

Abstract
B-cell lymphoma/leukaemia 11A (BCL11A) is a modulator of foetal-to-adult globin switching and is involved in brain development and normal lymphopoiesis. The three-dimensional structure of BCL11A and its structural domains had not yet been completely determined; hence, this study aimed to elucidate the structural domains of BCL11A. Molecular modelling and dynamics simulation studies were conducted using in silico tools with the templates selected based on Basic Local Alignment Search Tool (BLAST) searches of the Protein Data Bank (PDB). Ten protein models were generated using the MODELLER software, and the best model was selected according to the Discrete Optimised Protein Energy (DOPE) score and validated using the RAMPAGE server by evaluation of the Ramachandran plot. More than 93% of the amino acid residues of the best model of BCL11A were found to be in the favoured and allowed regions. The best model was validated using a 100-ns time span molecular dynamics simulation. The root-mean-square deviation, root-mean-square fluctuation, and radius of gyration values were found to explain the stability of the best BCL11A protein molecular model generated in the study. The validated best model of the BCL11A protein may be useful for effective modulator studies on foetal-to-adult globin switching and related research.
Keywords: BCL11A, homology modelling, molecular dynamics simulation, RAMPAGE, Phyre2 server
Introduction
B-cell lymphoma/leukaemia 11A (BCL11A) is a major modulator of haemoglobin switching and foetal haemoglobin (HbF) silencing, has a major role in brain development, and is essential for postnatal development and normal lymphopoiesis. BCL11A is expressed in several haematopoietic tissues, including splenic B cells, T cells, and monocytes in bone marrow [1]. The BCL11A gene is located on Chromosome 2p16.1 and the UniProtKB database accession number of the protein is Q9H165. The BCL11A is a zinc-finger protein comprising 835 amino acids. It has six C2H2-type zinc finger domains and interacts with Kruppel-like factor 1 (KLF1), v-erbA-related protein 2 (EAR2), actin-related protein 1A (ARP1), protein inhibitor of activated STAT (PIAS3), and COUP transcription factor 1 (TFCOUP1).
BCL11A is associated with the level of HbF and was identified through a genome-wide association study (GWAS). A microdeletion syndrome caused by deletion of the 2p15-p16.1 region causes patients to exhibit conditions such as hypotonia, autism spectrum disorder (ASD), facial dysmorphism, and fine motor dysfunction. Other BCL11A variations, such as a 2.5-Mb deletion in the upstream region or a 3.5-Mb deletion in the distal downstream region, are associated with increased HbF levels in thalassemia patients [2-5]. A nonsense mutation in exon 4 of BCL11A has been identified in patients with ASD and intellectual disabilities (ID) [3]. BCL11A expression was significantly elevated in triple-negative breast cancer (TNBC), mainly in BLBC/IC10 tumours, with increases at both the protein and RNA levels [6]. Genetic variations disturb the expression of BCL11A, which regulates HbF production and modifies the severity of sickle cell disease and β-thalassemia disorders [7]. The three-dimensional (3D) structure of the BCL11A protein has not completely elucidated; thus, our objective was to visualise the major protein structural domains, the protein’s functions, and the effects of mutations on its biological functions. Determination of the BCL11A structure has proven difficult in X-ray crystallography and nuclear magnetic resonance (NMR) studies, as it is a multi-domain protein. Homology modelling is the most suitable and widely accepted method for predicting the three-dimensional (3D) structure of the BCL11A protein. The generated 3D structure of BCL11A was subjected to molecular dynamics (MD) simulations using the Groningen MAchine for Chemical Simulation (GROMACS) and structural quality assessment.
Materials and methods
Homology modelling
Target selection
A homology modelling approach was used to predict the 3D structure of the BCL11A protein using FASTA sequences obtained from the UniProtKB database with accession number Q9H165.
Template selection
The 3D crystal structure of the template molecule (PDB ID: 5VTB) [8] was retrieved from the RCSB-Protein Data Bank (PDB) based on the similarity and identity index from the significant hits list acquired using The National Center for Biotechnology Information’s Basic Local Alignment Search Tool (NCBI-BLAST) [9].
3D Structure prediction and validation
Homology modelling was achieved using MODELLER software, version 9.19 [10,11]. The selected 3D structure of the protein generated from homology modelling was validated using various structural parameters and comparative evaluation tools, such as PROCHECK software, ProSA software, the discrete optimised protein energy (DOPE) [12-15] score, construction of a Ramachandran plot using the RAMPAGE server (http://mordred.bioc.cam.ac.uk/~rapper/rampage.php), and the SWISS-MODEL structure assessment software (https://swissmodel.expasy.org).
Docking simulation study
The BCL11A model protein was selected and ligands were chosen from databases such as DrugBank (which contains > 4100 drug entries) and the PubChem database, which were used to retrieve structure files of ligands and compounds to use for the docking [16,17]. Lipinski’s rule of five was used. The chosen ligand was docked using the PatchDock docking server [18]. PatchDock is a geometry-based, rigid molecular docking algorithm that operates by object recognition and image segmentation techniques that are used in Computer Vision. The recommended clustering root-mean-square deviation (RMSD) of 4.0A° was used as the default setting for PatchDock analysis. The data of 500 docked complexes were chosen from the solution list.
Molecular dynamics
MD simulation is a powerful technique and a useful molecular modelling method for exploring the symmetry structure, dynamics, and transport properties of bio-molecules in dynamic states. MD simulation provides substantial insights into the time-dependent fluctuations and conformational changes that occur in macro molecules and other systems, which is useful for understanding biological functions. In the present study, the 3D model of the BCL11A protein was developed using in silico homology modelling. The best model was constructed based on a 100-ns time span MD simulation study. The MD simulation was conducted using GROMACS software version 5.1.2 [19]. For all ionisable residues, the protonation states were fixed to their normal states at pH 7. For all complexes, the simulations were carried out using the CHARMM27 tool [20] with the all-atom force field of the GROMACS version 5.0.6 software package installed with the Ubuntu 11.10 Linux package in desktop mode. In the MD simulation, the protein was kept in the centre of a cubic box, and all protein molecules were surrounded by a cubic box of single-point charge 3 (SPC3) water molecules. The distance between any atom of the protein and the boundary of the cubic box was kept at a minimum of 10Å. The periodic boundary conditions (PBC) were applied in all directions. Na+ and Cl- counter ions played the role of replacing the water molecules to make the system neutral. The steepest descent algorithm was used to minimise each system for 10,000 steps. Each system was considered for 100-ps position-restrained MD simulations. The MD simulation was performed for a 100-ns production with a time step of 2 fs at constant pressure (1 atm) and temperature (300 K). The RMSD, root-mean-square fluctuation (RMSF), and radius of gyration were recorded to analyse the behaviour of each system [21,22].
Results and discussion
Secondary and tertiary structure
The secondary structures of the BCL11A protein were stabilised by hydrogen bonds, alpha helices, and beta sheets. The secondary structure and disorder prediction were achieved using the Phyre2 server, which exhibited alpha helix (16%), beta strand (9%), TM helix (2%), and disorder prediction (42%) (Figure 1).
Figure 1.
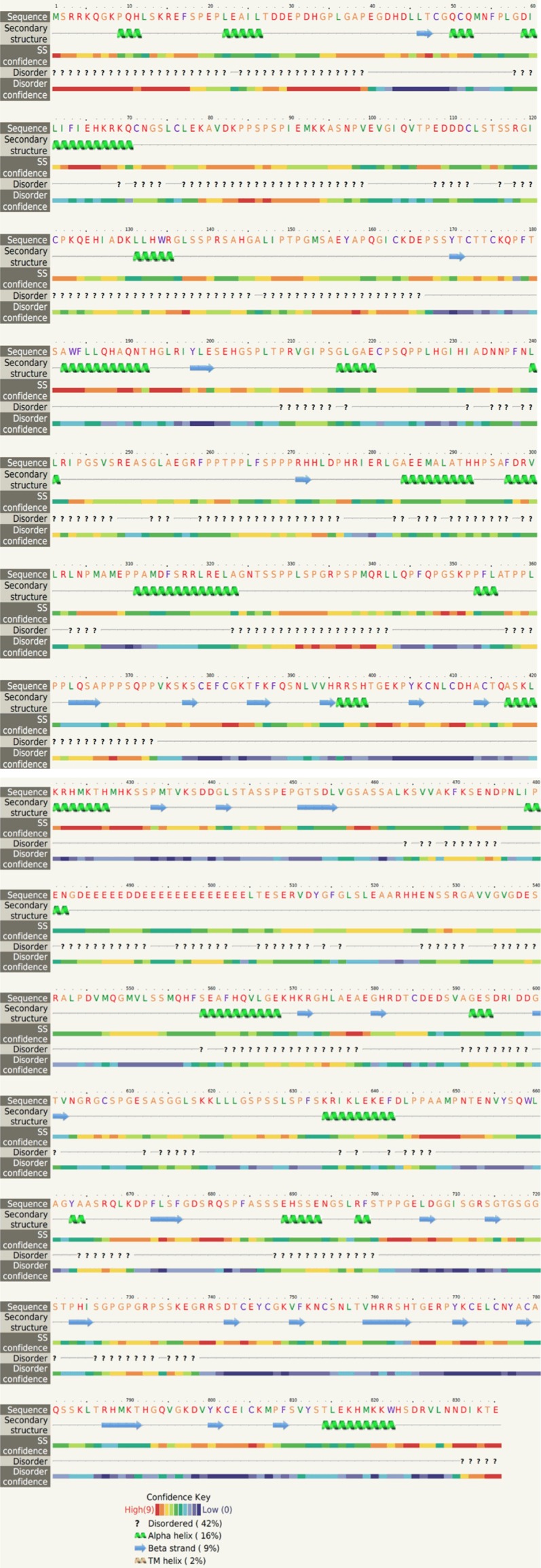
Secondary structure of BCL11A and disorder prediction by phyre2.
From the input sequence and the given template molecule, a total of 10 3D protein molecules were generated. MODELLER software was used to automatically calculate the model containing all non-hydrogen atoms using the given template. With the help of de novo loop modelling, structure optimisation clustering, screening of sequence databases, protein structure assessment, and other analyses, the MODELLER software was used to predict the best structures that resulted in fulfilment of spatial restraints. The DOPE score of the 10 best models selected are provided in Table 1.
Table 1.
DOPE score of best 10 models of BCL11A
| Model | DOPE score |
|---|---|
| Model 1 | -42197.797 |
| Model 2 | -41692.445 |
| Model 3 | -40916.637 |
| Model 4 | -43254.223 |
| Model 5 | -42189.961 |
| Model 6 | -41169.457 |
| Model 7 | -42919.227 |
| Model 8 | -42316.207 |
| Model 9 | -41614.074 |
| Model 10 | -43936.098 |
As shown in Table 1, Model 10 had the highest score (-43936.098), and it was designated as the best model for further validation. The 3D structure of the best model of the BCL11A protein is given in Figure 2.
Figure 2.
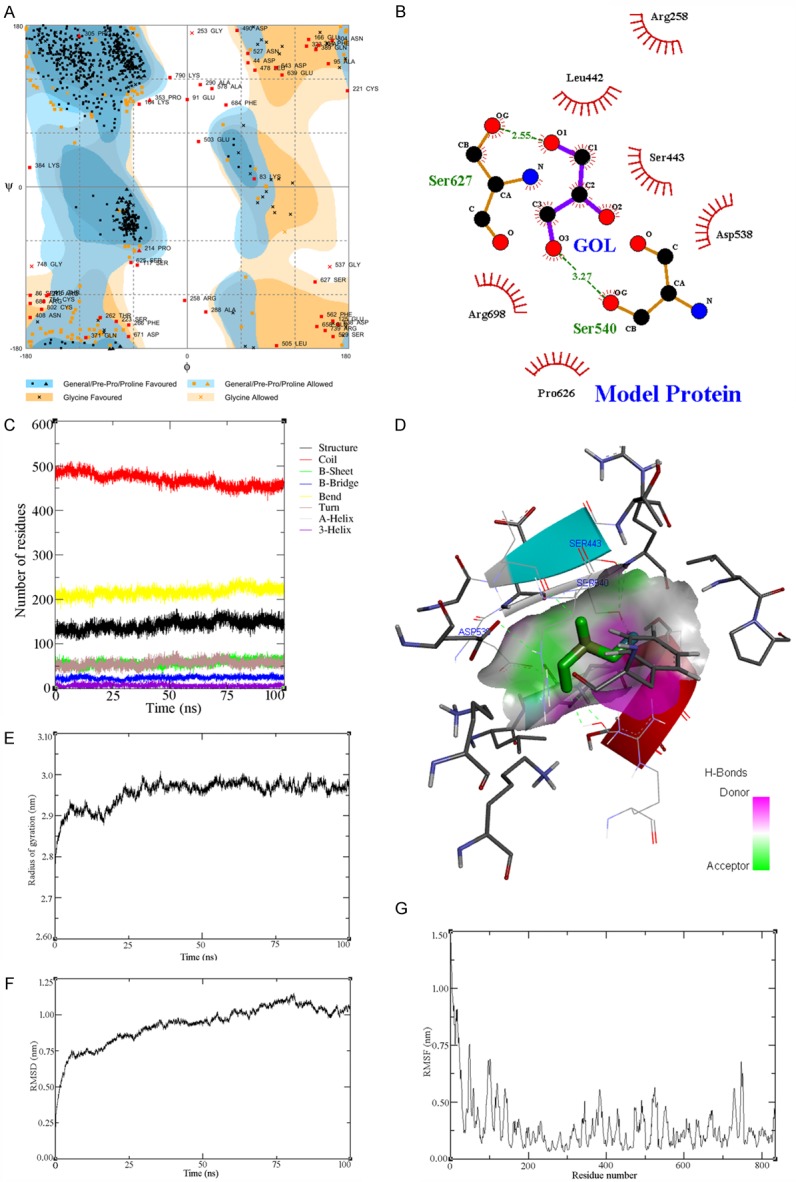
A: Ramachandran plot of model 10 of BCL11A from RAMPAGE. B: Three-dimensional view of Protein and ligand binding. C: Interpretation of the secondary structure. D: Ligplot of GOL (purple in color) interactions with human BCL11A protein model. Polar interactions are shown in green dotted line. Other interacting residues are shown in red lines. Carbon atoms are shown in black, oxygen atoms are shown in red, and nitrogen atoms are shown in blue color. E: RMSF vs. residue number of best model of BCL11A. F: RMSD vs. Time of the best model of BCL11A. G: Rg. vs. time of the best model of BCL11A.
Model 10 was further validated using RAMPAGE geometric evaluations to prepare a Ramachandran plot (Figure 2A). The Ramachandran plot of the best model is depicted in Figure 2A. Analysis of the plot revealed a total of 706 (84.80%) amino acid residues in the favoured region, 71 (8.50%) in the allowed region, and 56 (6.70%) in the outlier region. More than 93% of the amino acid residues were found to be in the allowed region. Similarly, analysis using the ProSA Web Server generated satisfactory scores for the amino acid residues [23].
Thus, the above results demonstrate the good quality of the selected model. An MD simulation of the best model was performed to conduct further validation by exploring the stability of the molecule.
Docking
As discussed above, the model built using SWISS-MODEL and MODELLER software was used for the molecular docking studies with glycerol (GOL) as a free ligand. The ligand was docked with the BCL11A model protein using PatchDock software. A LIGPLOT of the GOL (purple in colour) interactions with the human BCL11A protein model is shown in Figure 2B. Several docked structures for the BCL11A-GOL complex were obtained from PatchDock and were ranked based on their geometric shape complementary score, interface area, and atomic contact energy. The highest-ranked structure had a geometric shape complementarity score of 2,366, approximate interface area of 253.20 Å, and atomic contact energy (ACE) of the approximate interface area of -106.05 kcal/mol, as shown in Figure 2F and 2G. This docked complex demonstrates hydrogen bonding interactions between the hydrogen atom of the -SH group of the active site amino acid residues ser627 and ser540 with inter-atomic distances 2.55 and 3.27 Å, respectively. The docked complex was simulated for 20 ns. The analysis of the structure obtained after the 20-ns simulation demonstrated a strengthened hydrogen bonding interaction between the active site residues ser627 and ser540 of the BCL11A model protein.
Molecular dynamics
To assess the stability of the selected model, a 100-ns MD simulation was performed using the GROMACS software version 5.1.2. The stability of the molecules was examined based on the RMSF and Rg values. The average, maximum and minimum RMSD, RMSF, and Rg are presented in Table 2.
Table 2.
Maximum, minimum and average RMSD, RMSF and Rg of best model of BCL11A
| RMSD (nm) | RMSF (nm) | Rg (nm) | |
|---|---|---|---|
| Maximum | 1.153 | 1.903 | 3.009 |
| Minimum | 0.0005 | 0.064 | 2.682 |
| Average | 0.932 | 0.290 | 2.2957 |
The RMSD values of each frame corresponding to time are given in Figure 2F. An initial sharp increase in the RMSD values indicated that, for a period of up to 10 ns, the model was unstable. Afterward, the RMSD values rose gradually and finally achieved equilibrium at around 70 ns. After 80 ns, the values decreased slightly and equilibrated. The difference between the highest and lowest RMSD values was 1.154 nm, which was very close to the average value (0.932 nm), demonstrating that the backbone of the protein molecule did not fluctuate much.
The fluctuation of the individual amino acid residues can be explained based on the RMSF values obtained from the 100-ns MD simulation. The maximum, minimum, and average RMSF values were 1.903, 0.064, and 0.290 nm, respectively. The low average RMSF value suggested that individual amino acid residues exhibited stability in the dynamic state of the protein during the MD simulation. A plot of the RMSF values vs. the amino acid residue number is shown in Figure 2F and 2G. The figure indicates that amino acid residues at the positions of about 1~180 and those at about 710~750 fluctuated somewhat relative to the others. The remaining amino acid residues were found to be quite stable during the MD simulation.
Finally, the rigidity of the protein system was examined using Rg values. The data in Table 2 show that the average Rg value (2.957 nm) was very close to the maximum (30.009 nm) and minimum (2.682) Rg values, indicating that the protein system retained its stability throughout the 100-ns time span of the MD simulation [24]. The Rg values corresponding to the simulation shown in Figure 2E were used to facilitate the interpretation of the secondary structure. As shown in Figure 2E, the Rg value increased until the 25-ns time point, indicating that the stability of the system was initially disturbed. After 25 ns the plot showed no fluctuation at all, demonstrating that the protein system retained its stability.
Conclusions
Molecular modelling studies were used to highlight the advantages of comparative in silico interaction studies using the BCL11A protein employing a template selected based on a BLAST search of the PDB. The top 10 protein models were generated for homology modelling using MODELLER software, and the best model was selected according to its DOPE score. Furthermore, the model was validated using the RAMPAGE server by evaluating the Ramachandran plot. More than 93% of the amino acid residues of the protein were found to be in the favoured and allowed regions. Finally, the model was validated using a 100-ns MD simulation. The RMSD, RMSF, and Rg values explained the stability of the protein molecule. The validated model of the BCL11A protein will be useful for the study of effective modulators of foetal-to-adult globin switching and similar research.
Acknowledgements
This study was supported by the Deanship of Scientific Research, Imam Abdulrahman Bin Faisal University, Saudi Arabia (Grant no: 2017-101-IRMC). The author thanks, the Dean, Institute for Research and Medical Consultations (IRMC), Imam Abdulrahman Bin Faisal University, Dammam, Saudi Arabia for her continuous support and encouragement. Author would like to thank Dr. J. Francis Borgio for critical comments.
Disclosure of conflict of interest
None.
References
- 1.Satterwhite E, Sonoki T, Willis TG, Harder L, Nowak R, Arriola EL, Liu H, Price HP, Gesk S, Steinemann D, Schlegelberger B. The BCL11 gene family: involvement of BCL11A in lymphoid malignancies. Blood. 2001;98:3413–20. doi: 10.1182/blood.v98.12.3413. [DOI] [PubMed] [Google Scholar]
- 2.Basak A, Hancarova M, Ulirsch JC, Balci TB, Trkova M, Pelisek M, Vlckova M, Muzikova K, Cermak J, Trka J, Dyment DA. BCL11A deletions result in fetal hemoglobin persistence and neurodevelopmental alterations. J Clin Invest. 2015;125:2363–8. doi: 10.1172/JCI81163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cai T, Chen X, Li J, Xiang B, Yang L, Liu Y, Chen Q, He Z, Sun K, Liu PP. Identification of novel mutations in the HbF repressor gene BCL11A in patients with autism and intelligence disabilities. Am J Hematol. 2017;92:E653–E656. doi: 10.1002/ajh.24902. [DOI] [PubMed] [Google Scholar]
- 4.Menzel S, Garner C, Gut I, Matsuda F, Yamaguchi M, Heath S, Foglio M, Zelenika D, Boland A, Rooks H, Best S. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet. 2007;39:1197–9. doi: 10.1038/ng2108. [DOI] [PubMed] [Google Scholar]
- 5.Sedgewick AE, Timofeev N, Sebastiani P, So JC, Ma ES, Chan LC, Fucharoen G, Fucharoen S, Barbosa CG, Vardarajan BN, Farrer LA. BCL11A is a major HbF quantitative trait locus in three different populations with β-hemoglobinopathies. Blood Cells Mol Dis. 2008;41:255–258. doi: 10.1016/j.bcmd.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khaled WT, Lee SC, Stingl J, Chen X, Ali HR, Rueda OM, Hadi F, Wang J, Yu Y, Chin SF, Stratton M. BCL11A is a triple-negative breast cancer gene with critical functions in stem and progenitor cells. Nat Commun. 2015;6:5987. doi: 10.1038/ncomms6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu N, Hargreaves VV, Zhu Q, Kurland JV, Hong J, Kim W, Sher F, Macias-Trevino C, Rogers JM, Kurita R, Nakamura Y. Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell. 2018;173:430–42. doi: 10.1016/j.cell.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moody RR, Lo MC, Meagher JL, Lin CC, Stevers NO, Tinsley SL, Jung I, Matvekas A, Stuckey JA, Sun D. Probing the interaction between the histone methyltransferase/deacetylase subunit RBBP4/7 and the transcription factor BCL11A in epigenetic complexes. J Biol Chem. 2018;293:2125–2136. doi: 10.1074/jbc.M117.811463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 10.Webb B, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Bioinformatics. 2016;54:5.6.1–5.6.37. doi: 10.1002/cpbi.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berendsen HJ, van der Spoel D, van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Computer Physics Communications. 1995;91:43–56. [Google Scholar]
- 12.Shen MY, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15:2507–24. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abdulazeez S, Sultana S, Almandil NB, Almohazey D, Bency BJ, Borgio JF. The rs61742690 (S783N) single nucleotide polymorphism is a suitable target for disrupting BCL11A-mediated foetal-to-adult globin switching. PLoS One. 2019;14:e0212492. doi: 10.1371/journal.pone.0212492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abdulazeez S. Homology-based structure prediction of the human Kruppel-like factor 1 protein. Int J Curr Microbiol App Sci. 2018;7:2169–2176. [Google Scholar]
- 15.Borgio JF, Al-Madan MS, AbdulAzeez S. Mutation near the binding interfaces at α-hemoglobin stabilizing protein is highly pathogenic. Am J Transl Res. 2016;8:4224. [PMC free article] [PubMed] [Google Scholar]
- 16.Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–72. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, Li Q, Shoemaker BA, Thiessen PA, Yu B, Zaslavsky L, Zhang J, Bolton EE. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 2019;47:D1102–1109. doi: 10.1093/nar/gky1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mashiach E, Schneidman-Duhovny D, Peri A, Shavit Y, Nussinov R, Wolfson HJ. An integrated suite of fast docking algorithms. Proteins. 2010;78:3197–204. doi: 10.1002/prot.22790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25. [Google Scholar]
- 20.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AD Jr. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem. 2010;31:671–90. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hünenberger PH, Mark AE, Van Gunsteren WF. Fluctuation and cross-correlation analysis of protein motions observed in nanosecond molecular dynamics simulations. J Mol Biol. 1995;252:492–503. doi: 10.1006/jmbi.1995.0514. [DOI] [PubMed] [Google Scholar]
- 22.Wang J, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? Journal of Computational Chemistry. 2000;21:1049–1074. [Google Scholar]
- 23.Bowie JU, Luthy R, Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991;253:164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- 24.Azam SS, Abro A, Raza S, Saroosh A. Structure and dynamics studies of sterol 24-C-methyltransferase with mechanism based inactivators for the disruption of ergosterol biosynthesis. Mol Biol Rep. 2014;41:4279–93. doi: 10.1007/s11033-014-3299-y. [DOI] [PubMed] [Google Scholar]