

Abstract
Cytosolic sulfotransferases (SULTs), including SULT1A, SULT1B, SULT1E, and SULT2A isoforms, play noteworthy roles in xenobiotic and endobiotic metabolism. We quantified the protein abundances of SULT1A1, SULT1A3, SULT1B1, and SULT2A1 in human liver cytosol samples (n = 194) by liquid chromatography–tandem mass spectrometry proteomics. The data were analyzed for their associations by age, sex, genotype, and ethnicity of the donors. SULT1A1, SULT1B1, and SULT2A1 showed significant age-dependent protein abundance, whereas SULT1A3 was invariable across 0–70 years. The respective mean abundances of SULT1A1, SULT1B1, and SULT2A1 in neonatal samples was 24%, 19%, and 38% of the adult levels. Interestingly, unlike UDP-glucuronosyltransferases and cytochrome P450 enzymes, SULT1A1 and SULT2A1 showed the highest abundance during early childhood (1 to <6 years), which gradually decreased by approx. 40% in adolescents and adults. SULT1A3 and SULT1B1 abundances were significantly lower in African Americans compared with Caucasians. Multiple linear regression analysis further confirmed the association of SULT abundances by age, ethnicity, and genotype. To demonstrate clinical application of the characteristic SULT ontogeny profiles, we developed and validated a proteomics-informed physiologically based pharmacokinetic model of acetaminophen. The latter confirmed the higher fractional contribution of sulfation over glucuronidation in the metabolism of acetaminophen in children. The study thus highlights that the ontogeny-based age-dependent fractional contribution (fm) of individual drug-metabolizing enzymes has better potential in prediction of drug-drug interactions and the effect of genetic polymorphisms in the pediatric population.
Introduction
The human cytosolic sulfotransferases (SULTs) are important phase II drug-metabolizing enzymes (DMEs) that catalyze sulfate conjugation by transferring a sulfonate (SO3) group from 3′-phosphoadenosine-5′-phosphosulfate to the hydroxyl or amino group of xenobiotic or endobiotic substrates. Several SULT isoforms, i.e., SULT1A1, SULT1A3, SULT1B1, SULT1E1, and SULT2A1, play important roles in the metabolism of drugs, environmental toxins, and endogenous steroids. For example, SULT1A1 is involved in the biotransformation of acetaminophen, minoxidil, 4-hydroxytamoxifen, oxymorphone, nalbuphine, nalorphine, naltrexone, isoflavones, estradiol, and iodothyronines (Coughtrie et al., 1994; Nishiyama et al., 2002; Nowell and Falany, 2006; Kurogi et al., 2014; Marto et al., 2017). Likewise, SULT1A3 is known to metabolize catecholamines, serotonin, salbutamol, ritodrine, and troglitazone (Eisenhofer et al., 1999; Honma et al., 2002; Hui and Liu, 2015; Bairam et al., 2018); SULT1B1 plays a role in elimination of iodothyronines, thyroxine, and 1-naphthol (Fujita et al., 1997; Wang et al., 1998; Gamage et al., 2006); SULT1E1 metabolizes raloxifene and estrogens (Falany et al., 1995; Schrag et al., 2004, 2006; Cubitt et al., 2011); and SULT2A1 assists in metabolism of ciprofloxacin, desipramine, metoclopramide, dehydroepiandrosterone (DHEA), several bile acids, and 25-hydroxyvitamin D3 (Falany et al., 1994; Meloche et al., 2002, 2004; Cook et al., 2009; Nakamura et al., 2009; Senggunprai et al., 2009; Huang et al., 2010; Wong et al., 2018). Because several of these substrates are relevant to children, it is important to characterize age-dependent abundances of these enzymes.
Unlike cytochrome P450 enzymes (CYPs), phase II drug-metabolism pathways are not well characterized for age-dependent activity and expression owing to the nonavailability of probe substrates, specific inhibitors, and antibodies. Recently, we performed selective quantitative proteomics analysis of UDP-glucuronosyltransferases (UGTs) in human liver samples from 137 pediatric and 37 adult samples, in which we observed distinct patterns of ontogeny for various UGTs (Bhatt et al., 2018, 2019). For example, UGT2B17 expression was rarely observed in children of age <9 years, whereas it sharply increased during teenage years. We also observed that UGT1A1 and UGT2B15 were the major neonatal UGTs, whereas UGT1A4 and UGT2B7 were the major adult isoforms. These ontogeny data were used by us to explain age-dependent pharmacokinetics (PK) of UGT substrates in children (Bhatt et al., 2019).
Some reports in literature indicate that the activity of SULTs is higher than that of UGTs in children, and the phenomenon reverses in adults. For example, acetaminophen glucuronide–to–sulfate metabolite ratio is reported to increase from 0.34 in newborns to 0.75 in children 3–9 years of age, compared with 1.80 in adults (Miller et al., 1976; Behm et al., 2003).
Such nonmonotonic development profiles of DMEs pose a challenge for predicting the fractional metabolism by individual enzymes (fm) for a given population, e.g., children versus adults. The parameter fm indicates clinical significance of a drug-metabolism pathway, i.e., a drug with high fm for a particular DME can display more drug-drug interactions (DDIs) and more pronounced in vivo variability owing to genetic polymorphism (Salem et al., 2013; Prasad and Unadkat, 2015; Umehara et al., 2017). Because fm is proportional to the relative abundances of DMEs, variable developmental trajectories for individual enzymes may lead to differential fm with age, eventually resulting in differential metabolic pathways.
Interestingly, several drugs are metabolized by CYPs, UGTs, and SULTs. However, data are sparse on SULT activity in children, and the age-dependent abundance of individual SULT isoforms is not well characterized. Our present study was targeted to fill this important knowledge gap by investigating protein abundances of SULT1A1, SULT1A3, SULT1B1, and SULT2A1 by a robust liquid chromatography–tandem mass spectrometry (LC-MS/MS) proteomics methodology (Bhatt and Prasad, 2018). We made use of cytosolic fractions prepared from the same human livers for which UGT ontogeny data had been reported by us earlier (Bhatt et al., 2019).
To demonstrate additionally the utility of the SULT ontogeny data generated in this study, we developed a proteomics-informed physiologically based pharmacokinetic (PBPK) model of acetaminophen for predicting age-dependent metabolic switching in its elimination. In adult human liver, acetaminophen is mainly metabolized by conjugation through glucuronidation (52%–57% by UGT1A1, UGT1A6, UGT1A9, and UGT2B15), with sulfation next in importance (30%–44% by SULT1A1, SULT1A3, SULT1E1, and SULT2A1), and a minor contribution by oxidation (5%–10% by CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4) (Prescott, 1983; Clements et al., 1984; Critchley et al., 1986, 2005). A PBPK model was reported in the literature to describe acetaminophen PK in children, including neonates and infants (Jiang et al., 2013). However, because selective ontogeny data were not available for the UGTs and SULTs, the reported model used few assumptions regarding DME ontogeny. To address this knowledge deficit, we used the ontogeny data of UGTs (Bhatt et al., 2019), SULTs (described here), and CYPs (unpublished data) to develop and validate a refined acetaminophen pediatric PBPK model.
Materials and Methods
Chemicals and Reagents.
Iodoacetamide, dithiothreitol, mass spectrometry-grade trypsin, bovine serum albumin, and synthetic heavy labeled peptides were purchased from Thermo Fisher Scientific (Rockford, IL). Purified SULT1A1 and SULT2A1 protein standards were procured from Abnova (Walnut, CA). Chloroform, ethyl ether, MS-grade acetonitrile (99.9% purity), methanol (>99.5% purity), formic acid (≥99.5% purity), and ammonium bicarbonate (98% purity) were purchased from Fischer Scientific (Fair Lawn, NJ).
Human Liver Cytosol Samples.
One hundred, ninety-four human liver samples (137 pediatric and 57 adults), a majority of which were previously used by our group to determine abundances of UGTs (Bhatt et al., 2019), carboxylesterases (Boberg et al., 2017), and aldehyde and alcohol dehydrogenases (Bhatt et al., 2017), were used in this study. Detailed donor demographic information on these samples was reported in the aforementioned studies. Of the 137 pediatric liver samples, 129 were provided by Children’s Mercy Kansas City (Kansas City, MO), which were originally procured from various sources, including the National Institute of Child Health and Human Development (NICHD) Brain and Tissue Bank for Developmental Disorders at the University of Maryland (Baltimore, MD); Liver Tissue Cell Distribution System at the University of Minnesota (Minneapolis, MN); University of Pittsburgh (Pittsburgh, PA); Vitron (Tucson, AZ); and XenoTech LLC (Lenexa, KS). The remaining 8 pediatric and 57 adult human liver samples were obtained from the University of Washington School of Pharmacy (Seattle, WA) liver bank. The use of these samples was approved and determined as nonhuman subject research by the institutional review boards of the Children’s Mercy Kansas City and the University of Washington. Information regarding the procurement and storage of these liver samples is described in previous reports (Prasad et al., 2016; Shirasaka et al., 2016; Boberg et al., 2017). The samples were categorized into following groups based on age, sex, and ethnicity: 1) Age: neonatal (0–27 days; n = 4), infancy (28–364 days; n = 17), toddler/early childhood (1 to <6 years; n = 30), middle childhood (6 to <12 years; n = 38), adolescence (12–18 years; n = 48), and adulthood (>18 years; n = 57); 2) Sex: male (n = 116), female (n = 76), and unknown (n = 2); and 3) Ethnicity: Caucasian (n = 123), African American (n = 29), Hispanic (n = 4), Native American (n = 1), Pacific Islander (n = 1), Asian (n = 1), and unknown (n = 35).
DNA Isolation and Genotype and Copy Number Variation Analyses.
Genomic DNA was isolated from liver tissues according to established protocols. Genotyping was performed using PGRN-SeqV1 (Gordon et al., 2016) or the DMET Plus Array, as stated in the manufacturer’s protocol (Affymetrix, Santa Clara, CA). SULT gene copy number variation (CNV) was determined using a quantitative multiplex polymerase chain reaction assay described previously (Gaedigk et al., 2012) for the samples provided by the Children’s Mercy Kansas City.
Protein Extraction, Trypsin Digestion, and Sample Preparation.
Human liver cytosol (HLC) fractions were isolated from liver tissues by a differential centrifugation method previously described (Pearce et al., 2016). Total cytosolic protein concentration was determined using the bicinchoninic acid protein assay kit. Three HLC aliquots (approx. 2 mg/ml) were prepared, separately digested, processed, and analyzed by LC-MS/MS by a previously reported protocol (Bhatt et al., 2017), which is detailed in the Supplemental Methodology. Surrogate peptides of SULT proteins were selected according to an optimized approach (Vrana et al., 2017).
LC-MS/MS Instrument and Quantitative Analysis.
The LC-MS system consisted of an Acquity UPLC (Waters Technologies, Milford, MA) coupled to Sciex Triple Quadrupole 6500 MS system (Framingham, MA). The mass spectrometer was operated in multiple reaction monitoring mode using positive ion electrospray ionization for targeted peptide analysis. Peak integration and quantification were performed using Skyline software (University of Washington).
The peptides were separated employing Waters Acquity UPLC column (HSS T3, 100 × 2.1 mm, 1.8 μm). The mobile phase was run in a gradient mode, composition of which is described in Supplemental Table 1. Optimized mass instrument parameters for analysis of surrogate peptides of SULTs, along with information on peptide sequences, and their types, are given in Supplemental Table 2. Data analysis utilized a three-step normalization process (Bhatt and Prasad, 2018) to ensure technical robustness. The absolute abundances of SULT1A1 and SULT2A1 were determined with commercially available purified protein standards which were used as calibrators. However, owing to nonavailability of protein standards of SULT1A3 and SULT1B1, their quantification was relative and acquired by normalization to total protein.
Statistical Analysis of LC-MS/MS Proteomics Data.
The distribution of age, ethnicity, and sex-dependent protein expression data were subjected to normal distribution tests (Kolmogorov-Smirnov and Shapiro-Wilk) employing GraphPad Prism 5 software (San Diego, CA). Because the data for all studied proteins were not normally distributed, nonparametric tests were applied for the statistical analysis. For age- and genotype-dependent protein abundance data, analyses were performed using the Kruskal-Wallis test followed by Dunn’s multiple comparison test. The effect of sex- and ethnicity-dependent protein abundance was evaluated using the Mann-Whitney rank order U test (using GraphPad Prism software), considering P values of < 0.05 as statistically significant. Jonckheere-Terpstra (JT) test was used for the trend analysis, whereas principal component analysis (PCA) was used to evaluate robustness of sample handling and storage, and also to identify unique patterns in protein abundances, as was done in our earlier studies (Bhatt and Prasad, 2018). Multiple linear regression was performed to rule out confounding effects of multiple covariates (e.g., age vs. ethnicity) during data analysis. Protein-protein correlation of the studied SULT isoforms was analyzed by Spearman correlation. RStudio (version 1.0.136) was used for JT (clinfun package; jonckheere.test function), PCA (prcomp function and ggbiplot package; ggbiplot function), multiple linear regression (lm function), and Spearman correlation (PerformanceAnalytics package; chart.Correlation function) analysis. Wherever applicable, a nonlinear allosteric sigmoidal eq. 1 was used to fit the ontogeny data (Bhatt et al., 2019), as age and enzyme abundance relationship was not expected to be linear.
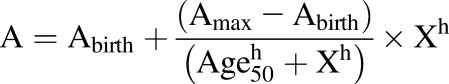 |
(1) |
where A is the enzyme abundance at age X; Abirth is the enzyme abundance at birth; Amax is the maximum average enzyme abundance; Age50 is the age in years at which 50% enzyme abundance is reached; X is the age in years; and h is the Hill coefficient.
Acetaminophen PBPK Model Development and Validation in Adults.
Acetaminophen PK data in the literature were mostly available either as concentration-time graphs or in the form of tables. The data from plasma concentration-time profiles were extracted with the GetData Graph Digitizer (http://www.getdata-graph-digitizer.com/index.php). Additional information, i.e., route of administration, dose strength, dosing regimen, and demographic details such as age and weight, was also collected. A whole-body PBPK model of acetaminophen for the adult population was developed using GastroPlus v9.6 (Simulations Plus Inc., Lancaster, CA, USA). For the purpose, reported values of adult plasma clearance after intravenous administration (CLIV) and steady-state volume of distribution (Vss) from existing PBPK model were used (Jiang et al., 2013). For adult physiology data, the Population Estimates for Age Related (PEAR) physiology module of GastroPlus was used and the considered parameters were for a healthy male, Caucasian, aged 30 years, and 70 kg body weight. Additionally, in vitro experimental enzyme kinetic, biochemical, and physicochemical data were collated from peer-reviewed articles (Chen et al., 1998; Mutlib et al., 2006; Adjei et al., 2008; Laine et al., 2009; Jiang et al., 2013; Villiger et al., 2016; Zurlinden and Reisfeld, 2016). The same are listed in Supplemental Table 3. The tissue partition coefficients were estimated using the default Lukacova method embedded in the PBPKPlus module, which considers all organs as perfusion limited tissues (Supplemental Table 3).
In vivo unbound total intrinsic hepatic clearance of acetaminophen (CLuint,H, l/h) was back-calculated using the well stirred model (Yang et al., 2007) as mentioned in eq. 2.
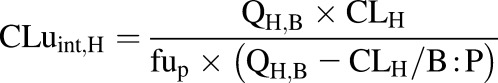 |
(2) |
The observed hepatic plasma clearance (CLH, 18.58 l/h) was obtained from CLiv (19.7 l/h) after subtracting renal plasma clearance (CLR, 1.12 l/h). It considered the hepatic blood flow (QH,B) as the default GastroPlus value of 84.36 l/h for 70 kg body weight, unbound fraction in plasma (fup) of 0.82, and blood-to-plasma drug concentration ratio (B:P) of 1.58 (Supplemental Table 3).
In vivo unbound intrinsic clearance for individual DME isoforms (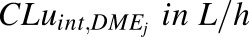 ) was calculated from eq. 3, which used fraction of acetaminophen metabolized by individual DME isoform (
) was calculated from eq. 3, which used fraction of acetaminophen metabolized by individual DME isoform (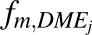 , such as
, such as 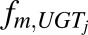 ,
,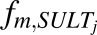 ,
,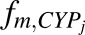 ), fraction of drug cleared through hepatic metabolism (
), fraction of drug cleared through hepatic metabolism (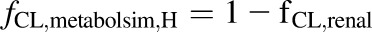 ), CLuint,H values (Supplemental Table 3) and fCL,renal or fe, which is the fraction of unchanged drug cleared renally.
), CLuint,H values (Supplemental Table 3) and fCL,renal or fe, which is the fraction of unchanged drug cleared renally.
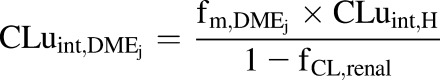 |
(3) |
In vitro intrinsic clearance of individual DME isoforms 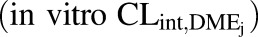 was back-calculated using eq. 4, with
was back-calculated using eq. 4, with 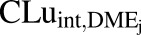 as a basis.
as a basis.
 |
(4) |
where MPPGL is milligrams of microsomal protein per gram of adult liver weight (default GastroPlus value of 38), CPPGL is milligrams of cytosolic protein per gram of adult liver weight (default GastroPlus value of 80), liver weight is in grams (default GastroPlus value of 1637.7), and a factor of 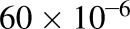 is for unit conversions.
is for unit conversions.
Thereafter, 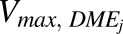 , which is maximum velocity of the kinetic reaction for individual DME isoforms (picomoles per minute per milligram protein) (i.e., UGT1A1, UGT1A9, UGT2B15, SULT1A1, SULT1A3, SULT1E1, SULT2A1, CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4) were calculated as the products of the in vitro
, which is maximum velocity of the kinetic reaction for individual DME isoforms (picomoles per minute per milligram protein) (i.e., UGT1A1, UGT1A9, UGT2B15, SULT1A1, SULT1A3, SULT1E1, SULT2A1, CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4) were calculated as the products of the in vitro 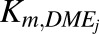 , which is Michaelis-Menten constant for individual DME isoforms (μM), unbound fraction in microsomes (fumic) (default GastroPlus value of 1), and in vitro
, which is Michaelis-Menten constant for individual DME isoforms (μM), unbound fraction in microsomes (fumic) (default GastroPlus value of 1), and in vitro 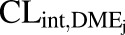 (microliters per minute per milligram protein) (values in Supplemental Table 3) in accordance with eq. 5.
(microliters per minute per milligram protein) (values in Supplemental Table 3) in accordance with eq. 5.
| (5) |
The model was applied to simulate the PK profile of various intravenous dosing regimens of acetaminophen. After validating the disposition model across different clinical data sets, an absorption model was established by integrating oral absorption parameters, such as permeability, solubility, diffusion coefficient, particle size, etc. using “Human-Fasted” gut physiology model of GastroPlus. On the basis of literature, the role of intestinal metabolism of acetaminophen was considered negligible (Clements et al., 1984). Likewise, oral PBPK models of acetaminophen were validated with available clinical data for different dosing regimens, i.e., oral doses of 500–2000 mg.
The predictive performance of the developed models was evaluated by comparing the simulated exposure parameters with literature-based clinical data, in accordance with the criteria suggested in the literature for comparison of area under the plasma concentration-time curve (AUC) and maximum plasma concentration (Cmax) (Abduljalil et al., 2014; Huang et al., 2017). The following criteria were considered: 1) bioequivalence criterion wherein the simulated AUC and Cmax values were required to be within 1.25-fold of the observed clinical data; 2) 2-fold criterion, which allows for a 0.5- to 2-fold variability between simulated and observed data, and 3) population-based criterion, wherein the basis of the fold change boundary is the corresponding observed values. In the latter, acceptance criteria were calculated with consideration of sample size (N) and percent coefficient of variation (%CV) (reported studies lacking N and %CV were excluded). The acceptance ranges of the mean Cmax and AUC were thence calculated by eqs. 6–8 (Abduljalil et al., 2014; Huang et al., 2017).
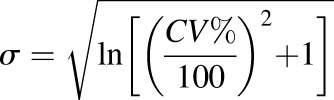 |
(6) |
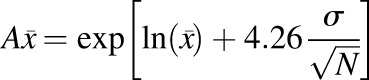 |
(7) |
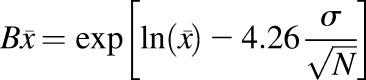 |
(8) |
wherein A and B are the upper and lower boundary limits for simulated data, respectively; 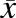 is the mean of Cmax or AUC of acetaminophen; and
is the mean of Cmax or AUC of acetaminophen; and  is the S.D. calculated from the %CV of the Cmax or AUC.
is the S.D. calculated from the %CV of the Cmax or AUC.
Once the PBPK model for acetaminophen was validated, PBPK models for its metabolites were developed using parameters described in Supplemental Tables 4 and 5. Because mechanistic elimination parameters (e.g., active efflux clearance) were not available for the metabolites, we assumed that they were eliminated unchanged in urine and the metabolite kinetics was formation-rate limited. Accordingly, the predicted total amount of metabolites eliminated in urine (Ae) was compared with the observed data.
Development of Pediatric Acetaminophen PBPK Model.
Following the development of adult acetaminophen PBPK model, we integrated ontogeny data (mean and 95% CI of protein abundance) of SULTs (from this study), UGTs (Bhatt et al., 2019), and CYPs (unpublished data) with pediatric physiologic parameters determined from the PEAR physiology module of GastroPlus software. The intention was to predict acetaminophen PK profile in the pediatric population. Besides major SULTs quantified in this study, published data of SULT1E1 (Duanmu et al., 2006) was also used for the model development. The pediatric population models were built for five age groups representing the mean of neonatal (14 days and 3.7 kg body weight), infancy (6 months and 8.23 kg body weight), early childhood (4 years and 17.34 kg body weight), middle childhood (9 years and 34.45 kg body weight), and adolescence (15 years and 63.69 kg body weight). Further, on the basis of the availability of clinical data, we also simulated data for three additional age-groups: infants (1 year and 10.23 kg body weight), children (7 years and 26.54 kg body weight), and adolescents (14 years and 58.74 kg body weight). Vss was estimated on the basis of the approach explained in Supplemental Table 3. fup was adjusted by the software to account for both age-related differences in plasma protein level as well as binding to plasma lipids.
Similar protein abundance values were considered for adult and pediatric samples by the software; however, to address the enzyme ontogeny, we adjusted 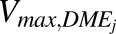 (picomoles per minute per milligram protein) values for individual enzymes on the basis of age-dependent protein abundances of SULTs, UGTs, and CYPs (Supplemental Table 3) using eq. 9. Adjusted Vmax,DMEj values were also used as an input in the Enzymes and Transporter module of GastroPlus.
(picomoles per minute per milligram protein) values for individual enzymes on the basis of age-dependent protein abundances of SULTs, UGTs, and CYPs (Supplemental Table 3) using eq. 9. Adjusted Vmax,DMEj values were also used as an input in the Enzymes and Transporter module of GastroPlus.
| (9) |
where, scale factor (SF) was generated for calculating the age-dependent abundances of each DME isoforms and MPPGL by eqs. 10 and 11, respectively. The resultant data are listed in Supplemental Table 6.
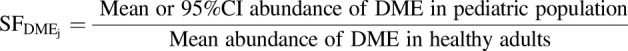 |
(10) |
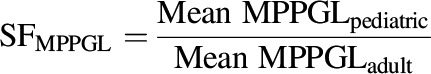 |
(11) |
Thus, age-dependent MPPGL values were integrated for CYPs and UGTs (Calvier et al., 2018). However, the CPPGL value was considered similar for adults and pediatrics as it was observed to be age-independent (unpublished data).
Further, a scaled 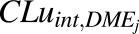 value was obtained through in vitro-in vivo extrapolation with eq. 12.
value was obtained through in vitro-in vivo extrapolation with eq. 12.
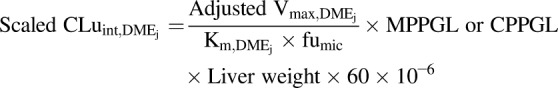 |
(12) |
Default GastroPlus liver weight values for each age group were input in the above-mentioned equation to account for the age-dependent change, viz., neonatal (123.44 g at 14 days), infancy (228.01 g at 6 months), infancy (325.11 g at 1 year), early childhood (592.12 g at 4 years), children (726.23 g at 7 years), middle childhood (906.24 g at 9 years), adolescence (1391.9 g at 14 years), and adolescence (1482.7 g at 15 years).
An age-dependent 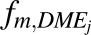 value was obtained from the DDI module of GastroPlus as well as from a scaled
value was obtained from the DDI module of GastroPlus as well as from a scaled 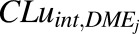 value as per eq. 13.
value as per eq. 13.
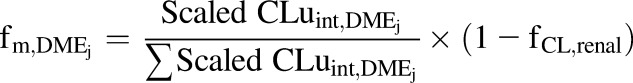 |
(13) |
Since fCL,renal or fe is age-independent (Miller et al., 1976), the fCL,renal value was considered to be constant (approx. 0.057) among all the age groups (Supplemental Table 3).
Cumulative fm,SULT, fm,UGT, and fm,CYP values were derived from the total of individual isoforms of SULT, UGT, and CYP, respectively, using eq. 14.
| (14) |
The scaled model was then used to predict acetaminophen PK in children and the predictions were compared with the reported observed data. The model was further extended to predict the dosing regimen in neonates and infants, which was validated by comparison with the dose adjustments recommended by the United States Food and Drug Administration (USFDA). Evaluation of the pediatric model was done by application of the same approach as discussed for the adult model. Also similar to the adults, prediction of Ae of the metabolites and unchanged drug was made for the pediatric groups.
Results
Abundances and Variability of Human SULT Enzymes.
The mean cytosolic protein abundances of SULT1A1, SULT1A3, SULT1B1, and SULT2A1 in the neonates (0–27 days) were 24%, 47%, 19%, and 38%, respectively, of the mean values for the adults (>18 years) (Fig. 1; Table 1). In the infants (28–364 days), the values for these SULT isoforms were 80%, 76%, 41%, and 111% of the adult levels, respectively. SULT1A1 protein abundance in early childhood (1 to <6 years) was approx.7-fold higher compared with the neonatal and approx. 2-fold higher than the adults. The association of categorical SULT1A3 abundance data with age was not significant (Fig. 1). SULT1B1 protein abundance in the adults was found to be approx. 5.4- and 2.4-fold higher than in the neonates (P value < 0.05) and the infants (P value < 0.0001), respectively. Likewise, SULT2A1 protein abundance in early childhood was approx. 4- and 2-fold higher compared with the neonatal and adolescence (12–18 years), respectively.
Fig. 1.

Age-dependent abundances (categorical) of SULT1A1 (A), SULT1A3 (B), SULT1B1 (C), and SULT2A1 (D) across six developmental periods, i.e., neonatal (0–27 days); infancy (28–364 days), toddler/early childhood (1 to <6 years), middle childhood (6 to <12 years), adolescence (12–18 years), and adulthood (>18 years). Statistical analysis for intercomparison of enzyme abundance across different age groups was performed using Kruskal-Wallis test followed by Dunn’s multiple comparison test. The number of samples in each age category is indicated in parentheses on the x-axis. Data for SULT1A1 and SULT2A1 represent absolute protein levels (determined using protein standard calibrator), whereas SULT1A3 and SULT1B1 data are presented as relative data (normalized by pooled quality control values). *, **, and ***P values < 0.05, <0.01, and <0.001, respectively.
TABLE 1.
Hepatic SULT protein abundance data with demographic details.
The number of samples in each category is indicated in parentheses. Median and mean values for SULT1A1 and SULT2A1 are expressed in picomoles per milligram of cytosol protein, whereas those for SULT1B1 and SULT1A3 are expressed in relative value (normalized by the pooled quality control values). Mean adult abundance value was taken as 1 to calculate SF for all the SULTs. Only SNPs with significant association with protein abundance are tabulated here (see also Supplemental Figs. 3 and 4 for SNPs and CNV, respectively.
| Median | Min | Max | Mean | S.D. | %CV | S.E. | SF | Lower 95% CI | Higher 95% CI | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SULT1A1 | All samples (190) | 387.0 | 27.1 | 1658.0 | 445.2 | 281.1 | 63.1 | 20.4 | NA | 404.9 | 485.4 |
| Neonatal (4) | 99.1 | 57.6 | 126.8 | 95.7 | 28.8 | 30.1 | 14.4 | 0.24 | 49.8 | 141.5 | |
| Infancy (17) | 189.2 | 52.2 | 824.0 | 323.1 | 254.1 | 78.6 | 61.6 | 0.80 | 192.4 | 453.7 | |
| Toddler/early childhood (29) | 610.3 | 113.5 | 1658.0 | 639.2 | 333.4 | 52.2 | 61.9 | 1.57 | 512.4 | 766.1 | |
| Middle childhood (37) | 512.0 | 30.8 | 1046.0 | 506.4 | 283.5 | 56.0 | 46.6 | 1.25 | 411.9 | 601.0 | |
| Adolescence (46) | 338.7 | 27.1 | 1153.0 | 397.7 | 306.3 | 77.0 | 45.2 | 0.98 | 306.7 | 488.7 | |
| Adulthood (57) | 370.9 | 147.6 | 1116.0 | 405.9 | 163.7 | 40.3 | 21.7 | 1.00 | 362.5 | 449.3 | |
| Female (73) | 394.4 | 30.8 | 1240.0 | 450.6 | 258.7 | 57.4 | 30.3 | NA | 390.2 | 510.9 | |
| Male (115) | 376.9 | 27.1 | 1658.0 | 442.8 | 297.6 | 67.2 | 27.8 | NA | 387.8 | 497.8 | |
| African American (29) | 313.2 | 78.6 | 792.3 | 348.7 | 233.9 | 67.1 | 43.4 | NA | 259.7 | 437.6 | |
| Caucasian (120) | 358.7 | 27.1 | 1116.0 | 386.3 | 231.4 | 59.9 | 21.1 | NA | 344.4 | 428.1 | |
| CNV 1 (5) | 219.2 | 121.4 | 376.4 | 245.1 | 108.2 | 44.1 | 48.4 | NA | 110.7 | 379.5 | |
| CNV 2 (82) | 387.0 | 27.1 | 1153.0 | 434.6 | 298.0 | 68.6 | 32.9 | NA | 369.2 | 500.1 | |
| CNV 3 (28) | 577.3 | 72.6 | 1240.0 | 534.4 | 347.6 | 65.0 | 65.7 | NA | 399.6 | 669.1 | |
| CNV 4 (7) | 506.4 | 146.8 | 1658.0 | 573.1 | 536.0 | 93.5 | 202.6 | NA | 77.3 | 1069.0 | |
| SULT1A3 | All samples (190) | 95.7 | 16.9 | 484.7 | 98.1 | 60.7 | 61.9 | 4.4 | NA | 89.4 | 106.8 |
| Neonatal (4) | 40.9 | 30.0 | 81.5 | 48.3 | 23.1 | 47.7 | 11.5 | 0.47 | 11.6 | 85.0 | |
| Infancy (17) | 40.9 | 27.2 | 256.8 | 77.8 | 75.2 | 96.6 | 18.2 | 0.76 | 39.2 | 116.5 | |
| Toddler/early childhood (29) | 109.2 | 25.7 | 330.2 | 116.2 | 74.2 | 63.9 | 13.8 | 1.14 | 87.9 | 144.4 | |
| Middle childhood (37) | 95.5 | 16.9 | 229.0 | 101.5 | 54.8 | 54.0 | 9.0 | 0.99 | 83.3 | 119.8 | |
| Adolescence (46) | 77.2 | 22.6 | 484.7 | 90.5 | 80.8 | 89.2 | 11.9 | 0.88 | 66.5 | 114.5 | |
| Adulthood (57) | 101.6 | 71.5 | 181.2 | 102.3 | 18.2 | 17.7 | 2.4 | 1.00 | 97.5 | 107.1 | |
| Female (75) | 97.8 | 16.9 | 330.2 | 98.2 | 53.0 | 54.0 | 6.1 | NA | 86.0 | 110.4 | |
| Male (113) | 92.9 | 22.6 | 484.7 | 98.3 | 66.0 | 67.1 | 6.2 | NA | 86.0 | 110.6 | |
| African American (28) | 39.3 | 23.6 | 128.5 | 55.6 | 32.8 | 59.0 | 6.2 | NA | 42.9 | 68.4 | |
| Caucasian (119) | 94.6 | 22.6 | 282.8 | 94.6 | 49.8 | 52.6 | 4.6 | NA | 85.5 | 103.6 | |
| SULT1B1 | All samples (191) | 99.9 | 16.7 | 364.7 | 109.1 | 65.6 | 60.1 | 4.7 | NA | 99.7 | 118.4 |
| Neonatal (3) | 19.2 | 16.7 | 28.2 | 21.4 | 6.0 | 28.3 | 3.5 | 0.19 | 6.3 | 36.4 | |
| Infancy (17) | 40.8 | 20.6 | 123.5 | 47.4 | 26.6 | 56.1 | 6.5 | 0.41 | 33.7 | 61.1 | |
| Toddler/early childhood (30) | 110.1 | 26.4 | 338.0 | 128.5 | 75.0 | 58.4 | 13.7 | 1.12 | 100.5 | 156.5 | |
| Middle childhood (37) | 117.8 | 19.4 | 269.1 | 114.9 | 57.2 | 49.8 | 9.4 | 1.00 | 95.8 | 134.0 | |
| Adolescence (47) | 90.9 | 19.8 | 364.7 | 113.2 | 85.2 | 75.3 | 12.4 | 0.99 | 88.2 | 138.3 | |
| Adulthood (57) | 114.7 | 43.9 | 222.2 | 114.6 | 39.5 | 34.4 | 5.2 | 1.00 | 104.1 | 125.1 | |
| Female (75) | 99.5 | 19.2 | 338.0 | 109.7 | 56.5 | 51.5 | 6.5 | NA | 96.7 | 122.7 | |
| Male (114) | 104.6 | 16.7 | 364.7 | 108.9 | 71.6 | 65.8 | 6.7 | NA | 95.6 | 122.2 | |
| African American (28) | 55.3 | 16.7 | 134.2 | 60.0 | 32.4 | 54.0 | 6.1 | NA | 47.4 | 72.5 | |
| Caucasian (121) | 98.7 | 19.2 | 231.3 | 102.2 | 53.6 | 52.5 | 4.9 | NA | 92.6 | 111.9 | |
| rs11249460_C/C (26)# | 122.3 | 68.7 | 222.2 | 127.6 | 41.6 | 32.6 | 8.2 | NA | 110.8 | 144.3 | |
| rs11249460_C/T (29)# | 108.4 | 43.9 | 186.1 | 103.3 | 33.8 | 32.7 | 6.3 | NA | 90.4 | 116.2 | |
| rs11249460_T/T (4)# | 99.7 | 67.6 | 123.9 | 97.7 | 23.2 | 23.8 | 11.6 | NA | 60.7 | 134.7 | |
| SULT2A1 | All samples (183) | 1065.0 | 80.0 | 4085.0 | 1290.0 | 829.0 | 64.3 | 61.3 | NA | 1169.0 | 1411.0 |
| Neonatal (4) | 453.3 | 158.4 | 617.0 | 420.5 | 193.8 | 46.1 | 96.9 | 0.38 | 112.1 | 728.8 | |
| Infancy (17) | 1012.0 | 80.0 | 4085.0 | 1249.0 | 987.3 | 79.0 | 239.5 | 1.11 | 741.3 | 1757.0 | |
| Toddler/early childhood (29) | 1885.0 | 160.4 | 3714.0 | 1832.0 | 1019.0 | 55.6 | 189.2 | 1.63 | 1444.0 | 2220.0 | |
| Middle childhood (37) | 1209.0 | 230.1 | 3949.0 | 1460.0 | 997.0 | 68.3 | 163.9 | 1.30 | 1128.0 | 1793.0 | |
| Adolescence (42) | 882.3 | 128.7 | 2885.0 | 1082.0 | 726.9 | 67.2 | 112.2 | 0.97 | 855.2 | 1308.0 | |
| Adulthood (54) | 1060.0 | 260.9 | 2071.0 | 1121.0 | 359.5 | 32.1 | 48.9 | 1.00 | 1023.0 | 1219.0 | |
| Female (74) | 1035.0 | 160.4 | 3949.0 | 1306.0 | 801.6 | 61.4 | 93.2 | NA | 1120.0 | 1492.0 | |
| Male (107) | 1080.0 | 80.0 | 4085.0 | 1284.0 | 857.7 | 66.8 | 82.9 | NA | 1120.0 | 1449.0 | |
| African American (28) | 853.5 | 80.0 | 3066.0 | 956.6 | 687.2 | 71.8 | 129.9 | NA | 690.1 | 1223.0 | |
| Caucasian (117) | 1036.0 | 95.6 | 4085.0 | 1185.0 | 747.9 | 63.1 | 69.1 | NA | 1048.0 | 1322.0 |
Min, minimum; Max, maximum; S.D., standard deviation; S.E., standard error; SF, scale factor; %CV, percent coefficient variation; 95% CI, 95% confidence interval; NA, not applicable.
The developmental trajectory of each SULT isoform was also assessed using age as a continuous variable (Supplemental Fig. 1). Data for both SULT1A3 and SULT1B1 could be adequately characterized by a nonlinear allosteric sigmoidal model justified by the fact that the age and enzyme-abundance relationship was not linear. The latter is consistent with the literature regarding ontogeny of DMEs (Johnson et al., 2006; Upreti and Wahlstrom, 2016; Boberg et al., 2017; Emoto et al., 2018). In both cases, the age50 value (the age at which protein expression reached 50% of the maximum adult abundance) was determined to be 0.91 years, i.e., approx. 11 months (Supplemental Fig. 1; Supplemental Table 7). The high biologic variability in SULT1A3 and SULT1B1 resulted in poor confidence in the age50 calculation, yet age-dependent abundances of these enzymes was supported by other statistical methods (Kruskal-Wallis test, multiple linear regression analysis, and JT trend analysis). However, this ontogeny model and statistical tests were not appropriate for SULT1A1 and SULT2A1 data, in which the developmental trajectories were characterized by protein abundance reaching maximum values in the toddler/early childhood group, and subsequently declining to the adult values.
Association of Ethnicity, Single-Nucleotide Polymorphisms, Copy Number Variations, and Sex with SULT Abundance.
The protein abundances of SULT1A3 and SULT1B1 was significantly higher in Caucasians compared with African Americans (P value < 0.0001) (Fig. 2; Table 1). But an age-ethnicity interplay made it difficult to draw a precise conclusion in the first instance. Therefore, multiple linear regression analysis was relied upon, as it considered the effect of age versus ethnicity independently. On the basis of the results, it could be concluded that ethnicity was indeed one of the key covariates in the abundances of SULT1A3 and SULT1B1. Additionally, the Mann-Whitney test for association of ethnicity with age groups <12 and ≥12 years also supported the conclusion drawn by the multiple linear regression analysis. A significant association (trend analysis; JT test) of SULT1B1 protein abundance with single-nucleotide polymorphism (SNP) rs11249460 (TT < CT < CC) was also observed. Detailed data analyses in this context are provided in Supplemental Figs. 2 and 3 and Supplemental Tables 8 and 9.
Fig. 2.

Association of ethnicity with human hepatic SULT1A1 (A), SULT1A3 (B), SULT1B1 (C), and SULT2A1 (D) protein levels. Statistical analysis for intercomparison of abundance between the two ethnic groups was performed through Mann-Whitney test. The number of samples in each ethnicity category is indicated in parentheses on the x-axis. ***P value < 0.001.
Also, trend (JT test) and multiple linear regression analyses confirmed significantly (P value = 0.042) higher median SULT1A1 abundance with increasing copy number (CN1 to CN4). No test revealed any relationship with ethnicity and sex in this case. The Mann-Whitney test indicated no association of ethnicity and SULT2A1 abundance, perhaps because of age-related variability. On the other hand, multiple linear regression analysis concluded that ethnicity was one of the key variables in the abundance of SULT2A1. Interestingly, the multiple linear regression analysis showed a modest, but significantly higher abundance of SULT2A1 in females than in males (P value < 0.05). No significant association of protein abundance was observed for other high-frequency SNPs, i.e., rs982861 (SULT1A1); rs11569731, rs11731028, rs11731028, rs1604741 (SULT1B1); and rs296365 (SULT2A1). The statistical results for this portion are summarized in Supplemental Figs. 4 and 5 and Supplemental Tables 8 and 9.
Protein-Protein Correlation.
This study, which was done with an objective to look for coregulation of SULT proteins, highlighted strong correlation between abundance levels of SULT1A1 and SULT2A1 (r2 = 0.60, P value < 0.001; Supplemental Fig. 6). Likewise, SULT1A3 showed correlation with SULT1B1 (r2 = 0.61 and P value < 0.05).
PCA Analysis of Proteomics Data.
The PCA analysis, which was done to evaluate robustness of sample handling and storage, and to identify unique patterns in protein abundances, highlighted that there was no degradation of samples before and during processing. As shown in our previous report (Bhatt and Prasad, 2018), data for degraded samples clustered distinctly toward the left lower side of PC1 vs. PC2 plots. In the present case, there was no clustering in the indicated region (Supplemental Fig. 7), suggesting that our sample quality was not compromised overall.
Regarding identification of unique patterns, the circled areas in the PCA plot (Supplemental Fig. 7) confirmed higher variability in the pediatric groups (0–18 years) for all SULT enzymes, compared with the adults.
Prediction of Age-Dependent Fractional Contribution of Sulfation over Glucuronidation in Acetaminophen Metabolism.
Considering that demonstration of the application of the ontogeny data was a key objective of this study, other population covariates were not considered for overall interindividual variability prediction (%CI), meaning that the predictions were made solely on the basis of mean and 95% CI data of protein abundance. As shown in Figs. 3 and 4 and Table 2, the observed versus predicted AUC and Cmax values of acetaminophen for both intravenous and oral administration in fasted- and fed-states were within the acceptance criteria. It is clearly evident from the data in Table 2 that 2-fold and population-based criteria yielded higher numbers of results within acceptable limits than the bioequivalence criterion. This confirmed that the latter was stringent in comparison, a reason why it is rarely used in PBPK modeling. Hence, our results relied only on 2-fold and population-based criteria. As 2-fold criterion is most commonly used for PBPK model validation, it was considered as the reference criterion for model acceptance in this study. But as it is also an arbitrary method and does not consider biologic variability in the data, additional analysis of predictive performance of the PBPK model was done using the population-based criterion.
Fig. 3.

Observed vs. predicted dose-normalized acetaminophen plasma concentration-time profiles for: intravenous infusion (2 hours) (A), oral solution-fasted state (B), oral tablet-fasted state (C), and oral tablet-fed state (D) dosing in adults. These profiles were generated by dividing the observed or predicted plasma concentrations by the dose. The gastric emptying time considered for the fed-state was 1 hour, whereas for the fasted state, oral tablet and oral solution, it was considered to be 15 and 6 minutes, respectively. The symbols represent observed data, and the solid lines indicate the model-predicted mean profile. The dotted and dashed lines represent the predicted profiles when lower and higher 95% CI of protein abundances of UGTs, SULTs, and CYPs (age >18 years) were considered for metabolism-related interindividual variability in adults. Abbreviations used in the illustration represent the following: L1 (5 mg/kg, predicted-fasted state); L2 [5 mg/kg (Clements et al., 1984)]; L3 [20 mg/kg (Clements et al., 1984)]; L4 (1000 mg, predicted-fasted state); L5 [1000 mg (Kamali et al., 1992)]; L6 [1000 mg (Prescott et al., 1989)]; L7 [12 mg/kg (Prescott, 1980)]; L8 (5 mg/kg, predicted-fasted state); L9 [500 mg (Rawlins et al., 1977)]; L10 [1000 mg (Rawlins et al., 1977)]; L11 [2000 mg (Rawlins et al., 1977)]; L12 [1000 mg (Singla et al., 2012)]; L13 [1000 mg (Zapater et al., 2004)); L14 [1000 mg (Rostami-Hodjegan et al., 2002)]; L15 (1000 mg, predicted-fed state), and L16 [1000 mg (Rostami-Hodjegan et al., 2002)].
Fig. 4.

Observed vs. predicted plasma concentration-time profiles of acetaminophen in neonates and infants for the mentioned doses. The drug was delivered as an intravenous infusion for 15 minutes in the case of all the figures (A, B, and D-F), except (C) in which the duration of infusion was 50 minutes. Also, separately indicated as bar diagrams is the comparison of mean predicted (without and with proteomics-based ontogeny data) and observed (L20) Cmax (G), and AUC0–6 (H) values for 15 mg/kg acetaminophen administered as intravenous infusion for 15 minutes to neonates and infants. In this case, the average age values for neonates and infants were considered as 14 days and 1 year, respectively, in accordance with US-FDA label (https://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/022450Orig1s000ClinPharmR.pdf). Abbreviations used in the illustration represent the following literature references: L17 (Zuppa et al., 2011), L18 (Cook et al., 2016), L19 (Allegaert et al., 2013), and L20 (https://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/022450Orig1s000ClinPharmR.pdf).
TABLE 2.
Comparison of acetaminophen PBPK model predicted and observed PK data
Clinical studies were used as the training and validation sets. The observed and the predicted Cmax (µg/ml) and AUC (µg⋅h/ml) (mean, lower and higher 95% CI of the proteomics data of DMEs) for these studies are shown for all pediatric and adult populations and for the intravenous and oral routes of administration. The simulated mean Cmax (µg/ml) and AUC (µg⋅/ml) values were compared with the observed data and the acceptance criteria [on the basis of bioequivalence criterion (1.25-fold), 2-fold criterion, and population-based criterion] was determined. Study ID in the table represent the following: 1 (Clements et al., 1984); 2 (Singla et al., 2012); 3 (Rawlins et al., 1977); 4 (Perucca and Richens, 1979); 5 (Prescott, 1980); 6 (Zapater et al., 2004); 7 (Rostami-Hodjegan et al., 2002); 8 (Manyike et al., 2000); 9 (Kamali et al., 1992); 10 (Prescott, 1980); 11 (https://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/022450Orig1s000ClinPharmR.pdf), and 12 (Rømsing et al., 2001).
| Study ID | Dose | Parameters | Mean Observed Value (O) | Acceptance Range | Mean Predicted Value (P) | P/O Ratio | Compliance to Model Evaluation Criteria |
|||
|---|---|---|---|---|---|---|---|---|---|---|
| Bioequivalence criterion | 2-Fold criterion | Population criterion | ||||||||
| Intravenous (adults) | 1 | 5 mg/kg | AUC (0–inf, h) | 18.38 | 15.50–21.80 | 18.42 | 1.00 | Yes | Yes | Yes |
| 1 | 20 mg/kg | AUC (0–inf, h) | 82.48 | 77.85–87.38 | 74.83 | 0.91 | Yes | Yes | Yes | |
| 2 | 1000 mg | Cmax | 21.6 | 15.86–29.42 | 30.01 | 1.39 | No | Yes | No | |
| 2 | 1000 mg | AUC (0–inf, h) | 42.5 | 31.96–56.52 | 42.22 | 0.99 | Yes | Yes | Yes | |
| 3 | 1000 mg | AUC (0–inf, h) | 50.5 | 31.50–80.96 | 53.42 | 1.06 | Yes | Yes | Yes | |
| 4 | 1000 mg | Cmax | 55.3 | 47.78–64.00 | 44.99 | 0.81 | No | Yes | No | |
| 5 | 12 mg/kg | AUC (0–inf, h) | 36.7 | 34.23–39.35 | 44.76 | 1.22 | No | Yes | No | |
| Oral tablet (adults) | 2 | 1000 mg | Cmax | 12.3 | 6.14–24.63 | 11.15 | 0.91 | Yes | Yes | Yes |
| 2 | 1000 mg | AUC (0–6 h) | 29.4 | 13.32–64.89 | 35.80 | 1.22 | Yes | Yes | Yes | |
| 3 | 500 mg | AUC (0–inf, h) | 15.6 | 6.53–37.27 | 22.93 | 1.47 | No | Yes | Yes | |
| 3 | 1000 mg | AUC (0–inf, h) | 44 | 30.87–62.72 | 46.40 | 1.05 | Yes | Yes | Yes | |
| 3 | 2000 mg | AUC (0–inf, h) | 87.6 | 48.32–158.81 | 94.54 | 1.08 | Yes | Yes | Yes | |
| 6 | 1000 mg | Cmax | 15.9 | 11.20–22.57 | 11.15 | 0.70 | No | Yes | No | |
| 6 | 1000 mg | AUC (0–6 h) | 38.8 | 32.48–46.35 | 35.80 | 0.92 | Yes | Yes | Yes | |
| 7 | 1000 mg | Cmax | 18 | 11.86–27.33 | 11.15 | 0.62 | No | Yes | No | |
| 7 | 1000 mg | AUC (0–inf, h) | 54.78 | 45.35–66.17 | 46.40 | 0.85 | Yes | Yes | Yes | |
| 8 | 500 mg | Cmax | 5.65 | 3.38–9.46 | 5.54 | 0.98 | Yes | Yes | Yes | |
| 8 | 500 mg | Cmax | 4.7 | 3.64–6.06 | 5.54 | 1.18 | Yes | Yes | Yes | |
| Oral tablet (adults)-fed state | 7 | 1000 mg | Cmax | 11 | 8.87–13.65 | 7.47 | 0.68 | No | Yes | No |
| 7 | 1000 mg | AUC (0–inf, h) | 51.92 | 43.51–61.95 | 46.44 | 0.89 | Yes | Yes | Yes | |
| Oral solution (adults) | 9 | 1000 mg | Cmax | 17.5 | 9.22–33.23 | 13.70 | 0.78 | Yes | Yes | Yes |
| 10 | 1000 mg | Cmax | 20 | 11.62–34.42 | 13.70 | 0.69 | No | Yes | Yes | |
| 10 | 1000 mg | AUC (0–24 h) | 45 | 32.53–62.25 | 46.37 | 1.03 | Yes | Yes | Yes | |
| 4 | 1000 mg | AUC (0–inf, h) | 49.4 | 42.31–57.68 | 46.51 | 0.94 | Yes | Yes | Yes | |
| 5 | 12 mg/kg | AUC (0–inf, h) | 28 | 18.09–43.33 | 38.93 | 1.39 | No | Yes | Yes | |
| Intravenous (neonates) | 11 | 15 mg/kg | Cmax | 25 | 8.88–70.35 | 24.75 | 0.99 | Yes | Yes | Yes |
| 11 | 15 mg/kg | AUC (0–6 h) | 62 | 52.91–72.65 | 68.66 | 1.11 | Yes | Yes | Yes | |
| Intravenous (infants) | 11 | 15 mg/kg | Cmax | 29 | 10.64–79.07 | 27.78 | 0.96 | Yes | Yes | Yes |
| 11 | 15 mg/kg | AUC (0–6 h) | 57 | 40.93–79.38 | 53.51 | 0.94 | Yes | Yes | Yes | |
| Intravenous (children) | 11 | 15 mg/kg | Cmax | 29 | 23.32–36.07 | 28.31 | 0.98 | Yes | Yes | Yes |
| 11 | 15 mg/kg | AUC (0–6 h) | 38 | 32.80–44.02 | 47.33 | 1.25 | Yes | Yes | No | |
| Intravenous (adolescents) | 11 | 15 mg/kg | Cmax | 31 | 25.43–37.79 | 28.60 | 0.92 | Yes | Yes | Yes |
| 11 | 15 mg/kg | AUC (0–6 h) | 41 | 33.81–49.71 | 52.83 | 1.29 | No | Yes | No | |
| Oral tablet (middle childhood) | 12 | 22.5 mg/kg | Cmax | 12.7 | 8.56–18.84 | 19.75 | 1.55 | No | Yes | No |
| 12 | 22.5 mg/kg | AUC (0–4 h) | 33.13 | 22.14–49.58 | 52.02 | 1.57 | No | Yes | No | |
The validated adult PBPK model was employed for prediction of PK in the pediatric population because it considered all the relevant information gathered. There was under-prediction of acetaminophen AUC for the pediatric population when DME ontogeny data were not considered (Fig. 4). The fm, ratio values (e.g., fm,UGT/fm,SULT) predicted by the ontogeny-based model were 0.46, 0.56, and 1.71 in neonates, children, and adults, respectively (Fig. 5; Table 3). These predicted fm and corresponding Ae data were in agreement with those observed (Fig. 5). Data in Supplemental Table 10 show that model-predicted pediatric PK data even correlated well with the FDA-recommended dose adjustments.
Fig. 5.

Ontogeny-based predicted fm values of acetaminophen-metabolizing enzymes across different age-groups (neonatal to adulthood) after oral administration of 10 mg/kg drug solution, as estimated in this study (shown by pie charts A–F). The pie charts are shown in different diameters to represent the magnitude of apparent clearance (CL/F = Dose/AUC0–inf). The predicted fm values were further confirmed by comparison of observed and predicted urinary recoveries (Ae, mmol) of acetaminophen (APAP), acetaminophen-sulfate (APAP-S), and acetaminophen-glucuronide (APAP-G) across different age-groups (shown as bar diagrams G–L). The predicted Ae data were generated in respective age groups after consideration of mean (bar) and 95% CI (error bars) protein abundance data. Dots indicate observed urinary elimination data. Abbreviations used in the illustration represent the following: L21 (Miller et al., 1976); L22 (Alam et al., 1977); L23 (Clements et al., 1984); L24 (Critchley et al., 1986), and L25 (Critchley et al., 2005).
TABLE 3.
Ontogeny-based predicted UGT/SULT fm values of acetaminophen across different age groups.
Mean, lower, and higher 95% CI of the proteomics data for DMEs were considered for prediction of fm of DMEs.
| Age Groups (Mean, Range) | UGT/SULT fm |
||
|---|---|---|---|
| Mean | Lower 95% CI | Higher 95% CI | |
| Neonatal (14 day, 0–27 day) | 0.46 | 0.12 | 0.55 |
| Infancy (6 mo, 28–364 day) | 0.54 | 0.62 | 0.50 |
| Infancy (1 yr, 29 to <2 yr) | 0.44 | 0.49 | 0.42 |
| Toddler/early childhood (4 yr, 1 to <6 yr) | 0.47 | 0.45 | 0.48 |
| Middle childhood (9 yr, 6 to <12 yr) | 0.64 | 0.62 | 0.64 |
| Children (7 yr, 2 to <12 yr) | 0.56 | 0.56 | 0.56 |
| Adolescence (14 yr, 12–16 yr) | 1.02 | 1.18 | 0.91 |
| Adolescence (15 yr, 12–18 yr) | 0.97 | 1.08 | 0.90 |
| Adulthood (30 yr, > 18 yr) | 1.71 | 1.60 | 1.86 |
95% CI, 95% confidence interval.
Discussion
Some data exist in the literature regarding the ontogeny of SULT1A1, SULT1A3, SULT2A1, and SULT1E1 (Brashear et al., 1988; Barker et al., 1994; Richard et al., 2001; Behm et al., 2003; Pacifici, 2005; Stanley et al., 2005; Duanmu et al., 2006; Ekström and Rane, 2015). However, several limitations are associated with these studies, for example: 1) lack of specific antibodies for the Western blotting, 2) use of nonselective in vitro or in vivo probe substrates for the activity, 3) smaller numbers of samples, and 4) inconsistent mRNA data that show poor correlation with functional activity. Hence, a comprehensive investigation of the ontogeny of SULT enzymes using an LC-MS/MS proteomics approach was performed in this study.
Among the notable findings, we observed that the ontogeny of hepatic SULTs are opposite to the trend of ontogeny of UGTs and most CYPs that are poorly expressed in fetal and early neonatal ages but are abundant in adult (Pacifici et al., 1982, 1993; Choonara et al., 1989; McCarver and Hines, 2002; Hines, 2007; Upreti and Wahlstrom, 2016). Using the same donor samples, we recently reported that, compared with adults, UGT levels were 3%–40% in the neonates, 24%–60% in the infants, and 37%–72% in the childhood age (Bhatt et al., 2019). These data are consistent with the literature, in which adult to fetal ratios of UGT and SULT activities are shown to be 114 and 3.5, respectively (Pacifici et al., 1989). Likewise, SULT1A3-mediated dopamine sulfation activity is shown to be 3-fold higher in the fetal liver compared with the adult liver, whereas an opposite trend was observed in SULT1A1 4-nitrophenol sulfation activity (Cappiello et al., 1991). Ritodrine, a tocolytic agent for the management of preterm labor, is inactivated by sulfation and glucuronidation. The ratio of ritodrine sulfate to glucuronide in urine was found to be higher in newborns, whereas the ratio was equal in maternal urine (Brashear et al., 1988; Pacifici et al., 1993). Such observed differential DME ontogeny has a direct significance in vivo for drugs metabolized by multiple DMEs (e.g., SULTs, UGTs, CYPs, etc.).
Our data do not agree with other reports in a few instances. For example, we observed that SULTs (particularly SULT1A1 and SULT2A1) are expressed at higher levels in childhood age (1–12 years) compared with the adolescents and adults. This is not in agreement with the observations by Duanmu et al. (2006), who found no difference in hepatic SULT1A1 and SULT2A1 abundances in postnatal samples. A limited number of pediatric samples from children between 2 and 10 years could be a potential reason for this discrepancy. Likewise, although SULT1A3 was detected in all age groups in our study, this enzyme was only detected in fetal and neonatal livers by others (Richard et al., 2001). We believe this indicates a better sensitivity of our method. Further, LC-MS/MS proteomics allowed discrimination of the highly homologous SULT proteins compared with conventional antibody- or activity-based methods.
Practically nothing was known previously regarding SULT1B1 ontogeny. Our data show a significant and gradual age-dependent increase in SULT1B1 abundance during the first year of life, unlike SULT1A1 and SULT2A1 enzymes, but similar to CYPs and UGTs.
The mechanisms that regulate the age-dependent abundances of SULTs remain unclear. However, transcription and environmental factors could be the potential regulators of SULT expression during development. The differential tissue and cross-species expression are considered to be regulated by aryl hydrocarbon receptor, constitutive androstane receptor, pregnane X receptor (PXR), liver X receptor, farnesoid X receptor (FXR), peroxisome proliferator-activated receptors (PPARs), and vitamin D receptor (Dubaisi et al., 2018). As the expression of some of these transcriptional factors, e.g., PXR, PPARα, or PPARγ, is age-dependent (Balasubramaniyan et al., 2005), one can anticipate their role in corresponding developmental change in SULT abundance. In particular, the expression of SULT2A1 is reported to increase 2-fold in fetal hepatocyte culture by agonists of PPARα (GW7647) or PPARγ (rosiglitazone) and suppressed by the FXR agonist (GW4064) (Dubaisi et al., 2018). Steroidogenic factor 1 and GATA-binding factor 6 are also reported to be involved in regulation of SULT2A1 in the adrenals (Saner et al., 2005). Likewise, because SULT2A1 is the major DHEA-metabolizing enzyme and the levels of urinary DHEA and hydroxylated DHEA (representing sulfate conjugates) transform during early to late childhood age, one can postulate that DHEA is involved in the regulation of SULT2A1 during the pubertal development (Rainey et al., 2002; Remer et al., 2005). This is also supported by the fact that adrenal expression of SULT2A1 increases with the gradual growth of adrenal zona reticularis (Nakamura et al., 2009).
In accordance with our objective, we applied the differential ontogeny data of SULTs, UGTs, and CYPs to estimate the effect of age on fm values of these enzymes in acetaminophen metabolism. The major reason to select this drug was the availability of extensive reported in vitro and PK data in the neonates, infants, and children (https://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/022450Orig1s000ClinPharmR.pdf; https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/204767Orig1s000TOC.cfm). The predicted fm and corresponding metabolite Ae data were in good agreement with the observed data (Fig. 5). For example, the ratio of UGT/SULT fm for acetaminophen was simulated to be 0.46, 0.56, and 1.71 compared with the observed values of the excreted fraction of glucuronide/sulfate metabolite of 0.34, 0.75, and 1.8 in the neonates, children, and adults, respectively (Miller et al., 1976). Acetaminophen is also transformed to N-acetyl-p-benzoquinone imine (NAPQI), an hepatotoxic metabolite mainly formed through oxidative mechanism via CYP2E1. Accurate fm prediction for CYP-mediated bioactivation of acetaminophen is important for predicting toxicity in children. With higher contribution of SULT in the clearance of various drugs in children, including acetaminophen, it is hypothesized that SULT-mediated DDIs or food-drug interactions may be significant in the pediatric population.
Our proteomics-informed PBPK predictions of acetaminophen PK in neonates and infants are consistent with the FDA label doses for acetaminophen injection. For example, FDA suggests a dose reduction of 50% and 33% for the neonates and infants, respectively, to produce PK exposure similar to that of children. The origin of this dose reduction is related to age-dependent in vivo clearance of the drug (Zuppa et al., 2011). The extent of acetaminophen dose reduction in pediatrics is reasonably captured by our model (Supplemental Table 10).
No significant association of ethnicity has been previously reported for sulfation clearance of acetaminophen in adults (African Americans and European Americans) (Court et al., 2017). Rather, we found that SULT1A3 and SULT1B1 abundances were significantly lower in African Americans compared with Caucasians, which represented a majority of pediatric samples. Although the mechanisms leading to these differences are unknown, genetic polymorphisms or environmental factors could be tested in the future as potential contributors.
No association of sex with enzyme abundances was observed across all age groups and ethnicity for SULT1A1, SULT1A3, and SULT1B1, albeit multiple linear regression analysis showed a modest, but significantly higher abundance of SULT2A1 in females consistent with reported mouse data (Kocarek et al., 2008) and perhaps owing to the role of SULT2A1 in androgen disposition.
Drugs such as clomiphene, danazol, imipramine, chlorpheniramine, spironolactone, chlorpromazine, amitriptyline, and propranolol are potent inhibitors of SULT enzymes (Coughtrie et al., 1994; Marto et al., 2017) and can produce greater DDIs with SULT substrates in children. A risk of SULT-mediated drug-food interactions also exists in children (Nishimuta et al., 2007), as constituents of certain beverages can inhibit SULT enzymes. Various endogenous molecules, such as bile acid, estrogen, thyroid hormones, catecholamines, DHEA, etc., can be differentially affected by modulators of SULT activity, depending on age (Coughtrie et al., 1994; Coughtrie, 2002). The ontogeny data of SULTs presented here can prove useful in interpreting these data.
Regarding limitations of this study, we were unable to quantify SULT1E1 owing to a higher limit of quantification of its surrogate peptide. Further, data for SULT2A1 CNV and SNPs including rs296361, which have previously been shown to affect protein abundance (Ekström and Rane, 2015; Wong et al., 2018), were not available for all the samples. Nevertheless, our large sample size precludes a potential confounding effect of genetic variability on conclusions regarding the ontogeny. Although we have not reported any activity data in this study, we recently showed that SULT2A1 activity correlated well with the abundance data quantified by the same LC-MS/MS method (Wong et al., 2018). The absolute levels of SULT1A1 and SULT2A1 were quantified using the commercially available purified protein standards; however, no further purification and characterization of these standards were conducted and the purity was assumed to be >95%. Further, the PBPK model accurately predicted AUC as well as Cmax and Tmax for intravenous dosing. However, the predicted values of Cmax and Tmax for oral dosing were not accurate. This may be explained by a highly variable absorption rate of acetaminophen, which is affected by food and formulation type, including excipients (e.g., sodium bicarbonate) (Rostami-Hodjegan et al., 2002). In the fed-state, where the gastric emptying time is approx. 1 hour, compared with 15 minutes for the fasting state, significantly decreased Cmax and delayed Tmax were predicted (Fig. 3).
In summary, in this first comprehensive report of its kind, we successfully established ontogeny of SULT1A1, SULT1A3, SULT1B1, and SULT2A1 enzymes in a large group of donor human livers (n = 194) using a quantitative LC-MS/MS proteomics approach. This study is a typical case of pediatric drug-metabolism prediction in situations when multiple metabolic phase I and phase II pathways are involved. The ontogeny data were applied to predict age-dependent fm values for DMEs (SULTs, UGTs, and CYPs) involved in acetaminophen metabolism. The age-dependent fm data can be further applied to predict DDIs and drug-food interactions and for predicting variability caused by genetic variation.
Acknowledgments
The authors thank Aravind Rachapally, Sarang Mishra, and Priyanka Bobe for assisting in PBPK modeling and simulation. We thank Simulations Plus (Lancaster, CA) for providing licensed access to GastroPlus v9.6 software.
Abbreviations
- Ae
predicted total amount of metabolites eliminated in urine
- AUC
area under the plasma concentration-time curve
- Cmax
maximum plasma concentration
- CNV
copy number variation
- DDIs
drug-drug interactions
- DHEA
dehydroepiandrosterone
- DMEs
drug-metabolizing enzymes
- fm
fractional metabolism by individual enzymes
- JT
Jonckheere-Terpstra
- PBPK
physiologically based pharmacokinetic
- PCA
principal component analysis
- PK
pharmacokinetics
- %CV
percent coefficient of variation
- SNPs
single-nucleotide polymorphisms
- SULTs
human cytosolic sulfotransferases
- UGTs
UDP-glucuronosyltransferases
Authorship Contributions
Participated in research design: Ladumor, Bhatt, Leeder, Singh, Prasad.
Conducted experiments: Ladumor, Bhatt, Gaedigk, Pearce, Sharma, Thakur, Prasad.
Performed data analysis: Ladumor, Bhatt, Gaedigk, Sharma, Thakur, Bolger, Prasad.
Wrote or contributed to the writing of the manuscript: Ladumor, Bhatt, Gaedigk, Sharma, Thakur, Pearce, Leeder, Bolger, Singh, Prasad.
Footnotes
The proteomics and genotyping work was funded by a grant from the Eunice Kennedy Shriver National Institute of Child Health and Human Development [R01.HD081299]. The National Institute of Child Health and Human Development Brain and Tissue Bank for Developmental Disorders at the University of Maryland is funded by the National Institutes of Health [contract N01-HD-9-0011/HHSN275200900011C], and the Liver Tissue Cell Distribution System are funded by NIH [contract N01-DK-7- 0004/HHSN267200700004C]. Financial assistance for the Ph.D. fellowship to M.K.L. for PBPK modeling work was provided by Bristol-Myers Squibb [NIPER SP-215].
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Abduljalil K, Cain T, Humphries H, Rostami-Hodjegan A. (2014) Deciding on success criteria for predictability of pharmacokinetic parameters from in vitro studies: an analysis based on in vivo observations. Drug Metab Dispos 42:1478–1484. [DOI] [PubMed] [Google Scholar]
- Adjei AA, Gaedigk A, Simon SD, Weinshilboum RM, Leeder JS. (2008) Interindividual variability in acetaminophen sulfation by human fetal liver: implications for pharmacogenetic investigations of drug-induced birth defects. Birth Defects Res A Clin Mol Teratol 82:155–165. [DOI] [PubMed] [Google Scholar]
- Alam SN, Roberts RJ, Fischer LJ. (1977) Age-related differences in salicylamide and acetaminophen conjugation in man. J Pediatr 90:130–135. [DOI] [PubMed] [Google Scholar]
- Allegaert K, Naulaers G, Vanhaesebrouck S, Anderson BJ. (2013) The paracetamol concentration-effect relation in neonates. Paediatr Anaesth 23:45–50. [DOI] [PubMed] [Google Scholar]
- Bairam AF, Rasool MI, Alherz FA, Abunnaja MS, El Daibani AA, Gohal SA, Kurogi K, Sakakibara Y, Suiko M, Liu M-C. (2018) Sulfation of catecholamines and serotonin by SULT1A3 allozymes. Biochem Pharmacol 151:104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramaniyan N, Shahid M, Suchy FJ, Ananthanarayanan M. (2005) Multiple mechanisms of ontogenic regulation of nuclear receptors during rat liver development. Am J Physiol Gastrointest Liver Physiol 288:G251–G260. [DOI] [PubMed] [Google Scholar]
- Barker EV, Hume R, Hallas A, Coughtrie WH. (1994) Dehydroepiandrosterone sulfotransferase in the developing human fetus: quantitative biochemical and immunological characterization of the hepatic, renal, and adrenal enzymes. Endocrinology 134:982–989. [DOI] [PubMed] [Google Scholar]
- Behm M, Abdel‐Rahman S, Leeder J, Kearns G. (2003) Ontogeny of phase II enzymes: UGT and SULT. Clin Pharmacol Ther 73:P29. [Google Scholar]
- Bhatt DK, Basit A, Zhang H, Gaedigk A, Lee SB, Claw KG, Mehrotra A, Chaudhry AS, Pearce RE, Gaedigk R, et al. (2018) Hepatic abundance and activity of androgen- and drug-metabolizing enzyme UGT2B17 are associated with genotype, age, and sex. Drug Metab Dispos 46:888–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt DK, Gaedigk A, Pearce RE, Leeder JS, Prasad B. (2017) Age-dependent protein abundance of cytosolic alcohol and aldehyde dehydrogenases in human liver. Drug Metab Dispos 45:1044–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt DK, Mehrotra A, Gaedigk A, Chapa R, Basit A, Zhang H, Choudhari P, Boberg M, Pearce RE, Gaedigk R, et al. (2019) Age- and genotype-dependent variability in the protein abundance and activity of six major uridine diphosphate-glucuronosyltransferases in human liver. Clin Pharmacol Ther 105:131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt DK, Prasad B. (2018) Critical issues and optimized practices in quantification of protein abundance level to determine interindividual variability in DMET proteins by LC‐MS/MS proteomics. Clin Pharmacol Ther 103:619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boberg M, Vrana M, Mehrotra A, Pearce RE, Gaedigk A, Bhatt DK, Leeder JS, Prasad B. (2017) Age-dependent absolute abundance of hepatic carboxylesterases (CES1 and CES2) by LC-MS/MS proteomics: application to PBPK modeling of oseltamivir in vivo pharmacokinetics in infants. Drug Metab Dispos 45:216–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brashear WT, Kuhnert BR, Wei R. (1988) Maternal and neonatal urinary excretion of sulfate and glucuronide ritodrine conjugates. Clin Pharmacol Ther 44:634–641. [DOI] [PubMed] [Google Scholar]
- Calvier EAM, Krekels EHJ, Yu H, Välitalo PAJ, Johnson TN, Rostami-Hodjegan A, Tibboel D, van der Graaf PH, Danhof M, Knibbe CAJ. (2018) Drugs being eliminated via the same pathway will not always require similar pediatric dose adjustments. CPT Pharmacometrics Syst Pharmacol 7:175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappiello M, Giuliani L, Rane A, Pacifici GM. (1991) Dopamine sulphotransferase is better developed than p-nitrophenol sulphotransferase in the human fetus. Dev Pharmacol Ther 16:83–88. [PubMed] [Google Scholar]
- Chen W, Koenigs LL, Thompson SJ, Peter RM, Rettie AE, Trager WF, Nelson SD. (1998) Oxidation of acetaminophen to its toxic quinone imine and nontoxic catechol metabolites by baculovirus-expressed and purified human cytochromes P450 2E1 and 2A6. Chem Res Toxicol 11:295–301. [DOI] [PubMed] [Google Scholar]
- Choonara IA, McKay P, Hain R, Rane A. (1989) Morphine metabolism in children. Br J Clin Pharmacol 28:599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JA, Critchley JA, Prescott LF. (1984) The role of sulphate conjugation in the metabolism and disposition of oral and intravenous paracetamol in man. Br J Clin Pharmacol 18:481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook IT, Duniec-Dmuchowski Z, Kocarek TA, Runge-Morris M, Falany CN. (2009) 24-hydroxycholesterol sulfation by human cytosolic sulfotransferases: formation of monosulfates and disulfates, molecular modeling, sulfatase sensitivity, and inhibition of liver x receptor activation. Drug Metab Dispos 37:2069–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook SF, Stockmann C, Samiee-Zafarghandy S, King AD, Deutsch N, Williams EF, Wilkins DG, Sherwin CM, van den Anker JN. (2016) Neonatal maturation of paracetamol (acetaminophen) glucuronidation, sulfation, and oxidation based on a parent-metabolite population pharmacokinetic model. Clin Pharmacokinet 55:1395–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughtrie MW. (2002) Sulfation through the looking glass--recent advances in sulfotransferase research for the curious. Pharmacogenomics J 2:297–308. [DOI] [PubMed] [Google Scholar]
- Coughtrie MW, Bamforth KJ, Sharp S, Jones AL, Borthwick EB, Barker EV, Roberts RC, Hume R, Burchell A. (1994) Sulfation of endogenous compounds and xenobiotics--interactions and function in health and disease. Chem Biol Interact 92:247–256. [DOI] [PubMed] [Google Scholar]
- Court MH, Zhu Z, Masse G, Duan SX, James LP, Harmatz JS, Greenblatt DJ. (2017) Race, gender, and genetic polymorphism contribute to variability in acetaminophen pharmacokinetics, metabolism, and protein-adduct concentrations in healthy African-American and European-American volunteers. J Pharmacol Exp Ther 362:431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchley JA, Critchley LA, Anderson PJ, Tomlinson B. (2005) Differences in the single-oral-dose pharmacokinetics and urinary excretion of paracetamol and its conjugates between Hong Kong Chinese and Caucasian subjects. J Clin Pharm Ther 30:179–184. [DOI] [PubMed] [Google Scholar]
- Critchley JA, Nimmo GR, Gregson CA, Woolhouse NM, Prescott LF. (1986) Inter-subject and ethnic differences in paracetamol metabolism. Br J Clin Pharmacol 22:649–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubitt HE, Houston JB, Galetin A. (2011) Prediction of human drug clearance by multiple metabolic pathways: integration of hepatic and intestinal microsomal and cytosolic data. Drug Metab Dispos 39:864–873. [DOI] [PubMed] [Google Scholar]
- Duanmu Z, Weckle A, Koukouritaki SB, Hines RN, Falany JL, Falany CN, Kocarek TA, Runge-Morris M. (2006) Developmental expression of aryl, estrogen, and hydroxysteroid sulfotransferases in pre- and postnatal human liver. J Pharmacol Exp Ther 316:1310–1317. [DOI] [PubMed] [Google Scholar]
- Dubaisi S, Barrett KG, Fang H, Guzman-Lepe J, Soto-Gutierrez A, Kocarek TA, Runge-Morris M. (2018) Regulation of cytosolic sulfotransferases in models of human hepatocyte development. Drug Metab Dispos 46:1146–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhofer G, Coughtrie MW, Goldstein DS. (1999) Dopamine sulphate: an enigma resolved. Clin Exp Pharmacol Physiol Suppl 26:S41–S53. [PubMed] [Google Scholar]
- Ekström L, Rane A. (2015) Genetic variation, expression and ontogeny of sulfotransferase SULT2A1 in humans. Pharmacogenomics J 15:293–297. [DOI] [PubMed] [Google Scholar]
- Emoto C, Johnson TN, Neuhoff S, Hahn D, Vinks AA, Fukuda T. (2018) PBPK model of morphine incorporating developmental changes in hepatic OCT1 and UGT2B7 proteins to explain the variability in clearances in neonates and small infants. CPT Pharmacometrics Syst Pharmacol 7:464–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falany CN, Krasnykh V, Falany JL. (1995) Bacterial expression and characterization of a cDNA for human liver estrogen sulfotransferase. J Steroid Biochem Mol Biol 52:529–539. [DOI] [PubMed] [Google Scholar]
- Falany CN, Wheeler J, Oh TS, Falany JL. (1994) Steroid sulfation by expressed human cytosolic sulfotransferases. J Steroid Biochem Mol Biol 48:369–375. [DOI] [PubMed] [Google Scholar]
- Falany JL, Macrina N, Falany CN. (2004) Sulfation of tibolone and tibolone metabolites by expressed human cytosolic sulfotransferases. J Steroid Biochem Mol Biol 88:383–391. [DOI] [PubMed] [Google Scholar]
- Falany JL, Pilloff DE, Leyh TS, Falany CN. (2006) Sulfation of raloxifene and 4-hydroxytamoxifen by human cytosolic sulfotransferases. Drug Metab Dispos 34:361–368. [DOI] [PubMed] [Google Scholar]
- Fujita K, Nagata K, Ozawa S, Sasano H, Yamazoe Y. (1997) Molecular cloning and characterization of rat ST1B1 and human ST1B2 cDNAs, encoding thyroid hormone sulfotransferases. J Biochem 122:1052–1061. [DOI] [PubMed] [Google Scholar]
- Gaedigk A, Twist GP, Leeder JS. (2012) CYP2D6, SULT1A1 and UGT2B17 copy number variation: quantitative detection by multiplex PCR. Pharmacogenomics 13:91–111. [DOI] [PubMed] [Google Scholar]
- Gamage N, Barnett A, Hempel N, Duggleby RG, Windmill KF, Martin JL, McManus ME. (2006) Human sulfotransferases and their role in chemical metabolism. Toxicol Sci 90:5–22. [DOI] [PubMed] [Google Scholar]
- Gordon AS, Fulton RS, Qin X, Mardis ER, Nickerson DA, Scherer S. (2016) PGRNseq: a targeted capture sequencing panel for pharmacogenetic research and implementation. Pharmacogenet Genomics 26:161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines RN. (2007) Ontogeny of human hepatic cytochromes P450. J Biochem Mol Toxicol 21:169–175. [DOI] [PubMed] [Google Scholar]
- Honma W, Shimada M, Sasano H, Ozawa S, Miyata M, Nagata K, Ikeda T, Yamazoe Y. (2002) Phenol sulfotransferase, ST1A3, as the main enzyme catalyzing sulfation of troglitazone in human liver. Drug Metab Dispos 30:944–949. [DOI] [PubMed] [Google Scholar]
- Huang J, Bathena SP, Tong J, Roth M, Hagenbuch B, Alnouti Y. (2010) Kinetic analysis of bile acid sulfation by stably expressed human sulfotransferase 2A1 (SULT2A1). Xenobiotica 40:184–194. [DOI] [PubMed] [Google Scholar]
- Huang W, Nakano M, Sager J, Ragueneau-Majlessi I, Isoherranen N. (2017) Physiologically based pharmacokinetic model of the CYP2D6 probe atomoxetine: extrapolation to special populations and drug-drug interactions. Drug Metab Dispos 45:1156–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui Y, Liu M-C. (2015) Sulfation of ritodrine by the human cytosolic sulfotransferases (SULTs): effects of SULT1A3 genetic polymorphism. Eur J Pharmacol 761:125–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang XL, Zhao P, Barrett JS, Lesko LJ, Schmidt S. (2013) Application of physiologically based pharmacokinetic modeling to predict acetaminophen metabolism and pharmacokinetics in children. CPT Pharmacometrics Syst Pharmacol 2:e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TN, Rostami-Hodjegan A, Tucker GT. (2006) Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin Pharmacokinet 45:931–956. [DOI] [PubMed] [Google Scholar]
- Kamali F, Edwards C, Rawlins MD. (1992) The effect of pirenzepine on gastric emptying and salivary flow rate: constraints on the use of saliva paracetamol concentrations for the determination of paracetamol pharmacokinetics. Br J Clin Pharmacol 33:309–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocarek TA, Duanmu Z, Fang H-L, Runge-Morris M. (2008) Age- and sex-dependent expression of multiple murine hepatic hydroxysteroid sulfotransferase (SULT2A) genes. Biochem Pharmacol 76:1036–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurogi K, Chepak A, Hanrahan MT, Liu M-Y, Sakakibara Y, Suiko M, Liu M-C. (2014) Sulfation of opioid drugs by human cytosolic sulfotransferases: metabolic labeling study and enzymatic analysis. Eur J Pharm Sci 62:40–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine JE, Auriola S, Pasanen M, Juvonen RO. (2009) Acetaminophen bioactivation by human cytochrome P450 enzymes and animal microsomes. Xenobiotica 39:11–21. [DOI] [PubMed] [Google Scholar]
- Manyike PT, Kharasch ED, Kalhorn TF, Slattery JT. (2000) Contribution of CYP2E1 and CYP3A to acetaminophen reactive metabolite formation. Clin Pharmacol Ther 67:275–282. [DOI] [PubMed] [Google Scholar]
- Marto N, Morello J, Monteiro EC, Pereira SA. (2017) Implications of sulfotransferase activity in interindividual variability in drug response: clinical perspective on current knowledge. Drug Metab Rev 49:357–371. [DOI] [PubMed] [Google Scholar]
- McCarver DG, Hines RN. (2002) The ontogeny of human drug-metabolizing enzymes: phase II conjugation enzymes and regulatory mechanisms. J Pharmacol Exp Ther 300:361–366. [DOI] [PubMed] [Google Scholar]
- Meloche CA, Sharma V, Swedmark S, Andersson P, Falany CN. (2002) Sulfation of budesonide by human cytosolic sulfotransferase, dehydroepiandrosterone-sulfotransferase (DHEA-ST). Drug Metab Dispos 30:582–585. [DOI] [PubMed] [Google Scholar]
- Miller RP, Roberts RJ, Fischer LJ. (1976) Acetaminophen elimination kinetics in neonates, children, and adults. Clin Pharmacol Ther 19:284–294. [DOI] [PubMed] [Google Scholar]
- Mutlib AE, Goosen TC, Bauman JN, Williams JA, Kulkarni S, Kostrubsky S. (2006) Kinetics of acetaminophen glucuronidation by UDP-glucuronosyltransferases 1A1, 1A6, 1A9 and 2B15. Potential implications in acetaminophen-induced hepatotoxicity. Chem Res Toxicol 19:701–709. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Gang HX, Suzuki T, Sasano H, Rainey WE. (2009) Adrenal changes associated with adrenarche. Rev Endocr Metab Disord 10:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimuta H, Ohtani H, Tsujimoto M, Ogura K, Hiratsuka A, Sawada Y. (2007) Inhibitory effects of various beverages on human recombinant sulfotransferase isoforms SULT1A1 and SULT1A3. Biopharm Drug Dispos 28:491–500. [DOI] [PubMed] [Google Scholar]
- Nishiyama T, Ogura K, Nakano H, Ohnuma T, Kaku T, Hiratsuka A, Muro K, Watabe T. (2002) Reverse geometrical selectivity in glucuronidation and sulfation of cis- and trans-4-hydroxytamoxifens by human liver UDP-glucuronosyltransferases and sulfotransferases. Biochem Pharmacol 63:1817–1830. [DOI] [PubMed] [Google Scholar]
- Nowell S, Falany CN. (2006) Pharmacogenetics of human cytosolic sulfotransferases. Oncogene 25:1673–1678. [DOI] [PubMed] [Google Scholar]
- Pacifici GM. (2005) Sulfation of drugs and hormones in mid-gestation human fetus. Early Hum Dev 81:573–581. [DOI] [PubMed] [Google Scholar]
- Pacifici GM, Franchi M, Giuliani L, Rane A. (1989) Development of the glucuronyltransferase and sulphotransferase towards 2-naphthol in human fetus. Dev Pharmacol Ther 14:108–114. [PubMed] [Google Scholar]
- Pacifici GM, Kubrich M, Giuliani L, de Vries M, Rane A. (1993) Sulphation and glucuronidation of ritodrine in human foetal and adult tissues. Eur J Clin Pharmacol 44:259–264. [DOI] [PubMed] [Google Scholar]
- Pacifici GM, Säwe J, Kager L, Rane A. (1982) Morphine glucuronidation in human fetal and adult liver. Eur J Clin Pharmacol 22:553–558. [DOI] [PubMed] [Google Scholar]
- Pearce RE, Gaedigk R, Twist GP, Dai H, Riffel AK, Leeder JS, Gaedigk A. (2016) Developmental expression of CYP2B6: a comprehensive analysis of mRNA expression, protein content and bupropion hydroxylase activity and the impact of genetic variation. Drug Metab Dispos 44:948–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perucca E, Richens A. (1979) Paracetamol disposition in normal subjects and in patients treated with antiepileptic drugs. Br J Clin Pharmacol 7:201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad B, Gaedigk A, Vrana M, Gaedigk R, Leeder JS, Salphati L, Chu X, Xiao G, Hop C, Evers R, et al. (2016) Ontogeny of hepatic drug transporters as quantified by LC‐MS/MS proteomics. Clin Pharmacol Ther 100:362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad B, Unadkat JD. (2015) The concept of fraction of drug transported (ft) with special emphasis on BBB efflux of CNS and antiretroviral drugs. Clin Pharmacol Ther 97:320–323. [DOI] [PubMed] [Google Scholar]
- Prescott LF. (1980) Kinetics and metabolism of paracetamol and phenacetin. Br J Clin Pharmacol 10 (Suppl 2):291S–298S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott LF. (1983) Paracetamol overdosage. Pharmacological considerations and clinical management. Drugs 25:290–314. [DOI] [PubMed] [Google Scholar]
- Prescott LF, Speirs GC, Critchley JA, Temple RM, Winney RJ. (1989) Paracetamol disposition and metabolite kinetics in patients with chronic renal failure. Eur J Clin Pharmacol 36:291–297. [DOI] [PubMed] [Google Scholar]
- Rainey WE, Carr BR, Sasano H, Suzuki T, Mason JI. (2002) Dissecting human adrenal androgen production. Trends Endocrinol Metab 13:234–239. [DOI] [PubMed] [Google Scholar]
- Rawlins MD, Henderson DB, Hijab AR. (1977) Pharmacokinetics of paracetamol (acetaminophen) after intravenous and oral administration. Eur J Clin Pharmacol 11:283–286. [DOI] [PubMed] [Google Scholar]
- Remer T, Boye KR, Hartmann MF, Wudy SA. (2005) Urinary markers of adrenarche: reference values in healthy subjects, aged 3-18 years. J Clin Endocrinol Metab 90:2015–2021. [DOI] [PubMed] [Google Scholar]
- Richard K, Hume R, Kaptein E, Stanley EL, Visser TJ, Coughtrie MW. (2001) Sulfation of thyroid hormone and dopamine during human development: ontogeny of phenol sulfotransferases and arylsulfatase in liver, lung, and brain. J Clin Endocrinol Metab 86:2734–2742. [DOI] [PubMed] [Google Scholar]
- Rømsing J, Østergaard D, Senderovitz T, Drozdziewicz D, Sonne J, Ravn G. (2001) Pharmacokinetics of oral diclofenac and acetaminophen in children after surgery. Paediatr Anaesth 11:205–213. [DOI] [PubMed] [Google Scholar]
- Rostami-Hodjegan A, Shiran MR, Ayesh R, Grattan TJ, Burnett I, Darby-Dowman A, Tucker GT. (2002) A new rapidly absorbed paracetamol tablet containing sodium bicarbonate. I. A four-way crossover study to compare the concentration-time profile of paracetamol from the new paracetamol/sodium bicarbonate tablet and a conventional paracetamol tablet in fed and fasted volunteers. Drug Dev Ind Pharm 28:523–531. [DOI] [PubMed] [Google Scholar]
- Salem F, Johnson TN, Barter ZE, Leeder JS, Rostami-Hodjegan A. (2013) Age related changes in fractional elimination pathways for drugs: assessing the impact of variable ontogeny on metabolic drug-drug interactions. J Clin Pharmacol 53:857–865. [DOI] [PubMed] [Google Scholar]
- Saner KJ, Suzuki T, Sasano H, Pizzey J, Ho C, Strauss JF, III, Carr BR, Rainey WE. (2005) Steroid sulfotransferase 2A1 gene transcription is regulated by steroidogenic factor 1 and GATA-6 in the human adrenal. Mol Endocrinol 19:184–197. [DOI] [PubMed] [Google Scholar]
- Schrag ML, Cui D, Rushmore TH, Shou M, Ma B, Rodrigues AD. (2004) Sulfotransferase 1E1 is a low km isoform mediating the 3-O-sulfation of ethinyl estradiol. Drug Metab Dispos 32:1299–1303. [DOI] [PubMed] [Google Scholar]
- Senggunprai L, Yoshinari K, Yamazoe Y. (2009) Selective role of sulfotransferase 2A1 (SULT2A1) in the N-sulfoconjugation of quinolone drugs in human. Drug Metab Dispos 37:1711–1717. [DOI] [PubMed] [Google Scholar]
- Shirasaka Y, Chaudhry AS, McDonald M, Prasad B, Wong T, Calamia JC, Fohner A, Thornton TA, Isoherranen N, Unadkat JD, et al. (2016) Interindividual variability of CYP2C19-catalyzed drug metabolism due to differences in gene diplotypes and cytochrome P450 oxidoreductase content. Pharmacogenomics J 16:375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singla NK, Parulan C, Samson R, Hutchinson J, Bushnell R, Beja EG, Ang R, Royal MA. (2012) Plasma and cerebrospinal fluid pharmacokinetic parameters after single-dose administration of intravenous, oral, or rectal acetaminophen. Pain Pract 12:523–532. [DOI] [PubMed] [Google Scholar]
- Stanley EL, Hume R, Coughtrie MW. (2005) Expression profiling of human fetal cytosolic sulfotransferases involved in steroid and thyroid hormone metabolism and in detoxification. Mol Cell Endocrinol 240:32–42. [DOI] [PubMed] [Google Scholar]
- Umehara KI, Huth F, Gu H, Schiller H, Heimbach T, He H. (2017) Estimation of fractions metabolized by hepatic CYP enzymes using a concept of inter-system extrapolation factors (ISEFs) - a comparison with the chemical inhibition method. Drug Metab Pers Ther 32:191–200. [DOI] [PubMed] [Google Scholar]
- Upreti VV, Wahlstrom JL. (2016) Meta-analysis of hepatic cytochrome P450 ontogeny to underwrite the prediction of pediatric pharmacokinetics using physiologically based pharmacokinetic modeling. J Clin Pharmacol 56:266–283. [DOI] [PubMed] [Google Scholar]
- Villiger A, Stillhart C, Parrott N, Kuentz M. (2016) Using physiologically based pharmacokinetic (PBPK) modelling to gain insights into the effect of physiological factors on oral absorption in paediatric populations. AAPS J 18:933–947. [DOI] [PubMed] [Google Scholar]
- Vrana M, Whittington D, Nautiyal V, Prasad B. (2017) Database of optimized proteomic quantitative methods for human drug disposition‐related proteins for applications in physiologically based pharmacokinetic modeling. CPT Pharmacometrics Syst Pharmacol 6:267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Falany JL, Falany CN. (1998) Expression and characterization of a novel thyroid hormone-sulfating form of cytosolic sulfotransferase from human liver. Mol Pharmacol 53:274–282. [DOI] [PubMed] [Google Scholar]
- Wong T, Wang Z, Chapron BD, Suzuki M, Claw KG, Gao C, Foti RS, Prasad B, Chapron A, Calamia J, et al. (2018) Polymorphic human sulfotransferase 2A1 mediates the formation of 25-hydroxyvitamin D3-3-O-sulfate, a major circulating vitamin D metabolite in humans. Drug Metab Dispos 46:367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Jamei M, Yeo KR, Rostami-Hodjegan A, Tucker GT. (2007) Misuse of the well-stirred model of hepatic drug clearance. Drug Metab Dispos 35:501–502. [DOI] [PubMed] [Google Scholar]
- Zapater P, Lasso de la Vega MC, Horga JF, Such J, Frances R, Esteban A, Palazòn JM, Carnicer F, Pascual S, Pérez-Mateo M. (2004) Pharmacokinetic variations of acetaminophen according to liver dysfunction and portal hypertension status. Aliment Pharmacol Ther 20:29–36. [DOI] [PubMed] [Google Scholar]
- Zuppa AF, Hammer GB, Barrett JS, Kenney BF, Kassir N, Mouksassi S, Royal MA. (2011) Safety and population pharmacokinetic analysis of intravenous acetaminophen in neonates, infants, children, and adolescents with pain or Fever. J Pediatr Pharmacol Ther 16:246–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurlinden TJ, Reisfeld B. (2016) Physiologically based modeling of the pharmacokinetics of acetaminophen and its major metabolites in humans using a Bayesian population approach. Eur J Drug Metab Pharmacokinet 41:267–280. [DOI] [PubMed] [Google Scholar]