


Abstract
The universally conserved N6-threonylcarbamoyladenosine (t6A) modification of tRNA is essential for translational fidelity. In bacteria, t6A biosynthesis starts with the TsaC/TsaC2-catalyzed synthesis of the intermediate threonylcarbamoyl adenylate (TC–AMP), followed by transfer of the threonylcarbamoyl (TC) moiety to adenine-37 of tRNA by the TC-transfer complex comprised of TsaB, TsaD and TsaE subunits and possessing an ATPase activity required for multi-turnover of the t6A cycle. We report a 2.5-Å crystal structure of the T. maritima TC-transfer complex (TmTsaB2D2E2) bound to Mg2+-ATP in the ATPase site, and substrate analog carboxy-AMP in the TC-transfer site. Site directed mutagenesis results show that residues in the conserved Switch I and Switch II motifs of TsaE mediate the ATP hydrolysis-driven reactivation/reset step of the t6A cycle. Further, SAXS analysis of the TmTsaB2D2-tRNA complex in solution reveals bound tRNA lodged in the TsaE binding cavity, confirming our previous biochemical data. Based on the crystal structure and molecular docking of TC–AMP and adenine-37 in the TC-transfer site, we propose a model for the mechanism of TC transfer by this universal biosynthetic system.
INTRODUCTION
Position 37 in the anticodon stem–loops of many tRNAs decoding ANN codons (N is any nucleotide) is modified with N6-threonylcarbamoyladenosine (t6A), one of few tRNA modifications found in all domains of life (1–3). t6A is required for the fidelity of ribosomal translation by promoting correct selection of start codons and preventing aberrant frameshifting (1,2,4–7), and its deficiency in humans is linked to renal-neural disease (8,9).
t6A biosynthesis starts with the TsaC/C2-catalyzed synthesis of the pathway intermediate threonylcarbamoyl adenylate (TC–AMP) from L-threonine, bicarbonate and adenosine triphosphate (ATP) (10–12) (Figure 1). The threonylcarbamoyl (TC) moiety of TC–AMP is then transferred onto A37 of tRNA by the TC-transfer complex, composed of TsaB, TsaD and TsaE in bacteria (known as YeaZ, YgjD and YjeE in Escherichia coli and Salmonella tymphimurium; and YdiC, YdiE and YdiB in Bacillus subtilis) (12–14). In archaea and the cysotols of eukarya, TC-transfer is carried out by the KEOPS complex, composed of the TsaD ortholog Kae1, protein kinase Bud32 and small proteins Cgi121 and Pcc1 (and Gon7 in eukarya only) (15–19). In mitochondria, TC-transfer is performed by the TsaD ortholog Qri7 (20). The TsaD/Kae1/Qri7 protein family carries the active site for catalysis of the TC-transfer reaction. TsaB, TsaD and TsaE form an essential interaction network in vivo (21–23). Biochemical and structural studies showed that TsaB and TsaD are homodimeric proteins (14,24–28) that associate to form a TsaB/TsaD hetero complex with either a stoichiometry of 1:1 as in the cases of the E. coli and S. typhimurium systems (EcTsaBD and StTsaBD, respectively) (25,26), or a stoichiometry of 2:2 where TsaB acts as a dimerization module, as seen for the Thermotoga maritima system (TmTsaB2D2) (14).
Figure 1.

The bacterial t6A biosynthesis cycle, shown here for T. maritima. tRNA and ATP-bound TsaE bind to separate sites on the TmTsaB2D2 heterocomplex (1). TC–AMP, synthesized by TsaC2, is then delivered to the active site of the tRNA-bound TsaD subunit (2), and the threonylcarbamoyl (TC) moiety is transferred to A37 of tRNA, forming t6A (3). Following dissociation of the t6A-modified tRNA from the complex (4), ATP hydrolysis resets the system to the initial state and TsaE is released (5), thus allowing the next substrate tRNA to bind for the next cycle. The two halves of the complex are reset and utilized in alternating cycles. Halos indicate a pre-‘reset’ state for a given half.
In the presence of ATP or a nonhydrolyzable ATP analog, TsaE binds to the TsaB/TsaD complex to form a tripartite complex that was shown in E. coli and S. typhimurium to have a stoichiometry of 1:1:1 (EcTsaBDE and StTsaBDE) (25,26). Later, Small-Angle X-ray Scattering (SAXS) analysis, aided by a 5.0-Å crystal structure, revealed that the T. maritima TsaB/TsaD/TsaE complex is a symmetric complex with a stoichiometry of 2:2:2 (TmTsaB2D2E2) (14). In both the EcTsaBDE and TmTsaB2D2E2 complexes, TsaE binds to a concave surface formed by the interface between TsaB and TsaD (14,26). The TmTsaB2D2 complex (as well as EcTsaBD) was shown to constitute the minimal tRNA binding platform that carries out the TC transfer reaction (14). TmTsaB2D2 binds a single tRNA molecule and can then accommodate one TsaE subunit, and binding of a second TsaE subunit (forming TmTsaB2D2E2) blocks tRNA binding (14).
TsaE is an atypical ATPase that hydrolyzes ATP to ADP and belongs to the P-loop NTPase structural superfamily characterized by the P-loop NTPase hydrolase fold, and harbors the Walker A motif (GX4GKT/S, also known as the P-loop) and Walker B motif (h4D/E, where h is a hydrophobic residue), required for nucleotide binding and hydrolysis, respectively (29). It also contains the Switch I and Switch II loops known in P-loop NTPases to act in transmitting the signal of ATP hydrolysis to remote sites in the enzyme (30,31). The ATPase activity of TsaE is activated in the presence of TsaB and TsaD (13,14), and this activity is required for cell viability (32). Early experiments on the E. coli system suggested that ATP-mediated formation of the EcTsaBDE complex is required for t6A biosynthesis on tRNA (26). Later, we showed in T. maritima that the TmTsaB2D2 complex alone can catalyze TC transfer in the absence of TsaE, however ATP hydrolysis in the presence of TsaE – not TsaE binding per se - is required for multiple turnover of the t6A synthesis reaction (14). Furthermore, we also showed that product tRNA is released before the binding of TsaE and before ATP hydrolysis occurs. This led us to propose a model of the t6A catalytic cycle in which TC transfer is catalyzed by the active site of the TsaD subunit (the TC-transfer site) of tRNA-bound TmTsaB2D2, rendering it inactive. ATP hydrolysis in the ATPase site then ‘resets’ the system to the active state for the next cycle (Figure 1).
Recently, the crystal structure of the TmTsaB2D2E2 complex was determined at 3.14 Å by Missoury et al. in the presence of the nonhydrolyzable ATP analog AMPCPP and MgCl2 (33). The structure revealed the TsaE subunit positioned at the entrance of the TC-transfer site in TsaD, and blocking access of tRNA to the site, consistent with previous biochemical and SAXS data (14,26). The structure also revealed the interactions of the enzyme with AMPCPP and Mg2+ bound at a novel, interfacial ATPase site between the TsaE and TsaD subunits. In this structure, the TC-transfer site was found empty and disordered and the S61PTF64 motif of TmTsaE dipped into the site, which led to the proposal that binding of TmTsaE partially melts the active site of TmTsaD, causing loss of its essential metal binding site, hence its inactivation. Further, because the S61PTF64 motif of TmTsaE is involved in a hydrogen bond-network with the γ-phosphate of bound AMPPCP, it was suggested that this motif may act as a communicator between the ATPase site and the TC-transfer site.
Here, we report a 2.5 Å crystal structure of the TmTsaB2D2E2 complex bound to ATP in the ATPase site and to carboxy-AMP (as a TC–AMP mimic) in a fully ordered TC-transfer site and an intact and Zn2+-occupied metal binding site, together with extensive structure-guided mutagenesis of these sites and of residues in the conserved Switch I and Switch II loops of the TsaE subunit. From this data we identify a portion of the communication path through which ATP hydrolysis in the ATPase site is communicated to the TmTsaB2D2 platform, and confirm the predictions by Missoury et al. of a role of the Switch loops in such communication (33). Finally, we elucidate the mode of tRNA binding to TmTsaB2D2 based on SAXS data of the ribonucleoprotein complex in solution. The structural and mutagenesis results confirm our previous predictions that tRNA binds at the interface between the TsaB and TsaD subunits and occupies the same site as TsaE. The results also shed light on the mechanism of the reset step of the catalytic cycle, and the mechanism of the TsaD-catalyzed TC transfer reaction.
MATERIALS AND METHODS
Cloning and mutagenesis
Cloning of T. maritima tsa genes (tsaB, tsaC, tsaD and tsaE) has been described previously (14). Point mutagenesis was carried out on pET28a-TsaE and pET45b-TsaD using the Q5® Site-Directed Mutagenesis Kit (NEW ENGLAND Biolabs Inc.) and the primers listed in Supplementary Table S1. DNAs encoding the deletion mutant of TmTsaB (TmTsaBΔ189-206) were amplified from the pET28A plasmid harboring TmTsaB using primers listed in Supplementary Table S1 and cloned into the NheI and XhoI restriction sites of pET28a in frame with the N-terminal polyhistidine tag (Novagen, San Diego, CA, USA). All constructs were verified by sequencing.
Protein and tRNA preparation
All recombinant wild-type and mutant proteins were overexpressed in E. coli and purified by Ni-NTA and gel filtration chromatography as described previously (14). Purity was verified by SDS-PAGE to be >95%. The TmTsaB2D2E2 complex was prepared for crystallization as described previously (14). Briefly, 10 μM each T. maritima TsaB (TmTsaB) and T. maritima TsaD (TmTsaD) were mixed with excess T. maritima TsaE (TmTsaE) in buffer containing 50 mM Tris–HCl (pH 7.5), 50 mM NaCl, 1 mM ATP and 1 mM MgCl2, followed by purification of the complex by size exclusion chromatography. The final TmTsaB2D2E2 complex was concentrated to 4 mg/ml using an Amicon filtration device, flash cooled in liquid nitrogen and stored at −80°C until use. Escherichia coli tRNAThrCGU and tRNALysUUU transcripts used for binding studies and radiochemical incorporation assays were produced by in vitro transcription and purified as described previously (14).
Crystallization and X-ray data collection
High throughput screening for crystallization conditions of TmTsaB2D2E2 was carried out using the vapor diffusion method. Sitting drops were set up in 96-well microplates using a Mosquito crystallization robot (TTP Labteck, Melbourn, UK), incubated at 20°C and monitored over several weeks using a Rock Imager crystal imaging system (Formulatrix, Bedford, MA, USA). Initial crystals appeared in several conditions from the JCSG-plus crystallization screening kit (Qiagen Inc., Valencia, CA, USA) at a protein concentration of 3 mg/ml. Conditions were further refined manually and diffraction quality crystals were grown at 20°C in vapor-diffusion sitting drops by combining 2 μl sample solution containing 3.1 mg/ml protein, 50 mM Tris (pH 7.5), 50 mM NaCl, 1 mM ATP and 0.1 mM MgCl2 with 2 μl reservoir solution containing 8% polyethylene glycol (PEG) 400, 100 mM KCl, 50 mM MES (pH 6.0) and 0.8 mM MgCl2. Crystals with size 0.1–0.2 mm appeared in four days and were cryoprotected by direct addition of 25% PEG 400 to the crystal mother liquor and flash cooling in liquid nitrogen. X-ray diffraction data were collected on beamline 14-1 at the Stanford Synchrotron Radiation Lightsource (Menlo Park, CA, USA). All data were processed and scaled using the HKL3000 program suite (34). Data collection statistics are summarized in Table 1.
Table 1.
X-ray data collection parameters and structure refinement statistics
| Parameter | Value |
|---|---|
| Data collection | |
| Beamline | SSRL BL14-1 |
| Space group | P 31 2 1 |
| Unit cell (Å) | 124.05, 124.05, 119.04 |
| Resolution (Å) | 42.95–2.50 (2.54–2.50) |
| Wavelength (Å) | 1.19499 |
| Unique Reflections | 37 514 (1824) |
| Completeness (%)a | 99.9 (98.2) |
| Redundancy | 10.5 (6.3) |
| Rmeas | 0.127 (3.262) |
| Rpim | 0.038 (1.275) |
| CC1/2 | 0.992 (0.170) |
| Average I/σ(I) | 17.1 (0.53) |
| Refinement statistics | |
| Resolution range (Å) | 42.94–2.50 (2.56–2.50) |
| Rcrsyt | 0.129 (0.400) |
| Rfreeb | 0.207 (0.404) |
| R.m.s. deviation from ideality | |
| Bond length (Å) | 0.003 |
| Bond angle (°) | 0.802 |
| Average B factor (Å2) | 78.27 |
| Ramachandran plot: | |
| Preferred (%) | 94.0 |
| Allowed (%) | 4.17 |
| Outlier (%) | 1.9 |
aValues in parentheses are for the highest-resolution shell.
b R free is monitored with 5% of the reflections excluded from the refinement.
Crystal structure determination and refinement
The crystal structure of TmTsaB2D2E2 in complex with Mg2+-ATP and carbxy-AMP was determined by molecular replacement using the program Phase-MR (35) in the Phenix program suite (35,36). Rotation and translation searches were conducted using a single subunit of TmTsaB from the crystal structure of the TmTsaB homodimer (PDB ID: 2A6A, (24)), and homology models of TmTsaD and TmTsaE (generated using the Phyre2 web portal (37)) as search models. The rotation and translation searches using the TmTsaB search model alone yielded a solution with Log-Likelihood Gain (LLG) = 130, representing one TmTsaB subunit in the asymmetric unit. Subsequent rotation and translation searches with the TmTsaD and TmTsaE search models resulted in a unique solution with LLG = 342, placing one copy of each subunit in the asymmetric unit. At that point, the assembled model had a crystallographic R-factor of 0.374 (R-free = 0.46) and accounted for 98% of the expected residues in the asymmetric unit. Iterative cycles of model building in COOT (38) and refinement using REFMAC5 (39) yielded the final structure with the statistics summarized in Table 1. All figures were prepared using the PyMOL Molecular Graphics System (Version 2.0, Schrödinger, LLC).
ATPase activity of the TC-transfer complex
The initial velocity conditions for the ATPase reaction catalyzed by the wild-type T. maritima TC-transfer complex were determined by conducting time-course ATPase assays in which ATP hydrolysis reactions were carried out at 37°C in 40 μl volumes using 2.5 μM each pre-isolated wild-type TmTsaB2D2 and wild-type TmTsaE, 50 μM [γ-32P]ATP (22 × 109 DPM/μmol, PerkinElmer), 25 mM Tris–HCl (pH 7.5), 50 mM KCl and 2 mM MgCl2. Two microliters of the reaction mix were taken at increasing time points (1–60 min) and spotted on a PEI-cellulose thin layer chromatography (TLC) plate. A reaction mixture lacking enzyme was likewise monitored as a control. The radioactive nucleotide and product [γ-32P]Pi were separated on the plate using 0.5 M KH2PO4 (pH 3.5) as solvent, visualized by phosphorimaging and quantified by densitometry. The percentage of ATP hydrolyzed was calculated as quantity of [γ-32P]Pi/quantities of ([γ-32P]Pi + unhydrolyzed [γ-32P]ATP) × 100. Time-course measurements from four independent reactions were averaged. These experiments demonstrated that for the wild-type system <10% of the ATP was consumed within the first 3 min of the reaction, and that 3 min single time-point assays provided accurate estimates of initial velocities when screening mutants exhibiting robust activity.
The ATPase activities of TC-transfer complexes containing TmTsaE mutants TsaEE11A, TsaET43A, TsaEF64A, TsaER83A and TsaER134A were assessed under initial velocity conditions as follows. ATP hydrolysis was carried out at 37°C for 3 min in 40 μl reactions containing 2.5 μM each pre-isolated wild-type TmTsaB2D2 and mutant TmTsaE, 50 μM [γ-32P]ATP (22 × 109 DPM/μmol, PerkinElmer), 25 mM Tris–HCl (pH 7.5), 50 mM KCl and 2 mM MgCl2. Similarly, for the TmTsaD mutants TsaDS134A, TsaDD165A and TsaDD182A, the reactions contained 2.5 μM each wild-type TmTsaE and TmTsaB2D2 made with mutant TsaD and were carried out at the same initial velocity conditions as described above. For the mutants TsaES61A, TsaET63A, TsaED80A, TsaEY82A and TsaEW109A, which exhibited severely compromised activity, the reactions contained 5 μM each wild-type TmTsaB2D2 complex and mutant TsaE and were carried out for 60 minutes. Similarly, for the mutants TsaDK166A and TsaDK213A, the reactions contained 5 μM each wild-type TmTsaE and TmTsaB2D2 made with mutant TsaD and were carried out for 60 minutes. Control reactions contained either wild-type TmTsaE alone, wild-type TmTaB2D2 alone, wild-type TmTsaB2D2E2 or no enzyme. The reactions were then analyzed by TLC and quantified by densitometry as described above for the time-course ATPase assay. For all reactions, the initial velocity was calculated using the formula:
Initial velocity (μM / min) = fraction of ATP hydrolyzed × total ATP concentration (μM)/reaction time (min),
where fraction of ATP hydrolyzed = quantity of [γ-32P]Pi/quantities of ([γ-32P]Pi + unhydrolyzed [γ-32P]ATP).
t6A biosynthesis assay
Time-course enzyme assays to measure the activity of TmTsaD and TmTsaE mutants in t6A synthesis were executed in 275 μl-reactions containing 100 mM Tris–HCl (pH 8.0), 300 mM KCl, 5.0 mM DTT, 2 mM ATP, 20 mM MgCl2, 50 mM NaHCO3, 250 μM l-[U-14C]-threonine (36 μCi/μmol), 20 μM E. coli tRNAThrCGU or E. coli tRNALysUUU, 2 μM each TmTsaB, TmTsaC2 and TmTsaDWT/Mut and 1 μM TmTsaEWT/Mut. Assays were incubated at 37°C and 50 μl aliquots were taken at 15, 30, 60, 90, 120, 180 min. RNA was precipitated by the addition of 500 μl 10% TCA and cooling on ice for 15 min. RNA was then collected by filtration through Whatman GF/C filters, and the filters were subsequently washed with 100% cold ethanol. The dried filters were combined with Econo-Safe scintillation cocktail (RPI™) and counted in an LS 6500 Multi-purpose scintillation counter (Beckman Coulter, Inc.). The initial velocity for each reaction was estimated from the initial slope (first derivative) of the concentration of modified tRNA product as a function of time. Note that because the goal of the experiments was to discriminate between single- and multiple-turnover regimes of the enzyme complex, full steady-state kinetic analysis was unnecessary and the assays with mutants of TsaD and TsaE were designed as time-courses at a single concentration of substrates.
Assessment of formation of TmTsaB2D2E2 and TmTsaB2D2 complexes with mutant proteins using analytical gel filtration
Analytical gel filtration experiments were performed on a calibrated Enrich SEC 650 10 × 300 mm column (Bio-Rad, Hercules, CA, USA) using a Bio-Rad NGC chromatography system, as described previously (14). Briefly, 500 μl of sample containing 10 μM each TmTsaBWT/Mut, TmTsaDWT/Mut and TmTsaEWT/Mut in buffer containing 50 mM Tris–HCl pH 7.5, 50 mM KCl, 1 mM DTT, 1 mM ATP and 1 mM MgCl2 were loaded on the column and run using buffer containing 50 mM Tris (pH 7.5) and 50 mM KCl, at a flow rate of 0.3 ml/min, with detection at 280 nm. To assess formation of TmTsaB2D2 complexes, the same process was followed but TmTsaE, MgCl2 and ATP were excluded from the mixture.
Assessment of tRNA binding by electrophoretic mobility shift assay (EMSA)
To assess the tRNA binding properties of TmTsaB2D2 complexes containing mutant TmTsaD or mutant TmTsaB, EMSA assays were performed as described previously (14). The binding reactions were carried out in 10 μl solutions containing 0–10 μM pre-isolated TmTsaB2D2WT/Mut, 100 nM 32P-labeled E. coli tRNAThrCGU transcript, 50 mM Tris–HCl (pH 7.5), 50 mM NaCl, 5 mM MgCl2, 5% glycerol, 1 mM DTT and 0.1 unit of RNasin®.
Small angle X-ray scattering (SAXS) data acquisition of TmTsaB2D2-tRNA complex
SAXS data were collected on the Bio-SAXS beamline BL4-2 at the Stanford Synchrotron Research Laboratory using a Rayonix MX225-HE CCD detector (Rayonix, Evanston, IL, USA) with a sample-to-detector distance of 1.7 m and a beam energy of 11 keV (λ = 1.127 Å). The momentum transfer (scattering vector) q was defined as q = 4π sin(θ)/λ, where 2θ is the scattering angle. All data were collected to a maximum q of 0.5 Å−1. Before data collection, 5 μM of pre-pre-isolated TmTsaB2D2 were mixed with 5 μM of E. coli tRNAThrCGU transcript in 40 μl solution containing 50 mM Tris–HCl (pH 7.5), 50 mM NaCl, 5 mM MgCl2, 5% glycerol, 1 mM DTT and 0.1 unit RNasin®. Scattering data were collected at a wavelength of 1.3 Å for ten consecutive 2-second exposures from the buffer alone and subtracted from the total protein-tRNA complex solution scattering. Data were processed using PRIMUS (40). Radii of gyration (Rg), extrapolated from the Guinier region of the Guinier plot, were computed using PRIMUS. P(r) functions were calculated using the program Datgnom (41). Theoretical scattering curves were computed from different structural models and compared to experimental scattering curves using the program FoXS (42). The molecular mass was determined using DATMOW (41) and SAXS-MoW (HTTP://www.if.sc.usp.br/∼saxs/). Ab initio shape determination was performed using the programs Dammif followed by DAMAVER (41). The structural parameters derived from the SAXS data are summarized in Table 2.
Table 2.
Parameters calculated from SAXS data of the TmTsaB2D2-EctRNAThr complex
| Structural Parameters | |
| R g (Å) from Guinier plot | 53.50 |
| R g (Å) from P(r) | 53.34 |
| Dmax (Å) | 185 |
| Molecular Mass (kDa) | |
| From Datmow | 148 |
| From DAMMIN models | 142 |
| Theoretical protein sequence | 153 |
| Modeling parameters | |
| DAMAVER (10 models) NSD | 0.84 ± 0.072 |
To generate the structural model for the protein-tRNA complex, the TmTsaB2D2 complex was first extracted from our crystal structure and the polypeptide segment Ser31–His50 of one TsaD subunit was subjected to homology modeling based on its orientation in the crystal structure of E. coli TsaBD (PDB ID 4WOS, (26)), resulting in a TmTsaB2D2 model representing the putative TsaE-free state on one half of the tRNA binding platform. A molecule of E. coli tRNAThrCGU (extracted from the crystal structure of E. coli threonyl-tRNA synthetase in complex with tRNA, PDB ID 1QF6 (43), and in which all modifications were removed) was then docked using the HADDOCK server (version 2.2) (44). In the docking protocol, TmTsaD residues Ser134, His137, Lys166 and Lys213, and A37 of tRNA were designated as active residues to apply distance restraints.
Docking of TC–AMP
Docking of TC–AMP on the crystal structure of TmTsaB2D2E2 was carried out in Maestro (Schrödinger, LLC, New York, NY, USA). The threonyl moiety was built into the structure of bound carboxy-AMP using the 3D builder and the initial model of the complex was prepared with the protein preparation wizard in Maestro. Following optimization of the protonation states of protein side chains in PROPKA, structure minimization was performed using OPLS2005 and a r.m.s.d cutoff of 0.3 Å. The resulting model was used to dock adenine-37 manually onto the Phe64 position, followed by structure minimization.
RESULTS
Overall structure
T. maritima TsaB2D2E2 (TmTsaB2D2E2) was isolated by size-exclusion chromatography in the presence of ATP and MgCl2, and its crystal structure was determined by molecular replacement at a resolution of 2.5 Å (Table 1, Figure 2A). The crystal asymmetric unit consists of one half of the complex (one subunit each TsaB, TsaD and TsaE), and the full complex is generated by application of the crystallographic 2-fold symmetry to the asymmetric unit. The structure contains an ATP molecule bound in the ATPase site at the interface between the TsaD and TsaE subunits (Figure 2B), and a carboxy-AMP molecule bound in the TC-transfer site of the TsaD subunit (Figure 2C), with an 18-Å distance between the two sites. Also, an ADP molecule is seen bound in a remote site on the outer surface of the TsaD subunit, but this site appears to be unrelated to any catalytic activity and may represent a binding pocket of an adenine nucleotide on the tRNA substrate. The overall structure of the protein complex is similar to the recently reported structure bound to the nonhydrolyzable ATP analog AMPCPP in the ATPase site (PDB ID 6FPE, r.m.s.d. 1.58 Å over 482 atoms) (33), but with an important difference. Unlike the previous structure in which the active site in the TsaD subunit (the TC-transfer site) is disordered and lacks bound metal, the present structure reveals a perfectly ordered TC-transfer site containing a fully coordinated Zn2+ and occupied by a carboxy-AMP molecule. In both TmTsaB2D2E2 structures, the observation that a nonhydrolyzed nucleotide is bound in the ATPase site identifies these structures as pre-‘reset’ structures in the proposed course of the t6A cycle, i.e. representing the inactivated half of the complex in proposed states 1, 2, 3 and 4 in Figure 1.
Figure 2.

Overall structure of TmTsaB2D2E2 in complex with ATP and carboxy-AMP. (A) Ribbon diagram of the complex showing locations of the ATPase site and TC-transfer site. Subunits TsaB, TsaD and TsaE are colored in two shades of blue, magenta and cyan, respectively. Bound Zn2+ and Mg2+ ions are shown as gray and red spheres, respectively, and bound nucleotides as stick model and labeled. (B) Stereoview of the omit Fo-Fc electron density map (2.5 Å, contour level 4.5σ; nucleotide, ion, and water molecules were omitted from the refinement and phase calculation) of ATP and Mg2+ bound in the interfacial ATPase site between TsaD and TsaE. (C) Stereoview of the omit Fo– Fc electron density map (2.5 Å, contour level 3.2σ; nucleotide, water, metal and surrounding residues were omitted from the refinement and phase calculation) in the TC-transfer site showing bound carboxy-AMP and Zn2+ and an ordered active site. Metal coordination spheres and the intraloop hydrogen bond between Ser134 and His137 are indicated with dashed lines.
TsaE-induced conformational changes in the tRNA binding platform TsaB2D2
As was previously shown for the structure of the complex with AMPPCP (33), comparison with the crystal structure of EcTsaBD (PDB ID 4YDU (26), r.m.s.d. = 2.0 Å over 408 Cα atoms) highlights the TsaE-induced conformational changes in the tRNA binding platform TmTsaB2D2 (Figure 3). The main changes are the concerted movements of the C-terminal tail (Leu191-Glu206) of the TsaB subunit and the helical hairpin α1-α2 (Ser31-His50) in the TsaD subunit, allowing the Switch I loop of TsaE to lodge near the TC-transfer site. In the absence of TsaE, as seen in the EcTsaBD structure (26), these two parts of the complex are pinned together. The movement of the C-terminal tail of TsaB, and its refolding from its extended conformation seen in EcTsaBD to a 3-turn helix was seen in the structure in complex with AMPPCP (33). However, due to disorder in that structure, the helical hairpin α1–α2 in TsaD was not resolved. In the present structure, the N-terminal 2.5 turns of the 6-turn helix α2 of TsaD unfolds, and the resulting loop and preceding helix α1 (altogether we name ‘the lever arm’) rotate by 67° away from the TsaB subunit (Figure 3). The hinge residues around which the lever arm rotates are Ser31 and Arg49, both invariant in TsaD proteins (Supplementary Figure S1), and the two residues are stabilized by specific interactions with TsaE (vida infra) that are only resolved in this structure.
Figure 3.
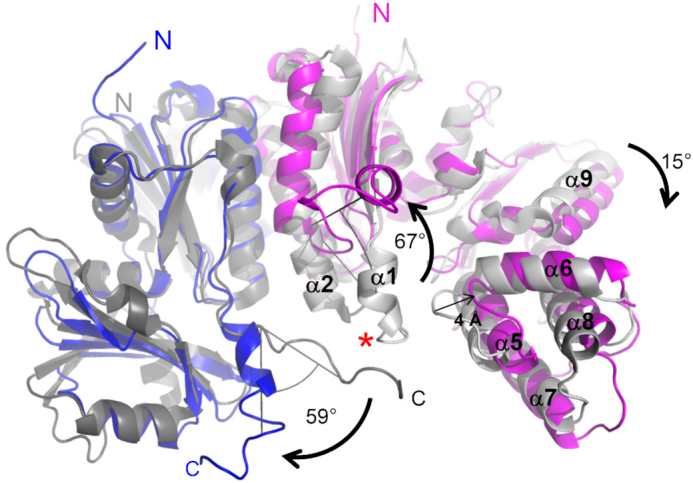
TsaE-induced conformational changes in the TsaB2D2 platform. Superposition of the crystal structures of TmTsaB2D2E2 (colors) and EcTsaBD (gray) showing TsaE-induced separation of the TsaB C-terminal tail and ‘lever arm’ of TsaD. The TsaB and TsaD subunits of TmTsaB2D2E2 are in blue and magenta, respectively. The corresponding subunits of EcTsaBD are in dark grey and light grey, respectively. For clarity, only half the TmTsaB2D2E2 complex is shown, and the TsaE subunit is not shown. The red asterisk marks the position of the center of mass of TmTsaE in TmTsaB2D2E2. The angles of rotation of the C-terminal tail of TsaB and the lever arm of TsaD (α1–α2), and the shift in the 5-helical bundle of the C-terminal domain of TsaD (α5–α9) are indicated.
Although these conformational changes result in 21% reduction in the protein surface area buried in the TsaB/TsaD intersubunit interface upon TsaE binding (2170 Å2 in TmTsaB2D2E2 versus 2758 Å2 in EcTsaBD), the additional surface area buried by TsaE increases the total area buried within the complex by 2-fold (5942 Å2 in TmTsaB2D2E2 versus 2758 Å2 in EcTsaBD). This explains the stabilizing effect, previously observed by SAXS, of TsaE on the structure of the tRNA binding platform (14). Lastly, as in the structure in complex with AMPPCP (33), TsaE pushes apart the two domains of TsaD through a 15° rotation in the C-terminal domain, resulting in expansion of the cavity available for TsaE binding by another 4 Å, relative to its size in EcTsaBD. Collectively, the conformational changes seen in the tRNA binding platform are consistent with an induced-fit mechanism for binding of the TsaE subunit.
Mutagenesis rationale and functional characterization of mutants
The high resolution of the present structure allows accurate visualization of the enzyme–nucleotide interactions, water structure, and metal coordination spheres in both the ATPase and TC-transfer sites. To test functional and mechanistic hypotheses offered by the structure, single mutations were introduced in TmTsaE and TmTsaD at residues that directly or indirectly interact with the ligands in both active sites, as well as invariant or highly conserved residues located between but outside these sites. The mutants were functionally characterized using several assays as follows. Each mutant was tested for its ability to make a stable TmTsaB2D2E2 complex using gel filtration chromatography, and the TmTsaD mutants were additionally tested for ability to form stable TmTsaB2D2 complexes and to support tRNA binding using EMSA. Each mutant was also tested for ability to support ATP hydrolysis in the presence of all other wild-type proteins (except TmTsaC2) using a quantitative 32P TLC assay conducted under initial velocity conditions. Initial velocity conditions were first determined by monitoring the reaction progress of ATP hydrolysis by an equimolar mixture of wild-type TmTsaB2D2 and TmTsaE. In the presence of 2.5 μM enzyme, <10% hydrolysis is achieved in 3 minutes, and complete hydrolysis is reached after 90 min (Supplementary Figure S2). Therefore, the ATPase activities of highly active TmTsaE and TmTsaD mutants were evaluated using 3-minute reactions at an enzyme concentration of 2.5 μM, whereas inactive or weakly active mutants were assessed in 60-min reactions at an enzyme concentration of 5.0 μM.
Finally, each mutant was tested for ability to support multiple-turnover t6A synthesis activity on tRNA in the presence of all other wild-type Tsa proteins using a quantitative time-course 14C radioactivity incorporation assay (14). In this assay, single-turnover activity is detected when the production of t6A-modified tRNA plateaus at the concentration of TsaB2D2 in the reaction.
Interactions and mutagenesis of the ATPase site
The Mg2+ ion in the ATPase site is coordinated in an octahedral geometry in which the equatorial ligands are the TsaE Walker B motif residues Thr42 and Glu108, the ATP γ-phosphate O3 atom, and a water molecule, while the axial ligands are the ATP β-phosphate O2 atom and a second water molecule that in turn is coordinated by Ser61 and Asp80 from the Switch I and Switch II motifs, respectively (Figure 2B and Figure 4A). Mutating any of these invariant side chains (Thr42 and Glu108 in previously published work (14), and Ser61 and Asp80 in this study) to alanine results in mutants that support only a single turnover of the t6A cycle (Figure 4C) with initial velocities similar to that of the wild-type TmTsaB2D2 (Supplementary Table S2). The loss of multiple-turnover t6A synthesis activity appears to be due to loss of ATP hydrolysis as reflected by their extremely low rates in the ATPase assay (even lower than that of wild-type TmTsaE alone, Figure 4B, Supplementary Figure S3A), even though the mutant proteins still make stable complexes with TmTsaB2D2 (Supplementary Figure S4A).
Figure 4.

Interactions and mutagenesis of the ATPase site. (A) Schematic of the interactions with Mg2+-ATP bound in the ATPase site at the interface between the TsaB and TsaD subunits, as seen in the crystal structure of TmTsaB2D2E2. The nucleotide, Mg2+ and water molecules are shown in ball-and-stick representation and colored. Protein residues are labeled, their percent conservation in 250 inspected sequences, and the subunits to which they belong are indicated in parentheses. Where relevant for TsaE residues, the conserved TsaE motif is indicated in brackets. Residues required for multiple-turnover t6A synthesis are indicated with red asterisks. Hydrogen bonds are shown as dashed line. The figure was made in LIG-PLOT (45). (B) Effect of mutations in the ATPase site on the ATP hydrolysis activity of the TC-transfer complex. Shown are the initial velocities of ATP hydrolysis reactions catalyzed by equimolar mixtures of pre-isolated TmTsaB2D2 and TmTsaE harboring ATPase-site mutations either in the TsaE or the TsaD subunit. Data represent averages of duplicate measurements from three independent reactions. (C) Effect of mutations in the ATPase site on the turnover of the t6A biosynthesis reaction, assessed using the time-course radiochemical t6A assay measuring the incorporation of [14C]-L-threonine into tRNA. Assays contained either mutant TmTsaE or mutant TmTsaD harboring mutations in the ATPase site, and the other three wild-type Tsa proteins. Multiple-turnover activity in the presence of all wild-type proteins (solid black line) and single-turnover activity in the absence of TmTsaE (dashed black line) are also shown. Data represent duplicate measurements from at least three independent reactions.
The adenine and ribose rings of ATP interact solely with TsaE, with the adenine stacked between the TsaE side chains Glu11 and Arg134 (Figure 4A). Glu11 in turn makes a hydrogen bond with the ribose 2′-OH, while Arg134 is positioned perhaps for a weak interaction with the ribose O4′. Arg134 also makes a hydrogen bond with Tyr218 of TsaD, sealing the adenine binding pocket. None of these interactions with the adenine and ribose is required for ATP hydrolysis, as indicated by the robust ATPase activities of the TsaEGlu11Ala and TsaEArg134Ala mutants in the presence of wild-type TmTsaB2D2 (Figure 4B and Supplementary Figure S3B). However, the interaction with Arg134, an invariant residue in TsaE, is required for efficient t6A synthesis, as the TsaEArg134Ala mutant only supports multiple turnovers of t6A synthesis at a very slow rate (Figure 4C and Supplementary Table S2). This effect could be explained by the elimination of the Arg134(TsaE)–Tyr218(TsaD) hydrogen bond, resulting in weaker affinity of the mutant to TmTsaB2D2 (partial destabilization of the protein complex as detected by size exclusion chromatography, Supplementary Figure S4A). Further, the fact that the TsaEGlu11Ala mutant supports almost wild-type level ATPase and t6A synthesis activities (Figures 4B and 4C, respectively) suggests that deoxy-ATP (dATP) may too act as a substrate in the ATP hydrolysis step of the t6A synthesis cycle. This hypothesis was tested using the ATPase assay and [γ-32P]-dATP, and the result shows robust hydrolysis of dATP by the TC-transfer complex (Supplementary Figure S5).
The triphosphate of ATP is coordinated by the Mg2+ ion, residues from the conserved P-loop (Walker A motif Gly35-Thr43) of TsaE, and by Lys166 and Lys213 from TsaD (Figure 4A). The role of some of these side chains was tested by site-directed mutagenesis. Unlike Thr43 of TsaE, which also contributes a backbone interaction to the β-phosphate and is only partially conserved as Thr/Ser, alanine substitutions of the invariant residues Lys166 and Lys213 of TsaD dramatically affect the ATP-dependent activities of the complex. Specifically, these substitutions abolish multi-turnover t6A synthesis activity (Figure 4C, Supplementary Table S2) due to loss of ATP hydrolysis (Figure 4B and Supplementary Figure S6A). They also exhibit reduced tRNA binding by the mutant TmTsaB2D2 complexes (vida infra Figure 7A and Supplementary Figure S7), consistent with the reduced rate of t6A synthesis (Supplementary Table S2). Notably, the TsaDLys166Ala also exhibits reduced ability to form a stable TC-transfer complex (Supplementary Figure S4C), consistent with the known requirement of ATP binding for the formation of a high-affinity complex (14,26).
Figure 7.

tRNA binding determinants on TmTsaB2D2 and SAXS model of the tRNA-bound complex. (A) tRNAThrCGU binding by pre-isolated wild-type TmTsaB2D2 (open circles, Kd = 1.46 ± 0.08 μM) and TmTsaB2D2K166A (triangles), TmTsaB2D2K213A (squares), and TmTsaB2Δ189-206D2 (inverted triangles) as measured by EMSA (see autoradiograms in Supplementary Figure S7C), n = 2. (B) Comparison of the experimental solution SAXS curve (black circles) and theoretical scattering curves calculated from various models (colored lines), and associated chi values. (C) Docking model of the TmTsaB2D2–tRNA complex superposed on the ab initio reconstruction of the molecular envelope (gray surface) calculated using Dammif. The C-terminal helix and tail region of the TsaB subunit is boxed.
Further, the invariant Walker B motif residue Trp109 of TsaE makes a water-mediated hydrogen bond with the γ-phosphate of ATP. This water molecule (W5 in Figure 4A) is well positioned to act as the nucleophile for attack on Pγ in the ATPase reaction. Consistent with this, alanine substitution of Trp109 abolishes the ATPase activity (Figure 4B and Supplementary Figure S3A) and multi-turnover t6A synthesis activity of the complex (Figure 4C and Supplementary Table S2).
Interactions and mutagenesis of the TC-transfer site
Clear electron density is seen for a bound carboxy-AMP molecule and a Zn2+ ion in the TC-transfer site in the TsaD subunit (Figure 2C). The identity of the bound metal was confirmed by X-ray fluorescence (Supplementary Figure S8), and it occupies the same site as Fe in EcTsaD (26) and Zn2+ in StTsaD (25). It is not clear what the origin of the bound carboxy-AMP is; it may form in the crystal by TsaD from ATP or ADP (generated by the ATPase activity of the crystallized complex) and bicarbonate (amply present in the crystallization solutions). Also, while residues of the Mg2+ site seen in EcTsaD and StTsaD and shown to be required for t6A biosynthesis activity (25,26) are conserved in TmTsaD (Asp11 and Glu12), there is no electron density for any bound Mg2+ ion in the TC-transfer site in this structure, even though magnesium was present in the crystallization buffer.
Zn2+ is octahedrally coordinated by TsaD residues His109, His113 and two water molecules as equatorial ligands, and His137 and Asp296 as axial ligands (Figure 2C and Figure 5A). Mutating any of these residues to alanine yielded insoluble and misfolded proteins (data not shown), suggesting a structural role for the metal. Unlike the case in nucleotide-bound EcTsaBD (26) and StTsaBD (25) where the metal is coordinated by the nucleotide α- and β-phosphates, carboxy-AMP in this structure does not interact with the metal. The bound carboxy-AMP adopts a similar anti-conformation as that seen for ADP bound in the TC-transfer site of EcTsaBD (26) and for AMP/ADP/ATPγS bound to StTsaBD (25), and makes similar interactions via its purine and ribose groups, but different interactions via the carboxy-phosphate moiety. Specifically, the adenine moiety is lodged in a hydrophobic pocket in the C-terminal domain of TsaD, between helix α6 and the glycine-rich loop-310-helix η2 harboring the conserved sequence motif V262GGVXXN268 (X is a small or hydrophobic residue, Supplementary Figure S1, and Figure 5A). Helices α6 and η2 also contribute Asp182 and Asn268, respectively, as hydrogen bonding ligands to the adenine, as well as Gly264 as a backbone ligand to the α-phosphate (Figure 5A). The ribose moiety of carboxy-AMP is held by hydrogen bonding of its 2′- and 3′-OH groups with Asp165. The ligand carboxy moiety, which mimics the carbamoyl moiety of the TC–AMP substrate, is anchored via backbone hydrogen bonds to the β-turn between strands β6 and β7 (Figure 2C and Figure 5A). This β-turn carries the conserved glycine-rich sequence V133SGGH137 (Supplementary Figure S1), and is stabilized by an internal hydrogen bond between the side chain OH of Ser134 and backbone NH of the Zn2+-ligand His137 (Figure 2C). Alanine substitutions of these residues, all of which are conserved in the TsaD protein family, yielded mutants that form stable TmTsaB2D2 complexes (Supplementary Figure S4B) that bind tRNA with apparent affinity of the wild-type complex (Supplementary Figure S7A and B), and support binding of TmTsaE (Supplementary Figure S4C). The mutant TsaDSer134Ala supports the ATPase activity of the complex (Figure 5B and Supplementary Figure S6B) but exhibits extremely low activity for t6A synthesis (Figure 5C and Supplementary Table S2), consistent with a role of the glycine-rich beta-turn (specifically Gly135 and Gly136) in TC transfer to tRNA. Similarly, the TsaDAsp165Ala mutant supports the ATPase activity and exhibits low t6A synthesis activity, most likely due to weak TC–AMP binding in the TC-transfer site. Finally, the TsaDAsp182Ala mutant exhibits slow multi-turnover t6A synthesis activity, consistent with a modest role of Asp182 in TC–AMP recognition.
Figure 5.

Interactions and mutagenesis of the TC-transfer site. (A) Schematic of the interactions with carboxy-AMP bound in the TC-transfer site. The ligand, Zn2+ and water molecules are shown in ball-and-stick representation and colored. Protein residues are labeled, hydrogen bonds are shown as dashed line, and van der Waals interactions as half circles. The figure was made in LIG-PLOT (45). (B) Effect of mutations of the TC-transfer site on the ATP hydrolysis activity of the TC-transfer complex. Shown are initial velocities of ATP hydrolysis reactions catalyzed by a mixture of wild-type TmTsaE and TmTsaB2D2 containing either wild-type or mutant TmTsaD harboring mutations in the TC-transfer site. Data represent averages of duplicate measurements from three independent reactions. (C) Effect of mutations in the TC-transfer site on the turnover of the t6A biosynthesis reaction, assessed using the time-course radiochemical t6A assay measuring the incorporation of [14C]-threonine into tRNAThrCGU. Assays contained wild-type TmTsaC2, TmTsaB, TmTsaE and mutant TmTsaD harboring mutations in the TC-transfer site (colored lines). Multiple-turnover activity catalyzed by a mixture of all wild-type proteins (solid black line), and single-turnover activity catalyzed in the absence of TmTsaE (dashed black line) are shown. Data represent duplicate measurements from three independent reactions.
Residues in the conserved Switch I and Switch II motifs of TsaE mediate communication between the ATPase site and the TC-transfer site
TsaE contains two loops, Switch I and Switch II (30), conserved in P-loop NTPases and known to play a major role as mediators of signal transduction by amplifying and transferring conformational changes occurring at the nucleotide hydrolysis site to distal parts of the molecule (31). In the structure, as in the structure in complex with AMPPCP (33), Switch I and Switch II, which bear the consensus sequences SPT/SF/Y (S61PTF64 in TmTsaE) and HXDXY/FR (78HLDLYR83 in TmTsaE), respectively, are characteristically located adjacent to the ATP binding site (Figure 6A). To address the potential role of these loops in conformational changes associated with the t6A catalytic cycle we targeted residues in Switch I and II that lie outside the ATP binding pocket, i.e. that do not interact directly or via water with the metal or nucleotide in the ATPase site. These are Thr63 and Phe64 in Switch I, and Tyr82 and Arg83 in Switch II. Mutating Thr63 or Tyr82 to alanine results in complete loss of ATP hydrolysis activity (Figure 6B, Supplementary Figure S3A). As expected, the loss of ATPase activity is accompanied by the loss of multi-turnover t6A synthesis (Figure 6C and Supplementary Table S2) by these mutants. Significantly, mutating Phe64 or Arg83 to alanine maintains formation of a stable TC-transfer complex (Supplementary Figure S4A) with a robust ATPase activity (Figure 6B, Supplementary Figure S3B), but compromises t6A synthesis, with the TsaEPhe64Ala mutant exhibiting only single-turnover activity, and TsaEArg83Ala exhibiting multiple turnovers but at a very slow rate (Figure 6C and Supplementary Table S2). These results are consistent with a role for Phe64 and Arg83 in post ATP-hydrolysis signal transduction that leads to ‘resetting’ the TsaB2D2 platform for the next catalytic cycle.
Figure 6.
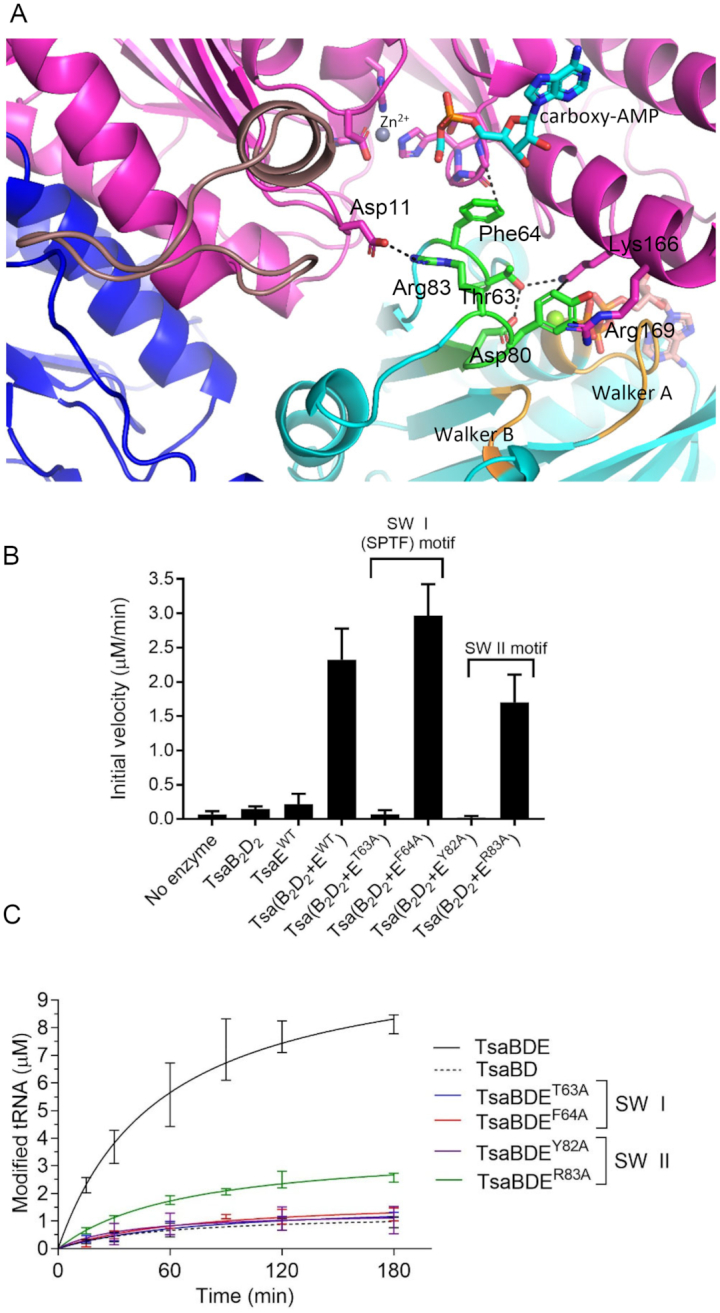
Role of Switch I and Switch II residues of TmTsaE in signal transduction between the ATPase site and TC-transfer site. (A) Interactions of Thr63 and Phe64 in Switch I loop, and Tyr82 and Arg83 in Switch II loop of TsaE and their locations relative to the bound nucleotides in the ATPase and TC-transfer sites. (B) Effect of mutations in Switch I and Switch II motifs on the ATPase activity of the TC-transfer complex. Shown are initial velocities of ATP hydrolysis reactions catalyzed by an equimolar mixture of pre-isolated wild-type TmTsaB2D2 and mutant TmTsaE proteins harboring mutations in Switch I or Switch II. Data represent averages of duplicate measurements from three independent reactions. (C) Effect of mutations in Switch I and Switch II motifs on the turnover of the t6A biosynthesis reaction, assessed using the time-course radiochemical t6A assay measuring the incorporation of [14C]-threonine into tRNAThrCGU. Assays contained mutant TmTsaE and the other three wild-type Tsa proteins (colored lines). Multiple-turnover activity catalyzed by a mixture of all wild-type proteins (solid black line), and single-turnover activity catalyzed in the absence of TmTsaE (dashed black line) are shown. Data represent duplicate measurements from three independent reactions.
In the structure, the phenyl ring of Phe64 stacks against (within 4 Å) and stabilizes a rigid conformation of the glycine-rich β-turn S134GGH137 in TsaD (Figure 6A), which appears to stabilize the binding pocket for the carbamoyl moiety of TC–AMP. Given that TsaE is not bound during TC-tranfser, this interaction may recapitulate an interaction with a tRNA base during the reaction. Indeed, we propose that this is the site for A37 during TC-transfer (vide infra). In contrast, Arg83 of TsaE makes a salt bridge with Asp11 of TsaD at the N-terminal hinge of the lever arm, thus stabilizing it in the up position (Figure 6A). The observation that Arg83 is required for efficient multi-turnover t6A synthesis is consistent with movement of the lever arm of TsaD comprising at least part of the reset mechanism.
tRNA binding determinants on TmTsaB2D2 and SAXS model of the tRNA-bound complex
Binding studies demonstrate that tRNA and TsaE share a common or overlapping binding site(s) on TmTsaB2D2 (14). The crystal structure clearly reveals a set of conserved basic residues that line the TsaE binding cavity that could be involved in tRNA binding. These include the ATP binding residues Lys166 and Lys213 of TsaD, and the lysine- and arginine-rich C-terminal tail of TsaB, which has a conserved overall basic character in the TsaB family. To test the role of these residues in tRNA binding, a TmTsaB construct was generated that lacks the C-terminal helix and tail Leu189-Gly206 (TmTsaBΔ189-206) and EMSA experiments were conducted on the pre-isolated TmTsaB2D2 complexes made with TmTsaDK166A, TmTsaDK213A or TmTsaBΔ189-206, using 32P-labeled E. coli tRNAThrCGU as substrate. The results show that while these mutants form stable TmTsaB2D2 complexes with their wild-type partners (Supplementary Figures S4B and D), the complexes exhibit significantly reduced binding affinity for tRNA (Figure 7A and Supplementary Figure S7C), consistent with the TsaE binding cavity comprising at least part of the tRNA binding site.
Based on these results, a docking model of tRNA onto the TmTsaB2D2 complex was built using proximity of the tRNA anticodon loop to TsaD residues Lys166 and Lys213, and residues Ser134, His137 in the TC-transfer site, and the model was used to fit SAXS data of the TmTsaB2D2-tRNA complex. The results show an excellent fit to the experimental SAXS data (χ2 = 2.6) (Figure 7B) with an experimental molecular weight of 148 kDa (Table 2), close to the theoretical molecular weight of 151 kDa. Additionally, the ab initio shape created based on the experimental data shows extra features on one side of TmTsaB2D2, indicating the presence of only one bound tRNA molecule (Figure 7C), in agreement with previous binding studies (14). In this model, the anticodon stem–loop of tRNA, containing the substrate A37, is lodged in the positively charged TsaE binding cavity and contacts the C-terminal tail of the TsaB subunit.
Docking model of TC–AMP and insights into the mechanism of threonylcarbamoyl transfer
The crystal structure presented here is the only structure of a TC-transfer complex bound to a ligand that closely mimics TC–AMP. This enabled us to build a mechanistically meaningful model of the TC–AMP bound complex by directly docking a TC–AMP molecule in place of carboxy-AMP and applying energy minimization to the docked structure (Figure 8A). The model reveals a putative binding pocket for the TC moiety of TC–AMP where the backbone COO- is appropriately positioned to interact with the Zn2+, replacing one of the two equatorial water ligands. Further, the model shows that the only space available near the TC moiety for the binding of Adenine-37 (Ade37) of tRNA is the space occupied by Ph64 of TsaE, consistent with the requirement that TsaE must be unbound in order for the anticodon stem–loop of tRNA to occupy the TsaE binding cavity, as seen by SAXS (vida supra). When docking adenine in the position of Phe64 of TsaE (Figure 8B) to mimic Ade37 of the tRNA, the N6 atom of adenine is 4.6 Å from the carbamoyl carbon atom of TC–AMP, well-positioned for nucleophilic attack. Furthermore, the carbamoyl carbonyl oxygen of TC–AMP is 5.6 Å from the Zn2+, which may serve to stabilize the formation of the resulting tetrahedral intermediate either directly if the actual distances are shorter with tRNA bound, or indirectly via the bound water molecule, which is 3.6 Å away.
Figure 8.
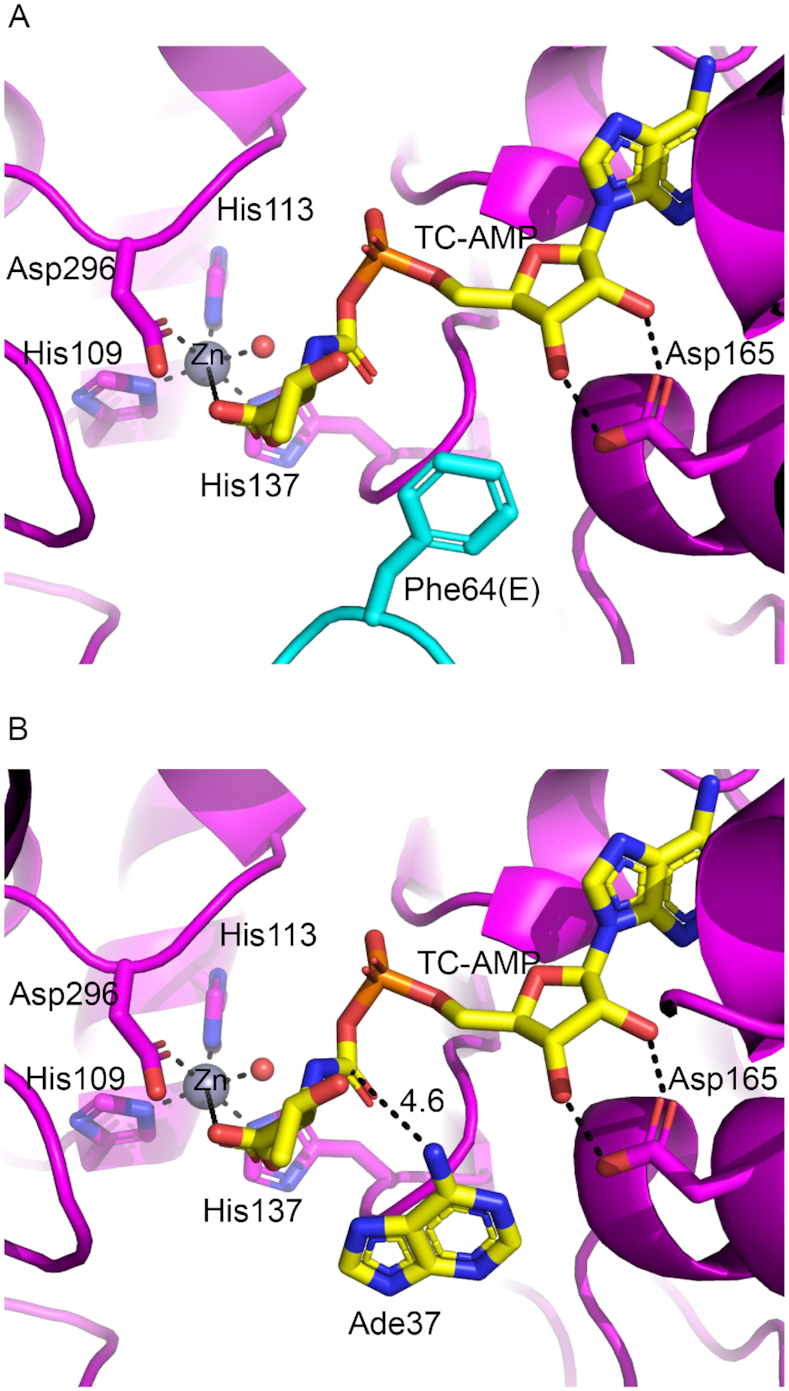
Mechanistic insights into the TC transfer reaction. (A) Docking model of TC–AMP in the TC-transfer site of TmTsaB2D2E2 based on its crystal structure in complex with carboxy-AMP. All atoms in the adenine, ribose, phosphate, and carbamoyl moieties of TC–AMP occupy the same positions as the corresponding atoms in carboxy-AMP. (B) Docking of adenine-37 in the position of Phe64 of TsaE showing the N6 atom in proximity to the carbamoyl group of TC–AMP. Putative interaction distances (Å) and the Zn2+ coordination sphere are indicated with dashed lines.
DISCUSSION
Based on prior results in which we demonstrated that TmTsaB2D2 alone was capable of a single turnover of t6A synthesis, and that multiple-turnover catalysis required the participation of TsaE mediated ATP hydrolysis, we proposed that the full catalytic cycle involved an ATP-dependent structural change in the TmTsaB2D2 complex (14). Furthermore, because t6A-modified product tRNA readily dissociates from the TmTsaB2D2 complex, we proposed that this structural change likely involved a conformational ‘reset’ that occurred after the release of product tRNA. While the details of the structural changes associated with the ‘reset’ remain obscure, the data reported here are consistent with this proposal (Figure 1), and provide additional insight into the nature and timing of these changes.
First, mutagenesis of the ATP binding site of TsaE, and demonstration that loss of ATPase activity is always accompanied by a loss of multi-turnover t6A activity, further establishes the coupling between ATPase activity and multi-turnover (i.e. catalytic) synthesis of t6A-modified tRNA. Second, the observation that ATP is bound in the ATPase site of TsaE in the TmTsaB2D2E2 structure identifies this as a pre-‘reset’ structure in the proposed catalytic cycle. Third, the presence of a TC–AMP mimic (carboxy-AMP) bound in the TsaD active site (TC-transfer site) of the TmTsaB2D2E2 structure suggests that the TC–AMP binding site is largely intact throughout the catalytic cycle. This is not surprising given that the active site contains a Zn2+ that partly plays a structural role, but this result is at odds with the recent report by Missoury et al., who describe a crystal structure of TmTsaB2D2E2 (3.1 Å resolution) in which the TsaD active site was empty and disordered (33). This led the authors to propose that the ‘reset’ step of the catalytic cycle involved partial denaturation or melting of the TC-transfer site as a consequence of TsaE binding, perturbing the integrity of the TsaD metal-binding center and resulting in an inactive conformation of TsaD. Given that our data reveal an ordered TC-transfer site with a fully-coordinated Zn2+ and a bound carboxy-AMP molecule that mimics the natural reaction intermediate TC–AMP, this proposal appears to be untenable. Fourth, the data from mutagenesis of the Switch I/Switch II residues Phe64 and Arg83 demonstrate uncoupling of ATPase activity from t6A activity when the structural changes generated by ATP hydrolysis cannot be communicated throughout the complex. Given the large conformational changes associated with the movement of the lever arm, it is not unreasonable to propose that ATP hydrolysis might power that structural change, although our data do not directly address that question.
CONCLUSIONS
The results presented here have enabled us to build for the first-time a mechanistically meaningful model of the TC-transfer step that is consistent with all of the data currently available for this system, and provides detailed insight into how TC-transfer to tRNA might be accomplished. However, there remain a number of important unanswered questions regarding the full catalytic cycle of the system. For example, it remains unclear what all of the structural changes associated with TsaE-catalyzed hydrolysis of ATP are, how these structural changes get communicated throughout the complex, and how they govern the single- vs. multiple-turnover regimes observed with the TCT complex. It is also unclear what role, if any, TsaC2 plays in the reset step, and if it binds before or after tRNA. The answers to these and other key questions await further structural and mechanistic studies.
DATA AVAILABILITY
Atomic coordinates and structure factors for TmTsaB2D2E2 have been deposited with the Protein Data Bank under accession number 6N9A.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Thomas Weiss for help with the SAXS data collection, and Rick McClintock for assistance with the FPLC equipment.
Notes
Present address: Amit Luthra, Department of Medicine, University of Connecticut Health Center, 263 Farmington Ave., Farmington, CT 06030.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [GM110588 to M.A.S. and D.I.-R.]; California Metabolic Research Foundation. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences [DE-AC02-76SF00515]; The SSRL Structural Molecular Biology Program is supported by the Department of Energy Office of Biological and Environmental Research; National Institutes of Health; and the National Institute of General Medical Sciences [P41GM103393]. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health or the National Institute of General Medical Sciences. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Thiaville P.C., El Yacoubi B., Kohrer C., Thiaville J.J., Deutsch C., Iwata-Reuyl D., Bacusmo J.M., Armengaud J., Bessho Y., Wetzel C. et al.. Essentiality of threonylcarbamoyladenosine (t6A), a universal tRNA modification, in bacteria. Mol. Microbiol. 2015; 98:1199–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. El Yacoubi B., Bailly M., de Crécy-Lagard V.. Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu. Rev. Genet. 2012; 46:69–95. [DOI] [PubMed] [Google Scholar]
- 3. Grosjean H., de Crécy-Lagard V., Marck C.. Deciphering synonymous codons in the three domains of life: co-evolution with specific tRNA modification enzymes. FEBS Lett. 2010; 584:252–264. [DOI] [PubMed] [Google Scholar]
- 4. El Yacoubi B., Hatin I., Deutsch C., Kahveci T., Rousset J.P., Iwata-Reuyl D., Murzin A.G., de Crécy-Lagard V.. A role for the universal Kae1/Qri7/YgjD (COG0533) family in tRNA modification. EMBO J. 2011; 30:882–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Murphy F.V.4th., Ramakrishnan V., Malkiewicz A., Agris P.F.. The role of modifications in codon discrimination by tRNA(Lys)UUU. Nat. Struct. Mol. Biol. 2004; 11:1186–1191. [DOI] [PubMed] [Google Scholar]
- 6. Agris P.F. Bringing order to translation: the contributions of transfer RNA anticodon-domain modifications. EMBO Rep. 2008; 9:629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thiaville P.C., Legendre R., Rojas-Benitez D., Baudin-Baillieu A., Hatin I., Chalancon G., Glavic A., Namy O., de Crécy-Lagard V.. Global translational impacts of the loss of the tRNA modification t6A in yeast. Microb. Cell. 2016; 3:29–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Edvardson S., Prunetti L., Arraf A., Haas D., Bacusmo J.M., Hu J.F., Ta-Shma A., Dedon P.C., de Crécy-Lagard V., Elpeleg O.. tRNA N6-adenosine threonylcarbamoyltransferase defect due to KAE1/TCS3 (OSGEP) mutation manifest by neurodegeneration and renal tubulopathy. Eur. J. Hum. Genet. 2017; 25:545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Braun D.A., Rao J., Mollet G., Schapiro D., Daugeron M.C., Tan W., Gribouval O., Boyer O., Revy P., Jobst-Schwan T. et al.. Mutations in KEOPS-complex genes cause nephrotic syndrome with primary microcephaly. Nat. Genet. 2017; 49:1529–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thiaville P.C., Iwata-Reuyl D., de Crécy-Lagard V.. Diversity of the biosynthesis pathway for threonylcarbamoyladenosine (t6A), a universal modification of tRNA. RNA Biol. 2014; 11:1529–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. El Yacoubi B., Lyons B., Cruz Y., Reddy R., Nordin B., Agnelli F., Williamson J.R., Schimmel P.R., Swairjo M.A., de Crécy-Lagard V.. The universal YrdC/Sua5 family is required for the formation of threonylcarbamoyladenosine in tRNA. Nucleic Acids Res. 2009; 37:2894–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lauhon C.T. Mechanism of N6-threonylcarbamoyladenonsine (t6A) biosynthesis: isolation and characterization of the intermediate threonylcarbamoyl-AMP. Biochemistry. 2012; 51:8950–8963. [DOI] [PubMed] [Google Scholar]
- 13. Deutsch C., El Yacoubi B., de Crécy-Lagard V., Iwata-Reuyl D.. Biosynthesis of threonylcarbamoyl adenosine (t6A), a universal tRNA nucleoside. J. Biol. Chem. 2012; 287:13666–13673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luthra A., Swinehart W., Bayooz S., Phan P., Stec B., Iwata-Reuyl D., Swairjo M.A.. Structure and mechanism of a bacterial t6A biosynthesis system. Nucleic Acids Res. 2018; 46:1395–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Perrochia L., Guetta D., Hecker A., Forterre P., Basta T.. Functional assignment of KEOPS/EKC complex subunits in the biosynthesis of the universal t6A tRNA modification. Nucleic Acids Res. 2013; 4:9484–9499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Srinivasan M., Mehta P., Yu Y., Prugar E., Koonin E.V., Karzai A.W., Sternglanz R.. The highly conserved KEOPS/EKC complex is essential for a universal tRNA modification, t6A. EMBO J. 2011; 30:873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Daugeron M.-C., Lenstra T.L., Frizzarin M., El Yacoubi B., Liu X., Baudin-Baillieu A., Lijnzaad P., Decourty L., Saveanu C., Jacquier A. et al.. Gcn4 misregulation reveals a direct role for the evolutionary conserved EKC/KEOPS in the t6A modification of tRNAs. Nucleic Acids Res. 2011; 39:6148–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Perrochia L., Crozat E., Hecker A., Zhang W., Bareille J., Collinet B., van Tilbeurgh H., Forterre P., Basta T.. In vitro biosynthesis of a universal t6A tRNA modification in Archaea and Eukarya. Nucleic Acids Res. 2013; 41:1953–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wan L.C., Maisonneuve P., Szilard R.K., Lambert J.P., Ng T.F., Manczyk N., Huang H., Laister R., Caudy A.A., Gingras A.C. et al.. Proteomic analysis of the human KEOPS complex identifies C14ORF142 as a core subunit homologous to yeast Gon7. Nucleic Acids Res. 2017; 45:805–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wan L.C., Mao D.Y., Neculai D., Strecker J., Chiovitti D., Kurinov I., Poda G., Thevakumaran N., Yuan F., Szilard R.K. et al.. Reconstitution and characterization of eukaryotic N6-threonylcarbamoylation of tRNA using a minimal enzyme system. Nucleic Acids Res. 2013; 41:6332–6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Msadek T. Grasping at shadows: revealing the elusive nature of essential genes. J. Bacteriol. 2009; 191:4701–4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Handford J.I., Ize B., Buchanan G., Butland G.P., Greenblatt J., Emili A., Palmer T.. Conserved network of proteins essential for bacterial viability. J. Bacteriol. 2009; 191:4732–4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Butland G., Peregrin-Alvarez J.M., Li J., Yang W., Yang X., Canadien V., Starostine A., Richards D., Beattie B., Krogan N. et al.. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature. 2005; 433:531–537. [DOI] [PubMed] [Google Scholar]
- 24. Xu Q., McMullan D., Jaroszewski L., Krishna S.S., Elsliger M.-A., Yeh A.P., Abdubek P., Astakhova T., Axelrod H.L., Carlton D. et al.. Structure of an essential bacterial protein YeaZ (TM0874) from Thermotoga maritima at 2.5 Å resolution. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2010; 66:1230–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nichols C.E., Lamb H.K., Thompson P., Omari K.E., Lockyer M., Charles I., Hawkins A.R., Stammers D.K.. Crystal structure of the dimer of two essential Salmonella typhimurium proteins, YgjD & YeaZ and calorimetric evidence for the formation of a ternary YgjD–YeaZ–YjeE complex. Protein Sci. 2013; 22:628–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang W., Collinet B., Perrochia L., Durand D., van Tilbeurgh H.. The ATP-mediated formation of the YgjD–YeaZ–YjeE complex is required for the biosynthesis of tRNA t6A in Escherichia coli. Nucleic Acids Res. 2015; 43:1804–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aydin I., Saijo-Hamano Y., Namba K., Thomas C., Roujeinikova A.. Structural analysis of the essential resuscitation promoting factor YeaZ suggests a mechanism of nucleotide regulation through dimer reorganization. PLoS One. 2011; 6:e23245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vecchietti D., Ferrara S., Rusmini R., Macchi R., Milani M., Bertoni G.. Crystal structure of YeaZ from Pseudomonas aeruginosa. Biochem. Biophys. Res. Commun. 2016; 470:460–465. [DOI] [PubMed] [Google Scholar]
- 29. Koonin E.V. A common set of conserved motifs in a vast variety of putative nucleic acid-dependent ATPases including MCM proteins involved in the initiation of eukaryotic DNA replication. Nucleic Acids Res. 1993; 21:2541–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Teplyakov A., Obmolova G., Tordova M., Thanki N., Bonander N., Eisenstein E., Howard A.J., Gilliland G.L.. Crystal structure of the YjeE protein from Haemophilus influenzae: a putative Atpase involved in cell wall synthesis. Proteins. 2002; 48:220–226. [DOI] [PubMed] [Google Scholar]
- 31. Wittinghofer A., Vetter I.R.. Structure-function relationships of the G domain, a canonical switch motif. Annu. Rev. Biochem. 2011; 80:943–971. [DOI] [PubMed] [Google Scholar]
- 32. Karst J.C., Foucher A.E., Campbell T.L., Di Guilmi A.M., Stroebel D., Mangat C.S., Brown E.D., Jault J.M.. The ATPase activity of an ‘essential’ Bacillus subtilis enzyme, YdiB, is required for its cellular function and is modulated by oligomerization. Microbiology. 2009; 155:944–956. [DOI] [PubMed] [Google Scholar]
- 33. Missoury S., Plancqueel S., Li de la Sierra-Gallay I., Zhang W., Liger D., Durand D., Dammak R., Collinet B., van Tilbeurgh H.. The structure of the TsaB/TsaD/TsaE complex reveals an unexpected mechanism for the bacterial t6A tRNA-modification. Nucleic Acids Res. 2018; 46:5850–5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Minor W., Cymborowski M., Otwinowski Z., Chruszcz M.. HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 2006; 62:859–866. [DOI] [PubMed] [Google Scholar]
- 35. McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J.. Phaser crystallographic software. J. Appl. Crystallogr. 2007; 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. et al.. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kelley L.A., Mezulis S., Yates C.M., Wass M.N., Sternberg M.J.E.. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015; 10:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Emsley P., Cowtan K.. COOT: Model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004; D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 39. Murshudov G.N., Skubak P., Lebedev A.A., Pannu N.S., Steiner R.A., Nicholls R.A., Winn M.D., Long F., Vagin A.A.. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011; 67:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Konarev P.V., Volkov V.V., Sokolova A.V., Koch M.H.J., Svergun D.I.. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003; 36:1277–1282. [Google Scholar]
- 41. Franke D., Petoukhov M.V., Konarev P.V., Panjkovich A., Tuukkanen A., Mertens H.D.T., Kikhney A.G., Hajizadeh N.R., Franklin J.M., Jeffries C.M. et al.. ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J Appl Crystallogr. 2017; 50:1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schneidman-Duhovny D., Hammel M., Tainer J.A., Sali A.. FoXS, FoXSDock and MultiFoXS: Single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Res. 2016; 44:W424–W429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sankaranarayanan R., Dock-Bregeon A.C., Romby P., Caillet J., Springer M., Rees B., Ehresmann C., Ehresmann B., Moras D.. The structure of threonyl-tRNA synthetase-tRNA(Thr) complex enlightens its repressor activity and reveals an essential zinc ion in the active site. Cell. 1999; 97:371–381. [DOI] [PubMed] [Google Scholar]
- 44. van Zundert G.C., Rodrigues J.P., Trellet M., Schmitz C., Kastritis P.L., Karaca E., Melquiond A.S., van Dijk M., de Vries S.J., Bonvin A.M.. The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016; 428:720–725. [DOI] [PubMed] [Google Scholar]
- 45. Wallace A.C., Laskowski R.A., Thornton J.M.. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995; 8:127–134. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for TmTsaB2D2E2 have been deposited with the Protein Data Bank under accession number 6N9A.