

Abstract
Pharmacokinetic studies in rats and dogs were performed to characterize the in vivo performance of a novel prodrug, fosciclopirox. Ciclopirox olamine (CPX-O) is a marketed topical antifungal agent with demonstrated in vitro and in vivo preclinical anticancer activity in several solid tumor and hematologic malignancies. The oral route of administration for CPX-O is not feasible due to low bioavailability and dose-limiting gastrointestinal toxicities. To enable parenteral administration, the phosphoryl-oxymethyl ester of ciclopirox (CPX), fosciclopirox (CPX-POM), was synthesized and formulated as an injectable drug product. In rats and dogs, intravenous CPX-POM is rapidly and completely metabolized to its active metabolite, CPX. The bioavailability of the active metabolite is complete following CPX-POM administration. CPX and its inactive metabolite, ciclopirox glucuronide (CPX-G), are excreted in urine, resulting in delivery of drug to the entire urinary tract. The absolute bioavailability of CPX following subcutaneous administration of CPX-POM is excellent in rats and dogs, demonstrating the feasibility of this route of administration. These studies confirmed the oral bioavailability of CPX-O is quite low in rats and dogs compared with intravenous CPX-POM. Given its broad-spectrum anticancer activity in several solid tumor and hematologic cancers and renal elimination, CPX-POM is being developed for the treatment of urothelial cancer. The safety, dose tolerance, pharmacokinetics, and pharmacodynamics of intravenous CPX-POM are currently being characterized in a United States multicenter first-in-human Phase 1 clinical trial in patients with advanced solid tumors (NCT03348514).
Introduction
Ciclopirox (CPX) is a broad spectrum antifungal agent (Gupta, 2001). CPX is believed to act as a fungicidal agent by chelating polyvalent metal cations such as Fe3+ and Al3+, resulting in the inhibition of peroxide degradation (Gupta and Plott, 2004) and mitochondrial electron transport processes and energy production (Gupta et al., 1994; Sakurai et al., 1978). Several topical drug products containing CPX free acid or CPX olamine (CPX-O), including a cream, lotion, gel, nail lacquer, and shampoo, are currently marketed in the United States and other countries. Consistent with its iron chelation properties, researchers at the Ontario Cancer Institute demonstrated that CPX inhibited ribonucleotide reductase in vitro in leukemia, myeloma, and a number of solid tumor cancer cells, resulting in decreased cell growth and viability at concentrations associated with cell death (Eberhard et al., 2009). Oral administration of CPX-O decreased tumor weight and volume in mouse xenograft models of leukemia and lymphoma and also prevented engraftment of primary acute myeloid leukemia cells in mice (Eberhard et al., 2009). Song et al. (2011) subsequently showed that CPX inhibited Wnt signaling through its iron chelation effects, suggesting a possible mechanism of action for CPX’s anti-cancer activity.
We collaborated with Ontario Cancer Institute and The Leukemia and Lymphoma Society to translate these findings to patients with advanced hematologic malignancies. The safety, pharmacokinetics, and pharmacodynamics of CPX-O were characterized in patients with relapsed or refractory hematologic malignancies (Minden et al., 2014), representing the first oral, multiple dose CPX-O trial in humans. In relapsed and refractory leukemia and lymphoma patients, circulating CPX and CPX-G appeared to increase proportionately with increasing oral dose across a dose range of 5–80 mg/m2 once daily (Minden et al., 2014). Plasma CPX concentrations were quite low, however, while the major metabolite, CPX-G, circulated in plasma at concentrations at least 10-fold higher than those of CPX (Minden et al., 2014). Although repression of survivin expression was observed in two patients, administration of 80 mg/m2 CPX-O four times daily to patients was associated with grade 3 dose-limiting gastrointestinal toxicities (Minden et al., 2014).
These data, combined with results from an oral [14C]-CPX-O human metabolism and mass balance study demonstrating complete absorption (Kellner et al., 1981), led us to conclude the drug undergoes significant presystemic metabolism following oral administration. Low oral bioavailability, coupled with the dose-limiting gastrointestinal toxicities observed in patients, resulted in discontinuation of efforts to repurpose oral CPX-O for the treatment of hematologic malignancies.
Our CPX drug repurposing research efforts turned to developing a drug product that could be administered parenterally to cancer patients. Unfortunately, CPX and CPX-O are sparingly soluble in water (PENLAC, 2012) creating challenges in developing a suitable injectable formulation for this drug. We were successful in employing a prodrug strategy, synthesizing the phosphoryl-oxymethyl (POM) derivative (Stella, 1996; Tanol and Weir, 2013, 2015, 2016) of CPX. Fosciclopirox (CPX-POM) possesses outstanding aqueous solubility and is readily formulated into an injectable drug product. Herein, we have described the plasma and urine pharmacokinetics of CPX-POM, its active metabolite, CPX, and major inactive metabolite, ciclopirox glucuronide (CPX-G), in rats and dogs following intravenous and subcutaneous administration. A United States multicenter first-in-human Phase 1 clinical trial is ongoing to characterize the safety, dose tolerance, pharmacokinetics, and pharmacodynamics of intravenous CPX-POM in patients with advanced solid tumors (NCT03348514).
Materials and Methods
Test Articles.
Disodium ((6-cyclohexyl-4-methyl-2-oxopyridin-1(2H)-yl) oxy) methyl phosphate heptahydrate (CPX-POM), the structure of which is illustrated in Fig. 1, has a proposed International Nonproprietary Name of fosciclopirox. Fosciclopirox has a molecular formula of C13H32NNa2O13P and molecular weight of 487.35 g/mol. CPX-POM exists as a white solid, is water soluble, and possesses solution stability for parenteral administration. CPX-POM and CPX-O were formulated as sterile injectable solutions in 25 mM phosphate buffer, pH 7, with 50 mM Captisol (Ligand, San Diego, CA) and stored under refrigerated conditions at 2–8°C. CPX-O was administered orally in Orasweet SF (Perrigo, Allegan, MI) and water. All test article nominal dosing parameters for following studies are summarized in Table 1.
Fig. 1.
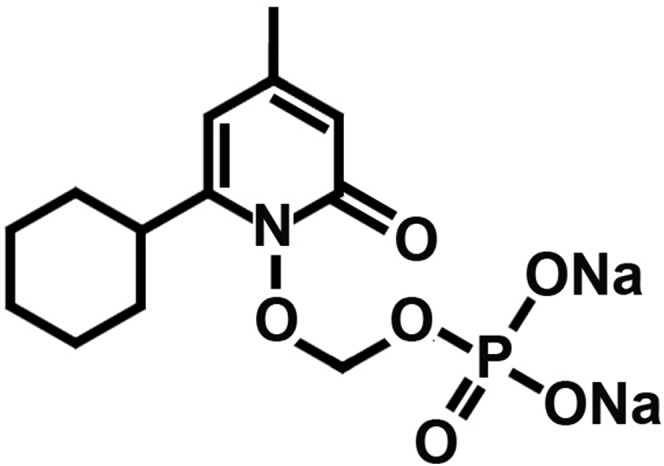
Chemical structure of fosciclopirox ((6-cyclohexyl-4-methyl-2-oxopyridin-1(2H)-yl) oxy) methyl phosphate heptahydrate (CPX-POM).
TABLE 1.
Treatments administered to rats and dogs including test article, test article strength, dose volume, formulation description, test article dose, and ciclopirox equivalent dose
| Study | Species | Route | Test Article | Test Article Strength (mg/ml) | Dose Volume (ml/kg) | Formulation Description | Test Article Dose (mg/kg) | Ciclopirox Equivalent Dose (mg/kg) |
|---|---|---|---|---|---|---|---|---|
| 1 | Rat | i.v. | CPX-O | 6.47 | 2.0 | 25 mM Phosphate Buffer, pH 7, 50 mM Captisol | 13.0 | 10.0 |
| i.v. | CPX-POM | 8.72 | 2.0 | 25 mM Phosphate Buffer, pH 7, 50 mM Captisol | 17.5 | 7.4 | ||
| 2 | Dog | i.v. | CPX-O | 1.94 | 2.0 | 25 mM Phosphate Buffer, pH 7, 50 mM Captisol | 4.0 | 3.1 |
| i.v. | CPX-POM | 2.61 | 2.0 | 25 mM Phosphate Buffer, pH 7, 50 mM Captisol | 5.4 | 2.3 | ||
| 3 | Rat | i.v. | CPX-POM | 23.53 | 2.1 | 25 mM Phosphate Buffer, pH 7, 50 mM Captisol | 47.0 | 20.0 |
| s.c. | CPX-POM | 23.53 | 3.0 | 25 mM Phosphate Buffer, pH 7, 50 mM Captisol | 70.6 | 30.0 | ||
| Oral | CPX-O | 29.39 | 1.3 | Orasweet SF, Water | 38.8 | 30.0 | ||
| 4 | Dog | i.v. | CPX-POM | 3.53 | 2.0 | 25 mM Phosphate Buffer, pH 7, 50 mM Captisol | 7.3 | 3.1 |
| s.c. | CPX-POM | 3.53 | 6.0 | 25 mM Phosphate Buffer, pH 7, 50 mM Captisol | 21.8 | 9.3 | ||
| Oral | CPX-O | 1.94 | 6.0 | Orasweet SF, Water | 12.2 | 9.3 |
Animal Care and Use Statement.
All studies in animals have been carried out at Xenometrics LLC, Stilwell, KS, a US-based Good Laboratory Practice (GLP) compliant, United States Department of Agriculture registered, Association for Assessment and Accreditation of Laboratory Animal Care accredited nonclinical contract laboratory, in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the US National Institutes of Health and were approved by the Institution’s Animal Care and Use Committee.
Absolute Bioavailability of CPX following Intravenous CPX-POM in Rats.
Plasma and urine pharmacokinetics of CPX were characterized following intravenous administration of CPX-O and CPX-POM to 16 fasted male Sprague-Dawley rats. In this study, CPX-O served as the reference treatment to characterize the absolute bioavailability of CPX following intravenous CPX-POM. A single intravenous dose of CPX-O was administered to six rats employing the intravenous formulation described above. In a second group of six male rats, a single intravenous dose of CPX-POM was administered. Intravenous doses of CPX-O and CPX-POM were administered directly into the tail vein using a syringe and needle at a volume of 2 ml/kg. Intravenous doses of 13.0 mg/kg CPX-O and 17.5 mg/kg CPX-POM doses were administered using intravenous formulation concentrations of 6.47 and 8.72 mg/ml, respectively. Dose volumes were adjusted for the weight of the animal on the morning of drug administration. Serial blood (plasma) samples were collected using a syringe via jugular vein catheter prior to and 0.083, 0.167, 0.25, 0.5, 0.75, 1, 1.5, 2, 3 and 4 hours postdose. In a third group of four male rats, complete urine was collected at predose and at 0- to 8- and 8- to 24-hour collection intervals postdose following intravenous CPX-O and CPX-POM. The same four animals were dosed with both test articles in a nonrandomized, complete crossover fashion with 72 hours separating treatments. Only urine was collected in this treatment group. Each treatment was administered under fasting conditions.
Absolute Bioavailability of CPX following Intravenous CPX-POM in Dogs.
Plasma and urine pharmacokinetics of CPX were characterized following intravenous administration of CPX-O and CPX-POM to four fasted male beagle dogs. In this study, CPX-O served as the reference treatment to characterize the absolute bioavailability of CPX following intravenous CPX-POM.
Single intravenous doses of 4 mg/kg CPX-O and 5.4 mg/kg CPX-POM were administered under fasting conditions on separate occasions in a nonrandomized, complete crossover fashion with 72 hours separating treatments. Intravenous doses were administered through an indwelling catheter in the cephalic vein at a volume of 2 ml/kg. CPX-O and CPX-POM were formulated at strengths of 1.94 and 2.61 mg/ml, respectively. Dose volumes were adjusted for the weight of the animal on the morning of drug administration. Each treatment was administered under fasting conditions. Serial blood (plasma) samples were collected using a syringe via jugular vein prior to and 0.083, 0.167, 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, and 8 hours postdose. Complete urine was also collected at predose and at 0- to 8- and 8- to 24-hour collection intervals postdose following intravenous CPX-O and CPX-POM.
Absolute Bioavailability of CPX following Subcutaneous CPX-POM and Oral CPX-O in Rats.
Plasma and urine pharmacokinetics of CPX were characterized following subcutaneous CPX-POM, oral CPX-O, and intravenous CPX-POM to 22 male Sprague-Dawley rats. In this study, intravenous CPX-POM served as the reference treatment. The feasibility of subcutaneous CPX-POM was determined by comparing the plasma pharmacokinetics of the active metabolite, CPX, following subcutaneous and intravenous CPX-POM. The absolute bioavailability of CPX following oral CPX-O (compared with intravenous CPX-POM) was determined to bridge previously reported GLP toxicology data generated in the rat (Kellner et al., 1981; Weir et al., 2018). A single intravenous dose of CPX-POM was administered to six rats, employing the intravenous formulation described above. In a second group of six male rats, a single subcutaneous dose of CPX-POM was administered. Intravenous doses of CPX-POM were administered directly into the tail vein using a syringe and needle. Subcutaneous doses of CPX-POM were administered in two different locations in the scapular region. Intravenous and subcutaneous CPX-POM doses of 47 and 70.6 mg/kg, respectively, were administered to two separate groups of six rats each, of using intravenous formulation concentrations of 23.53 mg/ml. In a third group of six male rats, a single oral dose of 38.8 mg/kg CPX-O was administered using the oral formulation described above at a concentration of 29.39 mg/ml. All dose volumes were adjusted for the weight of the animal on the morning of drug administration. Each treatment was administered under fasting conditions. Serial blood (plasma) samples were collected using a syringe via jugular vein catheter prior to and 0.083, 0.167, 0.25, 0.5, 0.75, 1, 1.5, 2, 3, and 4 hours postdose. In a fourth group of four male Sprague-Dawley rats, complete urine was collected at CPX-POM, subcutaneous CPX-POM, and oral CPX-O. The same four animals received all three treatments in a nonrandomized, complete crossover fashion with 72 hours separating treatments. Only urine was collected in this treatment group. Each treatment was administered under fasting conditions.
Absolute Bioavailability of CPX following Subcutaneous CPX-POM and Oral CPX-O in Dogs.
Plasma and urine pharmacokinetics of CPX were characterized following single subcutaneous CPX-POM, oral CPX-O, and intravenous CPX-POM dose administration under fasting conditions to four male beagle dogs. In this study, intravenous CPX-POM served as the reference treatment. The feasibility of subcutaneous CPX-POM was determined by comparing the plasma pharmacokinetics of the active metabolite, CPX, following subcutaneous and intravenous CPX-POM. The absolute bioavailability of CPX following oral CPX-O (compared with intravenous CPX-POM) was determined to bridge previously reported GLP toxicology data generated in the dog (Kellner et al., 1981; Weir et al., 2018). Intravenous and subcutaneous doses of CPX-POM were administered to dogs employing the intravenous formulation described above at a concentration of 3.5 mg/ml. Intravenous doses of CPX-POM were administered via an indwelling catheter into the cephalic vein at a volume of 2 ml/kg resulting in a dose of 7.3 mg/kg. Subcutaneous CPX-POM was administered to the back between the scapulae at a volume of 6 ml/kg, resulting in a dose of 21.8 mg/kg. CPX-O was administered directly into the stomach via gavage tube. Immediately following oral dose administration, the gavage tube was flushed with approximately 10 ml of water to assure complete delivery of the intended dose. Oral doses were administered at a volume of 6 ml/kg, resulting in an oral CPX-O dose of 12.2 mg/kg. All dose volumes were adjusted for the weight of the animal on the morning of drug administration. Each treatment was administered under fasting conditions. Serial blood (plasma) samples were collected using a syringe via jugular vein prior to and 0.083, 0.167, 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, and 8 hours postdose. Complete urine was also collected at predose and at 0- to 8- and 8- to 24-hour collection intervals postdose following intravenous CPX-POM, subcutaneous CPX-POM, and oral CPX-O.
Bioanalytical Sample Collection, Storage, and Shipment.
Blood samples were transferred to blood collection tubes containing K2EDTA, inverted several times to ensure adequate mixing of the blood with the anticoagulant, and placed on ice prior to processing. Blood samples were centrifuged at 3200 rpm for 10 minutes at approximately 5°C. Plasma samples were transferred into 1.1 ml 96-well plate tubes then stored at −20 ± 5°C until bioanalysis. Urine samples were collected frozen on dry ice into glass collection jars, thawed, transferred to tared plastic jars, and weighed. All urine was stored at −20 ± 5°C until bioanalysis. Plasma and urine samples were transferred from the animal facility to the bioanalytical laboratory on dry ice by same day courier.
Bioanalytical Methods in Rat and Dog Plasma and Urine.
Rat and dog plasma and urine bioanalytical methods were developed and validated under non-GLP conditions to quantitate concentrations of CPX-POM, CPX, and CPX-G. CPX-POM and CPX-G do not appear to be active based on in vitro studies in human high-grade urothelial cancer cell lines (unpublished data).
CPX-POM concentrations were determined in 20 µl sample volumes. Samples were placed in polypropylene centrifuge tubes followed by the addition of another phosphoryl-oxymethyl ester, SN38 prodrug, as the internal standard. An additional 50 µl of acetonitrile was added to each sample, as well as 60 µl of 0.1% formic acid. Samples were then vortexed and extracted with ethyl acetate. Samples were frozen and organic phase transferred into clean polypropylene tubes and evaporated to dryness. Plasma samples were reconstituted in 50 µl of 30% methanol in water containing 0.1% formic acid and 15 µl of each sample injected for analysis by liquid chromatography-mass spectrometry and tandem mass spectrometry (LC-MS/MS) for CPX-POM. The chromatographic separation of CPX-POM and internal standard was achieved on a Phenomenex Kinetex C18 column (100 × 2.1 mm, 2.6 µm) maintained at 40°C. The mobile phase gradient program with solvent A (95:5:0.1 deionized water/acetonitrile/formic acid) and solvent B (95:5:0.1 acetonitrile/deionized water/formic acid) was run at a flow rate of 0.25 ml/min. The gradient started at 25% solvent B and increased linearly to 95% over 6.5 minutes, then reduced linearly to the initial conditions within 0.1 minutes. Total run time was 10 minutes for each sample. An ABSciex 3200 Linear Ion TRAP quadrupole mass spectrometer was operated in the negative electrospray ionization (ESI) mode for mass detection and analysis of CPX-POM. Multiple reaction monitoring (MRM) was used to monitor the precursor/product ion transitions of 316.07/79.1 (CPX-POM) and 500.79/373.0 (SN38 prodrug, internal standard). All mass spectrometer settings were standardized to optimize detection and analysis of CPX-POM and internal standard. The lower limit of quantitation (LLOQ) for CPX-POM was 100 ng/ml, and the standard curve was linear between 100 and 5000 ng/ml, with a correlation coefficient of 0.998.
CPX concentrations were determined in 50 µl sample volumes. Samples were placed in polypropylene centrifuge tubes followed by the addition of internal standard (d3-methoxy-ciclopirox). An equal volume of 0.2 M sodium hydroxide was added to each tube and the sample vortexed. Samples were chilled until ice cold and then derivatized with the addition of 25 µl dimethyl sulfate. Samples were then heated in an oven at 37°C for 30 minutes. After the derivatization reaction, 25 µl of triethylamine was added to each sample to neutralize the reaction. Plasma samples were then extracted using methyl tert-butyl ether and frozen over into clean polypropylene tubes for evaporation and sample concentration. Plasma samples were reconstituted in 100 µl of 40% acetonitrile in water. Five microliters of each prepared sample were injected onto the LC-MS/MS for determination of CPX concentration. The chromatographic separation of CPX and internal standard was achieved on a Waters Sunfire C18 column (50 × 2 mm, 3.5 µm) that was maintained at 40°C. The mobile phase gradient program with solvent A (95:5:0.1 deionized water/acetonitrile/formic acid) and solvent B (95:5:0.1 acetonitrile/deionized water/formic acid) was run at a flow rate of 0.25 ml/min. The gradient started at 25% B and increased linearly to 95% over 6.5 minutes, then reduced linearly to the initial conditions within 0.1 minutes. Total run time for each sample was 10 minutes. An ABSciex 3200 Linear Ion TRAP quadrupole mass spectrometer was operated in the positive ESI mode for mass detection and analysis of CPX. MRM was used to monitor the precursor/product ion transitions of 222.2/136.0 (CPX) and 225.2/136.0 (d3-methoxy-ciclopirox, internal standard). All mass spectrometer settings were standardized to optimize detection and analysis of CPX and internal standard. The LLOQ for CPX was 25 ng/ml and the standard curve was linear between 25 and 5000 ng/ml, with a correlation coefficient of 0.998.
CPX-G concentrations were determined in 20 µl sample volumes. Samples were placed in polypropylene centrifuge tubes followed by the addition of ciclopirox-D11-β-glucuronide as the internal standard in 20 µl of 1:1 acetonitrile:deionized water. Samples were then vortexed followed by transfer of the supernatant to clean polypropylene tubes. Fifteen milliliter aliquots were injected for analysis by LC-MS/MS for CPX-G. For urine samples, an aliquot of urine was diluted 1:10 with 1% formic acid, internal standard added, and samples mixed well prior to injection onto the LC-MS/MS for CPX-G determination. The chromatographic separation of CPX-G and internal standard was achieved on a Zorbax Agilent column (50 × 2.1 mm, 2.6 µm) that was maintained at 15°C. The mobile phase gradient program with solvent A (95:5:0.1 deionized water/acetonitrile/formic acid) and solvent B (95:5:0.1 acetonitrile/deionized water/formic acid) was run at a flow rate of 0.40 ml/min. The gradient started at 15% solvent B and increased linearly to 95% over 1.0 minute beginning at 2.0 minutes, then reduced linearly at 5.1 minutes to the initial conditions at 6.3 minutes. Total run time for each sample was 10 minutes. An ABSciex 3200 Linear Ion TRAP quadrupole mass spectrometer was operated in the negative ESI mode for mass detection and analysis of CPX-G. MRM was used to monitor the precursor/product ion transitions of 384.1 (CPX-G) and 395.1 (ciclopirox-D11-β-glucuronide) internal standard. All mass spectrometer settings were standardized to optimize detection and analysis of CPX-G and internal standard. The LLOQ for CPX-G was 250 ng/ml, and the standard curve was linear between 100 and 5000 ng/ml, with a correlation coefficient of 0.998.
Pharmacokinetic Analysis.
Nonparametric plasma and urine pharmacokinetic parameters for CPX-POM, CPX, and CPX-G were generated from resultant concentration-time data using Phoenix WinNonlin (Version 6.3; Certara, Princeton, NJ) software. For plasma data, actual blood sample collection times were used. Predose plasma concentration values reported as below the LLOQ were designated as below the quantifiable limit and treated as zero in calculation. Below the quantifiable limit values observed in the terminal phase of the plasma concentration-time profiles were treated as missing values in determination of the apparent first-order elimination rate constant and area under the plasma concentration-time curve. Given the absence of quantifiable concentrations of CPX-POM in plasma and urine for all studies in rats and dogs, pharmacokinetic data analysis was not possible for parent drug.
Statistical Analysis.
Descriptive statistics (mean ± S.D.) were reported for the resultant plasma and urine pharmacokinetic parameters. Given the objective of the studies conducted, inferential statistical analysis was not performed.
Data Management.
The study protocol, in-life report including dosing and sample collection data, Excel bioanalytical data, pharmacokinetic data analysis, data tabulation, and graphics files are stored at CicloMed LLC, Kansas City, MO.
Results
The rationale for developing the injectable formulation of CPX-POM was based on pH, solubility, and the desired concentration of active pharmaceutical ingredient in the injectable formulation. The desired concentration of the injectable CPX-POM formulation for study in early phase clinical trials, based on the anticipated dose range being evaluated, was 76.8 mg/ml as CPX-POM disodium heptahydrate (which is equivalent to concentrations of 50 and 32.6 mg/ml CPX-POM and CPX, respectively). Formulating an injectable product containing CPX at this concentration required approximately 300 mg/ml Captisol and adjustment of the pH of the solution to between 9.5 and 10.0. In contrast, CPX-POM for injection is supplied as a sterile solution containing sterile water for injection, anhydrous dibasic sodium phosphate as a buffer, and sodium hydroxide and hydrochloric acid for pH adjustment. The much more desirable pH of the CPX-POM injectable formulation is between 7.5 and 8.0.
Treatments administered to rats and dogs in the four studies, including test article, formulation description, route of administration, dose volume administered, test article dose as well as CPX equivalent dose are summarized in Table 1. CPX-POM intravenous and subcutaneous doses chosen for these studies were selected as well-tolerated doses determined in non-GLP toxicology dose range finding studies conducted in rats and dogs. Oral and intravenous CPX-O doses were selected based on published data as well as available Freedom of Information data released following US Food and Drug Administration approval of topical CPX-containing antifungal drug products. Derived CPX plasma and urine pharmacokinetic parameters (e.g., clearance, absolute bioavailability) were calculated based on CPX equivalent doses. CPX has a pKa of approximately 6.8 for the phenolic hydrogen. The anionic form of CPX has excellent solubility, exceeding 500 mg/ml. The neutral protonated form of CPX, in contrast, is poorly soluble at approximately 0.3 mg/ml. Captisol, at 50 mM (108.15 mg/ml), was added to the CPX formulations used in the studies described herein to avoid potential precipitation of the neutral protonated phenolic form of CPX. The CPX-O intravenous dosing solution for the rat was formulated at pH 7.0 at a concentration of 6.47 mg/ml (5.0 mg/ml CPX). The intravenous CPX-O dosing solution for the dog was more dilute, 1.94 mg/ml CPX-O (1.5 mg/ml CPX). Although not required to solubilize CPX-POM, Captisol was used in the prodrug injectable formulation for rat and dog studies to control for any potential effects of this excipient on the pharmacokinetics of CPX.
The plasma and urine pharmacokinetics of CPX were characterized in the rat following intravenous administration of CPX-O and CPX-POM doses of 13.0 and 17.5 mg/kg, respectively. Intravenous CPX-O was used as the reference in characterizing the bioavailability of CPX following intravenous CPX-POM in this rodent toxicology species. Metabolism of CPX-POM was rapid and complete following intravenous administration of CPX-POM. Only a few blood samples, collected within minutes of intravenous bolus administration, contained plasma CPX-POM concentrations exceeding the assay LLOQ. Plasma CPX concentrations declined rapidly in a biexponential fashion following intravenous administration of both test articles as shown in Fig. 2. Resultant plasma CPX pharmacokinetic parameters are summarized in Table 2. Following intravenous administration of CPX-POM, the systemic availability of CPX was 97% compared with intravenous administration of CPX-O. The apparent elimination half-life and mean residence time values for the active metabolite were less than 1 hour and were similar for both treatments. The systemic clearance of CPX was 3326 ml/h/kg following intravenous CPX-POM. The apparent and steady-state volume of distribution values for CPX following intravenous CPX-POM were 2664 ml/kg and 1853 ml/kg, respectively. These values were comparable to those observed following intravenous CPX-O administration. CPX-POM was not detected in any urine samples collected. Urine CPX pharmacokinetic parameters obtained in rats following intravenous CPX-O and CPX-POM are presented in Table 3. Following single intravenous doses of CPX-POM and CPX-O, less than 1% of the dose was excreted as CPX while a significant portion of the administered dose, approximately 18%–34%, was eliminated as the glucuronide metabolite of CPX following both treatments.
Fig. 2.

Mean ± S.D. plasma CPX concentration-time profiles following single dose intravenous administration of 17.5 mg/kg CPX-POM and 13 mg/kg CPX-O to male Sprague-Dawley rats. Data are presented on rectilinear (A) and semilogarithmic (B) plots as mean ± S.D., n = 6 per treatment group. CPX, ciclopirox; CPX-O, ciclopirox olamine; CPX-POM, fosciclopirox; hr, hour; IV, intravenous.
TABLE 2.
Mean ± S.D. plasma CPX pharmacokinetic parameters in male Sprague-Dawley rats following single intravenous doses of CPX-POM and CPX-O
| Treatmenta | ||
|---|---|---|
| Parameter | CPX-POM | CPX-O |
| Body weight (kg) | 0.26 ± 0.01 | 0.27 ± 0.01 |
| CPX dose (mg/kg)b | 7.47 ± 0.12 | 10.02 ± 0.04 |
| Test article dose (mg/kg)c | 17.56 ± 0.28 | 12.96 ± 0.05 |
| C0 (ng/ml) | 9.310 ± 5251 | 25,883 ± 13,321 |
| Kel (h−1) | 1.580 ± 0.286 | 1.453 ± 0.634 |
| T1/2 (h) | 0.452 ± 0.087 | 0.615 ± 0.404 |
| AUC0∫n (ng⋅h/ml) | 2121 ± 501 | 3077 ± 427 |
| AUC0∫∞ (ng⋅h/ml) | 2161 ± 477 | 3141 ± 349 |
| AUMC0∫n (ng⋅h2/ml) | 827 ± 275 | 947 ± 608 |
| AUMC0∫∞ (ng⋅h2/ml) | 950 ± 229 | 1291 ± 1206 |
| MRT (h) | 0.455 ± 477 | 0.446 ± 0.474 |
| Cl (ml/h/kg) | 3326 ± 296 | 3225 ± 342 |
| Vdz (ml/kg) | 2664 ± 670 | 2983 ± 2319 |
| Vss (ml/kg) | 1853 ± 899 | 1563 ± 1853 |
| F (%) | 96.7 | |
AUC0∫n, area under the plasma CPX concentration versus time curve from time zero to the last measurable time point; AUC0∫∞, area under the plasma CPX concentration versus time curve from time zero to infinity; AUMC0∫n, CPX area under the first moment versus time curve from time zero to the last measurable time point; AUMC0∫∞, CPX area under the first moment versus time curve from time zero to infinity; C0, extrapolated plasma CPX concentration at time zero following intravenous bolus administration; Cl, CPX systemic drug clearance; CPX, ciclopirox; CPX-O, ciclopirox olamine; CPX-POM, fosciclopirox; F, CPX bioavailability; Kel, apparent first-order elimination rate constant; MRT, CPX mean residence time; T1/2, apparent elimination half-life, Vdz, CPX apparent volume of distribution; Vss, CPX apparent steady-state volume of distribution.
N = 6 rats per treatment.
Ciclopirox free acid dose.
CPX-POM disodium heptahydrate was formulated in 25 mM phosphate buffer pH 7 with 50 mM Captisol at a strength of 8.72 mg/ml (3.71 mg/ml ciclopirox free acid), CPX-O was formulated in 25 mM phosphate buffer pH 7 with 50 mM Captisol at a strength of 6.47 mg/ml (5.0 mg/ml ciclopirox free acid).
TABLE 3.
Mean ± S.D. urine CPX and CPX-G pharmacokinetic parameters in male Sprague-Dawley rats following single doses of intravenous CPX-POM and CPX-O
| Treatmenta | ||
|---|---|---|
| Parameter | CPX-POM | CPX-O |
| Body weight (kg) | 0.252 ± 0.006 | 0.238 ± 0.006 |
| CPX dose (mg/kg)b | 7.38 ± 0.01 | 9.98 ± 0.06 |
| Test article dose (mg/kg)c | 17.35 ± 0.03 | 12.91 ± 0.08 |
| UCPX 0–8 h (ng/ml) | 1182 ± 498 | 921 ± 112 |
| UCPX 8–24 h (ng/ml) | 198 ± 172 | 118 ± 45 |
| UCPX 0–8 h (μM) | 4.45 ± 0.54 | 8.79 ± 2.41 |
| UCPX 8–24 h (μM) | 0.57 ± 0.22 | 0.96 ± 0.83 |
| dXe/dtCPX 0–8 h (μg/h) | 0.23 ± 0.09 | 0.76 ± 0.26 |
| dXe/dtCPX 8–24 h (μg/h) | 0.07 ± 0.01 | 0.13 ± 0.04 |
| XeCPX (μg) | 2.88 ± 0.78 | 8.18 ± 2.90 |
| FeCPX (%) | 0.15 ± 0.04 | 0.34 ± 0.12 |
| FeCPX-G (%) | 34.07 ± 12.30 | 17.58 ± 2.46 |
CPX, ciclopirox; CPX-G, ciclopirox glucuronide metabolite; dXe/dtCPX, mass of CPX excreted per hour over × hours following drug administration; FeCPX, fraction of the administered dose excreted as CPX; FECPX-G, fraction of the administered dose excreted as CPX-G; UCPX, urine CPX concentration; XECPX, mass of CPX excreted in urine.
Four rats received both treatments in a complete, nonrandomized, crossover fashion.
Ciclopirox free acid dose.
CPX-POM disodium heptahydrate was formulated in 25 mM phosphate buffer pH 7 with 50 mM Captisol at a strength of 8.72 mg/ml (3.71 mg/ml ciclopirox free acid), CPX-O was formulated in 25 mM phosphate buffer pH 7 with 50 mM Captisol at a strength of 6.47 mg/ml (5.0 mg/ml ciclopirox free acid).
The plasma and urine pharmacokinetics of CPX were characterized in the dog following intravenous administration of 4 mg/kg CPX-O and 5.4 mg/kg CPX-POM. Intravenous CPX-O was used as the reference in characterizing the bioavailability of CPX following intravenous CPX-POM in this nonrodent toxicology species. Metabolism of CPX-POM was rapid and complete following intravenous administration of CPX-POM. Only a few blood samples, collected within minutes of intravenous bolus administration, contained plasma CPX-POM concentrations exceeding the LLOQ. Like the rat, plasma CPX concentrations declined biexponentially in the dog following intravenous administration of both test articles as illustrated in Fig. 3. As summarized in Table 4, the systemic availability of CPX was 105% following intravenous CPX-POM compared with intravenous administration of CPX-O. The apparent elimination half-life and mean residence time values for the active metabolite ranged from 2 to 4 hours following both treatments. The systemic clearance of CPX was 762 ml/h/kg following intravenous CPX-POM. The apparent and steady-state volume of distribution values for CPX were 4133 ml/kg and 3686 ml/kg, respectively, following intravenous CPX-POM. These values were comparable to those observed following intravenous CPX-O administration. Urine CPX pharmacokinetic parameters obtained in dogs following intravenous CPX-O and CPX-POM are summarized in Table 5. CPX-POM was not detected in any urine samples collected. Following single intravenous doses of CPX-POM and CPX-O, less than 1% of the dose was excreted as CPX. Approximately 63% of test article doses were recovered in urine as the glucuronide metabolite.
Fig. 3.

Mean ± S.D. plasma CPX concentration-time rectilinear profiles following a single dose intravenous administration of 5.4 mg/kg CPX-POM and 4.0 mg/kg CPX-O to male beagle dogs. Plasma CPX concentration-time profiles following single dose administration of 5.4 mg/kg intravenous CPX-POM and 4.0 mg/kg CPX-O to 4 male beagle dogs in a complete crossover fashion. Data are presented on rectilinear (A) and semilogarithmic (B) plots as mean ± S.D. CPX, ciclopirox; CPX-O, ciclopirox olamine; CPX-POM, fosciclopirox; hr, hour; IV, intravenous.
TABLE 4.
Mean ± S.D. plasma CPX pharmacokinetic parameters in male beagle dogs following single intravenous doses of CPX-POM and CPX-O
| Treatmenta | ||
|---|---|---|
| Parameter | CPX-POM | CPX-O |
| Body weight (kg) | 9.65 ± 0.38 | 9.64 ± 0.34 |
| CPX dose (mg/kg)b | 2.28 ± 0.01 | 3.10 ± 0.02 |
| Test article dose (mg/kg)c | 5.40 ± 0.02 | 4.01 ± 0.02 |
| C0 (ng/ml) | 2970 ± 969 | 5996 ± 1387 |
| Kel (h−1) | 0.242 ± 0.030 | 0.185 ± 0.019 |
| T1/2 (h) | 2.90 ± 0.329 | 3.77 ± 0.40 |
| AUC0∫n (ng⋅h/ml) | 2571 ± 436 | 3314 ± 655 |
| AUC0∫∞ (ng⋅h/ml) | 3075 ± 591 | 4009 ± 802 |
| AUMC0∫n (ng⋅h2/ml) | 5485 ± 1293 | 7293 ± 1520 |
| AUMC0∫∞ (ng⋅h2/ml) | 11,680 ± 3682 | 16,665 ± 3971 |
| MRT (h) | 3.74 ± 0.58 | 2.20 ± 0.09 |
| Cl (ml/h/kg) | 762 ± 158 | 804 ± 200 |
| Vdz (ml/kg) | 4133 ± 363 | 4253 ± 1266 |
| Vss (ml/kg) | 3686 ± 431 | 3218 ± 810 |
| F (%) | 105.4 ± 11.9 | |
AUC0∫n, area under the plasma CPX concentration versus time curve from time zero to the last measurable time point; AUC0∫∞, area under the plasma CPX concentration versus time curve from time zero to infinity; AUMC0∫n, CPX area under the first moment versus time curve from time zero to the last measurable time point; AUMC0∫∞, CPX area under the first moment versus time curve from time zero to infinity; C0, extrapolated plasma CPX concentration at time zero following intravenous bolus administration; Cl, CPX systemic drug clearance; CPX, ciclopirox; CPX-O, ciclopirox olamine; CPX-POM, fosciclopirox; F, CPX bioavailability; Kel, apparent first-order elimination rate constant; MRT, CPX mean residence time; T1/2, apparent elimination half-life; Vdz, CPX apparent volume of distribution; Vss, CPX apparent steady state volume of distribution.
N = 4 dogs receiving both treatments in a complete, nonrandomized, cross-over study design.
Ciclopirox free acid dose.
CPX-POM disodium heptahydrate was formulated in 25 mM phosphate pH 7 with 50 mM Captisol at a strength of 2.61 mg/ml (1.11 mg/ml ciclopirox free acid), CPX-O was formulated in 25 mM phosphate pH 7 with 50 mM Captisol at a strength of 1.94 mg/ml (1.50 mg/ml ciclopirox free acid).
TABLE 5.
Mean ± S.D. urine CPX pharmacokinetic parameters in male beagle dogs following single intravenous doses of CPX-POM and CPX-O
| Treatmenta | ||
|---|---|---|
| Parameter | CPX-POM | CPX-O |
| Body weight (kg) | 9.65 ± 0.38 | 9.64 ± 0.34 |
| CPX dose (mg/kg)b | 2.28 ± 0.01 | 3.10 ± 0.02 |
| Test article dose (mg/kg)c | 5.40 ± 0.02 | 4.01 ± 0.02 |
| UCPX 0–8 h (ng/ml) | 838 ± 149 | 1006 ± 370 |
| UCPX 8–24 h (ng/ml) | 112.0 ± 56.1 | 72.5 ± 54.5 |
| UCPX 0–8 h (μM) | 4.04 ± 0.72 | 4.86 ± 1.79 |
| UCPX 8–24 h (μM) | 0.541 ± 0.271 | 0.350 ± 0.263 |
| dXe/dtCPX 0–8 h (μg/h) | 9.88 ± 0.3.89 | 12.58 ± 5.61 |
| dXe/dtCPX 8–24 h (μg/h) | 0.97 ± 0.19 | 0.43 ± 0.42 |
| XeCPX (μg) | 94.5 ± 32.6 | 107.5 ± 39.9 |
| FeCPX (%) | 0.428 ± 0.138 | 0.358 ± 0.123 |
| Clr (ml/h/kg)d | 3.38 ± 1.80 | 2.82 ± 0.90 |
| FeCPX-G (%) | 63.46 ± 6.68 | 62.67 ± 12.15 |
CPX, ciclopirox; CPX-G, ciclopirox glucuronide metabolite; dXe/dtCPX, mass of CPX excreted per hour over × hours following drug administration; FeCPX, fraction of the administered dose excreted as CPX; FECPX-G, fraction of the administered dose excreted as CPX-G; IV, intravenous; SD, standard deviation; UCPX, urine CPX concentration; XECPX, mass of CPX excreted in urine.
N = 4 dogs receiving both treatments in a complete, nonrandomized, cross-over study design.
Ciclopirox free acid dose.
CPX-POM disodium heptahydrate was formulated in 25 mM phosphate pH 7 with 50 mM Captisol at a strength of 2.61 mg/ml (1.11 mg/ml ciclopirox free acid), CPX-O was formulated in 25 mM phosphate pH 7 with 50 mM Captisol at a strength of 1.94 mg/ml (1.50 mg/ml ciclopirox free acid).
Renal clearance calculated per kilogram body weight as Clr = Xecpx/AUC0∫∞.
The pharmacokinetics of CPX were characterized following intravenous administration of 47 mg/kg CPX-POM and subcutaneous administration of 70.6 mg/kg CPX-POM, as well as oral administration of 38.8 mg/kg of CPX-O in fasted rats. In this study, intravenous CPX-POM was used as the reference treatment to characterize the absolute bioavailability of subcutaneous CPX-POM as well as to establish a pharmacokinetic bridge between intravenous CPX-POM and oral CPX-O. Resultant mean plasma CPX concentration-time profiles following administration of intravenous and subcutaneous CPX-POM as well as orally administered CPX-O are illustrated in Fig. 4. Plasma CPX concentrations declined rapidly following intravenous administration of CPX-POM. Plasma concentrations of CPX following subcutaneous CPX-POM were consistently greater than those observed following intravenous CPX-POM over the terminal portion of the profile and clearly demonstrated an absorption phase. Comparatively, plasma CPX concentrations following oral administration of CPX-O were consistently quite low. Resultant plasma CPX pharmacokinetic parameters following administration of the three treatments are summarized in Table 6. Systemic clearance, apparent, and steady-state volume of distribution values for CPX following intravenous CPX-POM were 3696 ml/h/kg, 3160 ml/kg, and 1106 ml/kg, respectively. These values were comparable to those observed following intravenous CPX-POM administration to rats described above. The absolute bioavailability of CPX following subcutaneous CPX-POM was 80% with a relatively rapid absorption rate (Tmax = 0.26 hour). In contrast, the bioavailability of CPX following oral administration of CPX-O was 4.6% compared with intravenous CPX-POM. CPX-POM was not detected in any blood or urine sample collected in this study. Urine CPX pharmacokinetic parameters obtained in rats are summarized in Table 7. Less than 1% of the administered dose was recovered in the urine following subcutaneous CPX-POM and CPX-O administration as CPX. Approximately 3.5% and 9% of the dose was recovered in urine as CPX and CPX-G, respectively, following intravenous CPX-POM administration.
Fig. 4.

Mean ± S.D. plasma CPX concentration-time rectilinear profiles following administration of 47 mg/kg intravenous CPX-POM, 70.6 mg/kg subcutaneous CPX-POM, and 38.8 mg/kg CPX-O to 6 rats per treatment group. Plasma CPX concentration-time profiles following single dose administration of 47 mg/kg intravenous CPX-POM, 71 mg/kg subcutaneous CPX-POM, and 39 mg/kg CPX-O to male Sprague-Dawley rats. Data are presented on rectilinear (A) and semilogarithmic (B) plots as mean ± S.D., n = 6 per treatment group. CPX, ciclopirox; CPX-O, ciclopirox olamine; CPX-POM, fosciclopirox; hr, hour; IV, intravenous; PO, oral; SC, subcutaneous.
TABLE 6.
Mean ± S.D. plasma CPX pharmacokinetic parameters in male Sprague-Dawley rats following single doses of intravenous CPX-POM, SC CPX-POM, and oral CPX-O
| Treatmenta | |||
|---|---|---|---|
| Parameter | CPX-POM Intravenous | CPX-POM Subcutaneous | CPX-O Oral |
| Body weight (kg) | 0.314 ± 0.012 | 0.316 ± 0.004 | 0.314 ± 0.013 |
| CPX dose (mg/kg)b | 19.98 ± 0.10 | 30.04 ± 0.06 | 29.78 ± 0.16 |
| Test article dose (mg/kg)c | 47.01 ± 0.24 | 70.68 ± 0.15 | 38.55 ± 0.21 |
| C0 (ng/ml) | 33,569 ± 13,124 | ||
| Cmax (ng/ml) | 5005 ± 808 | 373 ± 158 | |
| Tmax (h) | 0.264 ± 0.123 | 0.250 ± 0.129 | |
| Kel (h−1) | 1.529 ± 0.680 | 1.039 ± 0.253 | 1.234 ± 0.742 |
| T1/2 (h) | 0.537 ± 0.238 | 0.700 ± 0.163 | 0.801 ± 0.558 |
| AUC0∫n (ng⋅h/ml) | 5449 ± 756 | 6548 ± 1007 | 211.2 ± 84.5 |
| AUC0∫∞ (ng⋅h/ml) | 5496 ± 756 | 6633 ± 1019 | 378.0 ± 140.4 |
| AUMC0∫n (ng⋅h2/ml) | 1534 ± 457 | ||
| AUMC0∫∞ (ng⋅h2/ml) | 1687 ± 478 | ||
| MRT (h) | 0.285 ± 0.092 | ||
| Cl (ml/h/kg) | 3696 ± 531 | ||
| Vdz (ml/kg) | 3160 ± 1696 | ||
| Vss (ml/kg) | 1106 ± 394 | ||
| F (%) | 80.3 | 4.61 | |
AUC0∫n, area under the plasma CPX concentration versus time curve from time zero to the last measurable time point; AUC0∫∞, area under the plasma CPX concentration versus time curve from time zero to infinity; AUMC0∫n, CPX area under the first moment versus time curve from time zero to the last measurable time point; AUMC0∫∞, CPX area under the first moment versus time curve from time zero to infinity; C0, extrapolated plasma CPX concentration at time zero following intravenous bolus administration; Cl, CPX systemic drug clearance; CPX, ciclopirox; CPX-O, ciclopirox olamine; CPX-POM, fosciclopirox; F, CPX bioavailability; Kel, apparent first-order elimination rate constant; MRT, CPX mean residence time; T1/2, apparent elimination half-life; Vdz, CPX apparent volume of distribution; Vss, CPX apparent steady-state volume of distribution.
N = 6 rats per treatment.
CPX free acid dose.
CPX-POM disodium heptahydrate was formulated in 25 mM phosphate buffer pH 7 with 50 mM Captisol at a strength of 23.53 mg/ml (10.0 mg/ml ciclopirox free acid); ciclopirox olamine was formulated in Orasweet SF and Water at a strength of 29.39 mg/ml (22.7 mg/ml ciclopirox free acid).
TABLE 7.
Mean ± S.D. urine CPX and CPX-G pharmacokinetic parameters in male Sprague-Dawley rats following single doses of intravenous CPX-POM, SC CPX-POM, and oral CPX-O
| Treatmenta | |||
|---|---|---|---|
| Parameter | CPX-POM Intravenous | CPX-POM Subcutaneous | CPX-O Oral |
| Body weight (kg) | 0.276 ± 0.008 | 0.293 ± 0.007 | 0.289 ± 0.010 |
| CPX dose (mg/kg)b | 20.02 ± 0.07 | 29.97 ± 0.18 | 38.89 ± 0.23 |
| Test article dose (mg/kg)c | 47.10 ± 0.16 | 70.53 ± 0.08 | 30.44 ± 0.18 |
| UCPX (ng/ml) 0–24 h | 20,623 ± 11,948 | 5931 ± 5689 | 1711 ± 1427 |
| UCPX (μM) 0–24 h | 99.63 ± 57.72 | 28.65 ± 27.48 | 8.27 ± 6.89 |
| XeCPX (μg) 0–24 h | 190.5 ± 103.0 | 85.44 ± 65.89 | 22.30 ± 16.65 |
| FeCPX (%) | 3.46 ± 1.90 | 0.98 ± 0.76 | 0.25 ± 0.18 |
| dXe/dtCPX (μg/h) 0–8 h | 18.36 ± 11.6 | 6.22 ± 6.50 | 0.63 ± 0.16 |
| dXe/dtCPX (μg/h) 8–24 h | 2.76 ± 1.79 | 2.22 ± 2.76 | 1.08 ± 1.11 |
| Clr (ml/h) | 34.6 | ||
| Clr (m/h/kg) | 126 | ||
| FeCPX-G (%) | 9.35 ± 9.05 | 13.73 ± 8.83 | 9.13 ± 4.28 |
Clr, renal clearance of the drug from plasma; CPX, ciclopirox; CPX-G, ciclopirox glucuronide metabolite; dXe/dtCPX, mass of CPX excreted per hour over × hours following drug administration; FeCPX, fraction of the administered dose excreted as CPX; FECPX, fraction of the administered dose excreted as CPX; UCPX, urine CPX concentration; XeCPX, mass of CPX excreted in urine.
N = 4 rats who received all three treatments in a complete, nonrandomized, crossover fashion.
CPX free acid dose.
CPX-POM disodium heptahydrate was formulated in 25 mM phosphate buffer pH 7 with 50 mM Captisol at a strength of 23.53 mg/ml (10.0 mg/ml ciclopirox free acid).
The pharmacokinetics of CPX were characterized following intravenous administration of 7.3 mg/kg CPX-POM and subcutaneous administration of 21.8 mg/kg CPX-POM, as well as the administration of 12.2 mg/kg CPX-O to four fasted beagle dogs. In this study, intravenous CPX-POM was used as the reference treatment to characterize the absolute bioavailability of subcutaneous CPX-POM as well as to establish a pharmacokinetic bridge between intravenous CPX-POM and oral CPX-O. Resultant plasma CPX pharmacokinetic parameters are summarized in Table 8. Plasma CPX concentrations declined rapidly following intravenous administration of CPX-POM as illustrated in Fig. 5. Plasma concentrations of CPX following subcutaneous CPX-POM were consistently greater than those observed following intravenous CPX-POM over the terminal portion of the profile, while plasma CPX concentrations following oral administration of CPX-O were comparatively quite low. The absolute bioavailability of CPX following subcutaneous CPX-POM was complete, 106%, in the dog based on area-under-the-plasma-CPX-concentration-time curve to the last sampling time (8 hours postdose). The rate of absorption from the injection site was dramatically slower, Tmax = 5.5 hours compared with the rat. The absolute bioavailability of CPX following oral CPX-O was approximately 17%. CPX-POM was not detected in any blood or urine sample collected. Systemic clearance, apparent, and steady-state volume of distribution values for CPX following intravenous CPX-POM were 615 ml/h/kg, 4486 ml/kg, and 3776 ml/kg, respectively. These values were comparable to those observed following intravenous CPX-POM administration to dogs described above. Urine CPX pharmacokinetic parameters obtained in dogs are summarized in Table 9. Less than 1% of the dose was excreted as CPX following each the three treatments while 37%–49% of the dose was eliminated as the glucuronide conjugate.
TABLE 8.
Mean ± S.D. plasma CPX pharmacokinetic parameters in male beagle dogs following single doses of intravenous CPX-POM, subcutaneous CPX- POM, and oral CPX-O
| Treatmenta | |||
|---|---|---|---|
| Parameter | CPX-POM Intravenous | CPX-POM Subcutaneous | CPX-O Oral |
| Body weight (kg) | 9.58 ± 0.49 | 9.91 ± 0.81 | 9.48 ± 0.64 |
| CPX dose (mg/kg)b | 3.10 ± 0.02 | 9.28 ± 0.09 | 9.39 ± 0.041 |
| Test article dose (mg/kg)c | 7.30 ± 0.04 | 21.88 ± 0.14 | 12.14 ± 0.05 |
| C0 (ng/ml) | 3770 ± 1280d | ||
| Cmax (ng/ml) | 1988 ± 374 | 1017 ± 271 | |
| Tmax (h) | 0.264 ± 0.122 | 0.250 ± 0.129 | |
| Kel (h−1) | 0.153 ± 0.072 | 0.137 ± 0.031 | 0.126 ± 0.009 |
| T1/2 (h) | 5.54 ± 2.92 | 5.27 ± 1.18 | 5.52 ± 0.39 |
| AUC0∫n (ng⋅h/ml) | 3622 ± 255 | 11,518 ± 2632 | 1901 ± 650 |
| AUC0∫∞ (ng⋅h/ml) | 5262 ± 1243 | 19,614 ± 6161 | 2697 ± 1013 |
| AUMC0∫n (ng⋅h2/ml) | 8836 ± 1186 | ||
| AUMC0∫∞ (ng⋅h2/ml) | 38,965 ± 29,492 | ||
| MRT (h) | 6.76 ± 3.76 | ||
| Cl (ml/h/kg) | 615 ± 145 | ||
| Vdz (ml/kg) | 4486 ± 1366 | ||
| Vss (ml/kg) | 3776 ± 1266 | ||
| F (%) | 106 ± 22.3e | 17.4 ± 5.65 | |
AUC0∫n, area under the plasma CPX concentration versus time curve from time zero to the last measurable time point; AUC0∫∞, area under the plasma CPX concentration versus time curve from time zero to infinity; AUMC0∫n, CPX area under the first moment versus time curve from time zero to the last measurable time point; AUMC0∫∞, CPX area under the first moment versus time curve from time zero to infinity; C0, extrapolated plasma CPX concentration at time zero following intravenous bolus administration; Cl, CPX systemic drug clearance; Cmax, maximum plasma CPX concentration following single dose administration; CPX, ciclopirox; CPX-O, ciclopirox olamine; CPX-POM, fosciclopirox; F, CPX bioavailability; Kel, apparent first-order elimination rate constant; MRT, CPX mean residence time; T1/2, apparent elimination half-life; Tmax, time to maximal plasma CPX concentration; Vdz, CPX apparent volume of distribution; Vss, CPX apparent steady state volume of distribution.
N = 4 dogs who received all three treatments in a nonrandomized, complete cross-over fashion.
Ciclopirox free acid dose.
CPX-POM disodium heptahydrate was formulated in 25 mM phosphate buffer pH 7 with 50 mM Captisol at a strength of 3.53 mg/ml (1.50 mg/ml ciclopirox free acid), CPX-O was formulated in 25 mM phosphate buffer pH 7 with 50 mM Captisol at a strength of 1.94 mg/ml (1.50 mg/ml ciclopirox free acid).
C0 corresponds to y-intercept value derived from non-parametric pharmacokinetic data analysis.
Absolute bioavailability for subcutaneous CPX-POM was determined using AUC0∫n given 39% of AUC0∫∞ was comprised of extrapolated AUC.
Fig. 5.

Mean ± S.D. plasma CPX concentration-time rectilinear profiles following intravenous administration of 7.3 mg/kg CPX-POM, SC administration of 21.8 mg/kg SC CPX-POM, and 12.2 mg/kg oral CPX-O to four male beagle dogs. Plasma CPX concentration-time profiles following single dose administration of 7.3 mg/kg intravenous CPX-POM, 21.8 mg/kg SC CPX-POM, and 12.2 mg/kg oral CPX-O to four male beagle dogs in a complete crossover fashion. Data are presented on rectilinear (A) and semilogarithmic (B) plots as mean ± S.D. CPX, ciclopirox; CPX-O, ciclopirox olamine; CPX-POM, fosciclopirox; hr, hour; IV, intravenous; PO, oral; SC, subcutaneous.
TABLE 9.
Mean ± S.D. urine CPX and CPX-G pharmacokinetic parameters in male beagle dogs following single doses of intravenous CPX-POM, SC CPX-POM, and oral CPX-O
| Treatmenta | |||
|---|---|---|---|
| Parameter | CPX-POM Intravenous | CPX-POM Subcutaneous | CPX-O Oral |
| Body weight (kg) | 9.58 ± 0.49 | 9.91 ± 0.81 | 9.48 ± 0.64 |
| CPX dose (mg/kg)b | 3.10 ± 0.02 | 9.28 ± 0.09 | 9.39 ± 0.04 |
| test article dose (mg/kg)c | 7.30 ± 0.04 | 21.88 ± 0.14 | 12.14 ± 0.05 |
| UCPX (ng/ml) 0–24 h | 957 ± 508 | 2530 ± 958 | 1602 ± 750 |
| UCPX (μM) 0–24 h | 4.62 ± 2.45 | 12.2 ± 4.6 | 7.74 ± 3.62 |
| XeCPX (μg) 0–24 h | 208 ± 117 | 339 ± 206 | 299 ± 208 |
| FeCPX (%) | 0.70 ± 0.38 | 0.38 ± 0.24 | 0.32 ± 0.21 |
| Xe/dtCPX (μg/h) 0–8 h | 19.8 ± 15.2 | 23.0 ± 16.6 | 31.6 ± 26.2 |
| Xe/dtCPX (μg/h) 8–24 h | 3.10 ± 3.33 | 9.67 ± 9.49 | 2.90 ± 1.70 |
| Clr (ml/h/kg)d | 4.47 + 3.35 | ||
| FeCPX-G (%) | 49.2 ± 34.47 | 43.0 ± 19.7 | 37.0 ± 18.7 |
CPX, ciclopirox; CPX-G, ciclopirox glucuronide metabolite; CPX-O, ciclopirox olamine; CPX-POM, fosciclopirox; dXe/dtCPX, mass of CPX excreted per hour over × hours following drug administration; FeCPX, fraction of the administered dose excreted as CPX; FECPX-G, fraction of the administered dose excreted as CPX-G; UCPX, urine CPX concentration; XECPX, mass of CPX excreted in urine.
N = 4 dogs who received all three treatments in a nonrandomized, complete crossover fashion.
Ciclopirox free acid dose.
CPX-POM disodium heptahydrate was formulated in 25 mM phosphate buffer pH 7 with 50 mM Captisol at a strength of 3.53 mg/ml (1.50 mg/ml ciclopirox free acid), CPX-O was formulated in 25 mM phosphate buffer pH 7 with 50 mM Captisol at a strength of 1.94 mg/ml (1.50 mg/ml ciclopirox free acid).
Renal clearance calculated per kilogram body weight as Clr = Xecpx/AUC0∫∞.
Discussion
Incorporation of the phosphoryl-oxymethyl (POM) prodrug moiety has been an effective approach to improving aqueous solubility and oxidative stability of successful commercial products such as fosphenytoin and fospropofol (Stella, 1996; Stella and Nti-Addae, 2007; Dhareshwar and Stella, 2010). In vivo conversion of POM prodrugs to their active metabolites is mediated by phosphatases (Dhareshwar and Stella, 2008). Following single intravenous and subcutaneous doses of CPX-POM to rats and dogs, rarely was the prodrug detected in plasma and never in urine. Furthermore, we demonstrated complete bioavailability of the active metabolite, CPX, following intravenous CPX-POM. This rapid and complete metabolism of the POM prodrug of CPX observed in rats and dogs is consistent with circulating phosphatases.
The rodent and nonrodent toxicology species evaluated in oral GLP toxicology studies conducted to support registration of CPX and CPX-O containing topical drug products were rats and dogs, respectively (Loprox, 2003). To support development of CPX-POM, these same species were selected for the studies described herein. Systemic clearance values for CPX determined in rats and dogs were quite comparable to reported hepatic blood flow rates (Davies and Morris, 1993), suggesting the active metabolite is a high clearance drug. Despite rapid clearance of the active metabolite, CPX-POM administered intravenous to rats and dogs at doses used in these studies, resulted in plasma CPX concentrations exceeding in vitro IC50 values in human leukemia and lymphoma cell lines several-fold (Eberhard et al., 2009; Song et al., 2011). CPX has been shown to be extensively metabolized via glucuronidation followed by renal excretion in rats, dogs, and humans (Kellner et al., 1981). The relatively low urinary recoveries of CPX observed in rats and dogs, along with renal clearance values of the active metabolite, are consistent with extensive hepatic metabolism. Despite reported protein binding of 96% (Kellner et al., 1981), CPX distributes beyond the central (blood) compartment in rats and dogs based on steady-state and apparent volume of distribution values observed in these studies.
The low absolute bioavailability of CPX following oral administration of CPX-O to rats and dogs compared with intravenous CPX-POM, is consistent with the drug or active metabolite undergoing significant presystemic metabolism. These data established a pharmacokinetic bridge between CPX-POM and the olamine salt of CPX contained in marketed antifungal agents, and was useful in designing intravenous CPX-POM GLP toxicology studies subsequently conducted in these species. Excellent bioavailability of CPX was observed following subcutaneous administration, demonstrating feasibility of this route of administration should CPX-POM treatment prove appropriate in an outpatient or ambulatory cancer treatment setting. CPX-POM is being developed for the treatment of nonmuscle invasive and muscle invasive bladder cancer.
Acknowledgments
We would like to thank KUCC’s Lead Development and Optimization Shared Resource for their screening and formulation expertise as well as Nancy Bella of The Gnomon Group LLC for her editorial support.
Abbreviations
- CPX
ciclopirox
- CPX-O
ciclopirox olamine
- CPX-POM
fosciclopirox
- ESI
electrospray ionization
- GLP
Good Laboratory Practice
- LLOQ
lower limit of quantitation
- MRM
multiple reaction monitoring
- POM
phosphoryl-oxymethyl
Authorship Contributions
Participated in research design: Weir, Wood, McKenna, McCulloch, Dalton, Jensen, Baltezor, Anant, Taylor.
Conducted experiments: Weir, Wood, Schorno, McKenna.
Contributed new reagents or analytic tools: Ramamoorthy, Heppert, Rajewski, Tanol, Reed, Baltezor, Anant, Taylor.
Performed data analysis: Weir, Wood, Brinker.
Wrote or contributed to the writing of the manuscript: Weir, Wood, Brinker, Ham, Reed, Jensen, Baltezor, Anant, Taylor.
Footnotes
Research reported in this presentation was supported by the National Institutes of Health National Cancer Institute Cancer Center Support Grant [Grant P30 CA168524] and by a public-private partnership between The Institute for Advancing Medical Innovation at the University of Kansas Medical Center and CicloMed LLC, Kansas City, MO.
References
- Davies B, Morris T. (1993) Physiological parameters in laboratory animals and humans. Pharm Res 10:1093–1095. [DOI] [PubMed] [Google Scholar]
- Dhareshwar SS, Stella VJ. (2008) Your prodrug releases formaldehyde: should you be concerned? No!. J Pharm Sci 97:4184–4193. [DOI] [PubMed] [Google Scholar]
- Dhareshwar SS, Stella VJ. (2010) A novel prodrug strategy for beta-dicarbonyl carbon acids: syntheses and evaluation of the physicochemical characteristics of C-phosphoryloxymethyl (POM) and phosphoryloxymethyloxymethyl (POMOM) prodrug derivatives. J Pharm Sci 99:2711–2723. [DOI] [PubMed] [Google Scholar]
- Eberhard Y, McDermott SP, Wang X, Gronda M, Venugopal A, Wood TE, Hurren R, Datti A, Batey RA, Wrana J, et al. (2009) Chelation of intracellular iron with the antifungal agent ciclopirox olamine induces cell death in leukemia and myeloma cells. [published correction appears in Blood (2010) 115:4006]. Blood 114:3064–3073. [DOI] [PubMed] [Google Scholar]
- Loprox (2003) Ciclopirox Shampoo 1% Pharmacology/Toxicology NDA 21-159 Review. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2003/21159_Loprox_pharmr.pdf H.H. Services.
- Gupta AK. (2001) Ciclopirox: an overview. Int J Dermatol 40:305–310. [DOI] [PubMed] [Google Scholar]
- Gupta AK, Plott T. (2004) Ciclopirox: a broad-spectrum antifungal with antibacterial and anti-inflammatory properties. Int J Dermatol 43 (Suppl 1):3–8. [DOI] [PubMed] [Google Scholar]
- Gupta AK, Sauder DN, Shear NH. (1994) Antifungal agents: an overview. Part I. J Am Acad Dermatol 30:677–698, quiz 698–700. [PubMed] [Google Scholar]
- Kellner HM, Arnold C, Christ OE, Eckert HG, Herok J, Hornke I, Rupp W. (1981) [Pharmacokinetics and biotransformation of the antimycotic drug ciclopiroxolamine in animals and man after topical and systemic administration]. Arzneimittelforschung 31 (8A):1337–1353. [PubMed] [Google Scholar]
- Minden MD, Hogge DE, Weir SJ, Kasper J, Webster DA, Patton L, Jitkova Y, Hurren R, Gronda M, Goard CA, et al. (2014) Oral ciclopirox olamine displays biological activity in a phase I study in patients with advanced hematologic malignancies. Am J Hematol 89:363–368. [DOI] [PubMed] [Google Scholar]
- Sakurai K, Sakaguchi T, Yamaguchi H, Iwata K. (1978) Mode of action of 6-cyclohexyl-1-hydroxy-4-methyl-2(1H)-pyridone ethanolamine salt (Hoe 296). Chemotherapy 24:68–76. [DOI] [PubMed] [Google Scholar]
- PENLAC Product Monograph (2012) Ciclopirox Topical Solution, 8% w/w Nail Lacquer. Valeant Canada LP. https://pdf.hres.ca/dpd_pm/00015736.PDF.
- Song S, Christova T, Perusini S, Alizadeh S, Bao RY, Miller BW, Hurren R, Jitkova Y, Gronda M, Isaac M, et al. (2011) Wnt inhibitor screen reveals iron dependence of β-catenin signaling in cancers. Cancer Res 71:7628–7639. [DOI] [PubMed] [Google Scholar]
- Stella V. (1996) A case for prodrugs: fosphenytoin. Adv Drug Deliv Rev 19:311–330. [Google Scholar]
- Stella VJ, Nti-Addae KW. (2007) Prodrug strategies to overcome poor water solubility. Adv Drug Deliv Rev 59:677–694. [DOI] [PubMed] [Google Scholar]
- Tanol M and Weir S (2013) inventors, USPTO, assignee. Prodrugs of 6-cyclohexyl-1-hydroxy-4-4methylyridin-2-(1H)-one and derivatives thereof. Patent US 8609637. US8609637B2 Filed Dec 2, 2011 and Issued Dec 7, 2013.
- Tanol M and Weir S (2015) inventors, JPO (Japan), assignee. Prodrugs of 6-cyclohexyl-1-hydroxy-4-4methylyridin2-(1H)-one and derivatives thereof. Japanese Patent No. 5853028. Filed Dec 2, 2011 and Issued Feb 9, 2016.
- Tanol M and Weir S (2016) inventors, WIPO (Austria, Belgium, Croatia, Czech Republic, Denmark, Finland, France, Germany, Greece, Hungary, Italy, Ireland, Luxembourg, Netherlands, Norway, Poland, Portugal, Serbia, Spain, Sweden, Switzerland, Turkey and United Kingdom), assignee. Prodrugs of 6-cyclohexyl-1-hydroxy-4-4methylyridin-2-(1H)-one and derivatives thereof. Patent PCT 2646035. Filed Dec 2, 2011 and Issued March 9, 2016.
- Weir SJ, Wood R, Ham T, Ranjarajan P, Ramamoorthy P, Rajewski L, Heppert K, Haslam J, Schorno K, Dalton M, et al. (2018) Preclinical development of ciclopirox prodrug for the treatment of non-muscle invasive and muscle invasive bladder cancer. J Clin Oncol 36:e14576. [Google Scholar]