

Abstract
Liver X receptor alpha (LXRα) plays important roles in lipid metabolism and inflammation. Therefore, it is essential for protection against atherosclerosis (AS). In AS plaques, the key cells involved in lipid metabolism and inflammation are macrophages. However, the mechanism by which LXRα regulates macrophage involvement in AS formation remains unclear. In this study, we first confirmed the effects of an LXRα agonist (T0901317) and antagonist (GSK2033) on foam cell formation and inflammation in vivo and in vitro. Indeed, T0901317 reduced the number of macrophages in AS plaques and decreased the number of migrated macrophages, as assessed using an in vitro transwell assay. Next, we investigated the relationship between the reduction in macrophages in AS plaques and cytokine levels or foam cell formation. The results show that T0901317 reduced the number of high cholesterol-induced M1 macrophages by converting them into M2 macrophages in vivo and in vitro. Due to this phenotypic transition of macrophages, the inflammatory response was alleviated, and lipid metabolism was enhanced in AS plaques. This effect was achieved by promoting the expression of reverse transporters (ATP-binding cassette transporter member 1 and ATP-binding cassette subfamily G member 1) and inhibiting the phosphorylation of nuclear factor-κB-mediated phosphorylation.
Keywords: Atherosclerosis, macrophages, LXRα, inflammation, plaques
Introduction
Cholesterol metabolism, including biosynthesis, catabolism, and reverse cholesterol transport (RCT), is closely related to a large number of diseases, such as atherosclerosis (AS). At the same time, an excessive inflammatory response also increases the risk of AS. RCT plays a key role in the formation of AS plaques and the necrotic core. In turn, cytokines and chemokines can damage endothelial cells and smooth muscle cells, thus leading to the increased recruitment of macrophages that promote AS formation. In addition to participating in RCT, macrophages play a role in the regulation of inflammation, which further contributes to the formation of AS [1,2].
LXRα belongs to a subgroup of nuclear receptors and is considered to play an important role in the regulation of lipid metabolism [3]. Target genes of LXRα have been shown to regulate the assimilation, transportation, and biosynthesis of lipids. LXRα is mainly distributed in the liver, intestine, and adipose tissue, and the lack of LXRα leads to the accumulation of cholesterol in the liver [4]. The biological functions of LXRα include the following: (1) regulation of cholesterol transport; (2) regulation of lipid absorption and transformation; (3) regulation of lipid synthesis; (4) modulation of inflammatory factor expression; (5) regulation of carbohydrate metabolism; and (6) autocatalytic function. The related functions of LXRα play an important role in regulating the occurrence of AS. Currently, the clinical treatment for AS is to directly induce the utilization of cholesterol by liver cells and to reduce the content of low-density lipoprotein (LDL) cholesterol. However, these drugs have many side effects. Recent studies have investigated the use of LXRα agonists to increase high-density lipoprotein (HDL) cholesterol levels and to lower plasma cholesterol levels [5,6]. However, these studies have mainly focused on role of LXRα in regulating blood lipids and liver cholesterol metabolism, and the role of LXRα in regulating macrophages in AS plaques remains poorly understood.
The ATP-binding cassette (ABC) protein family is directly related to cholesterol transport, as it can transport cholesterol from the cytosol to extracellular receptors. For example, ABC transporter member 1 (ABCA1) can transfer cytosolic cholesterol to apoprotein to form HDL cholesterol. ABCA1 is widely distributed in the tissue and is mainly expressed on the surface of macrophages and monocytes. Animal experiments have shown that mice fed LXRα agonists for 12 hours have significantly increased lipoprotein lipase expression levels in hepatocytes and macrophages [7,8]. The target genes of LXRα are involved in various processes, such as absorption, transport, transformation, and lipid synthesis. Furthermore, LXRα can bind to the promoter region of tumor necrosis factor alpha (TNFα) and promote the expression of TNFα in macrophages [9,10].
Based on the function of LXRα in regulating lipid metabolism and the inflammatory response, we decided to target macrophages in AS plaques and focused on whether LXRα affects the development of AS by regulating macrophage function. Overall, our results demonstrate that the LXRα agonist reduced the area of AS plaques and delayed the development of AS by inducing the conversion of M1 macrophages into M2 macrophages. This effect was achieved by promoting the expression of reverse transporters (ABCA1 and ABCG1) and inhibiting the phosphorylation of NF-κB. Therefore, our study aimed to investigate the role of macrophages in the development of AS and to assess the role of liver X receptor alpha (LXRα) in the regulation of RCT and inflammatory response at the macrophage level.
Materials and methods
Apoe-/- mice and LXRα agonists/antagonists
Eighty adult male Apoe-/- C57BL/6 mice (8 months old, 20-25 g) were used for the following experiments: western blot analysis and enzyme-linked immunosorbent assay (ELISA; n = 20), immunohistochemistry (n = 40), and transmission electron microscope studies (n = 20). C57BL/6 mice were obtained from the model animal research center of Nanjing University. Experimental animals were divided into four groups: Apoe-/- mice on a normal diet (control group), Apoe-/- mice on a high-fat diet (model group), T0901317-treated (Sigma, CA, USA) Apoe-/- mice on a high-fat diet (Sigma, CA, USA) (T0901317 group), and GSK2033-treated (Sigma, CA, USA) Apoe-/- mice on a high-fat diet (GSK2033 group). Drugs were administered via gavage through a stomach tube. The total treatment time was 3 months for each group, for the drug groups, mice simultaneously were given the drug while on the high-fat diet. Each drug was prepared as previously described [11]. Briefly, T0901317 was dissolved in dimethyl sulfoxide (DMSO), and the solution was diluted to a final concentration of 0.03% in double distilled water. T0901317 was administered at a dosage of 15 mg/kg mouse/day. GSK2033 was formulated in the same manner as T0901317 and was administered at a dosage of 10 mg/kg mouse/day [12]. All Apoe-/- mice were kept in the SPF grade animal facility at the animal center of the Shanghai University of Traditional Chinese Medicine. The room temperature was maintained at 24°C with a relative humidity of 50-60%, and a light/dark cycle of 12 hours/12 hours was utilized. All drug gavages and tissue extractions were approved by the Animal Care Committee of the Use of Laboratory Animals at Shanghai University of Traditional Chinese Medicine.
RAW264.7 cell culture
RAW264.7 cells (Cell Bank, Shanghai Institute for Biological Science, Shanghai, China) were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Thermo Fisher Scientific, MA, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Thermo Fisher Scientific, MA, USA) at 37°C in a humidified atmosphere of 95% air and 5% CO2. Cells were passaged according to the number of cells required for different experiments. On the second day after passage, the cells were incubated with 100 μg/ml ox-LDL (Yuanye Biotechnology, Shanghai, China) with or without 10 μM T0901317 or 10 μM GSK2033 to establish lipid oxidation and AS therapy models in vitro. Cell experiments were divided into four groups: (1) RAW264.7 cells incubated with 0.03% DMSO (control); (2) RAW264.7 cells incubated with 100 μg/ml ox-LDL (model treatment group); (3) RAW264.7 cells incubated with 100 μg/ml ox-LDL for 4 hours, followed by 10 μM T0901317 for 24 hours (T0901317 treatment group); and (4) RAW264.7 cells incubated with 100 μg/ml ox-LDL for 4 hours, followed by 10 μM GSK2033 for 24 hours (GSK2033 treatment group). For Oil Red O assays, RAW264.7 cells were cultured in 24-well plates and fixed with 4% paraformaldehyde (Servicebio, Wuhan, China) for 15 minutes, after which they were washed with 0.01 M phosphate-buffered solution (PBS) (pH 7.4) for three times prior to Oil Red O staining.
Hematoxylin and eosin (H&E) staining
Apoe-/- mice were anesthetized by intraperitoneal injections of sodium pentobarbital (80 mg/kg) and perfused with 20 ml of 0.9% saline, followed by 50 ml of 4% paraformaldehyde. After perfusion, the heart and aorta (aortic arch, thoracic aorta, abdominal aorta) were carefully removed, blocked, and post-fixed for an additional 12 hours in the same fixation solution at 4°C. The sample was then transferred to 20% sucrose in 0.01 M PBS, followed by 30% sucrose in 0.01 M PBS at 4°C until it stayed at the bottom of the container. The heart tissue was embedded in paraffin, and 5-μm sections of the left ventricular aortic outflow tract were obtained transversely using a paraffin slicer (Leica SYD-S2020, Bannockburn, IL). H&E staining was performed according to the manufacturer’s protocol (Servicebio, Wuhan, China). Briefly, after dewaxing paraffin sections with xylene and hydrating with gradient ethanol, tissue sections were incubated in sodium citrate antigen retrieval solution for 15 minutes at 100°C. Afterwards, they were stained with hematoxylin for 10 minutes and then stained with eosin for 1 minute at room temperature. The sections were then dehydrated, cleared, cover slipped, and examined using a light microscope (CX43 Biological Microscope, Olympus, Japan). Image-Pro Plus 6.0 software (Mediacybernetics, MD, USA) was used to measure the plaque area and left ventricular outflow tract area, and the ratio of plaque area to left ventricular outflow tract area was calculated. Ratios were compared among groups.
Oil red O staining
Heart tissues were embedded in tissue freezing medium (Tissue-Tek, Elkart, IN), and 15-μm frozen sections of the left ventricular aortic outflow tract were obtained using a cryostat (Leica CM1900, Bannockburn, IL) and thaw-mounted on gelatin-coated slides. Oil Red O staining was performed according to the manufacturer’s protocol (Abcam, Cambridge, UK). Briefly, slides were placed in propylene glycol for 2 minutes and then incubated in Oil Red O solution for 6 minutes. Afterwards, the section was incubated in 85% propylene glycol (diluted in distilled water) for 1 minute and rinsed with distilled water twice. Sections were then incubated in hematoxylin for 1-2 minutes, after which they were dehydrated, cleared, cover slipped, and examined using a light microscope (CX43 Biological Microscope, Olympus, Japan). Image Pro Plus 6.0 software (Mediacybernetics, MD, USA) was used to measure the Oil Red O-positive plaque area and left ventricular outflow tract area, and the ratio of positive plaque area to left ventricular outflow tract area was calculated. Ratios were compared among groups.
Transmission electron microscope analysis
Transmission electron microscope analysis was performed as previously described. Selected aorta tissues (mainly at the aortic arch) and RAW264.7 cells were processed, sectioned, and stained using standard techniques [13,14]. After 24 hours in the primary fixative (2.5% glutaraldehyde/0.1 M PBS, pH 7.3, 4°C), the aorta was cut along the long axis. The AS plaque was located and divided transversely into smaller pieces (1-2 mm3). RAW264.7 cells (5 × 106) were collected with 0.5% trypsin-EDTA (Thermo Fisher Scientific, MA, USA) and washed with 0.5 ml of 0.1 M PBS in an Eppendorf tube by centrifuge (500 × g, 5 minutes). Samples (tissues and cells) were both post-fixed in 2% osmium tetroxide for 2 hours, rinsed in water, stained with 1% uranyl acetate for 60 minutes, washed in distilled water, and dehydrated in graded concentrations of ethanol. Subsequently, the tissues or cells were embedded in EPON (TAAB 812 embedding resin, VWR-Bie and Berntsen A/S, Herlev, Denmark) and polymerized for 24 hours at 60°C. Sections (1 μm) were cut and stained with 0.5% toluidine blue. Ultrathin sectioning was performed using a diamond knife attached to an ultramicrotome. Sections (70-nm thick) were collected on copper grids coated with 2% parlodion and then stained with Reynold’s lead citrate. Imaging was conducted with a JEM-1011 Transmission Electron Microscope (JEOL, Tokyo, Japan).
Immunofluorescence labeling
We performed immunofluorescence single-labeling and immunofluorescence double-labeling (IDL) to identify the number of CD68-(Abcam, Cambridge, UK) positive macrophages, CD86-(Abcam, Cambridge, UK) and inducible nitric oxide synthase (iNOS)-(Abcam, Cambridge, UK) positive M1 macrophages, and CD206-(Abcam, Cambridge, UK) and Arginase 1 (Arg1)-(Abcam, Cambridge, UK) positive M2 macrophages, and the expression of LXRα (Abcam, Cambridge, UK), ABCA1 (Thermo Fisher Scientific, MA, USA), and ABCG1 (Thermo Fisher Scientific, MA, USA) in macrophages, phosphorylated (Phospho)-NF-κB (p-p65) (Cell Signaling Technology, MA, USA) into the nucleus of macrophages respectively. All tissue sections were frozen, and the left ventricular outflow tract was sliced. Briefly, after incubation with a blocking buffer (10% normal goat serum, 0.3% Triton × 100 in 0.01 M PBS) for 1 hour at room temperature, the sections were incubated with the primary antibodies against CD68 (1:200), CD86 (1:100), iNOS (1:100), CD206 (1:50), Arg1 (1:150), LXRα (1:100), ABCA1 (1:50), or ABCG1 (1:50), or p-p65 (1:50) overnight at 4°C. On the following day, the sections were incubated with goat anti-rabbit IgG (H+L) cross-adsorbed secondary antibody (Alexa Fluor 546) and/or rabbit anti-mouse IgG (H+L) cross-adsorbed secondary antibody (Alexa Fluor 488) (Thermo Fisher Scientific, MA, USA). Sections were washed, mounted, and examined using a light microscope (CX43 Biological Microscope, Olympus, Japan). Phospho-p65 fluorescence was observed under laser scanning confocal microscope (LSM 710 System, ZEISS, Germany). Nuclei were labeled with DAPI. Image Pro Plus 6.0 software (Mediacybernetics, MD, USA) was used to analyze the fluorescence intensity of p-p65 immune positive area in nucleus of RAW264.7 cells, and 10 fields were selected from each of the fluorescence stained slices.
ELISA
Total protein was extracted from the aorta, including the thoracic aorta and abdominal aorta, or cultured RAW264.7 cells using RIPA lysis buffer (Beyotime, Shanghai, China) with cocktail tablets (Roche, Germany). Protein supernatants from aorta tissues and cultured RAW264.7 cells were collected to measure the concentrations of vascular cell adhesion molecule 1 (Vcam1), monocyte chemoattractant protein 1 (Mcp1), macrophage inflammatory protein 1 alpha (Mip1α), TNFα, interleukin-1 beta (IL-1β), and IL-6. All ELISA kits were purchased from Abcam (Cambridge, UK), and the above cytokines were detected in accordance with the manufacturer’s protocol. Briefly, 50 μl of the diluted standard or samples were added to each well, and then 50 μl of the diluted Detector antibody were added to each well after washing the plate three times. Lastly, 50 μl of diluted horseradish peroxidase (HRP)-streptavidin solution and 100 μl of TMB substrate were added to each well for detection, and the plate was measured at 450 nm. Protein supernatants were diluted in different proportions (1:1, 1:5, 1:10), and the concentrations of the above cytokines in the aorta and cells were calculated based on protein standards.
Western blot analysis
Western blot analysis was performed as previously described [15]. Total protein was extracted from the aorta, including the thoracic aorta and abdominal aorta, or cultured RAW264.7 cells using RIPA lysis buffer (Beyotime, Shanghai, China) with cocktail tablets (Roche, Germany). Equal amounts of protein were subjected to SDS-PAGE and electro-transferred to 0.22 μm aperture nitrocellulose filter membranes (Millipore, MA, USA). The membranes were blocked with 5% skimmed milk powder in Tris-HCl buffered saline (pH 7.5) for 30 minutes at room temperature and incubated with primary antibodies against LXRα (Abcam, Cambridge, UK), ABCA1 (Thermo Fisher Scientific, MA, USA), ABCG1 (Thermo Fisher Scientific, MA, USA), nuclear factor-kappaB (p65), phosphorylated p65 (Cell Signaling Technology, MA, USA), or glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Genscript, Nanjin, China); the dilution of all primary antibodies was 1:1000. Afterwards, the membranes were incubated with secondary HRP-conjugated goat anti-rabbit/mouse immunoglobulin (Ig) G antibodies (1:3,000; Genscript) for 1 hour at 37°C. Signals were detected by the electro-chemiluminescence (ECL) substrate (Thermo Fisher Scientific, MA, USA) and quantified using Image-Pro Plus 6.0 software (Media Cybernetics, MD, USA).
Cholesterol/cholesteryl ester quantitation assay
The total cholesterol and cholesteryl ester levels in RAW264.7 cells were quantified using the Cholesterol/Cholesteryl Ester Quantitation Assay kit (Abcam, Cambridge, UK). Briefly, after harvesting the amount of cells (> 1 × 106) necessary for each assay and washing with cold PBS, lipids were extracted by resuspending the sample in 200 µl of chloroform:isopropanol:NP-40 solution (7:11:0.1) in a microhomogenizer. Then, the extraction was centrifuged for 5-10 minutes at 15,000 × g, and the liquid, organic phase was transferred to a new tube. Chloroform and trace organic solvent were removed by air drying at 50°C and vacuuming for 30 minutes, respectively. After dissolving the dried lipids by sonicating or vortexing with 200 µl of assay buffer, reaction wells and reaction mix were set up according to the manufacturer’s protocol. The output was then measured on a microplate reader (Molecular Devices, CA, USA) at 570 nm.
Cell-counting kit 8 (CCK-8) assay for cell viability
The viability of RAW264.7 cells was evaluated with the CCK-8 kit (Beyotime Biotechnology, China). Cells (5 × 103/well) were individually cultured in 96-well plates for 12 hours. After adding 100 μg/ml ox-LDL with or without 10 μM T0901317 or 10 μM GSK2033 into the culture medium, cells were respectively incubated with 20 μl CCK-8 for 1 hour at 37°C, according to the manufacturer’s instructions. Cell viability rate was analyzed using a microplate reader to determine the absorbance at 450 nm (Molecular Devices, CA, USA). The assay was repeated three times and averaged for comparisons between groups.
Chemotactic migration assay
The chemotactic migration of RAW264.7 cells (Chinese Academy of Sciences Cell Bank, Shanghai, China) was measured using a transwell chamber with 6.5-mm polycarbonate membrane (8-μm pore size) (Corning, NY, USA). Normal RAW264.7 cells with fresh medium containing 100 μg/ml ox-LDL, 10 μM T0901317, and/or 10 μM GSK2033 were plated on the upper chamber. DMEM containing 10% FBS was added into the lower chamber. The cells were loaded at 5 × 104 cells/well to the upper chambers and allowed to migrate for 16 hours at 37°C. Then, the cells were fixed with 4% paraformaldehyde for 15 minutes, washed with PBS for three times, and stained with crystal violet for 1 hour at 37°C. Non-migrating cells on the upper surface of the membrane were removed using a cotton swab. Cells that had migrated to the lower surface of the membrane were counted under a microscope. The average of 10 fields was calculated in each group, and numbers were compared among groups.
Statistical analysis
Statistical analyses were performed using SPSS 17.0 software (SPSS Inc., Chicago, USA). Data are presented as the mean ± standard deviation of three independent experiments. One-way analysis of variance tests, followed by Tukey’s post-hoc test, were performed for comparisons of two or more groups, and a p-value less than 0.05 was considered statistically significant.
Results
LXRα agonist inhibits AS plaque formation by preventing the formation of foam cells
To determine the role and mechanism of LXRα in AS plaque formation, 8-month-old male Apoe-/- mice were treated with a high-fat diet in the presence and absence of an LXRα agonist (T0901317) or antagonist (GSK2033). T0901317 treatment decreased the plaque area at the left ventricular outflow tract of Apoe-/- mice (Figure 1A). We hypothesized that T0901317 promotes cholesterol transport in macrophages in AS plaques. Indeed, there were fewer Oil Red O-stained foam cells in the AS plaque in T0901317-treated mice (Figure 1B). Subsequently, results from transmission electron microscopy confirmed that T0901317 inhibited foam cell formation in AS plaques in vivo (Figure 1C). To determine whether the high expression of LXRα in macrophages was the only cause of AS plaque reduction, we first examined the number of CD68-positive macrophages in AS plaques. The number of CD68-positive macrophages in AS plaques was significantly lower in the T0901317-treated group, as compared to the other groups (Figure 2A). Therefore, we examined the expression of macrophage-associated adhesion and chemokines in the aortas of Apoe-/- mice. Vcam1 and Mcp1 levels decreased with the stimulation of T0901317 (Figure 2B), indicating that a high level of LXRα can inhibit macrophage migration in the AS plaque. Next, we examined the expression of cytokines in the aorta to detect inflammation in the AS plaques and found that T0901317 decreased levels of pro-inflammatory cytokines (Mip1α, TNFα, IL-1β, and IL-6) in the aorta (Figure 2C). This confirmed that the high expression of LXRα not only decreased the number of foam cells, but also inhibited the migration of macrophages and the inflammation in the AS plaque. These combined effects promoted the reduction in AS plaque area.
Figure 1.
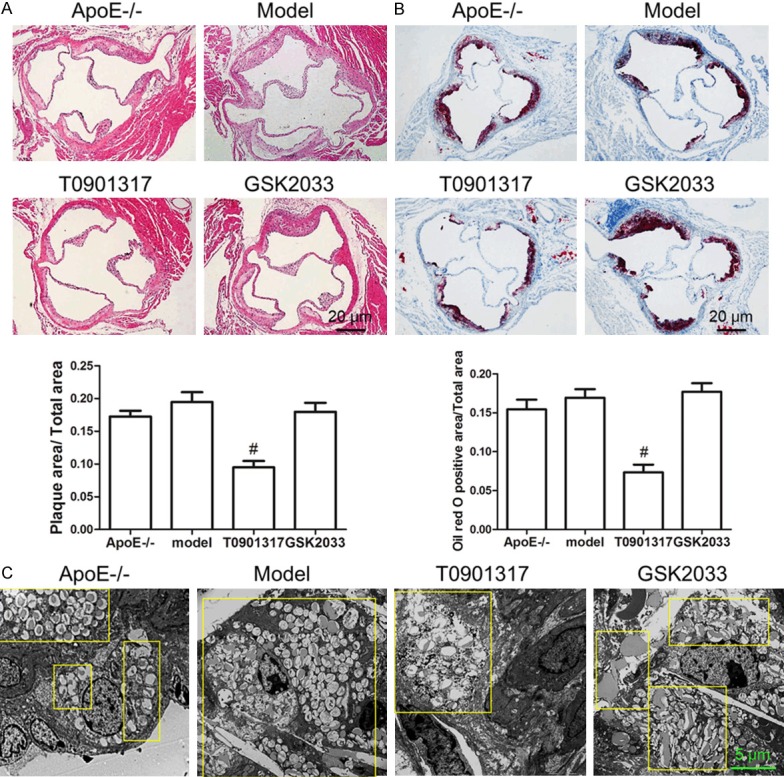
LXRα agonist T0901317 inhibits AS plaques and foam cell formation. Eight-month old male Apoe-/- mice were treated with a high-fat diet in the presence and absence of an LXRα agonist (T0901317) or antagonist (GSK2033) for 3 months. HE staining showed that T0901317 significantly decrease the plaque area at the left ventricular outflow tract of Apoe-/- mice contrast to normal chow diet (ApoE-/-) or high fat diet (Model) (A), Oil red O staining showed that there were fewer Oil Red O-stained foam cells in the AS plaque in T0901317-treated mice (B), lastly, transmission electron microscopy technology was used to confirm that fewer foam cells were found in T0901317 treated Apoe-/- high fat diet mice than in other groups (C). #P < 0.05 vs. model. Scale bar, 20 μm or 5 μm.
Figure 2.
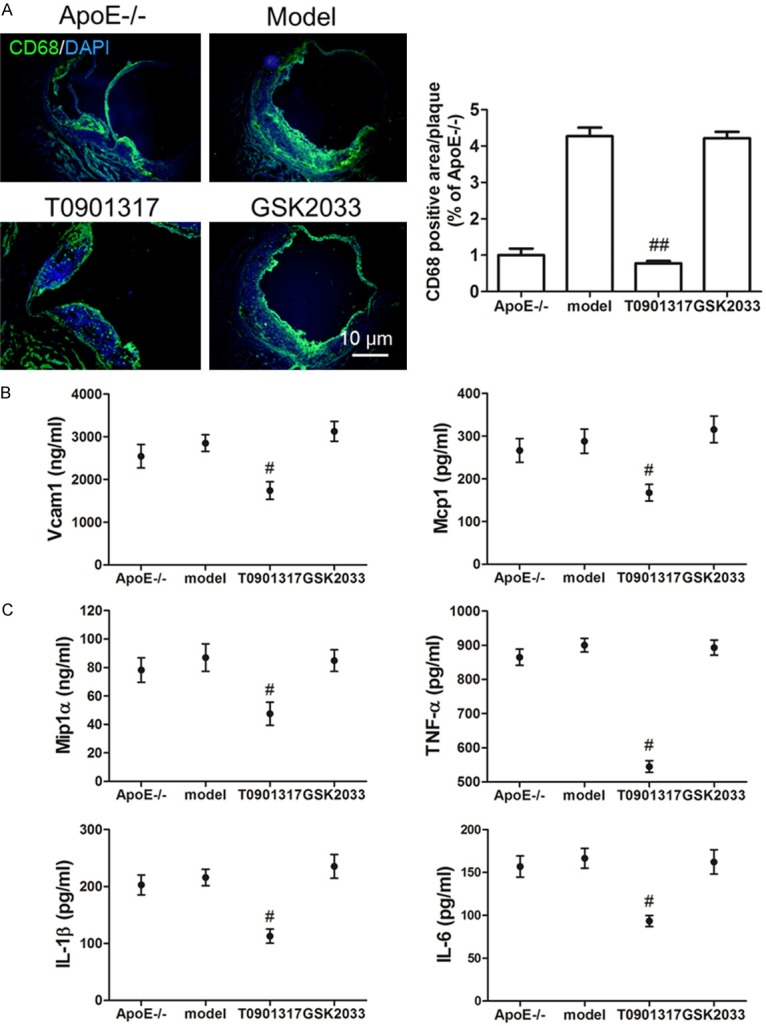
LXRα agonist inhibits macrophages infiltration and inflammation. In order to determine whether LXRα agonist indeed reduces the number of macrophages in AS plaque, we first examined the number of CD68-positive macrophages in AS plaques. CD68 immunofluorescence staining displays the decreased CD68 positive macrophages in AS plaque of T0901317 treated Apoe-/- high fat diet mice, statistics analysis shows that there is a significant difference between the T0901317 group and the model group (A). Then we examined the expression of macrophage-associated adhesion and chemokines in the aortas of Apoe-/- mice, to confirm LXRα agonist reduces the number of macrophages in AS plaque with indeed. Elisa results showed that Vcam1 and Mcp1 level were both decreased with the stimulation of T0901317 (B), and the pro-inflammatory cytokines Mip1α, TNFα, IL-1β and IL-6 were all decreased in the aorta of T0901317 treated Apoe-/- high fat diet mice (C). #P < 0.05 vs. model, ##P < 0.01 vs. model. Scale bar, 10 μm.
LXRα agonist decreases the number of foam cell by promoting the expression of reverse transporters and macrophage conversion in AS plaques
Next, we would detect causes of foam cell reduction in AS plaques in T0901317-treated mice. First of all, we confirmed whether T0901317 increased the expression of LXRα in macrophages. IDL was used to detect the expression of LXRα in macrophages. Figure 3A shows that T0901317 increases LXRα expression in the plaque, compared to those in Apoe-/- mice on normal and high-fat diets. In contrast, the LXRα antagonist decreased LXRα expression in the plaque. Consequently, we examine whether T0901317 promote the expression level of ABCA1 and ABCG1, which mainly participate in RCT in macrophages by reducing the amount of cholesterol in the cell membrane. Treatment with T0901317 increased ABCA1 and ABCG1 expression in macrophages simultaneously, and GSK2033 treatment partially inhibited ABCA1 expression (Figure 3B) and completely inhibited ABCG1 expression (Figure 3C). This may explain why the foam cells were decreased and the plaque area was smaller in the T0901317 group.
Figure 3.

LXRα agonist promotes the expression of LXRα, ABCA1 and ABCG1 in macrophages in AS plaque. In order to find out the cause of the reduction of foam cells and AS plaque area, we firstly confirm T0901317 can promote the expression of LXRα, then the RCT protein (ABCA1 and ABCG1), IDL was used to detect the expression of LXRα in CD68 positive macrophages, the yellow area of LXRα and CD68 double positive staining is indicated by the arrows, as shown, T0901317 stimulated more LXRα expression in macrophages (A), and the IDL assay also indicate that T0901317 promote more ABCA1 (B) and ABCG1 (C) expression in macrophages. Scale bar, 10 μm.
Whether the reduction of macrophages is a major factor in the reduction of inflammatory response remains unclear. Therefore, we examined M1 and M2 macrophages in AS plaques. CD86 and iNOS, which are markers of M1 macrophages, were highly expressed in Apoe-/- mice on a high-fat diet but were both expressed at low amounts in T0901317-treated Apoe-/- mice. CD206 and Arg1, which are markers of M2 macrophages, were highly expressed in T0901317-treated Apoe-/- mice but were expressed at low levels in Apoe-/- mice on a high-fat diet (Figure 4A). These results indicate that T0901317 reduces the number of M1 macrophages involved in the inflammatory response and promotes the number of protective M2 macrophages. This may play a role in its effects on inhibiting inflammation and plaque formation. What is the molecular mechanism regulating phenotypic conversion of macrophages? We examined phosphorylation of the transcription factor NF-κB, which is involved in the inhibition of inflammatory factor secretion and macrophage phenotype switching, we found that the level of phosphorylated NF-κB of Apoe-/- mice on a high-fat diet is higher than that of normal diet mice, T0901317 could inhibit the phosphorylation of NF-κB in Apoe-/- mice with high fat diet. The reason of reduced phosphorylation of NF-κB is T0901317 inhibits the phosphorylation of IκBα firstly, it leads to excessive IκBα binding to NF-κB in order to inhibit phosphorylation of NF-κB, low phosphorylated NF-κB level can inhibit inflammatory factor secretion, as well as promote M1 macrophage convert to M2 macrophage (Figure 4B).
Figure 4.

LXRα agonist converts M1 macrophages to M2 macrophages by inhibiting NF-κB phosphorylation. In order to detect whether the reduction of macrophages is a major factor in the reduction of inflammatory response, we also examine macrophage subtype in AS plaque. CD68, CD86, iNOS, CD206 and Arg1 antibody was used to examine the M1 or M2 type macrophage. IDL shows that M1 type macrophage marker CD86 and iNOS were both highly expressed in Apoe-/- mice with high fat diet (model group), M2 type macrophage marker CD206 and Arg1 were both highly expressed in T0901317 treated Apoe-/- high fat diet mice (T0901317 group) (A). Next, we examine the level of NF-κB phosphorylation to confirm whether T0901317 promotes the conversion of M1 to M2 macrophages by regulating NF-κB phosphorylation. We found that the level of phosphorylated NF-κB of Apoe-/- mice on a high-fat diet is higher than that of normal diet mice, T0901317 could inhibit the phosphorylation of NF-κB in Apoe-/- mice with high fat diet. The reason of reduced phosphorylation of NF-κB is T0901317 inhibits the phosphorylation of IκBα firstly, it leads to excessive IκBα binding to NF-κB in order to inhibit phosphorylation of NF-κB (B). *P < 0.05 vs. control, **P < 0.01 vs. control, ##P < 0.01 vs. model. Scale bar, 10 μm.
LXRα agonist inhibits foam cell formation and inflammation in vitro
Subsequently, we used the macrophage cell line, RAW264.7, to confirm the role of LXRα on cholesterol metabolism, cell migration, and inflammation. The expression of LXRα was increased and decreased by T0901317 and GSK2033, respectively (Figure 5A). T0901317 completely inhibited the formation of foam cells (cholesteryl ester/cholesterol < 50%), and GSK2033 greatly promoted the formation of foam cells (cholesteryl ester/cholesterol > 50%) (Figure 5B). At the same time, T0901317 partly rescued RAW264.7 cell viability, which was damaged by ox-LDL (Figure 5C). These experimental results suggest that T0901317 has a protective effect on cell function. We also used the Oil Red O assay to examine the number of foam cells in the presence of ox-LDL. T0901317 significantly decreased the number of Oil Red O-positive cells (Figure 5D), and the expression levels of ABCA1 and ABCG1 were both increased (Figure 5E). Transmission electron microscopy studies confirmed that T0901317 decreased the number of lipid droplets in RAW264.7 cells (Figure 5F). Overall, these results show that LXRα inhibited intracellular lipid deposition and prevented the formation of foam cells by promoting the expression of ABCA1 and ABCG1. Thus, in the Apoe-/- AS mouse model, foam cell reduction may play a role in the reduction of AS plaque area.
Figure 5.

LXRα agonist inhibits foam cells formation in vitro. We used the macrophage cell line, RAW264.7, to confirm the role of LXRα in foam cells formation, we found that T0901317 promotes the expression of LXRα in cultured RAW264.7 cells (A). We use cholesteryl ester/cholesterol assay kit to confirm the foam cells formation, the data shows that T0901317 totally inhibited the foam cells formation (Cholesteryl Ester/Cholesterol < 50%) (B). Then, CCK8 assay was used to detect the cell viability treated with ox-LDL (model), in the presence of T0901317 and GSK2033, cells was treated with phosphate buffer as control group, the result shows that T0901317 partly rescued RAW264.7 cells viability which damaged by ox-LDL (C). We used oil red O staining to observe the morphology of foam cells and counted the number of foam cells, we found thatT0901317 significantly decreased the oil red O positive cells (D), this effect of T0901317 is achieved by promoting the expression of ABCA1 and ABCG1 (E). Transmission electron microscopy technology was used to confirm the number of the foam cells in cultured RAW264.7 cells treated with ox-LDL, data shows that fewer foam cells found in T0901317 treated mice than in other groups (F). *P < 0.05 vs. control, **P < 0.01 vs. control. #P < 0.05 vs. model, ##P < 0.01 vs. model. Scale bar, 50 μm or 2 μm.
Next, we used in vitro cell migration experiments to confirm that LXRα regulates macrophage migration. T0901317 treatment inhibited RAW264.7 migration (Figure 6A) by suppressing the expression of macrophage-associated adhesion molecules (Vcam1 and Mcp1). Furthermore, T0901317 inhibited the release of inflammatory factors (Mip1α, TNFα, IL-1β, and IL-6) without reducing the number of macrophages (Figure 6B).
Figure 6.

LXRα agonist inhibits macrophages migration and inflammation in vitro. We used in vitro cell migration experiments to confirm that LXRα regulates macrophage migration. Chemotactic migration assay shows that T0901317 reduce the number of migrated cells and inhibit RAW264.7 migration (A). The results of Elisa assay show that macrophage-associated adhesion molecules (Vcam1, Mcp1) and cytokines (Mip1α, TNFα, IL-1β, and IL-6) are all decreased in cultured RAW264.7 cells treated with T0901317 (B). **P < 0.01 vs. control. #P < 0.05 vs. model, ##P < 0.01 vs. model. Scale bar, 50 μm.
Similarly, we examined the effect of T0901317 on ox-LDL-stimulated RAW264.7 cells. Ox-LDL-stimulated RAW264.7 cells highly expressed CD86 and iNOS, indicating that these M1 macrophages actively participated in the inflammatory response. Addition of T0901317 inhibited the expression of both CD86 and iNOS and promoted the expression of CD206 and Arg1. This suggests that T0901317 inhibits the inflammatory response by reducing the number of M1 macrophages while promoting lipid metabolism by increasing the number of M2 macrophages (Figure 7A). To determine how T0901317 can convert M1 macrophages into M2 macrophages, we focused on the phosphorylation of NF-κB (p65), which is a pro-inflammatory molecule that is associated with macrophage phenotype conversion. Indeed, T0901317 inhibited the phosphorylation of p65 (Figure 7B). To further confirm the effect of NF-κB phosphorylation on macrophage phenotype conversion, we tested the nuclear translocation of p-p65 with or without T0901317, we found that a large amount of p-p65 translocated into the nucleus with the stimulation of ox-LDL (model) in contrast to the control group, however, T0901317 inhibits most nuclear translocation of p-p65 in contrast to the model group (Figure 7C). These findings indicate that the LXRα agonist inhibits the inflammatory response and promotes lipid metabolism by regulating macrophage phenotypes. Furthermore, the LXRα agonist increased the number of M2 macrophages by inhibiting the phosphorylation of NF-κB. Lastly, the agonist promoted the expression of RCT proteins, ABCA1 and ABCG1, in M2 macrophages, thereby inhibiting the formation of AS plaques.
Figure 7.

M1 macrophages and M2 macrophages conversion and phosphorylation of NF-κB were detected in cultured RAW264.7 cells. We also examined the effect and mechanism of T0901317 on macrophage conversion in RAW264.7 cells. We found that T0901317 inhibits the expression of CD86 and iNOS, while promotes the expression of CD206 and Arg1, it indicates that T0901317 can convert M1 macrophages to M2 macrophages (A). Indeed, T0901317 inhibits the phosphorylation of NF-κB (p65) (B) and inhibits p-p65 from translocating into the nucleus (C) is the main cause of macrophage conversion. **P < 0.01 vs. control. ##P < 0.01 vs. model. Scale bar, 20 μm or 25 μm.
Discussion
Cardiovascular disease is the most common cause of morbidity and mortality worldwide, and studies have shown that LXRα plays a cardio-protective role in maintaining cholesterol homeostasis [16]. LXRα also has many other functions, including suppressing inflammation [17], decreasing oxidation and apoptosis [18], and suppressing insulin resistance [19] and hypertrophy [20]. Cholesterol metabolism is severely impaired in Lxr-knockout mice [21], which exhibit increased aortic foam cell accumulation after 18 months on a normal chow diet [22]. In contrast, LXR agonists have been shown to reduce plasma cholesterol levels and raise plasma HDL levels in mice [23]. LXRα acts as a systemic cholesterol sensor, and its activation helps to prevent AS. Based on the function of LXRα in regulating lipid metabolism and the inflammatory response, we targeted macrophages in AS plaques and focused on exploring whether LXRα affects the development of AS by regulating macrophage function. In addition to lipid accumulation and plaque development in the vasculature, chronic inflammation of the vasculature is a major cause of AS progression [24].
The anti-inflammatory effect of LXRs is mainly achieved by inhibiting the NF-κB signaling pathway [25]. Studies have found that T0901317 reduces lipopolysaccharide-stimulated inflammatory factor expression in macrophages lacking ABCA1 and ABCG1 in vitro; these cytokines include IL-1β, IL-6, Mcp-1, and Mip-1α. Moreover, LXR activation decreases AS and macrophage infiltration in Ldlr-/- mice that received transplanted ABCA1-/- ABCG1-/- bone marrow [17]. Our study also found that treatment with T0901317 reduced macrophage infiltration and decreased the expression of Vcam1, Mcp1, Mip1α, TNFα, IL-1β, and IL-6 in mouse aortas and cultured RAW264.7 cells. These findings suggest that LXRα agonists mediate inflammatory trans-repression without directly targeting cholesterol efflux. However, whether there is a correlation between LXRα-regulated inflammatory trans-repression and cholesterol efflux remains unknown.
Macrophages are a homogeneous cell population presenting a continuum phenotypic spectrum, with the classically M1 macrophage phenotypes and alternatively M2 macrophage phenotypes. M1 macrophages are characterized by high production of nitric oxide and reactive oxygen intermediates and participate in promoting inflammation. M2 macrophages are mainly functional in tissue remodeling, angiogenesis, and tumor progression; they are also involved in immuno-regulation and allergic reactions [26]. Macrophages infiltrating AS plaques in 20-week-old Apoe-/- mice fed a chow diet show the alternative Arg1-positive M2 phenotype. During disease progression, a switch from the M2 to M1 phenotype is observed [27]. In humans, the macrophage subtypes are found in different locations in the plaque [28,29]: M1 macrophages are mainly found in rupture-prone zones, whereas M2 macrophages are more commonly found in stable plaques. There are no dominant phenotypes in the fibrous cap. Arg1 expression in macrophages has been shown to be inversely correlated with AS progression and is markedly decreased in foam cells in the AS plaque. Moreover, Arg1 expression is enhanced in CD68+ cells in a murine aortic arch transplant model. In cultured macrophages, ligand-activated LXRα markedly enhances basal and IL-4-induced Arg1 mRNA and protein expression, as well as promoter activity, and this LXRα-enhanced Arg1 expression correlates with a reduction in nitric oxide levels [30]. In human macrophages, LXRα is expressed at relatively low levels in IL-4 polarized M2 macrophages, which show a decreased capacity to handle and efflux excess cholesterol [31]. These results suggested that LXRα could stabilize AS plaques by increasing the number of M2 macrophages, delaying AS progression. Therefore, we examined the distribution of M1 macrophages and M2 macrophages in the left ventricular outflow tract of 20-week-old Apoe-/- mice fed a high-fat diet with or without T0901317 treatment. In Apoe-/- mice fed a high-fat diet, a large amount of M1 macrophages were located at the plaque. T0901317 treatment converted most of the M1 macrophages to M2 macrophages. Similar results were observed in cultured macrophages in vitro, although T0901317 only reduced the expression levels of CD86 and iNOS and could not completely convert M1 macrophages into M2 macrophages. Due to the specificity and sensitivity of the primary CD86 and iNOS antibodies, we did not observe the same phenomenon in IDL experiments in vivo.
Overall, the findings in the current study show that treatment of Apoe-/- mice with an LXRα agonist ameliorated the formation of high-fat diet-driven AS plaques. This was attributed to the ability of T0901317 to convert M1 macrophages into M2 macrophages and to reverse the excess cholesterol efflux of foam cells and inflammation in AS plaques. Our results also suggest that it might be important to regulate the macrophage phenotype in the early stage of AS, such that the occurrence and development of AS can be delayed.
Acknowledgements
We would like to thank Zhihua Yu (Department of Pharmacology and Chemical Biology, Shanghai Jiao Tong University School of Medicine) for her helpful comments on the manuscript. The present study was supported by grants from Natural Science Foundation of China (program no. 81704129), from the Shanghai Health Bureau Youth Fund (program no. 201640232 and program no. ZY (2018-2020)-CCCX-4004) and from the projects sponsored by the development fund for Shanghai talents (2017054).
Disclosure of conflict of interest
None.
References
- 1.Favari E, Chroni A, Tietge UJ, Zanotti I, Escola-Gil JC, Bernini F. Cholesterol efflux and reverse cholesterol transport. Handb Exp Pharmacol. 2015;224:181–206. doi: 10.1007/978-3-319-09665-0_4. [DOI] [PubMed] [Google Scholar]
- 2.Rohatgi A, Khera A, Berry JD, Givens EG, Ayers CR, Wedin KE, Neeland IJ, Yuhanna IS, Rader DR, de Lemos JA, Shaul PW. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med. 2014;371:2383–2393. doi: 10.1056/NEJMoa1409065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonamassa B, Moschetta A. Atherosclerosis: lessons from LXR and the intestine. Trends Endocrinol Metab. 2013;24:120–128. doi: 10.1016/j.tem.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Repa JJ, Mangelsdorf DJ. The liver X receptor gene team: potential new players in atherosclerosis. Nat Med. 2002;8:1243–1248. doi: 10.1038/nm1102-1243. [DOI] [PubMed] [Google Scholar]
- 5.Joseph SB, Tontonoz P. LXRs: new therapeutic targets in atherosclerosis? Curr Opin Pharmacol. 2003;3:192–197. doi: 10.1016/s1471-4892(03)00009-2. [DOI] [PubMed] [Google Scholar]
- 6.Fievet C, Staels B. Liver X receptor modulators: effects on lipid metabolism and potential use in the treatment of atherosclerosis. Biochem Pharmacol. 2009;77:1316–1327. doi: 10.1016/j.bcp.2008.11.026. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y, Repa JJ, Gauthier K, Mangelsdorf DJ. Regulation of lipoprotein lipase by the oxysterol receptors, LXRalpha and LXRbeta. J Biol Chem. 2001;276:43018–43024. doi: 10.1074/jbc.M107823200. [DOI] [PubMed] [Google Scholar]
- 8.Ulven SM, Dalen KT, Gustafsson JA, Nebb HI. LXR is crucial in lipid metabolism. Prostaglandins Leukot Essent Fatty Acids. 2005;73:59–63. doi: 10.1016/j.plefa.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 9.Kiss M, Czimmerer Z, Nagy L. The role of lipid-activated nuclear receptors in shaping macrophage and dendritic cell function: from physiology to pathology. J Allergy Clin Immunol. 2013;132:264–286. doi: 10.1016/j.jaci.2013.05.044. [DOI] [PubMed] [Google Scholar]
- 10.Jennelle LT, Dandekar AP, Magoro T, Hahn YS. Immunometabolic signaling pathways contribute to macrophage and dendritic cell function. Crit Rev Immunol. 2016;36:379–394. doi: 10.1615/CritRevImmunol.2017018803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carter AY, Letronne F, Fitz NF, Mounier A, Wolfe CM, Nam KN, Reeves VL, Kamboh H, Lefterov I, Koldamova R. Liver X receptor agonist treatment significantly affects phenotype and transcriptome of APOE3 and APOE4 Abca1 haplo-deficient mice. PLoS One. 2017;12:e0172161. doi: 10.1371/journal.pone.0172161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Griffett K, Burris TP. Promiscuous activity of the LXR antagonist GSK2033 in a mouse model of fatty liver disease. Biochem Biophys Res Commun. 2016;479:424–428. doi: 10.1016/j.bbrc.2016.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beaufrere H, Nevarez JG, Holder K, Pariaut R, Tully TN, Wakamatsu N. Characterization and classification of psittacine atherosclerotic lesions by histopathology, digital image analysis, transmission and scanning electron microscopy. Avian Pathol. 2011;40:531–544. doi: 10.1080/03079457.2011.607427. [DOI] [PubMed] [Google Scholar]
- 14.Birck MM, Saraste A, Hyttel P, Odermarsky M, Liuba P, Saukko P, Hansen AK, Pesonen E. Endothelial cell death and intimal foam cell accumulation in the coronary artery of infected hypercholesterolemic minipigs. J Cardiovasc Transl Res. 2013;6:579–587. doi: 10.1007/s12265-013-9463-2. [DOI] [PubMed] [Google Scholar]
- 15.Dou F, Huang L, Yu P, Zhu H, Wang X, Zou J, Lu P, Xu XM. Temporospatial expression and cellular localization of oligodendrocyte myelin glycoprotein (OMgp) after traumatic spinal cord injury in adult rats. J Neurotrauma. 2009;26:2299–2311. doi: 10.1089/neu.2009.0954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong C, Tontonoz P. Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat Rev Drug Discov. 2014;13:433–444. doi: 10.1038/nrd4280. [DOI] [PubMed] [Google Scholar]
- 17.Kappus MS, Murphy AJ, Abramowicz S, Ntonga V, Welch CL, Tall AR, Westerterp M. Activation of liver X receptor decreases atherosclerosis in Ldlr(-)/(-) mice in the absence of ATP-binding cassette transporters A1 and G1 in myeloid cells. Arterioscler Thromb Vasc Biol. 2014;34:279–284. doi: 10.1161/ATVBAHA.113.302781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He Q, Pu J, Yuan A, Lau WB, Gao E, Koch WJ, Ma XL, He B. Activation of liver-X-receptor alpha but not liver-X-receptor beta protects against myocardial ischemia/reperfusion injury. Circ Heart Fail. 2014;7:1032–1041. doi: 10.1161/CIRCHEARTFAILURE.114.001260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He Q, Pu J, Yuan A, Yao T, Ying X, Zhao Y, Xu L, Tong H, He B. Liver X receptor agonist treatment attenuates cardiac dysfunction in type 2 diabetic db/db mice. Cardiovasc Diabetol. 2014;13:149. doi: 10.1186/s12933-014-0149-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuipers I, Li J, Vreeswijk-Baudoin I, Koster J, van der Harst P, Sillje HH, Kuipers F, van Veldhuisen DJ, van Gilst WH, de Boer RA. Activation of liver X receptors with T0901317 attenuates cardiac hypertrophy in vivo. Eur J Heart Fail. 2010;12:1042–1050. doi: 10.1093/eurjhf/hfq109. [DOI] [PubMed] [Google Scholar]
- 21.Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med. 2003;9:213–219. doi: 10.1038/nm820. [DOI] [PubMed] [Google Scholar]
- 22.Schuster GU, Parini P, Wang L, Alberti S, Steffensen KR, Hansson GK, Angelin B, Gustafsson JA. Accumulation of foam cells in liver X receptor-deficient mice. Circulation. 2002;106:1147–1153. doi: 10.1161/01.cir.0000026802.79202.96. [DOI] [PubMed] [Google Scholar]
- 23.Joseph SB, McKilligin E, Pei L, Watson MA, Collins AR, Laffitte BA, Chen M, Noh G, Goodman J, Hagger GN, Tran J, Tippin TK, Wang X, Lusis AJ, Hsueh WA, Law RE, Collins JL, Willson TM, Tontonoz P. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci U S A. 2002;99:7604–7609. doi: 10.1073/pnas.112059299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Im SS, Osborne TF. Liver x receptors in atherosclerosis and inflammation. Circ Res. 2011;108:996–1001. doi: 10.1161/CIRCRESAHA.110.226878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fessler MB. The challenges and promise of targeting the Liver X Receptors for treatment of inflammatory disease. Pharmacol Ther. 2018;181:1–12. doi: 10.1016/j.pharmthera.2017.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Colin S, Chinetti-Gbaguidi G, Staels B. Macrophage phenotypes in atherosclerosis. Immunol Rev. 2014;262:153–166. doi: 10.1111/imr.12218. [DOI] [PubMed] [Google Scholar]
- 27.Khallou-Laschet J, Varthaman A, Fornasa G, Compain C, Gaston AT, Clement M, Dussiot M, Levillain O, Graff-Dubois S, Nicoletti A, Caligiuri G. Macrophage plasticity in experimental atherosclerosis. PLoS One. 2010;5:e8852. doi: 10.1371/journal.pone.0008852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stoger JL, Gijbels MJ, van der Velden S, Manca M, van der Loos CM, Biessen EA, Daemen MJ, Lutgens E, de Winther MP. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis. 2012;225:461–468. doi: 10.1016/j.atherosclerosis.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 29.Cho KY, Miyoshi H, Kuroda S, Yasuda H, Kamiyama K, Nakagawara J, Takigami M, Kondo T, Atsumi T. The phenotype of infiltrating macrophages influences arteriosclerotic plaque vulnerability in the carotid artery. J Stroke Cerebrovasc Dis. 2013;22:910–918. doi: 10.1016/j.jstrokecerebrovasdis.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 30.Pourcet B, Feig JE, Vengrenyuk Y, Hobbs AJ, Kepka-Lenhart D, Garabedian MJ, Morris SM Jr, Fisher EA, Pineda-Torra I. LXRalpha regulates macrophage arginase 1 through PU.1 and interferon regulatory factor 8. Circ Res. 2011;109:492–501. doi: 10.1161/CIRCRESAHA.111.241810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chinetti-Gbaguidi G, Baron M, Bouhlel MA, Vanhoutte J, Copin C, Sebti Y, Derudas B, Mayi T, Bories G, Tailleux A, Haulon S, Zawadzki C, Jude B, Staels B. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARgamma and LXRalpha pathways. Circ Res. 2011;108:985–995. doi: 10.1161/CIRCRESAHA.110.233775. [DOI] [PMC free article] [PubMed] [Google Scholar]