

Abstract
Acute kidney injury (AKI) is a common clinical syndrome with high morbidity and mortality, which is mostly caused by acute tubular necrosis (ATN). AKI is associated with many factors, including cell senescence, inflammatory infiltration, apoptosis and excessive accumulation of reactive oxygen species (ROS). P16INK4a (hereafter termed p16) inhibits cell cycle, and the absence of p16 can significantly slow the progression of cell senescence. We found that the expression of p16 was significantly increased after ATN. To determine whether p16 could exacerbate ATN degree and whether p16 deletion had protective effects against the ATN and renal dysfunction in AKI progression, glycerol-rhabdomyolysis-induced ATN was performed in eight-week-old p16 knockout and wild-type (WT) littermates. Their ATN phenotypes were analyzed; the levels of serum creatinine and serum urea nitrogen were detected; inflammation, cell apoptosis, ROS level and ROS signaling pathway molecules were examined using histopathological and molecular techniques. We found that compared to WT mice, p16 deletion has protective effects against the ATN phenotype and renal dysfunction in AKI progression through ameliorating inflammatory infiltration and proinflammatory factor expression by inhibiting NF-κB proinflammatory pathway, decreasing cell apoptosis by balancing the expressions between pro-apoptotic and anti-apoptotic molecules, and reducing ROS levels and downregulating ROS signaling pathway molecules including AIF, PGAM5 and KEAP1. Thus, p16 deletion or inhibition and p16 positive cell clearance would be a novel strategy for preventing ATN in AKI progression.
Keywords: p16INK4a, acute kidney injury, acute tubular necrosis, NF-κB, renal cell apoptosis, reactive oxygen species
Introduction
Acute kidney injury (AKI) is a common clinical syndrome with high morbidity and mortality characterized by a decrease in glomerular filtration rate. Acute tubular necrosis (ATN) is the main pathological feature of acute kidney injury [1]. Thus, studying how to minimize the ATN and reduce AKI-related mortality is urgent. It has been reported that AKI progression is strongly connected with renal cell senescence mediated by oxidative stress and proinflammatory factors secretion [2,3]. Renal tubular epithelial cells show a senescent phenotype during renal reparation after ATN. Cell aging promotes interstitial fibrosis, tubular atrophy, and a sparse capillary network, which impedes the morphological and functional recovery of the kidney after AKI [2].
P16 locates on the short arm 2, region 1 of chromosome 9, which encodes p14ARF and p16 proteins. P16 protein mainly interacts with CDK4/6 in the nucleus, which prevents the binding of the cyclin D to CDK4/6, and maintains Rb dephosphorylation. The dephosphorylated Rb binds to the cell cycle-associated transcription factor E2F blocking transcriptional activation domain, thereby inhibiting expressions of downstream genes required for entering S phase from G1 phase. This leads to cell cycle arrest in G1 phase and growth arrest, thereby inducing cell senescence. P16 deletion can significantly delay cell senescence and inhibit senescence-associated secretory phenotype (SASP) [4]. Our previous study find that knocking out the p16 in premature aging mice can significantly correct chronic renal tubular interstitial fibrosis [5]. Studies have shown that in AKI, there will be an imbalance between oxidative stress and antioxidant defense system, which is manifested as significantly increased ROS level [6]. Excessive ROS accumulation activates the apoptosis pathway and accelerates the apoptosis of epithelial cells by damaging the mitochondrial membrane [7]. At the same time, ROS can also significantly promote the activation of NF-κB pathway and inflammatory response, which further aggravates the occurrence and development of ATN [8,9]. P16 contributes to development of renal postischemic interstitial fibrosis and tubular atrophy [10]. However, it is unclear whether p16 could exacerbate ATN degree and whether p16 deletion has protective effects against the ATN and renal dysfunction in AKI progression.
The pathogenic mechanism of glycerol-rhabdomyolysis-induced ATN in mouse model closely mimics rhabdomyolysis-induced clinical ATN in human [11]. It is widely studied that ATN is induced by a single intramuscular injection of 50% glycerol in 0.9% saline [12-14]. Therefore, we induced ATN in eight-week-old p16 knockout (p16KO) and wild-type (WT) littermates and then analyzed their phenotype using histopathological and molecular techniques. Inflammation, cell apoptosis, ROS level and ROS signaling molecules were examined after ATN.
Results
P16 protein was increased in WT mice after ATN
To observe whether the expression of p16 protein was changed in the WT ATN mice and control mice, we used western blot to detect p16 protein expression in the kidneys. The expression of p16 protein was significantly higher in the WT ATN mice than that in the WT control mice at 24 h, 48 h and 72 h. The expression of p16 protein at 48 h after model establishment was significantly higher than that at 24 h and 72 h. The expression of p16 protein at 24 h was similar to that at 72 h after ATN modeling (Figure 1A, 1B).
Figure 1.
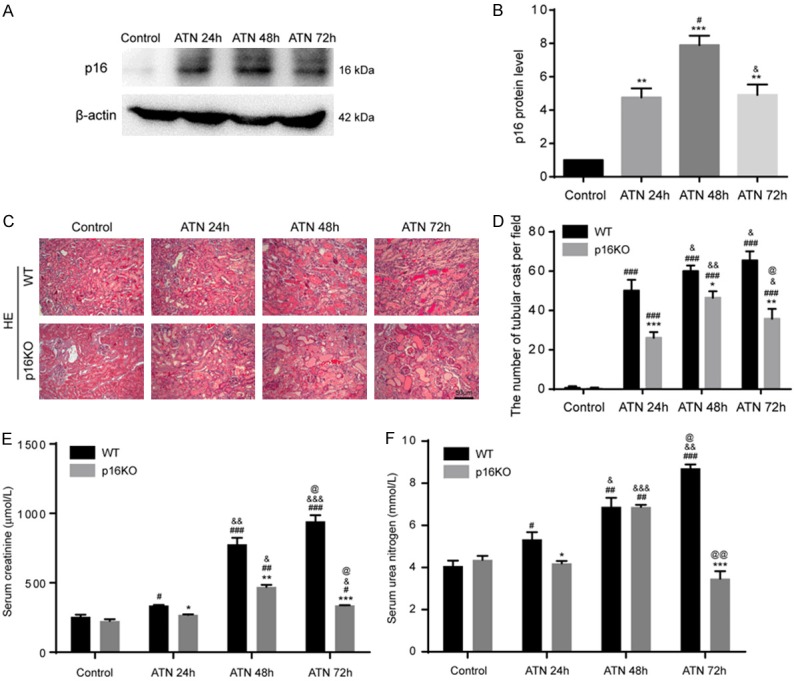
P16 was increased in WT mice after ATN, and its deletion ameliorated ATN phenotype and renal function. A, B. Western blots of renal extracts showing p16 and β-actin was the loading control. Values are mean ± SEM of six determinations per group. **P < 0.01, ***P < 0.001 compared with control mice; #P < 0.05 compared with ATN-24 h group mice. C. Representative micrographs of paraffin-embedded renal cortex and medulla stained with HE from WT ATN mice and p16KO ATN mice at 24 h, 48 h, 72 h and control mice after the second dehydrated. D. Tube cast number was determined in HE-stained sections. E, F. Serum creatinine levels and serum urea nitrogen concentrations were determined by spectrophotometry. Values are mean ± SEM of six determinations per group. *P < 0.05, **P < 0.01, ***P < 0.001 compared with WT mice at the same time after ATN; #P < 0.05, ##P < 0.01, ###P < 0.001 compared with control at the same genotyped mice; &P < 0.05, &&P < 0.01, &&&P < 0.001 compared with ATN-24 h group at the same genotyped mice; @P < 0.05, @@P < 0.01 compared with ATN-48 h at the same genotyped mice.
P16 deletion ameliorated ATN phenotype and renal function
Eight-week-old p16KO male mice and WT male littermate mice were used for ATN modeling. HE staining revealed that compared with the control group, the ATN model mice (24 h, 48 h and 72 h after ATN modeling) showed degeneration and necrosis of renal tubular epithelial cells and the formation of renal tubular cast. Compared with WT group, p16KO mice showed mild renal pathological changes after ATN modeling (24 h, 48 h and 72 h) (Figure 1C, 1D). The serum creatinine of WT mice and p16KO mice was measured and found to be lower in the p16KO mice than in the same group of WT mice at 24 h, 48 h and 72 h after ATN modeling. The serum urea nitrogen was measured and found to be lower in the p16KO mice than in the corresponding WT mice at 24 h and 72 h after ATN modeling (Figure 1E, 1F).
P16 deletion reduced inflammation infiltration after ATN
To investigate the inflammatory infiltration of the kidney after ATN, we used immunohistochemistry to detect the percentage of CD3 or F4/80 positive cells in WT and p16KO mice after ATN modeling. Based on the results of immunohistochemistry, we found that the percentages of CD3-positive and F4/80-positive cells were significantly increased in renal tissues from WT mice and p16KO mice after ATN modeling (24 h, 48 h and 72 h). The percentages of CD3-positive and F4/80-positive cells were significantly decreased compared with the same group of WT mice (Figure 2A-C). These results indicated that the loss of p16 could inhibit inflammatory infiltration induced by ATN modeling.
Figure 2.
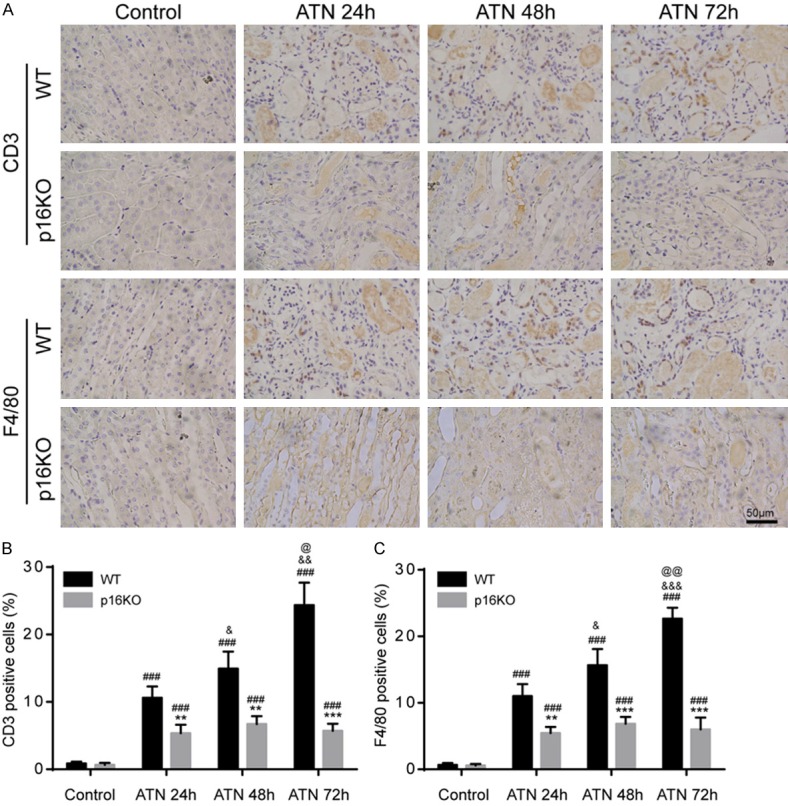
P16 deletion reduced inflammation infiltration after ATN. Eight-week-old p16KO male mice and WT male littermate mice were used for ATN modeling. After ATN for 24 h, 48 h, and 72 h. A. Representative micrographs of paraffin-embedded renal sections immunohistochemically for CD3 and F4/80. B, C. Percentage of CD3 or F4/80 positive cells relative to total cells. Values are mean ± SEM of six determinations per group. **P < 0.01, ***P < 0.001 compared with WT mice at the same time after ATN; ###P < 0.001 compared with control at the same genotyped mice; &P < 0.05, &&P < 0.01, &&&P < 0.001 compared with ATN-24 h group at the same genotyped mice; @P < 0.05, @@P < 0.01 compared with ATN-48 h at the same genotyped mice.
P16 deletion reduced the production of inflammatory factors after ATN
Since the inflammatory infiltration of p16KO mice improved after ATN, we hypothesized that p16 had an effect on the production of inflammatory factors. Therefore, we used immunohistochemistry to detect the expression of the inflammatory factor IL-1β, IL-6 or TNF-α in renal tissue of WT mice and p16KO mice after AKI. We found that the percentages of IL-1β-positive, IL-6-positive and TNF-α-positive cells or areas in p16KO mice were significantly decreased in renal tissues compared with WT mice after ATN modeling (24 h, 48 h and 72 h) (Figure 3A-F). Next, we used western blot to detect the expression of pro-IL-1β, IL-1β, and TNF-α associated with inflammation. We found that after ATN modeling, the expression of inflammation-related proteins in the kidneys of p16KO mice was obviously decreased compared with WT mice, consistent with the previously observed immunohistochemistry results (Figure 3G-J). These data indicated that p16 deletion could inhibit the production of inflammatory factors.
Figure 3.

P16 deletion reduced the production of inflammatory factors after ATN. A-C. Representative micrographs of paraffin-embedded renal sections immunohistochemically stained for IL-1β, IL-6 and TNF-α. D-F. Percentage of cells positive for IL-1β, IL-6 or TNF-α or positive areas relative to total cells or areas. Values are mean ± SEM of six determinations per group. *P < 0.05, **P < 0.01, ***P < 0.001 compared with WT mice at the same time after ATN; ###P < 0.001 compared with control at the same genotyped mice; &P < 0.05 compared with ATN-24 h group at the same genotyped mice. G. Western blots of renal extracts showing pro-IL-1β, IL-1β and TNF-α, and β-actin was the loading control. H-J. Protein levels relative to β-actin were assessed by densitometric analysis. Values are mean ± SEM of six determinations per group. *P < 0.05, **P < 0.01 compared with WT mice at the same time after ATN; #P < 0.05, ##P < 0.01 compared with control at the same genotyped mice; &P < 0.05, &&P < 0.01 compared with ATN-24 h group at the same genotyped mice; @P < 0.05, @@P < 0.01 compared with ATN-48 h at the same genotyped mice.
P16 deletion inhibited the NF-κB proinflammatory pathway after ATN
The NF-κB pathway is considered as a typical proinflammatory signaling pathway. We observed that p16KO mice have a marked improvement in inflammation after ATN. Because the NF-κB pathway is closely related to inflammation, we hypothesized that deletion of p16 might inhibit this pathway. To test our hypothesis, we used immunohistochemistry to detect the expression of p-p65 (Ser536) and found that the percentage of p-p65 (Ser536) positive cells in the kidneys of p16KO mice was decreased compared with WT mice after ATN modeling (24 h, 48 h and 72 h) (Figure 4A, 4B). We used western blot to detect the expression of p-p65 (Ser536) and p65 proteins and obtained consistent results (Figure 4C, 4D). These results indicated that p16 deletion could inhibit the expression of inflammatory factors, reduce inflammatory infiltration and proinflammatory response by inhibiting the NF-κB pathway after ATN modeling.
Figure 4.

P16 deletion inhibited the NF-κB proinflammatory pathway after ATN. A. Representative micrographs of paraffin-embedded renal sections immunohistochemically for p-p65 (Ser536). B. Percentage of cells positive for p-p65 (Ser536) or positive areas relative to total cells or areas. Values are mean ± SEM of six determinations per group. **P < 0.01 compared with WT mice at the same time after ATN; ###P < 0.001 compared with control at the same genotyped mice; &P < 0.05 compared with ATN-24 h group at the same genotyped mice; @P < 0.05 compared with ATN-48 h at the same genotyped mice. C. Western blots of renal extracts showing p-p65 (Ser536) and p65, and β-actin was the loading control. D. Protein levels relative to β-actin were assessed by densitometric analysis. Values are mean ± SEM of six determinations per group. *P < 0.05, **P < 0.01, ***P < 0.001 compared with WT mice at the same time after ATN; #P < 0.05 compared with control at the same genotyped mice; &&&P < 0.001 compared with ATN-24 h group at the same genotyped mice; @@@P < 0.001 compared with ATN-48 h at the same genotyped mice.
P16 deletion inhibited renal cell apoptosis after ATN
We used the TUNEL assay to detect apoptosis in the kidneys of p16KO mice and WT mice and found that the percentage of apoptotic cells in p16KO mice was obviously decreased compared with WT mice after ATN modeling (24 h, 48 h and 72 h) (Figure 5A, 5B). We used western blot to detect protein markers associated with apoptosis and found that the expression of the pro-apoptosis-related proteins p53, Bax and cleaved caspase-3 was decreased in the kidneys of p16KO mice after ATN modeling (24 h, 48 h and 72 h) compared with those of WT mice, and the expression of the anti-apoptosis-related protein Bcl-2 was increased in p16KO mice compared with WT mice (Figure 5C-G), indicating that the loss of p16 could inhibit apoptosis induced by ATN modeling.
Figure 5.

P16 deletion could reduce apoptosis. A. Representative micrographs of paraffin-embedded renal sections stained with TUNEL. Values are mean ± SEM of six determinations per group. B. Percentage of cells positive for TUNEL relative to total cells. *P < 0.05 compared with WT mice at the same time after ATN; ##P < 0.01, ###P < 0.001 compared with control at the same genotyped mice; &P < 0.05, &&P < 0.01, &&&P < 0.001 compared with ATN-24 h group at the same genotyped mice; @P < 0.05 compared with ATN-48 h at the same genotyped mice. C. Western blots of renal extracts showing p53, Bcl-2, Bax and cleaved caspase3, and β-actin was the loading control. D-G. Protein levels relative to β-actin were assessed by densitometric analysis. Values are mean ± SEM of six determinations per group. *P < 0.05, **P < 0.01, ***P < 0.001 compared with WT mice at the same time after ATN; #P < 0.05, ##P < 0.01, ###P < 0.001 compared with control at the same genotyped mice; &P < 0.05, &&P < 0.01, &&&P < 0.001 compared with ATN-24 h group at the same genotyped mice; @P < 0.05, @@P < 0.01 compared with ATN-48 h at the same genotyped mice.
P16 deletion inhibited ROS accumulation and its signaling pathway after ATN
Studies have shown that the mechanism of ATN is also related to ROS. We used flow cytometry to detect ROS levels in p16KO mice after ATN. Results showed that the ROS levels produced by renal cells in p16KO mice were lower than those in the WT group (Figure 6A, 6B). We also used western blot to detect the expression of ROS signaling pathway molecules AIF, PGAM5 and KEAP1, and found that the expression of AIF, PGAM5 and KEAP1 in p16KO mice was decreased compared to WT mice after ATN (Figure 6C-F), indicating that the loss of p16 could inhibit the ROS excessive accumulation and its signaling pathway.
Figure 6.

P16 deletion inhibited ROS accumulation and its signaling pathway after ATN. A, B. ROS was detected by flow cytometry. Values are mean ± SEM of six determinations per group. *P < 0.05, **P < 0.01, ***P < 0.001 compared with WT mice at the same time after ATN; ###P < 0.001 compared with control at the same genotyped mice; &P < 0.05, &&&P < 0.001 compared with ATN-24 h group at the same genotyped mice; @@P < 0.01 compared with ATN-48 h at the same genotyped mice. C. Western blots of renal extracts showing AIF, KEAP1 and PGAM5, and β-actin was the loading control. D-F. Protein levels relative to β-actin were assessed by d ensitometric analysis. *P < 0.05, **P < 0.01, ***P < 0.001 compared with control at the same genotyped mice; #P < 0.05, ##P < 0.01 compared with WT+ATN group; &P < 0.05, &&P < 0.01 compared with ATN 24 h at the same genotyped and treatment mice; @P < 0.05, @@P < 0.01 compared with ATN 48 h at the same genotyped and treatment mice.
Discussion
This study demonstrated that p16 deletion had protective effects against the ATN phenotype and renal dysfunction in AKI progression through ameliorating inflammatory infiltration and proinflammatory factor expression by inhibiting NF-κB proinflammatory pathway, decreasing cell apoptosis by balancing the expressions between pro-apoptotic and anti-apoptotic molecules, and reducing ROS levels and signaling pathway molecules including AIF, PGAM5 and KEAP1.
The aim of this study was to investigate the effects of p16 in promoting ATN and its possible underlying mechanisms. P16 protein is a key kinesin of cell senescence [4,5]. Several lines of evidence demonstrate that AKI is accompanied with renal cell senescence aggravated mainly by oxidative stress and SASP [2,3]. The elimination of senescent cells and an understanding of the role of cell senescence in AKI may potentially contribute to the treatment of AKI. However, the mechanism underlying the function of the aging-driven key protein p16 in AKI has not been studied. To this end, we conducted a preliminary exploration. We found that p16 deletion could alleviate the formation and necrosis of renal tubular type caused by AKI, reduce the content of creatinine and serum urea nitrogen, and enhance the recovery of kidney morphology and function after injury. Furthermore, p16 deletion attenuated the inflammatory infiltration, and inhibited the secretion of inflammatory factors and the activation of the NF-κB proinflammatory pathway that occurs after ATN. In addition, we observed that after the occurrence of ATN, p16 deletion inhibited apoptosis, reduced the level of ROS, and effectively prevented further damage of the renal tubules. These results indicated that p16 played an important role in preventing the progression of AKI. Consistent with this result is a previous observation that the absence of p16 and p19 has a protective effect on renal function after ischemia-reperfusion injury through significantly inhibiting cell apoptosis, and promoting cell proliferation and microvascular repair [15], suggesting that p16 plays an adverse role in different renal disease models and could be used as a target for treating renal diseases.
We established ATN model by intramuscular injection of glycerol closely mimics rhabdomyolysis-induced clinical ATN in human, measured serum creatinine and urea nitrogen levels to assess renal function and observed morphology changes of kidney [11,14,16]. Studies have reported that the phenotype associated with senescent cells is one of the important causes of inflammatory senescence [17-19]. During the process of cell senescence, a large number of inflammatory factors, such as IL-1β, IL-6 and TNF-α, are released and mediate inflammation [19,20]. P16 can drive cell aging and promote aging-related proinflammatory secretion. We found that p16 null mice showed less inflammatory cell infiltration and less senescence-related proinflammatory factors secretion, such as IL-1β, IL-6 and TNF-α, during AKI, suggesting that p16 might aggravate the development of AKI disease by increasing the proinflammatory factors secretion of senescent cells. The NF-κB pathway is a key upstream signaling molecule that mediates the SASP pathway. We found that NF-κB signaling pathway was significantly downregulated in p16 null mice, suggesting that the positive effect of p16 on senescent-associated proinflammatory factors secretion may be achieved by upregulating the NF-κB signaling pathway. In addition, we also examined the apoptosis of renal cells for assessing the protective effect of p16 deletion on the kidney. Previous studies have reported that apoptosis is involved in the development of glycerol-induced AKI and is regulated by proapoptotic or antiapoptotic molecules [21,22]. Consistent with the results observed in these studies, we found that the absence of p16 attenuated apoptosis for reducing further renal damage, thereby improving the prognosis of ATN.
ROS is a key signaling molecule that mediates cellular senescence. ROS activation is closely related to the SASP mediated mainly by NF-κB signaling pathway [23]. Studies have shown that excessive ROS accumulation plays a significant adverse role in AKI and mediates proinflammatory responses and massive apoptosis of tubular epithelial cells [21,22]. We found that the absence of p16 reduced ROS levels during ATN process. In AKI, KEAP1, PGAM5 and AIF protein were significantly reduced, suggesting that p16 enhanced the activation of ROS signaling pathway. Excessive accumulation of ROS may be one of the key factors by which p16 protein causes SASP and epithelial apoptosis in ATN. Our previous study reported that knocking out p16 could significantly meliorate renal tubular interstitial fibrosis in premature aging mice and reduce aging-related proinflammatory factors secretion in kidney, showing that p16 plays an important role in promoting progression of renal diseases [5]. However, the exact mechanism underlying p16 protein-mediated ROS accumulation in ATN is still unclear and remains to be further investigated.
Recent studies have found that ROS mediates a cell death pattern being called oxeiptosis [24]. Our study showed that p16 protein seemed to have a certain correlation with key proteins such as PGAM5 in this death mode, suggesting that p16 might be involved in this important cell death process. Recent study finds that antiapoptotic signaling pathway is activated during cell aging, and a large number of senescent cells can not be normally apoptotic or cleared, resulting in excessive accumulation of senescent cells that causes SASP. Improvements in a variety of mouse disease models and longevity have been observed by using senescent cell clearance drugs such as ABT263 [25]. Consistent with this, we found that p16 null downregulated the cell apoptosis and inhibited SASP through decreasing aging cells in AKI. As a new pathway to cell death, the question is whether oxeiptosis provides a new way to eliminate senescent cells? There is a certain correlation between p16 protein and new modes of cell death, such as oxeiptosis. Whether the accumulation of p16-positive senescent cells is related to oxeiptosis and is involved in the specific role played by the p16 protein remains to be further studied. These in-depth studies may provide new targets for the clearance of aging cells and for the treatment of a variety of aging-related diseases.
In conclusion, p16 promoted the development of ATN, including increasing inflammatory infiltration and proinflammatory factor expression by activating NF-κB proinflammatory pathway, promoting cell apoptosis by imbalance between pro-apoptotic and anti-apoptotic molecules, and increasing ROS levels and upregulating ROS signaling pathway, thereby aggravating the damaging effects of AKI. P16 deletion could ameliorate development of ATN. Thus, p16 deletion or inhibition and p16 positive cell clearance would be a novel strategy for preventing ATN in AKI progression.
Materials and methods
Mice
Exon 1α of p16 was deleted. P16 heterozygote male and female mice of the FVB N2 background were mated to generate eight-week-old p16KO mice and WT littermates genotyped as described previously [26,27]. All experiments were carried out according to the guidelines of the Experimental Animal Research Institute of Nanjing Medical University. The program was approved by the Nanjing Animal Experimental Ethics Committee (Permit Number: IACUC-1903001).
Acute tubular necrosis model
Rhabdomyolysis was induced by intramuscular injection of glycerol, causing acute tubular necrosis, thereby constructing an AKI model, as previously described [11,14,16,28]. Mice were anesthetized with 3% pentobarbital sodium (40 mg/kg). Adult eight-week-old male p16KO mice and WT littermates were intramuscularly injected with glycerol (purity > 99%) (Sigma-Aldrich) (50% solution, 0.9% saline, 8 ml/kg), and each upper hind limb was given an equal dose. Control mice were intramuscularly injected with an equal volume of saline in the same manner. Adult male C57BL/6J mice were given a standard diet, hydrated for 16 h before glycerol intramuscular injection, and after glycerol intramuscular injection, they were hydrated for 8 h to enhance the toxicity of glycerol [16].
Kidney processing
Mice were anesthetized with 3% pentobarbital sodium (40 mg/kg) and perfused with 100 ml of saline (0.01 mM PO4 3-, pH 7.4). Kidneys were separated and fixed with periodate-lysine-paraformaldehyde (PLP) solution [26,29]. The samples were cut into two identical pieces along the coronal axis according to the previous method and fixed in PLP solution (for histochemistry or immunohistochemistry) overnight at 4°C [26,29]. For hematoxylin and eosin (HE) or immunohistochemical staining, kidney sections were dehydrated in a series of gradient ethanol solutions and embedded in paraffin, and 5 μm sections were cut on a paraffin sectioner (Leica Microsystems Nussloch GmbH, Nucloch, Germany) as previously described [29].
Western blot analysis
The method for detecting protein in the kidney by western blot is as previously described [26,29]. Primary antibodies against p16 (ab211542), IL-1β (ab9722), AIF (ab32516), KEAP1 (ab119403), and PGAM5 (ab126534) were purchased from Abcam (Cambridge, MA, USA); p65 (#8242), p-p65 (Ser536) (#3033), p53 (#2524), Bcl-2 (#3498), Bax (#2772), and cleaved caspase3 (#9664) were purchased from Cell Signaling Technology (Beverly, MA, USA); β-actin (60008-1-Ig) was purchased from Proteintech (Rosemont, IL, USA); TNF-α (NBP1-19532) was purchased from Novus Biologicals (Centennial, CO, USA). HRP-conjugated Affinipure Goat Anti-Rabbit IgG (H+L) and HRP-conjugated Affinipure Goat Anti-Mouse IgG (H+L) were purchased from Proteintech (USA).
Immunohistochemical staining
Immunohistochemical staining was performed according to the previously described methods [26,29]. Serial paraffin sections were subjected to antigen retrieval, cooked in antigen retrieval solution for 20 min, then inactivated endogenous peroxidase (3% H2O2) and blocked with serum for 1 h, followed by incubation with primary antibodies against CD3 (SC-20047, Santa Cruz Biotechnology Inc., Dallas, TX, USA), F4/80 (SC-377009, Santa Cruz Biotechnology Inc.), IL-6 (SC-57315, Santa Cruz Biotechnology Inc.), IL-1β (ab9722, Abcam), TNF-α (NBP1-19532, Novus Biologicals), and p-p65 (Ser536) (#3033, Cell Signaling Technology). After washing, sections were incubated with secondary antibody for 1 h, washed and treated with SABC-POD kit (SA2001, Boster, China). Then, the sections were counterstained with hematoxylin and fixed with biomount medium [26,29,30].
Determination of serum urea nitrogen and serum creatinine
Mice were anesthetized with 3% sodium pentobarbital (40 mg/kg) and depilated in the abdominal region, and blood was drawn from the mouse heart with a 1 ml syringe. The method was as previously described, and the serum was separated for measurement of serum urea nitrogen (SUN) (C013-2 SUN test kit) and serum creatinine (SCr) (C011 Cr detection kit) according to the manufacturer’s instructions (Nanjing Jiancheng Institute of Bioengineering, China) [26].
TUNEL assay
Continuous paraffin sections were dewaxed, hydrated and stained with an apoptosis detection kit (YFXCA05, Yi Fei Xue Biotechnology, China) according to the manufacturer’s instructions.
Intracellular ROS detection
Renal cells were isolated from kidneys of 8-week-old mice, incubated in 5 mM diacetyldichlorofluorescein (DCFDA) (Invitrogen Inc., Carlsbad, CA, USA) and then placed in a shaker for 30 min at 37°C, immediately followed by flow cytometry analysis in a FACScalibur flow cytometer (Becton Dickinson, Germany) [26,29].
Statistical analysis
Measurement data were described as the mean ± SEM fold-change over the control and analyzed by Student’s t-test and one-way ANOVA to compare differences among groups. Qualitative data were described as percentages and analyzed using chi-square tests as indicated. All analyses were performed using SPSS software (Version 19.0; SPSS Inc., Chicago, IL, USA) or GraphPad Prism software (Version 6.07; GraphPad Software Inc., San Diego, CA, USA) as previously described. A value of P < 0.05 was considered statistically significant [26,29].
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 81571371 & No. 81871097 to J.J., No. 81170102 & No. 81441011 & No. 81670328 to Z.Y.), the Jiangsu Government Scholarship for Overseas Studies (JS-2017-095 to J.J.). the National High Technology Research and Development Program of China (863 Program to Z.Y.), the Doctoral Scientific Fund Project of the Ministry of Education of China (20123234110015 to Z.Y.), the Fourth Period Project “333” of Jiangsu Province (BRA2012207 to Z.Y.), the Chinese Medical Association of the Sunlight Foundation (SCRFCMDA201217 to Z.Y.), and the Collaborative Innovation Center of Nanjing Medical University (to Z.Y.).
Disclosure of conflict of interest
None.
References
- 1.Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol. 2005;16:3365–3370. doi: 10.1681/ASN.2004090740. [DOI] [PubMed] [Google Scholar]
- 2.Andrade L, Rodrigues CE, Gomes SA, Noronha IL. Acute kidney injury as a condition of renal senescence. Cell Transplant. 2018;27:739–753. doi: 10.1177/0963689717743512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson AC, Zager RA. Plasma and urinary p21: potential biomarkers of AKI and renal aging. Am J Physiol Renal Physiol. 2018;315:F1329–F1335. doi: 10.1152/ajprenal.00328.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vandenberk B, Brouwers B, Hatse S, Wildiers H. p16INK4a: a central player in cellular senescence and a promising aging biomarker in elderly cancer patients. Journal of Geriatric Oncology. 2011;2:259–269. [Google Scholar]
- 5.Jin J, Tao J, Gu X, Yu Z, Wang R, Zuo G, Li Q, Lv X, Miao D. P16 (INK4a) deletion ameliorated renal tubulointerstitial injury in a stress-induced premature senescence model of Bmi-1 deficiency. Sci Rep. 2017;7:7502. doi: 10.1038/s41598-017-06868-8. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6.Palipoch S. A review of oxidative stress in acute kidney injury: protective role of medicinal plants-derived antioxidants. Afr J Tradit Complement Altern Med. 2013;10:88–93. doi: 10.4314/ajtcam.v10i4.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meng XM, Ren GL, Gao L, Yang Q, Li HD, Wu WF, Huang C, Zhang L, Lv XW, Li J. NADPH oxidase 4 promotes cisplatin-induced acute kidney injury via ROS-mediated programmed cell death and inflammation. Lab Invest. 2018;98:63–78. doi: 10.1038/labinvest.2017.120. [DOI] [PubMed] [Google Scholar]
- 8.Shen WC, Liang CJ, Huang TM, Liu CW, Wang SH, Young GH, Tsai JS, Tseng YC, Peng YS, Wu VC, Chen YL. Indoxyl sulfate enhances IL-1beta-induced E-selectin expression in endothelial cells in acute kidney injury by the ROS/MAPKs/NFkappaB/AP-1 pathway. Arch Toxicol. 2016;90:2779–2792. doi: 10.1007/s00204-015-1652-0. [DOI] [PubMed] [Google Scholar]
- 9.Zhang H, Chen MK, Li K, Hu C, Lu MH, Situ J. Eupafolin nanoparticle improves acute renal injury induced by LPS through inhibiting ROS and inflammation. Biomed Pharmacother. 2017;85:704–711. doi: 10.1016/j.biopha.2016.11.083. [DOI] [PubMed] [Google Scholar]
- 10.Braun H, Schmidt BM, Raiss M, Baisantry A, Mircea-Constantin D, Wang S, Gross ML, Serrano M, Schmitt R, Melk A. Cellular senescence limits regenerative capacity and allograft survival. J Am Soc Nephrol. 2012;23:1467–1473. doi: 10.1681/ASN.2011100967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lv X, Yu Z, Xie C, Dai X, Li Q, Miao D, Jin J. Bmi-1 plays a critical role in the protection from acute tubular necrosis by mobilizing renal stem/progenitor cells. Biochem Biophys Res Commun. 2017;482:742–749. doi: 10.1016/j.bbrc.2016.11.105. [DOI] [PubMed] [Google Scholar]
- 12.Perin L, Sedrakyan S, Giuliani S, Da Sacco S, Carraro G, Shiri L, Lemley KV, Rosol M, Wu S, Atala A, Warburton D, De Filippo RE. Protective effect of human amniotic fluid stem cells in an immunodeficient mouse model of acute tubular necrosis. PLoS One. 2010;5:e9357. doi: 10.1371/journal.pone.0009357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nara A, Yajima D, Nagasawa S, Abe H, Hoshioka Y, Iwase H. Evaluations of lipid peroxidation and inflammation in short-term glycerol-induced acute kidney injury in rats. Clin Exp Pharmacol Physiol. 2016;43:1080–1086. doi: 10.1111/1440-1681.12633. [DOI] [PubMed] [Google Scholar]
- 14.Sagrinati C, Netti GS, Mazzinghi B, Lazzeri E, Liotta F, Frosali F, Ronconi E, Meini C, Gacci M, Squecco R, Carini M, Gesualdo L, Francini F, Maggi E, Annunziato F, Lasagni L, Serio M, Romagnani S, Romagnani P. Isolation and characterization of multipotent progenitor cells from the Bowman’s capsule of adult human kidneys. J Am Soc Nephrol. 2006;17:2443–2456. doi: 10.1681/ASN.2006010089. [DOI] [PubMed] [Google Scholar]
- 15.Lee DH, Wolstein JM, Pudasaini B, Plotkin M. INK4a deletion results in improved kidney regeneration and decreased capillary rarefaction after ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2012;302:F183–191. doi: 10.1152/ajprenal.00407.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim JH, Lee SS, Jung MH, Yeo HD, Kim HJ, Yang JI, Roh GS, Chang SH, Park DJ. N-acetylcysteine attenuates glycerol-induced acute kidney injury by regulating MAPKs and Bcl-2 family proteins. Nephrol Dial Transplant. 2010;25:1435–1443. doi: 10.1093/ndt/gfp659. [DOI] [PubMed] [Google Scholar]
- 17.He S, Sharpless NE. Senescence in health and disease. Cell. 2017;169:1000–1011. doi: 10.1016/j.cell.2017.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watanabe S, Kawamoto S, Ohtani N, Hara E. Impact of senescence-associated secretory phenotype and its potential as a therapeutic target for senescence-associated diseases. Cancer Sci. 2017;108:563–569. doi: 10.1111/cas.13184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xia S, Zhang X, Zheng S, Khanabdali R, Kalionis B, Wu J, Wan W, Tai X. An update on inflamm-aging: mechanisms, prevention, and treatment. J Immunol Res. 2016;2016:8426874. doi: 10.1155/2016/8426874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, Mazula DL, Brooks RW, Fuhrmann-Stroissnigg H, Pirtskhalava T, Prakash YS, Tchkonia T, Robbins PD, Aubry MC, Passos JF, Kirkland JL, Tschumperlin DJ, Kita H, LeBrasseur NK. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. 2017;8:14532. doi: 10.1038/ncomms14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Y, Feng X, Hu X, Sha J, Li B, Zhang H, Fan H. Dexmedetomidine ameliorates acute stress-induced kidney injury by attenuating oxidative stress and apoptosis through inhibition of the ROS/JNK signaling pathway. Oxid Med Cell Longev. 2018;2018:4035310. doi: 10.1155/2018/4035310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dennis JM, Witting PK. Protective role for antioxidants in acute kidney disease. Nutrients. 2017;9 doi: 10.3390/nu9070718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pardo A, Selman M. Lung fibroblasts, aging, and idiopathic pulmonary fibrosis. Ann Am Thorac Soc. 2016;13(Suppl 5):S417–S421. doi: 10.1513/AnnalsATS.201605-341AW. [DOI] [PubMed] [Google Scholar]
- 24.Holze C, Michaudel C, Mackowiak C, Haas DA, Benda C, Hubel P, Pennemann FL, Schnepf D, Wettmarshausen J, Braun M, Leung DW, Amarasinghe GK, Perocchi F, Staeheli P, Ryffel B, Pichlmair A. Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat Immunol. 2018;19:130–140. doi: 10.1038/s41590-017-0013-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, Luo Y, Wang X, Aykin-Burns N, Krager K, Ponnappan U, Hauer-Jensen M, Meng A, Zhou D. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016;22:78–83. doi: 10.1038/nm.4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jin J, Lv X, Chen L, Zhang W, Li J, Wang Q, Wang R, Lu X, Miao D. Bmi-1 plays a critical role in protection from renal tubulointerstitial injury by maintaining redox balance. Aging Cell. 2014;13:797–809. doi: 10.1111/acel.12236. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, Wu EA, Horner JW, DePinho RA. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature. 2001;413:86–91. doi: 10.1038/35092592. [DOI] [PubMed] [Google Scholar]
- 28.Lochhead K, Kharasch E, Zager R. Anesthetic effects on the glycerol model of rhabdomyolysis-induced acute renal failure in rats. J Am Soc Nephrol. 1998;9:305–309. doi: 10.1681/ASN.V92305. [DOI] [PubMed] [Google Scholar]
- 29.Xie C, Jin J, Lv X, Tao J, Wang R, Miao D. Anti-aging effect of transplanted amniotic membrane mesenchymal stem cells in a premature aging model of bmi-1 deficiency. Sci Rep. 2015;5:13975. doi: 10.1038/srep13975. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Jin J, Zhao Y, Tan X, Guo C, Yang Z, Miao D. An improved transplantation strategy for mouse mesenchymal stem cells in an acute myocardial infarction model. PLoS One. 2011;6:e21005. doi: 10.1371/journal.pone.0021005. [DOI] [PMC free article] [PubMed] [Google Scholar]