


Abstract
In-frame stop codons mark the termination of translation. However, post-termination ribosomes can reinitiate translation at downstream AUG codons. In mammals, reinitiation is most efficient when the termination codon is positioned close to the 5′-proximal initiation site and around 78 bases upstream of the reinitiation site. The phenomenon was studied mainly in the context of open reading frames (ORFs) found within the 5′-untranslated region, or polycicstronic viral mRNA. We hypothesized that reinitiation of translation following nonsense mutations within the main ORF of p53 can promote the expression of N-truncated p53 isoforms such as Δ40, Δ133 and Δ160p53. Here, we report that expression of all known N-truncated p53 isoforms by reinitiation is mechanistically feasible, including expression of the previously unidentified variant Δ66p53. Moreover, we found that significant reinitiation of translation can be promoted by nonsense mutations located even 126 codons downstream of the 5′-proximal initiation site, and observed when the reinitiation site is positioned between 6 and 243 bases downstream of the nonsense mutation. We also demonstrate that reinitiation can stabilise p53 mRNA transcripts with a premature termination codon, by allowing such transcripts to evade the nonsense mediated decay pathway. Our data suggest that the expression of N-truncated proteins from alleles carrying a premature termination codon is more prevalent than previously thought.
INTRODUCTION
As manifested in the central dogma of molecular biology, ribosome-mediated mRNA translation is the last step in the flow of genetic information (1). The fundamental and complicated cyclical mechanism of translation consists of initiation, elongation, termination and ribosome recycling phases. According to the linear scanning model, in eukaryotes, the pre-initiation complex scans the 5′-untranslated region (UTR) of the mRNA in the 5′ to 3′ direction until the first (proximal) AUG codon is recognized via base pairing with Met-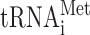 (reviewed in (2,3)). Following GTP hydrolysis-dependent structural and conformational rearrangements of the pre-initiation complex and recruitment of the 60S subunit, the resulting 80S complex enters the elongation stage (4), which terminates when the ribosome encounters an in-frame stop codon (UAA, UAG or UGA; reviewed in ref. (5)). That said, translation in eukaryotes may not always adhere to this oversimplified description (6,7). For example, following stop codon-dependent termination of translation, a fraction of 40S post-termination complexes can resume scanning and reinitiate translation at an AUG codon located downstream to the stop codon (4,8–12). Hence, reinitiation of translation defines at least two open reading frames (ORFs) in a single mRNA, namely an upstream ORF (uORF) that begins with the 5′-proximal AUG, and a downstream ORF that begins with an AUG codon located downstream to the stop codon.
(reviewed in (2,3)). Following GTP hydrolysis-dependent structural and conformational rearrangements of the pre-initiation complex and recruitment of the 60S subunit, the resulting 80S complex enters the elongation stage (4), which terminates when the ribosome encounters an in-frame stop codon (UAA, UAG or UGA; reviewed in ref. (5)). That said, translation in eukaryotes may not always adhere to this oversimplified description (6,7). For example, following stop codon-dependent termination of translation, a fraction of 40S post-termination complexes can resume scanning and reinitiate translation at an AUG codon located downstream to the stop codon (4,8–12). Hence, reinitiation of translation defines at least two open reading frames (ORFs) in a single mRNA, namely an upstream ORF (uORF) that begins with the 5′-proximal AUG, and a downstream ORF that begins with an AUG codon located downstream to the stop codon.
The efficiency of reinitiation in eukaryotes (and consequently, translation of the downstream ORF) is dependent on the mRNA context around the downstream AUG codon, the duration of the 5′-proximal ORF translation process, and the intercistronic distance (6,9–11,13). Since the reinitiation rate is usually lower than the initiation rate, uORFs located within the 5′-UTR of human mRNA transcripts were suggested to play a regulatory role in the translation of the downstream main ORF (5,14–22). In principle, mechanisms that enable reinitiation of translation downstream of an ORF within the 5′-UTR should also enable the reinitiation of translation from an AUG codon located downstream of a nonsense mutation within the main ORF (23–29). Hence, a nonsense mutation upstream to an AUG codon could artificially turn a monocistronic ORF into a polycistronic sequence and counterintuitively lead to the expression of an N-truncated protein (in addition to the C-truncated protein). However, reinitiation of translation is less studied in the context of nonsense mutations found within the main ORF.
The tumor suppressor protein p53 plays a pivotal role in the prevention of cancer development, with mutations in TP53 being the underlying cause of over 50% of human cancers (30–32). There are currently three known N-truncated isoforms of p53: Δ40p53, Δ133p53 and Δ160p53 (Figure 1A). These isoforms are produced through alternative translation initiation sites (Δ40p53, Δ160p53), alternative splicing (Δ40p53), and alternative promoter usage (Δ133p53, Δ160p53) (33–36). According to the IARC TP53 database (version R19, August 2018), there are hundreds of identified nonsense mutations upstream of the codon for Met160 (37). These mutated alleles are usually considered as null alleles. However, we hypothesized that the nonsense mutation-dependent expression of Δ40, Δ133 and Δ160p53 variants by reinitiation, is mechanistically feasible. Using p53 cDNA and minigene in transiently and stably transfected cell lines, together with amber suppression technology, we found significant nonsense mutation-dependent increase in the expression of N-truncated p53 variants by reinitiation of translation. We also describe, for the first time, the expression of the Δ66p53 variant by reinitiation of translation. Our data suggest that expression of N-truncated proteins by reinitiation is an underestimated phenomenon, implying that some alleles, currently regarded as null alleles due to nonsense mutations (including those introduced by CRISPR-Cas9 genome editing), may, in fact, enable the expression of N-truncated proteins.
Figure 1.

N-truncated variants of p53. (A) Potential p53 N-truncated isoforms. Full-length p53 is expressed when translation begins at the first AUG codon. N-truncated p53 isoforms can be expressed by translation initiation or reinitiation at downstream AUG codons at positions 40, 66, 133 and 160 to produce Δ40p53, Δ66p53, Δ133p53 and Δ160p53, respectively. (B) Main TP53 mutants used in the current study. In-frame stop codon mutations (TAG) were introduced at the indicated positions of wild type TP53, as well as the codon for position K120′ of M133′L p53 mutant. (C) Western blot of total cell extracts from HEK293T cells expressing WT or TAG mutants of p53. Cells were transfected with a plasmid carrying the indicated construct and expression was detected using antibodies against the C-terminal HA-tag. ‡ N-truncated p53 that might be produced by initiation of translation at non-AUG codons (39), or by post-translational proteolytic cleavage (40). (D) Expression levels of N-truncated p53 variants, displayed relative to WT p53. Values were obtained by densitometric analyses of Western blot results (panel C), and normalized to actin expression (n = 5, ± SD). Statistical analyses were performed using one-way ANOVA with Dunnett’s post-test. *P < 0.05, **P < 0.01, ***P < 0.001. See Supplementary Figure S1 for a more detailed presentation of the data.
MATERIALS AND METHODS
General
Unless stated otherwise, chemicals and DNA oligomers were ordered from Sigma Aldrich (Darmstadt, Germany) and used without further purification. Nε-acetyl lysine was purchased from Chem-Impex International (Wood Dale, IL, USA). Enzymes for molecular cloning (i.e. restriction, ligation and Phusion High-Fidelity DNA Polymerase for PCR reactions) were purchased from NEB (Ipswich, MA, USA) and used according to the manufacturer’s instructions. DNA was purified using spin columns from Macherey Nagel (Düren, Germany). Escherichia coli DH10B cells (Life technologies, Carlsbad, CA, USA) were used for molecular cloning and plasmid propagation. Escherichia coli BL21(DE3) cells (NEB) were used for recombinant protein expression. During cloning steps, the bacteria were incubated in liquid LB media or plated on LB-agar plates supplemented with the required antibiotic (32 µg/ml zeocin or 50 µg/ml ampicillin). Anti-6×His (#G020) and anti-HA tag (#G036) antibodies were purchased from abm (Richmond, ON). Anti actin antibody (#4967) was purchased from Cell Signaling (Danvers, MA, USA). Anti-hUPF1 antibody (#ab109363) was purchased from Abcam (Cambridge, UK). Secondary horseradish peroxidase-conjugated anti-mouse IgG (#ab7068) and anti-rabbit IgG (#ab92080) were purchased from Abcam.
Vector construction
All genes for protein expression in mammalian cells were cloned into the commercially available pBudCE4.1 vector (Invitrogen-Thermo Fisher Scientific, Waltham, MA, USA). DNA encoding N′-6×His- and C′-HA-tagged WT p53 was amplified using primers 1/2 or primers 3/4 (Supplementary Table S1), and cloned between the KpnI and NotI or BamHI and EcoRI restriction sites downstream of the EF1α or CMV promoters, respectively. Point mutations were introduced by site-directed mutagenesis using primers 5–34. The DNA for N′-6×His- and C′-HA-tagged p53 Q52′TAG mini gene (IDT, Leuven, Belgium) was cloned between KpnI and NotI restriction sites downstream of the EF1α promoter. The point mutation M66′L was introduced using primers 35/36. DNA for C′-HA-tagged WT Eno1 was amplified from plasmid HsCD00000030 (DNASU plasmid repository) using primers 37/38 and primers 2/37, and cloned between the KpnI and NotI restriction sites downstream of the EF1α promoter. In-frame TAG mutations at positions encoding K80 or G156 of Eno1 were introduced using primers 39/40 or 41/42, respectively. Similarly, DNA encoding C′-HA-tagged WT PFK (IDT) was amplified using primers 43/44 and 2/43, and cloned between KpnI and NotI restriction sites downstream of the EF1α promoter. In-frame TAG mutation at position K25 was introduced using primers 45 and 46. For the expression of p53 variants in bacteria, WT and truncated TP53 were amplified using primers 47–53, and cloned between the NdeI and XhoI restriction sites of the commercially available pRSETb vector (Thermo Fisher Scientific).
Cell culture
Cells were incubated at 37°C in a humidified chamber under a 5% CO2 atmosphere. HEK293T and HCT116-p53(–/–) cells were maintained in DMEM (Biological Industries, Beit Haemek, Israel) and McCoy’s medium (Sigma Aldrich), respectively. Culture media were supplemented with 10% (v/v) heat-inactivated fetal bovine serum, 2 mM l-glutamine, 1 mM sodium pyruvate, 100 units/ml penicillin G sodium, 0.1 mg/ml streptomycin sulfate and 1.25 units/ml nystatin (Biological Industries). For Western blot or RT-qPCR analyses, cells were seeded 24 h before transfection. HEK293T cells were transfected with Lipofactamine 2000 (Invitrogen-Thermo Fisher Scientific) or LipoD293 (Signagen Laboratories, Rockville, MD, USA), while HCT116-p53(–/–) cells were transfected with FuGENE (Promega, Madison, WI, USA). Transfections were performed at 3:1 to 2.5:1 transfection reagent:DNA ratios, according to the manufacturer’s protocol.
Stable cell lines
HEK293T or HCT116-p53(–/–) cells were seeded in 12-well plates and transfected with indicated pBudCE4.1-based plasmids, as described above for transient transfection. Twenty-four hours post-transfection, cells were transferred to a 10 cm plate, and incubated in media supplemented with 25 µg/ml zeocin. Cells were incubated for at least 14 days under selection conditions for polyclonal stable cell lines (HEK293T) or for monoclonal stable cell lines (HCT116). HCT116 monoclonal stable cell lines were isolated using cloning discs.
Western blotting
Twenty-four hours post-transfection, cells were lysed with ice-cold RIPA lysis buffer (50 mM Tris buffer pH 8.0, 150 mM NaCl, 1% (v/v) Triton X-100, 0.5% (w/v) sodium deoxycholate and 0.1% (w/v) SDS) supplemented with protease inhibitors (1.2 mg/ml leupeptin, 1 mM pepstatin A, 100 mM PMSF and 1 mg/ml aprotinin). Total protein concentration in clear lysates was determined using a BCA assay kit (Thermo Fisher Scientific) and samples were normalized accordingly. The cleared whole-cell lysate was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred to a 0.2 µm nitrocellulose membrane using a semi-dry transfer apparatus (Trans-Blot Turbo, BioRad, Hercules, CA, USA). Membranes were blocked with Tris-buffered saline containing 0.05% (v/v) Tween-20 (TBST) and 5% (w/v) non-fat dry milk and incubated over night with primary antibodies diluted in 5% (w/v) bovine serum albumin in TBST, at 4°C. Following washing with TBST, the membranes were incubated with secondary antibodies at room temperature for 1 h. Membranes were washed with TBST before antibody binding was visualized using ECL reagent (GE Healthcare, Chicago, IL, USA). Immunoblot intensities were quantified with ImageJ (38).
RNA interference
HEK293T or HCT116-p53(–/–)-derived stable cell lines were seeded 24 h before treatment at ∼15% confluency. Cells were transfected with specific siRNA directed against hUPF1 or with control siRNA (IDT) using Oligofectamine (Invitrogen), according to the manufacturer’s instructions. The cells were harvested 48 h post transfection. hUPF1-specific siRNA sequence: FW: 5′-GUGACGAGUUUAAAUCACAAAUCGA-3′, Rev: 5′-UCGAUUUGUGAUUUAAACUCGUCACCA-3′. Nonspecific control siRNA sequence: FW: 5′-CGUUAAUCGCGUAUAAUACGCGUA-3′, Rev: 5′-AUACGCGUAUUAUACGCGAUUAACGAC-3′.
Real time quantitative PCR
Total RNA was extracted from HCT116-p53(–/–) or HEK293T cells using a NucleoSpin RNA kit (Macherey Nagel) according to manufacturer’s instructions. Total RNA was subjected to reverse transcription using the iScript cDNA synthesis kit (BioRad). Real time quantitative PCR (RT-qPCR) was carried out using the UPL probe library system or SYBR green PCR master Mix (Roche, Basel, Switzerland). Samples were amplified in triplicates in a 384-well plate format using the following cycling conditions. UPL probe library: 10 min at 95°C, 45 cycles of 10 s at 95°C, 30 s at 60°C and 1 s at 72°C, followed by 30 s at 40°C. SYBR green: 5 min at 95°C, 45 cycles of 10 s at 95°C, 10 s at 60°C and 10 s at 72°C, followed by melting curve of 5 s at 95°C, 1 min 65–97°C, and a final step of 30 s at 40°C. mRNA levels were normalized to GAPDH. Analyses were performed using primers described in Supplementary Table S2.
Expression of truncated p53 variants in bacteria
Different p53 variants (full length, Δ40, Δ66, Δ133, Δ160 and Δ169) were expressed in E. coli BL21(DE3) cells grown in 2×TY medium. At OD600 = 0.6, protein expression was induced with 1 mM isopropyl β-d-1-thiogalactoside (IPTG) and the bacteria were incubated over night at 22°C. Cells from the over night culture were pelleted by centrifugation and lysed in 1×Laemmli sample buffer by heating to 95°C for 5 min, mixing and repeating the heating step. Cleared lysates were diluted as required before the sizes of expressed proteins were analysed by SDS-PAGE followed by Western blotting.
RESULTS
In-frame nonsense mutations promote the expression of N-truncated p53 variants
To study the effects of in-frame nonsense mutations on the expression of N-truncated p53, we introduced stop codon (TAG) mutations at different positions along TP53 fused to C-terminal hemagglutinin (HA) and N-terminal 6×His tags (Figure 1B).[To remain coherent with WT p53 amino acid numbering, we ignored the additional six histidine residues at the N-terminus of p53. The relevant numbers are marked by apostrophe, such that p53 residues 40′, 50′ and 60′ refer to positions 46, 56 and 66 of expressed p53, respectively.] To identify possible effects of p53 transcriptional activity, we also cloned the transcriptionally inactive R273′H p53 mutant (41,42). All variants were first cloned downstream of an EF1α promoter.
According to Western blot analysis using antibodies against the C-terminal HA tag, full-length p53 and Δ40′p53 were the two main variants expressed from WT and R273′H TP53, in addition to Δ133′p53 and Δ160′p53 that were expressed at lower levels (Figure 1C and D, and Supplementary Figure S1) (43). These results are expected, since Δ40p53, Δ133p53 and Δ160p53 are known transcription variants of p53 (33–36,44). However, in-frame nonsense mutations changed this expression pattern. When an in-frame TAG mutation was introduced at position Q38′ (Q38′TAG), expression levels of Δ40′p53, Δ66′p53 and Δ133′p53 increased significantly (Figure 1D and Supplementary Figure S1). The same phenomenon was observed when an in-frame TAG mutation was introduced at position Q52′ (Q52′TAG), with increased expression levels of Δ66′p53 and Δ133′p53, relative to WT and R273′H TP53. When the TAG mutation was introduced at position K120′ (K120′TAG), Δ133′ and Δ160′p53 were expressed at levels similar to those measured with Q52′TAG. This can be attributed to the fact that reinitiation downstream of 57 codons long uORF (Q52′TAG) is expected to be more efficient than reinitiation downstream of 125 codons long uORF (K120′TAG) (5,10). However, when M133′ was mutated to leucine (K120′TAG M133′L), a significant increase in the expression of Δ160′p53 was observed. Such an expression pattern cannot be explained by out-of-frame AUG codons (Supplementary Figure S2), and the sizes of all truncated variants were verified using bacterially-expressed N-truncated p53 (Supplementary Figure S3). Similar to p53, an in-frame stop codon in position G156 of α-enolase promoted the expression of Δ165 enolase (Supplementary Figure S4A), and an in-frame stop codon in position K25 of human platelet-specific isoform of phosphofructokinase (PFK) promoted the expression of Δ39 PFK (Supplementary Figure S4B). These data demonstrate that in our expression system, nonsense mutation-dependent expression of N-truncated proteins is not limited to p53. Furthermore, these results clearly show that in-frame nonsense mutations can promote the expression of N-truncated proteins translated from AUG codons positioned ∼160 codons downstream of the initiation site.
Nonsense mutation-dependent N-truncation is observed with different cell lines, promoters, and stop codons
To explore the generality of N-truncated protein expression, we considered the effects of the cell line and promoter on the expression of N-truncated p53. The same expression pattern was found when WT and TAG-mutants of TP53 were cloned downstream of the cytomegalovirus (CMV) promoter in the same expression vector (Figure 2A), or when expressed in the p53-null HCT116 cell line (Supplementary Figure S5) (45). Moreover, to determine whether the observed nonsense mutation-dependent expression was unique to the TAG codon, mutations to other stop codons (TGA and TAA) were introduced at positions Q38′ and Q52′ of TP53. The three stop codons equally promoted the expression of N-truncated p53 variants starting at AUG codons downstream of the nonsense mutation (Figure 2B and Supplementary Figure S6). Therefore, our data show that nonsense mutation-dependent expression of N-truncated p53 variants is not limited to a specific promoter, cell line or type of in-frame stop codon.
Figure 2.

Expression of N-truncated p53 by nonsense mutation-dependent reinitiation. (A) Expression of WT and N-truncated p53 from a TP53 cDNA cloned downstream of the CMV promoter (compared to genes cloned downstream of the EF1α promoter portrayed in Figure 1C). (B) Expression of N-truncated p53 as a function of premature stop codon type. Indicated stop codon mutations (TAG, TGA or TAA) were introduced at codons for positions Q38′ or Q52′, and expression of N-truncated p53 was evaluated by Western blotting using antibodies against the C-terminal HA-epitope. (C) The ratio between 5′ and 3′ p53 mRNA levels. For each indicated p53 variant, mRNA levels were quantified by RT-qPCR using primers recognizing a sequence at the 5′ (p-5′1, p-5′2) or 3′ (p-3′) end of p53 mRNA. The ratios between 5′ to 3′ mRNA levels are plotted relative to those measured for WT p53. (D) Expression of p53 variants detected using antibodies against the N-terminal 6×His-tag. Expression of p53 Q52′TAG and K120′TAG mutants of p53 yielded C-truncated p53(1–51′) and p53(1–119′), respectively. The unstable p53(1–37′) peptide was not detected. (E) Co-expression of p53 K120TAG together with the amber suppression machinery. The use of an evolved acetyl lysine synthetase and its cognate amber suppressor tRNA enabled the incorporation of the non-canonical amino acid Nε-acetyl lysine in response to the in-frame TAG mutation encoding position 120. (F) The effect of frame-shift mutation on expression of N-truncated p53 variants evaluated by western blotting using antibodies against the C-terminal HA-tag.
mRNA transcripts of p53 cDNA with an in-frame nonsense mutation are not subjected to 5′-mRNA degradation
Next, we asked whether the expression of N-truncated p53 variants was the result of mRNA degradation. To answer this question, we measured p53 mRNA levels by real-time quantitative PCR (RT-qPCR) using specific primers designed to amplify a sequence close to the 3′-end of p53 mRNA (Figure 2C, p-3′) and two sets of primers designed to amplify different sequences close to the 5′-end of p53 mRNA (p-5′1 and p-5′2). We then calculated the ratio between mRNA levels (i.e. p-5′/p-3′) measured for WT p53 and p53 Q38′TAG (using p-5′1) or WT p53 and p53 K120′TAG (using p-5′2). In both cases, the normalized p-5′/p-3′ ratio calculated for the TAG mutants was similar to the ratio measured for WT p53, indicating that no mutation-dependent degradation of 5′-mRNA had occurred. Furthermore, total levels of p53 mRNA transcripts carrying an in-frame nonsense mutation were similar to each other, although lower than the levels of WT p53 mRNA transcript (Supplementary Figure S7). Thus, the expression of N-truncated p53 variants was enabled at the level of translation and was not regulated at the transcriptional level, nor was the result of mRNA degradation.
N-truncated p53 variants are expressed by nonsense mutation-dependent reinitiation of translation
Given the data presented above, we sought to describe the mechanism underlying nonsense mutation-dependent expression of N-truncated p53. Leaky scanning may contribute to the expression of N-truncated proteins, but the positive correlation between expression levels and the presence of an upstream nonsense mutation suggests that another mechanism was responsible for the observed nonsense mutation-dependent expression of N-truncated variants. One possible mechanism is initiation of translation at the 5′-proximal AUG codon, followed by termination at an in-frame stop codon and reinitiation of translation at downstream AUG codons. In such cases, the N-terminal of the protein (i.e. C-truncated p53) should be expressed. To asses the expression of C-truncated p53, we performed Western blot analysis using antibodies against the N-terminal 6×His-tag (Figure 2D). As expected, strong bands observed at ∼53 kDa when WT and R273′H p53 were expressed (lanes 1 and 2), indicative of the expression of full-length p53. As the N-terminal region of p53 is natively unfolded and unstable, p53(1–37′) could not be detected (lane 3). However, weak bands of C-truncated p53 were detected, reflecting the expression of p53(1–51′) and p53(1–119′) from Q52′TAG and K120′TAG mutants of p53, respectively (lanes 4 and 5). Hence, during the expression of p53 Q52′TAG or K120′TAG mutants, translation was initiated at the 5′-proximal AUG codon and terminated at the in-frame stop codon at position 52′ or 120′, respectively. In addition, we co-expressed the p53 K120TAG mutant with amber suppression machinery for the incorporation of Nε-acetyl-lysine at position 120 by an aminoacylated amber suppressor tRNA (Figure 2E) (46,47). Suppression of the in-frame stop codon enabled the expression of full-length K120-acetylated p53, indicating that the translation of p53 mRNA with an in-frame UAG codon at position K120 was initiated at the first AUG codon. Finally, we introduced a frame-shift between the TAG mutation at position 38′, 52′ or 120′, and the downstream ATG codon at position 40′, 66′ or 133′, respectively. In all cases, the frame-shift mutation had no effect on N-truncated p53 expression levels, supporting a mechanism of ribosome disassembly and re-assembly (Figure 2F). Taken together, our data show that translation of the studied N-truncated p53 variants initiated at the 5′-proximal AUG codon, terminated at an in-frame stop codon and then reinitiated at downstream AUG codons. Hence, we conclude that the underlying mechanism of in-frame nonsense mutation-dependent expression of N-truncated p53 is reinitiation of translation. Therefore, known N-truncated isoforms of p53 may be expressed by mechanism of reinitiation, in addition to the previously described alternative initiation from internal AUG codons.
Reinitiation from the AUG codon at position 66′ is mechanistically feasible despite the long uORF and short intercistronic distance
Previous studies found that reinitiation decreases significantly when the uORF length increases beyond 13–18 codons (11), and that reinitiation is most efficient when the intercistronic distance is 80–90 nucleotides (9). However, we found that expression levels of Δ66′p53 from Q52′TAG were ∼80% of WT p53 (Figure 1D), although in this construct the length of the uORF was 58 codons and the intercistronic distance was 42 nucleotides. Therefore, we decided to check if the relatively high expression level of Δ66′p53 from Q52′TAG is indeed a unique example of reinitation—suggesting that the previously determined constraints on reinitiation are too strict—or maybe other factors affect the reinitiation from the AUG codon at position 66′. Specifically, one may argue that the known alternative translation initiation site at position 40′ promotes the reinitiation from position 66′. According to this hypothetical model, the AUG codon at position 40′ can serve as an additional initiation site (pseudo initiation site), thereby creating an uORF between codons 40′ and 52′, with an ideal length of 12 codons, upstream to the AUG codon at position 66′. To verify that the AUG codon at position 40′ (or 44′) is not a pseudo initiation site, we mutated Met40′ and Met44′ to leucines in both WT and Q52′TAG p53. We quantified Δ66′p53 expression levels and found that these mutations had no negative effect on reinitiation of translation from the AUG codon at position 66′ of the Q52′TAG construct (Figure 3A and Supplementary Figure S8). Moreover, we found that M40′L and M44′L mutations in WT p53 had a small effect on expression levels of Δ66′p53 (Figure 3A, lanes 1–3), suggesting only a minor contribution of leaky scanning or alternative initiation to the expression of Δ66′p53. Thus, the AUG codon at position 66′ is a bona fide reinitiation site and the codon for Met40′ (or Met44′) does not serve as a pseudo-upstream initiation site. Hence, nonsense mutation-dependent expression of Δ66′p53 is mechanistically feasible, and efficient reinitiation may occur when the uORF is longer than 13–18 codons and the intercistronic distance is shorter than 80–90 nucleotides.
Figure 3.

A premature termination codon at position Q52′ enables the expression of Δ66′p53 and stabilises the mRNA transcript. (A) The effect of initiation from the AUG codons at positions 40′ and 44′ on the level of reinitiation from the AUG codon at position 66′. Western blot analysis of total cell lysates of HEK293T cells expressing the indicated variants of WT p53 (lanes 1–3) or Q52′TAG p53 (lanes 4–6). (B) The effect of intron 5 on the expression of Δ66′p53. Expression of N-truncated p53 variants in HEK293T cells, transiently (left) or stably (right) transfected with p53 Q52′TAG cDNA (–intron 5, lane 1) or Q52′TAG minigene (+intron 5, lane 2), were evaluated by Western blot analysis using antibodies against the C-terminal HA tag. (C) Knockdown of hUPF1. HEK293T cells stably expressing Q52′TAG minigene with either Met or Leu at position 66′ were treated with siRNA directed against hUPF1 or control siRNA (cont.). Protein expression levels were evaluated by immunoblotting using a specific antibody against hUPF1. (D) Left: Levels of SC35 1.7, CARS, SMG1 and p53 mRNA transcripts measured by RT-qPCR, following transfection of cells with sequence-specific or control siRNA, as described in panel C. Data represent the fold change in mRNA levels of the indicated transcript, relative to control. Right: Relative levels of mRNA transcripts measured in cells stably expressing the indicated Q52′TAG minigene and treated with hUPF1-specific or control siRNA. Data are displayed relative to mRNA levels of Q52′TAG minigene in cells treated with control siRNA. Values represent the mean ± SD of at least three independent experiments, each measured in triplicates.
Premature termination codon-dependent reinitiation of translation stabilises mRNA transcripts by circumventing the NMD
The data presented thus far was collected using TP53 cDNA with an in-frame nonsense mutation. However, mRNA of genomic TP53 with a premature termination codon (PTC) is expected to be recognized and degraded by the nonsense-mediated mRNA decay (NMD) pathway. According to current models, an in-frame nonsense mutation can be identified as a PTC, if positioned upstream to an exon-exon junction. However, if reinitiation is enabled by an AUG codon positioned between the PTC and the downstream exon-exon junction, the protein complex that marks the exon-exon junction should be displaced from the mRNA by the translating ribosome during the pioneering round of translation. Consequently, the in-frame nonsense mutation may not be recognized as a PTC by the NMD. Thus, reinitiation may allow mRNA transcripts with a PTC to escape degradation by the NMD pathway and thereby stabilises such transcripts (13,23,27,48–51).
To study the interplay between NMD-mediated degradation of mRNA transcripts with a PTC and nonsense mutation-dependent reinitiation of translation, we created a p53 minigene with intron 5 downstream of the nonsense mutation at position Q52′ (Q52′TAG minigene). We found that expression levels of Δ66′p53 were not affected by the addition of intron 5, when HEK293T cells were transiently transfected with Q52′TAG p53 (–intron 5) or Q52′TAG minigene (+intron 5; Figure 3B, left blot). Moreover, the addition of intron 5, which defines the UAG codon at position 52′ as a PTC, had no effect on expression levels of Δ66′p53 in HEK293T cell lines stably transfected with the same constructs (Figure 3B, right blot). Thus, p53 mRNA transcripts with a PTC upstream of the exon 4–exon 5 junction allow for the expression of Δ66′p53.
Next, we asked if TP53 mRNA transcripts carrying a PTC at position Q52′ are substrates of the NMD pathway, and if reinitiation affects NMD efficiency. To answer these questions, we first created another HEK293T cell line, stably transfected with M66′L mutant of the Q52′TAG minigene (Q52′TAG M66′L minigene). In this mutant, reinitiation from the AUG codon at position 66′ and subsequent mRNA stabilisation, is not expected. We then inhibited the NMD using siRNA directed against hUPF1, an essential factor of the NMD pathway (52). Western blot analyses showed a significant downregulation of hUPF1 in cells transfected with sequence specific siRNA, compared to random siRNA (Figure 3C). NMD inhibition following hUPF1 knockdown was also confirmed by the measured increase in mRNA levels of several known physiological NMD substrates: SC35 1.7, CARS and SMG1 (Figure 3D, left) (53,54). Importantly, NMD inhibition resulted in a significant increase in p53 mRNA levels, with a stronger effect on mRNA transcripts of the M66′L mutant. Moreover, similar effects of NMD inhibition on mRNA levels were measured in three monoclonal HCT116p53(–/–) cell lines stably transfected with Q52′TAG minigene (Supplementary Figure S9). These data show that Δ66′p53 can be expressed via nonsense mutation-dependent reinitiation, although p53 transcripts with a PTC at position 52′ are subjected to degradation by the NMD pathway.
In agreement with current models, our data show that reinitiation stabilises mRNA transcripts with a PTC, by circumventing the NMD. NMD inhibition in cells expressing Q52′TAG minigene resulted in an ∼1.5-fold increase in p53 mRNA levels, while a 1.9-fold increase in mRNA levels was measured in cells expressing Q52′TAG M66′L minigene (Figure 3D, right). Similarly, p53 mRNA levels in cells expressing Q52′TAG M66′L minigene were lower, compared to their levels in cells expressing Q52′TAG minigene. Hence, mRNA transcripts of Q52′TAG minigene that enable reinitiation from position 66′ were less sensitive to NMD inhibition and accumulated to higher levels, compared to mRNA transcripts of the Q52′TAG M66′L minigene with no reinitiation site between the PTC and the downstream exon-exon junction.
DISCUSSION
Reinitiation of translation was first reported by Marilyn Kozak (8,9). Here, we provided three examples for nonsense mutation-dependent reinitiation of translation and more rigorous studies are required in order to understand how general this observation is. The phenomenon and its underlying mechanisms in mammalian cells were mainly studied in the context of a regulatory element following translation of an uORF within the 5′-UTR. The stress-dependent expression of GCN4 in Saccharomyces cerevisiae (or its mammalian homologue ATF4) via eIF2α phosphorylation-dependent reinitiation of translation is one such example (16,17,55,56). That said, in the Exac database (57), which currently includes the exomes of 60,706 individuals, there are over 30,000 valid stop codon mutations, of which, thousands are located within the first 150 codons. Alleles with PTCs are usually considered null alleles. However, in line with our data, such alleles may enable the expression of N-truncated proteins by reinitiation of translation from a downstream AUG codon, and may even ‘rescue’ patients from potentially deleterious effects of a null allele. For example, the E12X and Y27X mutations in the aristaless-related homeobox gene (ARX) promoted the expression of Δ41ARX using the Met41 codon as reinitiation site (58). Patients carrying the R37X mutation in the ATRX gene escape the rather lethal ATRX-null phenotype, due to expression of Δ40ATRX (59). Moreover, as demonstrated here, reinitiation can stabilise mRNA transcripts with a PTC by rescuing them from degradation by the NMD, thereby allowing such transcripts to accumulate in the cell. Hence, nonsense mutation-dependent reinitiation within the main ORF has the potential for either protecting against disease by enabling the expression of truncated—yet functional—proteins, or intensifying the disease phenotype by allowing expression of potentially harmful N- as well as C-truncated proteins. While nonsense mutation-dependent reinitiation was suggested to increase when the reinitiation site is close to the 5′-end of the main ORF, we found significant reinitiation of translation from AUG codons positioned over 160 codons downstream of the first initiation site. Thus, our data suggest that when referring to genes with PTCs, nonsense mutation-dependent expression of N-truncated proteins should be considered, in addition to the default assumption of a null allele.
Our results are also important for studies in cultured mammalian cells involving genetic code expansion technology, where a non-canonical amino acid is encoded by an in-frame stop codon. As also demonstrated in yeast (24), an introduced stop codon mutation can promote the expression of N-truncated proteins, which must be considered in such experiments. In addition, in recent years, we have witnessed a surge in the use of CRISPR/Cas9 technology. One of the common strategies for knocking-out specific genes using this genome editing approach is to introduce a PTC within the first exon. Given the data presented here, it may be important to confirm whether an introduced PTC promotes the expression of an N-truncated protein.
It is still not clear why nonsense mutation-dependent reinitiation of translation is mechanistically feasible. Is PTC-dependent reinitiation of translation an evolved mechanism with specific functions in the eukaryotic cell? Does it provide an evolutionary advantage? Is reinitiation of translation in mutated proteins regulated, and if so, by which mechanism(s)? We hope that future research will address these and other questions related to reinitiation of translation downstream of a PTC.
Supplementary Material
Notes
Present address: Noa Deshe, Department of Genetics, Silberman Institute of Life Sciences, Edmond J. Safra Campus, The Hebrew University, Jerusalem, Israel.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Research Council (ERC) under the European Union Horizon 2020 research and innovation programme [678461 to E.A.]; Israel Science Foundation [807/15 to E.A.]. Funding for open access charge: H2020 ERC grant.
Conflict of interest statement. None declared.
REFERENCES
- 1. Crick F. Central dogma of molecular biology. Nature. 1970; 227:561–563. [DOI] [PubMed] [Google Scholar]
- 2. Hinnebusch A.G. Structural insights into the mechanism of scanning and start codon recognition in eukaryotic translation initiation. Trends Biochem. Sci. 2017; 42:589–611. [DOI] [PubMed] [Google Scholar]
- 3. Hinnebusch A.G. The scanning mechanism of eukaryotic translation initiation. Annu. Rev. Biochem. 2014; 83:779–812. [DOI] [PubMed] [Google Scholar]
- 4. Kozak M. Initiation of translation in prokaryotes and eukaryotes. Gene. 1999; 234:187–208. [DOI] [PubMed] [Google Scholar]
- 5. Jackson R.J., Hellen C.U.T., Pestova T.V.. Termination and Post-termination Events in Eukaryotic Translation. Advances in Protein Chemistry and Structural Biology. 2012; 86:1st edn. Elsevier Inc; 45–93. [DOI] [PubMed] [Google Scholar]
- 6. Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986; 44:283–292. [DOI] [PubMed] [Google Scholar]
- 7. Kozak M. A short leader sequence impairs the fidelity of initiation by eukaryotic ribosomes. Gene Expression. 1991; 1:111–115. [PMC free article] [PubMed] [Google Scholar]
- 8. Kozak M. Selection of initiation sites by eucaryotic ribosomes: effect of inserting AUG triplets upstream from the coding sequence for preproinsulin. Nucleic Acids Res. 1984; 12:3873–3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kozak M. Effects of intercistronic length on the efficiency of reinitiation by eucaryotic ribosomes. Mol. Cell Biol. 1987; 7:3438–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Luukkonen B.G., Tan W., Schwartz S.. Efficiency of reinitiation of translation on human immunodeficiency virus type 1 mRNAs is determined by the length of the upstream open reading frame and by intercistronic distance. J. Virol. 1995; 69:4086–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kozak M. Constraints on reinitiation of translation in mammals. Nucleic Acids Res. 2001; 29:5226–5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Skabkin M., Skabkina O., Hellen C.T., Pestova T.. Reinitiation and other unconventional posttermination events during eukaryotic translation. Mol. Cell. 2013; 51:249–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pereira F.J., Teixeira A., Kong J., Barbosa C., Silva A.L., Marques-Ramos A., Liebhaber S.A., Romão L.. Resistance of mRNAs with AUG-proximal nonsense mutations to nonsense-mediated decay reflects variables of mRNA structure and translational activity. Nucleic Acids Res. 2015; 43:6528–6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Andreev D.E., O’Connor P.B., Fahey C., Kenny E.M., Terenin I.M., Dmitriev S.E., Cormican P., Morris D.W., Shatsky I.N., Baranov P.V.. Translation of 5′ leaders is pervasive in genes resistant to eIF2 repression. eLife. 2015; 4:e03971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Starck S.R., Tsai J.C., Chen K., Shodiya M., Wang L., Yahiro K., Martins-Green M., Shastri N., Walter P.. Translation from the 5′ untranslated region shapes the integrated stress response. Science. 2016; 351:aad3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lu P.D., Harding H.P., Ron D.. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 2004; 167:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mueller P.P., Hinnebusch A.G.. Multiple upstream AUG codons mediate translational control of GCN4. Cell. 1986; 45:201–207. [DOI] [PubMed] [Google Scholar]
- 18. Calvo S.E., Pagliarini D.J., Mootha V.K.. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:7507–7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee S., Liu B., Lee S., Huang S.-X., Shen B., Qian S.-B.. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:E2424–E2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ingolia N.T., Ghaemmaghami S., Newman J. R.S., Weissman J.S.. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009; 324:218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Barbosa C., Peixeiro I., Romão L.. Gene expression regulation by upstream open reading frames and human disease. PLoS Genet. 2013; 9:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hinnebusch A.G., Ivanov I.P., Sonenberg N.. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science. 2016; 352:1413–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stump M.R., Gong Q., Packer J.D., Zhou Z.. Early LQT2 nonsense mutation generates N-terminally truncated hERG channels with altered gating properties by the reinitiation of translation. J. Mol. Cell Cardiol. 2012; 53:725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kalstrup T., Blunck R.. Reinitiation at non-canonical start codons leads to leak expression when incorporating unnatural amino acids. Scientific Rep. 2015; 5:11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Puel A., Reichenbach J., Bustamante J., Ku C.-L., Feinberg J., Döffinger R., Bonnet M., Filipe-Santos O., de Beaucoudrey L., Durandy A. et al.. The NEMO mutation creating the most-upstream premature stop codon is hypomorphic because of a reinitiation of translation. Am. J. Hum.Genet. 2006; 78:691–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Paulsen M., Lund C., Akram Z., Winther J.R., Horn N., Møller L.B.. Evidence that translation reinitiation leads to a partially functional Menkes protein containing two copper-binding sites. Am. J. Hum. Genet. 2006; 79:214–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Buisson M., Anczuków O., Zetoune A.B., Ware M.D., Mazoyer S.. The 185delAG mutation (c.68-69delAG) in the BRCA1 gene triggers translation reinitiation at a downstream AUG codon. Hum. Mutat. 2006; 27:1024–1029. [DOI] [PubMed] [Google Scholar]
- 28. Melis J.P.M., Hoogervorst E.M., van Oostrom C. T.M., Zwart E., Breit T.M., Pennings J.L.A., de Vries A., van Steeg H.. Genotoxic exposure: novel cause of selection for a functional ΔN-p53 isoform. Oncogene. 2011; 30:1764–1772. [DOI] [PubMed] [Google Scholar]
- 29. Gunišová S., Hronová V., Mohammad M.P., Hinnebusch A.G., Valášek L.S.. Please do not recycle! Translation reinitiation in microbes and higher eukaryotes. FEMS Microbiol. Rev. 2018; 42:165–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bieging K.T., Attardi L.D.. Deconstructing p53 transcriptional networks in tumor suppression. Trends Cell Biol. 2012; 22:97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vousden K.H., Prives C.. Blinded by the light: the growing complexity of p53. Cell. 2009; 137:413–431. [DOI] [PubMed] [Google Scholar]
- 32. Menendez D., Inga A., Resnick M.a.. The expanding universe of p53 targets. Nat. Rev. Cancer. 2009; 9:724–737. [DOI] [PubMed] [Google Scholar]
- 33. Surget S., Khoury M.P., Bourdon J.-C.. Uncovering the role of p53 splice variants in human malignancy: a clinical perspective. OncoTargets Ther. 2013; 7:57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sharathchandra A., Katoch A., Das S.. IRES mediated translational regulation of p53 isoforms. Wiley Interdiscipl. Rev.: RNA. 2014; 5:131–139. [DOI] [PubMed] [Google Scholar]
- 35. Khoury M.P., Bourdon J.-C.. p53 isoforms: an intracellular microprocessor?. Genes Cancer. 2011; 2:453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Courtois S., Verhaegh G., North S., Luciani M.-G., Lassus P., Hibner U., Oren M., Hainaut P.. ΔN-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene. 2002; 21:6722–6728. [DOI] [PubMed] [Google Scholar]
- 37. Bouaoun L., Sonkin D., Ardin M., Hollstein M., Byrnes G., Zavadil J., Olivier M.. TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum. Mutat. 2016; 37:865–876. [DOI] [PubMed] [Google Scholar]
- 38. Schneider C.A., Rasband W.S., Eliceiri K.W.. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012; 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Peabody D.S. Translation initiation at non-AUG triplets in mammalian cells. J. Biol. Chem. 1989; 264:5031–5035. [PubMed] [Google Scholar]
- 40. Okorokov A.L., Ponchel F., Milner J.. Induced N- and C-terminal cleavage of p53: a core fragment of p53, generated by interaction with damaged DNA, promotes cleavage of the N-terminus of full-length p53, whereas ssDNA induces C-terminal cleavage of p53. EMBO J. 1997; 16:6008–6017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Magrini R., Russo D., Fronza G., Inga A., Menichini P.. The kinetics of p53-binding and histone acetylation at target promoters do not strictly correlate with gene expression after UV damage. J. Cell Biochem. 2007; 100:1276–1287. [DOI] [PubMed] [Google Scholar]
- 42. Ang H.C., Joerger A.C., Mayer S., Fersht A.R.. Effects of common cancer mutations on stability and DNA binding of full-length p53 compared with isolated core domains. J. Biol. Chem. 2006; 281:21934–21941. [DOI] [PubMed] [Google Scholar]
- 43. Toledo F., Wahl G.M.. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat. Rev. Cancer. 2006; 6:909–923. [DOI] [PubMed] [Google Scholar]
- 44. Ray P.S., Grover R., Das S.. Two internal ribosome entry sites mediate the translation of p53 isoforms. EMBO Rep. 2006; 7:404–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bunz F. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998; 282:1497–1501. [DOI] [PubMed] [Google Scholar]
- 46. Neumann H., Hancock S.M., Buning R., Routh A., Chapman L., Somers J., Owen-Hughes T., van Noort J., Rhodes D., Chin J.W.. A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Molecular Cell. 2009; 36:153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cohen S., Arbely E.. Single-plasmid-based system for efficient noncanonical amino acid mutagenesis in cultured mammalian cells. ChemBioChem. 2016; 17:1008–1011. [DOI] [PubMed] [Google Scholar]
- 48. Zhang J., Maquat L.E.. Evidence that translation reinitiation abrogates nonsense-mediated mRNA decay in mammalian cells. EMBO J. 1997; 16:826–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Harries L.W., Bingham C., Bellanne-Chantelot C., Hattersley A.T., Ellard S.. The position of premature termination codons in the hepatocyte nuclear factor - 1 beta gene determines susceptibility to nonsense-mediated decay. Hum. Genet. 2005; 118:214–224. [DOI] [PubMed] [Google Scholar]
- 50. Neu-Yilik G., Amthor B., Gehring N.H., Bahri S., Paidassi H., Hentze M.W., Kulozik A.E.. Mechanism of escape from nonsense-mediated mRNA decay of human β-globin transcripts with nonsense mutations in the first exon. RNA. 2011; 17:843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lindeboom R.G.H., Supek F., Lehner B.. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat. Genet. 2016; 48:1112–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mendell J.T. Separable roles for rent1/hUpf1 in altered splicing and decay of nonsense transcripts. Science. 2002; 298:419–422. [DOI] [PubMed] [Google Scholar]
- 53. Sureau A., Gattoni R., Dooghe Y., Stévenin J., Soret J.. SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EMBO J. 2001; 20:1785–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mendell J.T., Sharifi N.A., Meyers J.L., Martinez-Murillo F., Dietz H.C.. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat. Genet. 2004; 36:1073–1078. [DOI] [PubMed] [Google Scholar]
- 55. Hinnebusch A.G. Translational regulation of GCN4 and the general amino acid control of yeast. Annu. Rev. Microbiol. 2005; 59:407–450. [DOI] [PubMed] [Google Scholar]
- 56. Vattem K.M., Wek R.C.. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:11269–11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B. et al.. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016; 536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Moey C., Topper S., Karn M., Johnson A.K., Das S., Vidaurre J., Shoubridge C.. Reinitiation of mRNA translation in a patient with X-linked infantile spasms with a protein-truncating variant in ARX. Eur. J. Hum. Genet. 2016; 24:681–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Howard M.T., Malik N., Anderson C.B., Voskuil J.L.A., Atkins J.F., Gibbons R.J.. Attenuation of an amino-terminal premature stop codon mutation in the ATRX gene by an alternative mode of translational initiation. J. Med. Genet. 2004; 41:951–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.