

Abstract

Two sulfated diterpene glycosides featuring a highly substituted and sterically encumbered cyclopropane ring have been isolated from the marine red alga Peyssonnelia sp. Combination of a wide array of 2D NMR spectroscopic experiments, in a systematic structure elucidation workflow, revealed that peyssonnosides A–B (1–2) represent a new class of diterpene glycosides with a tetracyclo [7.5.0.01,10.05,9] tetradecane architecture. A salient feature of this workflow is the unique application of quantitative interproton distances obtained from the rotating frame Overhauser effect spectroscopy (ROESY) NMR experiment, wherein the β-d-glucose moiety of 1 was used as an internal probe to unequivocally determine the absolute configuration, which was also supported by optical rotatory dispersion (ORD). Peyssonnoside A (1) exhibited promising activity against liver stage Plasmodium berghei and moderate antimethicillin-resistant Staphylococcus aureus (MRSA) activity, with no cytotoxicity against human keratinocytes. Additionally, 1 showed strong growth inhibition of the marine fungus Dendryphiella salina indicating an antifungal ecological role in its natural environment. The high natural abundance and novel carbon skeleton of 1 suggests a rare terpene cyclase machinery, exemplifying the chemical diversity in this phylogenetically distinct marine red alga.
Introduction
Diterpenoids including diterpene glycosides are one of the most widely distributed and structurally diverse groups of compounds.1,2 Although diterpene glycosides and natural products with cyclopropane rings are each reported frequently, molecules with highly substituted cyclopropanes embedded in a complex polycyclic system are rare.3,4 Their structural novelty and bioactivity is valuable for medicinal chemistry, biosynthetic studies, computational studies, and encourage the development of new synthetic methods.5
In recent years, the determination of absolute configuration in natural products has advanced substantially because of the availability of accurate, high-speed computational prediction of spectroscopic properties. Electronic circular dichroism has become a hallmark for absolute configuration determination of molecules with chromophores. On the other hand, although used infrequently, vibrational circular dichroism (VCD), Raman optical activity, and optical rotatory dispersion (ORD) experiments are gaining popularity, particularly for molecules devoid of chromophores.6,7 Similarly, a wide array of chiral derivatizing agents are available with Mosher’s ester for secondary alcohols being the most widely applied.8 Despite these advancements, absolute configuration analysis of natural products is often challenging, especially when molecules lack chromophores and are unstable toward derivatization.
Recently, monosaccharides were reported as chiral probes in NOE-based NMR experiments for the determination of absolute configuration of secondary alcohols.9 The authors showed that a qualitative speculation of interproton cross-peaks between the monosaccharide and aglycone can be used to predict absolute configuration if a molecule assumes one major conformation in solution. However, because NOE cross-peaks represent the average of the inverse sixth power of internuclear distance between conformations, a qualitative evaluation of cross-peak intensity can be ambiguous and even misleading when a molecule assumes more than one conformation, as a small Boltzmann population of a minor conformer can have pronounced effects on the intensity of relevant cross-peaks.10 Herein, we address this issue by employing quantitative interproton distances, where the known configuration of a monosaccharide is used as an internal probe, such that the cross-peaks observed between the sugar and aglycone signals were quantified and compared with calculated interproton distances obtained from a Boltzmann population of conformers for the putative diastereomers under study. The method is noteworthy for being nondestructive and applicable to molecules that are sensitive to chemical modification.
The structural elucidation of natural products is an inverse problem, where all solutions besides the most logical one must be carefully ruled out based on observations made from empirical data.11 Consequently, it is imperative that conclusions derived in each step of the process be supported with multiple analytical or computational tools. Along these lines, we have undertaken a systematic multifaceted approach, wherein the planar structure, relative and absolute configuration, as determined with extensive NMR-based methods, were sequentially supported with computer-assisted structure elucidation (CASE), density functional theory (DFT)-based prediction of 13C chemical shifts, and simulation of experimental ORD data (Figure 1).
Figure 1.
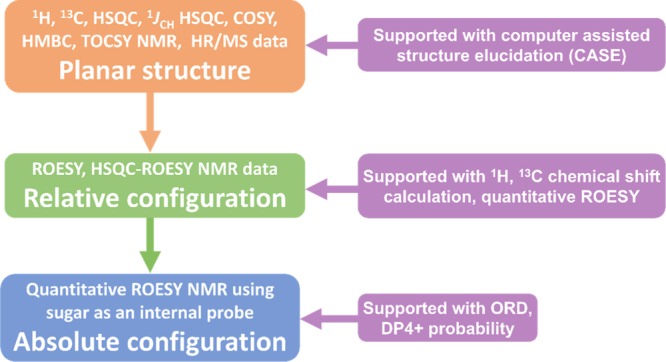
Overview of the systematic structure determination workflow.
Result and Discussion
Peyssonnosides A–B (1–2) (Figure 2), were isolated from the mid-polarity fraction of the Peyssonnelia sp. extract, guided by antibacterial activity against MRSA. HRESIMS data of 1 (m/z 531.2625 [M]−) indicated a molecular formula of C26H43O9S. 1H, 13C, and HSQC NMR spectra of 1 (Table 1) suggested the presence of five methyls, eight aliphatic methylenes, nine aliphatic methines, and four quaternary centers. The molecular formula along with the absence of sp2 carbons in the 13C NMR spectrum indicated five rings, accounting for the calculated degrees of unsaturation.
Figure 2.
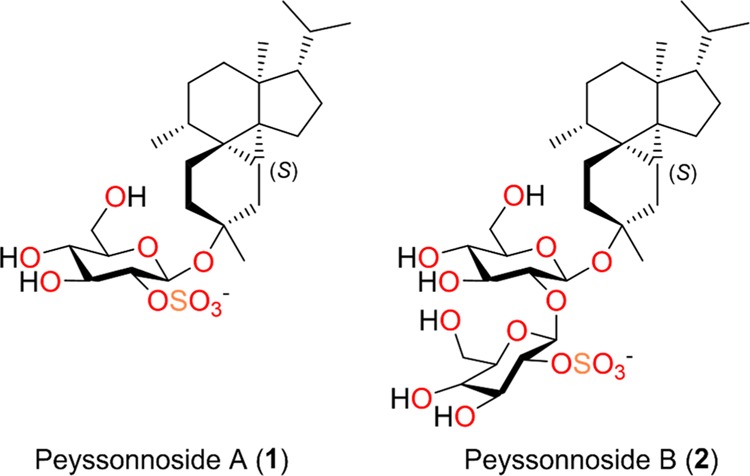
Novel diterpene glycosides peyssonnosides A–B (1–2) from the marine red alga Peyssonnelia sp.
Table 1. NMR Spectral Data for 1 in DMSO-d6 Acquired with a 700 MHz Instrumenta.
| position | δC | δH(mult,J,Hz) | COSY | HMBC | ROESY |
|---|---|---|---|---|---|
| 1 | 13.9 (CH) | 0.69 dd (9.6, 2.2) | H-2a, H-2b | C-3, C-5, C-6, C-7, C-10, C-11, C-12 | H-2b, H-19, 6′-OH |
| 2a | 29.8 (CH2) | 0.85 m | H-1 | C-1, C-11 | H-12 |
| 2b | 2.16 ddd (15.2, 9.6, 2.2) | H-1, H-4b | C-1, C-3, C-4, C-11 | H-1, H-2a, H-20, H-1′, H-5′, 6′-OH | |
| 3 | 72.5 (C) | ||||
| 4a | 33.9 (CH2) | 0.77 m | H-5a, H-5b | C-2, C-5, C-20, C-1′ | H-12 |
| 4b | 1.35 m | H-2b, H-5b | C-2, C-3, C-5, C-6 | H-5b, H-20, H-1′ | |
| 5a | 21.3 (CH2) | 1.15 m | H-4a | C-1, C-3, C-4, C-6, C-11 | H-4b, H-5b, H-12 |
| 5b | 1.96 ddd (13.8, 12.4, 7.5) | H-4a, H4b | C-3, C-4, C-6, C-7 | H-4a, H-4b, H-5a, H-19, H-1′, H-2′ | |
| 6 | 24.2 (C) | ||||
| 7 | 35.0 (CH) | 1.35 m | H-8a, H-19 | C-1, C-8, C-9, C-19 | H-9a |
| 8a | 26.8 (CH2) | 0.75 m | H-7, H-9a | C-6, C-7, C-9, C-10, C-19 | H-9b |
| 8b | 1.16 m | H-9b | C-10 | H-9b | |
| 9a | 39.2 (CH2) | 0.91 m | H-8a | C-7, C-10, C-18 | H-7 |
| 9b | 1.47 m | H-8b | C-7, C-8, C-10, C-11, C-18 | H-8a, H-8b, H-18 | |
| 10 | 41.6 (C) | ||||
| 11 | 36.2 (C) | ||||
| 12 | 25.1 (CH2) | 1.27 m | H-13a, H-13b | C-1, C-6, C-10, C-11, C-13, C-14 | H-2a, H-4a, H-13b, H-18 |
| 13a | 27.9 (CH2) | 1.19 m | H-12 | C-12, C-14 | H-13b, H-18 |
| 13b | 1.73 m | H-12, H-14 | C-10, C-11, C-12, C-14, C-15, C-18 | H-12, H-13a, H-14, H-17 | |
| 14 | 61.4 (CH) | 1.19 m | H-13b, H-15 | C-10, C-13, C-15, C-18 | H-13b |
| 15 | 28.5 (CH) | 1.56 o (6.7) | H-14, H-16, H-17 | C-10, C-13, C-14, C-16, C-17 | H-16, H-17, H-18 |
| 16 | 23.3 (CH3) | 0.91 d (6.6) | H-15 | C-10, C-14, C-15, C-17 | H-15, H-18 |
| 17 | 23.0 (CH3) | 0.87 d (6.6) | H-15 | C-14, C-15, C-16 | H-13b, H-15 |
| 18 | 19.0 (CH3) | 0.64 s | C-1, C-9, C-10, C11, C-14 | H-9b, H-12, H-13a, H-15, H-16 | |
| 19 | 19.1 (CH3) | 1.02 d (6.4) | H-7 | C-6, C-7, C-8 | H-1, H-5b, H-2′, H-4′ |
| 20 | 25.8 (CH3) | 1.00 s | C-1, C-2, C-3, C-4, C-5 | H-2b, H-4b, H-1′ | |
| 1′ | 94.7 (CH) | 4.39 d (7.7) | H-2′ | C-3, C-2′, C-3′, C-5′ | H-2b, H-4b, H-5b, H-20, H-3′, H-5′ |
| 2′ | 79.3 (CH) | 3.65 dd (8.8, 7.7) | H-1′, H-3′ | C-1′, C-3′ | H-19, H-5b, 3′-OH |
| 3′ | 76.7 (CH) | 3.44 t (8.8) | H-2′, H-4′ | C-1′, C-2′, C-4′, C-5′ | H-1′, 3′-OH, 4′-OH |
| 4′ | 70.7 (CH) | 3.00 td (9.3, 5.3) | H-3′, H-5′ | C-3′, C-5′, C-6′ | H-19, 3′-OH, 4′-OH, 6′-OH |
| 5′ | 76.1 (CH) | 3.13 ddd (9.5, 6.8, 2.3) | H-4′, H-6′a, H6′b | C-1′, C-3′, C-4′, C-6′ | H-2b, H-1′, 4′-OH |
| 6′a | 61.8 (CH2) | 3.37 m | H-5′ | C-4′, C-5′ | H-6′b |
| 6′b | 3.72 ddd (11.2, 4.7, 2.3) | H-5′ | C-4′, C-5′ | H-6′a, 4′-OH, 6′-OH | |
| 3′-OH | 5.63 s | C-2′, C-3′, C-4′ | H-2′, H-3′, H-4′ | ||
| 4′-OH | 4.97 d (5.4) | C-3′, C-4′, C-5′ | H-3′, H-4′, H-5′, H-6′b | ||
| 6′-OH | 4.22 t (5.2) | C-5′, C-6′ | H-1, H-2b, H-4′ |
δC, δH—experimental carbon and proton chemical shifts, mult—multiplicity, J—coupling constant, s—singlet, d—doublet, m—multiplet, t—triplet, o—octet.
A sharp doublet at δH 4.39 (d, 3JH-1′,H-2′ = 7.7 Hz, H-1′, a β anomeric proton) and downfield proton signals between δH 2.9 and 5.7 were indicative of a monosaccharide. Starting from the anomeric proton H-1′, vicinal COSY correlations among H-1′, H-2′, H-3′, H-4′, H-5′, and H-6′ revealed the sugar moiety. Large vicinal couplings of >7 Hz were observed for all methine protons in the sugar spin system, characteristic of β-glucose. The sulfur atom along with the remaining four oxygens were suggestive of a sulfate moiety which was assigned to C-2′ based on its relatively downfield chemical shift (δC = 79.3) and was also supported by predicted 13C chemical shifts (Figure 3, Table 3). The specific optical rotation of the sulfated sugar from acid-hydrolyzed 1 measured a positive value of [α]D27 = +60.6 consistent with β-d-glucose.12
Figure 3.
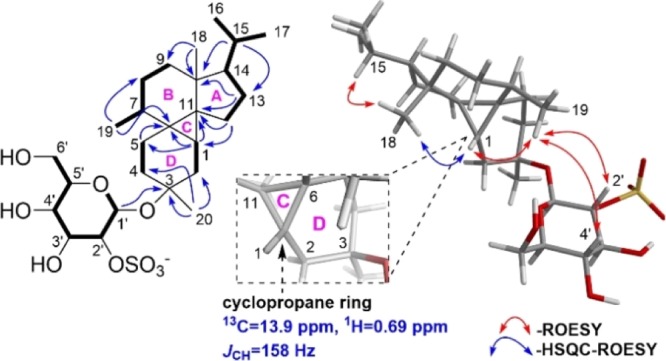
Left: Key COSY (bold lines), HMBC (blue arrows), Right: HSQC-ROESY (blue arrow), and ROESY (red arrows) correlations for 1.
Table 3. Comparison of Experimental and Predicted (Calculated) NMR Chemical Shifts for 1a,b.
| 1′S,2′R,3′S,4′S,5′R,1S,3R,6S,7R,10S,11S,14S—1 |
1′S,2′R,3′S,4′S,5′R,1R,3S,6R,7S,10R,11R,14R—1 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| position | δC expt | δH expt | δC calc | ΔδC | δH calc | ΔδH | δC calc | ΔδC | δH calc | ΔδH |
| 1 | 13.9 | 0.69 | 15.8 | 1.9 | 0.51 | –0.18 | 15.9 | 2.0 | 0.44 | –0.25 |
| 2a | 29.8 | 0.85 | 28.2 | –1.6 | 0.99 | 0.14 | 32.5 | 2.7 | 1.16 | 0.31 |
| 2b | 2.16 | 1.97 | –0.19 | 1.84 | –0.32 | |||||
| 3 | 72.5 | 75.8 | 3.3 | 75.8 | 3.3 | |||||
| 4a | 33.9 | 0.77 | 34.1 | 0.2 | 0.90 | 0.13 | 31.0 | –2.9 | 0.81 | 0.04 |
| 4b | 1.35 | 1.33 | –0.02 | 1.40 | 0.05 | |||||
| 5a | 21.3 | 1.15 | 21.7 | 0.4 | 1.32 | 0.17 | 22.3 | 1.0 | 1.25 | 0.10 |
| 5b | 1.96 | 1.90 | –0.06 | 2.06 | 0.10 | |||||
| 6 | 24.2 | 26.0 | 1.8 | 26.2 | 2.0 | |||||
| 7 | 35 | 1.35 | 36.8 | 1.8 | 1.26 | –0.09 | 35.6 | 0.6 | 1.27 | –0.08 |
| 8a | 26.8 | 0.75 | 26.5 | –0.3 | 0.82 | 0.07 | 27.0 | 0.2 | 0.81 | 0.06 |
| 8b | 1.16 | 1.15 | –0.01 | 1.19 | 0.03 | |||||
| 9a | 39.2 | 0.91 | 38.7 | –0.5 | 0.91 | 0.00 | 38.6 | –0.6 | 0.84 | –0.07 |
| 9b | 1.47 | 1.42 | –0.05 | 1.48 | 0.01 | |||||
| 10 | 41.6 | 44.5 | 2.9 | 45.3 | 3.7 | |||||
| 11 | 36.2 | 38.5 | 2.3 | 37.0 | 0.8 | |||||
| 12a | 25.1 | 1.27 | 25.5 | 0.4 | 1.21 | –0.06 | 25.4 | 0.3 | 1.26 | –0.01 |
| 12b | 1.27 | 1.34 | 0.07 | 1.29 | 0.02 | |||||
| 13a | 27.9 | 1.19 | 30.2 | 2.3 | 1.27 | 0.08 | 27.6 | –0.3 | 1.32 | 0.13 |
| 13b | 1.73 | 1.73 | 0.00 | 1.58 | –0.15 | |||||
| 14 | 61.4 | 1.19 | 62.4 | 1.0 | 1.00 | –0.19 | 61.4 | 0.0 | 1.04 | –0.15 |
| 15 | 28.5 | 1.56 | 31.9 | 3.4 | 1.45 | –0.11 | 31.0 | 2.5 | 1.52 | –0.04 |
| 16 | 23.3 | 0.91 | 19.5 | –3.8 | 0.93 | 0.02 | 20.4 | –2.9 | 0.92 | 0.01 |
| 17 | 23 | 0.87 | 21.6 | –1.4 | 0.89 | 0.02 | 20.9 | –2.1 | 0.84 | –0.03 |
| 18 | 19 | 0.64 | 15.4 | –3.6 | 0.70 | 0.06 | 15.8 | –3.2 | 0.68 | 0.04 |
| 19 | 19.1 | 1.02 | 17.2 | –1.9 | 0.90 | –0.12 | 17.0 | –2.1 | 0.92 | –0.10 |
| 20 | 25.8 | 1 | 23.3 | –2.5 | 1.00 | 0.00 | 23.7 | –2.1 | 1.05 | 0.05 |
| 1′ | 94.7 | 4.39 | 94.0 | –0.7 | 4.30 | –0.09 | 92.9 | –1.8 | 4.31 | –0.08 |
| 2′ | 79.3 | 3.65 | 78.7 | –0.6 | 3.60 | –0.05 | 78.0 | –1.3 | 3.66 | 0.01 |
| 3′ | 76.7 | 3.44 | 76.6 | –0.1 | 3.37 | –0.07 | 75.8 | –0.9 | 3.44 | 0.00 |
| 4′ | 70.7 | 3 | 69.1 | –1.6 | 3.46 | 0.46 | 73.4 | 2.7 | 3.28 | 0.28 |
| 5′ | 76.1 | 3.13 | 73.8 | –2.3 | 3.08 | –0.05 | 72.4 | –3.7 | 3.18 | 0.05 |
| 6′a | 61.8 | 3.72 | 61.2 | –0.6 | 3.67 | –0.05 | 63.8 | 2.0 | 3.65 | –0.07 |
| 6′b | 3.37 | 3.53 | 0.16 | 3.46 | 0.09 | |||||
| rmsec | 2.0 | 0.1 | 2.1 | 0.1 | ||||||
| DP4+ 1H datad | 21.3% | 78.7% | ||||||||
| DP4+ 13C datad | 99.2% | 0.8% | ||||||||
| DP4+ all datad | 97.2% | 2.8% | ||||||||
δC expt, δH expt—experimental carbon and proton chemical shifts, δC calc, δH calc—calculated carbon and proton chemical shifts, ΔδC—difference between calculated and experimental carbon chemical shifts, ΔδH—difference between calculated and experimental proton chemical shifts. Bold represents large deviations |ΔδH| > 0.2, |ΔδC| > 3.0.
δ values were calculated at the B3LYP/6-311++G(2d,2p) level of theory with PCM model using DMSO as solvent.
Root-mean-square error.
DP4+ probability.26
The 20 carbons of the aglycone including five upfield methyls in the 1H NMR spectrum and the remaining four degrees of unsaturation were strongly indicative of a tetracyclic diterpene core for 1. Two methyls H3-16 (δH 0.91) and H3-17 (δH 0.87) showed COSY correlations with H-15 (δH 1.56), reminiscent of an isopropyl group. COSY correlations were also observed for H-7/H3-19, H-7/H2-8/H2-9, and H2-12/H2-13/H-14. HMBC correlations from H3-19 to C-6, C-7, and C-8 established the position of C-7 between C-6 and C-8. Taken together, HMBC correlations from H-15 to C-10, C-13, C-14, from H3-18 to C-9, C-10, C-11, C-14, and from H2-12 to C-6, C-10, C-11 established the planar structure of rings A and B for 1 (Figure 3, Table 1).
COSY correlations were observed for H-1/H2-2 and H2-4/H2-5 in 1. HMBC correlations from H3-20 to C-2, C-3, and C-4 as well as from H-1′ to C-3 confirmed the position of the sugar at C-3 between spin systems H-1/H2-2 and H2-4/H2-5. Finally, the cyclopropyl H-1 (with characteristic upfield chemical shifts of δH 0.69 and δC 13.9) acted as a lynchpin, with HMBC correlations to C-5, C-6, C-7, C-10, C-11, and C-12 revealing the complex tetracyclic core of 1 (Figure 3, Table 1). The cyclopropane was further confirmed by a J-coupled HSQC NMR signal with a diagnostic 1JCH of 158 Hz (Figure 4).13 The planar structure of the aglycone, representing a novel carbon skeleton, was additionally supported with MestReNova CASE, whereby the proposed aglycone of 1 stood out as the top hit.14 The remaining possibilities generated by CASE were readily ruled out with additional NMR spectroscopic data (Figure 5).
Figure 4.

Left: J-coupled HSQC showing a large 1JCH of 158 Hz, characteristic of a cyclopropane. Right: HSQC-ROESY cross-peak suggesting cis-fusion of rings A, B for 1.
Figure 5.
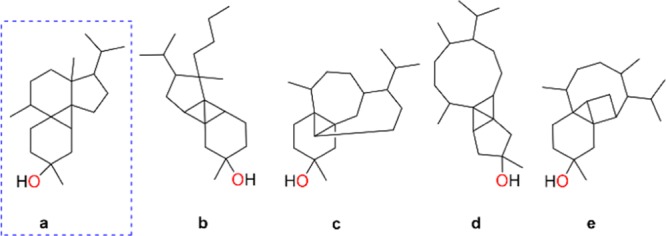
Top five structural hits for the diterpene core of 1 generated by MestReNova CASE. The structure a in the hatched box represents the top hit.
With the planar structure of 1 established, ROESY NMR was applied to study the relative configuration. As the relationship between H-1 and H3-18 could not be deduced by 1H–1H ROESY because of cross-peak overlap with the diagonal, the HSQC-ROESY spectrum was employed to reveal a strong correlation between H-1 and H3-18 (Figures 3 and 4) suggesting a cis-fusion between rings A and B at C-10 and C-11. ROESY cross-peaks for H-1/H3-19 and H3-18/H-15 implied an α-orientation for H3-19 at C-7 and the isopropyl at C-14, respectively. Similarly, correlations between H3-19 and both H-2′ and H-4′ revealed the relative configuration at C-3 (Figure 3). The proposed planar and relative configuration of 1 was supported by comparison of experimental and DFT-predicted 13C chemical shifts (Table 3).
At this point, there were two possible diastereomers for 1: 1′S,2′R,3′S,4′S,5′R,1S,3R,6S,7R,10S,11S,14S, and 1′S,2′R,3′S,4′S,5′R,1R,3S,6R,7S,10R,11R,14R (Figures S9 and S10). Attempts to cleave the glycosidic bond using acid-catalyzed hydrolysis of 1 (so that the diterpene structure could be determined using VCD) were hindered by the generation of a mixture of products. Enzymatic hydrolysis of 1 was unsuccessful too, most likely because of the presence of a sulfated sugar linked to the sterically encumbered tertiary center of the diterpene. VCD spectral analysis of the intact molecule showed poor correlation with computationally predicted data (Figure S13). Similarly, attempts at crystallization of the p-bromobenzoylated product of 1 were unfruitful. Instead, we turned to ROESY NMR spectroscopy again, envisioning that the multiple ROESY cross-peaks observed between the sugar and diterpene could, if quantified as interproton distances, be compared with calculated interproton distances obtained from a Boltzmann population of conformers for the two diastereomers, which could reveal the absolute configuration.
Molecular mechanics force fields (MMFF)-based conformational search15,16 and final structural optimization with DFT calculations at the B3LYP/6-311++G(2d,2p) level17 using a polarizable continuum model (PCM)18 to represent DMSO as the solvent, gave five conformations for each of the two possible diastereomers of 1.19 However, overlaid conformations of each diastereomer were very similar, with differences primarily observed in the orientation of glycosidic hydroxyls. Therefore, for both diastereomers, the relative conformation between the sugar and diterpene exhibited one major orientation attributed to steric encumbrance at C-3 (Figures S9–S11).
| 1 |
Experimental interproton distances were obtained via eq 1, from a 2D quantitative ROESY NMR spectrum employing peak amplitude normalization for improved cross-relaxation (PANIC), where the intensity of the irradiated peaks were kept constant in every slice of the 2D ROESY spectrum (Figures S29–S37).20,21 Distance between i and j is represented by rij, whereas rref is a reference distance between methylene protons. Similarly, aij represents the cross-peak intensity between i and j, whereas aref is the reference cross-peak between methylene protons. Tropps equation was used to account for the protons in a methyl group using r–3 averaging.22
Comparison of quantitative interproton distances between the β-d-glucose moiety and the diterpene core obtained via ROESY with those obtained from the lowest energy conformations clearly supported 1′S,2′R,3′S,4′S,5′R,1S,3R,6S,7R,10S,11S,14S as the absolute configuration of 1. Interproton distance differences greater than 1, 1.7, and 2.7 Å between experimental and calculated values for H-1′/H-4b, H-1′/H-2b, and H-2b/H-5′, respectively, and a poor R2 of 0.2947, strongly disfavored the 1′S,2′R,3′S,4′S,5′R,1R,3S,6R,7S,10R,11R,14R configuration (Table 2).
Table 2. Experimental (from ROESY) and DFT-Calculated Interproton Distances for the Two Possible Diastereomers of 1.
| distance between | Expt. distance (Å) | 3R—1a | 3S—1b |
|---|---|---|---|
| H-1′, H3-20 | 2.69 | 2.54 | 2.56 |
| H-2′, H3-19 | 3.55 | 4.04 | 4.13 |
| H-4′, H3-19 | 4.65 | 4.72 | 4.67 |
| H-1′, H-4b | 3.61 | 4.16 | 2.57 |
| H-1′, H-2b | 2.45 | 2.61 | 4.18 |
| H-2′, H-5b | 4.24 | 4.12 | 4.54 |
| H-5′, H-2b | 3.53 | 3.44 | 6.27 |
| H-1′, H-5b | 4.48 | 4.82 | 3.78 |
| H-1, H3-19 | 3.48 | 3.27 | 3.23 |
| H-15, H3-18 | 2.84 | 2.67 | 2.75 |
| H-5b, H3-19 | 2.67 | 2.65 | 2.67 |
| H-1′, H-3′ | 2.55 | 2.63 | 2.66 |
| H-1′, H-5′ | 2.26 | 2.35 | 2.35 |
| R2 | 0.9249 | 0.2947 |
Calculated for 1′S,2′R,3′S,4′S,5′R,1S,3R,6S,7R,10S,11S,14S—1.
Calculated for 1′S,2′R,3′S,4′S,5′R,1R,3S,6R,7S,10R,11R,14R—1.
The computational modeling and quantitative ROESY data enabled further confirmation of the relative configuration analysis performed earlier. Comparison of calculated and experimental distances for H-15/H3-18 supported the previously asserted α orientation of the isopropyl group at C-14. A β-oriented isopropyl would imply a H-15/H3-18 distance of 4.12 Å, inconsistent with observed ROESY data (Table 2, Figure S12). Quantitative interproton distances for H-1/H3-19, H-15/H3-18, and H-5b/H3-19 matched the proposed relative configuration of the diterpene core. Some unusual 1H NMR spectral features were explained by the lowest energy conformers of 1: i. Geometric alignment (W-coupling) of H-2b and H-4b was evident from the 4 bond COSY correlation observed between these protons.23ii. A significant downfield chemical shift for H-2b (δH 2.16) compared with H-2a (δH 0.85) was likely due to steric compression arising from its eclipsed conformation with H-1 and proximity to H3-20 (Figure 6).24,25
Figure 6.
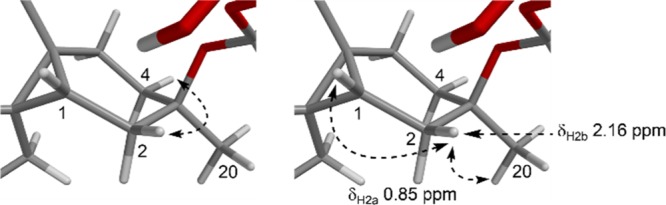
W-coupling (left) and interactions causing steric compression of H-2b (right) for 1.
Literature precedent has shown that computation of specific optical rotation for sugars is reliable when major conformations in solution are well predicted.27 Encouraged by the extensive conformational search performed using two different programs (Spartan, Conflex) and quantitative ROESY data clearly supporting one diastereomer, we embarked on analyzing experimental and DFT-predicted ORD for 1. A close agreement was observed between the sign of the experimental ORD curve with that computed at the B3LYP/6-311++G(2d,2p) level for 1′S,2′R,3′S,4′S,5′R,1S,3R,6S,7R,10S,11S,14S—1 (Figure 7). Finally, when DP4+ probabilistic comparison of NMR spectroscopic data with calculated values was applied for the two candidate diastereomers of 1, the 1′S,2′R,3′S,4′S,5′R,1S,3R,6S,7R,10S,11S,14S isomer was highly favored (Table 3). DP4+ probabilities for this isomer generated 99.2% confidence for 13C NMR chemical shifts and 97.2% confidence for combined 13C and 1H NMR chemical shifts. 1H NMR chemical shifts on their own produced poor probabilities for either isomer.26
Figure 7.
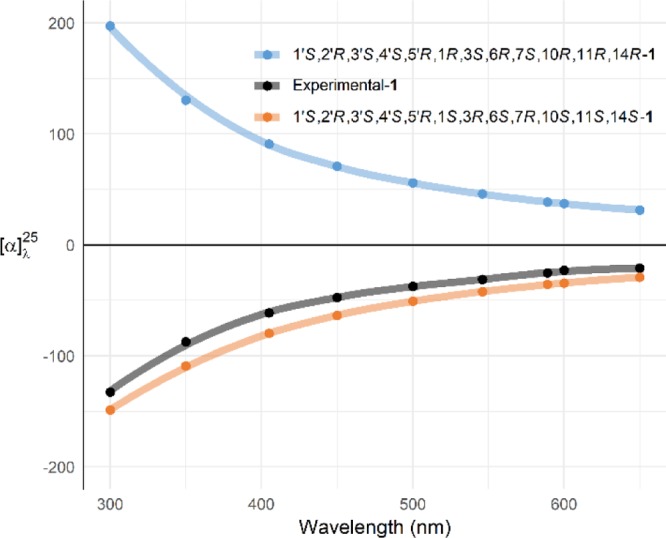
Comparison of experimental and calculated ORD curves for 1.
Peyssonnoside B (2) was isolated as an amorphous white powder with a HRESIMS m/z of 693.3162 [M]− (C32H53O14S). 1H and 13C NMR spectroscopic data of 2 showed close similarities with 1 indicating an identical diterpene core with an additional sugar moiety (Table S10). TOCSY correlations clearly differentiated the spin systems associated with the two sugars (Figure S27), whereas an HMBC correlation from H-1″ to C-2′ confirmed that the second sugar moiety was attached at C-2′. The structure was further confirmed with a combination of COSY, HMBC, and ROESY NMR spectral data (Table S10).
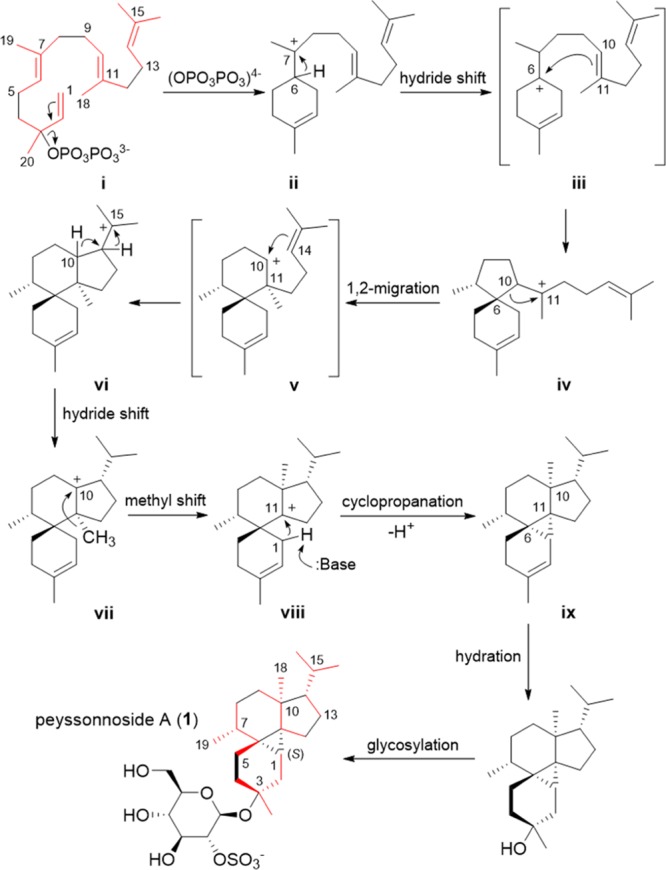
Although the genus Peyssonnelia is phylogenetically diverse (Figure S2), its chemistry is understudied with only a handful of biosynthetic classes reported, for example: sterol glycosides, sesquiterpene hydroquinones, and enediyne oxylipins.28−30 Therefore, it is not entirely surprising that examination of a previously undescribed Peyssonnelia would yield new chemistry. More specifically, the complex diterpene core along with the sulfated β-d-glucose units of the peyssonnosides provides evidence for a distinctive biosynthetic mechanism (Scheme 1). The biosynthetic pathway for 1–2 is proposed to begin with geranylgeranyl diphosphate via the formation of the six-membered cyclohexene ring as shown in intermediate ii. Subsequent hydride shift is envisioned as a concerted process with a ring closing reaction and would form intermediate iv.31,32 The cyclopentane ring in iv would undergo a Wagner–Meerwein ring expansion reaction leading to the formation of the secondary carbocation v which consequently forms the cyclized intermediate vi. It is important to note that although secondary carbocations have been proposed in reaction coordinates of isoprenoid biosynthetic pathways, they often are not intermediates that exist as energy minima.33,34 A series of hydride and methyl shifts is expected to form a tertiary carbocation at C-11 (intermediate viii), which would be intercepted by an incipient allylic carbanion at C-1, leading to a highly substituted cyclopropane ring.35 It is plausible to speculate that the sterically encumbered cis-fusion of rings A and B at C-10 and C-11 in ix arises because the carbanion formed at C-1 in intermediate viii is conformationally constrained and consequently can attack C-11 from only one direction. As shown in Scheme 1, only structure a shown in Figure 5 adequately accounts for a reasonable diterpene biosynthetic pathway.
Scheme 1. Proposed Biosynthetic Mechanism for 1, in Which Isoprenoid Units are Indicated in Red, as Incorporated into Geranylgeranyl Diphosphate and the Final Product.
Chemical ecology experiments to determine the role of 1 in coral reef environments revealed that although it does not deter feeding by at least one potential consumer (the hermit crab Clibanarius striolatus, Figure S5), 1 does exhibit strong antifungal activity against Dendryphiella salina (IC50 = 0.14 μM) leading to morphological changes in fungal growth (Figure S4). Taken together with the high natural abundance of 1 (0.42% of dry weight) along with the phylogenetically distinct characteristics of this marine red alga (Figure S2), it is strongly suggestive that 1 is a novel antimicrobial chemical defense.
The antifungal activity appears to be targeted, as the human pathogen amphotericin B-resistant Candida albicans is not inhibited (IC50 > 50 μM) by 1. Additionally, 1 exhibited promising low micromolar pharmacological activity against the liver stage of the malarial parasite Plasmodium berghei and moderate MRSA activity with no cytotoxicity against human keratinocytes (Table 4). Interestingly, 1 did not inhibit blood stage Plasmodium falciparum, suggesting that the compound is either liver-specific or the selectivity is greater for Plasmodium sporozoites. Additional bioassay data are reported in Table S1.
Table 4. Pharmacological and Ecological Activities of 1–2.
| assay | 1 | 2 |
|---|---|---|
| P. berghei EC50 (μM)a | 2.4 | 5.8 |
| MRSA MIC90 (μg/mL) | 16.7 ± 0.3 | >50 |
| HaCaT toxicity (μM)b | >50 | >50 |
| D. salina IC50 (μM) | 0.14 | NT |
Exoerythrocytic (liver) stage of P. berghei.
Human skin keratinocytes, NT—not tested.
Conclusions
In summary, the reported isolation, structure elucidation, and bioactivity of 1–2 open exciting avenues for further exploration. The moderate and selective activity of 1 against the liver stage of malaria with no detected cytotoxicity holds promise for mechanism of action studies. Strong inhibition of the marine fungus D. salina provides evidence for the ecological relevance of 1 in protecting the Peyssonnelia sp. The unusual cyclopropane containing tetracyclic diterpene core of 1–2 suggests a unique diterpene cyclase machinery in this marine red alga, interesting for biosynthetic, mechanistic, and computational studies on terpene cyclisation. Peyssonnosides A–B (1–2) represent a valuable molecular scaffold for synthetic method development and total synthesis. The reported application of quantitative ROESY/NOESY-based interproton distance measurement for absolute configuration determination is a promising tool for cases in which absolute configuration of a certain molecular portion is understood and can be used as a probe to deduce the global absolute configuration.
Experimental Procedures
General Experimental Procedures
NMR spectra including 1H, 13C, COSY, HSQC, HSQC-ROESY, 1JCH-coupled HSQC, ROESY, and TOCSY, were acquired on 18.8 T (800 MHz for 1H and 200 MHz for 13C) Bruker Advance IIIHD instrument equipped with a 3 mm triple resonance cryoprobe and a 16.4 T (700 MHz for 1H and 175 MHz for 13C) Bruker Advance IIIHD 5 mm indirect broadband cryoprobe. Spectra were recorded in DMSO-d6 and referenced to solvent residual peaks (δH and δC). All spectra were processed and analyzed using MestReNova 11.0.4.
Preparative chromatographic separation was performed using Silicycle (20 × 20 cm, 200 μm thickness) thin-layer chromatography (TLC) plates. HPLC separation was conducted using a Waters 1525 binary pump and a Luna 5 μm silica (100 Å, 250 × 4.6 mm) column and was monitored with an Altech ELSD 800 detector. High-resolution mass spectrometric data were acquired in a Thermo Scientific Q Extractive Mass Spectrometer and LTQ Orbitrap XL.
A Jasco J-815 instrument was used to measure UV–vis absorbance and ORD in 20% aqueous methanol and DMSO, respectively, using a 1 cm pathlength cuvette at a concentration of 0.2 mg/mL for UV–vis and 12 mg/mL for ORD measurement. Optical rotation for the sulfated sugar obtained from hydrolysis of 1 was measured on a Jasco DIP-360 Digital Polarimeter (589 nm Na lamp) with water as solvent, whereas DMSO was used for 1–2. VCD measurement for 1 was carried out using a commercial Chiral IR instrument using a fixed pathlength (100 μm) SL3 cell at concentrations of 40 and 80 mg/mL using DMSO-d6 as solvent.
Specimen Collection and Species Identification
Peyssonnelia sp. (G-1163) was collected on March 17, 2012 from Singi Locale, Tetepare Island, Solomon Islands (S 8°42.561′, E 157°26.500′), and Peyssonnelia sp. (G-1588) was collected on June 17, 2017 off Uepi Island, Solomon Islands (S 8°26.549′, E 157°56.695′). The algae were found growing on reef slopes and on underhangs at depths of 3-25 m. Morphologically, the algae were red in color with intermittent tinges of yellow with a hard, brittle, and calcified texture (Figure S1). Voucher specimens preserved in aqueous formaldehyde and ethanol were stored at the University of the South Pacific and Georgia Institute of Technology. Bulk algal sample was stored at −80 °C at Georgia Institute of Technology.
Morphological comparison of G-1163 with red algae reported in the literature suggested that G-1163 belongs to the genus Peyssonnelia.36 An attempt was made to characterize G-1163 using sequence analysis of nuclear, small subunit (SSU) ribosomal RNA (18S rRNA), however, this approach was not viable as genomic DNA could not be isolated from the ethanol-preserved specimen nor from the frozen bulk algal sample. As an alternative, collection of G-1588 was used for 18S rRNA analysis because of its morphological similarities to G-1163 (Figure S1). Additionally, comparison of the 1H NMR spectra for relevant fractions of these two collections revealed that both contained high concentrations of peyssonnoside A (1) and a small quantity of peyssonnoside B (2) (Figure S38). Taken together, we conclude that G-1588 and G-1163 belong to the same chemotype and genotype.
Genomic DNA from the ethanol-preserved G-1588 algal specimen was extracted using the innuPREP plant DNA kit (Analytik Jena, Germany) according to the manufacturer’s protocol. The four overlapping 18S rRNA gene fragments from genomic DNA were amplified via the polymerase chain reaction (PCR), in four separate reactions using (G01/G10, G02/G14, G04/G13, and G06/G07).37 Each PCR amplification was performed in a 25 μL reaction volume consisting of 5–50 ng of purified genomic DNA; 200 μM of each of the dNTPs; 1 μM of each of the oligonucleotide primer and 1.0 U Taq DNA Polymerase; and 1× Standard PCR reaction buffer (NEB, Ipswich, MA). All PCR amplifications were performed in a GeneAmp PCR system 2700 (Applied Biosystems, Foster City, CA) thermocycler using the following temperature cycling parameters: initial denaturation at 94 °C for 5 min followed by a total of 40 cycles of amplification in which each cycle consisted of denaturation at 94 °C for 40 s, primer annealing at 50 °C for 40 s and primer extension at 72 °C for 1 min. After amplification, final extension of the incompletely synthesized DNA was carried out at 72 °C for 7 min. The PCR fragments were analyzed by agarose gel electrophoresis (1% wt/vol). The gel was stained with ethidium bromide and visualized under a UV transilluminator. All the PCR fragments were either sequenced with forward or reverse primers, and sequences were manually edited and assembled using the CAP3 Sequence Assembly Program.38 The assembled G-1588 18S rRNA sequence (1808 bp) was submitted to GenBank (accession no. MK129455). The sequence similarity of the assembled contig of G-1588 18S rRNA to other known red algae from family Peyssonneliaceae was determined by comparing it with the nonredundant nucleotide database (NCBI) using the blastn program.39 The top ranked matches according to E-values and maximum scores revealed high similarities to multiple species within the order Gigartinales. A maximum-sequence similarity between G-1588 and previously reported Peyssonnelia capensis was 94% with 99% query coverage.
In order to determine the closest phylogenetically related species of G-1588 within the family Peyssonneliaceae (order Gigartinales), its 18S rRNA sequence was compared with those of known representatives from family Peyssonneliaceae and outgroup taxa Bonnemaisonia asparagoides and B. hamifera from the same subclass Rodymeniophycidae but different order (Bonnemaisoniales), obtained from GenBank. Phylogenetic analysis was conducted in MEGA X40 using the maximum likelihood method based on the Kimura 2-parameter model41 with 1000 bootstrap iterations. The evolutionary relationship revealed that G-1588 is genetically distant from other closely related species of the genus Peyssonnelia but belongs to the order Gigartinales because it does not cluster with other orders (data not shown) or the order Bonnemaisoniales of the outgroup taxa shown in the phylogenetic tree (Bonnemaisonia spp.) (Figure S2). The majority of the differences in nucleotides were detected in the variable region V2 of the 18S rRNA. Overall, morphological and phylogenetic analyses are consistent with identification of the red algae used in this study as a member of the genus Peyssonnelia, however, this species is genetically distinct from other known species represented in GenBank.
Isolation of Peyssonnosides A–B (1–2)
The frozen bulk sample of Peyssonnelia sp. sample G-1163 was thawed at room temperature and 300 g of wet material was exhaustively extracted (four times) with 500 mL methanol, combined and dried to yield 6.1 g of the crude extract. The extract was subjected to fractionation using Diaion HP20SS (120 g) first eluting with 200 mL water to remove salts. Four fractions were then collected, eluting 400 mL each of 50% aqueous methanol, 80% aqueous methanol, methanol, and acetone. The fractions were concentrated and dried in vacuo to yield: F1 (0.11 g), F2 (1.26 g), F3 (0.34 g), and F4 (0.16 g). F2 (1.02 g) was subjected to silica gel column chromatography, eluting with hexanes and ethyl acetate (33% → 100% ethyl acetate step gradient) followed by ethyl acetate and methanol (2% → 100% methanol step gradient) and finally with 80% aqueous methanol to produce 89 fractions. These fractions were pooled to 21 sub-fractions based on TLC analysis. Fractions 11–14 together yielded 244 mg of pure 1. Fraction 15 (92 mg) was subjected to preparative silica TLC (Silicycle 200 μm, 20 × 20 cm) using a 0.05:1:2.5 ratio of water, 0.1% trifluoroacetic acid (TFA) in methanol, and chloroform as a mobile phase resulting in two fractions, fraction 15.1 (Rf 0.62) and fraction 15.2 (Rf 0.37). Fraction 15.2 was subjected to HPLC purification using a normal phase silica column (Luna 5 μm, 250 × 4.6 mm) with ethyl acetate and 0.1% TFA in methanol (0% → 50% 0.1% TFA in methanol gradient in 20 min) as the mobile phase to furnish 2 (Rt = 17.5 min, 3.8 mg). Fraction 15.1 contained an undetermined quantity of 1. F2 was also subjected to quantitative 1H NMR analysis which showed that 1 accounted for 0.42% of the dry algal weight. As isolation of 1 was conducted without addition of acid, base, or inorganic salts, its counterion was almost certainly Na+, the most abundant cation in seawater. However, 2 was isolated using acidified solvents by reversed-phase HPLC and was therefore likely characterized as its conjugate acid.
Peyssonnoside A (1)
It was obtained as a white amorphous solid; [α]D25 −25 (c 1.2, DMSO); UV (80% MeOH/H2O): λmax (log ε) 208 sh (3.2); 1H and 13C NMR, see Table 1; HRMS (ESI) m/z: [M]− calcd for C26H43O9S, 531.2633; found, 531.2625.
Peyssonnoside B (2)
It was obtained as a white amorphous solid; [α]D27 −7 (c 1.7, DMSO); UV (80% MeOH/H2O): λmax (log ε) 207 sh (3.6); 1H and 13C NMR, see Table S10; HRMS (ESI) m/z: [M]− calcd for C32H53O14S, 693.3162; found, 693.3162.
Pharmacological Assays
Antimicrobial, antifungal, and antituberculosis assays were conducted as reported earlier, with the MIC being defined as the minimum concentration required to achieve ≥90% inhibition in comparison to the vehicle control.42,43 The compounds were tested against methicillin-resistant Staphylococcus aureus (MRSA, ATCC 33591), vancomycin-resistant Enterococcus faecium (VREF, ATCC 700221), Escherichia coli (EC, ATCC 25922), multidrug-resistant E. coli (MDREC, ATCC BAA-1743), and amphotericin B-resistant C. albicans (ARCA, ATCC 90873) as pathogens. The positive controls used were vancomycin for MRSA, chloramphenicol for VREF and EC, nitrofurantoin for MDREC and cycloheximide for ARCA. Antimycobacterial activity was tested against H37Rv using isoniazid as positive control.
Additional antimicrobial screening was performed against strains from the Centers for Disease Control and Prevention Antibiotic Resistance Bank: Enterobacter cloacae (CDC0008), Klebsiella pneumoniae (CDC0016), Acinetobacter baumannii (CDC0033), E. faecium (HM-959), and Pseudomonas aeruginosa (PA01). Gentamicin was used as a positive control for all strains. Tryptic Soy Broth was used to grow the overnight bacterial cultures. The Clinical and Laboratory Standards Institute (CLSI) method was used to determine the minimum inhibitory concentration (MIC) using cation-adjusted Mueller Hinton broth (CAMHB).44 Optical density (OD) of overnight cultures were adjusted to 5 × 105 cfu/mL and MIC values were determined as described earlier; where readings were taken at an OD600nm in a Cytation-3 multimode plate reader (Biotek). Cytotoxicity was assessed using human skin keratinocytes (HaCaT cell line) and lactate dehydrogenase test kit (G-Biosciences, St. Louis, MO) as described earlier.45
Activity against asexual blood-stage P. falciparum was assessed using the standard parasite proliferation assay with SYBR Green detection.46 In brief, cultures of the P. falciparum strain Dd2L (gift from Dr. David Fidock, Columbia University) or D10 (gift from Dr. Akhil Vaidya, Drexel University) to be used for screening were prepared with screening media (complete media without human serum but supplemented with 0.5% Albumax II) and fresh donation of O+ erythrocytes (TSRI Normal Blood Donation). Compounds were transferred via the Labcyte ECHO Acoustic Liquid Handler into the assay plates. Parasitized erythrocytes and fresh erythrocytes were prepared with screening media and dispensed (Multi-Flo; BioTek, VT) into the assay plates containing compound for a final parasitemia of 0.3 and 2.5% hematocrit. The assay plates were directly transferred and cultured in a gas chamber at 37 °C in the presence of a low oxygen gas mixture. After 72 h and daily gas exchanges, the assay plates were removed from the incubator and SYBR Green lysis buffer was added to each well using the Multi-Flo liquid dispenser. Plates were incubated for an additional 24 h at room temperature for optimal development of the fluorescence signal. Fluorescence intensity was read on an Envision Multimode Reader (PerkinElmer, MA).
Assessment of activity against liver-stage P. berghei was measured using the 48 h luminescence-based assay described by Swann et al. with minor modifications.47 Briefly, P. berghei-ANKA-GFP-Luc-SMCON (PbLuc) sporozoites were freshly dissected from infected Anopheles stephensi salivary glands (received from Dr. Ana Rodriguez, NYU School of Medicine), filtered twice through a 20 μm nylon net filter (Steriflip, Millipore, MA), counted in a hemocytometer, and adjusted to a final concentration of 200 sporozoites per 1 μL in the assay media (DMEM without Phenol Red (Life Technologies, CA), 5% FBS, and 5× Pen–Strep and glutamine (Life Technologies, CA)). Purified sporozoites, 1 × 103 sporozoites per well (5 μL), were dispensed using a 1 μL cassette in the Multi-Flo (BioTek, VT) to preseeded assay plates with HepG2 cells. After incubation at 37 °C for 48 h, the liver-stage growth were assessed by a bioluminescence measurement as follows: media was removed by spinning the inverted plates at 150 g for 30 s; 2 μL per well of BrightGlo (Promega, WI) for quantification of Pb-luc viability. Immediately after addition of the luminescence reagent, the luminescence was measured by the Envision Multilabel Reader (PerkinElmer, MA). Cytotoxicity against HEK293T cells was measured as previously described using mefloquine as a positive control.48 Malaria parasite assay data were analyzed in Genedata Screener (v13.0-Standard). In all cases, data were normalized to neutral controls minus inhibitors (mefloquine for asexual blood stage parasites, atovaquone for liver-stage parasites and puromycin for HepG2 cytotoxicity). Dose–response curves were fit with the Genedata Analyzer using the Smart Fit function and half maximal effective concentration (EC50) were determined.
Ecological Experiments
Antifungal Bioassay
Peyssonnoside A (1) was tested against the ecologically relevant saprophytic filamentous fungus D. salina.49 Fungal culture was grown at 24 °C in 790 By+ medium (1.0 g/L yeast extract, 1.0 g/L peptone, 5.0 g/L d-(+)-glucose, in 1 L of seawater). A tissue grinder was used to homogenize fungal hyphae before distributing D. salina in 790 By+ media to a sterile 96-well plate. The antifungal assay was performed with a 1:1 serial dilution of concentrations ranging from 0.94 to 0.0070 μM for 1, which is about three times lower than the natural concentration of 1 in Peyssonnelia sp., and 0.14–0.0010 μM for the positive control nystatin. Percent growth inhibition at each concentration was calculated relative to fungus exposed to solvent (DMSO). Growth measurements were taken using a BioTek ELx800 Absorbance Microplate Reader at 600 nm. Images were recorded using a dissecting microscope to confirm that elevated OD readings corresponded to fungal growth. The growth inhibition data were fit to a sigmoidal dose–response curve using GraphPad and the IC50 of 1 was calculated after 3 days incubation with D. salina.
Omnivore Feeding Assay
Using a standard field assay,50 peyssonnoside A (1) was evaluated for feeding deterrence against one generalist marine omnivore, the hermit crab C. striolatus. This compound was found to be ineffective at preventing consumption of artificial food impregnated with pure 1 by C. striolatus collected from an intertidal zone off Ono Island in Fiji (amount eaten: control 45%, treatment 45%; n = 16, P = 0.99; Figure S5). Artificial food was prepared using a mixture of molten agar (0.02 g/mL) and Ulva sp. (0.1 g/mL), a green alga palatable to a wide range of marine herbivores, incorporated into window screen. Peyssonnoside A (1) was dissolved in 0.48 mL methanol and added to 10 mL of the artificial food, resulting in a final concentration of 1 mg/mL which is approximately 20% of the natural concentration of 1. The full natural concentration was not used because of constraints on the total amount of compound available for ecological studies. Controls were prepared by using an equivalent amount of solvent in the artificial food mix. After approximately 50% of the control or treated food available to any given hermit crab was consumed, the food-containing window screens were removed and analyzed for the total amount consumed of each treatment by counting the number of empty squares versus total that started with food, analyzing with a two-tailed t-test. Replicates where crabs ate less than 10% or more than 90% of the total food available to them were excluded. Of the 36 crabs housed individually for the assay, 16 produced useable results.
Computational Procedures
3D models for the two putative diastereomers of peyssonnoside A (1) (1′S,2′R,3′S,4′S,5′R,1S,3R,6S,7R,10S,11S,14S and 1′S,2′R,3′S,4′S,5′R,1R,3S,6R,7S,10R,11R,14R) were generated using Spartan 10.15 A conformational search was performed using molecular mechanics force fields with the Monte Carlo algorithm implemented in Spartan 10. Similarly, conformational search was also performed using Conflex 7 software with built-in MMFF94S force field.16 Taken together, 4860 and 2372 conformations were generated for 1′S,2′R,3′S,4′S,5′R,1S,3R,6S,7R,10S,11S,14S and 1′S,2′R,3′S,4′S,5′R,1R,3S,6R,7S,10R,11R,14R respectively, within 10 kcal/mol. The geometries of all conformers were sequentially optimized using the Gaussian 09 package at PM6,51 PM752 level followed by B3LYP53−55 functional using 3-21G56,57 and 6-31G* basis sets,17 each time selecting the conformers within a 10 kcal/mol window for the geometry optimizations at a next higher level of calculation. Despite the use of PM6 and PM7 for initial conformational optimization58 which are comparatively low level calculations, the number of conformations analyzed at B3LYP/3-21G were substantially high: 1410 conformers for 1′S,2′R,3′S,4′S,5′R,1S,3R,6S,7R,10S,11S,14S, and 583 conformations for 1′S,2′R,3′S,4′S,5′R,1R,3S,6R,7S,10R,11R,14R. Boltzmann population distribution was obtained for the Gaussian optimized conformers mentioned above. Conformers with population greater than 1% at B3LYP/6-31G* level were subjected to further geometry optimization using B3LYP/6-311++G (2d,2p) level of theory (polarization continuum model with DMSO as solvent).17 Calculated frequencies were used to confirm that the optimized geometries were true minima (no imaginary frequency). The resulting geometries along with their respective Boltzmann population were used to calculate the weighted distance between protons of interest for comparison with interproton distances obtained from the ROESY spectrum, to calculate VCD, specific optical rotation and NMR shielding tensors using the GIAO method.59 The calculated 1H and 13C NMR shielding tensors were scaled against experimental 1H and 13C NMR chemical shift values.60
Experimental Interproton Distances from the 2D ROESY NMR Spectrum for Peyssonnoside A (1)
The 2D ROESY NMR spectrum for 1 was acquired using a spin lock of 200 ms and a relaxation delay of 10 s. Interproton distances were obtained from a 2D ROESY experiment using eq 1 mentioned in the main text where average of the cross-peak intensities between methylene protons H2-2 (δH = 0.85, 2.16 ppm) and H2-5 (δH = 1.15, 1.96 ppm) were used as aref. PANIC was used where the irradiated peak in each slice of the 2D ROESY NMR spectrum was set to 20 000 units (Figures S29–S37).
Interproton Distances for Lowest Energy Conformations Obtained from DFT Calculations for Peyssonnoside A (1)
Interproton distances for each conformer were weighted using the equation
where ri represents the interproton distance of interest in conformer i, and pi is the relative Boltzmann population of conformer i. Tropps equation was used to account for protons in a methyl group using r–3 averaging.20,22
Acknowledgments
This work was supported by International Cooperative Biodiversity Groups (ICBG) Grant U19-TW007401 from the U.S. National Institutes of Health and the Bill & Melinda Gates Foundation, Seattle, WA Grant OPP1107194. We thank the governments of the Solomon Islands and Fiji for allowing us to perform research in their territorial waters. M. E. Hay, S. Lavoie, J. Kelly, and C. Dell performed collections and photography of algal samples; L. T. Gelbaum provided NMR assistance; D. E. Bostwick and D. Gaul shared expertise for high resolution mass spectrometric analyses; S. Karunakaran helped with acquisition of ORD data; A. Burns and J. Mathew-Valayil performed selected antimicrobial assays; and S. G. Franzblau contributed data from a tuberculosis assay. Enterococcus faecium, strain 513, HM-959 was provided by BEI Resources, NIAID, NIH as part of the Human Microbiome Project.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.9b00884.
Species identification, isolation, pharmacological assays, ecological experiments, computational procedures, 1D, 2D NMR, HR/MS, and VCD data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Peters R. J. Two rings in them all: the labdane-related diterpenoids. Nat. Prod. Rep. 2010, 27, 1521–1530. 10.1039/c0np00019a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrué F.; McCulloch M. W. B.; Kerr R. G. Marine diterpene glycosides. Bioorg. Med. Chem. 2011, 19, 6702–6719. 10.1016/j.bmc.2011.06.083. [DOI] [PubMed] [Google Scholar]
- Le Bideau F.; Kousara M.; Chen L.; Wei L.; Dumas F. Tricyclic sesquiterpenes from marine origin. Chem. Rev. 2017, 117, 6110–6159. 10.1021/acs.chemrev.6b00502. [DOI] [PubMed] [Google Scholar]
- Fan Y.-Y.; Gao X.-H.; Yue J.-M. Attractive natural products with strained cyclopropane and/or cyclobutane ring systems. Sci. China: Chem. 2016, 59, 1126–1141. 10.1007/s11426-016-0233-1. [DOI] [Google Scholar]
- Ebner C.; Carreira E. M. Cyclopropanation strategies in recent total syntheses. Chem. Rev. 2017, 117, 11651–11679. 10.1021/acschemrev.6b00798. [DOI] [PubMed] [Google Scholar]
- Comprehensive Chiroptical Spectroscopy: Applications in Stereochemical Analysis of Synthetic Compounds, Natural Products, and Biomolecules; Berova N., Polavarapu P. L., Nakanishi K., Woody R. W., Eds.; Wiley: Hoboken, NJ, 2012; Vol. 2. [Google Scholar]
- Comprehensive Chiroptical Spectroscopy: Instrumentation, Methodologies, and Theoretical Simulations; Berova N., Polavarapu P. L., Nakanishi K., Woody R. W., Eds.; Wiley: Hoboken, NJ, 2012; Vol. 1. [Google Scholar]
- Seco J. M.; Quiñoá E.; Riguera R. The Assignment of Absolute Configuration by NMR. Chem. Rev. 2004, 104, 17–118. 10.1021/cr000665j. [DOI] [PubMed] [Google Scholar]
- Laskowski T.; Szwarc K.; Szczeblewski P.; Sowiński P.; Borowski E.; Pawlak J. Monosaccharides as potential chiral probes for the determination of the absolute configuration of secondary alcohols. J. Nat. Prod. 2016, 79, 2797–2804. 10.1021/acs.jnatprod.6b00471. [DOI] [PubMed] [Google Scholar]
- Butts C. P.; Jones C. R.; Song Z.; Simpson T. J. Accurate NOE-distance determination enables the stereochemical assignment of a flexible molecule - arugosin C. Chem. Commun. 2012, 48, 9023–9025. 10.1039/c2cc32144k. [DOI] [PubMed] [Google Scholar]
- Chhetri B. K.; Lavoie S.; Sweeney-Jones A. M.; Kubanek J. Recent trends in the structural revision of natural products. Nat. Prod. Rep. 2018, 35, 514–531. 10.1039/c8np00011e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peat S.; Bowker D. M.; Turvey J. R. Sulphates of monosaccharides and derivatives. Carbohydr. Res. 1968, 7, 225–231. 10.1016/s0008-6215(00)81196-9. [DOI] [Google Scholar]
- Dalisay D. S.; Morinaka B. I.; Skepper C. K.; Molinski T. F. A Tetrachloro Polyketide Hexahydro-1H-isoindolone, Muironolide A, from the Marine Sponge Phorbas sp. Natural Products at the Nanomole Scale. J. Am. Chem. Soc. 2009, 131, 7552–7553. 10.1021/ja9024929. [DOI] [PubMed] [Google Scholar]
- Mnova Structure Elucidation, Mnova 12.0.4, Mestrelab Research, Santiago de Compostela, Spain, www.mestrelab.com, 2019.
- Spartan′10; Wavefunction, Inc., Irvine, CA.
- Conflex , High Performance Conformational Analysis, www.conflex.net.
- Hehre W. J.; Radom L.; Schleyer P. v. R.; Pople J. A.. Ab Initio Molecular Orbital Theory; John Wiley: New York, 1986. [Google Scholar]
- Barone V.; Cossi M.; Tomasi J. Geometry optimization of molecular structures in solution by the polarizable continuum model. J. Comput. Chem. 1998, 19, 404–417. . [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Keith T.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas O.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford CT, 2013.
- Chini M. G.; Jones C. R.; Zampella A.; D’Auria M. V.; Renga B.; Fiorucci S.; Butts C. P.; Bifulco G. Quantitative NMR-Derived Interproton Distances Combined with Quantum Mechanical Calculations of 13C Chemical Shifts in the Stereochemical Determination of Conicasterol F, a Nuclear Receptor Ligand from Theonella swinhoei. J. Org. Chem. 2012, 77, 1489–1496. 10.1021/jo2023763. [DOI] [PubMed] [Google Scholar]
- Butts C. P.; Jones C. R.; Towers E. C.; Flynn J. L.; Appleby L.; Barron N. J. Interproton distance determinations by NOE - surprising accuracy and precision in a rigid organic molecule. Org. Biomol. Chem. 2011, 9, 177–184. 10.1039/c0ob00479k. [DOI] [PubMed] [Google Scholar]
- Neuhaus D.; Williamson M. P.. The Nuclear Overhauser Effect in Structural and Conformational Analysis, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 2000. [Google Scholar]
- Constantino M. G.; Lacerda V. Jr; da Silva G. V. J.; Tasic L.; Rittner R. Principal component analysis of long-range ‘W’ coupling constants of some cyclic compounds. J. Mol. Struct. 2001, 597, 129–136. 10.1016/s0022-2860(01)00591-9. [DOI] [Google Scholar]
- Cooper M. A.; Manatt S. L. Effect of steric compression on proton-proton, spin-spin coupling constants. Further evidence and mechanistic considerations. J. Am. Chem. Soc. 1970, 92, 4646–4652. 10.1021/ja00718a029. [DOI] [Google Scholar]
- Wang J.; Tong R. A NMR method for relative stereochemical assignments of the tricyclic core of cephalosporolides, penisporolides and related synthetic analogues. Org. Chem. Front. 2017, 4, 140–146. 10.1039/c6qo00556j. [DOI] [Google Scholar]
- Grimblat N.; Zanardi M. M.; Sarotti A. M. Beyond DP4: an Improved Probability for the Stereochemical Assignment of Isomeric Compounds using Quantum Chemical Calculations of NMR shifts. J. Org. Chem. 2015, 80, 12526–12534. 10.1021/acs.joc.5b02396. [DOI] [PubMed] [Google Scholar]
- da Silva C. O.; Mennucci B.; Vreven T. Density functional study of the optical rotation of glucose in aqueous solution. J. Org. Chem. 2004, 69, 8161–8164. 10.1021/jo049147p. [DOI] [PubMed] [Google Scholar]
- McPhail K. L.; France D.; Cornell-Kennon S.; Gerwick W. H. Peyssonenynes A and B, Novel Enediyne Oxylipins with DNA Methyl Transferase Inhibitory Activity from the Red Marine Alga Peyssonnelia caulifera. J. Nat. Prod. 2004, 67, 1010–1013. 10.1021/np0400252. [DOI] [PubMed] [Google Scholar]
- Lane A. L.; Mular L.; Drenkard E. J.; Shearer T. L.; Engel S.; Fredericq S.; Fairchild C. R.; Prudhomme J.; Le Roch K.; Hay M. E.; Aalbersberg W.; Kubanek J. Ecological leads for natural product discovery: Novel sesquiterpene hydroquinones from the red macroalga Peyssonnelia sp. Tetrahedron 2010, 66, 455–461. 10.1016/j.tet.2009.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A.-S.; Engel S.; Smith B. A.; Fairchild C. R.; Aalbersberg W.; Hay M. E.; Kubanek J. Structure and biological evaluation of novel cytotoxic sterol glycosides from the marine red alga Peyssonnelia sp. Bioorg. Med. Chem. 2010, 18, 8264–8269. 10.1016/j.bmc.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan M. M.; Wang Q.; Webster F. X.; Kiemle D.; Hong Y. J.; Tantillo D. J.; Coates R. M.; Wray A. T.; Askew W.; O’Donnell C.; Tokuhisa J. G.; Tholl D. Formation of the unusual semivolatile diterpene rhizathalene by the Arabidopsis class I terpene synthase TPS08 in the root stele is involved in defense against belowground herbivory. Plant Cell 2013, 25, 1108–1125. 10.1105/tpc.112.100057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantillo D. J. Recent excursions to the borderlands between the realms of concerted and stepwise: carbocation cascades in natural products biosynthesis. J. Phys. Org. Chem. 2008, 21, 561–570. 10.1002/poc.1320. [DOI] [Google Scholar]
- Tantillo D. J. The carbocation continuum in terpene biosynthesis-where are the secondary cations?. Chem. Soc. Rev. 2010, 39, 2847–2854. 10.1039/b917107j. [DOI] [PubMed] [Google Scholar]
- Tantillo D. J. Biosynthesis via carbocations: theoretical studies on terpene formation. Nat. Prod. Rep. 2011, 28, 1035–1053. 10.1039/c1np00006c. [DOI] [PubMed] [Google Scholar]
- Marcos I. S.; Moro R. F.; Gil-Mesón A.; Díez D. In Studies in Natural Products Chemistry; Rahman A. U., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; Vol. 48, pp 137–207. [Google Scholar]
- Littler D. S.; Littler M. M.. South Pacific Reef Plants: A Divers’ Guide to the Plant Life of South Pacific Coral Reefs; Offshore Graphic, Inc.: Washington, D.C., 2003. [Google Scholar]
- Harper J. T.; Saunders G. W. The application of sequences of the ribosomal cistron to the systematics and classification of the florideophyte red algae (Florideophyceae, Rhodophyta). Cah. Biol. Mar. 2001, 42, 25–38. 10.21411/CBM.A.ADC850A3. [DOI] [Google Scholar]
- Huang X.; Madan A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.; Gish W.; Miller W.; Myers E. W.; Lipman D. J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. 10.1006/jmbi.1990.9999. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Stecher G.; Li M.; Knyaz C.; Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. 10.1007/bf01731581. [DOI] [PubMed] [Google Scholar]
- Collins L.; Franzblau S. G. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob. Agents Chemother. 1997, 41, 1004–1009. 10.1128/aac.41.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubanek J.; Prusak A. C.; Snell T. W.; Giese R. A.; Hardcastle K. I.; Fairchild C. R.; Aalbersberg W.; Raventos-Suarez C.; Hay M. E. Antineoplastic Diterpene–Benzoate Macrolides from the Fijian Red Alga Callophycus serratus. Org. Lett. 2005, 7, 5261–5264. 10.1021/ol052121f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLSI . Performance Standards for Antimicrobial Susceptibility Testing; Twenty-Third Informational Supplement. CLSI Document M100-S23; Clinical and Laboratory Standards Institute: Wayne, PA, 2013. [Google Scholar]
- Quave C. L.; Lyles J. T.; Kavanaugh J. S.; Nelson K.; Parlet C. P.; Crosby H. A.; Heilmann K. P.; Horswill A. R. Castanea sativa (European Chestnut) leaf extracts rich in ursene and oleanene derivatives block Staphylococcus aureus virulence and pathogenesis without detectable resistance. PLoS One 2015, 10, e0136486 10.1371/journal.pone.0136486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plouffe D.; Brinker A.; McNamara C.; Henson K.; Kato N.; Kuhen K.; Nagle A.; Adrián F.; Matzen J. T.; Anderson P.; Nam T.-g.; Gray N. S.; Chatterjee A.; Janes J.; Yan S. F.; Trager R.; Caldwell J. S.; Schultz P. G.; Zhou Y.; Winzeler E. A. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 9059–9064. 10.1073/pnas.0802982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swann J.; Corey V.; Scherer C. A.; Kato N.; Comer E.; Maetani M.; Antonova-Koch Y.; Reimer C.; Gagaring K.; Ibanez M.; Plouffe D.; Zeeman A.-M.; Kocken C. H. M.; McNamara C. W.; Schreiber S. L.; Campo B.; Winzeler E. A.; Meister S. High-throughput luciferase-based assay for the discovery of therapeutics that prevent malaria. ACS Infect. Dis. 2016, 2, 281–293. 10.1021/acsinfecdis.5b00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M. S.; Beasley F. C.; Jumani R. S.; Wright T. M.; Chatterjee A. K.; Huston C. D.; Schultz P. G.; McNamara C. W. A high-throughput phenotypic screen identifies clofazimine as a potential treatment for cryptosporidiosis. PLoS Neglected Trop. Dis. 2017, 11, e0005373 10.1371/journal.pntd.0005373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmeyer J.; Kohlmeyer E.. Marine Mycology: The Higher Fungi: Academic Press; New York, 1979. [Google Scholar]
- Hay M. E.; Kappel Q. E.; Fenical W. Synergisms in plant defenses against herbivores: interactions of chemistry, calcification, and plant quality. Ecology 1994, 75, 1714–1726. 10.2307/1939631. [DOI] [Google Scholar]
- Stewart J. J. P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. 10.1007/s00894-007-0233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart J. J. P. Optimization of parameters for semiempirical methods VI: more modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. 10.1007/s00894-012-1667-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- Vosko S. H.; Wilk L.; Nusair M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can. J. Phys. 1980, 58, 1200–1211. 10.1139/p80-159. [DOI] [Google Scholar]
- Pietro W. J.; Francl M. M.; Hehre W. J.; DeFrees D. J.; Pople J. A.; Binkley J. S. Self-consistent molecular orbital methods. 24. Supplemented small split-valence basis sets for second-row elements. J. Am. Chem. Soc. 1982, 104, 5039–5048. 10.1021/ja00383a007. [DOI] [Google Scholar]
- Dobbs K. D.; Hehre W. J. Molecular orbital theory of the properties of inorganic and organometallic compounds. 6. Extended basis sets for second-row transition metals. J. Comput. Chem. 1987, 8, 880–893. 10.1002/jcc.540080615. [DOI] [Google Scholar]
- Raghavan V.; Johnson J. L.; Stec D. F.; Song B.; Zajac G.; Baranska M.; Harris C. M.; Schley N. D.; Polavarapu P. L.; Harris T. M. Absolute Configurations of Naturally Occurring [5]- and [3]-Ladderanoic Acids: Isolation, Chiroptical Spectroscopy, and Crystallography. J. Nat. Prod. 2018, 81, 2654–2666. 10.1021/acs.jnatprod.8b00458. [DOI] [PubMed] [Google Scholar]
- Cheeseman J. R.; Trucks G. W.; Keith T. A.; Frisch M. J. A comparison of models for calculating nuclear magnetic resonance shielding tensors. J. Chem. Phys. 1996, 104, 5497–5509. 10.1063/1.471789. [DOI] [Google Scholar]
- Lavoie S.; Sweeney-Jones A. M.; Mojib N.; Dale B.; Gagaring K.; McNamara C. W.; Quave C. L.; Soapi K.; Kubanek J. Antibacterial Oligomeric Polyphenols from the Green Alga Cladophora socialis. J. Org. Chem. 2019, 84, 5035–5045. 10.1021/acs.joc.8b03218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.