


Abstract
Ribosome-mediated transcription attenuation is a basic posttranscriptional regulation mechanism in bacteria. Liberated attenuator RNAs arising in this process are generally considered nonfunctional. In Sinorhizobium meliloti, the tryptophan (Trp) biosynthesis genes are organized into three operons, trpE(G), ppiD-trpDC-moaC-moeA, and trpFBA-accD-folC, of which only the first one, trpE(G), contains a short ORF (trpL) in the 5′-UTR and is regulated by transcription attenuation. Under conditions of Trp sufficiency, transcription is terminated between trpL and trpE(G), and a small attenuator RNA, rnTrpL, is produced. Here, we show that rnTrpL base-pairs with trpD and destabilizes the polycistronic trpDC mRNA, indicating rnTrpL-mediated downregulation of the trpDC operon in trans. Although all three trp operons are regulated in response to Trp availability, only in the two operons trpE(G) and trpDC the Trp-mediated regulation is controlled by rnTrpL. Together, our data show that the trp attenuator coordinates trpE(G) and trpDC expression posttranscriptionally by two fundamentally different mechanisms: ribosome-mediated transcription attenuation in cis and base-pairing in trans. Also, we present evidence that rnTrpL-mediated regulation of trpDC genes expression in trans is conserved in Agrobacterium and Bradyrhizobium, suggesting that the small attenuator RNAs may have additional conserved functions in the control of bacterial gene expression.
INTRODUCTION
Since the discovery of ribosome-mediated transcription attenuation in bacteria 40 years ago (1,2), it has been presumed that terminated attenuator RNAs have no own functions (3). Here, we show that, in Sinorhizobium meliloti, an attenuator RNA that is liberated under conditions of tryptophan (Trp) sufficiency acts in trans to destabilize Trp biosynthesis (trp) mRNA.
Transcription attenuation sensing specific uncharged tRNA is a sophisticated posttranscriptional mechanism that is used by bacteria to regulate their gene expression. A prime example is the regulation of the trpEDCBA operon in Escherichia coli. The structural genes of this operon are preceded by an mRNA leader that is able to form mutually exclusive secondary structures and contains a small open reading frame (sORF), trpL, with consecutive Trp codons (4). Ribosome pausing at the Trp codons prevents the formation of a terminator hairpin, resulting in structural genes transcription when cellular Trp concentrations are low. At high Trp levels, the leader peptide TrpL is translated efficiently, resulting in the formation of the terminator hairpin and, consequently, transcription termination, thus leading to the release of a small attenuator RNA. In this way, bacteria respond in a very fast and resource-saving manner to amino acid (aa) availability. Similar ribosome-dependent transcription attenuators are widespread (3,5,6). Many Gram-positive bacteria developed different strategies for transcription attenuation of trp operons. For example, Bacillus subtilis utilizes a Trp-activated RNA binding protein (TRAP) while other gram-positive bacteria, such as Streptococcus, harbor a tRNATrp sensing structure (T-box) in the leader of trp mRNA (3,7,8).
Thus, at least three different mechanisms for RNA-based regulation of trp operons evolved in bacteria, indicating the importance of strict posttrancriptional control of Trp biosynthesis for bacterial survival. Most probably, this is due to the fact that Trp is the most costly-to-synthesize amino acid (3). Despite this, many bacteria have split trp operons with only one of them being preceded by a transcription attenuator (3). This raises the question of whether the other trp operons are posttrancriptionally regulated by alternative mechanisms, for example by small RNAs. Small RNAs (sRNAs) regulate virtually all aspects of bacterial physiology, including metabolism and adaptation to changing environmental conditions. Mechanistically, they can exert their regulatory function by protein binding or by base pairing with mRNAs (9). Bacterial trans-acting and base-pairing sRNAs are highly versatile due to their capacity to target multiple mRNAs (10–12). Typically, such sRNAs display limited complementarity to their targets and need an RNA chaperone, such as Hfq or ProQ, for efficient mRNA binding (9,13). In most cases, the interaction between an sRNA and an mRNA prevents translation and/or destabilizes the mRNA target (9,14,15).
Most of the functionally characterized sRNAs have orphan genes. Additionally, 3′-UTRs of bacterial mRNAs are considered as established sRNA reservoirs (16,17). In contrast, functional sRNAs originating from 5′-UTRs were reported much less frequently and there are very few examples of riboregulators that act both in cis and trans (18). These include the S-adenosylmethionine riboswitches SreA and SreB which act as base-pairing sRNAs that control the expression of a virulence gene in Listeria monocytogenes (19). Further, in L. monocytogenes and Enterococcus faecalis, a coenzyme B12 (AdoCbl) riboswitch was shown to work as a protein-binding sRNA that sequesters a response regulator (20,21). To date, small trans-acting RNAs derived from ribosome-mediated transcription attenuation have not been described.
We are interested in Sinorhizobium meliloti, a soil-dwelling alpha-proteobacterium that is capable of fixing molecular nitrogen in symbiosis with some legume plants. In S. meliloti, the structural trp genes are organized into operons trpE(G), trpDC, and trpFBA (3,22), of which only trpE(G) is regulated by transcription attenuation (23). In the past decade, hundreds of sRNA candidates have been detected in S. meliloti (24) and, meanwhile, physiological roles based on direct binding to target mRNAs have been shown for several of them. For example, AbcR1, AbcR2 and NfeR1 were shown to regulate transporter genes (25,26), while RcsR1 was reported to down-regulate the autoinducer synthase encoding mRNA sinI (27), and EcpR1 and GspR were suggested to regulate the cell cycle (28,29). Given that sRNA RcsR1 is transcribed from a position upstream of trpE(G) and is identical to the trpL-derived attenuator RNA (23), a possible role of this particular RNA in the (co)regulation of the trp operons seemed plausible. In line with this hypothesis, RcsR1 was predicted to base-pair with several mRNAs including trpDC and constitutive ectopic RcsR1 overproduction was shown to result in lower trpC mRNA levels (27). However, it was not clear if this was a direct effect of RcsR1 on trpDC mRNA and if it had any functional implications for bacterial gene regulation in response to Trp availability.
Here, we demonstrate that the liberated attenuator sRNA of the Trp biosynthesis gene trpE(G) base-pairs with trpD and down-regulates trpDC under conditions of Trp sufficiency. Please note that, in this study, the sRNA RcsR1 was re-named rnTrpL to underline its biogenesis (in transcription attenuation) and its role in the posttranscriptional regulation of bacterial trp operons. The data obtained in this study provide an interesting example for a multifunctional mRNA leader with regulatory functions in both cis and trans.
MATERIALS AND METHODS
Cultivation of bacteria
Escherichia coli strains were grown in LB medium (30). Unless stated otherwise, Sinorhizobium (Ensifer) meliloti 2011 (31,32) and Agrobacterium tumefaciens (A. fabrum) NTL4 (pZLR4) (33,34) were grown in TY medium (35) as described (27). For analysis of gene expression at different Trp availability conditions, GMX medium (36) with the indicated Trp concentrations was used. Bradyrhizobium japonicum (B. diazoefficiens) 110spc4 (37,38) was grown in PSY medium (39). When appropriate, antibiotics were included at the following concentrations: ampicillin (200 μg/ml), streptomycin (250 μg/ml), tetracycline (20 μg/ml for S. meliloti and A. tumefaciens; B. japonicum was cultivated with 25 μg/ml Tc in liquid and 50 μg/ml Tc on plates), gentamycin (10 μg/ml; for S. meliloti on plates, 20 μg/ml were used); kanamycin (25 μg/ml for E. coli and 200 μg/ml for S. meliloti), spectinomycin (100 μg/ml for B. japonicum). IPTG was used at a final concentration of 1 mM.
Plasmid construction and conjugation
Cloning procedures were performed essentially as described (30). FastDigest Restriction Endonucleases and Phusion polymerase (Thermo Fischer Scientific) were used routinely for cloning in E. coli JM109 (40) or E. coli DH5α. When pJet1.2/blunt (CloneJet PCR Cloning Kit, Thermo Fischer Scientific) was used for cloning, the inserts were subcloned into the conjugative plasmids pRK4352 (41), pSRKGm, pSRKTc (42), or pK18mobsacB (43). Insert sequences were analyzed by Sanger sequencing with plasmid-specific primers (sequencing service by Microsynth Seqlab, Göttingen, Germany) prior to conjugation into S. meliloti. Plasmids used for conjugation in S. meliloti are listed in Supplementary Table S1 and oligonucleotides used in this study are listed in Supplementary Table S2. Oligonucleotides were synthesized by Eurofins Genomics, Ebersberg (Germany) and Microsynth, Balgach (Switzerland).
Plasmids pDrive-RcsR1 and pRK-SmelRcsR1 (from now on pDrive-rnTrpL and pRK-rnTrpL, respectively; see Supplementary Table S1) were described previously (27). In S. meliloti, pRK-rnTrpL leads to constitutive transcription of rnTrpL from a heterologous ribosomal RNA (rrn) promoter originating from the alpha-proteobacterium Rhodobacter shpaeroides (41). For constitutive overexpression of the B. japonicum rnTrpL homolog Bja-rnTrpL, the corresponding sequence was cloned in the chromosome integration plasmid pRJ-MCS (44) that was cleaved with KpnI and SpeI. Plasmid pRK-rnTrpL-AU1,2UA, which allows for constitutive transcription of an S. meliloti rnTrpL derivative that lacks a functional sORF (replacement of the trpL AUG codon with UAG), was constructed as follows. The rnTrpL-sequence was amplified with primers RcsR1-ATG/TAG-fw and pRKSmelRcsR1re and cloned into pRK4352. To construct pRK-rnTrpL-AU1,2UA-G44C, the same primers were used in a PCR with pRK-rnTrpL-G44C as the template and the amplicon was cloned into pRK4352. To construct pRK-rnTrpL-CG40,41GC and pRK-rnTrpL-GG46,47CC, site-directed mutagenesis of the rnTrpL-sequence in pDrive-rnTrpL was performed by inverse PCR with Phusion polymerase, followed by treatment with DpnI (Thermo Fischer Scientific). The mutated inserts were excised with BamHI and EcoRI, and cloned into pRK4352. To construct plasmid pRK-trpL-egfp, egfp was amplified with primers Bam-RcsR1-egfp-fw and Eco-egfp-re using pLK64 (45) as template, and the amplicon was cloned into pRK4352 using its BamHI and EcoRI restriction sites. The resulting translational rnTrpL::egfp fusion contains the first six codons of trpL fused to the third codon of egfp. Plasmids pSRKTc-rnTrpL and pSRKGm-rnTrpL for IPTG-inducible transcription of lacZ’-rnTrpL were constructed as follows. The rnTrpL-sequence of S. meliloti was amplified and ligated with NdeI/SpeI-digested pSRKTc or pSRKGm, resulting in an in-frame insertion of the trpL sORF to the ATG of NdeI. Similarly, plasmids pSRKTc-Atu-rnTrpL and pSRKTc-Ec-rnTrpL were constructed by cloning the rnTrpL homologs of A. tumefaciens and E. coli into pSRKTc. To construct pSRKTc-rnTrpL-CG40,41GC and pSRKTc-rnTrpL-GG46,47CC, mutated rnTrpL-sequences were amplified using as template pDrive-rnTrpL-CG40,42GC and pDrive-RcsR1-GG46,47CC, respectively, and cloned into pSRKTc cleaved with NdeI and SpeI. To construct pSRKGm-trpDC’-egfp, the trpDC’ sequence of S. meliloti was amplified with primers NdeI-trpD-fw and 5′-egfp-trpC-re. In parallel, egfp was amplified using pLK64 (45) as template and primers trpC-egfp-fw and XbaI-egfp-re. The two PCR products were mixed and used for overlap-PCR with the primers NdeI-trpD-fw and XbaI-egfp-re. The resulting amplicon was ligated with pJet1.2/blunt, resulting in pJet-trpDC’-egfp. The insert was subcloned into pSRKGm using NdeI and XbaI. The resulting plasmid pSRKGm-trpDC’-egfp allows for IPTG-inducible transcription of a bicistronic trpDC’::egfp mRNA encoding TrpD and the fusion protein TrpC’-EGFP. The fusion construct contains the first 16 trpC codons fused in frame to the third codon of egfp. To introduce compensatory mutations into the trpDC’::egfp reporter, pJet-trpDC’-egfp was subjected to site-directed mutagenesis as described above. The mutated inserts were subcloned into pSRKGm using NdeI and XbaI, resulting in pSRKGm-trpD-CG985,986GC-trpC’-egfp and pSRKGm-trpD-CC977,978GG-trpC’-egfp. Transconjugants containing one of the pSRK-plasmids were stored at –80°C. Double transconjugants (containing pSRK-plasmids with different antibiotic resistance genes) were used directly after the transfer of the second plasmid to S. meliloti. To construct plasmid pK18mobsacB-ΔtrpL for marker-less deletion of the rnTrpL-sequence in the chromosome of S. meliloti, regions located upstream and downstream of trpL in the chromosome were amplified separately. Overlap PCR was performed and the resulting amplicon was cloned into pK18mobsacB (43), which was cleaved with EcoRI and PstI. Plasmid pK18mobsacB-ΔtrpL was used to delete the chromosomal sequence ranging from the third nucleotide after the rnTrpL transcription start site to the last T in the TTTT stretch of the rnTrpL transcription terminator. Similarly, plasmid pK18mobsacB-ΔtrpC for marker-less in-frame deletion in gene trpC of S. meliloti was constructed. The in-frame deletion construct contains the first ten codons fused to the last nine codons of trpC.
Plasmid constructs were transferred to S. meliloti, A. tumefaciens or B. japonicum by diparental conjugation with E. coli S17–1 as the donor (46). Bacteria were mixed, washed in saline and spotted onto a sterile membrane filter, which was placed onto a TY plate without antibiotics. After incubation for at least 4 h at 30°C, serial dilutions were plated on TY agar with selective antibiotics.
Construction of S. meliloti 2011 deletion mutants
The deletion mutants 2011ΔtrpL, 2011ΔtrpC and 2011ΔtrpLΔtrpC were constructed as described (47), using TY solid media. For in-frame deletion of trpC, it was necessary to supplement the TY medium with 20 µg/ml L-tryptophan. Deletion mutant candidates were confirmed by PCR analysis of cell lysates using primers which correspond to sequences located outside the chromosomal region that was cloned in the pK18mobsacB-ΔtrpL and pK18mobsacB-ΔtrpC, respectively. The ΔtrpL deletion was additionally confirmed by Northern blot analysis in comparison to the parental strain.
TrpC’-EGFP fluorescence measurement
Double S. meliloti transconjugants containing pSRKGm- and pSRKTc- constructs for IPTG-induced expression of egfp reporter fusions and the sRNA rnTrpL of S. meliloti, respectively, were grown in TY with Gm (gentamycin) and Tc (tetracycline), but without IPTG, to an OD600nm = 0.5. Then, 1 mM IPTG was added to induce simultaneous transcription of the bicistronic egfp-fusion mRNA and the regulatory sRNA. Induced cultures were incubated for 20 min at 30°C with shaking. Thereafter, 300 μl of the cultures were transferred to a 96-well microtiter plate and fluorescence of the fusion protein TrpC’-EGFP was measured on a Tecan Infinite M200 reader. Values were normalized to the ODs measured on the Tecan.
RNA purification, Northern blot analysis, qRT-PCR and RNA half-life measurements
RNA was isolated from bacterial cultures grown to an OD600nm = 0.5. Unless stated otherwise, 15 ml S. meliloti or A. tumefaciens culture was filled into a tube with ice rocks and centrifuged. The pellet was resuspended in 250 ml TRIzol and glass beads (with a volume corresponding to the volume of the pellet) were added. Cells were lysed in a shacking mill (4°C), two times for 15 min, interrupted by a 10 min incubation at 65°C. Then 750 μl TRIzol was added and RNA was isolated according to the manufacturer instructions. After precipitation with isopropanol, the RNA was additionally purified by extraction with hot phenol and chloroform-isoamylalcohol to remove residual RNases. RNA from B. japonicum or E. coli DH5α was isolated using hot phenol. For RNA half-life measurements, S. meliloti RNA was isolated using RNeasy columns (Qiagen) as described (27). RNA concentration and purity was determined by measuring the absorbance at 260 and 280 nm. RNA integrity was controlled in a 10% polyacrylamide-urea gels stained with ethidium bromide.
For real-time RT-PCR (qRT-PCR), 10 μg RNA was treated with 1 μl TURBO-DNase for 30 min and DNA removal was confirmed by PCR with rpoB-specific primers for each sample. The qRT-PCR analysis was performed with Brilliant III Ultra Fast SYBR® Green QRT-PCR Mastermix (Agilent) according to the manufacturer manual. Each 10 μl reaction mixture contained 5 μl Master Mix (supplied), 0.1 μl DTT (100 mM, supplied), 0.5 μl Ribo-Block solution (supplied), 0.4 μl water, 1 μl of each primer (10 pmol/μl), and 2 μl RNA (20 ng/μl). For Figures 1D and 2C, both primers were added simultaneously before the cDNA synthesis step. For other Figures, strand-specific qRT-PCR was performed. For this, first only the primer needed for cDNA synthesis was added to the reaction mixture. After cDNA synthesis and incubation for 10 min at 96°C to inactivate the reverse transcriptase, the probes were cooled to 4°C, the second primer was added, and PCR was performed starting with 5 min incubation at 96°C. Used primer pairs and their efficiencies (as determined by PCR using serial two-fold dilutions of RNA) are listed in Supplementary Table S2. As a reference mRNA, rpoB was used when mRNA steady-state levels were compared. For half-life measurements, the stable 16S rRNA was used as a reference. For the real-time RT-PCR of this reference molecule, the RNA samples were additionally diluted till the quantification cycle (Cq) of mRNA and 16S rRNA was similar. Routinely, 2 μl RNA with a concentration of 0.002 ng/μl was used in a 10 μl reaction for real-time RT-PCR of 16S rRNA. The reactions were performed in a spectrofluorometric thermal cycle (Biorad). The Cq was set to a cycle at which the curvature of the amplification is maximal (48) using BioRad CFX Manager 3.0. When outlier were identified (e. g. technical duplicate range > 0.5 Cq), the qRT-PCR was repeated. No template-controls were always included. qPCR product specificity was validated by a melting curve after the qPCR-reaction and by gel electrophoresis. Fold changes of mRNA amounts were calculated with the Pfaffl formula (49). All qRT-PCR graphs show means and standard deviations from three independent experiments, each performed in technical duplicates. Northern blot hybridization, signal detection and sRNA bands quantification was performed as described (27,36).
Figure 1.

Ectopic overproduction of the attenuator sRNA rnTrpL decreases the level of the polycistronic trpDC mRNA in S. meliloti. (A) Schematic representation of the three trp operons and the attenuator RNA rnTrpL. ORFs are depicted as gray arrows, the RpoD-like promoter is shown as a gray rectangle, a transcription start site is indicated by a flexed arrow and a transcription terminator by a hairpin (according to 23 and 59). The terminated attenuator sRNA rnTrpL (110 nt; synonym RcsR1) is shown as a white box with start and stop codons indicated. Below this box, regions corresponding to stem-loops (SL) 1, 2 and 3 are indicated by convergent arrows. SL2 corresponds to the antiterminator and SL3 to the terminator of transcription. SL2 is preferentially formed in the presence of translating ribosomes that pause upon tRNATrp shortage. For the rnTrpL secondary structure, see Figure 2. The proposed binding site of rnTrpL in the trpD ORF is indicated by a white box. (B) Schematic representation of the plasmid pRK-rnTrpL. Transcription of rnTrpL from this plasmid is expected to be terminated at SL3 in TY medium (see Supplementary Figure S1). (C) Northern blot analysis of RNA isolated from the EVC strain 2011 (pRK4352) and the overexpressing strain 2011 (pRK-rnTrpL), grown in TY. The membrane was first hybridized with an rnTrpL-specific probe and subsequently hybridized with a 5S rRNA-specific probe (loading control). (D) Results of qRT-PCR analyses of the indicated mRNAs of the trpDC operon and trpE (control mRNA). Strain 2011 (pRK-rnTrpL) was compared to the EVC and log2(fold change) (log2FC) in mRNA steady-state levels were calculated. Shown are the results from three independent experiments, each performed in technical duplicates (mean values and standard deviations are indicated).
Figure 2.

The apical part of rnTrpL stem-loop 1 is critically involved in controlling the mRNA levels of the trpDC operon. (A) Chemical probing with DMS or CMCT of in vitro-transcribed RNA representing the rnTrpL of S. meliloti. DMS and CMCT modifications were identified by primer extension analysis. Shown is the autoradiogram of a representative denaturing polyacrylamide gel. Lanes: -, reaction performed in the absence of DMS; 1:2, 1:5, 1:10 and 1:20, reactions performed with the indicated dilutions of DMS; 2.5, 5, 10, 20 and 50 mg/ml, reactions performed with the indicated concentrations of CMCT; T, G, C, A sequencing reactions using the indicated dideoxynucleotides. (B) RNA secondary structure model of rnTrpL. Mutations characterized in this study as well as the SL1 and SL3 structures are indicated. The nucleotides highlighted by a gray box indicate the antitermination structure (SL2) reported previously (23). Nucleotides shown in boldface within the gray box indicate the stems S2a and S2b of SL2 as proposed previously (23). Dashed lines indicate regions involved in the formation of the respective stem structures according to the structural analyses shown here. Chemical modifications of specific nucleotides (highlighted by black circles) that were reproducibly detected in repeated experiments are indicated by arrowheads (DMS) and pinned arrowheads (CMCT), respectively. (C) qRT-PCR analysis of the trpDC operon in strains overexpressing wt rnTrpL or rnTrpL variants with the indicated mutations. The mRNA levels of the indicated ORFs in the overexpressing strains were compared to the levels in the EVC. Shown are results from three independent experiments, each performed in technical duplicates (mean values and standard deviations are indicated). For other details, see Figure 1.
The measurement of mRNA half-lives was performed as described (50) with modifications (27). In brief, transcription was terminated by adding rifampicin (600 μg/ml final concentration; stock concentration 60 mg/ml in methanol) to the S. meliloti culture at an OD600 = 0.5. Aliquots of 2 ml were withdrawn at time points 0, 2, 4 and 6 min and mixed with 4 ml RNAprotect Bacteria Reagent (Qiagen). After centrifugation, RNA was purified from the bacterial pellet and used in qRT-PCR analysis with 16S rRNA as a reference. The half-life of rnTrpL was determined as described (50). In brief, 15 μg total RNA was separated in 10% denaturing polyacrylamide gel and analyzed by Northern blot hybridization with the radioactively labeled oligonucleotide SmelRcsR1 as the probe. 5S rRNA was used as the loading control. Half-lives were calculated from linear-log graphs. The time after rifampicin addition was plotted against relative mRNA amounts.
In vitro transcription
A DNA template suitable for subsequent in vitro transcription of rnTrpL was obtained by PCR using the forward primer ivTr_RcsR1_f_GGGA, the reverse primer Random_RcsR1_Rev (Supplementary Table S2), and plasmid pRK-rnTrpL as a template. The primer Random_RcsR1_Rev contained an additional sequence at the 5′-end (cgattgtcgttatccattctcgttcatc), generating an artificial 3′-tail in the in vitro transcribed rnTrpL. This tail sequence did not affect the folding of SL1 and SL3 in the transcript and served as a binding site for the oligonucleotide used for primer extension. In this way, modifications in the entire rnTrpL sequence could be probed. To avoid transcription termination by T7 polymerase, the U-stretch of the SL3 transcription terminator was limited to three uridines (51).
The PCR product was purified using HiBind DNA spin-column of the E.Z.N.A DNA probe purification kit (Omega) and eluted in ultrapure water. A total amount of 500 ng of each purified PCR product was used for in vitro transcription using the MEGAshortscript T7 kit (Ambion) or the HiScribeTM T7 High Yield RNA Synthesis Kit (NEB). The reactions were performed according to the manufacturer's instructions (1x T7-polymerase buffer, 7.5 mM ATP, 7.5 mM CTP, 7.5 mM GTP, 7.5 mM UTP, 25 U T7-enzyme mix) for at least 5 h at 37°C. The DNA template was removed by digestion with 1 μl TURBO-DNase for 1 h at 37°C. The in vitro transcript was phenol-extracted, ethanol-precipitated and analyzed by electrophoresis in a 10% polyacrylamide-urea gel and subsequent staining with ethidium bromide.
rnTrpL structure probing
To analyze the structure of rnTrpL, RNA structure probing experiments were done as described (52–54). Typically, 0.6 μg of in vitro-synthesized RNA was heat denatured at 90°C for 1 min and then put on ice for 5 min. The RNA was renatured either in AN buffer (DMS probing, 50 mM sodium cacodylate, pH 7.5, 5 mM MgCl2, 60 mM KCl) or BN buffer (CMCT probing, 50 mM sodium borate, pH 8.0, 5 mM MgCl2, 60 mM KCl) for 20 min at room temperature. Next, the samples (total volume of 8 μl) were mixed with 1 μl of yeast tRNA (2 mg/ml, Ambion) and 1 μl of dimethyl sulfate (DMS; Sigma Aldrich) solution (diluted to 1/2, 1/5, 1/10 and 1/20, respectively, in 20% ethanol) or 1 μl of 1-cyclohexyl-3-(2-morpholinoethyl)-carbodiimide metho-p-toluenesulfonate (CMCT; Sigma Aldrich) solution (diluted to 2.5 mg/ml, 5 mg/ml, 10 mg/ml, 20 mg/ml and 50 mg/ml, respectively, in RNase/DNase-free water). Control reactions were done under equal conditions in the absence of DMS and CMCT. Following incubation for 5 min at room temperature, the reactions were terminated by ethanol precipitation in the presence of 1/10 volume 3 M sodium acetate (pH 5.2). Chemical modifications of specific nucleotides by DMS and CMCT, respectively, were determined by primer extension analysis. Briefly, reverse transcription reactions were performed using primer RcsR1-Random_Rev_PE. Aliquots of chemically modified RNAs were hybridized with 1–3 pmol of 5′-labeled primer (1–2 × 105 c.p.m.). Following a brief heating step (90°C, 2 min), the reaction was cooled slowly (5 min at 75°C, 10 min at 50°C, 5 min at 37°C, 10 min at room temperature). Next, the primer annealing mixture was used to set up a 20-μl reverse transcription reaction in 1 × SuperScript® III RTase reaction buffer supplemented with 170 units of SuperScript® III RTase (Invitrogen), 20 units RNaseOUT (Invitrogen) and 1 mM of each dNTP. The reaction was performed at 42°C for 50 min and then at 55°C for 60 min. Reactions were terminated by the addition of 1/10 vol of 3 M sodium acetate, pH 5.2, and 10 volumes of ice-cold ethanol. Following centrifugation, the pellets were washed with 70% ethanol. The dried pellets were resuspended in water and treated with DNase-free RNase A for 20 min at 37°C (0.2 mg/ml, Invitrogen). Next, PCR-grade proteinase K (Invitrogen) was added to a final concentration of 1 mg/ml and the reaction was incubated for another 15 min at 55°C. Reactions were stopped by adding Fu-mix (6 M urea, 80% deionized formamide, 1× TBE, 0.1% (w/v) Bromophenol blue, and 0.1% (w/v) Xylene cyanol). Reaction products were separated in TBE-buffered 10% polyacrylamide gels containing 7 M urea. Signals were visualized using a Typhoon 9200 imager (GE Healthcare) and analysed using Quantity One software (BioRad).
Bioinformatic methods
RNA structures were predicted by Mfold (55) and RNAfold (56) and visualized using VARNA (57). RNA-RNA interactions were predicted by IntaRNA (58) using the following input seed parameters: minimal number of base-pairs in seed, 6; maximal number of mismatches in seed, 2; maximal energy, 20; minimal unpaired probability 8 (each), 0; seed positions were not provided. For other parameters, default settings were used. For alignment of DNA and RNA sequences, T-Coffee (59) and LocaRNA (58) were used, respectively.
RESULTS
Overproduction of the attenuator sRNA rnTrpL decreases the level of the polycistronic trpDC mRNA
It was shown previously that, in RNA from S. meliloti 2011 cultures grown in the rich TY medium, the terminated attenuator sRNA rnTrpL can be detected in polyacrylamide gels as a 110-nt band (23,27). Since in E. coli, in medium containing an excess of Trp, approximately 15% of the transcription events proceed into the structural trp genes (60), we tested whether there is a trpLE(G) co-transcription in S. meliloti 2011 grown in TY. qRT-PCR analyses revealed co-transcription and suggested an approximately 45-fold higher steady-state level of rnTrpL when compared to trpLE(G) or trpE(G) (Supplementary Figure S1). Thus, in the TY medium routinely used in this work, transcription was efficiently terminated at the attenuator. As a result, the sRNA rnTrpL was accumulated, and was potentially available for regulation in trans, while the readthrough transcript was present at a low level only. To further characterize rnTrpL, we determined its stability in exponentially growing cultures in TY (Supplementary Figure S2). With a half-life of 7.6 ± 0.2 min, the decay of rnTrpL was faster than that of other S. meliloti sRNAs under comparable conditions (50).
Previously, constitutive rnTrpL overproduction from plasmid pRK-rnTrpL led to a decrease in the steady-state level of trpC mRNA (27). Here, we asked the question of whether or not rnTrpL overproduction affects the whole polycistronic operon ppiD-trpDC-moaC-moeA (from now on referred to as trpDC operon) (see Figure 1A). First, we confirmed higher levels for the sRNA rnTrpL in the overexpressing strain S. meliloti 2011 (pRK-rnTrpL) in comparison to the empty vector control strain (EVC) by Northern blot analysis (see Figure 1B and C). The length of the ectopically overproduced sRNA was similar to that of the native sRNA rnTrpL, which was transcribed from the chromosome. Then, we compared the mRNA levels of the trpDC operon in the overexpressing strain and the EVC by qRT-PCR and found that the levels of RT-PCR products covering the entire polycistronic mRNA were decreased (Figure 1D).
The apical part of rnTrpL stem-loop 1 is critically involved in controlling trpDC mRNA levels
Previous genetic analyses of the S. meliloti trp attenuator strongly suggested that it is able to form mutually exclusive RNA stem-loop structures: under conditions of efficient trpL translation or SL1 formation in the absence of translation, formation of the antiterminator SL2 is prevented, thus facilitating the formation of the terminator SL3 (23,61). However, there is currently no direct experimental support for the rnTrpL structure. We performed in vitro structural probing to determine experimentally the SL1 and SL3 structures that are expected to be formed in solution (Figure 2A). Based on the probing data, we provide a structure model (Figure 2B) with structural details for the terminator stem-loop. In addition, the model suggests that, with few structural rearrangements involving flexible regions, an alternative (anti-terminator) structure may be formed. According to this model, the sequence predicted to interact with trpD (from nt 21 to nt 55, see also Figure 4A) includes the apical region of SL1. In order to investigate the mechanism by which rnTrpL contributes to reducing trpDC mRNA levels, several mutations were introduced in this rnTrpL region.
Figure 4.
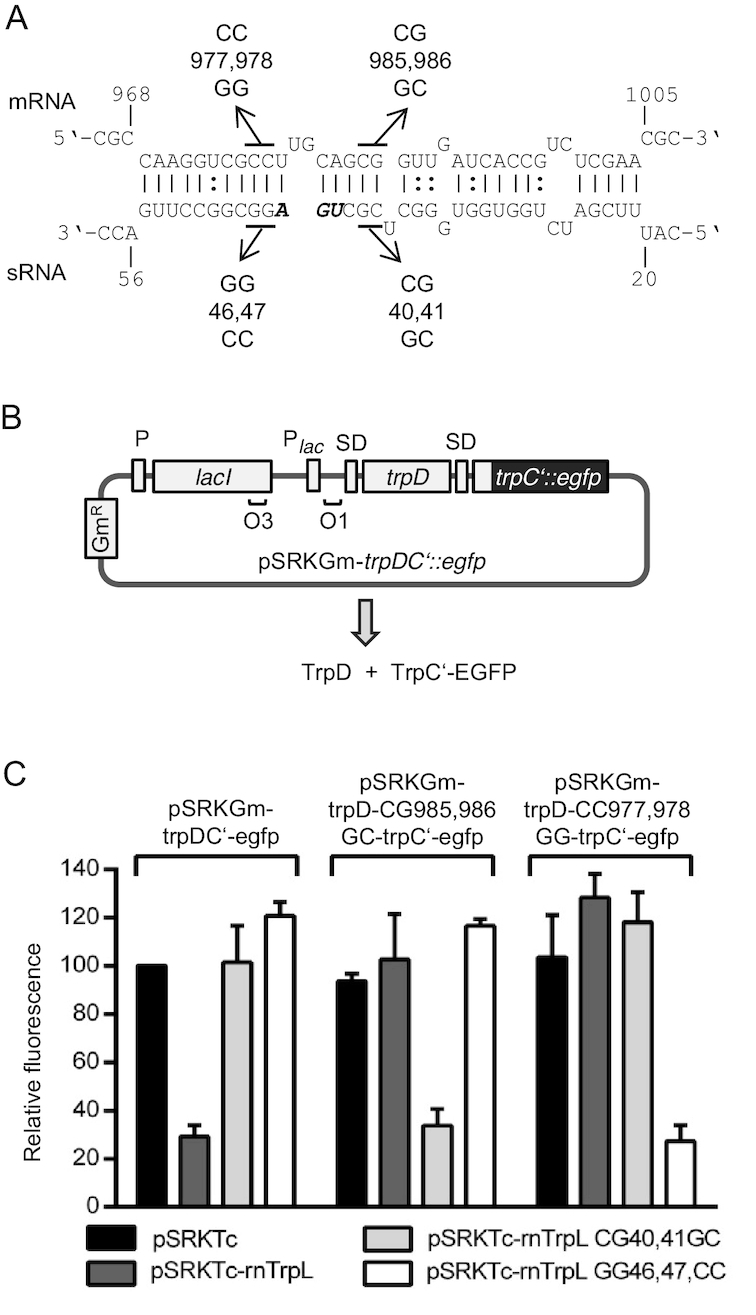
The attenuator sRNA rnTrpL base-pairs with trpD to down-regulate trpDC expression. (A) Scheme of the duplex structure predicted to be formed between trpD (mRNA) and rnTrpL (sRNA) (ΔG = –13.51 kcal/mol). rnTrpL mutations characterized in this experiment are given below the sRNA sequence. The trpL stop codon is shown in bold and italics. Compensatory trpD mutations used to restore base-pairing interactions (see C) are shown above the mRNA sequence. Nucleotide numbering starts at the translation start codon of the trpD mRNA and the sRNA harboring the trpL sORF, respectively. (B) Scheme of pSRKGm-trpDC’-egfp. The trpDC‘::egfp fusion contains the first 16 trpC codons fused to the third egfp codon. For more details, see Figure 3. (C) Analysis of possible base-pairing interactions between lacZ’-rnTrpL and the fusion mRNA trpDC’::egfp in strain 2011ΔtrpL. Plasmids used in this experiment are indicated. Fluorescence was measured at 20 min after induction with IPTG; the fluorescence obtained for strain 2011ΔtrpL(pSRKGm-trpDC’-egfp, pSRKTc) was set to 100% and used for normalization. Shown are the results from three independent experiments, each performed in duplicates (means and standard deviations are indicated).
The mutated rnTrpL derivatives were constitutively overproduced from pRK-plasmids in strain 2011. Mutations CG40,41GC and GG46,47CC located at or near the apical loop of SL1 (see Figure 2B) were not predicted to drastically change the secondary structure of rnTrpL, but were expected to weaken the predicted base-pairing interaction of SL1 with trpD (see Figure 4A). Also, we produced a mutant (AU1,2UA) in which the small ORF trpL (encoding the leader peptide) was eliminated. Using this mutant, we sought to address the question of whether translation of the liberated rnTrpL has any effect on the sRNA-mediated reduction of trpDC levels (see above) or whether the resulting 14-aa peptide translated from this sRNA (independently) affects trpDC mRNA levels (translation of the ectopically overproduced wild type (wt) rnTrpL was confirmed, see Supplementary Figure S3). A double mutant (AU1,2UA-G44C), in which the start and the stop codon of trpL were mutated, was also constructed. In addition, two mutant rnTrpL derivatives constructed previously (G44C and G67C, respectively) (27) were included in our analysis. All mutated rnTrpL derivatives and wt rnTrpL were confirmed to be overexpressed to similar levels (Supplementary Figure S3).
Next, we analyzed the effect of the mutated rnTrpL sRNAs on the steady-state level of the polycistronic trpDC mRNA using qRT-PCR. Figure 2C shows that all genes of the trpDC operon were affected in a similar manner. The sRNA rnTrpL-AU1,2UA and wt rnTrpL diminished the mRNA levels to a similar extent, suggesting that (under the conditions used in this experiment) ribosome occupancy of rnTrpL and/or the leader peptide were not important for the rnTrpL-mediated effects on trpDC mRNA. In contrast, all mutations that were predicted to weaken the base-pairing between rnTrpL and trpD (see Figure 4A), either abolished the sRNA-mediated downregulation or even led to increased mRNA levels (see the effect of the CG40,41GC mutation in Figure 2C). Further, rnTrpL-G67C, carrying a mutation in a region confirmed to specifically interact with sinI mRNA (27), had a readily detectable negative effect on trpC levels, very similar to that observed for the wt rnTrpL (Figure 2C), suggesting that this mutation did not impede the sRNA’s capability of affecting trpC levels. Altogether, these results support the idea that base-pairing interactions between the apical part of rnTrpL SL1 and trpDC mRNA are involved in mediating the observed decrease in the trpDC mRNA steady-state level upon rnTrpL overproduction.
Short-term overproduction of rnTrpL in a ΔtrpL background suggests a direct effect on trpDC
For further investigations in the absence of native rnTrpL RNA being transcribed from the chromosome, we constructed the deletion mutant 2011ΔtrpL, in which the original transcription start site (TSS) of trpLE(G) including the first two nucleotides (AT) was preserved (Figure 3A). To avoid secondary effects due to constitutive sRNA overproduction, we constructed a plasmid, pSRKGm-rnTrpL, for IPTG-inducible transcription of recombinant rnTrpL. In this plasmid, the trpL ORF of rnTrpL was fused in frame to a 38-nt lacZ mRNA leader harboring a ribosome-binding site, and transcription was terminated at SL3 of rnTrpL (Figure 3B). Figure 3C shows the induction of this rnTrpL derivative (designated lacZ’-rnTrpL) in strain 2011ΔtrpL (pSRKGm-rnTrpL).
Figure 3.

Short-term overproduction of recombinant lacZ’-rnTrpL transcript in ΔtrpL background decreases the level of trpDC mRNA. (A) Schematic representation of the trpE(G) locus in the deletion mutant 2011ΔtrpL. The original TSS including the first two nucleotides (AT) was preserved. (B) Scheme of pSRKGm-rnTrpL. The pSRK-based plasmids harbor the lac repressor gene lacI with its own promoter, the lacI-lacZ intergenic region, the lac-promoter Plac, and the 38-nt lacZ leader containing the Shine-Dalgarno sequence (SD), followed by an NdeI restriction site containing the ATG translation start codon. Positions of the LacI-binding operators O1 and O3, the proper spacing of which ensures tight regulation at Plac, are indicated (42). The rnTrpL sequence was cloned in frame in the NdeI site of pSRKGm. The resulting sRNA lacZ‘-rnTrpL contains the 38-nt lacZ-leader. The scheme shows the plasmid conferring resistance to gentamycin (Gm) (pSRKGm-rnTrpL). A similar plasmid conferring resistance to tetracycline (Tc) was also constructed. (C) Northern blot hybridization showing IPTG-induced lacZ’-rnTrpL transcription at 10 and 20 min post induction in strain 2011ΔtrpL (pSRKGm-rnTrpL) (lanes 3, 4 and 5). Data for control strains are shown in lanes 1 and 2. Also shown are the hybridization data using a 5S rRNA-specific probe (loading control). (D) qRT-PCR analysis of trpD, trpC and trpE (control) expression. At 10 min and 20 min post induction with IPTG, mRNA levels of the respective mRNA in 2011ΔtrpL (pSRKGm-rnTrpL) were measured and compared to the levels prior to induction. Bar C represents the mRNA levels in the constitutively overexpressing strain 2011 (pRK-rnTrpL) compared to those of the EVC. (E) qRT-PCR analysis of trpC and the control mRNA sinI in strain 2011 and the RNase mutant strains 2011rne and 2011Δrnc (indicated). Each strain contains the plasmid pSRKTc-rnTrpL. Changes in the levels of the respective mRNA were determined 10 min post induction with IPTG. For other details, see D). The graphs show results from three independent experiments, each performed in technical duplicates (mean values and standard deviations are indicated).
We also considered the possibility that the sRNA function of lacZ’-rnTrpL may be impaired due to an increased ribosome load compared to the leaderless rnTrpL transcribed from its native locus on the chromosome (23) or from pRK-rnTrpL (see Figure 1B and Supplementary Figure S3). However, we found that the level of trpDC mRNA was decreased in strain 2011ΔtrpL (pSRKGm-rnTrpL) already 10 min post induction, although the lacZ’-rnTrpL level was much lower than the rnTrpL level in the parental strain 2011 (Figure 3C and D). At 20 min post induction, the effect of lacZ’-rnTrpL was similar to the effect of constitutively overproduced rnTrpL. The induction of lacZ’-rnTrpL production had no detectable effect on the control mRNA trpE(G) (Figure 3D). This specific, short-term effect on trpDC provides additional support to the idea that trpDC mRNA is a direct target of rnTrpL.
To address the role of ribonucleases in the rnTrpL-mediated decrease of trpDC mRNA levels, the lacZ’-rnTrpL production was induced for 10 min in the RNase mutants 2011rne (27,62) and 2011Δrnc (63) using plasmid pSRKTc-rnTrpL, and changes in the levels of trpC mRNA were analyzed. In contrast to the parental strain 2011, in which trpC was confirmed to be decreased, the trpC levels were increased in both mutants (Figure 3E). As a control, we also analyzed the changes in sinI (Figure 3E). As expected, the sinI mRNA level was decreased upon rnTrpL induction in strain 2011 (27). A similar decrease was observed in the RNase III mutant 2011Δrnc, while an increase was detected in the RNase E mutant 2011rne (Figure 3E). The latter result is in line with the RNase E-dependent downregulation of sinI by rnTrpL (27). Together, these results support the importance of RNase E for posttranscriptional gene regulation by rnTrpL in trans, and suggest that RNase III may also participate in the regulation of trpDC, although it is dispensable for the regulation of sinI by rnTrpL.
sRNA rnTrpL base-pairs with trpD to destabilize the polycistronic trpDC mRNA
The predicted binding of rnTrpL to a region in the trpD coding sequence is shown in Figure 4A. To provide experimental evidence for the predicted base-pairing interaction, we performed in vivo assays in strain 2011ΔtrpL using bicistronic trpDC’::egfp reporter constructs and lacZ’-rnTrpL derivatives. The trpDC’::egfp fusions (Figure 4B) were expressed from pSRKGm (conferring Gmr) and challenged with wt or mutated lacZ’-rnTrpL transcribed from pSRKTc (conferring Tcr) (see Figure 3B). To avoid long-term effects, fluorescence was measured at 20 min after simultaneous induction of transcription of the reporter fusion construct and the sRNA.
Figure 4C shows that TrpC’-EGFP fluorescence derived from plasmid pSRKGm-trpDC’-egfp was strongly decreased if lacZ’-rnTrpL was coexpressed, supporting the idea that the sRNA binds to trpD and thereby induces a reduction of trpDC’::egfp mRNA levels. In contrast, if sRNA derivatives carrying CG40,41GC and CC46,47GG mutations, respectively, were co-expressed, no decrease in fluorescence was observed. To test whether this was due to a significantly reduced (or lack of) binding of the mutated sRNAs to trpD in the bicistronic reporter mRNA, appropriate mutations were introduced into the trpD binding site to restore the presumed base-pairing interactions (see Figure 4A). Indeed, a decrease in fluorescence produced by the pSRKGm-trpD-CG985,986GC-trpC’-egfp construct was (only) observed if the corresponding base-pairing sRNA lacZ’-rnTrpL-CG40,41GC was coexpressed. Consistent with this, the fluorescence produced by the pSRKGm-trpD-CC977,978GG-trpC’-egfp construct was (only) decreased if lacZ’-rnTrpL-GG46,47CC sRNA was coexpressed (Figure 4C). These results validate the base-pairing between rnTrpL and trpD and show that this interaction is responsible for the negative effect of rnTrpL on trpDC.
The negative effect of the sRNA rnTrpL on trpDC (a decrease in the steady-state level of the mRNA) can be explained by a destabilization of trpDC mRNA. To test whether rnTrpL affects the decay of this mRNA, half-life measurements were performed with strain 2011ΔtrpL (pSRKTc-rnTrpL). The stabilities of the polycistronic trpDC mRNA and the control mRNA trpE were measured without IPTG addition and 10 min post induction of lacZ’-rnTrpL overproduction. Figure 5 shows that, with primers specific for trpD and trpC, respectively, a decrease in the mRNA half-life upon rnTrpL induction was detectable, while the half-life of trpE mRNA remained unchanged. Similarly, mRNA destabilization upon rnTrpL induction was detected with primers specific for ppiD, moaC and moeA (Supplementary Figure S4). The ppiD- and trpD-specific primers bind upstream of the rnTrpL-binding site in the polycistronic mRNA, while the trpC-, moaC- and moeA-specific primers target downstream regions. Shorter half-lives of all transcript parts upon rnTrpL overproduction is consistent with the proposed destabilization of the polycistronic trpDC mRNA after base-pairing between rnTrpL and trpD.
Figure 5.
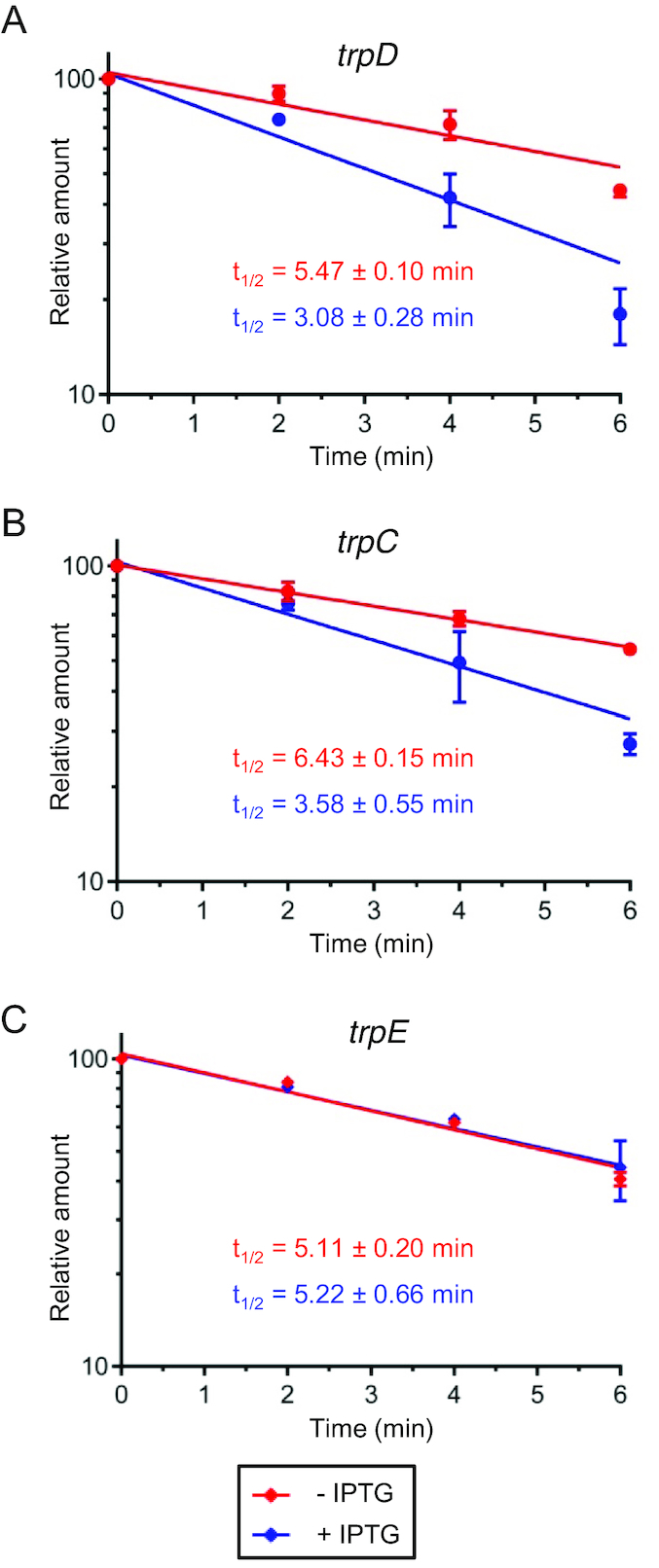
The sRNA rnTrpL destabilizes the polycistronic trpDC mRNA. At an OD600nm of 0.5, one half of a liquid culture of strain 2011 ΔtrpL (pSRKTc-rnTrpL) was supplemented with IPTG for 10 min to induce lacZ‘-rnTrpL production, while the other half was incubated without IPTG. Then, rifampicin was added to both cultures and RNA was isolated at time points 0, 2, 4 and 6 min after rifampicin addition. Relative mRNA levels in induced (blue) and uninduced cultures (red) were determined by qRT-PCR analysis and half-lives (t1/2, in min) were calculated from three independent experiments, each with two technical replicates. The analysis was performed with primers specific for trpD (A), trpC (B) and trpE (C). The analysis with ppiD-, moaC- and moeA-specific primers is shown in Supplementary Figure S4.
sRNA rnTrpL down-regulates trpDC expression under Trp sufficiency conditions
Finally, we addressed the question of whether the attenuator sRNA rnTrpL is involved in trpDC regulation in response to changes in cellular Trp availability. To assess transcription attenuation between trpL and trpE(G), we constructed a 2011ΔtrpC mutant to prevent cellular Trp production and used minimal medium with defined Trp concentrations. According to previous data (23), during growth in a medium supplemented with 20 μg/ml Trp (Trp sufficiency condition), transcription is regularly terminated between trpL and trpEG, and the sRNA rnTrpL is liberated. In contrast, during growth at 2 μg/ml Trp (Trp insufficiency), transcription termination is abolished and trpL is cotranscribed with trpE(G). Indeed, Figure 6A shows that the sRNA rnTrpL was readily detectable by Northern blot analysis when 2011ΔtrpC was grown at Trp sufficiency. Under these conditions, readthrough transcripts were present at a very low level (Supplementary Figure S1). Large trpLE(G) transcripts could not be detected because the protocol used in this experiment was optimized for efficient separation and blotting of small RNAs. When the cells were washed in medium without Trp and subsequently grown for 4 h in medium supplemented with 2 μg/ml Trp, the level of rnTrpL was steadily decreased. 10 min after restoring Trp sufficiency, the sRNA rnTrpL level was increased as expected (Figure 6A). Under these conditions, accumulation of a small degradation product of the trpLE(G) transcript was detected (see also Supplementary Figure S5).
Figure 6.

The sRNA rnTrpL down-regulates trpDC in response to Trp availability. (A) Northern blot analysis of rnTrpL, showing transcription attenuation in strain 2011ΔtrpC which was grown first in minimal medium (MM) containing 20 μg/ml Trp and then transferred to MM containing 2 μg/ml Trp for 0, 1, 2 and 4 h, respectively. Depletion of the cellular Trp pool causes cotranscription of trpL and trpE(G) and a decrease in the level of the sRNA rnTrpL. 4 h 10 min, following incubation with 2 μg/ml Trp for 4 h, the Trp concentration was increased to 20 μg/ml for 10 min. Increased Trp availability led to transcription termination at SL3 of rnTrpL and an immediate increase in the level of the sRNA rnTrpL. The band migrating slower than rnTrpL was identified as a degradation product (degr.) of the trpLE(G) cotranscript (see Supplementary Figure S5). On the left, RNA isolated from the indicated control strains grown in media with 20 μg/ml Trp was loaded. All lanes shown in this panel originate from the same blot. (B) Changes in trpE, trpD and rnTrpL RNA levels in response to Trp availability in the indicated strains. Relative trpD and trpE mRNA levels were measured by qRT-PCR using RNA samples from Trp availability experiments (as shown in panel A). The levels measured at the indicated time points were compared to the level determined at time point 0. (C) Changes in ppiD, moaC and moeA RNA levels in response to Trp availability in the indicated strains. For details, see B). (D) Changes in trpF, trpB and trpA mRNA levels in response to Trp availability in the indicated strains. For details, see B). All mRNA graphs show means and standard deviations from three independent experiments, each performed in duplicates. The rnTrpL graph in B) shows a quantification of the signals from panel A).
To analyze the role of rnTrpL in trans in the context of transcription attenuation, we additionally constructed a control strain, the double mutant 2011ΔtrpCΔtrpL. Strains 2011ΔtrpC and 2011ΔtrpCΔtrpL were used to detect changes in the levels of the polycistronic ppiD-trpD-ΔtrpC-moaC-moeA mRNA and trpE(G) mRNA in response to changes in cellular Trp availability. The experimental setup was as described above (transfer from minimal medium containing 20 μg/ml Trp to medium containing 2 μg/μl Trp, incubation for 4 h with sample collection at one-hour intervals and, at the end, incubation with 20 μg/ml Trp for another 10 min). Upon Trp insufficiency, when the level of the released sRNA rnTrpL was reduced, the level of the polycistronic ppiD-trpD-ΔtrpC-moaC-moeA mRNA was found to be increased in strain 2011ΔtrpC (Figure 6B and C). Importantly, 10 min after restoring Trp sufficiency, when the level of the sRNA rnTrpL was increased again, the level of the polycistronic mRNA was decreased (Figure 6B and C). Similar changes were observed for trpE (Figure 6B). Notably, such changes were not observed in the double mutant 2011ΔtrpCΔtrpL (Figure 6B and C), suggesting a posttranscriptional downregulation of the trpDC operon by the liberated sRNA rnTrpL in response to Trp availability. In addition, these data lead us to conclude that, similarly to trpE(G) (23), the mRNA level of the trpDC operon is not regulated at the level of transcription initiation in response to Trp availability, but posttranscriptionally by rnTrpL.
The trpFBA operon is not regulated by the sRNA rnTrpL
In contrast to the trpDC operon, trpFBA was not predicted to interact with rnTrpL in a previous analysis of putative conserved targets of this sRNA (27). Here, we analyzed trpB mRNA and found that its level was not changed 10 min post induction of lacZ’-rnTrpL in strain 2011ΔtrpL (log2FC = 0.12 ± 0.29), suggesting that trpFBA mRNA is not a target of rnTrpL.
Next, we asked the question of whether the expression of the trpFBA operon is regulated in response to Trp availability. A qRT-PCR analysis of trpF, trpB and trpA was performed with strains 2011ΔtrpC and 2011ΔtrpCΔtrpL grown in minimal medium containing different Trp concentrations as described above. Growth of strain 2011ΔtrpC in medium containing 2 μg/ml Trp for 4 h led to increasing trpFBA mRNA levels while, at 10 min after addition of 20 μg/ml Trp to this Trp-starved culture, the level of trpFBA was decreased (Figure 6D). These changes show that trpB is regulated in response to Trp availability. However, in contrast to what was observed for the trpE(G) and trpDC operons (Figure 6B and C), similar trpFBA changes were also detected in the double mutant 2011ΔtrpCΔtrpL. The trpF, trpB and trpA values obtained for the two strains 2011ΔtrpC and 2011ΔtrpCΔtrpL were not statistically different (Figure 6D), indicating that regulation of the trpFBA operon in response to Trp availability does not depend on the attenuator sRNA rnTrpL.
In summary, Figure 6 shows that all three trp operons are regulated in response to Trp availability. However, while the regulation of the mRNA levels of the two operons trpE(G) and trpDC was revealed to critically involve rnTrpL, the regulation of trpFBA proved to be independent of rnTrpL.
Conservation of the trans-regulating function of rnTrpL on the expression of trp genes
Many Alphaproteobacteria have three trp operons with gene orders that are identical or very similar to that seen in S. meliloti (3). Among them are the plant pathogen A. tumefaciens and the soybean symbiont B. japonicum. In the three alphaproteobacterial species S. meliloti, A. tumefaciens and B. japonicum, transcription of the attenuator sRNA (and of the bicistronic transcript trpLE(G)) starts with the ATG codon of trpL (64–66) (see Supplementary Figure S6). To address the question of whether trp genes are regulated in trans by the rnTrpL homolog in these species, the sequences corresponding to Atu-rnTrpL and Bj-rnTrpL, respectively, were cloned in pSRKTc and pRJ-MCS, respectively. The effect of sRNA overproduction in the corresponding homologous host on the mRNA levels of trpD and trpB were analyzed by qRT-PCR. We found decreased trpD mRNA levels and no change in the levels of trpB upon overexpression of the attenuator sRNA in both A. tumefaciens and B. japonicum (Figure 7A). This indicates that, similarly to what was observed in S. meliloti, trpDC is down-regulated by the attenuator sRNA in trans, while trpFBA is not regulated by this sRNA. To predict base-pairing interactions between A. tumefaciens trpDC and Atu-rnTrpL, as well as between B. japonicum trpDC and Bj-rnTrpL, we used IntaRNA (58). Potential interactions were predicted between SL1 of the respective attenuator RNA and the 3′-region of the respective trpD (Figure 7B; see also Supplementary Figures S7 and S8). Altogether, these results suggest that trpDC regulation by rnTrpL is conserved in the families Rhizobiaceae and Bradyrhizobiaceae, to which the studied Alphaproteobacteria belong.
Figure 7.
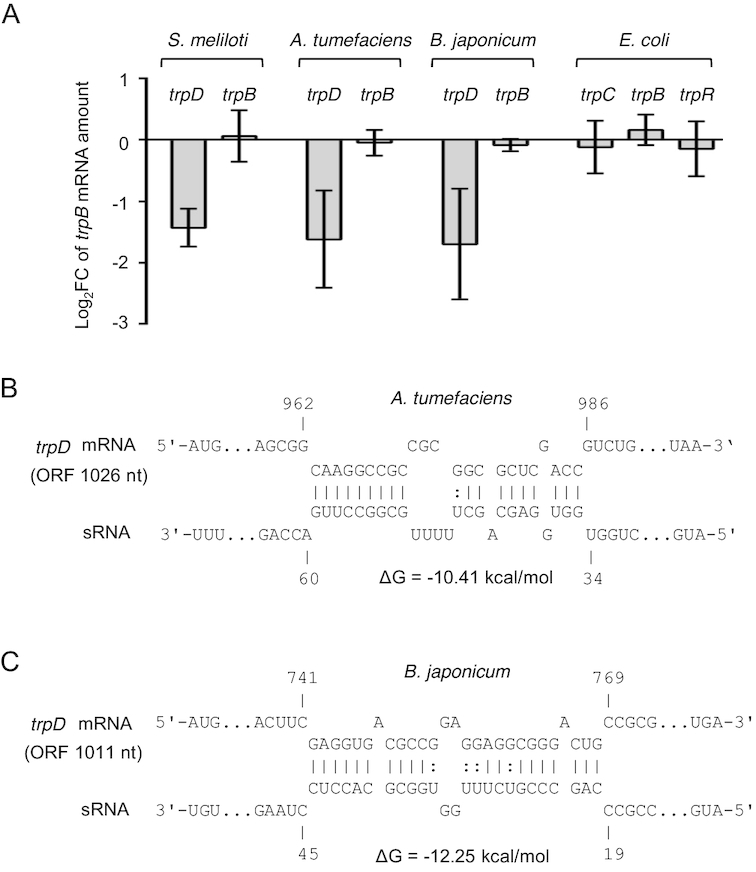
Conservation of trpDC regulation by rnTrpL in trans. (A) qRT-PCR analysis of the indicated mRNAs and bacterial species. For S. meliloti (pSRKTc-rnTrpL), A. tumefaciens (pSRKTc-Atu-rnTrpL) and E. coli (pSRKTc-Ec-rnTrpL), changes in the levels of the indicated mRNAs were analyzed 10 min post induction. In the constitutively overexpressing B. japonicum (pRJ-Bj-rnTrpL), the levels of trpD and trpB were compared to the EVC. (B) Schematic representations of the duplex structures predicted to be formed between trpDC and rnTrpL homologs in A. tumefaciens and B. japonicum. The numbering starts at the translation start codons of trpD mRNA and the trpL ORF in the sRNAs, respectively. Predicted secondary structures of the sRNAs Atu-rnTrpL and Bj-rnTrpL are shown in Supplementary Figures S7 and S8.
To address the conservation of rnTrpL function beyond Alphaproteobacteria, we constructed plasmid pSRKTc-Ec-rnTrpL and tested whether overproduction of the attenuator RNA Ec-rnTrpL for 10 min affects the level of trpC, trpB and of the separately transcribed Trp repressor mRNA trpR (3) in E. coli. The levels of trpC, trpB and trpR were not changed significantly upon Ec-rnTrpL overproduction (Figure 7A), suggesting that this sRNA does not regulate trp genes in trans.
In summary, our data show that in bacteria with trp genes organized in several operons, the attenuator sRNA rnTrpL acts in trans to regulate the trpDC operon posttranscriptionally (Figure 8).
Figure 8.
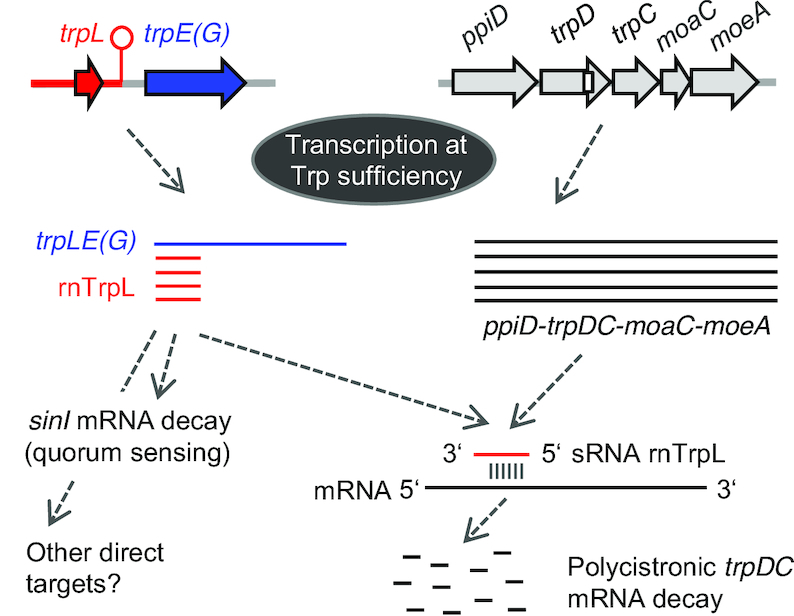
Model of gene regulation in trans by the attenuator sRNA rnTrpL in S. meliloti. At TrpL sufficiency, transcription is generally terminated between trpL and trpE(G). The liberated attenuator sRNA rnTrpL binds to trpD and destabilizes the polycistronic mRNA ppiD-trpDC-moaC-moeA. In this way, the trp attenuator posttranscriptionally coordinates the expression of the two trp operons trpE(G) and trpDC in response to Trp availability. In addition, the destabilization of the polycistronic trpDC mRNA by rnTrpL probably affects protein translocation through the outer membrane (ppiD) and MoCo biosynthesis (moaC-moeA). Moreover, the sRNA rnTrpL binds to and destabilizes sinI (27), thus probably linking quorum sensing to Trp availability.
DISCUSSION
In this study, we demonstrate that a bacterial sRNA derived by ribosome-dependent transcription attenuation acts by base-pairing in trans. According to our data, the liberated attenuator RNA of the trpE(G) operon down-regulates the expression of the trpDC operon in S. meliloti and other bacteria.
The gene trpE(G) encodes anthranilate synthase that catalyzes the first step of Trp biosynthesis starting with chorismate, while trpD and trpC encode anthranilate phosphoribosyltransferase and indole-3-glycerol phosphate synthase that catalyze the two subsequent reactions. The remaining enzymes of the pathway, phosphoribosylanthranilate isomerase and Trp synthase are encoded by the trpFBA operon. Our data show that ectopic oveproduction of rnTrpL does not influence trpFBA but decreases the steady-state level of the polycistronic trpDC mRNA, in line with the predicted base-pairing between rnTrpL and trpD. Consistent with a direct effect of rnTrpL on trpDC mRNA, a decrease of the trpC level was detected as early as 10 min after induction of lacZ’-rnTrpL in the ΔtrpL background (Figure 3). The results shown in Figure 4C and Figure 5 provide strong support for this hypothesis. Thus, for example, a base complementarity between specific rnTrpL and trpD sequences proved to be required for trpDC downregulation (Figure 4C). Furthermore, parts of the polycistronic mRNA located upstream (in ppiD and trpD) and downstream (in trpC, moaC and moeA) of the rnTrpL binding site were destabilized upon lacZ’-rnTrpL induction (Figure 5 and Supplementary Figure S4). The latter result shows that rnTrpL acts at the level of trpDC mRNA (rather than DNA, for example, by disturbing transcription). Based on the data presented in this study, we conclude that the attenuator sRNA rnTrpL base-pairs with the distal part of the trpD coding region in the polycistronic trpDC mRNA and thereby promotes degradation of the entire transcript (see the model in Figure 8).
Upon sRNA binding to an mRNA in gram-negative bacteria, an endoribonuclease, such as RNase III or RNase E, is recruited. After an initial endonucleolytic cleavage, RNA fragments are degraded by (other) exo- and endoribonucleases, leading to rapid decay of both the sRNA and the mRNA. Alternatively, inhibition of translation by the sRNA results in unprotected mRNA regions that are endonucleolytically cleaved, mostly by RNase E (9,67), a mechanism suggested for the action of rnTrpL on sinI mRNA (27). According to Figure 3E, both RNase E and RNase III are important for the rnTrpL-mediated downregulation of trpDC. Future analyses are needed to elucidate whether these RNases directly participate in the destabilization of the trpDC message by rnTrpL. Further, according to Figures 2 and 4, the apical region of SL1 of rnTrpL is critically involved in the base-pairing interaction with trpD, suggesting that rnTrpL may need an RNA chaperone for efficient binding to this target. Since rnTrpL was described as an Hfq-independent sRNA (27), and since S. meliloti does not harbor ProQ (13), another RNA-binding protein that interacts with rnTrpL may be identified in the future.
The physiological role of rnTrpL as an attenuator RNA in cis and base-pairing sRNA in trans is an intriguing example for the evolution of bacterial operons and their co-regulation. Although bacteria belonging to Escherichia, Salmonella, Vibrio and most members of Bacillales and Lactobacillales have all the trp genes in a single operon, most other bacteria (Alpha-, Beta-, Gamma-, Delta-, and Epsilonproteobacteria, Spirochaeta and most Actinobacteriadae) carry trp genes that are organized into several operons. It has been proposed that this allows for more flexibility in the regulation of the genes involved in each step of Trp synthesis since, in different bacteria, specific Trp precursors have additional functions and are linked to different metabolic pathways (3). However, as already mentioned in the introduction, the high costs of Trp synthesis could pose a selection pressure for evolving RNA-based mechanisms suitable to mediate an appropriate response to varying Trp concentrations in the cell. This is in an apparent contradiction to the observation that, in bacteria with split trp operons, only one of them (usually trpE) is equipped with an mRNA leader that mediates transcription attenuation. Our finding that, in Sinorhizobium, Agrobacterium and Bradyrhizobium, rnTrpL is capable to regulate trpDC in trans provides an example of how bacteria evolved posttranscriptional strategies to coordinate the expression of trp operons according to Trp availability (Figure 8). In contrast, we did not detect posttranscriptional regulation by ectopically overexpressed Ec-rnTrpL in E. coli where all the trp biosynthesis genes are co-transcribed (Figure 7A).
Our data show that the sRNA rnTrpL can downregulate the entire trpDC operon including ppiD, moaC and moeA (Figure 1, Figure 2, Figure 5 and Supplementary Figure S4) The gene ppiD encodes peptidyl-prolyl isomerase required for folding of outer membrane proteins/protein translocation (68,69), while moaC and moeA encode enzymes required for the first and the last step in the biosynthesis of the molibdenum cofactor MoCo (70). This pterin-based cofactor is the active compound present in the catalytic site of all molybdenum-containing enzymes except for the bacterial molybdenum nitrogenase (71). The MoCo biosynthesis is highly conserved in all domains of life and enzymes requiring this cofactor are necessary for carbon, nitrogen, and sulfur metabolism (72). Because the trpLE(G) and ppiD-trpDC-moaC-moeA operons are conserved in many Alphaproteobacteria and because rnTrpL overexpression downregulates trpD in A. tumefaciens and B. japonicum, we propose that the specific regulatory function of rnTrpL revealed in this study is widely conserved. It is tempting to suggest that, in bacteria with trp genes organized in several operons, the Trp sufficiency status may influence not only the cellular Trp biosynthesis but also other functions, such as protein translocation through the outer membrane and MoCo-dependent metabolic processes.
It is also noteworthy that, in S. meliloti, rnTrpL can downregulate the autoinducer synthase gene sinI (27), thereby probably connecting Trp availability to quorum sensing. According to our data, the sRNA rnTrpL is available for regulation in trans under conditions of Trp sufficiency. Under such conditions, destabilization of sinI mRNA by rnTrpL may impede the autoinducer accumulation and, as a consequence, quorum sensing may begin at higher population density than under conditions of Trp insufficiency. In line with this, it was recently shown that quorum sensing begins at higher population density, when S. meliloti cultures are supplied with water-soluble humic materials as additional nutrient sources (73). Indeed, quorum sensing is known to be influenced by environmental factors in addition to the population density (74). Previously, it was shown that, during growth in TY medium, the steady-state level of rnTrpL is influenced by temperature and salt (27). The effects of these factors on rnTrpL generation and/or decay as well as the regulation of the rnTrpL targets remain to be elucidated.
It was predicted that, in addition to sinI and trpD, mRNAs of several regulatory genes can base-pair with rnTrpL (27). Thus, it is possible that this attenuator sRNA is at the center of a regulatory network that responds to Trp availability as a marker indicating the nutrient status of the cell. Under conditions of Trp sufficiency, the signal (charged tRNA-Trp) is the critical regulator of ribosome-mediated transcription attenuation, while the liberated attenuator sRNA may get involved in regulatory functions in trans by base-pairing with specific mRNAs in addition to trpD (Figure 8).
As one of the rare examples of 5′-UTR-derived sRNAs, the trans-acting attenuator RNA rnTrpL characterized in this study highlights the regulatory potential and the versatility of bacterial mRNA leaders.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Marta Robledo and José I. Jiménez-Zurdo (Granada, Spain) for sending us the RNase III mutant strain, and Matthew McIntosh (Giessen, Germany) for fruitful discussions.
Authors contributions: Conceptualization, E.E.H., H.M.; Methodology, E.E.H., H.M., R.M.; Investigation, H.M., S.L., R.M., M.S., S.A., S.B.W., K.B.; Formal analysis, H.M., S.L., R.M; Writing – Original Draft, E.E.H.; Writing – Review and Editing, E.E.H., J.Z., H.M., R.M., S.L.; Visualization, E.E.H., H.M., M.S., S.L., R.M.; Supervision, E.E.H. and J.Z.; Funding acquisition, E.E.H.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Deutsche Forschungsgemeinschaft [Ev42/6–1 to E.E.H.; RTG 2355 to E.E.H; SFB1021, A01 to J.Z.]; China Scholarship Council [No. 2017080800082 to S.L.]. Funding for open access charge: Deutsche Forschungsgemeinschaft [Ev42/6-1] and Justus Liebig University.
Conflict of interest statement. None declared.
REFERENCES
- 1. Bertrand K., Korn L., Lee F., Platt T., Squires C.L., Squires C., Yanofsky C.. New features of the regulation of the tryptophan operon. Science. 1975; 189:22–26. [DOI] [PubMed] [Google Scholar]
- 2. Yanofsky C. Attenuation in the control of expression of bacterial operons. Nature. 1981; 289:751–758. [DOI] [PubMed] [Google Scholar]
- 3. Merino E., Jensen R.A., Yanofsky C.. Evolution of bacterial trp operons and their regulation. Curr. Opin. Microbiol. 2008; 11:78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lee F., Yanofsky C.. Transcription termination at the trp operon attenuators of Escherichia coli and Salmonella typhimurium: RNA secondary structure and regulation of termination. Proc. Natl. Acad. Sci. U.S.A. 1977; 74:4365–4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Keller E.B., Calvo J.M.. Alternative secondary structures of leader RNAs and the regulation of the trp, phe, his, thr, and leu operons. Proc. Natl. Acad. Sci. U.S.A. 1979; 76:6186–6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vitreschak A.G., Lyubetskaya E.V., Shirshin M.A., Gelfand M.S., Lyubetsky V.A.. Attenuation regulation of amino acid biosynthetic operons in proteobacteria: comparative genomics analysis. FEMS Microbiol. Lett. 2004; 234:357–370. [DOI] [PubMed] [Google Scholar]
- 7. Gollnick P., Babitzke P., Antson A., Yanofsky C.. Complexity in regulation of tryptophan biosynthesis in Bacillus subtilis. Annu. Rev. Genet. 2005; 39:47–68. [DOI] [PubMed] [Google Scholar]
- 8. Gutierrez-Preciado A., Jensen R.A., Yanofsky C., Merino E.. New insights into regulation of the tryptophan biosynthetic operon in Gram-positive bacteria. Trends Genet. 2005; 21:432–436. [DOI] [PubMed] [Google Scholar]
- 9. Waters L.S., Storz G.. Regulatory RNAs in bacteria. Cell. 2009; 136:615–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sharma C.M., Darfeuille F., Plantinga T.H., Vogel J.. A small RNA regulates multiple ABC transporter mRNAs by targeting C/A-rich elements inside and upstream of ribosome-binding sites. Genes Dev. 2007; 21:2804–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Storz G., Vogel J., Wassarman K.M.. Regulation by small RNAs in bacteria: expanding frontiers. Mol. Cell. 2011; 43:880–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Feng L., Rutherford S.T., Papenfort K., Bagert J.D., van Kessel J.C., Tirrell D.A., Wingreen N.S., Bassler B.L.. A qrr noncoding RNA deploys four different regulatory mechanisms to optimize quorum-sensing dynamics. Cell. 2015; 160:228–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Smirnov A., Wang C., Drewry L.L., Vogel J.. Molecular mechanism of mRNA repression in trans by a ProQ-dependent small RNA. EMBO J. 2017; 36:1029–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Altuvia S., Zhang A., Argaman L., Tiwari A., Storz G.. The Escherichia coli OxyS regulatory RNA represses fhlA translation by blocking ribosome binding. EMBO J. 1998; 17:6069–6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Massé E., Escorcia F.E., Gottesman S.. Coupled degradation of a small regulatory RNA and its mRNA targets in Escherichia coli. Genes Dev. 2003; 17:2374–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chao Y., Papenfort K., Reinhardt R., Sharma C.M., Vogel J.. An atlas of Hfq-bound transcripts reveals 3′ UTRs as a genomic reservoir of regulatory small RNAs. EMBO J. 2012; 31:4005–4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mellin J.R., Tiensuu T., Becavin C., Gouin E., Johansson J., Cossart P.. A riboswitch-regulated antisense RNA in Listeria monocytogenes. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:13132–13137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. De Lay N.R., Garsin D.A.. The unmasking of ‘junk’ RNA reveals novel sRNAs: from processed RNA fragments to marooned riboswitches. Curr. Opin. Microbiol. 2016; 30:16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Loh E., Dussurget O., Gripenland J., Vaitkevicius K., Tiensuu T., Mandin P., Repoila F., Buchrieser C., Cossart P., Johansson J.. A trans-acting riboswitch controls expression of the virulence regulator PrfA in Listeria monocytogenes. Cell. 2009; 139:770–779. [DOI] [PubMed] [Google Scholar]
- 20. DebRoy S., Gebbie M., Ramesh A., Goodson J.R., Cruz M.R., van Hoof A., Winkler W.C., Garsin D.A.. Riboswitches. A riboswitch-containing sRNA controls gene expression by sequestration of a response regulator. Science. 2014; 345:937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mellin J.R., Koutero M., Dar D., Nahori M.A., Sorek R., Cossart P.. Riboswitches. Sequestration of a two-component response regulator by a riboswitch-regulated noncoding RNA. Science. 2014; 345:940–943. [DOI] [PubMed] [Google Scholar]
- 22. Johnston A.W., Bibb M.J., Beringer J.E.. Tryptophan genes in Rhizobium - their organization and their transfer to other bacterial genera. Mol. Gen. Genet. 1978; 165:323–330. [DOI] [PubMed] [Google Scholar]
- 23. Bae Y.M., Crawford I.P.. The Rhizobium meliloti trpE(G) gene is regulated by attenuation, and its product, anthranilate synthase, is regulated by feedback inhibition. J. Bacteriol. 1990; 172:3318–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Becker A., Overlöper A., Schlüter J.P., Reinkensmeier J., Robledo M., Giegerich R., Narberhaus F., Evguenieva-Hackenberg E.. Riboregulation in plant-associated α-proteobacteria. RNA Biol. 2014; 11:550–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Torres-Quesada O., Millán V., Nisa-Martínez R., Bardou F., Crespi M., Toro N., Jiménez-Zurdo J.I.. Independent activity of the homologous small regulatory RNAs AbcR1 and AbcR2 in the legume symbiont Sinorhizobium meliloti. PLoS One. 2013; 8:e68147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Robledo M., Peregrina A., Millán V., García-Tomsig N.I., Torres-Quesada O., Mateos P.F., Becker A., Jiménez-Zurdo J.I.. A conserved α-proteobacterial small RNA contributes to osmoadaptation and symbiotic efficiency of rhizobia on legume roots. Environ. Microbiol. 2017; 19:2661–2680. [DOI] [PubMed] [Google Scholar]
- 27. Baumgardt K., Šmídová K., Rahn H., Lochnit G., Robledo M., Evguenieva-Hackenberg E.. The stress-related, rhizobial small RNA RcsR1 destabilizes the autoinducer synthase encoding mRNA sinI in Sinorhizobium meliloti. RNA Biol. 2016; 13:486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Robledo M., Frage B., Wright P.R., Becker A.. A stress-induced small RNA modulates alpha-rhizobial cell cycle progression. PLoS Genet. 2015; 11:e1005153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Robledo M., Schlüter J.P., Loehr L.O., Linne U., Albaum S.P., Jiménez-Zurdo J.I., Becker A.. An sRNA and cold shock protein homolog-based feedforward loop post-transcriptionally controls cell cycle master regulator CtrA. Front. Microbiol. 2018; 29:763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sambrook J., Fritsch E.F., Maniatis T.. Molecular Cloning: A Laboratory Manual. 2. 1989; NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 31. Casse F., Boucher C., Julliot J.S., Michel M., Denarie J.. Identification and characterization of large plasmids in Rhizobium meliloti using agarose-gel electrophoresis. Gen. Microbiol. 1979; 113:229–242. [Google Scholar]
- 32. Young J.M. The genus name Ensifer Casida 1982 takes priority over Sinorhizobium Chen et al. 1988, and Sinorhizobium morelense Wang et al. 2002 is a later synonym of Ensifer adhaerens Casida 1982. Is the combination ‘Sinorhizobium adhaerens’ (Casida 1982) Willems et al. 2003 legitimate? Request for an Opinion. Int. J. Syst. Evol. Microbiol. 2003; 53:2107–2110. [DOI] [PubMed] [Google Scholar]
- 33. Cha C., Gao P., Chen Y.C., Shaw P.D., Farrand S.K.. Production of acyl-homoserine lactone quorum-sensing signals by gram-negative plantassociated bacteria. Mol. Plant Microbe Interact. 1998; 11:1119–1129. [DOI] [PubMed] [Google Scholar]
- 34. Lassalle F., Campillo T., Vial L., Baude J., Costechareyre D., Chapulliot D., Shams M., Abrouk D., Lavire C., Oger-Desfeux C. et al.. Genomic species are ecological species as revealed by comparative genomics in Agrobacterium tumefaciens. Genome Biol. Evol. 2011; 3:762–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Beringer J.E. R factor transfer in Rhizobium leguminosarum. J. Gen. Microbiol. 1974; 84:188–198. [DOI] [PubMed] [Google Scholar]
- 36. Schlüter J.P., Reinkensmeier J., Daschkey S., Evguenieva-Hackenberg E., Janssen S., Jänicke S., Becker J.D., Giegerich R., Becker A.. A genome-wide survey of sRNAs in the symbiotic nitrogen-fixing alpha-proteobacterium Sinorhizobium meliloti. BMC Genomics. 2010; 11:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Regensburger B., Hennecke H.. RNA polymerase from Rhizobium japonicum. Arch. Microbiol. 1983; 135:103–109. [DOI] [PubMed] [Google Scholar]
- 38. Delamuta J.R., Ribeiro R.A., Ormeño-Orrillo E., Melo I.S., Martínez-Romero E., Hungria M.. Polyphasic evidence supporting the reclassification of Bradyrhizobium japonicum group Ia strains as Bradyrhizobium diazoefficiens sp. nov. Int. J. Syst. Evol. Microbiol. 2013; 63:3342–3351. [DOI] [PubMed] [Google Scholar]
- 39. Mesa S., Hauser F., Friberg M., Malaguti E., Fischer H.M., Hennecke H.. Comprehensive assessment of the regulons controlled by the FixLJ-FixK2-FixK1 cascade in Bradyrhizobium japonicum. J. Bacteriol. 2008; 190:6568–6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yanisch-Perron C., Vieira J., Messing J.. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 1985; 33:103–119. [DOI] [PubMed] [Google Scholar]
- 41. Mank N.N., Berghoff B.A., Hermanns Y.N., Klug G.. Regulation of bacterial photosynthesis genes by the small noncoding RNA PcrZ. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:16306–16311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Khan S.R., Gaines J., Roop R.M, Farrand S.K.. Broad-host-range expression vectors with tightly regulated promoters and their use to examine the influence of TraR and TraM expression on Ti plasmid quorum sensing. Appl. Environ. Microbiol. 2008; 74:5053–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schäfer A., Tauch A., Jäger W., Kalinowski J., Thierbach G., Pühler A.. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene. 1994; 145:69–73. [DOI] [PubMed] [Google Scholar]
- 44. Hahn J., Thalmann S., Migur A., von Boeselager R.F., Kubatova N., Kubareva E., Schwalbe H., Evguenieva-Hackenberg E.. Conserved small mRNA with an unique, extended Shine-Dalgarno sequence. RNA Biol. 2017; 14:1353–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McIntosh M., Krol E., Becker A.. Competitive and cooperative effects in quorum-sensing-regulated galactoglucan biosynthesis in Sinorhizobium meliloti. J. Bacteriol. 2008; 190:5308–5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Simon R., Priefer U., Pühler A.. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram-negative bacteria. Biotechnology. 1982; 1:784–791. [Google Scholar]
- 47. Mongiardini E.J., Quelas J.I., Dardis C., Althabegoiti M.J., Lodeiro A.R.. Transcriptional control of the lateral-flagellar genes of Bradyrhizobium diazoefficiens. J. Bacteriol. 2017; 199:e00253-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bustin S.A., Benes V., Garson J.A., Hellemans J., Huggett J., Kubista M., Mueller R., Nolan T., Pfaffl M.W., Shipley G.L. et al.. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009; 55:611–622. [DOI] [PubMed] [Google Scholar]
- 49. Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001; 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Voss B., Hölscher M., Baumgarth B., Kalbfleisch A., Kaya C., Hess W.R., Becker A., Evguenieva-Hackenberg E.. Expression of small RNAs in Rhizobiales and protection of a small RNA and its degradation products by Hfq in Sinorhizobium meliloti. Biochem. Biophys. Res. Commun. 2009; 390:331–336. [DOI] [PubMed] [Google Scholar]
- 51. Jeng S.T., Gardner J.F., Gumport R.I.. Transcription termination in vitro by bacteriophage T7 RNA polymerase. The role of sequence elements within and surrounding a rho-independent transcription terminator. J. Biol. Chem. 1992; 267:19306–19312. [PubMed] [Google Scholar]
- 52. Madhugiri R., Karl N., Petersen D., Lamkiewicz K., Fricke M., Wend U., Scheuer R., Marz M., Ziebuhr J.. Structural and functional conservation of cis-acting RNA elements in coronavirus 5′-terminal genome regions. Virology. 2018; 517:44–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ehresmann C., Baudin F., Mougel M., Romby P., Ebel J.P., Ehresmann B.. Probing the structure of RNAs in solution. Nucleic Acids Res. 1987; 15:9109–9128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Luo D., Condon C., Grunberg-Manago M., Putzer H.. In vitro and in vivo secondary structure probing of the thrS leader in Bacillus subtilis. Nucleic Acids Res. 1998; 26:5379–5387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003; 31:3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lorenz R., Bernhart S.H., Höner Zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Darty K., Denise A., Ponty Y.. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009; 25:1974–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Smith C., Heyne S., Richter A.S., Will S., Backofen R.. Freiburg RNA Tools: a web server integrating INTARNA, EXPARNA and LOCARNA. Nucleic Acids Res. 2010; 38:W373–W377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Notredame C., Higgins D.G., Heringa J.. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000; 302:205–217. [DOI] [PubMed] [Google Scholar]
- 60. Kolter R., Yanofsky C.. Genetic analysis of the tryptophan operon regulatory region using site-directed mutagenesis. J. Mol. Biol. 1984; 175:299–312. [DOI] [PubMed] [Google Scholar]
- 61. Bae Y.M., Stauffer G.V.. Genetic analysis of the attenuator of the Rhizobium meliloti trpE(G) gene. J. Bacteriol. 1991; 173:3382–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Baumgardt K., Charoenpanich P., McIntosh M., Schikora A., Stein E., Thalmann S., Kogel K.H., Klug G., Becker A., Evguenieva-Hackenberg E.. RNase E affects the expression of the acyl-homoserine lactone synthase gene sinI in Sinorhizobium meliloti. J. Bacteriol. 2014; 196:1435–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Saramago M., Robledo M., Matos R.G., Jiménez-Zurdo J.I., Arraiano C.M.. Sinorhizobium meliloti RNase III: Catalytic Features and Impact on Symbiosis. Front Genet. 2018; 9:350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schlüter J.P., Reinkensmeier J., Barnett M.J., Lang C., Krol E., Giegerich R., Long S.R., Becker A.. Global mapping of transcription start sites and promoter motifs in the symbiotic α-proteobacterium Sinorhizobium meliloti 1021. BMC Genomics. 2013; 14:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wilms I., Overlöper A., Nowrousian M., Sharma C.M., Narberhaus F.. Deep sequencing uncovers numerous small RNAs on all four replicons of the plant pathogen Agrobacterium tumefaciens. RNA Biol. 2012; 9:446–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Čuklina J., Hahn J., Imakaev M., Omasits U., Förstner K.U., Ljubimov N., Goebel M., Pessi G., Fischer H.M., Ahrens C.H. et al.. Genome-wide transcription start site mapping of Bradyrhizobium japonicum grown free-living or in symbiosis - a rich resource to identify new transcripts, proteins and to study gene regulation. BMC Genomics. 2016; 17:302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Belasco J.G. All things must pass: contrasts and commonalities in eukaryotic and bacterial mRNA decay. Nat. Rev. Mol. Cell. Biol. 2010; 11:467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dartigalongue C., Raina S.. A new heat-shock gene, ppiD, encodes a peptidyl-prolyl isomerase required for folding of outer membrane proteins in Escherichia coli. EMBO J. 1998; 17:3968–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fürst M., Zhou Y., Merfort J., Müller M.. Involvement of PpiD in Sec-dependent protein translocation. Biochim. Biophys. Acta. 2018; 1865:273–280. [DOI] [PubMed] [Google Scholar]
- 70. Iobbi-Nivol C., Leimkühler S.. Molybdenum enzymes, their maturation and molybdenum cofactor biosynthesis in Escherichia coli. Biochim. Biophys. Acta. 2013; 1827:1086–1101. [DOI] [PubMed] [Google Scholar]
- 71. Mendel R.R. The molybdenum cofactor. J. Biol. Chem. 2013; 288:13165–13172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Leimkühler S. Shared function and moonlighting proteins in molybdenum cofactor biosynthesis. Biol. Chem. 2017; 398:1009–1026. [DOI] [PubMed] [Google Scholar]
- 73. Xu Y.Y., Yang J.S., Liu C., Wang E.T., Wang R.N., Qiu X.Q., Li B.Z., Chen W.F., Yuan H.L.. Water-Soluble humic materials regulate quorum sensing in Sinorhizobium meliloti through a novel repressor of expR. Front. Microbiol. 2018; 9:3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. McIntosh M., Meyer S., Becker A.. Novel Sinorhizobium meliloti quorum sensing positive and negative regulatory feedback mechanisms respond to phosphate availability. Mol. Microbiol. 2009; 74:1238–1256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.