

Abstract
Epigenetic modifications via DNA methylation and long non-coding RNAs (lncRNAs) have been identified in bladder cancer (BC). However, DNA methylation of lncRNAs involved in BC has not been elucidated. Here, DNA immunoprecipitation-sequencing (MeDIP-seq) and RNA-sequencing (RNA-seq) were carried out using eight paired tumor and adjacent normal tissue samples from patients with BC. Differences in methylation patterns between tumors and controls were compared and the percentage of differentially methylated genes, including lncRNA genes, was calculated. RNA-seq data were subjected to gene ontology (GO), Kyoto encyclopedia of genes, and genomes (KEGG) analysis. The association between DNA methylation modification and lncRNA expression was determined by pairwise analysis of MeDIP-seq and RNA-seq data. The most enriched motifs in the promoter region, as well as the methylated density in the 3 kb region surrounding super-enhancers of lncRNA genes, were analyzed. A peak of 5mC methylation in the region 2 kb upstream of the transcription start site (TSS), with the lowest point in the TSS region, was observed. In total, 436 and 239 genes were identified to be hyper and hypomethylated, respectively, in BC tissue around the TSS region. RNA-seq revealed differentially expressed lncRNAs between tumor and normal tissues, many of which were cancer-associated lncRNAs based on GO and KEGG pathway analysis. Combined MeDIP-seq and RNA-seq analysis revealed that expression of 26 lncRNAs were candidates of 5mC controlled genes. The possible link between 5mC modification and differential lncRNAs may relate to enrichment of 5mC reads in the region surrounding super-enhancers of lncRNA. Survival analysis indicated that the methylated lncRNA, LINC00574, was associated with shorter overall survival time in patients with BC (HR = 1.7, p-value = 0.035). Taken together, these findings indicate that lncRNAs genes are under control of DNA methylation. Methylated lncRNA genes, which are transcripted to LINC00574, may serve as biomarkers for BC prognosis.
Keywords: DNA methylation, MeDIP-seq, bladder cancer, lncRNA
Introduction
Bladder cancer (BC) is one of the most common cancers of the urinary system. Risk factors for BC are known to include tobacco use, Schistosoma haematobium infection, industrial exposure, regular diet, and lifestyle trends. In 2012, approximately 430,000 new BC cases were diagnosed [1]. In western countries, it is estimated there are 118,000 newly diagnosed cases, which has led to a substantial health service burden [2,3]. In China, rates of BC have increased rapidly over the five-year period 2003-2008, with a higher increase in women than men [4]. Despite surgery, dissection, and various adjuvant treatments for BC, the five-year survival rate remains low (60%) and there is a high risk of recurrence. It is reported that 30-70% of tumors reoccur [5], and 30% of tumors may develop into muscle-invasive disease [6,7]. Therefore, better understanding of disease-causing molecules and mechanisms in BC is urgently needed to improve tumor prevention and control.
Epigenetics is the study of gene expression changes that do not involve alterations in the DNA sequence, such as DNA methylation, histone modifications, and non-coding RNA-mediated gene silencing. DNA methylation is a system of genetic text annotation, the most common form of which is to add a methyl group to the 5’ cytosine of C-G dinucleotides to form cytosine guanine dinucleotides (CpG), which provides information for gene transcription and mediating gene expression suppression or silencing [8]. Aberrant epigenetic modifications have been found in several types of cancers including BC [8].
Long non-coding RNAs (lncRNAs) are a class of RNA defined as transcripts > 200 nt in length transcribed from non-protein coding regions of the genome [9]. Multiple reports have demonstrated the expression of lncRNAs in mammals [10,11]. Many lncRNAs are expressed specifically in cancer cells [12], and changes to their expression are critical for tumor initiation and progression [13].
Recently, growing evidence has focused on the relationship between DNA methylation and non-coding genes in human cancer [14]. Aberrant epigenetic modifications of lncRNAs have been reported in several types of cancers, including BC [15-18]. However, understanding of DNA methylation patterns of lncRNAs genes in BC remains limited. In the present study, we carried out methylated DNA immunoprecipitation-sequencing (MeDIP-seq) and RNA-sequencing for BC-associated methylated lncRNA screening in eight pairs of BC and matched normal adjacent tissue samples. We then further analyzed the association between differentially expressed lncRNA controlled by DNA methylation and BC prognosis.
Methods and materials
Clinical sample
Eight paired tumor and adjacent normal tissues from patients with BC were collected at the People’s Hospital of Hainan Province from March 2014 to July 2016 before microscopic examination. BC tumor tissues were confirmed to be invasive urothelial bladder cancer by pathologists, with grades ranging from 1 to 4 and sizes ranging from 0.6 cm3 to 135 cm3. All tissue samples were immediately transferred into liquid nitrogen and stored at -80°C until genomic DNA extraction. This experiment was approved by the Committee on the Ethics of People’s Hospital of Hainan Province. All patients were fully informed about the study and provided informed consent.
MeDIP-seq
Genomic DNA from the eight tumor tissues and paired tumor-adjacent normal tissues was isolated using the MagMedIP kit (Diagenode, Denville, NJ, USA) according to the manufacturer’s instructions. Next, methylated DNA was separated from unmethylated fragments by immunoprecipitation using a monoclonal antibody against 5-methylcytidine (Diagenode, Denville, NJ, USA), as previously described [4]. Illumina sequencing libraries of the enriched methylated DNA were prepared using NEBNext reagents according to the manufacturer’s recommendations (New England Biolabs, MA, USA). After quantitative evaluation of the size distribution and concentration of the library using an Agilent Bioanalyzer 2100 (Agilent Technologies, CA, USA), 100 nucleotide (nt) sequencing runs were conducted on an Illumina HiSeq 2000 platform (Illumina, CA, USA).
Differential methylated gene profile analysis
Raw MeDIP-seq data were processed according to the standard procedures of the Beijing Genome Institute (BGI; Shenzhen, China). Reads were aligned to the human reference genome (hg19) using Bowtie2 (version 2.2.6), with default parameters (max mismatches in seed alignment = 0). We then used MACS software (MACS 1.4.0) to scan for methylation peaks (Peak) genome-wide. The read counts of tumors and normal groups were converted into peak values. We then extracted the distribution of peak values across genes, as well as regions 2 kb upstream and downstream. A heat map was created using R (version 3.4.3) to identify genes that were either hyper or hypomethylated based on a fold change of ≥ 1.5 and P < 0.05 (Student’s t-test). Hyper and hypomethylated reads were counted and sorted for the following gene regions: upstream 2 kb, 5’UTR, CDS, intron, 3’UTR, and downstream 2 kb. The average signals of DNA methylations were compared among genes derived from the coding regions and lncRNAc encoding regions.
RNA-seq for differential expression of lncRNAs
Total RNA was isolated from tumor and normal tissues using Trizol (Thermo Fisher Scientific, IL, USA), according to standard protocols. Ribosomal RNA (rRNA) removal was performed using the RiboMinus Eukaryote Kit (Qiagen, Hilden, Germany). To prepare the RNA library for Illumina sequencing, we used the NEB Next Ultra Directional RNA Library Prep Kit for Illumina (NEB, MA, USA), according to the manufacturer’s recommendations.
Libraries were assessed for quality with an Agilent 2100 assay and quantified by quantitative PCR prior to Illumina sequencing. After quality control, clean reads were obtained, filtered, and mapped to the human reference genome hg19 using TopHat (version 2.1.0) [19] with the default parameters (max mismatches in seed alignment = 0). The expression level of each gene was calculated, and the results were normalized using the RPKM (Reads Per Kilobase of exon model per Million mapped reads) method. Differentially expressed genes were identified according to established methods [20]. To address the issue of multiple comparisons, the threshold p-value was determined by controlling the false discovery rate (FDR, Benjamini-Hochberg method). In our analysis, differentially expressed genes were defined as those with FDR ≤ 0.001 and a more than two-fold expression change. Cluster analysis of differentially expressed genes was performed using R. We also used gene ontology (GO) annotation and the KEGG pathway analysis, with FDR ≤ 0.001 considered to be significantly enriched.
Prediction of super-enhancer and transcription factor binding enrichment
To analyze methylation modifications in lncRNA genes near the enhancer, transcription factor enriched motifs were identified on the 5mC gain (Up) and loss (Down) regions using HOMER (version 4.10), a de novo motif discovery algorithm. Hypo and hypermethylation enriched regions around the annotated transcription start sites (TSS) ± 3 kb with a signal density (normalized by the RPKM method) of reads above the background (P < 0.01, empirical test) were defined as candidate super-enhancers.
Survival analysis
Overall survival and disease-free survival time of patients with BC with different expression levels of LINC00574 and LINC00092 were analyzed using data obtained from the Cancer Genome Atlas (TCGA, https://cancergenome.nih.gov/). Survival analysis was analyzed on the GEPIA web server [21] (http://gepia.cancer-pku.cn/) with the default parameters. A log-rank test was used to compare differences in survival time between groups. P < 0.05 was considered to be statistically significant.
Results
Genome-wide distribution of 5mC in BC tissues
To visualize genome-wide changes in 5mC between tumor and normal tissues, MeDIP-seq was conducted. Sequencing reads matching tumor or normal groups were converted to density signals and depicted as a peak diagram on the basis of their genomic location. In total, MeDIP-seq identified 436 hypermethylated and 239 hypomethylated genes in BC tissues around the TSS region (Figure 1A). As shown in Figure 1B, the overall level of modification of intergenic regions was higher than the methylation level in regions 2 kb (Upstream 2 kb) upstream or downstream (Downstream 2 kb) of the transcriptional stop point. Notably, a sharp peak was observed in the 2 kb region upstream of the TSS (Figure 1B), which quickly dropped to its lowest point at the TSS. Determination of the distribution of 5mC methylation showed that 5mC was most enriched in the intron regions followed by CDS, but not in the UTR or gene body flanking regions (Figure 1D). Of the differentially 5mC methylated genes, > 70% were coding genes, whereas few (~20%) were lncRNAs coding genes (Figure 1C) in both tumor and normal tissues.
Figure 1.
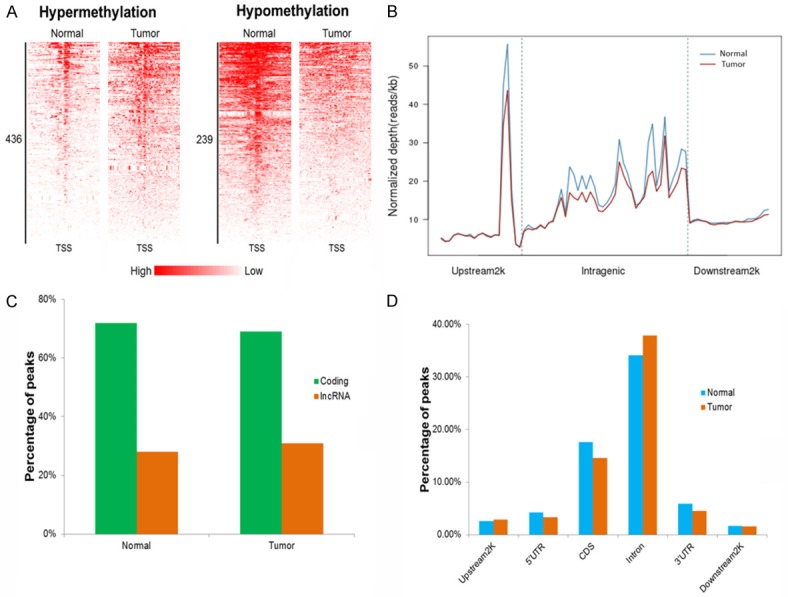
Differences in the pattern of DNA methylation between adjacent normal tissue (Normal) and BC tumor tissue (tumor). A. Heat maps show differences in the density profiles of hyper and hypomethylation between normal and tumor tissues. B. Distribution of reads obtained by MeDIP-seq in the intragenic gene region, and regions 2 kb upstream and downstream 2 kb of the genes. Upstream 2 kb, 2 kb region upstream of the transcription start site (TSS); intragenic, the entire gene from the TSS to the end of the transcript; Downstream 2 kb, 2 kb region downstream of the end of the transcript. C. Reads of MeDIP-seq matching to a coding gene or lncRNA gene in normal and tumor tissues. D. Statistical analysis of percentage of MeDIP-seq reads matching to regions of upstream2K, 5’UTR, CDS, intron, 3’UTR, and downstream2K.
Global change of lncRNA expression in BC tissue
Next, lncRNA expression profiles were analyzed in eight paired tumor and normal tissues by RNA-seq. As shown in Figure 2, 895 lncRNAs were differentially expressed in tumor tissue relative to normal tissue. Among these lncRNAs, 228 were upregulated and 667 were downregulated relative to normal tissue. The expression profile of tumor tissues thus differs from normal tissues (Figure 2A), as the expression signals in the heatmap show separate clustering of tumor and normal tissues. Furthermore, analysis of differentially expressed lncRNAs revealed that downregulated lncRNAs were distributed across all chromosomes in BC tissue, with the upregulated lncRNAs found on all but chromosomes 20 and 23 (Figure 2B). To provide an overview of the potential role of the differential lncRNAs, GO and KEGG enriched analyses were conducted. As shown in Figure 3A, the four most enriched GO terms were cell, cell part, cellular process and binding, with 1201, 1200, 1125, and 1106 genes enriched in each category, respectively (Figure 3A). KEGG enrichment analysis revealed the top five enriched pathways were the pathways in cancer, cGMP-PKG signaling pathway, PI3K-Akt signaling pathway, focal adhesion, and Rap1 signaling pathway (Figure 3B).
Figure 2.
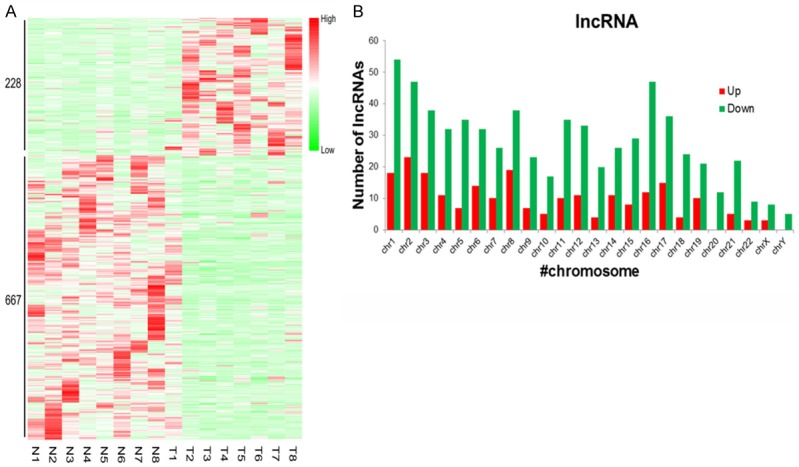
RNA-seq expression profiles of lncRNA between tumor and normal tissues in patients with BC. A. Heat map showing differential lncRNA expression patterns in BC tissue and adjacent normal tissue. B. Chromosome distribution of differentially expressed lncRNA between tumor and normal tissues. Upregulated lncRNA is indicated in red, while downregulated lncRNA is indicated in green.
Figure 3.

GO (A) and KEGG pathway enrichment (B) analysis of differentially expressed lncRNAs.
Expression of lncRNA is regulated by 5mC and associated with BC prognosis
To explore the potential link between change in lncRNA expression and differential methylation status in BC, the preference of 5mC modification and profile of 5mC state on the location around TSS were analyzed. Figure 4A lists the five most enriched motifs of gain-5mC (UP) and loss-5mC (Down) loci. Comparison of locus-specific changes in 5mC in the super-enhancer region of lncRNA genes between tumor and normal control showed a reduced signal of 5mC modification in tumor tissue (Figure 4B).
Figure 4.
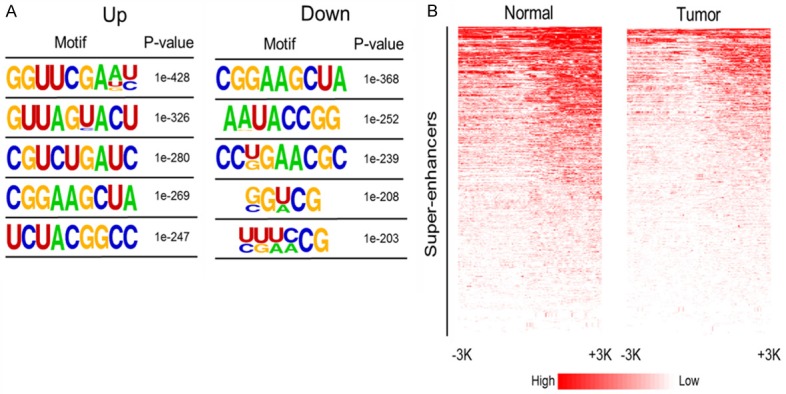
5mC is enriched at enhancer regions of lncRNA genes in patients with BC. A. Top five enriched motifs for gain-5mC (UP) and loss-5mC (Down) loci. B. Heat maps showing 5mC patterns in the 3 kb region surrounding the super-enhancers of lncRNA genes.
The combination of MeDIP-seq and lncRNA sequencing identified 26 lncRNAs that were consistently hypermethylated and downregulated or hypomethylated and upregulated across all tumor tissues from the eight patients with BC (Table 1). To determine if there was an association between these lncRNAs and BC outcome, lncRNAs were subjected to GEPIA analysis. Indeed, two lncRNAs were found to be associated with BC survival. As shown in Figure 5A, hypomethylated LINC00574 was associated with poor overall survival (HR = 1.7, p-value = 0.035) such that patients with BC with higher expression of LINC00574 displayed shorter overall survival time. However, no significant association was found between expression of LINC00574 and survival time in patients with BC (Figure 5A). In addition, hypomethylated LINC00092 showed no significant association with survival in patients with BC (Figure 5B). The association between lncRNA expression and progression of BC requires further verification.
Table 1.
Methylated lncRNAs showing differential expression in BC tumor tissue
| Methylation status | lncRNA expression | |
|---|---|---|
| LINC00839 | down | up |
| LINC00301 | down | up |
| LINC00111 | down | up |
| VENTXP1 | down | up |
| LINC00898 | down | up |
| LINC00887 | down | up |
| LINC00592 | down | up |
| LINC00669 | down | up |
| LINC00643 | up | down |
| LINC00473 | up | down |
| LINC00472 | up | down |
| SNHG9 | up | down |
| RBAKDN | up | down |
| LINC01056 | up | down |
| LINC01003 | up | down |
| LINC00626 | up | down |
| LINC00606 | up | down |
| LINC00574 | up | down |
| LINC00486 | up | down |
| LINC00474 | up | down |
| LINC00339 | up | down |
| LINC00303 | up | down |
| LINC00092 | up | down |
| LINC00028 | up | down |
| EPB41L4A-AS1 | up | down |
| C11orf95 | up | down |
Figure 5.
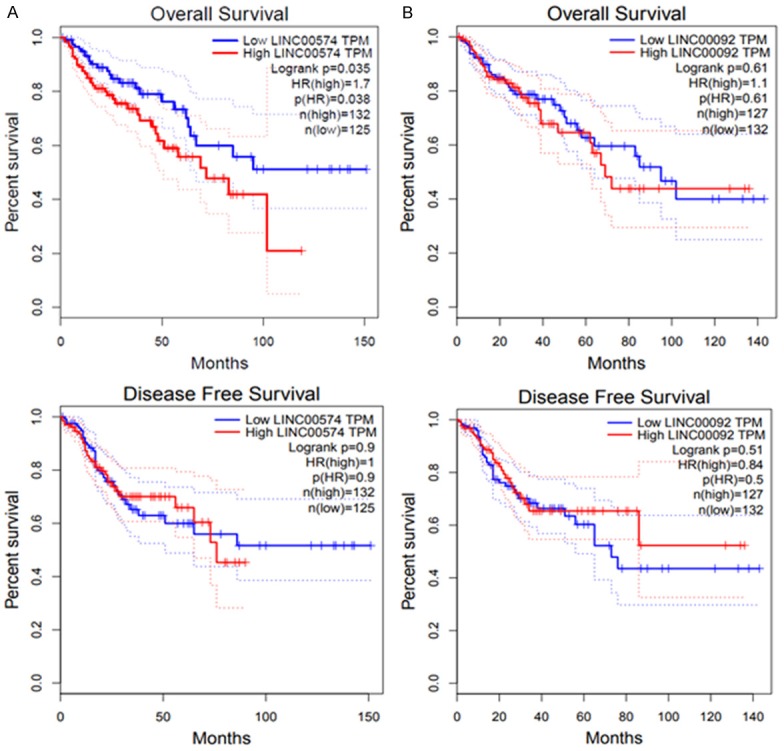
Two differentially expressed lncRNAs, namely LINC00574 and LINC00092, which were also differentially methylated in tumor tissues, were associated with poor overall survival or disease-free survival. A. Expression of LINC00574 with survival in patients with BC. B. Expression of LINC00092 with survival in patients with BC. Survival analysis was conducted using an online GEPIA tool (http://gepia.cancer-pku.cn/).
Discussion
DNA methylation and lncRNA are two important epigenetic regulatory factors in human cancer. In the present study, we investigated the association between DNA methylation and BC-related lncRNAs expression using MeDIP-seq and RNA-seq data in eight patients with BC.
Intragenic DNA methylation has been shown to play a role in several gene regulation processes [22]. Regions with increased CpG content (known as CpG islands) are known to overlap with TSS in about 60-70% of genes in the human genome. By contrast, regions with low CpG modification are frequently located regions other than the TSS [22]. DNA methylation primarily occurs in CpGs, but may also occur in non-CpG regions [23]. Our MeDIP-seq revealed that the lowest level of methylation was at the TSS site. At the same time, there was a signal of a significant DNA methylation peak in the 2 kb region near the TSS.
The function of lncRNA in regulating tumor cell behavior - including cell proliferation, apoptosis, migration, and invasion - has been widely reported [24-28]. In BC, lncRNA is particularly involved in the regulation of proliferation, metastasis, and drug resistance [29-32]. In the present study, we demonstrated the overall expression profile of lncRNA in control tissues and eight patients with BC tumor. KEGG enrichment analysis of differentially expressed lncRNAs in tumor and control tissues revealed that these lncRNAs were significantly enriched in pathways in cancer, cGMP-PKG signaling pathway, PI3K-Akt signaling pathway, focal adhesion, and the Rap1 signaling pathway. Among these, the PI3K-Akt signaling pathway plays a role in tumor proliferation and growth and is involved in lncRNA regulating the occurrence and progression of various tumors [33-35].
We also integrated the expression pattern of lncRNA with their 5mC modification levels. To investigate whether DNA methylation regulation is involved in regulating differentially expressed lncRNA, we calculated the 5mC acquisition and loss motif characteristics on the lncRNA gene. The results showed that certain motifs of DNA 5mC were preferentially gained and lost. At the same time, in the vicinity of TSS, the DNA methylation level of the lncRNA gene super-enhancer region decreased. These findings suggest that expression of BC-associated lncRNA is related to the DNA methylation status near the TSS region of the lncRNA gene. We also observed lncRNAs with opposite changes in lncRNA expression and DNA modification, which suggests the potential of DNA methylation in BC to regulate lncRNA. Previous studies have identified epigenetic modifications of lnRNAs that exert either beneficial effects on tumors, such as MINCR, or suppressed effects, such as MEG3 [36]. By contrast, the global profile of differentially methylated lncRNA in BC is poorly understood. The current study has thus provided novel insight into the methylation modifications that control BC-associated lncRNA candidates.
The functions of most lncRNAs have not been well characterized. However, LINC00643 has been reported to be differentially methylated and expressed in cancers [37] and serves as a tumor enhancer [38-40]. LINC00472 is regulated by DNA methylation in breast cancer and colorectal cancer, and is known to function as a tumor suppressor [41,42]. Several reports have provided evidence for the roles of SNHG9, LINC00574, and LINC00092 in cancer [43,44]. We further revealed that differential expression of LINC00574 in BC compared to normal tissue and a gain in methylation of 5mC in BC tissue are associated with reduced survival time. These findings suggest that LINC00574 is a biomarker for BC prognosis. LINC00574 has not been previously reported to have a role in BC, although it has been shown to be upregulated in breast cancer and gliomas [45,46]. In addition, its 5mC methylation status indicates a link between DNA methylation in regulating lncRNA expression and BC prognosis.
Taken together, the findings of the current study revealed a variety of methylation lncRNAs associated with BC. In particular, LINC00574 was associated with poor outcomes for patients with BC. These findings indicate a role for lncRNA regulation by 5mC methylation in BC prognosis.
Acknowledgements
This study was supported by the Finance Science and Technology Project of Hainan Province (Grant No. ZDYD2019163 and ZDKJ2017007), the Natural Science Foundation of Hainan Province (Grant No. 2017CXTD010), the National Natural Science Foundation of China (Grant No. 81760465, 81460450 and 81760461).
Disclosure of conflict of interest
None.
References
- 1.Antoni S, Ferlay J, Soerjomataram I, Znaor A, Jemal A, Bray F. Bladder cancer incidence and mortality: a global overview and recent trends. Eur Urol. 2017;71:96–108. doi: 10.1016/j.eururo.2016.06.010. [DOI] [PubMed] [Google Scholar]
- 2.Marcos-Gragera R, Mallone S, Kiemeney LA, Vilardell L, Malats N, Allory Y, Sant M. Urinary tract cancer survival in Europe 1999-2007: results of the population-based study EUROCARE-5. Eur J Cancer. 2015;51:2217–2230. doi: 10.1016/j.ejca.2015.07.028. [DOI] [PubMed] [Google Scholar]
- 3.Leal J, Luengo-Fernandez R, Sullivan R, Witjes JA. Economic burden of bladder cancer across the european union. Eur Urol. 2016;69:438–447. doi: 10.1016/j.eururo.2015.10.024. [DOI] [PubMed] [Google Scholar]
- 4.Li W, Huang Y, Sargsyan D, Khor TO, Guo Y, Shu L, Yang AY, Zhang C, Paredes-Gonzalez X, Verzi M, Hart RP, Kong AN. Epigenetic alterations in TRAMP mice: epigenome DNA methylation profiling using MeDIP-seq. Cell Biosci. 2018;8:3. doi: 10.1186/s13578-018-0201-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Babjuk M, Burger M, Zigeuner R, Shariat SF, van Rhijn BW, Comperat E, Sylvester RJ, Kaasinen E, Bohle A, Palou Redorta J, Roupret M. EAU guidelines on non-muscle-invasive urothelial carcinoma of the bladder: update 2013. Eur Urol. 2013;64:639–653. doi: 10.1016/j.eururo.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 6.Millan-Rodriguez F, Chechile-Toniolo G, Salvador-Bayarri J, Palou J, Algaba F, Vicente-Rodriguez J. Primary superficial bladder cancer risk groups according to progression, mortality and recurrence. J Urol. 2000;164:680–684. doi: 10.1016/s0022-5347(05)67280-1. [DOI] [PubMed] [Google Scholar]
- 7.Yuan L, Shu B, Chen L, Qian K, Wang Y, Qian G, Zhu Y, Cao X, Xie C, Xiao Y, Wang X. Overexpression of COL3A1 confers a poor prognosis in human bladder cancer identified by co-expression analysis. Oncotarget. 2017;8:70508–70520. doi: 10.18632/oncotarget.19733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harb-de la Rosa A, Acker M, Kumar RA, Manoharan M. Epigenetics application in the diagnosis and treatment of bladder cancer. Can J Urol. 2015;22:7947–7951. [PubMed] [Google Scholar]
- 9.Schmitt AM, Chang HY. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016;29:452–463. doi: 10.1016/j.ccell.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, Cabili MN, Jaenisch R, Mikkelsen TS, Jacks T, Hacohen N, Bernstein BE, Kellis M, Regev A, Rinn JL, Lander ES. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–1927. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, Barrette TR, Prensner JR, Evans JR, Zhao S, Poliakov A, Cao X, Dhanasekaran SM, Wu YM, Robinson DR, Beer DG, Feng FY, Iyer HK, Chinnaiyan AM. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet. 2015;47:199–208. doi: 10.1038/ng.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anastasiadou E, Jacob LS, Slack FJ. Non-coding RNA networks in cancer. Nat Rev Cancer. 2018;18:5–18. doi: 10.1038/nrc.2017.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Llinas-Arias P, Esteller M. Epigenetic inactivation of tumour suppressor coding and non-coding genes in human cancer: an update. Open Biol. 2017;7 doi: 10.1098/rsob.170152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo W, Liu S, Dong Z, Guo Y, Ding C, Shen S, Liang J, Shan B. Aberrant methylation-mediated silencing of lncRNA CTC-276P9.1 is associated with malignant progression of esophageal squamous cell carcinoma. Clin Exp Metastasis. 2018;35:53–68. doi: 10.1007/s10585-018-9881-2. [DOI] [PubMed] [Google Scholar]
- 16.Dong Z, Zhang A, Liu S, Lu F, Guo Y, Zhang G, Xu F, Shi Y, Shen S, Liang J, Guo W. Aberrant methylation-mediated silencing of lncRNA MEG3 functions as a ceRNA in esophageal cancer. Mol Cancer Res. 2017;15:800–810. doi: 10.1158/1541-7786.MCR-16-0385. [DOI] [PubMed] [Google Scholar]
- 17.Liao Q, Wang Y, Cheng J, Dai D, Zhou X, Zhang Y, Li J, Yin H, Gao S, Duan S. DNA methylation patterns of protein-coding genes and long non-coding RNAs in males with schizophrenia. Mol Med Rep. 2015;12:6568–6576. doi: 10.3892/mmr.2015.4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan W, Zhang N, Liu W, Liu J, Zhou L, Liu Y, Yang M. The long noncoding RNA GAS8-AS1 suppresses hepatocarcinogenesis by epigenetically activating the tumor suppressor GAS8. J Biol Chem. 2018;293:17154–17165. doi: 10.1074/jbc.RA118.003055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trapnell C, Salzberg SL. How to map billions of short reads onto genomes. Nat Biotechnol. 2009;27:455–457. doi: 10.1038/nbt0509-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–W102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kulis M, Queiros AC, Beekman R, Martin-Subero JI. Intragenic DNA methylation in transcriptional regulation, normal differentiation and cancer. Biochim Biophys Acta. 2013;1829:1161–1174. doi: 10.1016/j.bbagrm.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 23.Kim KD, El Baidouri M, Jackson SA. Accessing epigenetic variation in the plant methylome. Brief Funct Genomics. 2014;13:318–327. doi: 10.1093/bfgp/elu003. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Pitchiaya S, Cieslik M, Niknafs YS, Tien JC, Hosono Y, Iyer MK, Yazdani S, Subramaniam S, Shukla SK, Jiang X, Wang L, Liu TY, Uhl M, Gawronski AR, Qiao Y, Xiao L, Dhanasekaran SM, Juckette KM, Kunju LP, Cao X, Patel U, Batish M, Shukla GC, Paulsen MT, Ljungman M, Jiang H, Mehra R, Backofen R, Sahinalp CS, Freier SM, Watt AT, Guo S, Wei JT, Feng FY, Malik R, Chinnaiyan AM. Analysis of the androgen receptor-regulated lncRNA landscape identifies a role for ARLNC1 in prostate cancer progression. Nat Genet. 2018;50:814–824. doi: 10.1038/s41588-018-0120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun M, Nie F, Wang Y, Zhang Z, Hou J, He D, Xie M, Xu L, De W, Wang Z, Wang J. LncRNA HOXA11-AS promotes proliferation and invasion of gastric cancer by scaffolding the chromatin modification factors PRC2, LSD1, and DNMT1. Cancer Res. 2016;76:6299–6310. doi: 10.1158/0008-5472.CAN-16-0356. [DOI] [PubMed] [Google Scholar]
- 26.Xue X, Yang YA, Zhang A, Fong KW, Kim J, Song B, Li S, Zhao JC, Yu J. LncRNA HOTAIR enhances ER signaling and confers tamoxifen resistance in breast cancer. Oncogene. 2016;35:2746–2755. doi: 10.1038/onc.2015.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iguchi T, Uchi R, Nambara S, Saito T, Komatsu H, Hirata H, Ueda M, Sakimura S, Takano Y, Kurashige J, Shinden Y, Eguchi H, Sugimachi K, Maehara Y, Mimori K. A long noncoding RNA, lncRNA-ATB, is involved in the progression and prognosis of colorectal cancer. Anticancer Res. 2015;35:1385–1388. [PubMed] [Google Scholar]
- 28.Nie W, Ge HJ, Yang XQ, Sun X, Huang H, Tao X, Chen WS, Li B. LncRNA-UCA1 exerts oncogenic functions in non-small cell lung cancer by targeting miR-193a-3p. Cancer Lett. 2016;371:99–106. doi: 10.1016/j.canlet.2015.11.024. [DOI] [PubMed] [Google Scholar]
- 29.Liu D, Li Y, Luo G, Xiao X, Tao D, Wu X, Wang M, Huang C, Wang L, Zeng F, Jiang G. LncRNA SPRY4-IT1 sponges miR-101-3p to promote proliferation and metastasis of bladder cancer cells through up-regulating EZH2. Cancer Lett. 2017;388:281–291. doi: 10.1016/j.canlet.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 30.Feng SQ, Zhang XY, Fan HT, Sun QJ, Zhang M. Up-regulation of LncRNA MEG3 inhibits cell migration and invasion and enhances cisplatin chemosensitivity in bladder cancer cells. Neoplasma. 2018;65:925–932. doi: 10.4149/neo_2018_180125N55. [DOI] [PubMed] [Google Scholar]
- 31.Fan Y, Shen B, Tan M, Mu X, Qin Y, Zhang F, Liu Y. TGF-beta-induced upregulation of malat1 promotes bladder cancer metastasis by associating with suz12. Clin Cancer Res. 2014;20:1531–1541. doi: 10.1158/1078-0432.CCR-13-1455. [DOI] [PubMed] [Google Scholar]
- 32.Fan Y, Shen B, Tan M, Mu X, Qin Y, Zhang F, Liu Y. Long non-coding RNA UCA1 increases chemoresistance of bladder cancer cells by regulating Wnt signaling. FEBS J. 2014;281:1750–1758. doi: 10.1111/febs.12737. [DOI] [PubMed] [Google Scholar]
- 33.Dong Y, Liang G, Yuan B, Yang C, Gao R, Zhou X. MALAT1 promotes the proliferation and metastasis of osteosarcoma cells by activating the PI3K/Akt pathway. Tumour Biol. 2015;36:1477–1486. doi: 10.1007/s13277-014-2631-4. [DOI] [PubMed] [Google Scholar]
- 34.Xu S, Sui S, Zhang J, Bai N, Shi Q, Zhang G, Gao S, You Z, Zhan C, Liu F, Pang D. Downregulation of long noncoding RNA MALAT1 induces epithelial-to-mesenchymal transition via the PI3K-AKT pathway in breast cancer. Int J Clin Exp Pathol. 2015;8:4881–4891. [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang D, Sun G, Zhang H, Tian J, Li Y. Long non-coding RNA ANRIL indicates a poor prognosis of cervical cancer and promotes carcinogenesis via PI3K/Akt pathways. Biomed Pharmacother. 2017;85:511–516. doi: 10.1016/j.biopha.2016.11.058. [DOI] [PubMed] [Google Scholar]
- 36.Yang D, Wang Z, Yang B, Zhang M, Wu Z. Abstract 3495: the DNA methylation landscape of long noncoding RNAs in human cancer. Cancer Research. 2017;77:3495. [Google Scholar]
- 37.Maeda M, Yamashita S, Shimazu T, Iida N, Takeshima H, Nakajima T, Oda I, Nanjo S, Kusano C, Mori A, Moro H, Yamada H, Tsugane S, Sugiyama T, Sakai Y, Ushijima T. Novel epigenetic markers for gastric cancer risk stratification in individuals after Helicobacter pylori eradication. Gastric Cancer. 2018;21:745–755. doi: 10.1007/s10120-018-0803-4. [DOI] [PubMed] [Google Scholar]
- 38.Shi C, Yang Y, Yu J, Meng F, Zhang T, Gao Y. The long noncoding RNA LINC00473, a target of microRNA 34a, promotes tumorigenesis by inhibiting ILF2 degradation in cervical cancer. Am J Cancer Res. 2017;7:2157–2168. [PMC free article] [PubMed] [Google Scholar]
- 39.Wang L, Zhang X, Sheng L, Qiu C, Luo R. LINC00473 promotes the Taxol resistance via miR-15a in colorectal cancer. Biosci Rep. 2018;38 doi: 10.1042/BSR20180790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang W, Song Y. LINC00473 predicts poor prognosis and regulates cell migration and invasion in gastric cancer. Biomed Pharmacother. 2018;107:1–6. doi: 10.1016/j.biopha.2018.07.061. [DOI] [PubMed] [Google Scholar]
- 41.Shen Y, Wang Z, Loo LW, Ni Y, Jia W, Fei P, Risch HA, Katsaros D, Yu H. LINC00472 expression is regulated by promoter methylation and associated with disease-free survival in patients with grade 2 breast cancer. Breast Cancer Res Treat. 2015;154:473–482. doi: 10.1007/s10549-015-3632-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen L, Zhang W, Li DY, Wang X, Tao Y, Zhang Y, Dong C, Zhao J, Zhang L, Zhang X, Guo J, Zhang X, Liao Q. Regulatory network analysis of LINC00472, a long noncoding RNA downregulated by DNA hypermethylation in colorectal cancer. Clin Genet. 2018;93:1189–1198. doi: 10.1111/cge.13245. [DOI] [PubMed] [Google Scholar]
- 43.Zhang B, Li C, Sun Z. Long non-coding RNA LINC00346, LINC00578, LINC00673, LINC00671, LINC00261, and SNHG9 are novel prognostic markers for pancreatic cancer. Am J Transl Res. 2018;10:2648–2658. [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao L, Ji G, Le X, Wang C, Xu L, Feng M, Zhang Y, Yang H, Xuan Y, Yang Y, Lei L, Yang Q, Lau WB, Lau B, Chen Y, Deng X, Yao S, Yi T, Zhao X, Wei Y, Zhou S. Long noncoding RNA LINC00092 acts in cancer-associated fibroblasts to drive glycolysis and progression of ovarian cancer. Cancer Res. 2017;77:1369–1382. doi: 10.1158/0008-5472.CAN-16-1615. [DOI] [PubMed] [Google Scholar]
- 45.Li J, Peng W, Du L, Yang Q, Wang C, Mo YY. The oncogenic potentials and diagnostic significance of long non-coding RNA LINC00310 in breast cancer. J Cell Mol Med. 2018;22:4486–4495. doi: 10.1111/jcmm.13750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hassan A, Mosley J, Singh S, Zinn PO. A comprehensive review of genomics and noncoding RNA in gliomas. Top Magn Reson Imaging. 2017;26:3–14. doi: 10.1097/RMR.0000000000000111. [DOI] [PubMed] [Google Scholar]