


Abstract
DNA folding and dynamics along with major nuclear functions are determined by chromosome structural properties, which remain, thus far, elusive in vivo. Here, we combine polymer modeling and single particle tracking experiments to determine the physico-chemical parameters of chromatin in vitro and in living yeast. We find that the motion of reconstituted chromatin fibers can be recapitulated by the Rouse model using mechanical parameters of nucleosome arrays deduced from structural simulations. Conversely, we report that the Rouse model shows some inconsistencies to analyze the motion and structural properties inferred from yeast chromosomes determined with chromosome conformation capture techniques (specifically, Hi-C). We hence introduce the Rouse model with Transient Internal Contacts (RouseTIC), in which random association and dissociation occurs along the chromosome contour. The parametrization of this model by fitting motion and Hi-C data allows us to measure the kinetic parameters of the contact formation reaction. Chromosome contacts appear to be transient; associated to a lifetime of seconds and characterized by an attractive energy of –0.3 to –0.5 kBT. We suggest attributing this energy to the occurrence of histone tail-DNA contacts and notice that its amplitude sets chromosomes in ‘theta’ conditions, in which they are poised for compartmentalization and phase separation.
INTRODUCTION
The genome is organized into higher-order functional domains and territories (1) that contribute to the regulation of gene expression and to cell differentiation (2). In interphase of budding yeast cells cycle, chromosomes adopt a Rabl-like conformation (3), which is characterized by the positioning of centromeres and the nucleolus at opposite ends in the nucleoplasm. This large-scale organization has adequately been reproduced in silico by modeling chromosomes as homogeneous polymers (homopolymers) with some structural constraints associated to peripheral tethering of telomeres and centromeres (4–7). A detailed analysis of chromosome fluctuations with the Rouse model, which is a generic polymer model relevant to crowded environments, further corroborated the relevance of this homopolymer model in yeast (8). Indeed, the dynamics of chromosome loci, as measured by the temporal variation of the mean square displacement (MSD), appeared to follow an anomalous response characterized by:
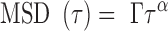 |
(1) |
with 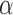 the diffusion exponent,
the diffusion exponent, 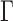 the amplitude, and
the amplitude, and 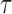 the time interval. The exponent
the time interval. The exponent 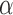 was in the range of 0.4–0.6 and the amplitude
was in the range of 0.4–0.6 and the amplitude 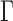 appeared to be homogeneous in the genome except for the proximity of telomeres and centromeres (9–16). Accordingly, the Rouse model predicts that the MSD should increase over time with an exponent of 0.5 and that the amplitude of fluctuations is determined by the local properties of chromosomes, specifically the bending flexibility and solvent friction resisting monomer displacements (17). Using the Kuhn length b, which is twice the persistence length of the polymer, as a proxy of the bending stiffness, and the monomer friction coefficient
appeared to be homogeneous in the genome except for the proximity of telomeres and centromeres (9–16). Accordingly, the Rouse model predicts that the MSD should increase over time with an exponent of 0.5 and that the amplitude of fluctuations is determined by the local properties of chromosomes, specifically the bending flexibility and solvent friction resisting monomer displacements (17). Using the Kuhn length b, which is twice the persistence length of the polymer, as a proxy of the bending stiffness, and the monomer friction coefficient 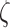 , the Rouse model predicts the MSD in 2D:
, the Rouse model predicts the MSD in 2D:
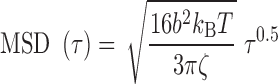 |
(2) |
with kBT the Boltzmann thermal energy. Note that this expression is obtained with the assumption that the chain contains a large number of monomers and behaves as a ‘phantom polymer’, i.e. without effects of volume exclusion between monomers. Taking the standard approximation that the friction coefficient 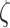 is proportional to the Kuhn length (17), the amplitude of MSD fluctuations
is proportional to the Kuhn length (17), the amplitude of MSD fluctuations 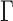 is expected to provide a means to infer the bending flexibility of chromosomes in vivo. However, direct adjustment with the Rouse theory showed that the in vivo value of
is expected to provide a means to infer the bending flexibility of chromosomes in vivo. However, direct adjustment with the Rouse theory showed that the in vivo value of 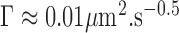 corresponded to a Kuhn length as small as 1–5 nm (9). This estimate appeared to be incompatible with structural and mechanical models of nucleosome fibers (18), as well as the recurrent detection of irregular 10-nm fibers by electron microscopy (EM) of thin nuclear sections (19–21). This inconsistency shows that the precise mechanisms underlying chromosome motion remain largely unclear.
corresponded to a Kuhn length as small as 1–5 nm (9). This estimate appeared to be incompatible with structural and mechanical models of nucleosome fibers (18), as well as the recurrent detection of irregular 10-nm fibers by electron microscopy (EM) of thin nuclear sections (19–21). This inconsistency shows that the precise mechanisms underlying chromosome motion remain largely unclear.
The study of chromosome higher-order structure based on chromosome conformation capture techniques (specifically, Hi-C) has recently shed light on the contribution of two essential mechanisms, which are not included in the simple Rouse model: active processes involving loop extrusion or transcription, and segregation through the random association of monomers along the chain (22–24). Theoretical studies have suggested that both mechanisms could modulate the amplitude of chromatin fluctuations (25,26). Consequently, we were motivated to clarify if and whether additional mechanisms had to be integrated into the Rouse model in order to recapitulate chromosome dynamics in yeast. In order to test polymer models in controlled settings, we first design a biomimetic system to validate the relevance of the Rouse model to analyze DNA and chromatin fluctuation in bulk. This study shows that fluctuation data are quantitatively fitted with the Rouse model parametrized with mechanical parameters of DNA and chromatin. Conversely, in living yeast, the inconsistency of the Rouse model to fit fluctuation and Hi-C data forced us to introduce an equilibrium polymer model, the RouseTIC model, in which random Transient Internal Contacts along the chromosome contour were considered. After a rigorous fitting of the kinetic parameters of the chromosome contact formation reaction, we show that this model recapitulates Hi-C and MSD measurements. Chromosome contacts appear to be transient, lasting a few seconds, and characterized by an interaction energy in the range of –0.3 to –0.5 kBT. We finally speculate that these labile interactions along the chromosome contour may originate from unspecific histone tail interactions and constitute a key mechanism for the formation of dynamic compartments in eukaryotes.
MATERIALS AND METHODS
DNA and chromatin preparation and characterization
Unless stated, chemical and biochemical reagents were purchased from Sigma-Aldrich or New England Biolabs, respectively. Chromosome fragments were purified from U2OS cells, which were synchronized at the beginning of S phase, then released in the presence of dUTP-Cy3 and scraped from glass surfaces in order to allow fluorescent nucleotide entry into the cells and incorporation into the genome during DNA replication (see (27) for details of the protocol). Cells were eventually embedded in agarose plugs, and treated with 5% SDS and 100 μg/ml proteinase-K during two days in order to purify genomic DNA. The resulting chromosome fragments were recovered from agarose plugs using β-Agarase treatment during 2 h. Nucleosome arrays were assembled with a reconstitution kit (Active Motif) using ∼10 ng of purified chromosome fragments mixed with 1 μg of unlabeled λ-DNA.
Structural characterization of chromatin fibers was carried out by electron microscopy using the protocol of (28). Briefly, 5 μl of reaction product diluted to ∼10−12 M in 10 mM Tris–HCl pH 7.5, 5 mM MgCl2, 50 mM, NaCl was deposited on a 600-mesh copper grid covered with a thin carbon film, activated by glow-discharge in the presence of pentylamine. Grids were washed with aqueous 2% (w/v) uranyl acetate, dried and observed in the annular dark-field mode, using a Zeiss 902 transmission electron microscope. Images were captured at a magnification of 85 000 or 140 000 with a Veletta CCD camera and analyzed with iTEM software (Olympus Soft Imaging Solution).
For imaging experiments, DNA or chromatin fibers were diluted 1000-fold in a low salt buffer (Tris–HCl 89 mM, 89 mM boric acid, 2 mM EDTA; pH 8.3) supplemented with 360 or 40 kDa PVP (Sigma-Aldrich). For DNA motion analysis, the viscosity was 5.4 or 2.3 mPa.s with the same mass:volume fraction of 2% of 360 or 40 kDa PVP, respectively. The 360 kDa PVP concentration was set to 3.2% and the viscosity to 15 mPa.s for chromatin loci tracking experiments. The overlapping concentration of these PVP solutions has been estimated in ref. (29). Note that very low concentrations of 100 nm carboxylated polystyrene fluorescent beads (Invitrogen) were added to the buffer in order to measure the viscosity by particle tracking.
Yeast culture
We used a fluorescent yeast strain containing one fluorescent locus with 224 tetO sequences to maximize signal-to-noise ratio located at genomic position 530 kb on chromosome XII (13). Cells were grown overnight at 30°C in YP media containing 2% glucose. They were then diluted at 106 cells/ml and harvested when OD600 reached 4 × 106 cells/ml. Cells were spread on slides coated with corresponding patch containing 2% agar and 2% glucose. Cover slides were sealed with ‘VaLaP’ (1/3 vaseline, 1/3 lanoline, 1/3 paraffin). Microscopy was performed during the first 10–20 min after the encapsulation of the cells in the chamber. Characterization of the signal-to-noise ratio of the fluorescent locus is reported in Supplementary Figure S1 with one example of a time lapse of a fluorescent focus.
Microscopy and trajectory analysis
DNA/chromatin or chromosome loci were tracked with a Zeiss AxioObserver or a Nikon TI-E/B inverted microscope, respectively. The Nikon status was equipped with an EM-CCD IxonULTRA DU897 camera (Andor), a 488 nm laser illumination (Sapphire 488, Coherent), and 100× oil immersion objective (numerical aperture = 1.4) with a 1.5× magnification placed ahead of the camera. Pixel size was 107 nm. The Zeiss status was equipped with an Zyla 4.2 sCMOS camera (Andor), a Sola light engine (Lumencor), a 40X oil immersion objective (NA = 1.4) placed ahead of the camera. Pixel size was 162.5 nm.
Trajectories with more than ∼100 consecutive positions were subsequently extracted with the TrackMate Plugin (30). We processed the MSD and the kurtosis 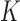 , as defined by (31):
, as defined by (31):
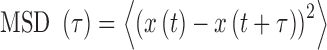 |
(3) |
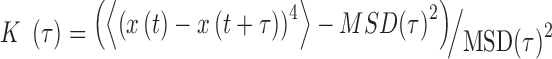 |
(4) |
with 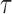 the time interval. We also extracted the step distribution function (SDF) for a given time interval
the time interval. We also extracted the step distribution function (SDF) for a given time interval 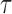 . For each trajectory, these MSD and kurtosis were computed for τ smaller than 30% of the total duration of the track in order to obtain statistically significant values in single cell datasets (32).
. For each trajectory, these MSD and kurtosis were computed for τ smaller than 30% of the total duration of the track in order to obtain statistically significant values in single cell datasets (32).
Monte Carlo Simulations of nucleosome arrays
The chromatin fiber was modelled as a string of coarse-grained DNA linkers and nucleosome core particles following the methodology described in (33). The nucleosomal DNA consists of 14 segments of 10.5 bp, anchored to the histone core by 14 minor groove locations, usually referred as Super Helix Locations (34). The linker DNA is made of an integer number of identical segments, each one containing a number of base pairs as close as possible to 10.5 bp. For example, we modeled a 22 bp linker with two segments of 11 bp or a 18 bp linker with two segments of 9 bp. Note that the length l of each linker segment is proportional to the number n of base pairs with 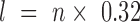 nm and the helical angle between consecutive base pairs is 34°. The articulation between linker DNA segments is modelled as a ball-and-socket joint with an energy penalty corresponding to the bending and twisting restoring torques of the DNA double helix. The bending energy is
nm and the helical angle between consecutive base pairs is 34°. The articulation between linker DNA segments is modelled as a ball-and-socket joint with an energy penalty corresponding to the bending and twisting restoring torques of the DNA double helix. The bending energy is 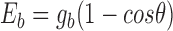 where
where 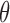 is the bending angle between two consecutive segments and gb is calculated from
is the bending angle between two consecutive segments and gb is calculated from 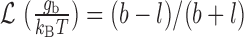 with b = 100 nm and
with b = 100 nm and 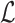 the Langevin function. The twisting energy is given by
the Langevin function. The twisting energy is given by 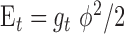 in the harmonic approximation where ϕ is the twist angle of two consecutive segments. The twisting energy constant
in the harmonic approximation where ϕ is the twist angle of two consecutive segments. The twisting energy constant 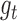 between two connected segments of length l is given by
between two connected segments of length l is given by 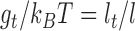 with lt = 95 nm the twist persistence length of naked DNA. At each step of the simulation, we chose one segment at random and rotated all the next segments around a random axis with a random angle (Pivot move). The resulting conformation was accepted following the Metropolis criterion.
with lt = 95 nm the twist persistence length of naked DNA. At each step of the simulation, we chose one segment at random and rotated all the next segments around a random axis with a random angle (Pivot move). The resulting conformation was accepted following the Metropolis criterion.
Extraction of chromatin Kuhn length from nucleosome array Monte Carlo simulations
We used the end-to-end distance distribution as readout of Monte-Carlo simulations to extract the Kuhn length of every nucleosome array configuration (see details in Supplementary Material 1). Briefly, we assumed that nucleosome arrays behaved according to the Worm-Like Chain model, which is defined by the persistence length 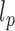 as half the Kuhn length and the contour length
as half the Kuhn length and the contour length 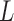 of the chain. We developed a specific approach to estimate these two parameters independently from the analysis of the end-to-end distance distribution based on (35). The second and fourth moments of the distribution can indeed be expressed as a function of
of the chain. We developed a specific approach to estimate these two parameters independently from the analysis of the end-to-end distance distribution based on (35). The second and fourth moments of the distribution can indeed be expressed as a function of 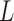 and
and 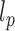 , and are the most relevant whenever the contour length
, and are the most relevant whenever the contour length 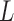 is comparable to
is comparable to 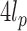 . Therefore, we simulated sufficiently, but not overly, long fibers with up to 100 nucleosomes and then, using an inversion method, extracted both
. Therefore, we simulated sufficiently, but not overly, long fibers with up to 100 nucleosomes and then, using an inversion method, extracted both 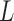 and
and 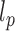 from the second and fourth moments. In order to limit end effects, we performed this extraction for central sub-chains containing 5 to 95 nucleosomes (Supplementary Figure S2). The stationary value of the persistence length detected for a contour length
from the second and fourth moments. In order to limit end effects, we performed this extraction for central sub-chains containing 5 to 95 nucleosomes (Supplementary Figure S2). The stationary value of the persistence length detected for a contour length 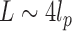 was used to estimate
was used to estimate 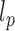 .
.
Kinetic Monte Carlo simulations of coarse-grained homopolymers with internal contacts
We used a coarse-grained self-avoiding homopolymer model for chromosome structure and fluctuations, as described in (36). Briefly, we modeled a typical chromosome containing 106 bp by a polymer chain of N monomers, each of size b and corresponding to n base pairs (N*n = 106). The dynamics of the chain were simulated on a face-centered-cubic lattice with periodic boundary conditions using a simple kinetic Monte-Carlo scheme with local moves: at each trial move, a monomer is randomly picked and randomly displaced to a nearest-neighbor (NN) site on the lattice. The move is accepted if the trial position is empty and maintains the chain connectivity. The density of monomer in the simulation box was set to 15% to effectively account for the presence of other chromosomes.
For the simulations of the Rouse model with transient internal contacts, we assumed that two monomers that occupied NN sites have the possibility to form a rigid pair at binding rate kb and that this complex can be disassembled with a rate of unbinding ku. We assumed that a monomer can be involved at most in only one pair. A paired monomer could then move only if it remained connected to its partner, i.e. only if its new location remained a NN site of its partner.
For each investigated situation, we initiated the system with random configurations and then let it reach equilibrium before measuring the average contact probabilities, the mean squared distances, mean squared displacements and kurtosis by averaging over 1000 trajectories. The time step of the simulation was 0.55 ms.
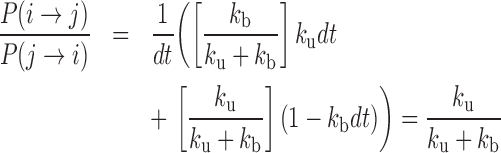
In order to evaluate the effective energy E0 of contact between monomers, we used the detailed balance of the Monte Carlo simulation between a micro-state i where two monomers were NN on the lattice and a micro-state j where they were not in contact. According to Boltzmann statistics, we have 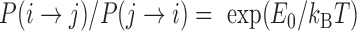 . Considering reaction rates kb and ku, the detailed balance yields during a time step dt:
. Considering reaction rates kb and ku, the detailed balance yields during a time step dt:
 |
(5) |
leading to 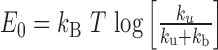 .
.
RESULTS
DNA flexibility can be determined from the analysis of its fluctuations
We first wished to validate our assumption that the bending stiffness of a polymer, equivalently its Kuhn length, can be inferred from the analysis of its spatial fluctuations with the Rouse model. Because DNA Kuhn length of b ∼100 to 110 nm in low salt conditions is well-documented in the literature (37), we used naked DNA as a model biopolymer. In order to generate long deproteinized DNA molecules containing randomly-distributed short fluorescent stretches, we purified chromosome fragments from human osteosarcoma cells treated with fluorescent nucleotides during DNA replication (see Materials and Methods). The size of the stretches of ∼50 kb and the contour length of the molecules of 500 kb or more have been characterized by DNA combing and optical mapping in nanochannels, respectively (27). Labeled DNA molecules were then dispersed in a crowded solution containing a high concentration of the neutral polymer poly-vinylpyrrolidone (PVP), equal to three times the overlapping concentration, i.e. the threshold concentration for which PVP chains become intermingled (Figure 1A, (29)). DNA loci movements were finally recorded during >60 images using wide field fluorescence microscopy at an inter-frame interval of 39 ms. The MSD in 2D was finally extracted for each trajectory (gray datasets in Figure 1B).
Figure 1.

Analysis of DNA fluctuations in vitro. (A) Schematics of the experiment: long DNA molecules with fluorescent stretches, which are shown with black and red segments, respectively, are dispersed in a crowded solution composed of a high concentration of PVP chains (gray lines). (B) The graph presents the temporal evolution of the MSD as a function of time for fluorescent DNA loci (gray dataset). The average response (red squares) is fitted with the Rouse model (black line), but not with the Zimm model (green line). (C) The graph shows the residuals of the fit with the Rouse model as a function of the Kuhn length. (D) The red dataset represents the average MSD over time for DNA loci dispersed in a solution without crowding. The green and black solid lines show the predictions of the Zimm and Rouse models, respectively, obtained by setting the Kuhn length to 100 nm and the viscosity to 2.3 mPa.s.
The average MSD response shown with the red dataset in Figure 1B was consistent with the Rouse model using anomalous diffusion parameters (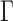 ,
,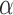 ) of (
) of (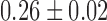 μm2/s0.53,
μm2/s0.53, 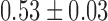 ), as plotted with the corresponding black curve, using 95% confidence intervals for the uncertainty. Taking b∼100 nm for DNA (37) and using the friction coefficient of a cylindrical monomer of diameter d ∼2 nm for DNA
), as plotted with the corresponding black curve, using 95% confidence intervals for the uncertainty. Taking b∼100 nm for DNA (37) and using the friction coefficient of a cylindrical monomer of diameter d ∼2 nm for DNA 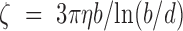 with
with 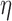 the solvent viscosity of 5.4 mPa.s, we deduce from equation (2) that
the solvent viscosity of 5.4 mPa.s, we deduce from equation (2) that  = 0.23 μm2/s0.5, in good agreement with experimental data. More quantitatively, we could estimate the Kuhn length by measuring the sum of the squared residuals between the fit and the MSD curve (Figure 1C), which reached a marked minimum for b = 119 nm. In addition, the same model enabled fitting the step distribution function (SDF) for three time intervals of 0.12, 0.23 and 0.35 s (Supplementary Figure S3).
= 0.23 μm2/s0.5, in good agreement with experimental data. More quantitatively, we could estimate the Kuhn length by measuring the sum of the squared residuals between the fit and the MSD curve (Figure 1C), which reached a marked minimum for b = 119 nm. In addition, the same model enabled fitting the step distribution function (SDF) for three time intervals of 0.12, 0.23 and 0.35 s (Supplementary Figure S3).
We next performed a series of experiments using a PVP concentration three times lower than the overlapping concentration (see Methods), i.e. in conditions where crowding becomes negligible because the PVP chains are no longer intermingled (38). For this, we used the same PVP concentration but the chains were characterized by a molecular weight 10 times smaller. The resulting viscosity of 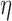 = 2.3 mPa.s was expectedly lower. In this regime where hydrodynamic interactions between DNA monomers contribute to the dynamics of the chain, the Zimm model is expected to be valid with the corresponding temporal variation of the MSD (39):
= 2.3 mPa.s was expectedly lower. In this regime where hydrodynamic interactions between DNA monomers contribute to the dynamics of the chain, the Zimm model is expected to be valid with the corresponding temporal variation of the MSD (39):
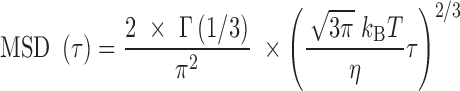 |
(6) |
with 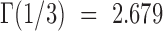 . Note that equation (6) is dependent on the viscosity
. Note that equation (6) is dependent on the viscosity 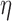 and unaffected by DNA persistence length. The Zimm model was in agreement with our measurements (green line in Figure 1D), whereas the Rouse model yielded an inconsistent prediction using a Kuhn length of 100 nm (black curve in Figure 1D). In addition, the Zimm model was not consistent with the dynamics of DNA in crowded conditions (green curve in Figure 1B). We note that the consistency of the Zimm model to analyze fluctuation data in ‘uncrowded’ conditions was already reported in published fluorescence correlation spectroscopy studies (40,41). We conclude that the motion of DNA loci can be accurately analyzed with polymer models, and that DNA bending stiffness can be inferred from real-time microscopy studies using the Rouse model in the case of crowded environments.
and unaffected by DNA persistence length. The Zimm model was in agreement with our measurements (green line in Figure 1D), whereas the Rouse model yielded an inconsistent prediction using a Kuhn length of 100 nm (black curve in Figure 1D). In addition, the Zimm model was not consistent with the dynamics of DNA in crowded conditions (green curve in Figure 1B). We note that the consistency of the Zimm model to analyze fluctuation data in ‘uncrowded’ conditions was already reported in published fluorescence correlation spectroscopy studies (40,41). We conclude that the motion of DNA loci can be accurately analyzed with polymer models, and that DNA bending stiffness can be inferred from real-time microscopy studies using the Rouse model in the case of crowded environments.
Nucleosome array fluctuations are consistent with the Rouse model
We then focused on the analysis of nucleosome array fluctuations in vitro. Because the Kuhn length of chromatin remains debated in the literature (18), we separated the presentation of experimental results from the analysis of the amplitude of spatial fluctuations with the Rouse model. We assembled nucleosome arrays on the same long DNA fragments with fluorescent stretches using human core histones and chromatin assembly factors to produce nucleosome arrays with a repeat length of ∼168 bp, as characterized by micrococcal nuclease digestion (Supplementary Figure S4). This nucleosome repeat length matches that of yeast chromatin, which is known to have an average nucleosome repeat of 167 bp that is equivalent to 20–22 bp of linker DNA (42). Electron microscopy revealed the characteristic pattern of 10 nm fibers, with gaps and random clustering of assembled chromatin (Figure 2A, see Methods). The resulting chromatin fibers were diluted in a crowded buffer, and the motion of chromatin loci was tracked by microscopy to measure the MSD and the SDF (Figure 2B, C). The average MSD plotted in red in Figure 2B was consistent with the Rouse model, as shown from the fit with an anomalous diffusion response characterized by parameters (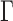 ,
,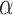 ) of (
) of (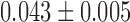 μm2/s0.54,
μm2/s0.54, 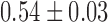 ). Furthermore, despite the broad variability of the MSD response from fiber to fiber (gray datasets in Figure 2B), possibly caused by the heterogeneous arrangements of nucleosome in single molecules and/or the residual random clustering observed in the electron micrographs (Figure 2A), the SDF appeared to adopt a Gaussian shape (solid lines in Figure 2C). This behavior is predicted by the Rouse model (31). More quantitatively, the width of the SDF is expected to be determined by the amplitude of the MSD:
). Furthermore, despite the broad variability of the MSD response from fiber to fiber (gray datasets in Figure 2B), possibly caused by the heterogeneous arrangements of nucleosome in single molecules and/or the residual random clustering observed in the electron micrographs (Figure 2A), the SDF appeared to adopt a Gaussian shape (solid lines in Figure 2C). This behavior is predicted by the Rouse model (31). More quantitatively, the width of the SDF is expected to be determined by the amplitude of the MSD:
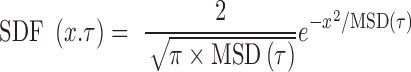 |
(7) |
Figure 2.

Nucleosome array fluctuations in vitro. (A) Electron micrographs of chromatin fibers reconstituted on λ-DNA molecules. (B) The graph shows the temporal evolution of the MSD of chromatin fibers dispersed in a crowded solution with individual trajectory reported in gray and the average response in red with the standard error. The viscosity of the solution was 15 mPa.s. The black curve is the fit with a power-law response characterized by an anomalous exponent of 0.54. (C) The plot shows the step distribution function extracted from the same set of data as in (B) for three consecutive time lags indicated in the caption. The corresponding Gaussian fits are directly expressed from equation (7).
By plugging the measured MSD in Figure 2B, we obtained a good fit between the experimental SDF and that predicted by the Rouse model (curves and corresponding fits in Figure 2C). Finally, exploiting the spontaneous destabilization of nucleosome arrays in diluted conditions (43), we compared the fluctuations of nucleosome arrays after assembly, and two days after their reconstitution, and detected a motion characterized by an amplitude  3.5-fold enhanced (data not shown). This response matched that of purified DNA molecules in these experimental conditions, showing that the amplitude of the fluctuations of nucleosome arrays are lower than that of DNA.
3.5-fold enhanced (data not shown). This response matched that of purified DNA molecules in these experimental conditions, showing that the amplitude of the fluctuations of nucleosome arrays are lower than that of DNA.
Inferring the mechanical properties of nucleosome arrays to test the Rouse model
In order to check the relevance of the Rouse model for chromatin, we had to determine the monomer friction coefficient 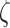 and the Kuhn length b from structural models of nucleosome arrays and integrate these parameters in equation (2). In order to sample the conformational space of nucleosome arrays thoroughly, we performed simulations with the nucleosome conformation of the crystal structure, i.e. with two turns of DNA wrapped around the histone core, hereafter referred to as the ‘closed negative’ structure (see Materials and Methods, Figure 3A, (44)). We also considered an ‘open’ nucleosome state, in which the most distal histone–DNA binding sites at super-helix location ±6.5 are disrupted (34), as detected in molecular biology assays (44) as well as single molecule techniques ((45), Figure 3B). For both nucleosome configurations, nucleosome arrays were simulated with variable lengths of DNA linkers in the range 18 to 22 bp, i.e. for nucleosome repeat lengths of ∼
and the Kuhn length b from structural models of nucleosome arrays and integrate these parameters in equation (2). In order to sample the conformational space of nucleosome arrays thoroughly, we performed simulations with the nucleosome conformation of the crystal structure, i.e. with two turns of DNA wrapped around the histone core, hereafter referred to as the ‘closed negative’ structure (see Materials and Methods, Figure 3A, (44)). We also considered an ‘open’ nucleosome state, in which the most distal histone–DNA binding sites at super-helix location ±6.5 are disrupted (34), as detected in molecular biology assays (44) as well as single molecule techniques ((45), Figure 3B). For both nucleosome configurations, nucleosome arrays were simulated with variable lengths of DNA linkers in the range 18 to 22 bp, i.e. for nucleosome repeat lengths of ∼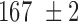 bp. Finally, in order to account for the structural defects of nucleosome arrays observed by electron microscopy (Figure 2A), we introduced some randomness in the positioning of nucleosomes associated to a deviation range of 3 or 5 bp, i.e. linkers of 17 to 23 bp or 15 to 25 bp, respectively (Figure 3C). For each condition, we simulated 1000 independent chromatin fibers structures containing 100 nucleosomes. Note that nucleosome-nucleosome interactions were neglected in the simulations because our experiments have been carried out in low salt conditions with minimal oligomerization or intramolecular contacts, as inferred from analytical sedimentation (46).
bp. Finally, in order to account for the structural defects of nucleosome arrays observed by electron microscopy (Figure 2A), we introduced some randomness in the positioning of nucleosomes associated to a deviation range of 3 or 5 bp, i.e. linkers of 17 to 23 bp or 15 to 25 bp, respectively (Figure 3C). For each condition, we simulated 1000 independent chromatin fibers structures containing 100 nucleosomes. Note that nucleosome-nucleosome interactions were neglected in the simulations because our experiments have been carried out in low salt conditions with minimal oligomerization or intramolecular contacts, as inferred from analytical sedimentation (46).
Figure 3.

Inferring the mechanical properties of nucleosome arrays. (A) Structure of a nucleosome array with closed negative nucleosomes and a linker length of 20 bp. Red spheres correspond to histone octamers. Gray segments represent DNA and the blue segments specifically show entry exit DNAs. (B) Structure of a nucleosome array with open nucleosomes and a linker length of 20 bp. (C) Structure of a nucleosome array with open nucleosomes and a variable linker length of 20 ± 5 bp. (D) The graph shows the end-to-end distance distribution as extracted from the simulation for a nucleosome array containing 20, 30, and 60 closed negative nucleosomes and a linker length of 20 bp. (E) The plot shows the contour length vs. Kuhn length deduced from the analysis of the moments of the end-to-end distance distribution. The colored dots correspond to the fit obtained with chromatin fibers containing 5–95 nucleosomes (plotted with dark blue to red colors, respectively). The three arrows show the results inferred from the analysis of the histograms shown in (D). The red line corresponds to the conditions for which the contour length is equal to two times the Kuhn length.
We then extracted the Kuhn length of each array using the end-to-end distance distribution as readout of the simulations (Figure 3D, see Materials and Methods). We found that the Kuhn length was larger for closed negative vs. open nucleosomes, altogether spanning 85–140 nm (third column of Table 1 and Supplementary table S2). Specifically, for regular arrays of negative nucleosomes with a repeat length of 165–169 bp, the Kuhn length was roughly constant in the range 120–140 nm, whereas fibers with open nucleosomes appeared to be slightly more flexible with a Kuhn length spanning 85–110 nm (Supplementary Figure S2 and Table S1). These values of the Kuhn length matched the predictions of other simulations for closed negative nucleosomes in the range of 102–154 nm for nucleosome repeat length of 169 and 168 bp, respectively (47,48). Measurements based on Hi-C in yeast also indicated that the Kuhn length was in a comparable size range of 110–230 nm (49). Last but not least, analytical models support the stiffness of fibers with short linkers of ∼20 bp with a Kuhn length of 100 nm or more (50).
Table 1.
Mechanical, structural and hydrodynamic modeling of nucleosome arrays by Monte Carlo simulations using different nucleosome conformations and repeat lengths. The last column corresponds to the amplitude of chromatin loci fluctuations according to the Rouse model
| Nucleosome configuration | Linker size (bp) | Kuhn length (nm) | Nucleosome per 11 nm | Hydrodynamic diameter (nm) | MSD amplitude Γ (μm2/s0.5) |
|---|---|---|---|---|---|
| Closed negative | 20 | 118 ± 6 | 2.6 | 215 ± 15 | 0.057 ± 0.03 |
| Open | 20 | 110 ± 3 | 1.8 | 96 ± 9 | 0.079 ± 0.06 |
| Closed negative | 20 ± 3 | 96 ± 5 | 3.0 | 228 ± 13 | 0.045 ± 0.03 |
| Open | 20 ± 3 | 106 ± 5 | 2.0 | 112 ± 9 | 0.071 ± 0.05 |
| Closed negative | 20 ± 5 | 92 ± 5 | 3.4 | 305 ± 14 | 0.037 ± 0.02 |
| Open | 20 ± 5 | 95 ± 3 | 2.4 | 145 ± 10 | 0.056 ± 0.05 |
These simulations also provided an estimate of the density of nucleosomes per 11 nm knowing the contour length and the number of nucleosomes for each fiber configuration (fourth column of Table 1). Nucleosome arrays with closed negative nucleosomes were more compact than fibers with open nucleosomes with a typical density in the range of 2.5–3.0 versus <2.0 nucleosomes/11 nm. Finally, we noticed that the addition of randomness in the positioning of nucleosomes tended to reduce the Kuhn length and to increase the density of nucleosomes (Table 1).
We subsequently inferred the monomer friction coefficient 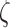 by performing simulations based on stochastic rotation dynamics (see details in Supplementary Material 2). These simulations are based on the explicit description of solvent particles, which transfer momentum to floating particles (51). They were performed at the level of one Kuhn segment, which was modeled as an ensemble of ‘spherical’ nucleosomes of 10 nm in diameter arranged in space according to the structural models shown in e.g. Figure 3A–C. We took three regularly repeated chromatin fibers with nucleosome densities of 1.2, 2.1, and 3.2 nucleosomes/11 nm. These simulations showed that the monomer friction coefficient
by performing simulations based on stochastic rotation dynamics (see details in Supplementary Material 2). These simulations are based on the explicit description of solvent particles, which transfer momentum to floating particles (51). They were performed at the level of one Kuhn segment, which was modeled as an ensemble of ‘spherical’ nucleosomes of 10 nm in diameter arranged in space according to the structural models shown in e.g. Figure 3A–C. We took three regularly repeated chromatin fibers with nucleosome densities of 1.2, 2.1, and 3.2 nucleosomes/11 nm. These simulations showed that the monomer friction coefficient 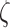 was proportional to the number of nucleosomes in one Kuhn segment for a low density of nucleosomes (Supplementary Table S2), whereas it was equal to half the number of nucleosomes for a high density of 3.2 nucleosomes/11 nm. Therefore, the friction coefficient appeared to decrease with the density of nucleosomes, confirming a trend documented for concentrated colloidal suspensions (52). Our results were finally extended to every simulation using linear interpolation of
was proportional to the number of nucleosomes in one Kuhn segment for a low density of nucleosomes (Supplementary Table S2), whereas it was equal to half the number of nucleosomes for a high density of 3.2 nucleosomes/11 nm. Therefore, the friction coefficient appeared to decrease with the density of nucleosomes, confirming a trend documented for concentrated colloidal suspensions (52). Our results were finally extended to every simulation using linear interpolation of 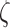 as a function of the nucleosome density. For clarity, we report the hydrodynamic diameter of the Kuhn segment as defined by
as a function of the nucleosome density. For clarity, we report the hydrodynamic diameter of the Kuhn segment as defined by 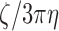 in the fifth column of Table 1.
in the fifth column of Table 1.
Using equation (2), we computed the MSD amplitude 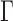 for every fiber structure (sixth column of Table 1). Given that Γ =
for every fiber structure (sixth column of Table 1). Given that Γ =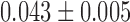 μm2/s0.5 in the experiments, the predictions of the simulations were in agreement with the fluctuation data if some degree of variability in the positioning of closed negative nucleosomes was allowed. The defects in nucleosome arrangements observed in Figure 2A may therefore account for the variability of MSD responses but also constitute an essential ingredient to recapitulate the amplitude of in vitro fluctuation data. Altogether, our results confirm that the mechanical parameters inferred from nucleosome array models integrated into the Rouse model recapitulate motion data in vitro.
μm2/s0.5 in the experiments, the predictions of the simulations were in agreement with the fluctuation data if some degree of variability in the positioning of closed negative nucleosomes was allowed. The defects in nucleosome arrangements observed in Figure 2A may therefore account for the variability of MSD responses but also constitute an essential ingredient to recapitulate the amplitude of in vitro fluctuation data. Altogether, our results confirm that the mechanical parameters inferred from nucleosome array models integrated into the Rouse model recapitulate motion data in vitro.
Inconsistency of the Rouse model for dynamic and Hi-C data in living cells
We next investigated if and whether the Rouse model could be applied to study chromosomes in living budding yeast cells. We studied chromatin dynamics by recording 110 trajectories (49 800 displacements) for a locus on chromosome XII at an inter-frame interval of 0.2 s. The average MSD shown in the red dataset of Figure 4A followed a temporal response consistent with the Rouse model associated to anomalous diffusion parameters (Γ,α) of (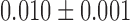 μm2/s0.54, 0.54) (black fit in Figure 4A), in agreement with our previous measurements (9). Interpreting this data with equation (2), as derived in ref. (9), leads to a Kuhn length of less than 5 nm. We tested the consistency of this estimate with respect to published Hi-C data in the form of probability of contact normalized at 20 kbp vs. genomic distance (black, blue, red and burgundy curves in Figure 4B). For this, we ran a coarse-grained lattice-based polymer simulation (see Methods) using n = 200 bp per monomer, a Kuhn length b = 5 nm, and the concentration of DNA set to 3 × 10−3 bp/nm3 as input parameters (53). We observed that the occurrence of distant contacts in the simulation was underestimated by more than one order of magnitude (cyan curve in Figure 4B). Furthermore, despite the contradiction with MSD data, we could run the simulation for a Kuhn length of b ∼ 50 nm for n = 2 kb per monomer, because this value is more consistent with in vitro data (49). Yet, this parametrization only marginally improved the fitting of Hi-C data (green curve in Figure 4B). Last, using our large ensemble of displacement measurements on chromosome XII, we compiled the SDF and detected a deviation to the Gaussian behavior expected for the Rouse model with a peaked distribution for time intervals of 0.4 and 0.8 s (Figure 4C, D). Overall, the Rouse model appeared to show some inconsistencies with structural and motion data in living yeast, suggesting that additional molecular parameters should be integrated in the model.
μm2/s0.54, 0.54) (black fit in Figure 4A), in agreement with our previous measurements (9). Interpreting this data with equation (2), as derived in ref. (9), leads to a Kuhn length of less than 5 nm. We tested the consistency of this estimate with respect to published Hi-C data in the form of probability of contact normalized at 20 kbp vs. genomic distance (black, blue, red and burgundy curves in Figure 4B). For this, we ran a coarse-grained lattice-based polymer simulation (see Methods) using n = 200 bp per monomer, a Kuhn length b = 5 nm, and the concentration of DNA set to 3 × 10−3 bp/nm3 as input parameters (53). We observed that the occurrence of distant contacts in the simulation was underestimated by more than one order of magnitude (cyan curve in Figure 4B). Furthermore, despite the contradiction with MSD data, we could run the simulation for a Kuhn length of b ∼ 50 nm for n = 2 kb per monomer, because this value is more consistent with in vitro data (49). Yet, this parametrization only marginally improved the fitting of Hi-C data (green curve in Figure 4B). Last, using our large ensemble of displacement measurements on chromosome XII, we compiled the SDF and detected a deviation to the Gaussian behavior expected for the Rouse model with a peaked distribution for time intervals of 0.4 and 0.8 s (Figure 4C, D). Overall, the Rouse model appeared to show some inconsistencies with structural and motion data in living yeast, suggesting that additional molecular parameters should be integrated in the model.
Figure 4.

Analysis of MSD and Hi-C data in yeast with the Rouse model. (A) The graph shows 110 MSD responses in gray, the average response in red with the standard error to the mean, and the corresponding fit with the Rouse model in black. (B) The plot presents normalized contact frequency vs. genomic distance inferred from Hi-C measurements by Mercy et al. in red (68), Duan et al. in black (69), Marie-Nelly et al. in blue (70) and Belton et al. in burgundy (71). The orange sector serves to define the limits of experimental responses. The green and cyan datasets are the results of simulations for one homopolymer freely diffusing in space and characterized by a Kuhn length of 50 or 5 nm, respectively. (C) The plot presents the SDF at four consecutive time lags, as indicated in the legend. The trajectories are the same as in A. (D) Same data as in (C) in log-lin.
Transient chromatin contacts account for chromosome dynamics and folding in yeast
Recent EM studies of thin nuclear sections of budding yeast have hinted to the existence of intra-chromosomal contacts (54). Although intramolecular contacts have not been thoroughly discussed in yeast, their contribution to higher-order chromosome folding in mammalian cells or drosophila has been proposed many times (23,55,56). We thus set out to explore if a Rouse model with transient internal contacts (RouseTIC) could fit chromosome structure and motion in vivo. We used the same simulations scheme, but introduced non-specific self-interaction in between the monomers (36). We defined kb and ku the binding and unbinding rates between monomers, respectively. In addition to these two kinetic parameters, the RouseTIC model relies on the definition of a concentration of DNA set to 3 × 10−3 bp/nm3, the Kuhn length and the characteristic time for the fluctuations.
In order to estimate kb and ku, we first performed simulations of the contact probability normalized at 20 kb versus genomic distance and compared them with Hi-C measurements. These simulations were performed setting the number of base-pair by Kuhn length to 2 kb and 50 nm, i.e. with a density of 2.6 nucleosomes/11 nm that roughly match the results of our simulations in Table 1 and those of experiments of ∼2.0 nucleosomes/11 nm based on Hi-C (49). Notably, Hi-C data map the steady-state conformation of a polymer, hence the occurrence of contacts can be recapitulated with one equilibrium kinetic parameter, namely the reaction constant kb/ku. These simulations were in good agreement with the experiments for kb/ku in the range 0.3–0.70 (Figure 5A). As an argument of self-consistency, we checked the validity of the RouseTIC model by comparing its predictions to distance measurements in between chromosome loci in fixed cells derived from ref. (57) (Figure 5B). For kb/ku spanning 0.3–0.7, we adjusted the Kuhn length and confirmed that its value of b =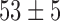 nm was compatible with the data.
nm was compatible with the data.
Figure 5.

Analysis of MSD and Hi-C data in yeast with the RouseTIC model. (A) The graph shows the comparison of experimental and simulated Hi-C contact frequency versus genomic distance. Experimental data are within the two orange limits defined in Figure 4B. The output of simulations fit experimental data for 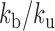 in the range 0.3 to 0.7, as indicated in the legend at the top. (B) The plot shows the Kuhn length as a function of
in the range 0.3 to 0.7, as indicated in the legend at the top. (B) The plot shows the Kuhn length as a function of 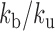 that best fits 3D mean square distance measurements with the RouseTIC model. The inset shows an example of fit for
that best fits 3D mean square distance measurements with the RouseTIC model. The inset shows an example of fit for 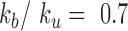 . Note the error bars for the determination of the Kuhn length corresponds to acceptable fits within the gray limits in the inset. (C) The orange dataset shows the experimental MSD versus time for nucleosome arrays in vitro, as reported in Figure 2B, and the corresponding black curve is obtained with the RouseTIC model for a simulation time step of 0.55 ms. (D) The plot presents the MSD as a function of time for chromosomes in living yeast. The dashed orange line is the experimental measurement of
. Note the error bars for the determination of the Kuhn length corresponds to acceptable fits within the gray limits in the inset. (C) The orange dataset shows the experimental MSD versus time for nucleosome arrays in vitro, as reported in Figure 2B, and the corresponding black curve is obtained with the RouseTIC model for a simulation time step of 0.55 ms. (D) The plot presents the MSD as a function of time for chromosomes in living yeast. The dashed orange line is the experimental measurement of 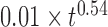 and the upper and lower gray lines correspond to limits characterized by
and the upper and lower gray lines correspond to limits characterized by 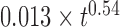 and
and 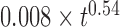 . The blue, black and green curves are obtained from the RouseTIC model with
. The blue, black and green curves are obtained from the RouseTIC model with 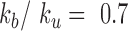 and different values of
and different values of 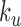 , as indicated in the legend.
, as indicated in the legend.
We then wished to analyze motion data. In order to adjust the time step of the simulation, we first ran simulations with no interaction and compared their predictions to our in vitro MSD measurements (Figure 5C). The temporal parameter of the simulation was 0.55 ms. We finally focused on the motion of chromosome loci in living yeast with the RouseTIC model and kb/ku in the range 0.3–0.70 (Figure 5D). Because the amplitude of fluctuations appeared to primarily depend on the unbinding rate ku, we could determine both reaction kinetic parameters. We proceeded by setting kb/ku in the range 0.3–0.7 and then varied ku to fit MSD data (Figure 5D and Supplementary Figure S5). In this parameter space, the typical unbinding time 1/ku varied from tens of seconds to seconds (inset of Figure 5D). Notably, the fit of MSD data was significantly better for kb/ku of 0.7 than 0.3 (see Supplementary Figure S5).
In order to reinforce the relevance of the RouseTIC model, we focused on higher-order moments of the SDF (Figure 4C, D). The anomaly of the SDF with respect to the Gaussian response can be measured from the kurtosis, which is computed from the fourth-order moment of the SDF (see Equation (4) in Materials and Methods). In the Rouse model, the kurtosis is equal to 2 ((31), dashed black line in Figure 6A), whereas an excess of kurtosis is detected for step distribution functions more peaked than a Gaussian function, as noticed for time intervals of 0.4 and 0.8 s in Figure 4C. Expectedly, the trajectory-averaged kurtosis appeared to be equal to 2.4 in the small time limit for chromosomes in vivo (blue solid line in Figure 6A). In addition, we noticed a clear temporal signature in the kurtosis, which decreased after 1 to 3 s. Conversely, the kurtosis measured for purified chromatin fibers was lower and equal to ∼1.75, and roughly constant over a narrower temporal window (black solid line in Figure 6A).
Figure 6.

Analysis of chromatin motion based on the kurtosis. (A) The kurtosis is plotted as a function of time for the locus on chromosome XII or for reconstituted nucleosome arrays (blue and black datasets, respectively). The dashed black line corresponds to the response for a Gaussian process, as in e.g. Brownian motion or the Rouse model (31). The dashed gray and blue curves are deduced from simulations of the RouseTIC model using kinetic parameters that fit Hi-C and MSD data, which are reported in the caption. (B) Same graph as in (A) with the two experimental datasets divided by 0.9.
We finally computed the kurtosis of the simulated trajectories with the RouseTIC model. We used the fitted parameters of 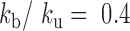 and 0.7 and
and 0.7 and 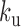 = 0.2 and 1 s−1, respectively, because these values fit MSD and Hi-C data. Simulations showed an excess of kurtosis equal to ∼2.7 in the small time limit (dashed lines in Figure 6A), and a decrease after a typical time defined by the unbinding reaction time scale
= 0.2 and 1 s−1, respectively, because these values fit MSD and Hi-C data. Simulations showed an excess of kurtosis equal to ∼2.7 in the small time limit (dashed lines in Figure 6A), and a decrease after a typical time defined by the unbinding reaction time scale 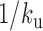 . These predictions were in qualitative agreement with our data, as could be clearly evidenced by the phenomenological normalization of in vitro and in vivo kurtosis data by the same factor of 0.9, and the resulting superposition of experiments and simulations (Figure 6B). Altogether, the analysis of the kurtosis tends to support the consistency of the RouseTIC model to reproduce structural and dynamic data of yeast chromosome in vivo with a steady-state polymer model that includes two kinetic parameters for the contact formation reaction.
. These predictions were in qualitative agreement with our data, as could be clearly evidenced by the phenomenological normalization of in vitro and in vivo kurtosis data by the same factor of 0.9, and the resulting superposition of experiments and simulations (Figure 6B). Altogether, the analysis of the kurtosis tends to support the consistency of the RouseTIC model to reproduce structural and dynamic data of yeast chromosome in vivo with a steady-state polymer model that includes two kinetic parameters for the contact formation reaction.
DISCUSSION
Origin of molecular interactions in RouseTIC
In this study, we monitored chromatin motion in vitro and in vivo by fluorescence microscopy, and analyzed experimental data with chromatin structure models and kinetic Monte Carlo simulations of self-interacting polymers. We propose that the Rouse model with transient internal contacts accounts for the in vivo dynamics of chromosomes (Figure 7A) and determines the kinetic parameters of the association reaction for the first time in living yeast. Internal contacts appear to be detectable from the occurrence of frequent contacts along the chromosome contour (Figure 4B) and the slow motion of chromatin loci with a deviation of the step distribution from the Gaussian response in the short time limit (Figure 4C). The latter consequence is qualitatively explained by the formation of transient contacts that tend to decrease the fraction of time a monomer freely diffuses, and restrain the displacement of freely diffusing monomers inside boundaries along the chromosome contour (25). Interestingly, the existence of these transient contacts is reminiscent of the mechanism of internal friction (58), which manifests itself if intramolecular conformational barriers lead to the dissipation of energy into internal degrees of freedom. Already proposed to be relevant for chromosomes (59), internal friction is known to lead to slow fluctuations, as observed in our datasets.
Figure 7.

Molecular scenario for the RouseTIC model. (A) The sketch represents the Rouse TIC model with the definition of the reaction rates ku and kb. The fiber displayed in the inset is obtained for ku/kb = 0.5. (B) Simulation of a chromatin fiber with a linker size of 20 bp and closed negative nucleosomes with a concentration of nucleosomes set to 3 × 10−3 bp/nm3. 127 nucleosomes are highlighted in green because they potentially form intramolecular contacts, as defined by an inter-nucleosome distance in trans of <15 nm, i.e. comparable to the typical length of histone tails, and a genomic distance in cis of more than 1650 bp (10 nucleosomes). (C) The same simulation is carried out for a fiber with linker size of 22 bp and open nucleosomes. 456 nucleosomes are marked in green.
Nevertheless, the molecular origin of the contacts in the RouseTIC model remains to be clarified. By simulating the structure of a fiber with 1000 nucleosomes at a volume density of 15% with the same structural models of chromatin as for deriving the Kuhn length, we suggest that numerous contacts are likely to pre-exist within chromosomes (Figure 7B, C), which could be stabilized by nucleosome interactions. By scaling the effective energy of the binding events, the RouseTIC model can help narrow down possible molecular origin of these interactions. In addition to the transient nature of binding events with a lifetime on the order of seconds, the effective energy E0 of contacts between monomers, as determined from the ratio of binding and unbinding rates kb/ku (see Equation (5)), is weak in the range of E0∼–0.3 to –0.5 kBT. Such strength for E0 is far smaller than the energy associated with ATP hydrolysis of ∼20 kBT, but also than the attractive stacking energy between nucleosomes of –2.7 kBT (60). Thus, this energy cannot be directly attributed to stacking between two nucleosomes through specific contacts, such as those described between the histone H4 N-terminal tail of one nucleosome with the ‘acidic patch’ of an adjacent one (61). Rather, we hypothesize that contacts predominantly correspond to transient, weak and labile interactions. Interestingly, an interaction energy E0 of –0.3 to –0.5 kBT compares to the conditions of equilibrium between histone–tail DNA attraction and DNA–DNA electrostatic repulsion in physiological salt conditions, as inferred from X-ray scattering of nucleosome core particles (62). Thus, contacts may originate from unspecific and dynamic histone tail–DNA contacts within the chromatin fiber as observed in recent cryo-EM studies on nucleosome core particles (63), potentially mediated or perturbed by chromatin-binding proteins. The higher chromatin density in the nucleus of living cells in comparison to in vitro experiments is expected to favor the formation of contacts. Yet the contribution of these contacts on chromatin fluctuations in vitro may be investigated by modulating the ionic strength of the solution. Using analytical centrifugation, it has indeed been shown that addition of 2 mM divalent salt enhances chromatin compaction and oligomerization mediated by histone tails interactions (46,64).
Relevance of contact reaction for the analysis of Hi-C data in metazoan
RouseTIC may also be relevant to chromatin compartment segregation, in which the occurrence of chromosome intramolecular contacts mediated by nucleosomes has been suggested (21). Compartments inferred from Hi-C contact maps could be modeled assuming limited attraction between the monomers (23,24), although the amplitude and origin of this interaction remains mostly elusive. The estimate of monomer-monomer interaction energy in the range of E0 ∼–0.3 to –0.5 kBT is notably close to the energy between monomers of –0.3 kBT for a polymer chain in theta conditions (65), i.e. with balanced monomer-monomer and monomer-solvent interactions. Because polymers in theta conditions are poised to fold into globules or coils whether monomer-monomer interaction increases or decreases, respectively, we suggest that the residual interactions between monomers can be modulated to trigger compartment segregation. This mechanism is likely necessary but not sufficient to model metazoan datasets due to the essential contribution of active processes involving loop extrusion or transcription (22–24). Nevertheless, our recent analysis of chromosome structure in drosophila appears to corroborate the globule to coil transition (data not shown), suggesting that nucleosome-nucleosome interactions stabilization by e.g. heterochromatin-specific proteins, such as HP1, could promote local collapse into globular state and phase separation (66,67). Despite a strong impact on chromosome large-scale organization, the number of proteins implicated in this structural transition could be limited. This hypothesis deserves further investigation using heteropolymer models (24) and combining them to chromatin structure models in order to reach a molecular description of segregation.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the French CNRS network GDR ADN for stimulating workshops, Serge Mazère for help in the FRAP experiments, as well as Vincent Dion, Julien Mozziconacci, Romain Koszul and Damian Erdel for critical reading of the manuscript. We gratefully acknowledge support from the PSMN (Pôle Scientifique de Modélisation Numérique) and from the CBP: IT Test Center (Centre Blaise Pascal) computing centers of ENS de Lyon. We completely used SIDUS, developed by Emmanuel Quemener in CBP, also used in PSMN.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
CSC for PhD fellowship (to R.W.); ANR-13-BSV5-0010 – ANDY, IDEX ATS NudGene, ANR-15-CE12-0006 EpiDevoMath and Fondation pour la Recherche Médicale [DEI20151234396 to K.B., O.G., D.J.]; CIMENT infrastructure (supported by the Rhône-Alpes region) Grant [CPER07_13 CIRA] for computing resources. Funding for open access charge: ANR funds.
Conflict of interest statement. None declared.
REFERENCES
- 1. Bickmore W.A. The spatial organization of the human genome. Annu. Rev. Genomics Hum. Genet. 2013; 14:67–84. [DOI] [PubMed] [Google Scholar]
- 2. Nozaki T., Imai R., Tanbo M., Nagashima R., Tamura S., Tani T., Joti Y., Tomita M., Hibino K., Kanemaki M.T. et al.. Dynamic organization of chromatin domains revealed by super-resolution live-cell imaging. Mol. Cell. 2017; 67:282–293. [DOI] [PubMed] [Google Scholar]
- 3. Wang R., Mozziconacci J., Bancaud A., Gadal O.. Principles of chromatin organization in yeast: relevance of polymer models to describe nuclear organization and dynamics. Curr. Opin. Cell Biol. 2015; 34:54–60. [DOI] [PubMed] [Google Scholar]
- 4. Cheng T.M., Heeger S., Chaleil R.A., Matthews N., Stewart A., Wright J., Lim C., Bates P.A., Uhlmann F.. A simple biophysical model emulates budding yeast chromosome condensation. eLife. 2015; 4:e05565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tjong H., Gong K., Chen L., Alber F.. Physical tethering and volume exclusion determine higher-order genome organization in budding yeast. Genome Res. 2012; 22:1295–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wong H., Marie-Nelly H., Herbert S.C., Blanc H., Koszul R., Fabre E., Zimmer C.. A predictive computational model of the dynamic 3D interphase nucleus. Curr. Biol. 2012; 22:1881–1890. [DOI] [PubMed] [Google Scholar]
- 7. Gehlen L.R., Gruenert G.M., Jones B., Rodley C.D., Langowski J., O’Sullivan J.M.. Chromosome positioning and the clustering of functionally related loci in yeast is driven by chromosomal interactions. Nucleus. 2012; 3:370–383. [DOI] [PubMed] [Google Scholar]
- 8. Tokuda N., Terada T.P., Sasai M.. Dynamical modeling of three-dimensional genome organization in interphase budding yeast. Biophys. J. 2012; 102:296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hajjoul H., Mathon J., Ranchon H., Goiffon I., Alber B., Gadal O., Bystricky K., Bancaud A.. High throughput chromatin motion tracking in living yeast reveals the flexibility of the fiber throughout the genome. Genome Res. 2013; 23:1829–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cabal G.G., Genovesio A., Rodriguez-Navarro S., Zimmer C., Gadal O., Lesne A., Buc H., Feuerbach-Fournier F., Olivo-Marin J.C., Hurt E.C.. SAGA interacting factors confine sub-diffusion of transcribed genes to the nuclear envelope. Nature. 2006; 441:770–773. [DOI] [PubMed] [Google Scholar]
- 11. Backlund M.P., Joyner R., Weis K., Moerner W.E.. Correlations of three-dimensional motion of chromosomal loci in yeast revealed by the double-helix point spread function microscope. Mol. Biol. Cell. 2014; 25:3619–3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Amitai A., Toulouze M., Dubrana K., Holcman D.. Analysis of single locus trajectories for extracting in vivo chromatin tethering interactions. PLoS Comput. Biol. 2015; 11:e1004433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Albert B., Mathon J., Shukla A., Saad H., Normand C., Villa D., Kamgoue A., Mozziconacci J., Wong H., Zimmer C. et al.. Systematic characterization of the conformation and dynamics of budding yeast chromosome XII. J. Cell Biol. 2013; 202:201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Amitai A., Seeber A., Gasser S.M., Holcman D.. Visualization of chromatin decompaction and break site extrusion as predicted by statistical polymer modeling of single-locus trajectories. Cell Rep. 2017; 18:1200–1214. [DOI] [PubMed] [Google Scholar]
- 15. Arbona J.-M., Herbert S., Fabre E., Zimmer C.. Inferring the physical properties of yeast chromatin through Bayesian analysis of whole nucleus simulations. Genome Biol. 2017; 18:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Herbert S., Brion A., Arbona J.M., Lelek M., Veillet A., Lelandais B., Parmar J., Fernández F.G., Almayrac E., Khalil Y. et al.. Chromatin stiffening underlies enhanced locus mobility after DNA damage in budding yeast. EMBO J. 2017; 36:2595–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Doi M., Edwards S.F.. The Theory of Polymer Dynamics. 1988; USA: Oxford University Press. [Google Scholar]
- 18. Schiessel H. The physics of chromatin. J. Phys. Condens. Matter. 2003; 15:R699–R774. [DOI] [PubMed] [Google Scholar]
- 19. Maeshima K., Ide S., Hibino K., Sasai M.. Liquid-like behavior of chromatin. Curr. Opin. Genet. Dev. 2016; 37:36–45. [DOI] [PubMed] [Google Scholar]
- 20. Maeshima K., Hihara S., Eltsov M.. Chromatin structure: does the 30-nm fibre exist in vivo. Curr. Opin. Cell Biol. 2010; 22:291–297. [DOI] [PubMed] [Google Scholar]
- 21. Ou H.D., Phan S., Deerinck T.J., Thor A., Ellisman M.H., O’Shea C.C.. ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science. 2017; 357:eaag0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rowley M.J., Nichols M.H., Lyu X., Ando-Kuri M., Rivera I.S.M., Hermetz K., Wang P., Ruan Y., Corces V.G.. Evolutionarily conserved principles predict 3D Chromatin Organization. Mol. Cell. 2017; 67:837–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nuebler J., Fudenberg G., Imakaev M., Abdennur N., Mirny L.A.. Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E6697–E6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jost D., Carrivain P., Cavalli G., Vaillant C.. Modeling epigenome folding: formation and dynamics of topologically associated chromatin domains. Nucleic Acids Res. 2014; 42:9553–9561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Indei T., Takimoto J.. Linear viscoelastic properties of transient networks formed by associating polymers with multiple stickers. J. Chem. Phys. 2010; 133:194902. [DOI] [PubMed] [Google Scholar]
- 26. Osmanović D., Rabin Y.. Dynamics of active Rouse chains. Soft Matter. 2017; 13:963–968. [DOI] [PubMed] [Google Scholar]
- 27. Lacroix J., Pélofy S., Blatché C., Pillaire M.-J., Huet S., Chapuis C., Hoffmann J.-S., Bancaud A.. Analysis of DNA replication by optical mapping in nanochannels. Small. 2016; 12:5963–5970. [DOI] [PubMed] [Google Scholar]
- 28. Dupaigne P., Lavelle C., Justome A., Lafosse S., Mirambeau G., Lipinski M., Piétrement O., Le Cam E.. Rad51 polymerization reveals a new chromatin remodeling mechanism. PLoS One. 2008; 3:e3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McFarlane N.L., Wagner N.J., Kaler E.W., Lynch M.L.. Poly(ethyleneoxide) (PEO) and poly(vinyl pyrolidone) (PVP) induce different changes in the colloid stability of nanoparticles. Langmuir. 2010; 26:13823–13830. [DOI] [PubMed] [Google Scholar]
- 30. Tinevez J.-Y., Perry N., Schindelin J., Hoopes G.M., Reynolds G.D., Laplantine E., Bednarek S.Y., Shorte S.L., Eliceiri K.W.. TrackMate: an open and extensible platform for single-particle tracking. Methods. 2017; 115:80–90. [DOI] [PubMed] [Google Scholar]
- 31. Tejedor V., Bénichou O., Voituriez R., Jungmann R., Simmel F., Selhuber-Unkel C., Oddershede L.B., Metzler R.. Quantitative analysis of single particle trajectories: mean maximal excursion method. Biophys. J. 2010; 98:1364–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saxton M.J. Lateral diffusion in an archipelago. Single-particle diffusion. Biophys J. 1993; 64:1766–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Carrivain P., Barbi M., Victor J.-M.. In silico single-molecule manipulation of DNA with rigid body dynamics. PLoS Comput. Biol. 2014; 10:e1003456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Luger K., Mader A.W., Richmond R.K., Sargent D.F., Richmond T.J.. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997; 389:251–260. [DOI] [PubMed] [Google Scholar]
- 35. Hamprecht B., Kleinert H.. End-to-end distribution function of stiff polymers for all persistence lengths. Phys. Rev. E. 2005; 71:031803. [DOI] [PubMed] [Google Scholar]
- 36. Olarte-Plata J.D., Haddad N., Vaillant C., Jost D.. The folding landscape of the epigenome. Phys. Biol. 2016; 13:026001. [DOI] [PubMed] [Google Scholar]
- 37. Brunet A., Tardin C., Salome L., Rousseau P., Destainville N., Manghi M.. Dependence of DNA persistence length on ionic strength of solutions with monovalent and divalent salts: a joint theory–experiment study. Macromolecules. 2015; 48:3641–3652. [Google Scholar]
- 38. Minton A.P. Molecular crowding: analysis of effects of high concentrations of inert cosolutes on biochemical equilibria and rates in terms of volume exclusion. Methods Enzym. 1998; 295:127–149. [DOI] [PubMed] [Google Scholar]
- 39. Teraoka I. Polymer Solutions: An Introduction to Physical Properties. 2002; John Wiley & Sons. [Google Scholar]
- 40. McHale K., Mabuchi H.. Precise characterization of the conformation fluctuations of freely diffusing DNA: beyond rouse and zimm. J. Am. Chem. Soc. 2009; 131:17901–17907. [DOI] [PubMed] [Google Scholar]
- 41. Lumma D., Keller S., Vilgis T., Rädler J.O.. Dynamics of large semiflexible chains probed by fluorescence correlation spectroscopy. Phys. Rev. Lett. 2003; 90:218301. [DOI] [PubMed] [Google Scholar]
- 42. Grigoryev S.A. Nucleosome spacing and chromatin higher-order folding. Nucleus. 2012; 3:493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McMurray C.T., van Holde K.E.. Binding of ethidium bromide causes dissociation of the nucleosome core particle. Proc. Natl. Acad. Sci. U.S.A. 1986; 83:8472–8476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Prunell A., Sivolob A.. Zlatanova J, Leuba SH. Paradox lost: nucleosome structure and dynamics by the DNA minicircle approach. Chromatin Structure and Dynamics: State-of-the-Art. 2004; 39:London: Elsevier; 45–73. [Google Scholar]
- 45. Bancaud A., Conde e Silva N., Barbi M., Wagner G., Allemand J.F., Mozziconacci J., Lavelle C., Croquette V., Victor J.M., Prunell A. et al.. Structural plasticity of single chromatin fibers revealed by torsional manipulation. Nat. Struct. Mol. Biol. 2006; 13:444–450. [DOI] [PubMed] [Google Scholar]
- 46. Hansen J.C. Conformational Dynamics of the chromatin fiber: determinants, mechanisms, and functions. Annu. Rev. Biophys. Biomol. Struct. 2002; 31:361–392. [DOI] [PubMed] [Google Scholar]
- 47. Clauvelin N., Lo P., Kulaeva O.I., Nizovtseva E.V., Diaz-Montes J., Zola J., Parashar M., Studitsky V.M., Olson W.K.. Nucleosome positioning and composition modulate in silico chromatin flexibility. J. Phys. Condens. Matter. 2015; 27:064112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kepper N., Foethke D., Stehr R., Wedemann G., Rippe K.. Nucleosome geometry and internucleosomal interactions control the chromatin fiber conformation. Biophys. J. 2008; 95:3692–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dekker J. Mapping in vivo Chromatin interactions in yeast suggests an extended chromatin fiber with regional in compaction. J. Biol. Chem. 2008; 283:34532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ben-Haïm E., Lesne A., Victor J.M.. Chromatin: a tunable spring at work inside chromosomes. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2001; 64:051921. [DOI] [PubMed] [Google Scholar]
- 51. Ceratti D.R., Obliger A., Jardat M., Rotenberg B., Dahirel V.. Stochastic rotation dynamics simulation of electro-osmosis. Mol. Phys. 2015; 113:2476–2486. [Google Scholar]
- 52. Nägele G., Baur P.. Long-time dynamics of charged colloidal suspensions: hydrodynamic interaction effects. Phys. Stat. Mech. Appl. 1997; 245:297–336. [Google Scholar]
- 53. Milo R., Jorgensen P., Moran U., Weber G., Springer M.. BioNumbers—the database of key numbers in molecular and cell biology. Nucleic Acids Res. 2010; 38:D750–D753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen C., Shi J., Lim H.H., Tamura S., Maeshima K., Surana U., Gan L.. Budding yeast chromatin is dispersed in a crowded nucleoplasm in vivo. Mol. Biol. Cell. 2016; 27:3357–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Szabo Q., Jost D., Chang J.-M., Cattoni D.I., Papadopoulos G.L., Bonev B., Sexton T., Gurgo J., Jacquier C., Nollmann M.. TADs are 3D structural units of higher-order chromosome organization in Drosophila. Sci. Adv. 2018; 4:eaar8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Giorgetti L., Galupa R., Nora E.P., Piolot T., Lam F., Dekker J., Tiana G., Heard E.. Predictive polymer modeling reveals coupled fluctuations in chromosome conformation and transcription. Cell. 2014; 157:950–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kimura H., Shimooka Y., Nishikawa J., Miura O., Sugiyama S., Yamada S., Ohyama T.. The genome folding mechanism in yeast. J. Biochem. 2013; 154:137–147. [DOI] [PubMed] [Google Scholar]
- 58. Soranno A., Buchli B., Nettels D., Cheng R.R., Müller-Späth S., Pfeil S.H., Hoffmann A., Lipman E.A., Makarov D.E., Schuler B.. Quantifying internal friction in unfolded and intrinsically disordered proteins with single-molecule spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:17800–17806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Poirier M.G., Marko J.F.. Effect of internal friction on biofilament dynamics. Phys. Rev. Lett. 2002; 88:228103. [DOI] [PubMed] [Google Scholar]
- 60. Funke J.J., Ketterer P., Lieleg C., Schunter S., Korber P., Dietz H.. Uncovering the forces between nucleosomes using DNA origami. Sci. Adv. 2016; 2:e1600974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tremethick D.J. Higher-Order structures of chromatin: the elusive 30 nm fiber. Cell. 2007; 128:651–654. [DOI] [PubMed] [Google Scholar]
- 62. Mangenot S., Leforestier A., Vachette P., Durand D., Livolant F.. Salt-Induced conformation and interaction changes of nucleosome core particles. Biophys. J. 2002; 82:345–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bilokapic S., Strauss M., Halic M.. Cryo-EM of nucleosome core particle interactions in trans. Sci. Rep. 2018; 8:7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Baumgärtner A. Statics and dynamics of the freely jointed polymer chain with Lennard-Jones interaction. J. Chem. Phys. 1980; 72:871–879. [Google Scholar]
- 65. Larson A.G., Elnatan D., Keenen M.M., Trnka M.J., Johnston J.B., Burlingame A.L., Agard D.A., Redding S., Narlikar G.J.. Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature. 2017; 547:236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Maison C., Almouzni G.. HP1 and the dynamics of heterochromatin maintenance. Nat. Rev. Mol. Cell Biol. 2004; 5:296–304. [DOI] [PubMed] [Google Scholar]
- 67. Erdel F., Rippe K.. Formation of chromatin subcompartments by phase separation. Biophys. J. 2018; 114:2262–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mercy G., Mozziconacci J., Scolari V.F., Yang K., Zhao G., Thierry A., Luo Y., Mitchell L.A., Shen M., Shen Y. et al.. 3D organization of synthetic and scrambled chromosomes. Science. 2017; 355:eaaf4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Duan Z., Andronescu M., Schutz K., McIlwain S., Kim Y.J., Lee C., Shendure J., Fields S., Blau C.A., Noble W.S.. A three-dimensional model of the yeast genome. Nature. 2010; 465:363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Marie-Nelly H., Marbouty M., Cournac A., Flot J.-F., Liti G., Parodi D.P., Syan S., Guillén N., Margeot A., Zimmer C.. High-quality genome (re) assembly using chromosomal contact data. Nat. Commun. 2014; 5:5695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Belton J.-M., Lajoie B.R., Audibert S., Cantaloube S., Lassadi I., Goiffon I., Baù D., Marti-Renom M.A., Bystricky K., Dekker J.. The conformation of yeast chromosome III is mating type dependent and controlled by the recombination enhancer. Cell Rep. 2015; 13:1855–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.