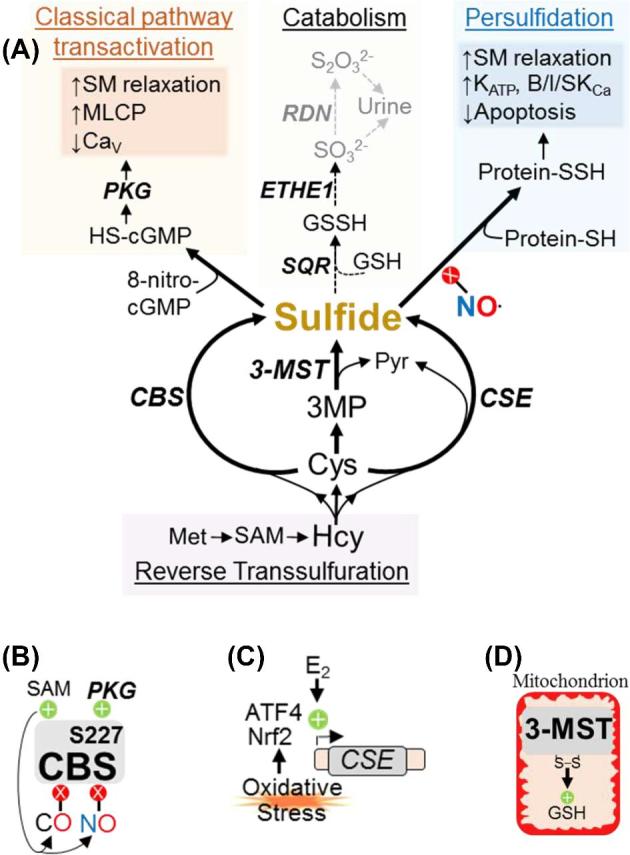
Sulfide metabolism and regulation. (A) Intermediates, enzymes (bold, italics), and biochemical effects (shaded boxes) of classical and persulfide-based sulfide signaling. B/I/SKCa: Ca2+-gated large, intermediate, and small conductance K+ channels. GSSH: GSH persulfide. Protein-SSH: Proteins with persulfidated cysteine residues. ROS: reactive oxygen species. (B–D) Transcriptional and post-translational regulation of CBS (B), CSE (C), and 3-MST (D).
Three enzymes synthesize sulfide by cysteine oxidation: cystathionine-β-synthase (CBS) and cystathionine-γ-lyase (CSE) which are primarily cytosolic, and 3-mercaptosulfurtransferase (3-MST) which is mitochondrial (Figure 3B–D) [89]. CBS and CSE can produce sulfide from numerous sulfur-containing amino acids, but cysteine and homocysteine (Hcy) are preferred substrates [96]. CBS is predominant in brain and kidney, whereas CSE is more abundant in liver and blood vessels [97]. CBS and CSE are also widely expressed as key enzymes in the reverse transsulfuration (RTS) pathway by which methionine (Met) is recycled to cysteine. 3-MST generates sulfide from 3-mercaptopyruvate (3-MP), a product of cysteine deamination. Expressed in all cell types, 3-MST is most abundant in liver, kidney, and brain [98].
Sulfide biosynthetic enzymes are subject to transcriptional and post-translational regulation. Oxidative stress stimulates ATF4- and Nrf2-dependent CSE transcription [99, 100], and estrogen (E2) promotes CSE activity in human osteoblasts and mouse liver and vasculature [101, 102]. Multiple allosteric mechanisms regulate CBS activity. CBS binds S-adenosyl methionine (SAM) with high affinity, increasing CBS activity and exposing an NO/CO sensitive inhibitory heme moiety [103, 104]. On the other hand, PKG phosphorylation of CBS S227 increases CBS activity [105]. These apparently opposing mechanisms suggest that CBS activity is maintained within an ideal range; high NO inhibits CBS to prevent overactivation of the classical pathway, while CBS phosphorylation enables sulfide synthesis to resume when NO decreases. Rat liver CBS translocates to mitochondria during hypoxia [106], which could bolster ATP production and prevent apoptosis by supplying sulfide as an alternate electron donor. CSE also translocates to mitochondria during hypoxia [107], and 3-MST may be an oxidant sensor because 3-MST cysteine oxidation increases mitochondrial GSH levels [108].
Sulfide catabolism requires a trifecta of mitochondrial enzymes. Sulfide-quinone reductase (SQR), sulfide dioxygenase (ETHE1), and rhodanese (RDN) utilize a soluble thiol (likely GSH) to oxidize sulfide to sulfite (SO32−) and thiosulfate (S2O32−), which are excreted in urine [109–111]. Since SQR, ETHE1, and RDN1 localize to mitochondria, alternate pathways exist in cells with few mitochondria. For example, erythrocytes couple methemoglobin reduction to sulfide oxidation [112].
Key points
NO, CO, and sulfide are gaseous paracrine and autocrine signaling molecules.
NO is produced by arginine oxidation or NO3− reduction. The NO metabolites GSNO and ONOO− mediate S-nitrosation and cytotoxicity, respectively.
CO is made via heme oxidation.
Sulfide is generated by cysteine oxidation. Sulfide augments the classical cGMP pathway and increases target protein activity by thiol persulfidation.
All three gasotransmitters activate the classical sGC-cGMP-PKG pathway. All three can modify ion channels by covalent modification or heme binding.
Gasotransmitter crosstalk
As discussed above, reactions among gas molecules can alter signaling (e.g. O2− and NO produce ONOO–). Gasotransmitters can also regulate each other's production or activity (e.g. CO reduces NO stimulated sGC activity) or metabolism (e.g. sulfide prolongs NO signals by inhibiting PDE5 activity). Other interactions likely exist among gasotransmitter metabolic pathways (Figure 4).
Figure 4.
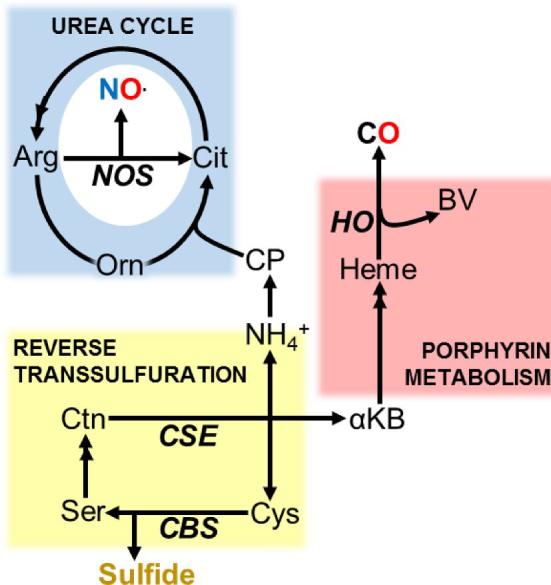
Integration of gasotransmitter metabolism. Sulfide synthesis via reverse transsulfuration generates NH4+ and αKB, which can produce NO and CO via the urea cycle and heme metabolism, respectively. αKB: α-ketobutyrate. CP: carbamoyl phosphate. Ctn: Cystathionine. Orn: ornithine. Ser: Serine. Double arrowheads indicate pathways in which multiple intermediates have been omitted for clarity
Having reviewed gasotransmitter biochemistry, in the next section we turn to specific roles of NO, CO, and sulfide in pregnancy.
Fueling pregnancy: gasotransmitters in conception, placentation, and vascular adaptation
Fallopian tube transit, decidualization, and implantation
Gasotransmitters and their biosynthetic enzymes change as the nascent conceptus travels to the uterus (Figure 5A). Women exhale twice as much CO in the luteal phase compared to the follicular phase [113]. The reason for this change is unknown but may reflect regulation of tubal transit or early systemic adaptations of pregnancy. In cattle, endothelins induce iNOS accumulation in the ampulla [114]. Human fallopian tubes express CBS, which produces sulfide to relax fallopian tube smooth muscle [115]. In mice, CBS inhibition causes morula retention in the tube. This suggests NO and sulfide can counteract procontractile endothelins to facilitate tubal peristalsis and embryo transit.
Figure 5.

Gasotransmitters regulate fallopian tube transit, decidualization, implantation, and placentation. (A) Fallopian tube transit. ET1 from the fallopian endothelium upregulates iNOS in the fallopian ampulla. Deficiencies in CBS hinder transit of fertilized embryos. Depending on its concentration, NO stimulates or inhibits pregnancy-sustaining progesterone (P4) by the corpus luteum (CL). (B) Decidualization and implantation. HO-1 drives proliferation of ectoplacental cone (EPC) cells. Decidual eNOS, nNOS, iNOS, and SOD1 increase in abundance as the endometrium decidualizes, and NOS activity facilitates implantation. ICM: inner cell mass, GC: Giant cells. (C) Developmental changes at the maternal-fetal interface (MFI). During pregnancy, CO facilitates remodeling of maternal spiral arteries (red circle) into low-resistance, high-flow canals (red patch) surrounding the placental villi (yellow, green, and black branched structure). Endothelial NOS potentiates placental amino acid transport from maternal circulation (red) to fetal circulation (black). On the fetal side, eNOS, both HO isoforms, CBS, and CSE facilitate dilation and angiogenesis of fetal vasculature (black). AAs: amino acids. SA: spiral arteries. Green circles and yellow layers represent cytotrophoblasts and the syncytiotrophoblast, respectively. (D) EVT motility. Wnt and PI3K pathways both activate iNOS, which inhibits EVT apoptosis and promotes cell motility. HO-1 and PPARγ inhibit EVT motility. Green +’s and red X’s denote up- and downregulation, respectively.
Decidualization is endometrial maturation under the influence of progesterone (P4) secreted by the corpus luteum (Figure 5B). It includes mesenchymal-to-epithelial transdifferentiation of endometrial fibroblasts into secretory stromal cells, invasion of leukocytes that assist tissue reorganization, and capillary angiogenesis to potentiate implantation [116]. Human decidua expresses both eNOS and iNOS in the vascular endothelium and glandular epithelium [117]. In nonhuman primates, decidualization enhances endometrial nNOS, iNOS, and eNOS expression [118], while in mice only iNOS and eNOS accumulate. In rats, the nonselective NOS inhibitor L-nitroarginine methyl ester (L-NAME) blocks decidualization [119, 120]. NO (probably from eNOS) also promotes secretory endometrial differentiation and endometrial vessel dilation to increase perfusion during early pregnancy. Additionally, first trimester decidua from women with spontaneous miscarriage shows higher prostaglandin F2α and lower Cu-Zn SOD (SOD1) than normal pregnancies [121]. This suggests SOD1 could suppress cytotoxic ONOO− production [46], preventing feed-forward synergy between rising NO and prostaglandin F2α.
The rigid glycocalyx surrounding the preimplantation embryo (the zona pellucida) is disassembled just before decidual implantation, a process called “hatching.” In mice, both NO donors and NOS inhibitors prevent hatching [122], suggesting hatching requires an optimal NO concentration. Implantation occurs when trophoblast cells of a hatched blastocyst attach to the decidualized endometrium [123] (Figure 5B). Trophoblast comprises multinucleated STB and mononucleated cytotrophoblasts (CTBs), some of which differentiate into extravillous trophoblasts (EVTs). In the preimplantation murine embryo, giant cells are analogous to human EVTs, and the ectoplacental cone is analogous to STBs and CTBs. If implantation is successful, EVT invasion extends chorionic villous STBs and CTBs deep into the decidua as the blastocyst inner cell mass undergoes gastrulation. Although giant cells and the ectoplacental cone express iNOS, and NOS inhibitors abolish implantation [119, 124], iNOS knockout mice have no pregnancy defects [125]. In contrast, conceptus-derived CO is crucial for implantation; HO-1 knockout embryos make fewer ectoplacental cone cells and implant less efficiently, a phenotype rescued by exogenous CO inhalation. In HO-1 heterozygous dams, 40% of HO-1 null fetuses die by E14, compared with only 20% of HO-1 heterozygous fetuses and 10% of wild-type fetuses [11]. CO upregulates and is upregulated by vascular endothelial growth factor (VEGF), a family of angiogenic signaling proteins [126]. However, CO likely promotes trophoblast proliferation independent from VEGF and angiogenesis since inhibition of HO enzymatic activity prevents differentiation of immature trophoblast stem cells into giant cells [11].
After ovarian follicle oocyte release, the corpus luteum forms from the remnant steroidogenic theca and granulosa cells. Luteal P4 sustains all mammalian pregnancies prior to placentation and is required for the entire pregnancy in rodents. In humans, by 10–12 weeks’ gestation the placenta replaces the corpus luteum as the main source of P4. Low concentrations of NO donors enhance luteal P4 secretion in horses, sheep, and rats, whereas high concentrations of NO donors decrease P4 [127–129]. High NO bolus doses (e.g. 0.1–1 mM) or NOS activators diminish P4 and increase apoptosis of corpora lutea obtained from eumenorrhoeic women [130, 131]. Similar to blastocyst hatching, intermediate NO levels may optimally prevent luteolysis.
Key points
Fallopian tube iNOS and CBS levels increase during embryo transit to the uterine cavity. Sulfide facilitates tubal peristalsis.
NO promotes decidualization, and eNOS increases in endometrial vascular endothelium and glandular epithelium from conception to placentation.
CO enhances trophoblast proliferation and aids embryo implantation and early survival.
Elevated NO prevents P4 synthesis and can be luteolytic in early pregnancy.
Feto-placental angiogenesis and hemodynamics
Primates, rodents, and lagomorphs exhibit hemochorial placentation, in which maternal and fetal blood are separated by placental CTB/STB and chorionic villus capillary endothelial cells (Figure 5C). Gas, nutrients, and wastes are transferred between maternal blood from spiral arteries (which become low-resistance conduits to the intervillous space after remodeling) and fetal blood in placental capillaries. Inadequate placental vascularization limits nutrient extraction (particularly O2) required for normal fetal growth. Abnormal placentation is associated with intrauterine growth restriction (IUGR), gestational diabetes, preeclampsia, and preterm birth [132].
Decidual invasion by EVTs is critical for normal pregnancy (Figure 5D), and in vitro iNOS stimulates EVT motility and invasiveness while inhibiting EVT apoptosis. Interdependent PI3K-Akt-mTORC1 and Wnt-β-catenin pathways activate EVT iNOS [133–135], and conversely iNOS and sGC inhibition inactivate Akt, destabilize β-catenin, and increase EVT apoptosis [136, 137]. The invasive EVT leading edge accumulates iNOS and matrix metalloprotease-9 (MMP9). Although MMP9 inhibition curtails EVT motility in cell culture models [138, 139], this has not been tested in vivo. Murine pregnancy does not require iNOS [125], but there is a correlation between stimuli that reduce trophoblast invasion and reduced placental iNOS and MMP9 [140]. On the other hand, HO-1 expression (and likely CO production) is inversely related to trophoblast invasiveness. By an unknown mechanism, HO-1 and PPARγ reduce EVT motility. HO-1 and PPARγ are decreased in first trimester EVTs, while overexpression or knockdown of HO-1 respectively enhances or suppresses PPARγ [141, 142], and HO-2 neutralizing antibodies inhibit CTB migration [143]. In contrast, iNOS-dependent EVT invasiveness is sensitive to CO activation of PPARγ [144].
Development of placental chorionic villi (composed of fetal vessels surrounded by CTB and STB) requires trophoblast remodeling (Figure 5C). Feto-placental angiogenesis and vasodilation occur throughout pregnancy to maintain fetal nutrient delivery; lower HO-2 in placental vascular endothelium correlates with preeclampsia and IUGR [145, 146]. Placental microvasculature is less developed in HO-1 knockout mice [147]. In rats, placenta-permeable HO inhibitors decrease placental growth factor (PLGF, a VEGF homolog [148]) and increase placental oxidative stress [149]. These effects are probably due to CO deficiency, as exogenous CO prevents HO inhibitor-induced giant cell apoptosis, and low dose inhaled CO in pregnancy rescues the HO-1 null placental structural defects [11]. Placental sulfide may also stimulate placental angiogenesis via miRNAs. Relative to term healthy placentas, CBS and CSE protein and VEGF transcript levels are lower and miRNAs 20a and 200c increase in term preeclamptic placentas. Sulfide donors rescue VEGF levels and decrease miR20a/200c and sFlt1. CBS or CSE siRNA knockdown in human placental explants also blocks sulfide donor effects [150, 151]. Further, CBS knockout dams resorb most CBS heterozygous embryos, underscoring the importance of maternal sulfide production [5]. Based on these data, CO and sulfide likely promote placental angiogenesis via PLGF and VEGF, respectively. Both VEGF [152] and villous trophoblast sulfide [153] stimulate Akt-dependent activation of eNOS, and eNOS-deficient mice exhibit placental hypoxia [4]. This suggests NO also mediates placental angiogenesis.
Along with placental angiogenesis, normal pregnancy requires regulated placental hemodynamics. NO donors dilate term human placental arteries in a sGC and BKCa-dependent manner [154]. VEGF increases eNOS expression in human umbilical vein endothelial cells and increases NO production from excised human umbilical vein [155, 156]. In IUGR placentas, NO effects are attenuated, and the proportion of phosphorylated eNOS S1177 (more active) decreases [157]. IUGR placental arteries also show eNOS promoter hypermethylation [158] and impaired Ca2+-dependent eNOS activation [159], suggesting that eNOS mediates placental NO production. Similarly, CO dilates human placental vessels via cGMP-PKG [160]. Although hypoxia does not induce HO-1 or HO-2 in term placental explants [161], HO inhibitors do reduce vasodilation and decrease placental perfusion [162–164]. Sulfide also regulates placental vascular tone. In human preeclamptic placentas, miRNA 20 suppresses CSE accumulation [165], and stem villus artery CSE loss is associated with human IUGR [166]. Placental explants subjected to hypoxia-reoxygenation show reduced actin and myosin, but sulfide donors prevent those changes [166]; thus, placental vascular sulfide may protect against IUGR. KATP channel and NOS may synergize to mediate placental sulfide signaling since glibenclamide (a KATP channel blocker) and L-NAME both block sulfide donor-dependent placental artery vasodilation [165]. Sulfide can augment classical signaling via PDE5-resistant 8-nitro-cGMP [3, 94, 95] and ONSS− stimulation of cGMP synthesis [41, 42]. These data show that gasotransmitters have both acute (vasodilation) and chronic (angiogenesis and cytoskeletal remodeling) effects on placental vasculature.
Key points
NO inhibits EVT apoptosis and stimulates EVT motility via Akt and Wnt pathways.
HO-1 and PPARγ reduce EVT motility.
HO enzymes and sulfide promote placental angiogenesis via PLGF and VEGF expression.
Endothelial NOS produces NO to dilate placental capillaries, and CSE produces sulfide to augment NO production and signaling and further relaxes placental vascular smooth muscle by activating KATP channels.
IUGR correlates with lower HO-1, HO-2, CBS, and CSE expression and decreased eNOS S1177 phosphorylation in placental vessels.
Maternal hemodynamics
Maternal vascular resistance decreases in pregnancy as cardiac output and blood volume increase, thus maintaining blood pressure. Central nervous system NO may regulate maternal hemodynamics. The hypothalamic paraventricular nucleus regulates autonomic sympathetic output [167, 168], which increases peripheral vascular resistance. In rats, paraventricular nNOS declines during pregnancy [169], and paraventricular expression of a dominant negative nNOS mutant increases vascular resistance [170]. Sympathetic neurotransmission also rises in pregnant women [171, 172], whereas parasympathetic output decreases [173–175]. This suggests decreased paraventricular nNOS in pregnancy disinhibits the sympathetic nervous system [169, 176] despite the overall decrease in vascular resistance during pregnancy. This is probably because of hormone-dependent increased NO production in the vasculature. Serum relaxin, a smooth muscle relaxant, and estrogen (E2) increase several hundred-fold during pregnancy [177–179]. Relaxin induces vasodilation via PI3K-Akt phosphorylation of eNOS S1177 [180]. E2 reduces paraventricular nNOS protein expression in rats [181] and increases eNOS transcription via E2 receptors (ERs) [181, 182]. E2 promotes phosphorylation of eNOS at S1177 by activating plasma membrane ERα-Gαi to stimulate a Src-PI3K-Akt pathway [183–185]. While ERα promotes activating eNOS S1177 phosphorylation, ERβ curtails inhibitory phosphorylation of eNOS T495 in pregnant bovine uterine artery endothelial cells [186]. Pregnant women express more uterine artery eNOS than age-matched nonpregnant women [187, 188], and E2 promotes uterine artery eNOS accumulation in ovariectomized sheep [189]. Moreover, eNOS knockout mice are hypertensive [190]. These studies suggest decreased paraventricular nNOS helps maintain vascular resistance to balance the increased peripheral vasodilation necessary for pregnancy (Figure 6A).
Figure 6.
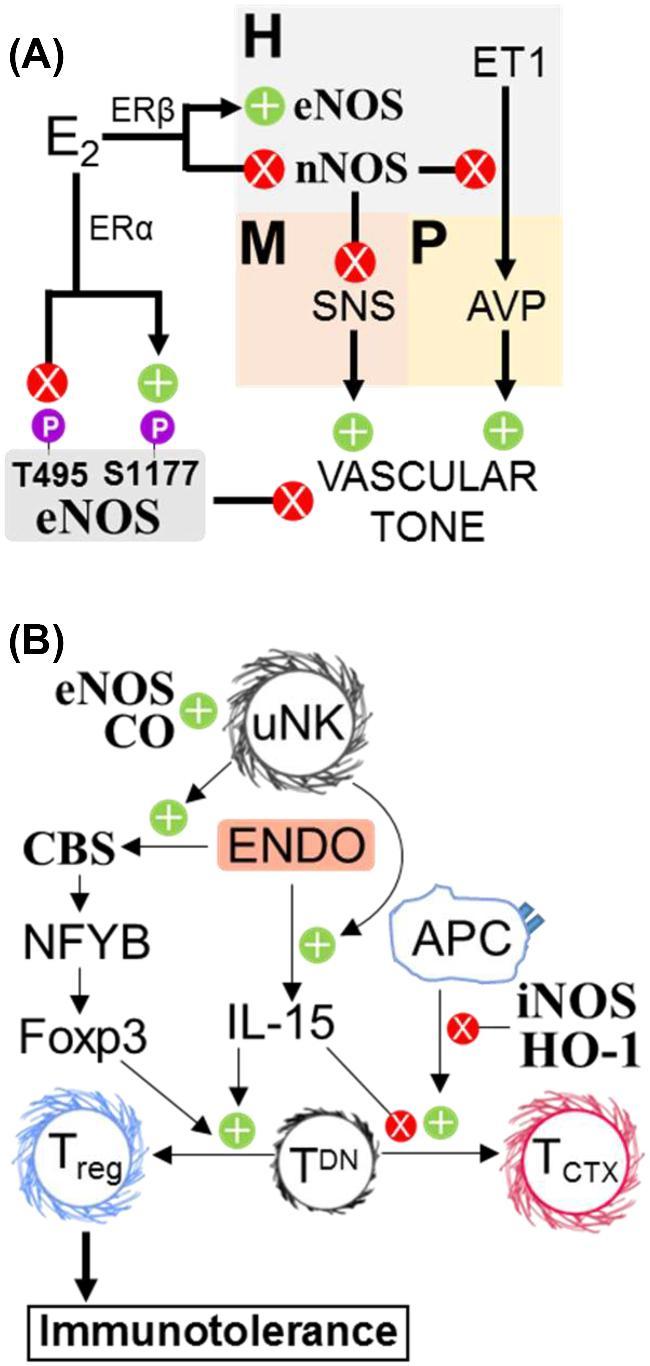
Gasotransmitters regulate maternal vascular tone and immunotolerance. (A) Prior to pregnancy, nNOS in the paraventricular nucleus of the hypothalamus (H) limits sympathetic activity in the rostral ventrolateral medulla (M) and inhibits endothelin 1 (ET1)-mediated release of vasopressin from the pituitary (P), both of which reduce peripheral vascular resistance. During pregnancy, rising E2 inhibits PVN nNOS and stimulates PVN eNOS expression via ERβ. ERα facilitates E2-mediated eNOS activation in the periphery by promoting S1177 and curtailing T495 phosphorylation. AVP: Vasopressin. (B) CO promotes recruitment of uNKs to the decidua, which enhance fetal alloimmune tolerance via endometrial IL-15 and CBS. Inducible NOS and HO-1 activity attenuate TCTX maturation, while persulfidation of NFYB enhances naïve TDN differentiation into Tregs. Endo: endometrium. TDN: double-negative (CD4− CD8−) naïve T cells.
CO and sulfide also regulate maternal vascular tone, and low respiratory and serum CO and sulfide are associated with hypertension in pregnancy. Exhaled CO is inversely correlated with gestational hypertension [191], while longer HO-1 promoters attenuate HO-1 expression and are predictive of preeclampsia [146]. In mice, HO inhibition at mid-pregnancy does not affect placental histology or fetal growth, but does increase maternal vascular resistance [192]. Therefore, while early placentation requires embryonic HO, maternal HO curtails vasoconstriction later in pregnancy. Uterine artery CBS increases during pregnancy [188], and decreased maternal serum sulfide and placental CBS and CSE protein levels correlate with preeclampsia [165, 193, 194]. CSE knockout mice exhibit reduced VEGF-dependent angiogenesis [195], and sulfide donors block sFlt1-induced hypertension in rats [196]. sFlt1 is a soluble VEGF receptor that reduces free serum VEGF, antagonizes VEGF signaling, and is linked to preeclampsia [196]. Sulfide may also regulate maternal vascular tone by directly persulfidating smooth muscle KATP channels [197, 198]. During pregnancy, KATP and CBS levels increase in human and ovine uterine artery smooth muscle cells [199, 200]. Endogenous sulfide stimulates KATP opening in rat mesenteric artery smooth muscle cells [201], and sulfide donors inhibit ATP-dependent Ca2+ entry into porcine vascular smooth muscle cells [202]. Hence, KATP persulfidation may increase uterine blood flow by antagonizing vasoconstriction.
Key points
E2 promotes maternal vascular eNOS transcription, protein accumulation, and NO production.
E2 downregulates hypothalamic nNOS protein expression in pregnancy. This increases sympathetic tone and thereby maintains blood pressure.
HO activity decreases maternal vascular resistance during pregnancy.
CSE- and CBS-produced sulfide enhance VEGF signaling in endothelial cells and KATP channel opening in vascular smooth muscle to lower vascular resistance.
Maternal angiogenesis and vascular remodeling
The maternal side of the maternal-placental interface undergoes vasodilation and angiogenesis akin to the fetal side to support adequate placental exchange. Uterine blood flow increases dramatically during pregnancy to about 20% of cardiac output by the third trimester. This is accommodated by increased myometrial radial artery angiogenesis, as well as remodeling of endometrial spiral arteries to low-resistance, high-flow canals that maximize maternal blood flow to the intervillous space (Figure 5C). Inadequate placental perfusion may result in placental ischemia and oxidative stress and promote the release of placental factors that cause preeclampsia [203]. Both NO and CO regulate uterine blood flow.
Although eNOS expression is similar in uterine artery endothelial cells from pregnant and virgin sheep, VEGF only stimulates uterine artery eNOS during pregnancy. VEGFR2 increases PI3K-dependent Ca2+ entry, which stimulates eNOS independently of ERK1/2, Akt, or eNOS S1177 phosphorylation [204, 205]. Pregnant eNOS knockout mice exhibit IUGR that is rescued by administration of an endothelin receptor A antagonist [190]. Similar to fallopian tube peristalsis, maternal uterine artery expansion could involve dynamic interplay between endothelin-dependent contraction and eNOS-dependent relaxation. Additionally, the placental basal plate expresses both HO isoforms, which influence placental perfusion [145, 162–164, 206]. HO-1 knockout mice exhibit reduced spiral artery remodeling, and human IUGR correlates with longer maternal HO-1 promoters (associated with decreased HO expression) but not with HO activity [11, 145, 146, 164]. CO inhalation (50 ppm) in early pregnancy nevertheless increases placental bed angiogenesis in HO-1 knockout mice [6, 11], so CO may drive uteroplacental vascular remodeling [149].
Spiral artery remodeling is associated with the appearance of decidual uterine natural killer cells (uNKs) in early pregnancy. The precise function of uNKs is still uncertain, but they mediate allorecognition and tolerance during placentation and also assist spiral artery remodeling [207]. They promote endometrial accumulation of VEGF-C that induces lymphangiogenesis [208, 209]. All three gasotransmitters regulate uNKs. Titers of uNKs are lower in mice with genetic deletion of eNOS, HO-1, or CBS. Decreased decidual uNKs in eNOS knockouts [4] is consistent with eNOS-dependent human NK resistance to apoptosis [210] and indicates NO autocrine or paracrine facilitation of uNK proliferation. Remarkably, early pregnancy CO inhalation (50 ppm) rescued uNK levels in pregnant HO-1 heterozygous mice and normalized the growth of HO-1 knockout conceptuses [6, 211]. CO, therefore, appears necessary for early pregnancy local immune cell changes but dispensable in late pregnancy. Uterine NKs upregulate decidual transcription of CBS and CSE [208], and early pregnant CBS knockout mice produce fewer uNKs [5]. Despite these connections, mechanisms are speculative since uNK origins are not clearly understood. While CO and NO cause uNK proliferation, CBS regulation of uNKs may be indirect.
Key points
VEGF induces eNOS to facilitate maternal uterine artery vasodilation.
HO promotes spiral artery remodeling.
CO increases uNKs in pregnancy that in turn upregulate endometrial CBS and CSE expression.
Immune adaptation
The developing conceptus is an allograft sharing only half of the maternal genes. Since maternal leukocytes can cross the placenta and enter fetal circulation [212] and may identify the placenta and fetus as foreign, maintaining maternal immune tolerance is critical for successful gestation. Pregnancy reshapes the maternal adaptive immune system (Figure 6B). Early in pregnancy, the ratio of T helper cell type-1 (Th1) cytokines (i.e. IFNγ) to Th2 cytokines (i.e. IL-4 and IL-10) decreases. Since Th2 cells transduce inflammatory signals less efficiently than Th1s [213], this shift likely contributes to maternal tolerance. In animal models, NOS and HO facilitate the Th1/Th2 shift. Lymphocyte NO synthesis increases during bovine pregnancy, and in pregnant goat serum NO3− correlates with Th2 cytokines [214, 215]. Maturation of rat naïve T cells into cytotoxic effectors (TCTX) requires downregulation of antigen-presenting cells' iNOS and HO-1 [216]. CO and sulfide also regulate T regulatory cell (Treg) suppression of TCTX maturation. HO inhibitors raise dendritic cell titers and induce embryo resorption in mice [217], but exogenous CO prevents antigen-presenting cell activation and TCTX maturation by a cGMP-independent pathway [218, 219]. Similarly, CBS- or CSE-produced sulfide persulfidates NFYB, driving Tet1/2 methylcytosine dioxygenase expression and de-repressing Foxp3 expression. Foxp3 (a forkhead transcription factor) commits CD4− CD8− progenitors to become Tregs [220]. Because CBS loss impairs murine fertility and uNK titers [5], CBS may regulate the uterine Treg niche.
Key points
Inducible NOS and HO-1 suppress naïve T-cell differentiation into TCTX.
HO-1 increases naïve T-cell differentiation to Tregs.
Sulfide increases Treg cells via NPYB persulfidation and increased Foxp3.
Rising to delivery: gasotransmitters in later pregnancy and parturition
Myometrial quiescence
The myometrium is the uterine visceral smooth muscle between the endometrium and the serosa. Endocrine signals (e.g. corticotrophin releasing hormone, P4 withdrawal) drive myometrial contractility through expression of contraction-associated proteins and altered ion channel activity. The myometrium is quiescent for about 90% of pregnancy until maternal-fetal-placental signals stimulate expression of Gq protein-coupled oxytocin receptor (OTR), prostaglandin F2α receptor, cyclooxygenase enzymes, and the gap junction protein Connexin 43 [221]. The OTR and prostaglandin F2α receptors activate phospholipase C to produce inositol triphosphate and diacylglycerol, which promote myocyte depolarization, voltage-sensitive L-type Cav channel activation, and regular intracellular Ca2+ spikes. Increased Ca2+ activates CaM-dependent myosin light chain phosphorylation and myosin-actin cross-bridge cycling to increase contractile force (Figure 7A). In contrast, relaxation occurs when the contractile phenotype has not been activated or when intracellular [Ca2+] is insufficient to activate MLCK. In addition to actin, myosin, and regulatory kinases and phosphatases, cytoskeletal accessory proteins can influence myocyte contractility. Connexin 43 increases electrochemical connections among myocytes, allowing depolarization to spread and amplify during labor [222]. About half of preterm labor leading to preterm delivery involves precocious activation of uterine contractions. To prolong pregnancy, investigators have studied mechanisms of tocolysis (the cessation of myometrial contraction). Exogenous NO and sulfide potently relax oxytocin-stimulated and spontaneous myometrial contractions [223, 224].
Figure 7.

Gasotransmitters in parturition. (A) Regulation of myometrial contractility (depicted cell is a uterine myocyte). NOS activity in myocytes or adjacent endothelium inhibits contraction by stimulating BKCa and raising GSNO levels. GSNO is tocolytic. Myocyte CBS curtails GPR109A activity, which reduces OTR signaling. CBS and/or CSE also activate KATP and potentiate an unidentified protein downstream of ClCa that induces tocolysis. Sulfide and GSNO produce ONSS−, which promotes uterine contractility. F2αR: receptor for prostaglandin F2α PLC: phospholipase C. (B) Cervical remodeling. In early pregnancy, P4 inhibits iNOS synthesis. More iNOS and less SOD1 near parturition allows ONOO− to accumulate, which stimulates prostaglandin F2α synthesis. Prostaglandin F2α in turn promotes PR-A accumulation and thus blocks P4 perception. (C) Rupture of fetal membranes. Inducible NOS activity drives formation of ONOO−, which accelerates membrane rupture. By an unknown mechanism, CBS activity promotes PGDH-dependent prostaglandin F2α degradation, which slows fetal membrane rupture.
Endothelial NOS may be the primary uterine NO source in pregnancy. In humans, E2 increases myometrial eNOS expression [225], while in mice both P4 and E2 increase uterine eNOS [226]. Human pregnant myometrium expresses vascular eNOS [225, 227] and myometrial eNOS [228, 229]. Since NO can freely diffuse between cells, localization may not be critical. Whereas rat uterine iNOS and eNOS increase until labor at E22, nNOS decreases by E18 [230, 231]. Like vascular and gastrointestinal smooth muscle, the myometrium contains autonomic (predominantly sympathetic) nerve endings. However, female puberty coincides with E2-dependent reduction of myometrial sympathetic innervation [232, 233], and uterine neuronal signals are nearly undetectable during pregnancy [234–236]. Collectively, there is little evidence for neuronal control of the myometrium, and nNOS is probably not a major quiescence mediator.
Pregnant myometrium expresses both HO isoforms as well as CBS and CSE. Myometrial explants from term laboring (TL) and term nonlaboring (TNL) pregnancy express more HO-1 and HO-2 and produce more CO than explants from nonpregnant women. Exogenous heme relaxes term human and E22 rat myometrial strips [237], but exogenous CO gas has no effect [238]. Hence, other HO products (Fe2+ or BV) are likely the tocolytic agent(s). TNL explants also express more CBS and CSE and lower levels of contraction-associated proteins (e.g. OTR) compared with TL tissue, and there is one report of endogenous cysteine-dependent relaxation of TL myometrium via CBS [239, 240]. Compared with wild-type animals, CBS+/– dams express OTR earlier and deliver earlier. Recent publications also report differential S-nitrosation of myometrial cytoskeletal proteins during pregnancy that may influence contractility [241–243].
There is some debate regarding molecular mechanisms of gasotransmitter-induced tocolysis. Classical NO signaling is insufficient to explain NO tocolysis because pharmacological inhibition of cGMP pathways does not inhibit uterine NO effects [223, 244]. Nonclassical NO and sulfide myometrial targets fall into three groups: cytoskeleton-associated proteins, ion channels, and membrane receptors. MLCK, vinculin, and galectin-1 SNOs are less abundant in preterm laboring human myometrium than in TNL tissue [244], while pregnant guinea pig myometrium exhibits increased desmin, vimentin, and transgelin SNOs relative to nonpregnant myometrium [38]. Myometrial GSNOR expression is higher in women with preterm labor than TNL [245], suggesting myometrial SNOs maintain uterine quiescence. Active site cysteine SNOs decrease the activity of numerous enzymes, including GAPDH [246], SIRT1 [247], and PDK1 [248], but effects of noncatalytic cysteine SNOs on actin-myosin accessory proteins are complex. S-nitrosation decreases skeletal muscle myosin cross-bridge cycling velocity and increases myosin stall force [249], and a cofilin-1 SNO depolymerizes actin fibers [250]. Myometrial protein SNOs correlate with pregnancy status, but their relationship to contractility is uncertain. ONSS− promotes contraction of rat myometrium despite relaxing blood vessels [42]. This may be due to unique cell-specific signaling mechanisms in vascular and myometrial smooth muscle. Altered protein expression in the two muscle types has been described; for example, α-actin and vimentin contents are higher in vascular muscle compared to visceral muscle [251]. Whatever the underlying mechanism, there are clear difficulties with directly extrapolating vascular findings to uterine myometrial function.
Uterine smooth muscle BKCa and KATP channels are important mediators of uterine quiescence [252, 253]. Contractile stimuli induce membrane depolarization [254] that promotes opening of voltage-sensitive Nav and Cav channels. As Ca2+-CaM dependent contraction proceeds, Ca2+ activates BKCa to increase K+ outflow, hyperpolarize the membrane, and close Cav channels. Ca2+ pumps reduce cytosolic [Ca2+], and the smooth muscle cell relaxes [255, 256]. At rest, BKCa, Nav, Cav, and Ca2+-gated Cl− channels (ClCa) are closed, Kir channels are intermittently open, and the resting membrane potential matches the K+ reversal potential. NO donors and arginine increase BKCa channel opening, and PKG/NOS inhibitors block BKCa current in myometrial cells from pregnant women [257]. PKG agonists and cGMP analogs stimulate BKCa activity in pregnant myometrial myocytes, but not in nonpregnant myocytes [258]. KATP is active when the ATP:ADP ratio decreases, and increased K+ permeability maintains uterine myocyte hyperpolarization and quiescence [259, 260]. Cysteine and sulfide donors activate myometrial KATP via persulfidation, but tocolysis is sensitive to the KATP inhibitor glibenclamide [9, 239, 261]. The mechanism may be more complex, as antagonists of myometrial ClCa channels (which conduct outward Cl− current and are therefore depolarizing/pro-contractile) [262, 263] paradoxically inhibit sulfide relaxation of rat myometrium [9]. Sulfide may, therefore, act downstream of ClCa channels.
G protein-coupled receptors and receptor kinases are expressed throughout the myometrium (e.g. OTR, VEGF receptor) and can be regulated by gasotransmitters. Intraperitoneal sulfide reduces murine preterm birth induced by the Toll-like receptor agonist LPS [10, 264, 265], but the mechanism is not known. The G protein-coupled niacin receptor GPR109A promotes inflammatory pathways and accumulates in the placenta and uterus of pregnant CBS heterozygous dams [266, 267]. Intriguingly, GPR109A deletion rescues OTR overexpression and premature delivery in pregnant CBS heterozygous mice [266], suggesting sulfide antagonizes GPR109A-activated contractility. Complete understanding of uterine receptor modulation by gasotransmitters will require additional investigation.
Key points
Uterine eNOS, CBS, and CSE levels increase during pregnancy and decrease during labor.
The HO substrate heme is a tocolytic, but direct CO application is not.
NO activates both classical pathway and non-cGMP dependent uterine relaxation.
NO activates myometrial smooth muscle BKCa activity, and sulfide increases myometrial KATP activity.
S-nitrosation of cellular contractile proteins correlates with preterm labor.
Cervical remodeling
Cervical remodeling is the softening, shortening, and dilation of the uterine cervix before delivery. It begins in mid pregnancy with glycosaminoglycan degradation, decreased collagen fiber production, and neutrophil/macrophage invasion [268–270]. Inflammatory mediators such as IL-8, IL-1α, and prostaglandin F2 α augment cervical effacement (i.e. shortening) [271, 272] and relax the myocervical circular smooth muscle [273, 274]. NO promotes cervical remodeling.
Pregnant human cervix expresses all three NOS isoforms, and cervical fluid NO3− is lower in TNL women [275]. Cervical epithelia contain nNOS. Inducible NOS and eNOS accumulate in vascular endothelial cells, and cervical leukocytes also express iNOS. Cervical iNOS increases 2-fold between the first trimester and term [276], and human cervical fibroblasts treated with IL-1α upregulate iNOS transcription 16-fold. L-NAME blocks IL-1α stimulated secretion of matrix metalloprotease-1 [277], suggesting that iNOS can facilitate cervical ripening. P4 suppresses iNOS transcription in RAW macrophages [278] and prevents NO-stimulated prostaglandin E2 production in human cervical explants [279, 280]. In rodents, peripartum luteolysis reduces P4, which then permits iNOS accumulation [231]. P4 receptor (PR) antagonists accelerate cervical remodeling in humans and animals and increase iNOS transcription fourfold in pregnant rats [281]. P4 and cervical iNOS are inversely related near the end of pregnancy and intimately associated with inflammation signals prior to parturition. In humans, P4 levels do not decline, but PR switching before labor from active PR-B to truncated PR-A causes functional withdrawal of P4 signaling [282, 283].
Both classical and cytotoxic (ONOO−) NO signaling influence cervical remodeling. NO donors increase cervical cGMP [284] and promote human myocervical relaxation ex vivo [285–288]. Low concentrations of NO donors (10 μM) suppress and high concentrations (3 mM) promote uterine prostaglandin synthesis in mice [289]. Cervical cytokines and reactive oxygen species increase as labor approaches [271, 272, 290], while cervical SOD1 decreases. Thus, increased cervical iNOS producing NO along with increased O2− and oxidative stress causes ONOO− formation [43, 57, 276, 291], suggesting the cytotoxic pathway mediates cervical change [292] (Figure 7B). However, there is no preterm labor phenotype in iNOS knockout mice [125]. This may reflect compensatory upregulation of nNOS/eNOS.
Key points
P4 signaling, via PR switching, and SOD1 decrease in the cervix at parturition, promoting ONOO− production.
NO may stimulate prostaglandin synthesis via either classical or cytotoxic NO pathways.
There is no evidence for sulfide or CO regulating cervical remodeling.
Rupture of membranes
The fetal membranes comprise the fused inner amnion and outer chorion. They provide a structural barrier to infection and the maternal immune system and contain the amniotic fluid reservoir and fetus [293]. Membrane cells continue to multiply in late gestation, and membrane stretch accommodates rapid fetal growth. As telomeres shorten in the cells comprising fetal membranes, apoptosis ensues, and the membranes rupture [294–296]. This releases inflammatory cytokines to enhance uterine contractions [221]. Loss of amniotic fluid is a strong signal for delivery [297], and NO and sulfide may influence the timing of membrane rupture (Figure 7C).
As in the cervix, iNOS in fetal membranes correlates with labor. In humans, TL membranes after vaginal delivery express more iNOS than intact TNL membranes from cesarean delivery [298]. Oxytocin stimulates iNOS expression and ONOO− synthesis in human membrane explants [299]. Inducible NOS expression stimulates p38 MAPK-dependent chorion cell apoptosis [300], while NOS inhibitors delay membrane apoptosis [301]. ONOO− protein nitration and p38 MAPK signaling increase in mouse fetal membranes exposed to cigarette smoke extract [302], implicating the cytotoxic pathway as a mechanism for preterm premature rupture of membranes in smokers. In contrast, sulfide reportedly maintains chorion/amnion integrity. Human and rat fetal membranes express CBS and CSE. CBS is more abundant in rats [303]. CBS and CSE expression is lower in TL membranes compared with TNL cesarean controls [304]. Since sulfide attenuates oxidative stress and inhibits prostaglandin synthesis, CBS expression might reduce inflammation and delay membrane rupture [305].
Key points
Fetal membrane iNOS increases in late gestation and may facilitate rupture of membranes via the cytotoxic pathway.
Membrane sulfide may curtail inflammatory signaling, thereby delaying membrane rupture.
Uterine involution
After birth, the uterus must continue contracting to limit postpartum bleeding [306]. Over several weeks, uterine involution proceeds with the myometrium and vasculature returning to the pregravid state. In pigs, uterine iNOS increases from postnatal day 7 to 35 [307], which may be a return to baseline and/or may represent increased inflammation and phagocytosis. Inducible NOS-expressing uterine M1 macrophages, however, accumulate sharply at parturition and then decrease after delivery [308]. Thus, the source, direction, and function of iNOS/NO in uterine involution are unclear. In mice, eNOS-derived NO may prevent postpartum uterine blood vessel narrowing. Parous wild-type dams have uterine arteries with wider lumens than virgin mice, but eNOS knockout uterine arteries remain similar to nulligravid wild-type mice [7]. Whether this is due to defects in eNOS-dependent vascular remodeling during pregnancy or altered postpartum involution involving NO is uncertain. If NO does facilitate uterine involution, it is probably not via the classical pathway in myocytes since involution is a contractile process. Inflammation in the postpartum uterus is prominent, and cytotoxic NO signaling may occur if sufficient O2− is present to yield ONOO−. A mechanism by which sulfide promotes autophagy and apoptosis has been identified [309], but this mechanism has not been studied in the postgravid uterus.
Key points
Few studies have examined the mechanisms of uterine involution or the effect of gasotransmitters in the postpartum uterus.
Uterine iNOS increases postpartum and may be associated with general tissue inflammation.
The winds of tomorrow: gasotransmitters in perinatal research
We have summarized the evidence for gasotransmitter regulation of pregnancy from conception to postpartum involution. Where possible, we have described specific mechanisms. However, important gaps remain in the present literature. Here we describe some of the challenges, questions, and future opportunities for gasotransmitter pregnancy research.
Novel gasotransmitters and signaling pathways. New small gas transmitters have been proposed in recent years, but almost none have been studied in pregnancy. For example, brain, kidney, and liver produce ammonia (NH3), the abundance of which influences renal pH even in healthy people [310]. Methane (CH4), a stable but rare volatile, may protect against hypoxia-reperfusion injury [311]. Carbonyl sulfide (COS) is rapidly converted to sulfide and bicarbonate by carbonic anhydrase [312], and carbon disulfide (CS2) prevents NFκB-mediated inflammation [313]. The relevance of these gasses and the novel products of gasotransmitter combinations require exploration. The product of NO and sulfide, SSNO−, is both a uterotonin and a vasodilator. This finding complicates the conventional view of NO/sulfide synergy [165, 314]. Methylated sulfides such as trimethylsulfonium [315] relax smooth muscle but also activate specific antioxidant enzymes [316], suggesting persulfidation may not be the only mechanism of sulfide signal transduction. As our technical capabilities expand, time and attention will become limiting resources to measure and identify novel gasotransmitters.
Nongaseous products of gasotransmitter enzymes. The gasotransmitter synthetic reactions create other nonvolatile products with possible signaling roles. Citrulline (produced with NO by NOS enzymes) is a hydroxyl radical scavenger [317]. Homocysteinemia and cystathioninuria occur with CBS and CSE deficiency, respectively, because of altered RTS that changes intracellular cysteine levels, affects glutathione and taurine metabolism, and perturbs the intracellular redox state [318, 319]. Biliverdin produced by HO is an antioxidant that modulates oxidative damage [320], while free Fe2+ can generate reactive free radicals. These nongaseous co-products may mediate some effects of gasotransmitter enzyme activity or deficiency, which deserves consideration in future experiments.
Current enzyme tools and pharmacology. For many commercially available gasotransmitter enzyme agonists and antagonists, isoform selectivity is low. For example, the NOS inhibitors 7-nitroindazole (7-NI), 2-amino-5,6-dihydro-6-methyl-4H-1,3-thiazine (AMT), and diphenyleneiodonium (DPI) are reportedly selective for nNOS, iNOS, and eNOS. Unfortunately, the IC50s show considerable overlap, which risks incorrect conclusions for tissues expressing multiple NOS isoforms [321]. Similar issues exist for HO and CSE/CBS inhibitors. Genetic knockout models can be informative, but confounding effects require attention (e.g. dramatically increased cystathionine in CSE knockout mice may be a cause of the phenotype). Experimental design using multiple drugs or approaches will produce more robust findings.
Gasotransmitter measurement and reporters. Specific, real-time detection of gasotransmitters in living cells and tissues can yield artifact since many gasotransmitter-reactive dyes also interact with other redox mediators. In vitro enzymatic assays are reliable, but they do not assess bioactivity in the natural intracellular milieu. New genetically encoded fluorescent biosensors for NO [322], CO [323], and sulfide [324] are promising, but their selectivity in living cells requires verification. This issue is well illustrated by a sulfide biosensor that is 25 times more selective for sulfide than GSH [324], even though intracellular GSH is more than 1000 times more abundant than sulfide [325]. Chemiluminescence [326–328], amperometry [329–331], and stable metabolite quantification [332, 333] accurately quantify gasotransmitters [334], but implementation is complicated and limited in scale. Multiple approaches and reagents are probably needed in present gasotransmitter research.
Limited perinatal research. After the initial excitement surrounding NO in the 1990s, few labs have ongoing basic investigation of gasotransmitters in pregnancy. Indeed, many seminal discoveries regarding gasotransmitters in pregnancy still require independent verification. Although many lines of promising research show therapeutic potential for obstetrical syndromes, few reproductive sciences labs are active in this area. Devoting increased effort and resources to basic mechanisms, including expanding the number of laboratories and investigators, could better inform clinical trials and thereby advance therapeutic and diagnostic tools for pregnancy.
Key points
In different cells and tissues, gasotransmitters can antagonize or synergize with one another.
Novel gasotransmitters have been identified, but their function in reproductive biology has not been tested.
The important effects of by-products of gasotransmitter enzymatic activity deserve separate consideration from gasotransmitters themselves.
Experimental design should account for limitations of current enzyme inhibitors and activators and gasotransmitter tracers.
The most accurate and sensitive tools to measure NO, CO, and sulfide are ozone chemiluminescence, palladium-catalyzed fluorescence, and amperometry, respectively.
Study of gasotransmitters in reproductive biology, especially with independent confirmation of findings, will accelerate opportunities for accurate development of pregnancy therapies and diagnostics.
Conclusion
NO, CO, and sulfide influence multiple aspects of pregnancy physiology. In the decades since the earliest discoveries showing that NO potently relaxes the uterus, we have developed a deeper and more complex knowledge of gasotransmitter production, regulation, and interactions (Figure 8). NO regulates P4 secretion to maintain early pregnancy and augments endometrial decidualization. NO and sulfide affect fallopian tube peristalsis. NO and CO balance trophoblast invasion and proliferation during implantation, and CO promotes spiral artery remodeling. All three gasotransmitters facilitate placental angiogenesis and augment maternal uterine blood flow, together maximizing utero-placental transfer. All three also modulate maternal immune function in pregnancy, activating uNK and Treg populations while suppressing alloimmunity. As the myometrium acquires resistance to NO and sulfide quiescence at labor, NO weakens the fetal membranes preparing for rupture and promotes cervical remodeling. The precise role of gasotransmitters in specific perinatal pathologies is not well established, and recently recognized reactions among gasotransmitters and established second messengers are largely unexplored. There are many opportunities for further study, and we anticipate the development of new pregnancy therapies from increased understanding of perinatal gasotransmitter signaling.
Figure 8.
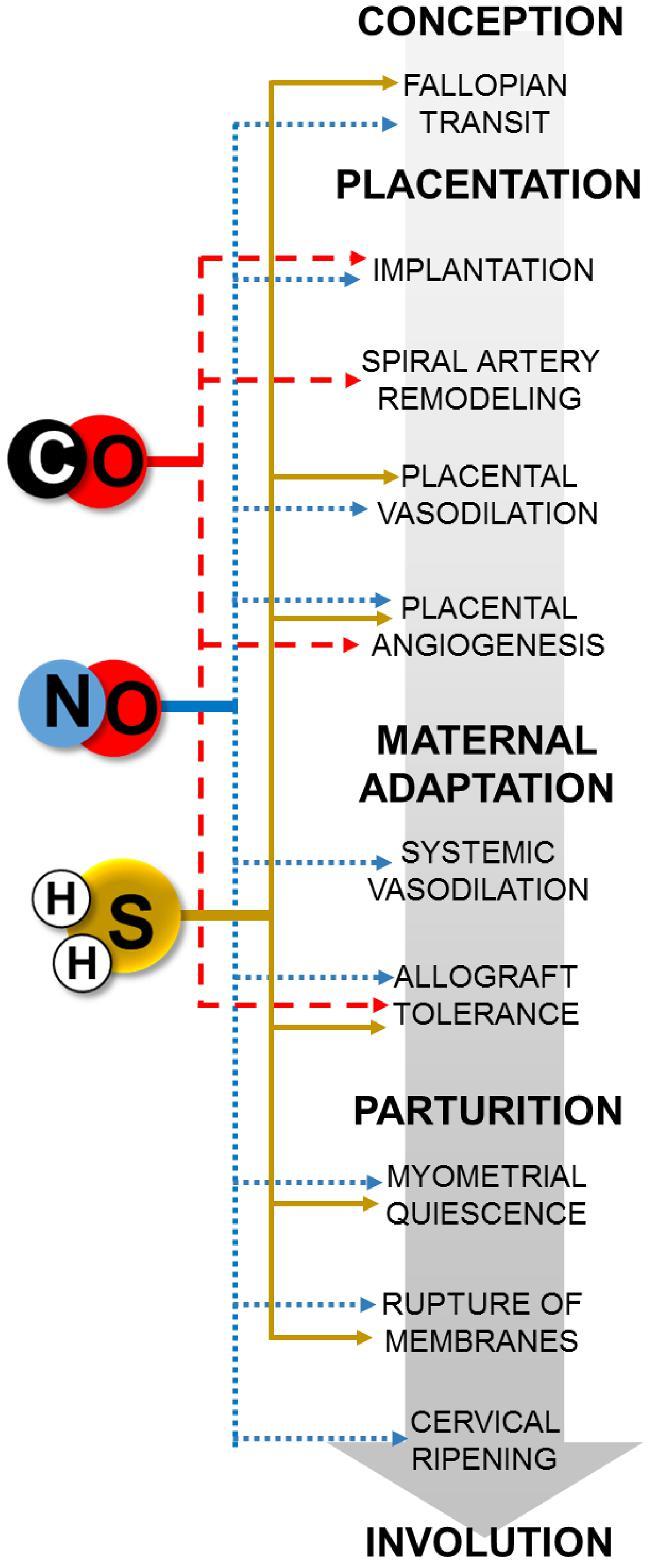
Summary schematic of gasotransmitter-mediated processes in pregnancy. Blue dotted, red dashed, and yellow solid lines denote NO, CO, and sulfide, respectively.
Acknowledgments
We are thankful for Charlotte Guerra's assistance with figure illustrations. Special appreciation to Thomas Jansson, Joshua Johnson, Nathen Bopp, and Daniel J. Guerra for their comments.
Footnotes
Grant Support: This work was supported by a National Institute of Child Health and Human Development Perinatology-Neonatology T32 training grant (5T32HD007186-37, to DDG) and a Society for Maternal Fetal Medicine/American Association of Obstetricians and Gynecologists Foundation Scholar Award (to KJH).
References
- 1. Friebe A, Koesling D. Mechanism of YC-1-induced activation of soluble guanylyl cyclase. Mol Pharmacol 1998; 53(1):123–127. [DOI] [PubMed] [Google Scholar]
- 2. Lewicki JA, Brandwein HJ, Mittal CK, Arnold WP, Murad F. Properties of purified soluble guanylate cyclase activated by nitric oxide and sodium nitroprusside. J Cyclic Nucleotide Res 1982; 8(1):17–25. [PubMed] [Google Scholar]
- 3. Nishida M, Sawa T, Kitajima N, Ono K, Inoue H, Ihara H, Motohashi H, Yamamoto M, Suematsu M, Kurose H, van der Vliet A, Freeman BA et al.. Hydrogen sulfide anion regulates redox signaling via electrophile sulfhydration. Nat Chem Biol 2012; 8(8):714–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kulandavelu S, Whiteley KJ, Qu D, Mu J, Bainbridge SA, Adamson SL. Endothelial nitric oxide synthase deficiency reduces uterine blood flow, spiral artery elongation, and placental oxygenation in pregnant mice. Hypertension 2012; 60(1):231–238. [DOI] [PubMed] [Google Scholar]
- 5. Nuño-Ayala M, Guillén N, Arnal C, Lou-Bonafonte JM, de Martino A, García-de-Jalón JA, Gascón S, Osaba L, Osada J, Navarro MA. Cystathionine β-synthase deficiency causes infertility by impairing decidualization and gene expression networks in uterus implantation sites. Physiol Genomics 2012; 44(14):702–716. [DOI] [PubMed] [Google Scholar]
- 6. Linzke N, Schumacher A, Woidacki K, Croy BA, Zenclussen AC. Carbon monoxide promotes proliferation of uterine natural killer cells and remodeling of spiral arteries in pregnant hypertensive heme oxygenase-1 mutant mice. Hypertension 2014; 63(3):580–588. [DOI] [PubMed] [Google Scholar]
- 7. van der Heijden OWH, Essers YP, Wijnands E, Mey JG, Peeters LL, van Eys GJ. Postpartum reversal of the pregnancy-induced uterine artery remodeling in young, aging, and eNOS-deficient mice. Reprod Sci 2009; 16(7):642–649. [DOI] [PubMed] [Google Scholar]
- 8. Hayden LJ, Goeden H, Roth SH, Growth and development in the rat during sub-chronic exposure to low levels of hydrogen sulfide. Toxicol Ind Health 1990; 6(3-4):389–401. [DOI] [PubMed] [Google Scholar]
- 9. Mijušković A, Kokić AN, Dušić ZO, Slavić M, Spasić MB, Blagojević D. Chloride channels mediate sodium sulphide-induced relaxation in rat uteri. Br J Pharmacol 2015; 172(14):3671–3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu W, Xu C, You X, Olson DM, Chemtob S, Gao L, Ni X. Hydrogen sulfide delays LPS-Induced preterm birth in mice via anti-inflammatory pathways. PLoS One 2016; 11(4):e0152838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zenclussen ML, Casalis PA, El-Mousleh T, Rebelo S, Langwisch S, Linzke N, Volk HD, Fest S, Soares MP, Zenclussen AC. Haem oxygenase-1 dictates intrauterine fetal survival in mice via carbon monoxide. J Pathol 2011; 225(2):293–304. [DOI] [PubMed] [Google Scholar]
- 12. Tanner FW. Studies on the bacterial metabolism of sulfur: I. formation of hydrogen sulfide from certain sulfur compounds under aerobic conditions. J Bacteriol 1917; 2(5):585–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Anson ML, Mirsky AE. On the combination of nitric oxide with haemoglobin. J Physiol 1925; 60(1-2):100–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hill AV. The combinations of haemoglobin with oxygen and with carbon monoxide. I. Biochem J 1913; 7(5):471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smythe CV. The utilization of cysteine and cystine by rat liver with the production of hydrogen sulfide. J Biol Chem 1942; 142(1):387–400. [Google Scholar]
- 16. Laskowski M, Fromageot C. Some properties of desulfurase. J Biol Chem 1941; 140:663–669. [Google Scholar]
- 17. Sjostrand T. The formation of carbon monoxide by the decomposition of haemoglobin in vivo. Acta Physiol Scand 1952; 26(4):338–344. [DOI] [PubMed] [Google Scholar]
- 18. Tenhunen R, Marver HS, Schmid R. Microsomal heme oxygenase: characterization of the enzyme. J Biol Chem 1969; 244(23):6388–6394. [PubMed] [Google Scholar]
- 19. Rodbell M, Birnbaumer L, Pohl SL, Krans HM. The glucagon-sensitive adenyl cyclase system in plasma membranes of rat liver. V. An obligatory role of guanylnucleotides in glucagon action. J Biol Chem 1971; 246(6):1877–1882. [PubMed] [Google Scholar]
- 20. Berthet J, Rall TW, Sutherland EW, The relationship of epinephrine and glucagon to liver phosphorylase. IV. Effect of epinephrine and glucagon on the reactivation of phosphorylase in liver homogenates. J Biol Chem 1957; 224(1):463–475. [PubMed] [Google Scholar]
- 21. Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980; 288(5789):373–376. [DOI] [PubMed] [Google Scholar]
- 22. Arnold WP, Mittal CK, Katsuki S, Murad F. Nitric oxide activates guanylate cyclase and increases guanosine 3':5'-cyclic monophosphate levels in various tissue preparations. Proc Natl Acad Sci USA 1977; 74(8):3203–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci USA 1987; 84(24):9265–9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Palmer RM, Ashton DS, Moncada S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature 1988; 333(6174):664–666. [DOI] [PubMed] [Google Scholar]
- 25. Brüne B, Ullrich V. Inhibition of platelet aggregation by carbon monoxide is mediated by activation of guanylate cyclase. Mol Pharmacol 1987; 32(4):497–504. [PubMed] [Google Scholar]
- 26. McFaul SJ, McGrath JJ. Studies on the mechanism of carbon monoxide-induced vasodilation in the isolated perfused rat heart. Toxicol Appl Pharmacol 1987; 87(3):464–473. [DOI] [PubMed] [Google Scholar]
- 27. Verma A, Hirsch DJ, Glatt CE, Ronnett GV, Snyder SH. Carbon monoxide: a putative neural messenger. Science 1993; 259(5093):381–384. [DOI] [PubMed] [Google Scholar]
- 28. Abe K, Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci 1996; 16(3):1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cheng Y, Ndisang JF, Tang G, Cao K, Wang R. Hydrogen sulfide-induced relaxation of resistance mesenteric artery beds of rats. Am J Physiol Heart Circ Physiol 2004; 287(5):H2316–H2323. [DOI] [PubMed] [Google Scholar]
- 30. Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science 2008; 322(5901):587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lancaster JR. Nitric oxide: a brief overview of chemical and physical properties relevant to therapeutic applications. Future Sci OA 2015; 1(1):FSO59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Francis SH, Busch JL, Corbin JD. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev 2010; 62(3):525–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Keszler A, Zhang Y, Hogg N. The reaction between nitric oxide, glutathione and oxygen in the presence and absence of protein: how are S-Nitrosothiols formed? Free Radic Biol Med 2010; 48(1):55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vitturi DA, Minarrieta L, Salvatore SR, Postlethwait EM, Fazzari M, Ferrer-Sueta G, Lancaster JR Jr, Freeman BA, Schopfer FJ. Convergence of biological nitration and nitrosation via symmetrical nitrous anhydride. Nat Chem Biol 2015; 11(7):504–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Seth D, Hess DT, Hausladen A, Wang L, Wang YJ, Stamler JS. A multiplex enzymatic machinery for cellular protein S-nitrosylation. Mol Cell 2018; 69(3):451–464.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen YY, Chu HM, Pan KT, Teng CH, Wang DL, Wang AH, Khoo KH, Meng TC. Cysteine S-nitrosylation protects protein-tyrosine phosphatase 1B against oxidation-induced permanent inactivation. J Biol Chem 2008; 283(50):35265–35272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sakakura M, Tamura M, Fujii N, Takeuchi T, Hatanaka T, Kishimoto S, Arata Y, Takahashi H. Structural mechanisms for the S-nitrosylation-derived protection of mouse galectin-2 from oxidation-induced inactivation revealed by NMR. FEBS J 2018; 285(6):1129–1145. [DOI] [PubMed] [Google Scholar]
- 38. Ulrich C, Quillici DR, Schegg K, Woolsey R, Nordmeier A, Buxton IL. Uterine smooth muscle S-nitrosylproteome in pregnancy. Mol Pharmacol 2012; 81(2):143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marino SM, Gladyshev VN. Structural analysis of cysteine S-nitrosylation: a modified acid-based motif and the emerging role of trans-nitrosylation. J Mol Biol 2010; 395(4):844–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xue Y, Liu Z, Gao X, Jin C, Wen L, Yao X, Ren J. GPS-SNO: Computational prediction of protein S-Nitrosylation sites with a modified GPS algorithm. PLoS One 2010; 5(6):e11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cortese-Krott MM, Fernandez BO, Santos JLT, Mergia E, Grman M, Nagy P, Kelm M, Butler A, Feelisch M. Nitrosopersulfide (SSNO−) accounts for sustained NO bioactivity of S-nitrosothiols following reaction with sulfide. Redox Biol 2014; 2:234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Berenyiova A, Grman M, Mijuskovic A, Stasko A, Misak A, Nagy P, Ondriasova E, Cacanyiova S, Brezova V, Feelisch M, Ondrias K. The reaction products of sulfide and S-nitrosoglutathione are potent vasorelaxants. Nitric Oxide 2015; 46:123–130. [DOI] [PubMed] [Google Scholar]
- 43. Szabo C, Ohshima H. DNA damage induced by peroxynitrite: subsequent biological effects. Nitric Oxide 1997; 1(5):373–385. [DOI] [PubMed] [Google Scholar]
- 44. Radi R. Peroxynitrite, a stealthy biological oxidant. J Biol Chem 2013; 288(37):26464–26472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Alvarez MN, Peluffo G, Piacenza L, Radi R. Intraphagosomal peroxynitrite as a macrophage-derived cytotoxin against internalized Trypanosoma cruzi: consequences for oxidative killing and role of microbial peroxiredoxins in infectivity. J Biol Chem 2011; 286(8):6627–6640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Miles AM, Bohle DS, Glassbrenner PA, Hansert B, Wink DA, Grisham MB. Modulation of superoxide-dependent oxidation and hydroxylation reactions by nitric oxide. J Biol Chem 1996; 271(1):40–47. [DOI] [PubMed] [Google Scholar]
- 47. Quijano C, Hernandez-Saavedra D, Castro L, McCord JM, Freeman BA, Radi R. Reaction of peroxynitrite with Mn-Superoxide dismutase. J Biol Chem 2001; 276(15):11631–11638. [DOI] [PubMed] [Google Scholar]
- 48. Blekkenhorst LC, Bondonno NP, Liu AH, Ward NC, Prince RL, Lewis JR, Devine A, Croft KD, Hodgson JM, Bondonno CP. Nitrate, the oral microbiome, and cardiovascular health: a systematic literature review of human and animal studies. Am J Clin Nutr 2018; 107(4):504–522. [DOI] [PubMed] [Google Scholar]
- 49. Peleli M, Zollbrecht C, Montenegro MF, Hezel M, Zhong J, Persson EG, Holmdahl R, Weitzberg E, Lundberg JO, Carlström M. Enhanced XOR activity in eNOS-deficient mice. Free Radic Biol Med 2016; 99:472–484. [DOI] [PubMed] [Google Scholar]
- 50. Forstermann U, Sessa WC, Nitric oxide synthases: regulation and function. Eur Heart J 2012; 33(7):829–837, 837a-837d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Oess S, Icking A, Fulton D, Govers R, Müller-Esterl W. Subcellular targeting and trafficking of nitric oxide synthases. Biochem J 2006; 396(3):401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bö L, Dawson TM, Wesselingh S, Mörk S, Choi S, Kong PA, Hanley D, Trapp BD. Induction of nitric oxide synthase in demyelinating regions of multiple sclerosis brains. Ann Neurol 1994; 36(5):778–786. [DOI] [PubMed] [Google Scholar]
- 53. Bon CLM, Garthwaite J. On the role of nitric oxide in hippocampal long-term potentiation. J Neurosci 2003; 23(5):1941–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Charles IG, Palmer RM, Hickery MS, Bayliss MT, Chubb AP, Hall VS, Moss DW, Moncada S. Cloning, characterization, and expression of a cDNA encoding an inducible nitric oxide synthase from the human chondrocyte. Proc Natl Acad Sci USA 1993; 90(23):11419–11423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Myatt L, Eis AL, Brockman DE, Greer IA, Lyall F. Endothelial nitric oxide synthase in placental villous tissue from normal, pre-eclamptic and intrauterine growth restricted pregnancies. Hum Reprod 1997; 12(1):167–172. [DOI] [PubMed] [Google Scholar]
- 56. Piech A, Massart PE, Dessy C, Feron O, Havaux X, Morel N, Vanoverschelde JL, Donckier J, Balligand JL. Decreased expression of myocardial eNOS and caveolin in dogs with hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol 2002; 282(1):H219–H231. [DOI] [PubMed] [Google Scholar]
- 57. Roe ND, Ren J. Nitric oxide synthase uncoupling: a therapeutic target in cardiovascular diseases. Vasc Pharmacol 2012; 57(5-6):168–172. [DOI] [PubMed] [Google Scholar]
- 58. Venema RC, Sayegh HS, Kent JD, Harrison DG. Identification, characterization, and comparison of the calmodulin-binding domains of the endothelial and inducible nitric oxide synthases. J Biol Chem 1996; 271(11):6435–6440. [DOI] [PubMed] [Google Scholar]
- 59. Silvagno F, Xia H, Bredt DS. Neuronal nitric-oxide synthase-, an alternatively spliced isoform expressed in differentiated skeletal muscle. J Biol Chem 1996; 271(19):11204–11208. [DOI] [PubMed] [Google Scholar]
- 60. Cheung WY. Calmodulin plays a pivotal role in cellular regulation. Science 1980; 207(4426):19–27. [DOI] [PubMed] [Google Scholar]
- 61. Hurt KJ, Sezen SF, Lagoda GF, Musicki B, Rameau GA, Snyder SH, Burnett AL. Cyclic AMP-dependent phosphorylation of neuronal nitric oxide synthase mediates penile erection. Proc Natl Acad Sci USA 2012; 109(41):16624–16629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, Power DA, Ortiz de Montellano PR, Kemp BE. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett 1999; 443(3):285–289. [DOI] [PubMed] [Google Scholar]
- 63. Rameau GA, Tukey DS, Garcin-Hosfield ED, Titcombe RF, Misra C, Khatri L, Getzoff ED, Ziff EB. Biphasic coupling of neuronal nitric oxide synthase phosphorylation to the NMDA receptor regulates AMPA receptor trafficking and neuronal cell death. J Neurosci 2007; 27(13):3445–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Michell BJ, Harris MB, Chen ZP, Ju H, Venema VJ, Blackstone MA, Huang W, Venema RC, Kemp BE. Identification of regulatory sites of phosphorylation of the bovine endothelial nitric-oxide synthase at serine 617 and serine 635. J Biol Chem 2002; 277(44):42344–42351. [DOI] [PubMed] [Google Scholar]
- 65. Matsubara M, Hayashi N, Jing T, Titani K. Regulation of endothelial nitric oxide synthase by protein kinase C. J Biochem (Tokyo) 2003; 133(6):773–781. [DOI] [PubMed] [Google Scholar]
- 66. Foster MW, Thompson JW, Forrester MT, Sha Y, McMahon TJ, Bowles DE, Moseley MA, Marshall HE. Proteomic analysis of the NOS2 interactome in human airway epithelial cells. Nitric Oxide 2013; 34:37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Matsumoto K, Nishiya T, Maekawa S, Horinouchi T, Ogasawara K, Uehara T, Miwa S. The ECS(SPSB) E3 ubiquitin ligase is the master regulator of the lifetime of inducible nitric-oxide synthase. Biochem Biophys Res Commun 2011; 409(1):46–51. [DOI] [PubMed] [Google Scholar]
- 68. Clapp KM, Peng HM, Jenkins GJ, Ford MJ, Morishima Y, Lau M, Osawa Y. Ubiquitination of neuronal nitric-oxide synthase in the calmodulin-binding site triggers proteasomal degradation of the protein. J Biol Chem 2012; 287(51):42601–42610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Stangl V, Lorenz M, Meiners S, Ludwig A, Bartsch C, Moobed M, Vietzke A, Kinkel HT, Baumann G, Stangl K. Long-term up-regulation of eNOS and improvement of endothelial function by inhibition of the ubiquitin-proteasome pathway. FASEB J 2004; 18(2):272–279. [DOI] [PubMed] [Google Scholar]
- 70. Helms C, Kim-Shapiro DB. Hemoglobin-mediated nitric oxide signaling. Free Radic Biol Med 2013; 61:464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Broniowska KA, Diers AR, Hogg N. S-nitrosoglutathione. Biochim Biophys Acta 2013; 1830(5):3173–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Guerra D, Ballard K, Truebridge I, Vierling E. S-Nitrosation of conserved cysteines modulates activity and stability of s-nitrosoglutathione reductase (GSNOR). Biochemistry 2016; 55(17):2452–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sharma VS, Magde D. Activation of soluble guanylate cyclase by carbon monoxide and nitric oxide: a mechanistic model. Methods 1999; 19(4):494–505. [DOI] [PubMed] [Google Scholar]
- 74. Imai T, Morita T, Shindo T, Nagai R, Yazaki Y, Kurihara H, Suematsu M, Katayama S. Vascular smooth muscle cell–directed overexpression of heme oxygenase-1 elevates blood pressure through attenuation of nitric oxide–induced vasodilation in mice. Circ Res 2001; 89(1):55–62. [DOI] [PubMed] [Google Scholar]
- 75. Yi L, Morgan JT, Ragsdale SW. Identification of a thiol/disulfide redox switch in the human BK channel that controls its affinity for heme and CO. J Biol Chem 2010; 285(26):20117–20127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Brazier SP, Telezhkin V, Mears R, Müller CT, Riccardi D, Kemp PJ. Cysteine residues in the C-terminal tail of the human BK(Ca)alpha subunit are important for channel sensitivity to carbon monoxide. Adv Exp Med Biol 2009; 648:49–56. [DOI] [PubMed] [Google Scholar]
- 77. Liang S, Wang Q, Zhang W, Zhang H, Tan S, Ahmed A, Gu Y. Carbon monoxide inhibits inward rectifier potassium channels in cardiomyocytes. Nat Commun 2014; 51:4676. [DOI] [PubMed] [Google Scholar]
- 78. Maitlis P, Haynes A. Chapter 4 syntheses based on carbon monoxide. In: Chiusoli GP, Maitlis P (eds.) Metal-catalysis in Industrial Organic Processes. The Royal Society of Chemistry: London, United Kingdom, 2006: 114–162. [Google Scholar]
- 79. Dennery PA. Signaling function of heme oxygenase proteins. Antioxid Redox Signal 2014; 20(11):1743–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ding Y, McCoubrey WK Jr, Maines MD. Interaction of heme oxygenase-2 with nitric oxide donors. Is the oxygenase an intracellular ‘sink’ for NO? Eur J Biochem 1999; 264(3):854–861. [DOI] [PubMed] [Google Scholar]
- 81. Wang J, Lu S, Moënne-Loccoz P, Ortiz de Montellano PR. Interaction of nitric oxide with human heme oxygenase-1. J Biol Chem 2003; 278(4):2341–2347. [DOI] [PubMed] [Google Scholar]
- 82. Alam J, Cook JL. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am J Respir Cell Mol Biol 2007; 36(2):166–174. [DOI] [PubMed] [Google Scholar]
- 83. Lee T-S, Chau L-Y. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med 2002; 8(3):240–246. [DOI] [PubMed] [Google Scholar]
- 84. Muñoz-Sánchez J, Chánez-Cárdenas ME. A review on hemeoxygenase-2: focus on cellular protection and oxygen response. Oxid Med Cell Longev 2014; 2014:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Fleischhacker AS, Sharma A, Choi M, Spencer AM, Bagai I, Hoffman BM, Ragsdale SW. The C-Terminal heme regulatory motifs of heme oxygenase-2 are redox-regulated heme binding sites. Biochemistry 2015; 54(17):2709–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Boehning D, Moon C, Sharma S, Hurt KJ, Hester LD, Ronnett GV, Shugar D, Snyder SH. Carbon monoxide neurotransmission activated by CK2 phosphorylation of heme oxygenase-2. Neuron 2003; 40(1):129–137. [DOI] [PubMed] [Google Scholar]
- 87. Wu L, Wang R. Carbon monoxide: endogenous production, physiological functions, and pharmacological applications. Pharmacol Rev 2005; 57(4):585–630. [DOI] [PubMed] [Google Scholar]
- 88. Balla J, Vercellotti GM, Jeney V, Yachie A, Varga Z, Jacob HS, Eaton JW, Balla G. Heme, heme oxygenase, and ferritin: how the vascular endothelium survives (and dies) in an iron-rich environment. Antioxid Redox Signal 2007; 9(12):2119–2138. [DOI] [PubMed] [Google Scholar]
- 89. Kimura H. Signaling molecules: hydrogen sulfide and polysulfide. Antioxid Redox Signal 2015; 22(5):362–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Sen N, Paul BD, Gadalla MM, Mustafa AK, Sen T, Xu R, Kim S, Snyder SH. Hydrogen sulfide-linked sulfhydration of NF-kappaB mediates its antiapoptotic actions. Mol Cell 2012; 45(1):13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Gao XH, Krokowski D, Guan BJ, Bederman I, Majumder M, Parisien M, Diatchenko L, Kabil O, Willard B, Banerjee R, Wang B, Bebek G et al.. Quantitative H2S-mediated protein sulfhydration reveals metabolic reprogramming during the integrated stress response. eLife 2015; 4:e10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK, Barodka VM, Gazi FK, Barrow RK, Wang R, Amzel LM, Berkowitz DE et al.. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res 2011; 109(11):1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Arimoto H, Takahashi D. 8-Nitro-cGMP: A novel protein-reactive cNMP and its emerging roles in autophagy. Handb Exp Pharmacol 2017; 238:253–268. [DOI] [PubMed] [Google Scholar]
- 94. Fusco F, di Villa Bianca R, Mitidieri E, Cirino G, Sorrentino R, Mirone V. Sildenafil effect on the human bladder involves the L-cysteine/hydrogen sulfide pathway: a novel mechanism of action of phosphodiesterase type 5 inhibitors. Eur Urol 2012; 62(6):1174–1180. [DOI] [PubMed] [Google Scholar]
- 95. Nalli AD, Bhattacharya S, Wang H, Kendig DM, Grider JR, Murthy KS. Augmentation of cGMP/PKG pathway and colonic motility by hydrogen sulfide. Am J Physiol Gastrointest Liver Physiol 2017; 313(4):G330–G341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Singh S, Padovani D, Leslie RA, Chiku T, Banerjee R. Relative contributions of cystathionine beta-synthase and gamma-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J Biol Chem 2009; 284(33):22457–22466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kabil O, Vitvitsky V, Xie P, Banerjee R. The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antioxid Redox Signal 2011; 15(2):363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Tomita M, Nagahara N, Ito T. Expression of 3-mercaptopyruvate sulfurtransferase in the mouse. Molecules 2016; 21(12):E1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Mistry RK, Murray TV, Prysyazhna O, Martin D, Burgoyne JR, Santos C, Eaton P, Shah AM, Brewer AC. Transcriptional regulation of cystathionine-γ-Lyase in endothelial cells by NADPH Oxidase 4-dependent signaling. J Biol Chem 2016; 291(4):1774–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Hassan MI, Boosen M, Schaefer L, Kozlowska J, Eisel F, von Knethen A, Beck M, Hemeida RA, El-Moselhy MA, Hamada FM, Beck KF, Pfeilschifter J. Platelet-derived growth factor-BB induces cystathionine γ-lyase expression in rat mesangial cells via a redox-dependent mechanism. Br J Pharmacol 2012; 166(8):2231–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Lambertini E, Penolazzi L, Angelozzi M, Grassi F, Gambari L, Lisignoli G, De Bonis P, Cavallo M, Piva R. The expression of cystathionine gamma-lyase is regulated by estrogen receptor alpha in human osteoblasts. Oncotarget 2017; 8(60):101686–101696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Li H, Mani S, Wu L, Fu M, Shuang T, Xu C, Wang R. The interaction of estrogen and CSE/H2S pathway in the development of atherosclerosis. Am J Physiol Heart Circ Physiol 2017; 312(3):H406–H414. [DOI] [PubMed] [Google Scholar]
- 103. Vicente JB, Colaço HG, Sarti P, Leandro P, Giuffrè A. S-Adenosyl-l-methionine modulates CO and NO• binding to the human H2S-generating enzyme cystathionine β-synthase. J Biol Chem 2016; 291(2):572–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ereño-Orbea J, Majtan T, Oyenarte I, Kraus JP, Martínez-Cruz LA. Structural insight into the molecular mechanism of allosteric activation of human cystathionine beta-synthase by S-adenosylmethionine. Proc Natl Acad Sci USA 2014; 111(37):E3845–E3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. d’Emmanuele di Villa Bianca R, Mitidieri E, Esposito D, Donnarumma E, Russo A, Fusco F, Ianaro A, Mirone V, Cirino G, Russo G, Sorrentino R. Human cystathionine-β-synthase phosphorylation on Serine227 modulates hydrogen sulfide production in human urothelium. PLoS One 2015; 10(9):e0136859. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 106. Teng H, Wu B, Zhao K, Yang G, Wu L, Wang R. Oxygen-sensitive mitochondrial accumulation of cystathionine β-synthase mediated by Lon protease. Proc Natl Acad Sci USA 2013; 110(31):12679–12684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Fu M, Zhang W, Wu L, Yang G, Li H, Wang R. Hydrogen sulfide (H2S) metabolism in mitochondria and its regulatory role in energy production. Proc Natl Acad Sci USA 2012; 109(8):2943–2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Nagahara N, Nagano M, Ito T, Suzuki H. Chapter thirteen - redox regulation of mammalian 3-mercaptopyruvate sulfurtransferase. In: Enrique C, Lester P (eds.) Methods in Enzymology. Academic Press: Amsterdam, Netherlands; 2015: 229–254. [DOI] [PubMed] [Google Scholar]
- 109. Libiad M, Yadav PK, Vitvitsky V, Martinov M, Banerjee R. Organization of the human mitochondrial hydrogen sulfide oxidation pathway. J Biol Chem 2014; 289(45):30901–30910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Landry AP, Ballou DP, Banerjee R. H2S oxidation by nanodisc-embedded human sulfide quinone oxidoreductase. J Biol Chem 2017; 292(28):11641–11649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Tiranti V, Viscomi C, Hildebrandt T, Di Meo I, Mineri R, Tiveron C, Levitt MD, Prelle A, Fagiolari G, Rimoldi M, Zeviani M. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat Med 2009; 15(2):200–205. [DOI] [PubMed] [Google Scholar]
- 112. Vitvitsky V, Yadav PK, Kurthen A, Banerjee R. Sulfide oxidation by a noncanonical pathway in red blood cells generates thiosulfate and polysulfides. J Biol Chem 2015; 290(13):8310–8320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Delivoria-Papadopoulos M, Coburn RF, Forster RE. Cyclic variation of rate of carbon monoxide production in normal women. J Appl Physiol 1974; 36(1):49–51. [DOI] [PubMed] [Google Scholar]
- 114. Kobayashi Y, Yoshimoto Y, Yamamoto Y, Kimura K, Okuda K. Roles of EDNs in regulating oviductal NO synthesis and smooth muscle motility in cows. Reproduction 2016; 151(6):615–622. [DOI] [PubMed] [Google Scholar]
- 115. Ning N, Zhu J, Du Y, Gao X, Liu C, Li J. Dysregulation of hydrogen sulphide metabolism impairs oviductal transport of embryos. Nat Commun 2014; 5(1):4107. [DOI] [PubMed] [Google Scholar]
- 116. Lima PD, Zhang J, Dunk C, Lye SJ, Croy BA. Leukocyte driven-decidual angiogenesis in early pregnancy. Cell Mol Immunol 2014; 11(6):522–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Telfer JF, Irvine GA, Kohnen G, Campbell S, Cameron IT. Expression of endothelial and inducible nitric oxide synthase in non- pregnant and decidualized human endometrium. Mol Hum Reprod 1997; 3(1):69–75. [DOI] [PubMed] [Google Scholar]
- 118. Sengupta J, Dhawan L, Lalitkumar PG, Ghosh D. Nitric oxide in blastocyst implantation in the rhesus monkey. Reproduction 2005; 130(3):321–332. [DOI] [PubMed] [Google Scholar]
- 119. Purcell TL, Given R, Chwalisz K, Garfield RE. Nitric oxide synthase distribution during implantation in the mouse. Mol Hum Reprod 1999; 5(5):467–475. [DOI] [PubMed] [Google Scholar]
- 120. Spencer F, Chi L, Zhu MX. Antiproliferative effects of inducible nitric oxide synthase inhibition on decidualization in pseudopregnant rats. Exp Biol Med 1998; 218(1):45–50. [DOI] [PubMed] [Google Scholar]
- 121. Sugino N, Nakata M, Kashida S, Karube A, Takiguchi S, Kato H. Decreased superoxide dismutase expression and increased concentrations of lipid peroxide and prostaglandin F2alpha in the decidua of failed pregnancy. Mol Hum Reprod 2000; 6(7):642–647. [DOI] [PubMed] [Google Scholar]
- 122. Pan X, Wang X, Wang X, Sun Z, Zhang X, Liang X, Li Z, Dou Z. Nitric oxide regulates blastocyst hatching in mice. Int J Clin Exp Med 2015; 8(5):6994–7001. [PMC free article] [PubMed] [Google Scholar]
- 123. Large MJ, DeMayo FJ. The regulation of embryo implantation and endometrial decidualization by progesterone receptor signaling. Mol Cell Endocrinol 2012; 358(2):155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Gagioti S, Scavone C, Bevilacqua E. Participation of the mouse implanting trophoblast in nitric oxide production during pregnancy. Biol Reprod 2000; 62(2):260–268. [DOI] [PubMed] [Google Scholar]
- 125. Laubach VE, Shesely EG, Smithies O, Sherman PA. Mice lacking inducible nitric oxide synthase are not resistant to lipopolysaccharide-induced death. Proc Natl Acad Sci USA 1995; 92(23):10688–10692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Choi YK, Kim CK, Lee H, Jeoung D, Ha KS, Kwon YG, Kim KW, Kim YM. Carbon monoxide promotes VEGF expression by increasing HIF-1alpha protein level via two distinct mechanisms, translational activation and stabilization of HIF-1alpha protein. J Biol Chem 2010; 285(42):32116–32125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Keator CS, Schreiber DT, Hoagland TA, McCracken JA. Luteotrophic and luteolytic effects of nitric oxide in sheep are dose-dependent in vivo. Domest Anim Endocrinol 2008; 35(1):74–80. [DOI] [PubMed] [Google Scholar]
- 128. Ferreira-Dias G, Costa AS, Mateus L, Korzekwa AJ, Galvão A, Redmer DA, Lukasik K, Szóstek AZ, Woclawek-Potocka I, Skarzynski DJ. Nitric oxide stimulates progesterone and prostaglandin E2 secretion as well as angiogenic activity in the equine corpus luteum. Domest Anim Endocrinol 2011; 40(1):1–9. [DOI] [PubMed] [Google Scholar]
- 129. Motta AB, Estevez A, Tognetti T, Gimeno MA, Franchi AM. Dual effects of nitric oxide in functional and regressing rat corpus luteum. Mol Hum Reprod 2001; 7(1):43–47. [DOI] [PubMed] [Google Scholar]
- 130. Vega M, Urrutia L, Iñiguez G, Gabler F, Devoto L, Johnson MC. Nitric oxide induces apoptosis in the human corpus luteum in vitro. Mol Hum Reprod 2000; 6(8):681–687. [DOI] [PubMed] [Google Scholar]
- 131. Johnson MC, Diaz HA, Stocco C, Palomino A, Devoto L, Vega M. Antisteroidogenic action of nitric oxide on human corpus luteum in vitro: mechanism of action. Endocrine 1999; 11(1):31–36. [DOI] [PubMed] [Google Scholar]
- 132. Dimasuay KG, Boeuf P, Powell TL, Jansson T. Placental responses to changes in the maternal environment determine fetal growth. Front Physiol 2016; 7:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Cartwright JE, Holden DP, Whitley GS. Hepatocyte growth factor regulates human trophoblast motility and invasion: a role for nitric oxide. Br J Pharmacol 1999; 128(1):181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Cartwright JE, Tse WK, Whitley GS. Hepatocyte growth factor induced human trophoblast motility involves phosphatidylinositol-3-kinase, mitogen-activated protein kinase, and inducible nitric oxide synthase. Exp Cell Res 2002; 279(2):219–226. [DOI] [PubMed] [Google Scholar]
- 135. Sonderegger S, Haslinger P, Sabri A, Leisser C, Otten JV, Fiala C, Knöfler M. Wingless (Wnt)-3A induces trophoblast migration and matrix metalloproteinase-2 secretion through canonical Wnt signaling and protein kinase B/AKT activation. Endocrinology 2010; 151(1):211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Dash PR, Whitley S, Ayling L-J, Johnstone AP, Cartwright JE. Trophoblast apoptosis is inhibited by hepatocyte growth factor through the Akt and β-catenin mediated up-regulation of inducible nitric oxide synthase. Cell Signal 2005; 17(5):571–580. [DOI] [PubMed] [Google Scholar]
- 137. Dash PR, Cartwright JE, Baker PN, Johnstone AP, Whitley GS. Nitric oxide protects human extravillous trophoblast cells from apoptosis by a cyclic GMP-dependent mechanism and independently of caspase 3 nitrosylation. Exp Cell Res 2003; 287(2):314–324. [DOI] [PubMed] [Google Scholar]
- 138. Harris LK, McCormick J, Cartwright JE, Whitley GS, Dash PR. S-nitrosylation of proteins at the leading edge of migrating trophoblasts by inducible nitric oxide synthase promotes trophoblast invasion. Exp Cell Res 2008; 314(8):1765–1776. [DOI] [PubMed] [Google Scholar]
- 139. Staun-Ram E, Goldman S, Gabarin D, Shalev E. Expression and importance of matrix metalloproteinase 2 and 9 (MMP-2 and -9) in human trophoblast invasion. Reprod Biol Endocrinol 2004; 2(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Silva JF, Ocarino NM, Serakides R. Maternal thyroid dysfunction affects placental profile of inflammatory mediators and the intrauterine trophoblast migration kinetics. Reproduction 2014; 147(6):803–816. [DOI] [PubMed] [Google Scholar]
- 141. Bilban M, Haslinger P, Prast J, Klinglmüller F, Woelfel T, Haider S, Sachs A, Otterbein LE, Desoye G, Hiden U, Wagner O, Knöfler M. Identification of novel trophoblast invasion-related genes: heme oxygenase-1 controls motility via peroxisome proliferator-activated receptor γ. Endocrinology 2009; 150(2):1000–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Levytska K, Drewlo S, Baczyk D, Kingdom J. PPAR- gamma regulates trophoblast differentiation in the BeWo cell model. PPAR Res 2014; 2014:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. McCaig D, Lyall F. Inhibitors of heme oxygenase reduce invasion of human primary cytotrophoblast cells in vitro. Placenta 2009; 30(6):536–538. [DOI] [PubMed] [Google Scholar]
- 144. Tsoyi K, Ha YM, Kim YM, Lee YS, Kim HJ, Kim HJ, Seo HG, Lee JH, Chang KC. Activation of PPAR-gamma by carbon monoxide from CORM-2 leads to the inhibition of iNOS but not COX-2 expression in LPS-stimulated macrophages. Inflammation 2009; 32(6):364–371. [DOI] [PubMed] [Google Scholar]
- 145. Barber A, Robson SC, Myatt L, Bulmer JN, Lyall F. Heme oxygenase expression in human placenta and placental bed: reduced expression of placenta endothelial HO-2 in preeclampsia and fetal growth restriction. FASEB J 2001; 15(7):1158–1168. [DOI] [PubMed] [Google Scholar]
- 146. Kaartokallio T, Klemetti MM, Timonen A, Uotila J, Heinonen S, Kajantie E, Kere J, Kivinen K, Pouta A, Lakkisto P, Laivuori H. Microsatellite polymorphism in the heme oxygenase-1 promoter is associated with nonsevere and late-onset preeclampsia. Hypertension 2014; 64(1):172–177. [DOI] [PubMed] [Google Scholar]
- 147. Zhao H, Azuma J, Kalish F, Wong RJ, Stevenson DK. Maternal heme oxygenase 1 regulates placental vasculature development via angiogenic factors in mice. Biol Reprod 2011; 85(5):1005–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Chau K, Hennessy A, Makris A. Placental growth factor and pre-eclampsia. J Hum Hypertens 2017; 31(12):782–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. George EM, Hosick PA, Stec DE, Granger JP. Heme oxygenase inhibition increases blood pressure in pregnant rats. Am J Hypertens 2013; 26(7):924–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Hu TX, Wang G, Guo XJ, Sun QQ, He P, Gu H, Huang Y, Gao L, Ni X. MiR 20a,-20b and -200c are involved in hydrogen sulfide stimulation of VEGF production in human placental trophoblasts. Placenta 2016; 39:101–110. [DOI] [PubMed] [Google Scholar]
- 151. Hu TX, Guo X, Wang G, Gao L, He P, Xia Y, Gu H, Ni X. MiR133b is involved in endogenous hydrogen sulfide suppression of sFlt-1 production in human placenta. Placenta 2017; 52:33–40. [DOI] [PubMed] [Google Scholar]
- 152. Liao WX, Feng L, Zheng J, Chen DB. Deciphering mechanisms controlling placental artery endothelial cell migration stimulated by vascular endothelial growth factor. Endocrinology 2010; 151(7):3432–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Chen DB, Feng L, Hodges JK, Lechuga TJ, Zhang H. Human trophoblast-derived hydrogen sulfide stimulates placental artery endothelial cell angiogenesis†. Biol Reprod 2017; 97(3):478–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Sand A, Andersson E, Fried G. Nitric oxide donors mediate vasodilation in human placental arteries partly through a direct effect on potassium channels. Placenta 2006; 27(2-3):181–190. [DOI] [PubMed] [Google Scholar]
- 155. Bouloumié A, Schini-Kerth VB, Busse R. Vascular endothelial growth factor up-regulates nitric oxide synthase expression in endothelial cells. Cardiovasc Res 1999; 41(3):773–780. [DOI] [PubMed] [Google Scholar]
- 156. Boeldt DS, Krupp J, Yi FX, Khurshid N, Shah DM, Bird IM. Positive versus negative effects of VEGF(165) on Ca(2+) signaling and NO production in human endothelial cells. Am J Physiol Heart Circ Physiol 2017; 312(1):H173–H181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Krause BJ, Carrasco-Wong I, Caniuguir A, Carvajal J, Farías M, Casanello P. Endothelial eNOS/arginase imbalance contributes to vascular dysfunction in IUGR umbilical and placental vessels. Placenta 2013; 34(1):20–28. [DOI] [PubMed] [Google Scholar]
- 158. Krause BJ, Costello PM, Muñoz-Urrutia E, Lillycrop KA, Hanson MA, Casanello P. Role of DNA methyltransferase 1 on the altered eNOS expression in human umbilical endothelium from intrauterine growth restricted fetuses. Epigenetics 2013; 8(9):944–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Krupp J, Boeldt DS, Yi FX, Grummer MA, Bankowski Anaya HA, Shah DM, Bird IM. The loss of sustained Ca(2+) signaling underlies suppressed endothelial nitric oxide production in preeclamptic pregnancies: implications for new therapy. Am J Physiol Heart Circ Physiol 2013; 305(7):H969–H979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Bainbridge SA, Farley AE, McLaughlin BE, Graham CH, Marks GS, Nakatsu K, Brien JF, Smith GN. Carbon monoxide decreases perfusion pressure in isolated human placenta. Placenta 2002; 23(8-9):563–569. [DOI] [PubMed] [Google Scholar]
- 161. McCaig D, Lyall F. Heme Heme oxygenase expression in human placental villous tissue in response to exposure to in vitro ischemia-reperfusion injury. Hypertens Pregnancy 2009; 28(3):256–272. [DOI] [PubMed] [Google Scholar]
- 162. Lyall F, Bulmer JN, Kelly H, Duffie E, Robson SC. Human trophoblast invasion and spiral artery transformation. Am J Pathol 1999; 154(4):1105–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Lyall F, Barber A, Myatt L, Bulmer JN, Robson SC. Hemeoxygenase expression in human placenta and placental bed implies a role in regulation of trophoblast invasion and placental function. FASEB J 2000; 14(1):208–219. [DOI] [PubMed] [Google Scholar]
- 164. McLaughlin BE, Lash GE, Smith GN, Marks GS, Nakatsu K, Graham CH, Brien JF. Heme oxygenase expression in selected regions of term human placenta. Exp Biol Med (Maywood) 2003; 228(5):564–567. [DOI] [PubMed] [Google Scholar]
- 165. Cindrova-Davies T, Herrera EA, Niu Y, Kingdom J, Giussani DA, Burton GJ. Reduced cystathionine γ-lyase and increased miR-21 expression are associated with increased vascular resistance in growth-restricted pregnancies: hydrogen sulfide as a placental vasodilator. Am J Pathol 2013; 182(4):1448–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Lu L, Kingdom J, Burton GJ, Cindrova-Davies T. Placental stem villus arterial remodeling associated with reduced hydrogen sulfide synthesis contributes to human fetal growth restriction. Am J Pathol 2017; 187(4):908–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Kenney MJ, Weiss ML, Haywood JR. The paraventricular nucleus: an important component of the central neurocircuitry regulating sympathetic nerve outflow. Acta Physiol Scand 2003; 177(1):7–15. [DOI] [PubMed] [Google Scholar]
- 168. Holbein WW, Blackburn MB, Andrade MA, Toney GM. Burst patterning of hypothalamic paraventricular nucleus-driven sympathetic nerve activity in ANG II-salt hypertension. Am J Physiol Heart Circ Physiol 2018; 314(3):H530–H541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Heesch CM, Zheng H, Foley CM, Mueller PJ, Hasser EM, Patel KP. Nitric oxide synthase activity and expression are decreased in the paraventricular nucleus of pregnant rats. Brain Res 2009; 1251:140–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Rossi NF, Black SM, Telemaque-Potts S, Chen H. Neuronal nitric oxide synthase activity in the paraventricular nucleus buffers central endothelin-1- induced pressor response and vasopressin secretion. J Cardiovasc Pharmacol 2004; 44(Supplement 1):S283–S288. [DOI] [PubMed] [Google Scholar]
- 171. Shi Z, Cassaglia PA, Gotthardt LC, Brooks VL. Hypothalamic paraventricular and arcuate nuclei contribute to elevated sympathetic nerve activity in pregnant rats. Hypertension 2015; 66(6):1191–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Jarvis SS, Shibata S, Bivens TB, Okada Y, Casey BM, Levine BD, Fu Q. Sympathetic activation during early pregnancy in humans. J Physiol 2012; 590(15):3535–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Kuo CD, Chen GY, Yang MJ, Lo HM, Tsai YS. Biphasic changes in autonomic nervous activity during pregnancy. Br J Anaesth 2000; 84(3):323–329. [DOI] [PubMed] [Google Scholar]
- 174. Mizuno T, Tamakoshi K, Tanabe K. Anxiety during pregnancy and autonomic nervous system activity: a longitudinal observational and cross-sectional study. J Psychosom Res 2017; 99:105–111. [DOI] [PubMed] [Google Scholar]
- 175. Avery ND, Wolfe LA, Amara CE, Davies GA, McGrath MJ. Effects of human pregnancy on cardiac autonomic function above and below the ventilatory threshold. J Appl Physiol 2001; 90(1):321–328. [DOI] [PubMed] [Google Scholar]
- 176. Zanzinger J, Czachurski J, Seller H. Neuronal nitric oxide reduces sympathetic excitability by modulation of central glutamate effects in pigs. Circ Res 1997; 80(4):565–571. [DOI] [PubMed] [Google Scholar]
- 177. Kristiansson P, Wang JX. Reproductive hormones and blood pressure during pregnancy. Hum Reprod 2001; 16(1):13–17. [DOI] [PubMed] [Google Scholar]
- 178. Goldsmith LT, Weiss G, Palejwala S, Plant TM, Wojtczuk A, Lambert WC, Ammur N, Heller D, Skurnick JH, Edwards D, Cole DM. Relaxin regulation of endometrial structure and function in the rhesus monkey. Proc Natl Acad Sci USA 2004; 101(13):4685–4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Conrad KP. G-Protein-coupled receptors as potential drug candidates in preeclampsia: targeting the relaxin/insulin-like family peptide receptor 1 for treatment and prevention. Hum Reprod Update 2016; 22(5):647–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. McGuane JT, Debrah JE, Sautina L, Jarajapu YP, Novak J, Rubin JP, Grant MB, Segal M, Conrad KP. Relaxin induces rapid dilation of rodent small renal and human subcutaneous arteries via PI3 kinase and nitric oxide. Endocrinology 2011; 152(7):2786–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Gingerich S, Krukoff TL. Estrogen modulates endothelial and neuronal nitric oxide synthase expression via an estrogen receptor beta-dependent mechanism in hypothalamic slice cultures. Endocrinology 2005; 146(7):2933–2941. [DOI] [PubMed] [Google Scholar]
- 182. MacRitchie AN, Jun SS, Chen Z, German Z, Yuhanna IS, Sherman TS, Shaul PW. Estrogen upregulates endothelial nitric oxide synthase gene expression in fetal pulmonary artery endothelium. Circ Res 1997; 81(3):355–362. [DOI] [PubMed] [Google Scholar]
- 183. Wyckoff MH, Chambliss KL, Mineo C, Yuhanna IS, Mendelsohn ME, Mumby SM, Shaul PW. Plasma membrane estrogen receptors are coupled to endothelial nitric-oxide synthase through Galpha(i). J Biol Chem 2001; 276(29):27071–27076. [DOI] [PubMed] [Google Scholar]
- 184. Haynes MP, Sinha D, Russell KS, Collinge M, Fulton D, Morales-Ruiz M, Sessa WC, Bender JR. Membrane estrogen receptor engagement activates endothelial nitric oxide synthase via the PI3-kinase-Akt pathway in human endothelial cells. Circ Res 2000; 87(8):677–682. [DOI] [PubMed] [Google Scholar]
- 185. Haynes MP, Li L, Sinha D, Russell KS, Hisamoto K, Baron R, Collinge M, Sessa WC, Bender JR. Src kinase mediates phosphatidylinositol 3-kinase/Akt-dependent rapid endothelial nitric-oxide synthase activation by estrogen. J Biol Chem 2003; 278(4):2118–2123. [DOI] [PubMed] [Google Scholar]
- 186. Pastore MB, Talwar S, Conley MR, Magness RR. Identification of differential ER-alpha versus ER-beta mediated activation of eNOS in ovine uterine artery endothelial cells. Biol Reprod 2016; 94(6):139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Nelson SH, Steinsland OS, Wang Y, Yallampalli C, Dong YL, Sanchez JM. Increased nitric oxide synthase activity and expression in the human uterine artery during pregnancy. Circ Res 2000; 87(5):406–411. [DOI] [PubMed] [Google Scholar]
- 188. Zhang HH, Chen JC, Sheibani L, Lechuga TJ, Chen DB. Pregnancy augments VEGF-Stimulated in vitro angiogenesis and vasodilator (NO and H2S) production in human uterine artery endothelial cells. J Clin Endocrinol Metab 2017; 102(7):2382–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Chen DB, Jia S, King AG, Barker A, Li SM, Mata-Greenwood E, Zheng J, Magness RR. Global protein expression profiling underlines reciprocal regulation of caveolin 1 and endothelial nitric oxide synthase expression in ovariectomized sheep uterine artery by estrogen/progesterone replacement therapy. Biol Reprod 2006; 74(5):832–838. [DOI] [PubMed] [Google Scholar]
- 190. Luo K, Thaete LG, Neerhof MG. Endothelin receptor a antagonism and fetal growth in endothelial nitric oxide synthase gene knockout maternal and fetal mice. Reprod Sci 2016; 23(8):1028–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Kreiser D, Baum M, Seidman DS, Fanaroff A, Shah D, Hendler I, Stevenson DK, Schiff E, Druzin ML. End tidal carbon monoxide levels are lower in women with gestational hypertension and pre-eclampsia. J Perinatol 2004; 24(4):213–217. [DOI] [PubMed] [Google Scholar]
- 192. Zhao H, Wong RJ, Doyle TC, Nayak N, Vreman HJ, Contag CH, Stevenson DK. Regulation of maternal and fetal hemodynamics by heme oxygenase in mice. Biol Reprod 2008; 78(4):744–751. [DOI] [PubMed] [Google Scholar]
- 193. Wang K, Ahmad S, Cai M, Rennie J, Fujisawa T, Crispi F, Baily J, Miller MR, Cudmore M, Hadoke PW, Wang R, Gratacós E et al.. Dysregulation of hydrogen sulfide producing enzyme cystathionine γ-lyase contributes to maternal hypertension and placental abnormalities in preeclampsia. Circulation 2013; 127(25):2514–2522. [DOI] [PubMed] [Google Scholar]
- 194. Holwerda KM, Bos EM, Rajakumar A, Ris-Stalpers C, van Pampus MG, Timmer A, Erwich JJ, Faas MM, van Goor H, Lely AT. Hydrogen sulfide producing enzymes in pregnancy and preeclampsia. Placenta 2012; 33(6):518–521. [DOI] [PubMed] [Google Scholar]
- 195. Yuan S, Pardue S, Shen X, Alexander JS, Orr AW, Kevil CG. Hydrogen sulfide metabolism regulates endothelial solute barrier function. Redox Biol 2016; 9:157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Holwerda KM, Burke SD, Faas MM, Zsengeller Z, Stillman IE, Kang PM, van Goor H, McCurley A, Jaffe IZ, Karumanchi SA, Lely AT. Hydrogen sulfide attenuates sFlt1-induced hypertension and renal damage by upregulating vascular endothelial growth factor. J Am Soc Nephrol 2014; 25(4):717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Gade AR, Kang M, Akbarali HI. Hydrogen sulfide as an allosteric modulator of ATP-sensitive potassium channels in colonic inflammation. Mol Pharmacol 2013; 83(1):294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Kang M, Hashimoto A, Gade A, Akbarali HI. Interaction between hydrogen sulfide-induced sulfhydration and tyrosine nitration in the K(ATP) channel complex. Am J Physiol Gastrointest Liver Physiol 2015; 308(6):G532–G539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Xiao D, Longo LD, Zhang L. Role of KATP and L-type Ca2+ channel activities in regulation of ovine uterine vascular contractility: effect of pregnancy and chronic hypoxia. Am J Obstet Gynecol 2010; 203(6):596.e6–596.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Sheibani L, Lechuga TJ, Zhang H, Hameed A, Wing DA, Kumar S, Rosenfeld CR, Chen DB. Augmented H2S production via cystathionine-beta-synthase upregulation plays a role in pregnancy-associated uterine vasodilation†. Biol Reprod 2017; 96(3):664–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Tang G, Wu L, Liang W, Wang R. Direct stimulation of K(ATP) channels by exogenous and endogenous hydrogen sulfide in vascular smooth muscle cells. Mol Pharmacol 2005; 68(6):1757–1764. [DOI] [PubMed] [Google Scholar]
- 202. Kloesch B, Steiner G, Mayer B, Schmidt K. Hydrogen sulfide inhibits endothelial nitric oxide formation and receptor ligand-mediated Ca2+ release in endothelial and smooth muscle cells. Pharmacol Rep 2016; 68(1):37–43. [DOI] [PubMed] [Google Scholar]
- 203. Roberts JM. Pathophysiology of ischemic placental disease. Semin Perinatol 2014; 38(3):139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Grummer MA, Sullivan JA, Magness RR, Bird IM, Vascular endothelial growth factor acts through novel, pregnancy-enhanced receptor signalling pathways to stimulate endothelial nitric oxide synthase activity in uterine artery endothelial cells. Biochem J 2009; 417(2):501–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Boeldt DS, Grummer MA, Magness RR, Bird IM. Altered VEGF-stimulated Ca2+ signaling in part underlies pregnancy-adapted eNOS activity in UAEC. J Endocrinol 2014; 223(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Morris NH, Sooranna SR, Learmont JG, Poston L, Ramsey B, Pearson JD, Steer PJ, Nitric oxide synthase activities in placental tissue from normotensive, pre‐eclamptic and growth retarded pregnancies. BrJ Obstet Gynaecol 1995; 102(9):711–714. [DOI] [PubMed] [Google Scholar]
- 207. Moffett A, Colucci F. Uterine NK cells: active regulators at the maternal-fetal interface. J Clin Invest 2014; 124(5):1872–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Gong X, Chen Z, Liu Y, Lu Q, Jin Z. Gene expression profiling of the paracrine effects of uterine natural killer cells on human endometrial epithelial cells. Int J Endocrinol 2014; 2014:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Schiessl B, Innes BA, Bulmer JN, Otun HA, Chadwick TJ, Robson SC, Lash GE. Localization of angiogenic growth factors and their receptors in the human placental bed throughout normal human pregnancy. Placenta 2009; 30(1):79–87. [DOI] [PubMed] [Google Scholar]
- 210. Furuke K, Burd PR, Horvath-Arcidiacono JA, Hori K, Mostowski H, Bloom ET. Human NK cells express endothelial nitric oxide synthase, and nitric oxide protects them from activation-induced cell death by regulating expression of TNF-alpha. J Immunol 1999; 163(3):1473–1480. [PubMed] [Google Scholar]
- 211. Zhao H, Kalish F, Wong RJ, Stevenson DK. Infiltration of myeloid cells in the pregnant uterus is affected by heme oxygenase-1. J Leukoc Biol 2017; 101(1):217–226. [DOI] [PubMed] [Google Scholar]
- 212. Erkers T, Stikvoort A, Uhlin M. Lymphocytes in placental tissues: immune regulation and translational possibilities for immunotherapy. Stem Cells Int 2017; 2017:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Sykes L, MacIntyre DA, Yap XJ, Ponnampalam S, Teoh TG, Bennett PR. Changes in the Th1-Th2 cytokine bias in pregnancy and the effects of the anti-inflammatory cyclopentenone prostaglandin 15-deoxy–prostaglandin. Mediat Inflamm 2012; 2012:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Chen Y, Lv W, Jia J, Wang J, Yang J. Evaluation of serum concentrations of tumor necrosis factor (TNF)-α, interleukin (IL)-2, IL-10, and nitric oxide (NO) during the estrous cycle, early pregnancy and abortion in goats. Anim Reprod Sci 2016; 174:73–79. [DOI] [PubMed] [Google Scholar]
- 215. Dixit VD, Parvizi N. Pregnancy stimulates secretion of adrenocorticotropin and nitric oxide from peripheral bovine lymphocytes. Biol Reprod 2001; 64(1):242–248. [DOI] [PubMed] [Google Scholar]
- 216. Chabannes D, Hill M, Merieau E, Rossignol J, Brion R, Soulillou JP, Anegon I, Cuturi MC. A role for heme oxygenase-1 in the immunosuppressive effect of adult rat and human mesenchymal stem cells. Blood 2007; 110(10):3691–3694. [DOI] [PubMed] [Google Scholar]
- 217. Schumacher A, Wafula PO, Teles A, El-Mousleh T, Linzke N, Zenclussen ML, Langwisch S, Heinze K, Wollenberg I, Casalis PA, Volk HD, Fest S et al.. Blockage of heme oxygenase-1 abrogates the protective effect of regulatory T cells on murine pregnancy and promotes the maturation of dendritic cells. PLoS One 2012; 7(8):e42301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Pae HO, Oh GS, Choi BM, Chae SC, Kim YM, Chung KR, Chung HT. Carbon monoxide produced by heme oxygenase-1 suppresses T cell proliferation via inhibition of IL-2 production. J Immunol 2004; 172(8):4744–4751. [DOI] [PubMed] [Google Scholar]
- 219. Rémy S, Blancou P, Tesson L, Tardif V, Brion R, Royer PJ, Motterlini R, Foresti R, Painchaut M, Pogu S, Gregoire M, Bach JM et al.. Carbon monoxide inhibits TLR-induced dendritic cell immunogenicity. J Immunol 2009; 182(4):1877–1884. [DOI] [PubMed] [Google Scholar]
- 220. Yang R, Qu C, Zhou Y, Konkel JE, Shi S, Liu Y, Chen C, Liu S, Liu D, Chen Y, Zandi E, Chen W et al.. Hydrogen sulfide promotes Tet1- and Tet2-Mediated Foxp3 demethylation to drive regulatory T cell differentiation and maintain immune homeostasis. Immunity 2015; 43(2):251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Menon R, Bonney EA, Condon J, Mesiano S, Taylor RN. Novel concepts on pregnancy clocks and alarms: redundancy and synergy in human parturition. Hum Reprod Update 2016; 22(5):535–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Smith R. Parturition. N Engl J Med 2007; 356(3):271–283. [DOI] [PubMed] [Google Scholar]
- 223. Buxton IL, Kaiser RA, Malmquist NA, Tichenor S. NO-induced relaxation of labouring and non-labouring human myometrium is not mediated by cyclic GMP. Br J Pharmacol 2001; 134(1):206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Kuenzli KA, Bradley ME, Buxton IL. Cyclic GMP-independent effects of nitric oxide on guinea-pig uterine contractility. Br J Pharmacol 1996; 119(4):737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Kakui K, Itoh H, Sagawa N, Yura S, Korita D, Takemura M, Miyamaoto Y, Saito Y, Nakao K, Fujii S. Augmented endothelial nitric oxide synthase (eNOS) protein expression in human pregnant myometrium: possible involvement of eNOS promoter activation by estrogen via both estrogen receptor (ER)α and ERβ. Mol Hum Reprod 2004; 10(2):115–122. [DOI] [PubMed] [Google Scholar]
- 226. Ogando D, Farina M, Ribeiro ML, Perez Martinez S, Cella M, Rettori V, Franchi A. Steroid hormones augment nitric oxide synthase activity and expression in rat uterus. Reprod Fertil Dev 2003; 15(5):269–274. [DOI] [PubMed] [Google Scholar]
- 227. Bartlett SR, Bennett PR, Campa JS, Dennes WJ, Slater DM, Mann GE, Poston L, Poston R. Expression of nitric oxide synthase isoforms in pregnant human myometrium. J Physiol 1999; 521(3):705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Aggelidou E, Hillhouse EW, Grammatopoulos DK. Up-regulation of nitric oxide synthase and modulation of the guanylate cyclase activity by corticotropin-releasing hormone but not urocortin II or urocortin III in cultured human pregnant myometrial cells. Proc Natl Acad Sci USA 2002; 99(5):3300–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Stuart-Smith K. Demystified. Nitric oxide. Mol Pathol 2002; 55(6):360–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Dong Y-L, Gangula PRR, Yallampalli C. Nitric oxide synthase isoforms in the rat uterus: differential regulation during pregnancy and labour. Reproduction 1996; 107(2):249–254. [DOI] [PubMed] [Google Scholar]
- 231. Buhimschi I, Ali M, Jain V, Chwalisz K, Garfield RE. Pregnancy: Differential regulation of nitric oxide in the rat uterus and cervix during pregnancy and labour. Hum Reprod 1996; 11(8):1755–1766. [DOI] [PubMed] [Google Scholar]
- 232. Chávez-Genaro R, Crutcher K, Viettro L, Richeri A, Coirolo N, Burnstock G, Cowen T, Brauer MM, Differential effects of oestrogen on developing and mature uterine sympathetic nerves. Cell Tissue Res 2002; 308(1):61–73. [DOI] [PubMed] [Google Scholar]
- 233. Barcena de Arellano ML, Oldeweme J, Arnold J, Schneider A, Mechsner S. Remodeling of estrogen-dependent sympathetic nerve fibers seems to be disturbed in adenomyosis. Fertil Steril 2013; 100(3):801–809.e2. [DOI] [PubMed] [Google Scholar]
- 234. Nakanishi H, Wood C. Cholinergic mechanisms in the human uterus. BrJ Obstet Gynaecol 1971; 78(8):716–723. [DOI] [PubMed] [Google Scholar]
- 235. Wikland M, Lindblom B, Dahlström A, Haglid KG. Structural and functional evidence for the denervation of human myometrium during pregnancy. Obstet Gynecol 1984; 64(4):503–509. [PubMed] [Google Scholar]
- 236. Morizaki N, Morizaki J, Hayashi RH, Garfield RE. A functional and structural study of the innervation of the human uterus. Am J Obstet Gynecol 1989; 160(1):218–228. [DOI] [PubMed] [Google Scholar]
- 237. Acevedo CH, Ahmed A. Hemeoxygenase-1 inhibits human myometrial contractility via carbon monoxide and is upregulated by progesterone during pregnancy. J Clin Invest 1998; 101(5):949–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Longo M, Jain V, Vedernikov YP, Saade GR, Goodrum L, Facchinetti F, Garfield RE. Effect of nitric oxide and carbon monoxide on uterine contractility during human and rat pregnancy. Am J Obstet Gynecol 1999; 181(4):981–988. [DOI] [PubMed] [Google Scholar]
- 239. You XJ, Xu C, Lu JQ, Zhu XY, Gao L, Cui XR, Li Y, Gu H, Ni X. Expression of cystathionine β-synthase and cystathionine γ-lyase in human pregnant myometrium and their roles in the control of uterine contractility. PLoS One 2011; 6(8):e23788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. You X, Chen Z, Zhao H, Xu C, Liu W, Sun Q, He P, Gu H, Ni X. Endogenous hydrogen sulfide contributes to uterine quiescence during pregnancy. Reproduction 2017; 153(5):535–543. [DOI] [PubMed] [Google Scholar]
- 241. Burgstaller G, Gregor M, Winter L, Wiche G. Keeping the vimentin network under control: cell–matrix adhesion–associated plectin 1f affects cell shape and polarity of fibroblasts. Mol Biol Cell 2010; 21(19):3362–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Paulin D, Li Z, Desmin: a major intermediate filament protein essential for the structural integrity and function of muscle. Exp Cell Res 2004; 301(1):1–7. [DOI] [PubMed] [Google Scholar]
- 243. Assinder SJ, Stanton J-AL, Prasad PD. Transgelin: An actin-binding protein and tumour suppressor. Int J Biochem Cell Biol 2009; 41(3):482–486. [DOI] [PubMed] [Google Scholar]
- 244. Ulrich C, Quilici DR, Schlauch KA, Buxton IL. The human uterine smooth muscle S-nitrosoproteome fingerprint in pregnancy, labor and preterm labor. Am J Physiol Cell Physiol 2013; 305(8):C803–C816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Barnett SD, Smith CR, Ulrich CC, Baker JE, Buxton ILO. S-nitrosoglutathione reductase underlies the dysfunctional relaxation to nitric oxide in preterm labor. Sci Rep 2018; 8(1):5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Padgett CM, Whorton AR. S-nitrosoglutathione reversibly inhibits GAPDH by S-nitrosylation. Am J Physiol Cell Physiol 1995; 269(3):C739–C749. [DOI] [PubMed] [Google Scholar]
- 247. Kornberg MD, Sen N, Hara MR, Juluri KR, Nguyen JV, Snowman AM, Law L, Hester LD, Snyder SH. GAPDH mediates nitrosylation of nuclear proteins. Nat Cell Biol 2010; 12(11):1094–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Liu JZ, Duan J, Ni M, Liu Z, Qiu WL, Whitham SA, Qian WJ. S-Nitrosylation inhibits the kinase activity of tomato phosphoinositide-dependent kinase 1 (PDK1). J Biol Chem 2017; 292(48):19743–19751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Evangelista AM, Rao VS, Filo AR, Marozkina NV, Doctor A, Jones DR, Gaston B, Guilford WH. Direct regulation of striated muscle myosins by nitric oxide and endogenous nitrosothiols. PLoS One 2010; 5(6):e11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Zhang HH, Lechuga TJ, Tith T, Wang W, Wing DA, Chen DB. S-nitrosylation of cofilin-1 mediates estradiol-17β-stimulated endothelial cytoskeleton remodeling. Mol Endocrinol 2015; 29(3):434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Gabbiani G, Schmid E, Winter S, Chaponnier C, de Ckhastonay C, Vandekerckhove J, Weber K, Franke WW. Vascular smooth muscle cells differ from other smooth muscle cells: predominance of vimentin filaments and a specific alpha-type actin. Proc Natl Acad Sci USA 1981; 78(1):298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Lorca RA, Prabagaran M, England SK. Functional insights into modulation of BKCa channel activity to alter myometrial contractility. Front Physiol 2014; 5:289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Benkusky NA, Fergus DJ, Zucchero TM, England SK. Regulation of the Ca2+-sensitive domains of the maxi-K channel in the mouse myometrium during gestation. J Biol Chem 2000; 275(36):27712–27719. [DOI] [PubMed] [Google Scholar]
- 254. Babenko AP, Aguilar-Bryan L, Bryan J. A view of sur/KIR6.X, KATP channels. Annu Rev Physiol 1998; 601:667–687. [DOI] [PubMed] [Google Scholar]
- 255. Kuo IY, Ehrlich BE. Signaling in muscle contraction. Cold Spring Harb Perspect Biol 2015; 7(2):a006023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, Park SP, Lee J, Lee B, Kim BM, Raouf R, Shin YK et al.. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 2008; 455(7217):1210–1215. [DOI] [PubMed] [Google Scholar]
- 257. Shimano M, Nakaya Y, Fukui R, Kamada M, Hamada Y, Maeda K, Aono T. Activation of Ca2+-activated K+ channels in human myometrium by nitric oxide. Gynecol Obstet Invest 2000; 49(4):249–254. [DOI] [PubMed] [Google Scholar]
- 258. Zhou X-B, Wang G-X, Ruth P, Hüneke B, Korth M. BKCa channel activation by membrane-associated cGMP kinase may contribute to uterine quiescence in pregnancy. Am J Physiol Cell Physiol 2000; 279(6):C1751–C1759. [DOI] [PubMed] [Google Scholar]
- 259. Hong SH, Kyeong KS, Kim CH, Kim YC, Choi W, Yoo RY, Kim HS, Park YJ, Ji IW, Jeong EH, Kim HS, Xu WX et al.. Regulation of myometrial contraction by ATP-sensitive potassium (K(ATP)) channel via activation of SUR2B and Kir 6.2 in mouse. J Vet Med Sci 2016; 78(7):1153–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Xu C, You X, Gao L, Zhang L, Hu R, Hui N, Olson DM, Ni X. Expression of ATP-sensitive potassium channels in human pregnant myometrium. Reprod Biol Endocrinol 2011; 9(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Robinson H, Wray S, A new slow releasing, H2S generating compound, GYY4137 relaxes spontaneous and oxytocin-stimulated contractions of human and rat pregnant myometrium. PLoS One 2012; 7(9):e46278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Bernstein K, Vink JY, Fu XW, Wakita H, Danielsson J, Wapner R, Gallos G. Calcium-activated chloride channels anoctamin 1 and 2 promote murine uterine smooth muscle contractility. Am J Obstet Gynecol 2014; 211(6):688.e1–688.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Song J, Zhang X, Qi Z, Sun G, Chi S, Zhu Z, Ren J, Qiu Z, Liu K, Myatt L, Ma RZ. Cloning and characterization of a calcium-activated chloride channel in rat uterus. Biol Reprod 2009; 80(4):788–794. [DOI] [PubMed] [Google Scholar]
- 264. Luo Z, Ren J, Huang Z, Wang T, Xiang K, Cheng L, Tang L. The role of exogenous hydrogen sulfide in free fatty acids induced inflammation in macrophages. Cell Physiol Biochem 2017; 42(4):1635–1644. [DOI] [PubMed] [Google Scholar]
- 265. Huang Z, Zhuang X, Xie C, Hu X, Dong X, Guo Y, Li S, Liao X. Exogenous hydrogen sulfide attenuates high glucose-induced cardiotoxicity by inhibiting NLRP3 inflammasome activation by suppressing TLR4/NF-kappaB pathway in H9c2 cells. Cell Physiol Biochem 2016; 40(6):1578–1590. [DOI] [PubMed] [Google Scholar]
- 266. Sonne SR, Bhalla VK, Barman SA, White RE, Zhu S, Newman TM, Prasad PD, Smith SB, Offermanns S, Ganapathy V. Hyperhomocysteinemia is detrimental to pregnancy in mice and is associated with preterm birth. Biochim Biophys Acta 2013; 1832(8):1149–1158. [DOI] [PubMed] [Google Scholar]
- 267. Xiao Y, Rungruang S, Hall LW, Collier JL, Dunshea FR, Collier RJ. Effects of niacin and betaine on bovine mammary and uterine cells exposed to thermal shock in vitro. J Dairy Sci 2017; 100(5):4025–4037. [DOI] [PubMed] [Google Scholar]
- 268. El Maradny E, Kanayama N, Kobayashi H, Hossain B, Khatun S, Liping S, Kobayashi T, Terao T. The role of hyaluronic acid as a mediator and regulator of cervical ripening. Hum Reprod 1997; 12(5):1080–1088. [DOI] [PubMed] [Google Scholar]
- 269. Gomez-Lopez N, Tanaka S, Zaeem Z, Metz GA, Olson DM. Maternal circulating leukocytes display early chemotactic responsiveness during late gestation. BMC Pregnancy Childbirth 2013; 13(Suppl 1):S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Hunter PJ, Sheikh S, David AL, Peebles DM, Klein N. Cervical leukocytes and spontaneous preterm birth. J Reprod Immunol 2016; 113:42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Kishore AH, Liang H, Kanchwala M, Xing C, Ganesh T, Akgul Y, Posner B, Ready JM, Markowitz SD, Word RA. Prostaglandin dehydrogenase is a target for successful induction of cervical ripening. Proc Natl Acad Sci USA 2017; 114(31):E6427–E6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Törnblom SA, Klimaviciute A, Byström B, Chromek M, Brauner A, Ekman-Ordeberg G. Non-infected preterm parturition is related to increased concentrations of IL-6, IL-8 and MCP-1 in human cervix. Reprod Biol Endocrinol 2005; 3(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Santos KC, Monte AP, Lima JT, Ribeiro LA, Palheta Junior RC. Role of NO–cGMP pathway in ovine cervical relaxation induced by Erythroxylum caatingae Plowman. Anim Reprod Sci 2016; 164:23–30. [DOI] [PubMed] [Google Scholar]
- 274. Vink JY, Qin S, Brock CO, Zork NM, Feltovich HM, Chen X, Urie P, Myers KM, Hall TJ, Wapner R, Kitajewski JK, Shawber CJ et al.. A new paradigm for the role of smooth muscle cells in the human cervix. Am J Obstet Gynecol 2016; 215(4):478.e1–478.e11. [DOI] [PubMed] [Google Scholar]
- 275. Väisänen-Tommiska M, Nuutila M, Ylikorkala O. Cervical nitric oxide release in women postterm. Obstet Gynecol 2004; 103(4):657–662. [DOI] [PubMed] [Google Scholar]
- 276. Ledingham M-A, Thomson AJ, Young A, Macara LM, Greer IA, Norman JE. Changes in the expression of nitric oxide synthase in the human uterine cervix during pregnancy and parturition. Mol Hum Reprod 2000; 6(11):1041–1048. [DOI] [PubMed] [Google Scholar]
- 277. Yoshida M, Sagawa N, Itoh H, Yura S, Korita D, Kakui K, Hirota N, Sato T, Ito A, Fujii S. Nitric oxide increases matrix metalloproteinase-1 production in human uterine cervical fibroblast cells. Mol Hum Reprod 2001; 7(10):979–985. [DOI] [PubMed] [Google Scholar]
- 278. Coughlan T, Gibson C, Murphy S. Modulatory effects of progesterone on inducible nitric oxide synthase expression in vivo and in vitro. J Neurochem 2005; 93(4):932–942. [DOI] [PubMed] [Google Scholar]
- 279. Denison FC, Calder AA, Kelly RW. The action of prostaglandin E2 on the human cervix: stimulation of interleukin 8 and inhibition of secretory leukocyte protease inhibitor. Am J Obstet Gynecol 1999; 180(3):614–620. [DOI] [PubMed] [Google Scholar]
- 280. Ledingham MA, Denison FC, Kelly RW, Young A, Norman JE. Nitric oxide donors stimulate prostaglandin F2alpha and inhibit thromboxane B2 production in the human cervix during the first trimester of pregnancy. Mol Hum Reprod 1999; 5(10):973–982. [DOI] [PubMed] [Google Scholar]
- 281. Marx SG, Wentz MJ, Mackay LB, Schlembach D, Maul H, Fittkow C, Given R, Vedernikov Y, Saade GR, Garfield RE. Effects of progesterone on iNOS, COX-2, and collagen expression in the cervix. J Histochem Cytochem 2006; 54(6):623–639. [DOI] [PubMed] [Google Scholar]
- 282. Nadeem L, Shynlova O, Matysiak-Zablocki E, Mesiano S, Dong X, Lye S. Molecular evidence of functional progesterone withdrawal in human myometrium. Nat Commun 2016; 7(1):11565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Condon JC, Jeyasuria P, Faust JM, Wilson JW, Mendelson CR. A decline in the levels of progesterone receptor coactivators in the pregnant uterus at term may antagonize progesterone receptor function and contribute to the initiation of parturition. Proc Natl Acad Sci USA 2003; 100(16):9518–9523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Ekerhovd E, Weijdegård B, Brännström M, Mattsby-Baltzer I, Norström A. Nitric oxide induced cervical ripening in the human: Involvement of cyclic guanosine monophosphate, prostaglandin F2α, and prostaglandin E2. Am J Obstet Gynecol 2002; 186(4):745–750. [DOI] [PubMed] [Google Scholar]
- 285. Ekerhovd E, Brännström M, Weijdegârd B, Norström A. Nitric oxide synthases in the human cervix at term pregnancy and effects of nitric oxide on cervical smooth muscle contractility. Am J Obstet Gynecol 2000; 183(3):610–616. [DOI] [PubMed] [Google Scholar]
- 286. Ekerhovd E, Bullarbo M, Andersch B, Norström A. Vaginal administration of the nitric oxide donor isosorbide mononitrate for cervical ripening at term: a randomized controlled study. Am J Obstet Gynecol 2003; 189(6):1692–1697. [DOI] [PubMed] [Google Scholar]
- 287. Bullarbo M, Orrskog ME, Andersch B, Granström L, Norström A, Ekerhovd E. Outpatient vaginal administration of the nitric oxide donor isosorbide mononitrate for cervical ripening and labor induction postterm: a randomized controlled study. Am J Obstet Gynecol 2007; 196(1):50.e1–50.e5. [DOI] [PubMed] [Google Scholar]
- 288. Agarwal K, Batra A, Batra A, Dabral A, Aggarwal A. Evaluation of isosorbide mononitrate for cervical ripening prior to induction of labor for postdated pregnancy in an outpatient setting. Int J Gynecol Obstet 2012; 118(3):205–209. [DOI] [PubMed] [Google Scholar]
- 289. Cella M, Farina MG, Dominguez Rubio AP, Di Girolamo G, Ribeiro ML, Franchi AM. Dual effect of nitric oxide on uterine prostaglandin synthesis in a murine model of preterm labour. Br J Pharmacol 2010; 161(4):844–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Venkatesh KK, Cantonwine DE, Ferguson K, Arjona M, Meeker JD, McElrath TF. Inflammatory and oxidative stress markers associated with decreased cervical length in pregnancy. Am J Reprod Immunol 2016; 76(5):376–382. [DOI] [PubMed] [Google Scholar]
- 291. Heng YJ, Di Quinzio MK, Permezel M, Rice GE, Georgiou HM. Temporal expression of antioxidants in human cervicovaginal fluid associated with spontaneous labor. Antioxidants & Redox Signaling 2010; 13(7):951–957. [DOI] [PubMed] [Google Scholar]
- 292. Landino LM, Crews BC, Timmons MD, Morrow JD, Marnett LJ. Peroxynitrite, the coupling product of nitric oxide and superoxide, activates prostaglandin biosynthesis. Proc Natl Acad Sci USA 1996; 93(26):15069–15074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Mitchell B, Sharma R. Chapter 2 - How do the placenta and fetal membranes form?. In: Mitchell B, Sharma R (eds.) Embryology (2nd edn), Churchill Livingstone: Amsterdam, Netherlands; 2009: 9–14. [Google Scholar]
- 294. Bonney EA, Krebs K, Saade G, Kechichian T, Trivedi J, Huaizhi Y, Menon R. Differential senescence in feto-maternal tissues during mouse pregnancy. Placenta 2016; 43:26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Menon R, Boldogh I, Hawkins HK, Woodson M, Polettini J, Syed TA, Fortunato SJ, Saade GR, Papaconstantinou J, Taylor RN. Histological evidence of oxidative stress and premature senescence in preterm premature rupture of the human fetal membranes recapitulated in vitro. Am J Pathol 2014; 184(6):1740–1751. [DOI] [PubMed] [Google Scholar]
- 296. Adams Waldorf KM, Singh N, Mohan AR, Young RC, Ngo L, Das A, Tsai J, Bansal A, Paolella L, Herbert BR, Sooranna SR, Gough GM et al.. Uterine overdistention induces preterm labor mediated by inflammation: observations in pregnant women and nonhuman primates. Am J Obstet Gynecol 2015; 213(6):830.e1–830.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Yudin MH, van Schalkwyk J, Eyk NV. Antibiotic therapy in preterm premature rupture of the membranes. J Obstet Gynaecol Can 2009; 31(9):863–867. [DOI] [PubMed] [Google Scholar]
- 298. Marinoni E, Di Iorio R, Villaccio B, Alberini A, Rota F, Cosmi EV. Amniotic fluid nitric oxide metabolite levels and nitric oxide synthase localization in feto-placental tissues are modified in association with human labor. Eur J Obstet Gynecol Reprod Biol 2000; 89(1):47–54. [DOI] [PubMed] [Google Scholar]
- 299. Nanetti L, Raffaelli F, Giulietti A, Sforza G, Raffaele Giannubilo S, Ciavattini A, Tranquilli AL, Mazzanti L, Vignini A. Oxytocin, its antagonist Atosiban, and preterm labor: a role for placental nitric oxide. J Matern Fetal Neonatal Med 2015; 28(5):611–616. [DOI] [PubMed] [Google Scholar]
- 300. Yuan B, Ohyama K, Takeichi M, Toyoda H. Direct contribution of inducible nitric oxide synthase expression to apoptosis induction in primary smooth chorion trophoblast cells of human fetal membrane tissues. Int J Biochem Cell Biol 2009; 41(5):1062–1069. [DOI] [PubMed] [Google Scholar]
- 301. Yuan B, Ohyama K, Bessho T, Toyoda H. Contribution of inducible nitric oxide synthase and cyclooxygenase-2 to apoptosis induction in smooth chorion trophoblast cells of human fetal membrane tissues. Biochem Biophys Res Commun 2006; 341(3):822–827. [DOI] [PubMed] [Google Scholar]
- 302. Polettini J, Richardson LS, Menon R. Oxidative stress induces senescence and sterile inflammation in murine amniotic cavity. Placenta 2018; 63:26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Patel P, Vatish M, Heptinstall J, Wang R, Carson RJ. The endogenous production of hydrogen sulphide in intrauterine tissues. Reprod Biol Endocrinol 2009; 71:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Sun Q, Chen Z, He P, Li Y, Ding X, Huang Y, Gu H, Ni X. Reduced expression of hydrogen sulfide–generating enzymes down-regulates 15-hydroxyprostaglandin dehydrogenase in chorion during term and preterm labor. Am J Pathol 2018; 188(1):63–71. [DOI] [PubMed] [Google Scholar]
- 305. Kwiatkoski M, Soriano RN, Araujo RM, Azevedo LU, Batalhao ME, Francescato HD, Coimbra TM, Carnio EC, Branco LG. Hydrogen sulfide inhibits preoptic prostaglandin E2 production during endotoxemia. Exp Neurol 2013; 240:88–95. [DOI] [PubMed] [Google Scholar]
- 306. Osol G, Mandala M. Maternal uterine vascular remodeling during pregnancy. Physiology (Bethesda) 2009; 24:58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Li Y, Zhou X, Wei QW, Huang RH, Shi FX. Cell-specific expression and immunolocalization of nitric oxide synthase isoforms and soluble guanylyl cyclase α and β subunits in postnatal porcine uteri. Acta Histochem 2014; 116(3):466–473. [DOI] [PubMed] [Google Scholar]
- 308. Zhang YH, He M, Wang Y, Liao AH. Modulators of the balance between M1 and M2 macrophages during pregnancy. Front Immunol 2017; 8:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Lu S, Gao Y, Huang X, Wang X. GYY4137, a hydrogen sulfide (H(2)S) donor, shows potent anti-hepatocellular carcinoma activity through blocking the STAT3 pathway. Int J Oncol 2014; 44(4):1259–1267. [DOI] [PubMed] [Google Scholar]
- 310. Weiner ID, Mitch WE, Sands JM. Urea and ammonia metabolism and the control of renal nitrogen excretion. Clin J Am Soc Nephrol 2015; 10(8):1444–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Boros M, Tuboly E, Mészáros A, Amann A. The role of methane in mammalian physiology—is it a gasotransmitter? J Breath Res 2015; 9(1):014001. [DOI] [PubMed] [Google Scholar]
- 312. Steiger AK, Marcatti M, Szabo C, Szczesny B, Pluth MD. Inhibition of mitochondrial bioenergetics by esterase-triggered COS/H2S donors. ACS Chem Biol 2017; 12(8):2117–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. DeMartino AW, Zigler DF, Fukuto JM, Ford PC. Carbon disulfide. Just toxic or also bioregulatory and/or therapeutic? Chem Soc Rev 2017; 46(1):21–39. [DOI] [PubMed] [Google Scholar]
- 314. Sitdikova GF, Fuchs R, Kainz V, Weiger TM, Hermann A. Phosphorylation of BK channels modulates the sensitivity to hydrogen sulfide (H2S). Front Physiol 2014; 5:431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Lajin B, Francesconi KA. The hydrogen sulfide metabolite trimethylsulfonium is found in human urine. Sci Rep 2016; 6(1):27038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Mijušković A, Oreščanin-Dušić Z, Nikolić-Kokić A, Slavić M, Spasić MB, Spasojević I, Blagojević D. Comparison of the effects of methanethiol and sodium sulphide on uterine contractile activity. Pharmacol Rep 2014; 66(3):373–379. [DOI] [PubMed] [Google Scholar]
- 317. Akashi K, Miyake C, Yokota A. Citrulline, a novel compatible solute in drought-tolerant wild watermelon leaves, is an efficient hydroxyl radical scavenger. FEBS Lett 2001; 508(3):438–442. [DOI] [PubMed] [Google Scholar]
- 318. Ishii I, Akahoshi N, Yamada H, Nakano S, Izumi T, Suematsu M. Cystathionine gamma-Lyase-deficient mice require dietary cysteine to protect against acute lethal myopathy and oxidative injury. J Biol Chem 2010; 285(34):26358–26368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. de Valk HW, van Eeden MK, Banga JD, van der Griend R, de Groot E, Haas FJ, Meuwissen OJ, Duran M, Smeitink JA, Poll-The BT, de Klerk JB, Wittebol-Post D et al.. Evaluation of the presence of premature atherosclerosis in adults with heterozygosity for cystathionine-beta-synthase deficiency. Stroke 1996; 27(6):1134–1136. [PubMed] [Google Scholar]
- 320. Ziberna L, Martelanc M, Franko M, Passamonti S. Bilirubin is an endogenous antioxidant in human vascular endothelial cells. Sci Rep 2016; 6(1):29240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Boer R, Ulrich WR, Klein T, Mirau B, Haas S, Baur I. The inhibitory potency and selectivity of arginine substrate site nitric-oxide synthase inhibitors is solely determined by their affinity toward the different isoenzymes. Mol Pharmacol 2000; 58(5):1026–1034. [PubMed] [Google Scholar]
- 322. Eroglu E, Charoensin S, Bischof H, Ramadani J, Gottschalk B, Depaoli MR, Waldeck-Weiermair M, Graier WF, Malli R. Genetic biosensors for imaging nitric oxide in single cells. Free Radic Biol Med 2018; 128:50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Wang J, Karpus J, Zhao BS, Luo Z, Chen PR, He C. A selective fluorescent probe for carbon monoxide imaging in living cells. Angew Chem Int Ed 2012; 51(38):9652–9656. [DOI] [PubMed] [Google Scholar]
- 324. Chen Z-J, Ai H-W. A highly responsive and selective fluorescent probe for imaging physiological hydrogen sulfide. Biochemistry 2014; 53(37):5966–5974. [DOI] [PubMed] [Google Scholar]
- 325. Velázquez-Moyado JA, Navarrete A. The detection and quantification, in vivo and in real time, of hydrogen sulfide in ethanol-induced lesions in rat stomachs using an ion sensitive electrode. J Pharmacol Toxicol Methods 2018; 89:54–58. [DOI] [PubMed] [Google Scholar]
- 326. Hetrick EM, Schoenfisch MH. Analytical chemistry of nitric oxide. Annual Rev Anal Chem 2009; 2(1):409–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Bailey TS, Pluth MD. Chemiluminescent detection of enzymatically produced hydrogen sulfide: substrate hydrogen bonding influences selectivity for H2S over biological thiols. J Am Chem Soc 2013; 135(44):16697–16704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Cao J, Lopez R, Thacker JM, Moon JY, Jiang C, Morris SN, Bauer JH, Tao P, Mason RP, Lippert AR. Chemiluminescent probes for imaging H2S in living animals. Chem Sci 2015; 6(3):1979–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Shin JH, Privett BJ, Kita JM, Wightman RM, Schoenfisch MH. Fluorinated xerogel-derived microelectrodes for amperometric nitric oxide sensing. Anal Chem 2008; 80(18):6850–6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Seto H, Kondo T, Yuasa M. Sensitive and selective electrochemical detection of carbon monoxide in saline at a Pt-Ru/Nafion/MnO2-modified electrode. Anal Sci 2012; 28(2):115–120. [DOI] [PubMed] [Google Scholar]
- 331. Doeller JE, Isbell TS, Benavides G, Koenitzer J, Patel H, Patel RP, Lancaster JR Jr, Darley-Usmar VM, Kraus DW. Polarographic measurement of hydrogen sulfide production and consumption by mammalian tissues. Anal Biochem 2005; 341(1):40–51. [DOI] [PubMed] [Google Scholar]
- 332. Kelm M. Nitric oxide metabolism and breakdown. Biochim Biophys Acta 1999; 1411(2-3):273–289. [DOI] [PubMed] [Google Scholar]
- 333. Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev 2012; 92(2):791–896. [DOI] [PubMed] [Google Scholar]
- 334. Hunter RA, Storm WL, Coneski PN, Schoenfisch MH. Inaccuracies of nitric oxide measurement methods in biological media. Anal Chem 2013; 85(3):1957–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]