

Abstract
We have previously demonstrated that anti-CD44s H4C4 or liposomal-delivered STAT3 inhibitor FLLL32 sensitized pancreatic cancer cells to radiotherapy through the elimination or inhibition of cancer stem cells (CSCs) and that HAb18G/CD147 promoted STAT3-mediated pancreatic tumor development by forming a signaling complex with CD44s. In this paper, we therefore explored whether anti-CD147 HAb18IgG sensitized pancreatic cancer cells to chemoradiotherapy via the targeting of CSCs. We tested the influence of HAb18IgG on the sensitivity of pancreatic cancer cells to chemoradiotherapy by clonogenic and MTT assays and on pancreatic CSCs by colony and sphere formation assays, flow cytometry, quantitative real-time RT-PCR (qRT-PCR) and stem cell transcription factors PCR array analysis. Changes in CD147 signaling were examined by immunoblot and reporter assays. We found that HAb18IgG sensitized pancreatic cancer cells to chemoradiotherapy by dose-dependently decreasing colony and sphere formation. Furthermore, HAb18IgG reduced the pancreatic CSC subpopulation and the expression of stem cell transcription factors OCT4, SOX2 and NANOG. Mechanistically, HAb18IgG inhibited CSCs by blocking CD44s-pSTAT3 signaling. The present findings indicated the promising therapeutic role of anti-CD147 HAb18IgG in suppressing pancreatic tumor initiation and overcoming post-chemoradiotherapy recurrence through the direct targeting of CSCs.
Keywords: CD147, antibody, HAb18IgG, pancreatic cancer, CSCs
Introduction
Emerging evidence suggests that cancer stem cells (CSCs), a minor population of cancer cells that display profound chemoradiotherapy resistance, play a critical role in tumor initiation, malignant progression, disease relapse and distant metastasis [1]. CSCs possess the capacity for self-renewal and multi-differentiation similar to stem cells. CSCs are also characterized by slow cell cycle kinetics, efficient disposal of chemotherapeutic agents by drug efflux transporters, increased aldehyde dehydrogenase 1 activity and altered mitochondrial metabolism [2,3]. Moreover, the microenvironment signals that mediate CSC plasticity accelerate the therapeutic inefficiency and onset of metastasis [4]. In various blood and solid tumors, including pancreatic cancer, the existence of CSCs has been verified [5,6]. Therefore, the current new therapeutic strategy has shifted towards targeting CSCs.
Compared to regularly proliferating cancer cells, CSCs have unique gene profiles and intracellular constitutions and express specific membrane markers. Therefore, therapeutic strategies targeting CSCs include targeting CSC surface/intrinsic markers or signaling pathways and targeting CSC metabolism or the microenvironment by means of antibodies, aptamers, peptide ligands, small molecules or RNA-based therapeutics [1,2,7]. Among these strategies, antibody therapeutics targeting CSC surface markers (CD44, CD47, EpCAM, EGFR, etc.) and small molecule inhibitors targeting CSC signaling pathways (STAT3, Notch, Wnt, etc.) are the most promising and have already been investigated in clinical trials [1,2,7].
CD44 is the most common CSC marker, and three antibody therapeutics against CD44 are currently in phase I clinical trials: anti-CD44v6 labeled with 186Re or conjugated with mertansine [8] and humanized anti-CD44 designed to inhibit CD44-HA interactions [9] or to target a glycosylated epitope [1]. In pancreatic cancer, we have reported that anti-CD44s H4C4 blocked tumor initiation and postradiotherapy recurrence via affecting both TICs and bulk tumor cells [10]. Unfortunately, CD44 is also present on normal stem cells and cancer cells, and CD44 has several alternative splicing and post-translational modifications. Moreover, resistance to anti-CD44 therapy was reported in the AML [11].
Recently, the targeting of signaling pathways shared by CSCs and non-CSCs, such as the STAT3 signaling pathway that is particularly hyperactivated in CSCs, has been shown to be effective in killing CSCs and non-CSCs and disrupting the interconversion between the two subpopulations. In our previous study, targeting pancreatic CSCs with STAT3 inhibitor FLLL32 blocked pancreatic tumor formation and overcame radioresistance [12]. In addition, the combination of a STAT3 inhibitor, napabucasin (BBI608), with paclitaxel or the FOLFIRI regimen is under investigation in a phase 3 clinical trial for treating non-small cell lung cancer (NSCLC) or metastatic colorectal carcinoma [1]. As STAT3 is activated by multiple factors, the most effective way to abrogate STAT3 activation could be the blockage of the STAT3 upstream signal. We have identified HAb18G/CD147 as a novel upstream activator of STAT3 signaling via interaction with CD44s and thus as a surrogate marker for STAT3-targeted therapies in pancreatic cancer [13].
CD147, also named EMMPRIN or HAb18G/CD147, has been reported to be linked with CSC characteristics, such as epithelial-mesenchymal transition (EMT) [14], anoikis resistance [15] and chemoradiotherapy resistance [16,17]. Anti-CD147 drug, metuximab (Licartin), has been successfully applied to prevent tumor recurrence of post liver transplantation or radiofrequency ablation in patients with advanced hepatocellular carcinoma [18,19]. However, the effect of anti-CD147 against pancreatic CSCs remains unclear.
In this paper, we demonstrated that anti-CD147 HAb18IgG sensitized pancreatic cancer cells to chemoradiotherapy by inhibiting the potential of CSCs and suppressing CSC CD44s-pSTAT3 signaling. Our data revealed a potential therapeutic application of the anti-CD147 drug metuximab for fatal and incurable pancreatic cancers.
Materials and methods
Antibodies and drugs
Anti-STAT3 and anti-phospho-STAT3 (Tyr705) were purchased from Cell Signaling Technology (Danvers, MA), and the anti-CD44s clone MEM-263 was purchased from Abnova (Walnut, CA). Goat anti-rabbit horseradish peroxidase (HRP), goat anti-mouse HRP and mouse IgG were purchased from Invitrogen (Carlsbad, CA). WP1066 was obtained from Calbiochem (Billerica, MA), and gemcitabine was obtained from Sigma (St. Louis, MO), genfitinib was obtained from MedChemExpress (Monmouth Junction, NJ). Anti-CD147 HAb18IgG was prepared as reported [13].
Cell culture and treatment
Human pancreatic cancer cell lines (MIA PaCa-2, CFPAC-1, PANC-1 and BxPC3) were purchased from American Type Culture Collection and cultured in DMEM containing 10% fetal bovine serum (HyClone). Cells were treated with 0-20 µg/ml HAb18IgG or mouse IgG (nIgG) and then used for the following experiments: MTT and clonogenic assays, cell growth and colony/sphere formation assays, ALDEFLUOR assay, stem cell transcription factors PCR array and quantitative real-time RT-PCR, immunoblotting assays and STAT3 reporter assay.
MTT assay
A total of 8 × 103 cells were seeded into 96-well plates and cultured for 24 hours. The next day, varying concentrations of gemcitabine and genfitinib with nIgG or HAb18IgG were added to the cells and incubated for 72 hours. The cell viability was determined by measuring the WST-8 dye absorbance at 450 nm and was presented as relative cell viability normalized to the individual nIgG controls. Chemo-sensitivity was expressed as IC50 values [12].
Colony formation and clonogenic assays
For the colony formation assay, cells were cultured in DMEM containing 10% FBS with 250 cells in each well of a 6-well plate for 7-10 days. For the clonogenic assay, different numbers of cells for various doses (200~10,000 cells/well) were subjected to X-ray radiation (0, 2, 4, 6, or 8 Gy) and then incubated for 2-3 weeks in 10% FBS supplemented DMEM. For both assays, the colonies were stained by 0.1% crystal violet and counted (> 50 cells) manually with the aid of an Olympus INT-2 inverted microscope. Data from radiation-treated cells were normalized to the nIgG treated cells. Plating efficiencies and survival fractions were calculated to obtain survival parameters and plot cell survival curves as described [10].
Cell growth assay
Cells were plated at a density of 1 × 105/ml with 2 ml per well in 6-well plates in complete media, collected and counted every 24 hours for 3 days using a haemocytometer. A cell growth curve was drawn according to the cell numbers at the specified incubation time.
Apoptosis assay
Cells were labeled with Annexin V-FITC and propidium iodide (PI) double staining kit (Trevigen, Gaithersburg, MD) and analyzed by a FACSCalibur flow cytometer, and the data were analyzed with CellQuest software (BD Biosciences, San Jose, CA).
Cell cycle analysis
A total of 1 × 106 cells were fixed with 70% cold ethanol at 4°C overnight and incubated in 200 μl of PBS containing 0.5 mg/ml RNase, 0.05% Triton X-100 and 10 μg/ml PI at 37°C for 1 hour, and the data were analyzed using a BD FACSCalibur flow cytometer.
Tumorsphere culture
The tumorsphere culture method and medium were described previously [10]. Briefly, cells were plated in 24-well ultralow attachment plates (Corning) at a concentration of 2,000 cells per well. Ten to 14 days later, spheres ≥50 µm in diameter were counted under a phase-contrast microscope. To quantify sphere size, spheres were collected with a 40 µm sieve (BD Biosciences) and disassociated into a single cell suspension with TrypLETM. Sphere size was then calculated as the number of cells in the total tumorsphere divided by the number of spheres.
ALDEFLUOR assay
The ALDEFLUOR kit (Stem Cell Technologies) was applied to analyze the population with ALDH enzymatic activity [12]. Briefly, cells were incubated in the ALDEFLUOR assay buffer containing ALDH substrate BAAA (1 µM per 1 × 106 cells) at 37°C for 45 min, whereas negative control cells were incubated with 50 mM of ALDH inhibitor diethylamino-benzaldehyde (DEAB) under the same conditions. BAAA-stained cells were analyzed using a FACSCalibur flow cytometer and CellQuest software.
Human stem cell transcription factors PCR array
Total RNA isolated from MIA PaCa-2 cells treated with 10 µg/mL HAb18IgG or nIgG was reverse transcribed into complementary DNA and then analyzed by the real-time RT2 Profiler PCR Array (QIAGEN, Cat. no. PAHS-501Z) in combination with RT2 SYBR® Green qPCR MasterMix (Cat. no. 330529).
Quantitative real-time RT-PCR (qRT-PCR)
Total RNA extraction and cDNA synthesis were carried out using a method described previously [20]. qRT-PCR was carried out in an ABI 7700® real-time PCR system (Applied Biosystems). The primers used for OCT4, SOX2, NANOG, and GAPDH are listed in Table 1. Individual genes of interest (GOIs) were normalized to the housekeeping gene GAPDH. Relative mRNA levels are calculated as 2-ΔCt = 2-(Ct (HKG) - Ct (GOI)).
Table 1.
The sequences of the oligonucleotide primers used for qRT-PCR
| Genes | Primer Sequence (5’-3’) | |
|---|---|---|
| OCT4 | Forward: | GGGCTCTCCCATGCATTCAAAC |
| Reverse: | ACCTTCCCTCCAACCAGTTGC | |
| SOX2 | Forward: | TGGACAGTTACGCGCACAT |
| Reverse: | CGAGTAGGACATGCTGTAGGT | |
| NANOG | Forward: | CCCCAGCCTTTACTCTTCCTA |
| Reverse: | CCAGGTTGAATTGTTCCAGGTC | |
| GAPDH | Forward: | TGATGACATCAAGAAGGTGGTGAAG |
| Reverse: | TCCTTGGAGGCCATGTGGGCCAT | |
Immunoblotting assay
Immunoblotting assay were performed as reported [13]. Proteins in the cellular lysates were equalized, and then separated and analyzed by probing with individual antibodies.
STAT3 reporter assay
STAT3 transcriptional activity was detected using the pSTAT3-TA-luc reporter plasmid (Beyotime). Briefly, cells were transfected with 0.5 μg pSTAT3-TA-luc reporter constructs or 0.5 μg of a pGL6-TA vector control using lipofectamine 2000. Sixteen hours after transfection, the cells were treated with 10 µg/mL HAb18IgG or nIgG for 1 hour. Luciferase activity was measured in the cell lysates by an Epoch™ microvolume spectrophotometer (BioTek) using the Firefly Luciferase Reporter Gene Assay Kit (Beyotime).
Statistical analysis
All data are shown as the mean ± SD of triplicate values from three independent experiments. The significance of the differences between groups was determined with t-tests and one-way ANOVA using GraphPad Prism 6.0 (GraphPad Software, http://www.graphpad.com). *P < 0.05, **P < 0.01 and ***P < 0.001 were considered to be statistically significant as indicated.
Results
HAb18IgG sensitized pancreatic cancer cells to chemoradiotherapy
As CD147 has been linked with CSC features such as chemoradiotherapy resistance [13,17], we first tested whether HAb18IgG, an anti-CD147 antibody, enhanced the growth inhibitory effect of radiation or gemcitabine/genfitinib on pancreatic cancer cells. As shown in clonogenic survival assay, compared to nIgG, HAb18IgG enhanced radiation-induced clonogenic cell death in a dose-dependent manner with a radiation dose enhancement ratio (ER) of 1.331 and 1.443 for 10 µg/mL and 20 µg/mL HAb18IgG, respectively (Figure 1A, 1B). Herein, the ER values are larger than 1.2 [12]; thus, HAb18IgG is considered to be able to sensitize pancreatic cancer cells to radiation. In MTT assay, with the HAb18IgG combinations, the IC50 value of gemcitabine greatly decreased by 34.6% and 45.4% in CFPAC-1 and MIA PaCa-2 cells, respectively, suggesting that HAb18IgG sensitized pancreatic cancer cells to gemcitabine (Figure 1C). Likewise, EGFR inhibitor genfitinib exhibited a similar extent reduction of IC50 values when combined with HAb18IgG and decreased the IC50 values both by 41.9% in CFPAC-1 and MIA PaCa-2 (Figure 1D). Together, these results demonstrate that HAb18IgG potentially sensitized pancreatic cancer cells to chemoradiotherapy.
Figure 1.
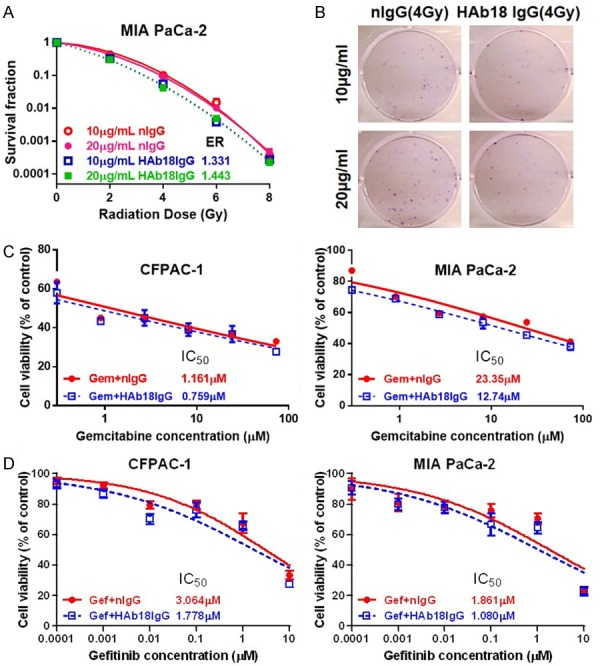
HAb18IgG sensitized pancreatic cancer cells to chemo-/radiotherapy. (A) Clonogenic survival assay of MIA PaCa-2 cells treated with 10 µg/ml or 20 µg/ml HAb18IgG or nIgG. Survival fractions were plotted, and the enhancement ratios (ER) were calculated by HAb18IgG vs. nIgG at the corresponding dose. (B) Respective images of MIA PaCa-2 cells treated with 4 Gy radiation combined with or without 10 µg/ml or 20 µg/ml HAb18IgG or nIgG. (C, D) Cytotoxicity of gemcitabine (C, Gem) or EGFR inhibitor genfitinib (D, Gef) in CFPAC-1 and MIA PaCa-2 cells combined with or without 10 µg/ml HAb18IgG or nIgG.
HAb18IgG suppressed cellular growth and colony formation
Our previous paper showed that CD147 promotes cellular and clonogenic growth in pancreatic cancer cells [13]. We then explored whether the enhancement effect of HAb18IgG on chemoradiotherapy was due to its inhibition of cellular and clonogenic growth (or colony formation). We found that HAb18IgG significantly decreased the growth of pancreatic cancer cells (Figure 2A). In addition, HAb18IgG suppressed the ability for colony formation by dose-dependently reducing the number of clones in MIA PaCa-2 and PANC-1 cells (Figure 2B). Thus, HAb18IgG sensitized pancreatic cancer cells to chemoradiotherapy by suppressing cellular growth and colony formation.
Figure 2.

HAb18IgG suppressed cellular growth and colony formation. A. Cellular growth assay in CFPAC-1 cells treated with 10 µg/ml HAb18IgG or nIgG for 3 days. B. Colony formation assay in MIA PaCa-2 and PANC-1 cells treated with 10 µg/ml or 20 µg/ml HAb18IgG or 20 µg/ml nIgG for 14 days. C. Apoptosis analysis in MIA PaCa-2 and PANC-1 cells treated with 10 µg/ml HAb18IgG or nIgG for 2 days. D. Cell cycle analysis in MIA PaCa-2 and PANC-1 cells treated with 10 µg/ml HAb18IgG or nIgG for 2 days.
Colony formation assay is designed to measure the cellular proliferative capacity and indirectly indicated cell death. To investigate whether the inhibition of HAb18IgG on cellular and clonogenic survival was the result of direct cell death, we examined cell apoptosis in HAb18IgG-treated cells. Unexpectedly, the apoptosis ratio in HAb18IgG-treated cells was almost the same as that in nIgG-treated cells (Figure 2C). Furthermore, the unchanged apoptosis level between HAb18IgG- and nIgG-treated cells coincided with a similar percentage of cells in the G0/G1 phase or in the S/G2 phase as determined by cell cycle analysis (Figure 2D). Therefore, the ability of HAb18IgG to inhibit colony formation may not derive from the decrease of cell death or cell proliferation in the total population.
HAb18IgG decreased the ratio and potential of CSCs
CSCs are the root of cancers and resisting chemoradiotherapy. Thus, we investigated whether the colony formation inhibition of HAb18IgG originated from its inhibition of CSCs using tumorsphere formation assay. Figure 3A and 3B showed that the number of spheres of HAb18IgG-treated cells was significantly lower than those of nIgG-treated cells and that the average sphere size of HAb18IgG-treated cells was smaller than that of nIgG-treated cells. Moreover, HAb18IgG decreased sphere number and size in a dose-dependent manner, with a maximal decrease at the concentration of 20 µg/mL. The abilities of chemoradiotherapy resistance and of colony and sphere formation are CSC features; thus, HAb18IgG affected the potentials of CSCs.
Figure 3.
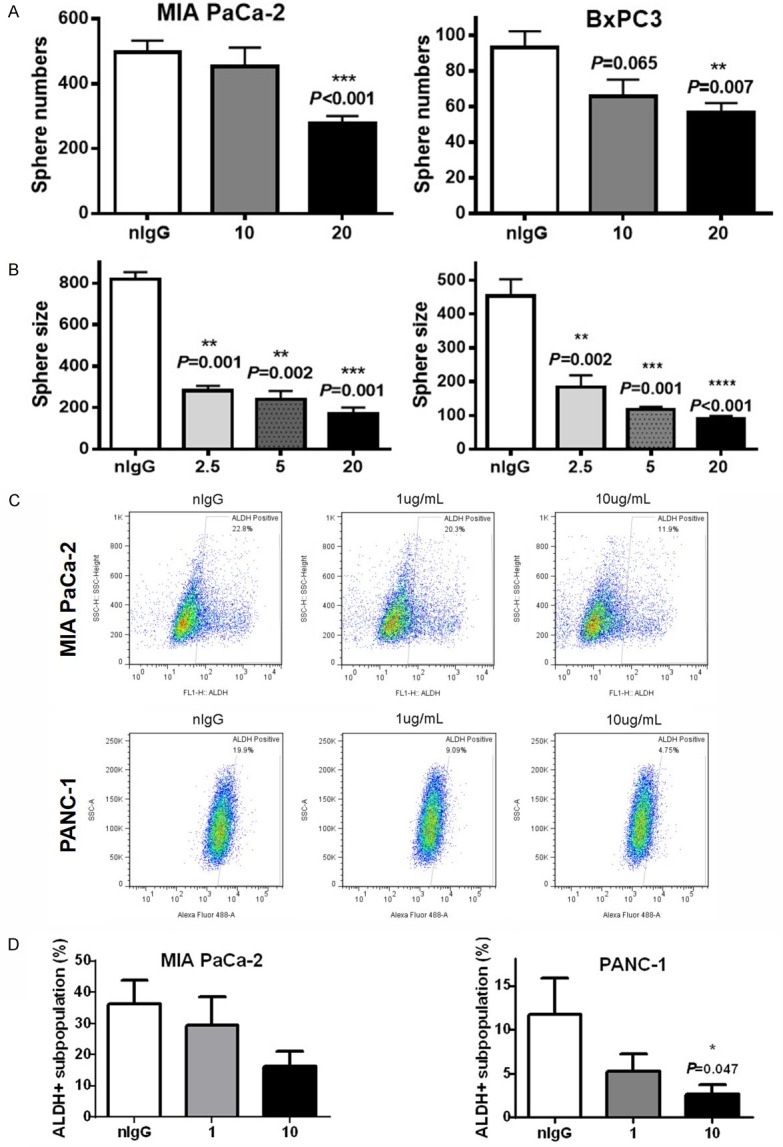
HAb18IgG decreased the ratio and potential of CSCs. (A, B) Sphere numbers (A) and sphere size (B) in 2.5, 5, 10, or 20 µg/ml HAb18IgG- or 20 µg/ml nIgG-treated cells. (C, D) ALDH+ subpopulation ratios in MIA PaCa-2 and PANC-1 cells treated with 1 µg/ml or 10 µg/ml HAb18IgG or 10 µg/ml nIgG for 2 days.
We further determined the alteration of CSC numbers upon HAb18IgG treatment using the ALDEFLUOR assay. As shown in Figure 3C and 3D, incubation with HAb18IgG significantly reduced the ratio of ALDH+ subpopulations in MIA PaCa-2 (average 54.9% reduction by 10 µg/mL HAb18IgG) and PANC-1 (average 77.6% reduction by 10 µg/mL HAb18IgG) compared to that of nIgG. As HAb18IgG reduced the number and potentials of CSCs, HAb18IgG could be a potential therapeutic agent for eliminating pancreatic CSCs.
To explore potential stem cell transcription factors that would be affected by HAb18IgG, we carried out the PCR array. In total, 27 of the 84 stem cell transcription factors screened (32.1%), including NANOG and STAT1, had altered expression (> 2-fold) after HAb18IgG treatment (Figure 4A; Table 2). As seen in qRT-PCR validation analysis, HAb18IgG reduced the mRNA levels of NANOG and the level of SOX2/OCT4 in pancreatic cancer cells (Figure 4B). As HAb18IgG reduced the level of stem cell transcription factors involving in pluripotent cell maintenance and differentiation, these data indicated HAb18IgG affected the CSCs differentiation.
Figure 4.
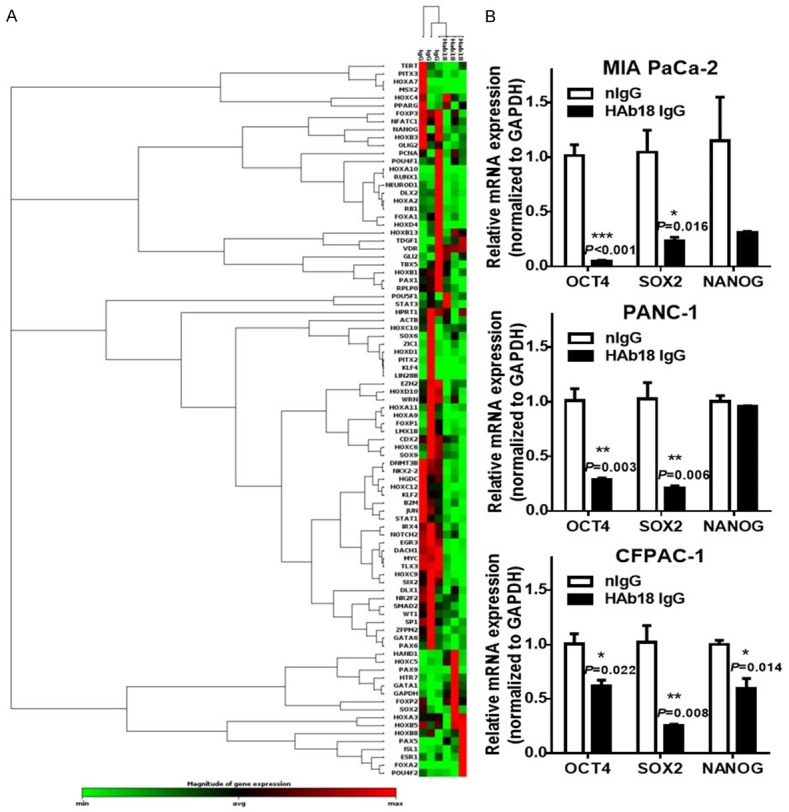
HAb18IgG inhibited the CSC stem cell transcription factors. A. Clustergram of genes expressed in MIA PaCa-2 cells treated with 10 µg/ml HAb18IgG or nIgG for 2 days. Data from non-supervised hierarchical clustering of the entire dataset to display a heat map with dendrograms indicating co-regulated genes across groups or individual samples. Changes in expression of individual genes are shown in Table 2. B. OCT4, SOX2 and NANOG mRNA expression in MIA PaCa-2, PANC-1 and CFPAC-1 cells treated with 10 µg/ml HAb18IgG or nIgG for 2 days. Levels were normalized to that of GAPDH.
Table 2.
Human stem cell transcription factors affected by HAb18IgG (QIAGEN RT2 Profiler™ PCR Array, Cat. no. PAHS-501Z)
| Position | Gene Symbol | Fold Regulation | p-Value |
|---|---|---|---|
| A1 | CDX2 | -3.54 | 0.023853 |
| A2 | DACH1 | -20.51 | 0.000957 |
| A5 | DNMT3B | -4.56 | 0.001173 |
| A6 | EGR3 | -10.65 | 0.002894 |
| A8 | EZH2 | -3.55 | 0.031319 |
| B1 | FOXP3 | -2.73 | 0.000197 |
| B2 | GATA1 | 11.40 | 0.015772 |
| B12 | HOXB1 | -3.28 | 0.022902 |
| C2 | HOXB3 | -4.25 | 0.003706 |
| C6 | HOXC12 | -6.05 | 0.008023 |
| C10 | HOXC9 | -8.05 | 0.012903 |
| C12 | HOXD10 | -4.35 | 0.028549 |
| D3 | IRX4 | -7.12 | 0.012293 |
| D5 | JUN | -2.19 | 0.009078 |
| D6 | KLF2 | -4.04 | 0.003574 |
| D11 | MYC | -5.23 | 0.000359 |
| D12 | NANOG | -2.47 | 0.044222 |
| E2 | NFATC1 | -2.95 | 0.000693 |
| E3 | NKX2-2 | -6.07 | 0.000268 |
| E4 | NOTCH2 | -3.29 | 0.011823 |
| E6 | OLIG2 | -5.35 | 0.018769 |
| E7 | PAX1 | -6.00 | 0.021686 |
| F8 | SIX2 | -14.29 | 0.026751 |
| G1 | SP1 | -2.96 | 0.029264 |
| G2 | STAT1 | -2.65 | 0.008213 |
| G7 | TLX3 | -7.23 | 0.000739 |
| G9 | WRN | -2.36 | 0.021091 |
HAb18IgG eliminated CSCs via CD44s-pSTAT3 signaling
We previously reported that CD147 promoted cellular and clonogenic growth in pancreatic cancer cells via CD44s-STAT3 signaling [13]. Additionally, targeting pancreatic CSCs with anti-CD44s H4C4 [10] or STAT3 inhibitor FLLL32 [12] blocked pancreatic tumor formation and overcame radioresistance. To explore whether HAb18IgG eliminated CSCs by inhibiting CD44s-STAT3 signaling, we performed western blotting. We observed that HAb18IgG greatly decreased the protein levels of CD44s, pSTAT3 and STAT3 in pancreatic cancer cells (Figure 5A). Furthermore, a functional reporter assay showed that HAb18IgG significantly decreased STAT3 transcriptional activity compared to nIgG, with more inhibition in MIA PaCa-2 cells (27.3%) with relatively high pSTAT3 expression and less inhibition in PANC-1 (17.9%) with relatively low pSTAT3 signaling (Figure 5B).
Figure 5.
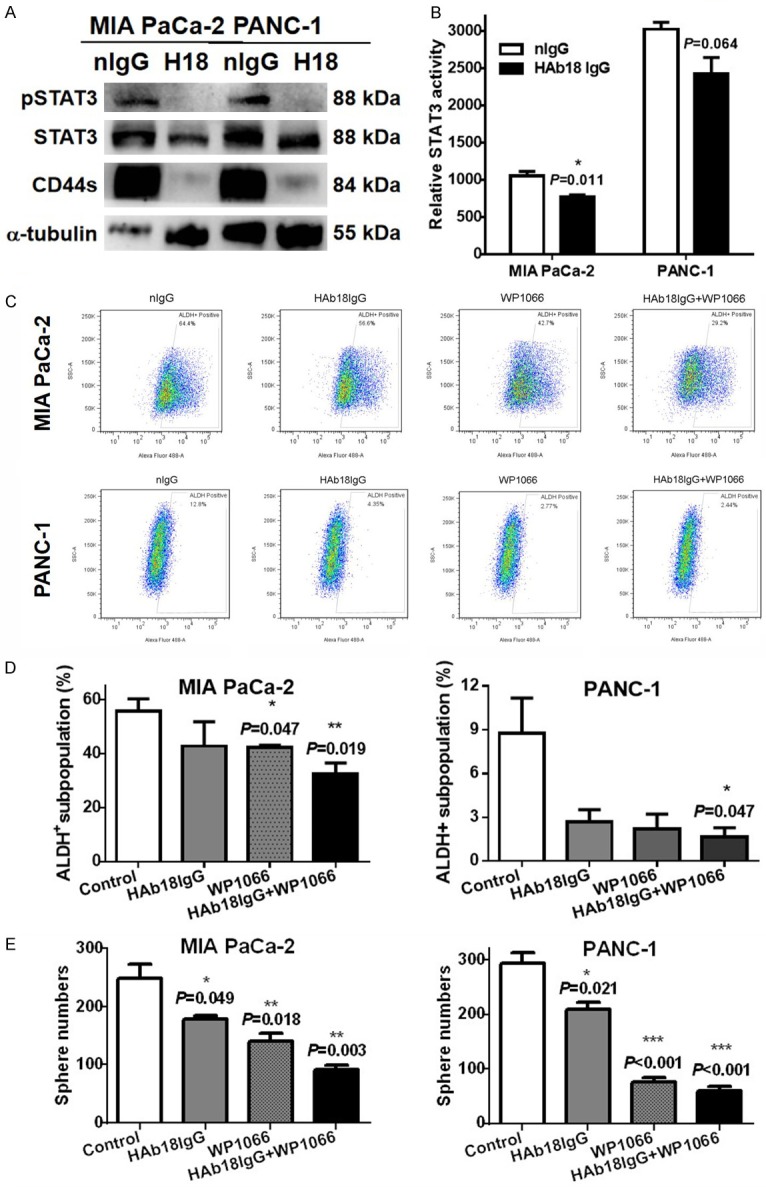
HAb18IgG eliminated CSCs via CD44s-STAT3 signaling. (A, B) CD44s, pSTAT3 and STAT3 protein levels (A) or relative STAT3 transcriptional activity (B) in MIA PaCa-2 and PANC-1 cells treated with 10 µg/ml HAb18IgG(H18) or nIgG for 2 days. (C-E) ALDH+ subpopulation ratios (C, D) or number of spheres (E) in MIA PaCa-2 and PANC-1 cells treated with HAb18IgG alone or in combination with STAT3 inhibitor WP1066 (5 µM).
Finally, we tested whether inhibiting STAT3 signaling could reinforce the inhibitory effect of HAb18IgG on the ratio of pancreatic CSCs. As shown in Figure 5C & 5D, WP1066, a specific STAT3 inhibitor that decreased the ratio of ALDH+ CSC subpopulations in MIA PaCa-2 alone, greatly enhanced this inhibition ratio from 23.13% in cells treated with HAb18IgG alone to 41.34% in cells treated with HAb18IgG plus WP1066. Similarly, WP1066 reinforced HAb18IgG-mediated the reduction of CSC subpopulations in PANC-1 cells, with an inhibition ratio that increased from 69.67% to 80.95%. Furthermore, STAT3 inhibition by WP1066 phenocopied the inhibitory effect of HAb18IgG into sphere formation and exhibited a significant decrease in the number of spheres compared with HAb18IgG alone (Figure 5E) (P=0.0009 and P=0.0004 for MIA PaCa-2 and PANC-1 cells, respectively). These results indicated that HAb18IgG eliminated pancreatic CSCs via the inhibition of CD44s-STAT3 signaling and STAT3 transcriptional activity.
Discussion
In this study, we showed that anti-CD147 HAb18IgG sensitized pancreatic cancer cells to chemoradiotherapy. Furthermore, HAb18IgG decreased colony and sphere formation and reduced CSC subpopulations and potentials by blocking CD44s-pSTAT3 signaling. Our data revealed a promising therapeutic role of HAb18IgG in suppressing pancreatic tumor initiation and overcoming post-chemoradiotherapy recurrence through the direct targeting of CSCs. Our study could widen the therapeutic application of anti-CD147 for fatal and incurable pancreatic cancer.
Due to its important roles in regulating CSC features and in promoting pSTAT3-mediated pancreatic tumor development, CD147 is an attractive therapeutic target. In our lab, anti-CD147 drug successfully prevented tumor recurrence post liver transplantation or radiofrequency ablation (metuximab) [18,19] and was demonstrated to enhance chemosensitivity in NSCLC (matuzumab) [21]. Although one study showed that the combined treatment with anti-EMMPRIN and gemcitabine had an antagonistic effect in hypovascular tumors [22], the other two studies supported that the combination of anti-EMMPRIN or 90Y-labeled 059-053 (fully human anti-CD147) with gemcitabine is a promising therapeutic option for pancreatic cancer [23,24]. In addition, the combination of anti-EMMPRIN and anti-DR5 showed an additive effect on tumor suppression [25]. These studies indicated the therapeutic potential of anti-CD147 in cancer. However, the therapeutic potential of anti-CD147 was not investigated in pancreatic CSCs. In this paper, we demonstrated the therapeutic role of anti-CD147 in pancreatic cancer by targeting CSCs for the first time. Our study provided further supporting evidence for anti-CD147 sensitizing pancreatic cancer cells to chemoradiotherapy via targeting of CSCs.
Targeting CSCs can be done through antibodies, aptamers and peptide ligands as well as small molecules, RNA-based therapeutics and CRISPR/Cas9 technology [2]. In comparisons, antibodies have shown good targeting specificity, affinity, and preferable therapeutic efficiency with minimal toxicity, while the peptide, short interfering RNA or microRNA approaches have delivery problems. Therefore, antibody therapeutic agents are powerful with clinically proven therapeutic value and a leading product within the biopharmaceutical market [26]. Unfortunately, the number of clinical trials targeting CSCs is quite low. Among the 86 clinical trials targeting CSCs, only 12 use antibodies as therapeutic agents [27]. A key reason for the low rate of using antibodies, apart from the lack of specific markers, is the plasticity of the CSCs, which is the transition of non-CSCs into CSCs [28]. Thus, antibodies that both recognize the surface antigen and have a functional influence on CSCs, such as the blockage of CSC plasticity transition, are urgently required. In addition to its linkage to CSC features, CD147 was recently found by us to promote the detachment-induced transversion of non-CSC into CSCs (data not shown). Then, the therapeutic effect of anti-CD147 is due not only to the direct elimination of CSCs but also to blocking the acquiring of the CSC properties.
Due to the heterogeneous of tumor and insufficient delivery, most antibodies alone are not necessarily curative. Therefore, an antibody-based strategy targeting CSCs, along with other chemotherapeutic drugs for the bulk of tumor cells, might be the best possible way to improve the outcome. In this paper, the therapeutic effects of chemoradiotherapy were improved further when the chemoradiotherapy agents were combined with HAb18IgG, although this result needs to be further studied before clinical application. Ideally, maintenance therapy with antibodies targeting CSCs when the residual tumor bulk is minimal would be the way forward in the long term.
In summary, we showed that anti-CD147 HAb18IgG sensitized pancreatic cancer cells to chemoradiotherapy by inhibiting the potential of CSCs and suppressing CD44s-pSTAT3 signaling. For application, HAb18IgG may be a promising therapeutic agent to suppress pancreatic tumor initiation and sensitize CSCs to chemoradiotherapy.
Acknowledgements
We wish to thank American Journal Experts for editing and proofreading this manuscript. This work was supported by the National Basic Research Program (2015CB553700), China National Science and Technology Major Project (2015ZX09501-009) and National Natural Science Foundation (31571469).
Disclosure of conflict of interest
None.
References
- 1.Annett S, Robson T. Targeting cancer stem cells in the clinic: current status and perspectives. Pharmacol Ther. 2018;187:13–30. doi: 10.1016/j.pharmthera.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Putzer BM, Solanki M, Herchenroder O. Advances in cancer stem cell targeting: how to strike the evil at its root. Adv Drug Deliv Rev. 2017;120:89–107. doi: 10.1016/j.addr.2017.07.013. [DOI] [PubMed] [Google Scholar]
- 3.Peixoto J, Lima J. Metabolic traits of cancer stem cells. Dis Model Mech. 2018;11 doi: 10.1242/dmm.033464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23:1124–1134. doi: 10.1038/nm.4409. [DOI] [PubMed] [Google Scholar]
- 5.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 6.Ji Q, Hao X, Zhang M, Tang W, Yang M, Li L, Xiang D, Desano JT, Bommer GT, Fan D, Fearon ER, Lawrence TS, Xu L. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS One. 2009;4:e6816. doi: 10.1371/journal.pone.0006816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agliano A, Calvo A, Box C. The challenge of targeting cancer stem cells to halt metastasis. Semin Cancer Biol. 2017;44:25–42. doi: 10.1016/j.semcancer.2017.03.003. [DOI] [PubMed] [Google Scholar]
- 8.Tijink BM, Buter J, de Bree R, Giaccone G, Lang MS, Staab A, Leemans CR, van Dongen GA. A phase I dose escalation study with anti-CD44v6 bivatuzumab mertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus. Clin Cancer Res. 2006;12:6064–6072. doi: 10.1158/1078-0432.CCR-06-0910. [DOI] [PubMed] [Google Scholar]
- 9.Birzele F, Voss E, Nopora A, Honold K, Heil F, Lohmann S, Verheul H, Le Tourneau C, Delord JP, van Herpen C, Mahalingam D, Coveler AL, Meresse V, Weigand S, Runza V, Cannarile M. CD44 isoform status predicts response to treatment with anti-CD44 antibody in cancer patients. Clin Cancer Res. 2015;21:2753–2762. doi: 10.1158/1078-0432.CCR-14-2141. [DOI] [PubMed] [Google Scholar]
- 10.Li L, Hao X, Qin J, Tang W, He F, Smith A, Zhang M, Simeone DM, Qiao XT, Chen ZN, Lawrence TS, Xu L. Antibody against CD44s inhibits pancreatic tumor initiation and postradiation recurrence in mice. Gastroenterology. 2014;146:1108–1118. doi: 10.1053/j.gastro.2013.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen P, Huang H, Wu J, Lu R, Wu Y, Jiang X, Yuan Q, Chen Y. Bone marrow stromal cells protect acute myeloid leukemia cells from anti-CD44 therapy partly through regulating PI3K/Akt-p27(Kip1) axis. Mol Carcinog. 2015;54:1678–1685. doi: 10.1002/mc.22239. [DOI] [PubMed] [Google Scholar]
- 12.Wu X, Tang W, Marquez RT, Li K, Highfill CA, He F, Lian J, Lin J, Fuchs JR, Ji M, Li L, Xu L. Overcoming chemo/radio-resistance of pancreatic cancer by inhibiting STAT3 signaling. Oncotarget. 2016;7:11708–11723. doi: 10.18632/oncotarget.7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li L, Tang W, Wu X, Karnak D, Meng X, Thompson R, Hao X, Li Y, Qiao XT, Lin J, Fuchs J, Simeone DM, Chen ZN, Lawrence TS, Xu L. HAb18G/CD147 promotes pSTAT3-mediated pancreatic cancer development via CD44s. Clin Cancer Res. 2013;19:6703–6715. doi: 10.1158/1078-0432.CCR-13-0621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu J, Ru NY, Zhang Y, Li Y, Wei D, Ren Z, Huang XF, Chen ZN, Bian H. HAb18G/CD147 promotes epithelial-mesenchymal transition through TGF-beta signaling and is transcriptionally regulated by Slug. Oncogene. 2011;30:4410–4427. doi: 10.1038/onc.2011.149. [DOI] [PubMed] [Google Scholar]
- 15.Ke X, Li L, Dong HL, Chen ZN. Acquisition of anoikis resistance through CD147 upregulation: a new mechanism underlying metastasis of hepatocellular carcinoma cells. Oncol Lett. 2012;3:1249–1254. doi: 10.3892/ol.2012.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang J, Guo YS, Zhang Y, Yu XL, Li L, Huang W, Li Y, Chen B, Jiang JL, Chen ZN. CD147 induces UPR to inhibit apoptosis and chemosensitivity by increasing the transcription of Bip in hepatocellular carcinoma. Cell Death Differ. 2012;19:1779–1790. doi: 10.1038/cdd.2012.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu J, Li Y, Dang YZ, Gao HX, Jiang JL, Chen ZN. HAb18G/CD147 promotes radioresistance in hepatocellular carcinoma cells: a potential role for integrin beta1 signaling. Mol Cancer Ther. 2015;14:553–563. doi: 10.1158/1535-7163.MCT-14-0618. [DOI] [PubMed] [Google Scholar]
- 18.Xu J, Shen ZY, Chen XG, Zhang Q, Bian HJ, Zhu P, Xu HY, Song F, Yang XM, Mi L, Zhao QC, Tian R, Feng Q, Zhang SH, Li Y, Jiang JL, Li L, Yu XL, Zhang Z, Chen ZN. A randomized controlled trial of Licartin for preventing hepatoma recurrence after liver transplantation. Hepatology. 2007;45:269–276. doi: 10.1002/hep.21465. [DOI] [PubMed] [Google Scholar]
- 19.Bian H, Zheng JS, Nan G, Li R, Chen C, Hu CX, Zhang Y, Sun B, Wang XL, Cui SC, Wu J, Xu J, Wei D, Zhang X, Liu H, Yang W, Ding Y, Li J, Chen ZN. Randomized trial of [131I] metuximab in treatment of hepatocellular carcinoma after percutaneous radiofrequency ablation. J Natl Cancer Inst. 2014;106 doi: 10.1093/jnci/dju239. [DOI] [PubMed] [Google Scholar]
- 20.Xu BQ, Fu ZG, Meng Y, Wu XQ, Wu B, Xu L, Jiang JL, Li L, Chen ZN. Gemcitabine enhances cell invasion via activating HAb18G/CD147-EGFR-pSTAT3 signaling. Oncotarget. 2016;7:62177–62193. doi: 10.18632/oncotarget.11405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feng F, Wang B, Sun X, Zhu Y, Tang H, Nan G, Wang L, Wu B, Huhe M, Liu S, Diao T, Hou R, Zhang Y, Zhang Z. Metuzumab enhanced chemosensitivity and apoptosis in non-small cell lung carcinoma. Cancer Biol Ther. 2017;18:51–62. doi: 10.1080/15384047.2016.1276126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim H, Rigell CJ, Zhai G, Lee SK, Samuel SL, Martin A, Umphrey HR, Stockard CR, Beasley TM, Buchsbaum DJ, Li LS, Boothman DA, Zinn KR. Antagonistic effects of anti-EMMPRIN antibody when combined with chemotherapy against hypovascular pancreatic cancers. Mol Imaging Biol. 2014;16:85–94. doi: 10.1007/s11307-013-0665-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shah N, Zhai G, Knowles JA, Stockard CR, Grizzle WE, Fineberg N, Zhou T, Zinn KR, Rosenthal EL, Kim H. (18)F-FDG PET/CT imaging detects therapy efficacy of anti-EMMPRIN antibody and gemcitabine in orthotopic pancreatic tumor xenografts. Mol Imaging Biol. 2012;14:237–244. doi: 10.1007/s11307-011-0491-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sugyo A, Tsuji AB, Sudo H, Koizumi M, Ukai Y, Kurosawa G, Kurosawa Y, Saga T, Higashi T. Efficacy evaluation of combination treatment using gemcitabine and radioimmunotherapy with 90Y-labeled fully human anti-CD147 monoclonal antibody 059-053 in a BxPC-3 xenograft mouse model of refractory pancreatic cancer. Int J Mol Sci. 2018;19 doi: 10.3390/ijms19102979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim H, Zhai G, Samuel SL, Rigell CJ, Umphrey HR, Rana S, Stockard CR, Fineberg NS, Zinn KR. Dual combination therapy targeting DR5 and EMMPRIN in pancreatic adenocarcinoma. Mol Cancer Ther. 2012;11:405–415. doi: 10.1158/1535-7163.MCT-11-0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elgundi Z, Reslan M, Cruz E, Sifniotis V, Kayser V. The state-of-play and future of antibody therapeutics. Adv Drug Deliv Rev. 2017;122:2–19. doi: 10.1016/j.addr.2016.11.004. [DOI] [PubMed] [Google Scholar]
- 27.Santamaria S, Delgado M, Kremer L, Garcia-Sanz JA. Will a mAb-based immunotherapy directed against cancer stem cells be feasible? Front Immunol. 2017;8:1509. doi: 10.3389/fimmu.2017.01509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sneha S, Nagare RP, Priya SK, Sidhanth C, Pors K, Ganesan TS. Therapeutic antibodies against cancer stem cells: a promising approach. Cancer Immunol Immunother. 2017;66:1383–1398. doi: 10.1007/s00262-017-2049-0. [DOI] [PMC free article] [PubMed] [Google Scholar]