


Abstract
The tumor suppressor protein 53BP1 plays key roles in response to DNA double-strand breaks (DSBs) by serving as a master scaffold at the damaged chromatin. Current evidence indicates that 53BP1 assembles a cohort of DNA damage response (DDR) factors to distinctly execute its repertoire of DSB responses, including checkpoint activation and non-homologous end joining (NHEJ) repair. Here, we have uncovered LC8 (a.k.a. DYNLL1) as an important 53BP1 effector. We found that LC8 accumulates at laser-induced DNA damage tracks in a 53BP1-dependent manner and requires the canonical H2AX-MDC1-RNF8-RNF168 signal transduction cascade. Accordingly, genetic inactivation of LC8 or its interaction with 53BP1 resulted in checkpoint defects. Importantly, loss of LC8 alleviated the hypersensitivity of BRCA1-depleted cells to ionizing radiation and PARP inhibition, highlighting the 53BP1-LC8 module in counteracting BRCA1-dependent functions in the DDR. Together, these data establish LC8 as an important mediator of a subset of 53BP1-dependent DSB responses.
INTRODUCTION
Our genome is continuously challenged by endogenous sources of DNA damage that arise during DNA replication and cellular metabolism, as well as from external factors including UV irradiation. DNA lesions, if left unrepaired or misrepaired, can result in genetic mutations and chromosomal aberrations, leading to cell death and on occasion, neoplastic transformation (1–3). The DNA Damage Response (DDR) pathway is an intricate network of proteins that protect genome integrity by repairing damaged DNA induced by genotoxic insult in a timely manner (4).
DNA double-strand breaks (DSBs) are primarily repaired by two pathways, homologous recombination (HR) and non-homologous end joining (NHEJ), which are tightly regulated throughout the cell cycle. HR repair is restricted to the S and G2 phases in the presence of sister chromatid while NHEJ can take place throughout the cell cycle, predominantly in during the G1 phase. One of the key DDR proteins, 53BP1 (TP53BP1), serves as a mediator to repair DNA breaks by promoting NHEJ repair and checkpoint signaling (5,6). Genetic studies have shown that 53BP1 restrains DNA end resection, and that loss of 53BP1 restores DNA end resection and HR repair in BRCA1-deficient cells (7–9). 53BP1 KO can also rescue embryonic lethality of BRCA1-deficient mice (10). Therefore, the balance between BRCA1 and 53BP1 is key in choice of DSBs repair pathway (11).
The roles of 53BP1 in DSB repair, including class switch recombination (CSR), V(D)J recombination, dysfunctional telomeres and suppression of HR repair, have been intensely investigated since its original report (5,6,11). 53BP1 is recruited to the damaged chromatin downstream of the canonical H2AX-MDC1-RNF8-RNF168 axis (12–18). The mechanism by which 53BP1 is recruited to DSBs has been comprehensively characterized (19), and involves its tudor domain (20–22), its ubiquitin dependent recruitment (UDR) domain (23,24) and its oligomerization motif (25). The tandem tudor domain and UDR domain recognize H2A/X K15ub and H4K20me2, histone marks that are catalyzed by RNF168 and SET8-SUV4-20H1, respectively (26–28). Due to the lack of putative enzymatic activity, 53BP1 was predicted to serve as a scaffold protein to form functional complexes with DSB-responsive factors at the damaged chromatin (5). Thus, identification of 53BP1 bona fide binding partners has become a major focus in an attempt to uncover the biological functions of 53BP1-dependent DSBs responses.
The 53BP1 N-terminus contains 28 Ser/Thr-Gln (SQ/TQ) sites that are potentially phosphorylated by the ATM kinase. These SQ/TQ sites are responsible for recruiting the downstream effector proteins RAP interacting factor (RIF1) (29–33) and PAX-transcription activation domain interacting protein (PTIP) (34,35). Through direct protein–protein interaction, RIF1 recruits REV7 (also known as MAD2L2) (36,37) and the Shieldin complex (C20or196/SHLD1, FAM35A/SHLD2 and CTC-534A2.2/SHLD3) (38–42) to damaged chromatin. Classical NHEJ is defective if this pathway is impaired, while CSR and DNA end resection in BRCA1-deficient cells is restored. The RIF1-Shieldin pathway acts exclusively during G1 phase by inhibiting BRCA1/CtIP accessibility to DSBs. On the other hand, another 53BP1 downstream protein, PTIP, recruits Artemis to promote cNHEJ independent of the cell cycle (43). Notably, the C-terminal BRCT domain of 53BP1 interacts with MUM1/EXPAND1 and may regulate repair of a subset of DSBs (44).
LC8 encodes a small subunit of the dynein motor complex and was previously identified as an 53BP1-interacting protein (45). Even though it was speculated that LC8 may participate in a variety of cellular functions independently of dynein (46), the role of LC8 in DNA repair and maintenance of genome stability has not been studied.
Here, we report the functional characterization of the 53BP1-LC8 module in the context of DDR control. While LC8 is recruited to laser-induced DNA damage tracks in a 53BP1-dependent manner, we found that LC8 is important in productive accumulation of 53BP1 at DNA damage foci. We show that 53BP1 constitutively interacts with LC8, and together associate on chromatin. LC8 KO cells show checkpoint defects and loss of LC8 attenuates hypersensitivity to ionizing radiation and PARP inhibition of BRCA1-depleted cells. Our finding uncovers a role of the 53BP1-LC8 axis in the DDR, and provides important insight into understanding 53BP1-dependent DSBs responses and in therapeutic interventions for clinical managements of human cancers with BRCA1 mutations.
MATERIALS AND METHODS
Plasmids and siRNAs
Human LC8 pENTR221 was purchased from Harvard PlasmID Database. HA-53BP1 and deletion mutant expression vectors were previously described (44). The LC8 cDNA was verified by Sanger sequencing and was subcloned into Gateway compatible destination vectors using Gateway cloning technology (Invitrogen). Mutations were created using QuikChange site-directed mutagenesis kit (Agilent Genomics) according to the manufacturer's instructions. siRNA SMARTpools were purchased from Dharmacon: siControl, siMDC1, siRNF8, siMRE11 and siNBS1. Specific siRNA sequences were used for targeting LC8 (5′-ACCCAGUGAUCCAUCCAAA-3′). siBRCA1 (5′-CUAGAAUCUGUUGUAUG-3′).
Cell culture
HEK293T, U2OS and HeLa cells were purchased from American Type Culture Collection and were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C with 5% CO2. Transfection was carried out using Polyethylenimine (PEI) (Sigma Aldrich) and Fugene HD (Promega) according to the manufacturer's instructions.
Antibodies
Primary antibodies used in this study were Flag M2 (Sigma, F1804), myc (Santa Cruz, sc-40), HA (Abcam, AB18181), LC8 (Hangzhou Labs, ET1612-64; Sigma Aldrich, HPA039954), 53BP1 (Novus Biologicals, NB100-304; BD Biosciences, 612523), BRCA1 (Santa Cruz Biotechnology, SC-6954), γH2AX (Cell Signaling, 2577; Millipore, 05-636), Chk1 (Santa Cruz Biotechnology, SC-8408), Chk1 oS317 (Cell Signaling, 2344), pChk1 S345 (Cell Signaling, 2348), RPA2 (Abcam, AB2175), pRPA2 S33 (Bethyl Laboratories, A300–246A), tubulin (Abcam, AB6046), actin (Santa Cruz Biotechnology, SC-47778), GFP (Invitrogen, A11122), BrdU (BD Biosciences, 347580), cyclin A (BD Biosciences, 611268), Histone H3 (Abcam, ab1791), phospho-histone H3 Ser10 (Cell Signaling, 3377S), RIF1 (Bethyl Laboratories, A300–596A), H2AX (Cell Signaling, 2595S), ub-H2A (Sigma, 05-678), H2A (Cell Signaling, 2578), RNF168 (EMD Millipore, ABE367). For western blotting, secondary antibodies HRP-linked anti-rabbit IgG and HRP-linked anti-mouse IgG were purchased from Cell Signaling (0704 and 0706). For immunofluorescence, Alexa Fluor 488 goat anti-rabbit and Alexa Fluor 594 goat anti-mouse were used (Invitrogen).
Tandem affinity purification
Tandem affinity purification on chromatin was performed as previously described (47). LC8 was subcloned into pMH-SFB (Addgene ID: 99391) to drive mammalian expression of S-protein-2XFlag-Streptavidin binding peptide (SFB)-tagged LC8 proteins. Briefly, SFB-LC8 HEK293T stable cells were harvested with NETN buffer (150 mM NaCl, 0.5 mM EDTA, 20 mM Tris–HCl at pH 8.0, 0.5% NP-40) with protease inhibitors for 20 min at 4°C. The supernatant was discarded and the pellet was washed with NETN and digested with Turbonuclease (Accelagen) to obtain the chromatin bound fraction. The chromatin cell lysate was incubated with streptavidin sepharose (GE Healthcare) for 1 h followed by washing with NETN buffer for three times and eluted with 2 mM biotin at 4°C. The eluent was then incubated with S-protein beads (EMD Millipore) overnight, washed with NETN buffer for three times and eluted with 1× Laemmli buffer. The immuno-complex was subjected to SDS-PAGE and excised for mass spectrometry analysis.
Streptavidin pull down assay and western blotting
Cells were expressed with SFB-fused proteins as indicated and harvested with NETN buffer with nuclease in 4°C for 30 min. The lysates were incubated with Streptavidin beads for 1 h at 4°C followed by washing with NETN buffer for three times. The immunoprecipitated complexes were eluted with 1× Laemmli buffer and were resolved by SDS-PAGE, transferred to PVDF membranes, immunoblotted with antibodies as indicated and imaged using BioRad ChemiDoc MP.
CRISPR/Cas9 mediated knockout
Knockout cells used in this study were generated using the CRISPR/Cas9 method (48). Two individual gRNAs were designed for each target gene (53BP1: gRNA1 – CATAATTTATCATCCACGTC; gRNA2 – ATAATTTATCATCCACGTCT, RNF168: gRNA1 – GCATAAACTCGCCTTTTCGA; gRNA2 – GGAAGTGGGTGAGTAAACCA; LC8: gRNA1 – CGGCTCATATGAAGAAGGTG; gRNA2 – GGTCGCACATGGTTACCGA A) and were subcloned into pSpCas9(BB)-2A-Puro or lentiCRISPR v2 (a gift from Feng Zhang – Addgene IDs: 48139 and 52961). Gene targeting was performed as previously described (48). Briefly, cells were transfected with mammalian expression vectors containing gRNAs using Fugene according to the manufacturer's instructions. Transfected cells were selected with puromycin for two days and seeded into 96-wells to obtain single colonies. The cells derived from single colonies were screened by western blotting, immunofluorescence and verified by sequencing. For lentiviral infection, lentiviral expression vectors, psPAX2 and pMD2.G were co-transfected into HEK293T in 4:3:1 ratio using PEI. Viral particles were collected 48 h after transfection and applied to target cell lines in the presence of 8 μg/ml polybrene (Sigma).
Laser-induced micro-irradiation
Cells were seeded on 35 mm glass bottom dishes and sensitized with 10 μM BrdU 24 h prior to experimentation. Laser-induced micro-irradiation was carried out using a Nikon Ti2 inverted fluorescent microscope and C2+ confocal system. Cells were damaged with a fixed wavelength 405-nm laser at 60% power. Live cell images were captured in 1-min intervals after damage. The fluorescence intensity of GFP-tagged protein in the damaged region was measured and normalized with the undamaged area in the same cell. Quantification analyses were done using ImageJ software.
Immunofluorescence confocal microscopy
Immunofluorescence was performed as previously described. Briefly, cells were seeded on poly-l-lysine coated coverslips (BD Biosciences) 24 h prior experiment. Coverslips were washed in PBS and fixed in 3% paraformaldehyde for 15 min at room temperature followed by permeabilization with 0.5% Triton X-100 solution. For LC8 and RPA2 foci detection, samples were pre-extracted with 0.5% Triton X-100 solution for 5 min prior paraformaldehyde fixation. Samples were incubated with the indicated primary antibodies for 2 h, washed and incubated with secondary antibodies and DAPI for 30 min at room temperature. Samples were then mounted onto glass slides with anti-fade solution (0.02% p-phenylenediamine [Sigma, P6001] in 90% glycerol in PBS). Samples were visualized and captured using a Ti-2 inverted C2+ confocal microscope. Recruitment intensity was analyzed by ImageJ software.
Double thymidine block
Cells were cultured to 30% confluence, thymidine was added to a final concentration of 2 mM and the cells were incubated at 37°C with 5% CO2 for 14 h. Cells were washed twice with PBS and incubated for 9 h in the absence of thymidine. Thymidine was added to a final concentration of 2 mM and cells were incubated for 14 h. Cells were harvested at various time points pre-release (asynchronous) or after thymidine release.
Mitotic index and cell cycle analysis
For mitotic index analysis, cells were treated with X-ray as indicated dose and time followed by nocodazole block. Cells were trypsinized and fixed in 80% ethanol overnight at 4°C. Cells were stained with H3pS10 antibodies for 2 h followed by goat anti-rabbit Alexa 488 secondary antibody for 30 min in the dark. Cell cycle analysis was performed by staining with 40 μg/ml propidium iodide containing 2 μg/ml RNase for 30 min at room temperature. Samples were analyzed by flow cytometry and data were processed with FlowJo software.
Colony formation survival assays
Cells were seeded at a density of 1000 cells per well in a six-well plate in triplicate. Cells were treated as indicated 24 h after plating. At 10–14 days, cells were fixed and stained with coomassie blue staining solution, washed, dried and followed by manual counting of visible colonies.
HR, NHEJ, SSA and aNHEJ DNA repair reporter assays
DNA repair reporter assays were performed as previously described (49). HeLa LC8 KO cells were transfected with reporter vectors of DR-GFP (HR), EJ5-GFP (NHEJ), SA-GFP (SSA) and EJ2-GFP (aNHEJ) and I-SceI expression vector (pCBASceI) as indicated by electroporation. Cells were allowed to recover for 48 h after transfection and were analyzed by flow cytometry.
Replication program
Replication program analysis was performed as previously described (50). Cells cultured on coverslips with or without siBRCA1 knockdown were incubated with 10 μM BrdU for 30 min. Cells were washed and incubated in fresh medium for 4 h followed by 10 μM EdU for 30 min. EdU was labeled using the 488 EdU Click Proliferation kit (BD 565455) according to manufacturer's protocol. Coverslips were incubated with 2 N HCl for 30 min, washed with PBS and incubated with anti-BrdU antibody and Alexa Fluor 594 goat anti-mouse secondary. Coverslips were then mounted onto glass slides and imaged as previously described in this paper. S-phase progression was analyzed by categorizing the stage of S-phase of BrdU and EdU as early, mid, or late S-phase based on the pattern and density of foci formation.
Statistical analysis
Data are represented as mean ± S.E.M. and S.D. as indicated from at least three independent experiments. Analyses were performed in GraphPad Prism by Student's t-test or Chi-square where indicated. Significance was reported starting at P < 0.05.
RESULTS
LC8 is a bona fide 53BP1-interacting protein
It has been reported that LC8 binds to 53BP1 fragment in vitro (45). However, there is no in vivo data that demonstrates the specific binding between LC8 and 53BP1. To investigate the role of LC8 on damaged chromatin, we first performed an unbiased proteomic analysis for the LC8 interactome in the chromatin fraction (Figure 1A) (Supplementary Table 1). We first pre-extracted non-chromatin bound proteins from SFB-tagged LC8 stable cells with NETN buffer, and subsequently resuspended and incubated the chromatin-enriched fraction with turbonuclease (Figure 1A). Employing an established chromatin-tandem affinity purification approach (47), we subjected the LC8 co-purifying proteins to mass spectrometric analysis and were able to recover 53BP1 as one of the top hits that co-purified with LC8 on chromatin (Figure 1B). Co-immunoprecipitation experiment confirmed that LC8 specifically binds to 53BP1. Importantly, deletion of the amino acids 1052–1302 from the 53BP1 polypeptide completely abolished its interaction with LC8 (Supplementary Figure S1A and B). We also verified the LC8 interaction of seven top proteins co-purified from the mass spectrometric analysis. None of them showed detectable binding to 53BP1, suggesting that these proteins do not form a complex with 53BP1-LC8 (Supplementary Figure S1C). LC8 encodes an evolutionarily conserved 10 kDa protein without any recognizable functional domain (Figure 1C). Previously structural studies have shown that LC8 forms a dimer with a secondary structure that provides a binding groove within its second and third beta sheets (Figure 1D) (51). Alanine substitution mutation of five amino acids on the third beta sheet (a.a. 62–67 FGSYV/AAAAA) that directly form hydrogen bonds to the putative Q residue of the target proteins impaired the LC8–53BP1 interaction (Figure 1E). Together, these data suggest that LC8 forms a complex with 53BP1 in vivo via LC8 canonical interaction.
Figure 1.

LC8 interacts with 53BP1 on chromatin. (A) Schematic diagram of chromatin interacting protein purification of SFB (S-protein, 2X flag, Streptavidin binding peptide) -tagged-LC8 in HEK293T cells. (B) The top ten list of proteins co-purified with LC8 from mass spectrometry analysis. (C) Protein sequence alignment of LC8 in different species. Protein regions were labeled with secondary structures. Region marked red represents the amino acid residues required for the canonical binding groove formation. (D) Dimerized LC8 structure illustration of the canonical binding groove located between beta 2 and beta 3 sheets. Beta 3 sequence FGSYV is required for directing binding to target protein. Green line represents the putative target binding peptide. (E) Streptavidin pull-down of SFB-LC8 wildtype and SFB-LC8 FGSYV/AAAAA mutant in HEK293T stable cell lines followed by western blotting analysis with Flag and 53BP1 (Novus Biologicals, NB100–304) antibodies.
LC8 DSB recruitment is dependent on 53BP1
To examine if LC8 plays a role in the DDR, we first determined whether LC8 may be recruited to DSBs. LC8 is localized in both the cytoplasm and the nucleus. In order to better visualize whether LC8 is enriched at DNA damage sites, we treated cells with 0.5% Triton X-100 prior to paraformaldehyde fixation. Flag-tagged LC8 form irradiation-induced foci (IRIF), and co-localized with DNA damage markers γH2AX and 53BP1 (Figure 2A and B). We also confirmed that LC8 is promptly recruited to DNA breaks within minutes upon laser-induced micro-irradiation (Figure 2C), exhibiting similar kinetics as 53BP1 recruitment. Since LC8 is recruited to DNA DSBs and interacts with 53BP1, we tested whether LC8 is recruited to DSBs via 53BP1. We genetically inactivated 53BP1 in U2OS cells using the CRISPR/Cas9 method (Supplementary Figure S2A). We also generated RNF168 KO cells in the same background to investigate the requirement for LC8 recruitment onto laser-induced micro-irradiated sites (Supplementary Figure S2B). Our data showed that LC8 DSBs recruitment is markedly suppressed in both 53BP1 and RNF168 KO cells (Figure 2D). Likewise, LC8 DSBs recruitment was impaired in cells pre-treated with siRNAs that targeted DSBs signal transducers MDC1 and RNF8 (Figure 2D), the upstream proteins required for RNF168 and 53BP1 IRIF formation (16,18). On the other hand, knockdown of the proteins that are required for DNA end resection initiation, including MRE11A and NBS1, did not affect LC8 DSBs recruitment (Figure 2D). Together, our data supported the notion that LC8 may play a role in the DDR pathway and acts downstream of the MDC1-RNF8-RNF168–53BP1 axis.
Figure 2.

LC8 is recruited to the DNA sites of damage. (A) LC8 localizes to DNA damage sites. U2OS cells with SFB-LC8 stable expression were plated on cover slips 24 h before irradiation with 10 Gy in a Faxitron and allowed to recover for 1 h. The samples were pre-extracted with 0.5% Triton X-100 prior to crosslinking with 3% paraformaldehyde. They were then incubated with Flag antibody to detect SFB-LC8 with γH2AX (Cell Signaling, 2577) co-staining followed by immunofluorescence analysis. (B) U2OS GFP-LC8 stable cells were treated as in A and stained with 53BP1 (BD Biosciences, 612523) followed by immunofluorescence analysis. GFP-LC8 was analyzed directly without incubation with antibody. (C) Recruitment kinetic analysis of GFP-LC8 live cell imaging. U2OS cells expressing the indicated GFP-tagged proteins were damaged and analyzed at indicated time point by confocal microscopy. Red arrows indicate the laser path. (D) Genetic analysis of LC8 recruitment dependency. GFP-LC8 expressed in U2OS cells in different genetic backgrounds or siRNA targeted specific knockdown were damaged and analyzed at 15 min by confocal microscopy. (E) LC8 chromatin association is dependent on 53BP1. U2OS cells were fractionated by NETN containing 150 mM NaCl (soluble fraction) and 0.2 M HCl acid extraction (chromatin fraction), tubulin and histone 3 were used as loading controls. Endogenous LC8 level was detected using a specific antibody (Hangzhou Labs, ET1612–64). LC8 chromatin loading oscillates during cell cycle as illustrated by double thymidine block and release. (F) U2OS cells were harvested and fractionated to obtain chromatin fraction. 53BP1 (Novus Biologicals, NB100–304) and LC8 (Hangzhou Labs, ET1612–64) expression were measured by Western blot. FACS analysis of cells after thymidine block and release. (G) LC8 chromatin association analysis throughout cell cycle. Double thymidine block and release followed by fractionation analysis as in C. (H) Immunofluorescence analysis of Flag-LC8 stable U2OS cells co-staining with cyclin A 1 h after 10 Gy irradiation. (I) Quantification of LC8 foci in cyclin A positive and negative cells as in H. N = 50.
LC8 associates with chromatin through 53BP1 interaction
LC8 interacts with 53BP1 on chromatin and recruitment of LC8 is 53BP1-dependent. We then tested whether 53BP1 is required for chromatin association of LC8. Cell fractionation analysis revealed that LC8 chromatin association is dramatically reduced in 53BP1 KO cells (Figure 2E). Since 53BP1 associates with chromatin, at least in part, via cell cycle-regulated H4K20 methylation marks, to test whether chromatin-associated LC8 is similar to 53BP1, we performed a double thymidine block to synchronize cells at the G1/S boundary, followed by removing thymidine from the media and collecting cell lysates at the indicated time points. We observed that LC8 chromatin loading oscillates throughout the cell cycle (Figure 2F). The level of LC8 on chromatin is relatively lower at early S-phase, and gradually increases as cells progress into mid-S phase. As expected, LC8 chromatin association after early S-phase is dramatically reduced in 53BP1 KO cells (Figure 2G). To examine whether LC8 foci formation is cell cycle-dependent, we performed immunofluorescence staining with Flag-LC8 with cyclin A. In order to eliminate the cytoplasmic LC8 expression to achieve distinct foci localization, cells were pre-extracted with 0.5% Triton X-100 solution prior fixation and antibodies incubation. However, we were not able to detect cyclin A staining in pre-extracted sample (Supplementary Figure S3A). Alternatively, even though we saw cytoplasmic LC8 expression, we were able to quantify LC8 foci in both cyclin A-positive and -negative cells without pre-extraction (Figure 2H and I). Our data showed that LC8 foci are not cell cycle-dependent. Together, our data suggests that LC8 associates with chromatin, possibly through direct constitutive 53BP1 interaction.
LC8 promotes efficient 53BP1 IRIF formation
53BP1 promotes chromatin association and DSBs enrichment of LC8. We then tested whether LC8 is involved in 53BP1 IRIF formation. In LC8 KO cell, we found that 53BP1 is still able to form IRIF upon DNA damage in LC8 KO cells (Figure 3A). However, the number of foci is moderately reduced in LC8 KO cells (Figure 3A and B). We then examined whether the reduction of 53BP1 foci is cell-cycle dependent by co-immunostaining with cyclin A, a cell cycle S-G2 phase marker. The reduction of 53BP1 foci in LC8 KO cells are not different in cyclin A-positive cells or cyclin A-negative cells suggesting that the effect of LC8 on 53BP1 foci formation is independent of the cell cycle (Figure 3C and D). To further investigate the effect of LC8 depletion in 53BP1-pathway, we quantified the IR-induced foci of the two major effectors (RIF1 and PTIP) as well as MUM1 and the localization of a recently identified 53BP1 binding protein, TIRR. We observed a significant reduction in both RIF1 and PTIP but not MUM1 foci (Supplementary Figure S3B-G). TIRR is not recruited to DSB and we did not observe any difference in TIRR localization between wildtype and LC8 KO cells (Supplementary Figure S3H–I).
Figure 3.
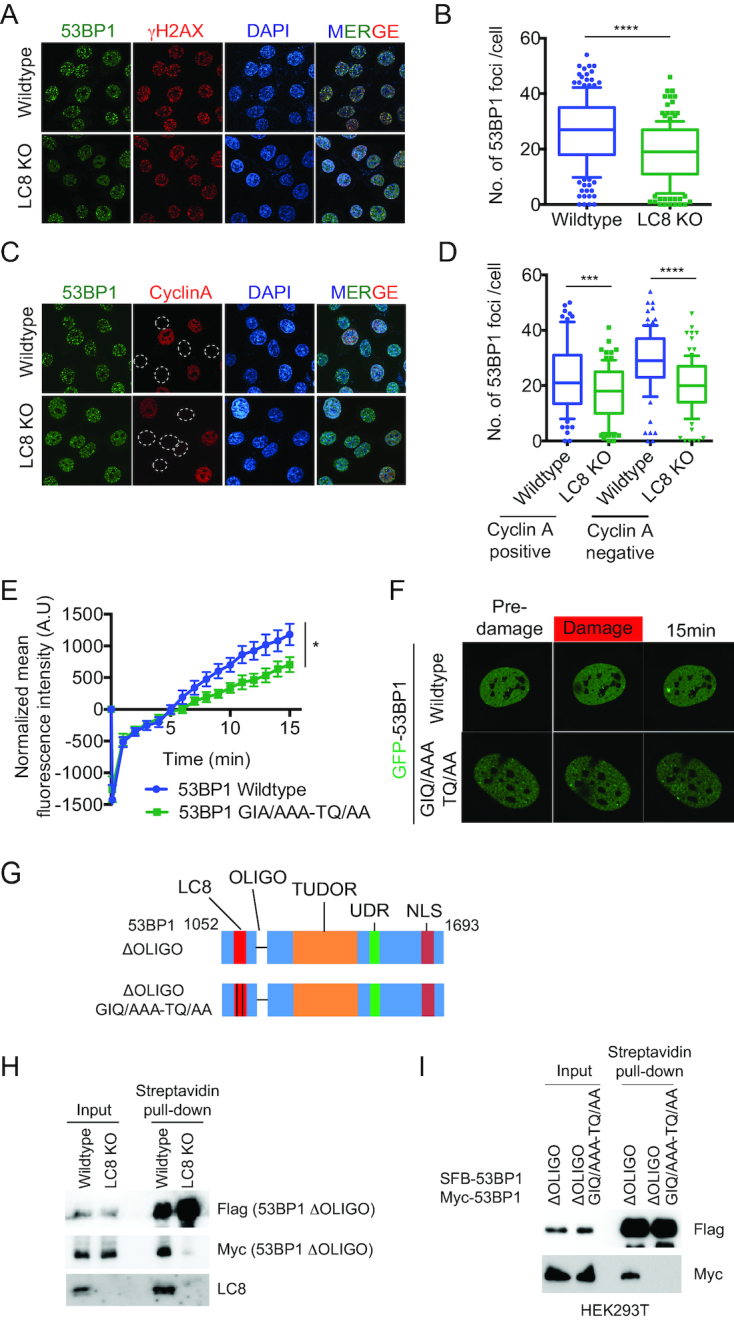
LC8 promotes 53BP1 irradiation induced foci formation. (A, B) 53BP1 foci is reduced in LC8 KO cells. U2OS cells were irradiated with a Faxitron with 10 Gy and allowed to recover for 1 h. Cells were then fixed, permeabilized and stained with 53BP1 (Novus Biologicals, NB100–304) and γH2AX (Millipore, 05-636) antibodies. Immunofluorescence was analyzed by confocal microscopy. Number of 53BP1 foci per cell were analyzed. (C, D) Cells were treated as in A and B, and stained with 53BP1 (Novus Biologicals, NB100–304) and cyclin A antibodies. Number of 53BP1 foci per cells in both cyclin A-positive group and cyclin A-negative group were analyzed. Yellow dash circle marked cyclin A negative cells. Data represent mean ± SD, ***P< 0.001, ****P< 0.0001. (E, F) U2OS with transient expression of GFP-53BP1 wildtype and GFP-53BP1 GIQ/AAA-TQ/AA were micro-irradiated and quantified. Data represent mean ± S.E.M., *P< 0.05. N = 10 (G) Schematic illustration of 53BP1 mutant fragments used in H and I. (H) Transient co-transfection of SFB-53BP1 ΔOLIGO and myc-53BP1 ΔOLIGO in HeLa wildtype and LC8 KO cells followed by streptavidin pull-down. (I) HEK293T cells were transiently co-transfected with SFB-53BP1 ΔOLIGO and myc-53BP1 ΔOLIGO or SFB-53BP1 ΔOLIGO-GIQ/AAA-TQ/AA and myc-53BP1 ΔOLIGO-GIQ/AAA-TQ/AA. Western blot analysis using antibodies as indicated.
We also confirmed the role of LC8 in 53BP1 DSBs recruitment using laser-induced micro irradiation. We expressed GFP-53BP1 and GFP-53BP1-GIQ/AAA-TQ/AA (LC8 defective binding) (45) in U2OS cells and imaged their recruitment onto DSBs following laser-induced micro-irradiation. Notably, DSBs recruitment intensity was moderately reduced in the GFP-53BP1-GIA/AAA-TQ/AA mutant when compared with wildtype 53BP1 (Figure 3E and F). To investigate the mechanism how LC8 modulates 53BP1 DSB recruitment, we hypothesize, based on the previously characterized role of LC8 as a dimerization hub (46), that LC8 may enhance 53BP1 dimerization and promote its DSB recruitment. In order to examine if LC8 can promote 53BP1 dimerization independent of the oligomerization domain, we generated an oligomerization domain deletion mutant (ΔOLIGO) and a double mutant with oligomerization domain deletion and LC8 binding defective mutant (ΔOLIGO GIQ/AAA-TQ/AA) (Figure 3G). We first performed a pull-down experiment by transiently expressing SFB-53BP1 ΔOLIGO and myc-53BP1 ΔOLIGO in HeLa wildtype and LC8 KO cells. SFB-53BP1 ΔOLIGO is able to pull down myc-53BP1 ΔOLIGO in wildtype cells but not LC8 KO cells. Consistently, SFB-53BP1 ΔOLIGO GIQ/AAA-TQ/AA failed to pull down myc-53BP1 ΔOLIGO GIQ/AAA-TQ/AA in HEK293T cells. These data suggest that LC8 is required for 53BP1 dimerization independent of the oligomerization domain. Together, LC8 may play a role in promoting 53BP1 dimerization and productive accumulation of 53BP1 at DSBs.
LC8 is required for proper checkpoint activation upon DNA damage
To examine the role of LC8 in the DDR signaling pathway, we pre-treated LC8-silenced cells as well as LC8 KO cells using either camptothecin (CPT) or hydroxyurea (HU). Consistently and reminiscent to 53BP1 KO cells, we found that LC8 inactivation attenuated Chk1 phosphorylation after CPT or HU treatment (Figure 4A–C; Supplementary Figure S4A and B) (52). Notably, Chk1 phosphorylation is reactivated by reintroducing GFP-LC8 into LC8 KO cells, suggesting a LC8 specific role in regulating Chk1 activation (Figure 4D). Moreover, we observed an increase in mitotic index in LC8 KO cells indicating that these cells failed to properly arrest at the G2/M checkpoint (Figure 4E). Even though we saw robust recruitment of LC8 to the damaged chromatin and a subtle increase in pRPA2 in CPT-treated LC8 depleted cells, LC8 depletion did not affect NHEJ repair, HR or SSA repair as measured by the established DSB repair reporter assays (Supplementary Figure S5A and B). Consistent with these data, we also did not observe a noticeable difference in RPA2 foci in CPT-treated LC8 KO cells in both wildtype and BRCA1 depleted groups (Supplementary Figure S5C and D).
Figure 4.

LC8 is required for proper checkpoint activation after DNA damage. (A, B) LC8 knockdown compromises Chk1 phosphorylation. U2OS with siRNA targeted LC8 knockdown cells were treated with CPT and HU for 1 h with indicated doses. (C) CRISPR/Cas9 mediated knockout of LC8 were treated with CPT and HU as indicated for 1 h followed by western blotting analysis using the antibodies as indicated. LC8 (Sigma Aldrich, HPA039954) γH2AX (Cell Signaling, 2577). (D) HeLa LC8 KO cells reconstituted with GFP-LC8 wildtype cDNA were treated with HU and analyzed by Western blotting using indicated antibodies. (E) Mitotic index was measured by Histone H3S10 phosphorylation in HeLa wildtype and LC8 KO cells at 1 and 3 h post-irradiation by 6 Gy using a Faxitron followed by immediate incubation of nocodazole (1 μg/ml). Data represent mean ± S.E.M., *P< 0.05, **P< 0.001. N = 6.
To test whether LC8–53BP1 interaction is required for the checkpoint defects observed in LC8 KO cells, we stably reconstituted LC8 KO cells with 53BP1 binding defective mutant, (GFP-LC8 FGSYV/AAAAA) and measured Chk1 phosphorylation level after HU treatment (Figure 5A). Importantly, we found that re-expression of the GFP-LC8 FGSYV/AAAAA mutant did not restore HU-induced Chk1 activation in LC8 KO cells (Figure 5A).
Figure 5.

53BP1-LC8 interaction mediates Chk1 activation and LC8 DSB recruitment. (A) HeLa LC8 KO cells reconstituted with GFP-LC8 FGSYV/AAAAA were treated with HU for 1 h. Samples were harvested and analyzed with Western blotting using indicated antibodies. (B) LC8 a.a. 62–67 FGSYV is required for DSBs recruitment. Cells stably expressing GFP-LC8 FGSYV/AAAAA mutant were micro-irradiated and analyzed using confocal microscopy live cell imaging. (C) U2OS 53BP1 KO cells with co-transfection of RFP-LC8 and GFP-53BP1 wildtype or GFP-53BP1 GIQ/AAA-TQ/AA mutants were subjected to laser-induced micro irradiation. Recruitment of GFP-53BP1 and RFP were examined in real time for 15 min after damage using a con-focal microscope.
We then examined whether 53BP1 binding is required for LC8 DSBs recruitment. With laser-induced micro-irradiation, we observed that the GFP-LC8 FGSYV/AAAAA mutant nucleus average intensity is greatly reduced from photo-bleaching (Figure 5B, bottom panel) compared to the specific track photo-bleaching occurred within seconds after micro-irradiation in the wildtype cells (Figure 5B, top panel). Even though the nucleus intensity is recovered to similar levels as to the wildtype cells, GFP-LC8 FGSYV/AAAAA mutant failed to accumulate at the defined micro-irradiated track. Together, these data suggest the LC8 interaction with 53BP1 is required for Chk1 phosphorylation and recruitment to damaged chromatin. We also performed laser-induced micro irradiation in cells transiently co-transfecting GFP-53BP1 wildtype or GIQ/AAA-TQ/AA (LC8 binding defective mutant) with RFP-LC8 in 53BP1 KO background. 53BP1 KO cells with GFP-53BP1 wildtype reconstitution restored the LC8 DSB recruitment. On the other hand, GFP-GIQ/TQT-TQ/AA reconstituted KO cells failed to rescue the LC8 DSB recruitment.
LC8 attenuates the IR and PARP inhibitor sensitivity in BRCA1-depleted cells
Since 53BP1 depletion can reverse the hypersensitivity to PARP inhibitor in BRCA1 mutant/depleted cells, we tested the hypothesis that LC8 may also be involved in regulating BRCA1-depletion induced PARP inhibitor hypersensitivity. Colony survival assays showed that BRCA1 knockdown sensitizes cells to X-Ray and PARP inhibitor treatment, whereas LC8 KO cells did not show significant changes in sensitivity to X-Ray and PARP inhibitor compared to wildtype cells (Figure 6A and B). Despite the fact that LC8 KO did not restore the HR repair efficiency (Supplementary Figure S5B), LC8 KO cells attenuated the DNA damage hypersensitivity in BRCA1 depleted cells (Figure 6A–C, Supplementary Figure S3C). LC8KO cells with LC8 wildtype, but not FGSYV/AAAAA reconstitution, re-sensitize the siBRCA1 cells to X-ray and PARP inhibitors (Figure 6D). These data suggested that LC8 may be involved in 53BP1 mediated resistance in BRCA1-depleted cells independent of DNA DSBs repair pathway choice. Interestingly, consistent with the LC8 depletion induced Chk1 activation defect, we observed an increase in sensitivity to HU in LC8 KO cells (Supplementary Figure S5E and F). Since LC8 is required for proper checkpoint activation, we then tested whether it is also involved in regulating replication dynamics. We labeled the cells with BrdU and chased for 4 h followed by EdU labeling. With immunofluorescence staining and Click-iT chemistry, we were able to trace the S-phase progression. We observed an increase in early-S phase in siBRCA1 cells. However, LC8 depletion did not significantly alter the replication program in both control and siBRCA1 cells (Supplementary Figure S6A-C). These data suggest that LC8 may not play a major role in regulating replication dynamics.
Figure 6.

LC8–53BP1 attenuates genotoxic hypersensitivity in BRCA1-depleted cells. LC8 KO attenuates BRCA1 depletion induced hypersensitivity to DNA damaging agents. (A) Quantification of survival of cells treated with Olaparib and X-ray with indicated dosages. (B) Wildtype HeLa and LC8 KO cells with BRCA1 knockdown were analyzed with Western blotting using indicated antibodies. (C) Representative pictures of colony formation assay. (D) Cell survival assays of GFP-LC8 and GFP-FGSYV/AAAAA reconstituted LC8 KO HeLa cells with BRCA1 knockdown. Quantification of cell survival with Olaparib and X-ray treated groups. Data represent mean ± S.E.M., *P< 0.05, **P< 0.01, ***P< 0.001. N = 6.
DISCUSSION
53BP1 is an important DNA repair protein that regulates DSBs repair pathway choice by suppressing DNA end resection to promote NHEJ, particularly during the G1 phase of the cell cycle when sister chromatids are absent. 53BP1 has been studied extensively since its discovery as a p53 binding protein two decades ago (53). The structural domains have been comprehensively characterized in the context of 53BP1 regulation as well as its function in recruiting downstream effector proteins to the damaged chromatin. It is suggested that the major 53BP1 downstream proteins RIF1-MAD2L2(REV7)-shieldins are responsible for CSR, promoting NHEJ during G1 phase of the cell cycle, and telomere dysfunction; whereas, PTIP-Artemis complex is largely involved in regulating repair pathway choice. Most of the 53BP1 binding partners are well characterized in the context of 53BP1 regulations and functions in DNA repair. Notably, LC8 has been identified as a 53BP1 binding protein more than a decade ago (45), yet the role of LC8 in DNA repair was overlooked.
In this study, we provide evidence that LC8 is involved in the DDR pathway. In particular, we have identified LC8 as downstream factor to 53BP1. LC8 constitutively interacts with 53BP1 on chromatin throughout the cell cycle. Our results showed that LC8 is recruited to DSBs by physically interacting with 53BP1 and is completely dependent on the H2AX-MDC1-RNF8-RNF168 and 53BP1 axis. In addition, loss-of-function studies showed that LC8 is required for checkpoint activation upon replication stress. Furthermore, LC8 depletion attenuates the IR and PARP inhibitor hypersensitivity in BRCA1-deficient cells. These data implicate that the function of LC8 is not restricted to G1 phase and LC8 is important in 53BP1-mediated DNA repair and counteracting the BRCA1-pathway, independent of 53BP1 functions in DNA end resection suppression.
An early study on LC8 and 53BP1 proposed that LC8 can import p53 to the nucleus through 53BP1 interaction (45). Nonetheless, the function of LC8 in the context of DDR was largely neglected. In addition to its role in the dynein motor complex, LC8 is also characterized as a hub protein that binds to a large number of proteins via the groove formed between the beta 2 and beta 3 sheets within the LC8 dimers to the putative TQT or GIQ motif on the target proteins (46,54–56). Consistent with previous in vitro study, our data showed that 53BP1 interacts with LC8 canonically in vivo. The putative bivalent LC8 interacting motifs of 53BP1 (a.a. 1158–1162 GIQTM and a.a. 1175–1180 ATQTIK) are located in the proximity of the oligomerization domain (45). Our data showed that LC8 is required for efficient 53BP1 IRIF formation possibly through promoting 53BP1 dimerization independent of its oligomerization domain. However, the reduction of 53BP1 DSB accumulation in LC8-depleted cells did not detectably affect DSB repair pathway choice.
A large portion of LC8 that associates with chromatin is 53BP1 dependent. LC8 is localized at DNA DSB independent of the cell cycle. Interestingly, we observed that 53BP1 and LC8 chromatin loading oscillates over the cell cycle during post-replication. Their chromatin association are relatively lower in early S-phase and gradually increase as the cell cycle progresses, which coincides with the re-establishment of H4K20 methylation state after replication (57,58) that is possibly required for 53BP1 chromatin loading (20). In Saccharomyces cerevisiae (Rad9) and Schizosaccharomyces pombe (Crb2), the orthologs of 53BP1, play an important role in the activation of Rad53 (Chk1 in humans) (59). Emerging evidence shows that 53BP1 is involved in ATR mediated checkpoint control and replication stress in mammalian cells (52,60) and the antagonism between 53BP1 and BRCA1 in replication restart is not dependent of DSBs repair. Our data on pRPA2 dysregulation, HU hypersensitivity and Chk1 activation defect of LC8 KO cells support the notion on the role of LC8 in 53BP1-mediated post-replication repair antagonism with BRCA1 independent of repair pathway choice. A recent study proposed a mechanism describing how PARP inhibition causes unstable replication forks by altering the replicating speed (50). Based on our observation, LC8 could possibly contribute to post-replicative 53BP1-mediated PARP inhibition hypersensitivity antagonism in BRCA1-deficient cells by modulating check-point control.
Chk1 phosphorylation is reduced in LC8 KO cells after CPT and HU treatment. It is unlikely that LC8 affects ATR activity since RPA2 S33 is also an ATR target substrate. One explanation for the ATR-mediated pChk1 and pRPA2 paradox is that LC8 may modulate the RAD17-RFC and 911 complex at the ssDNA/dsDNA junction, which is indirectly required for Chk1, but not RPA2, phosphorylation (61–64). Interestingly, we observed an increase in pRPA2 in HU-treated LC8 KO cells. It is possibly due to stalled replication fork and helicase-DNA polymerase uncoupling, which generate ssDNA on both the leading and lagging strand that is stably bound by RPA2 (65). We speculate that LC8 may be involved in resolving stalled replication fork upon replication stress. Further investigation is necessary to understand the role of LC8 in replication.
Although LC8 is recruited to DSBs, unlike RIF1 and PTIP KO cells, LC8 KO cells did not show a significant change in DNA repair reporter assays (i.e. HR, cNHEJ, aNHEJ and SSA). These data suggest that LC8 does not seem to suppress DSB resection. Interestingly, LC8 can promote 53BP1 DSB recruitment, yet showed no significant defect in NHEJ or X-Ray sensitivity. Several studies showed that an efficient depletion of 53BP1 confers about 30–40% reduction in the NHEJ (5EJ-GFP) reporter assay (21,66). It is formally possible that the moderate reduction of 53BP1 foci in LC8 KO cells may not have significantly effect in the NHEJ pathway. However, we do not exclude the possibility that there is an unidentified protein functionally redundant to LC8 in regulating the repair pathway choice. This observation also explains why LC8 knockout cells did not show sensitivity to genotoxic drugs. Two recent independent studies showed that LC8 depletion promotes PARP inhibitor survival in BRCA1 mutant cells. Specifically, He et al. proposed that LC8 inhibits MRE11 and end resection in BRCA1-deficient cells (67) while Becker et al. proposed that LC8 functions through the 53BP1-pathway (68), a proposition in line with our observations in this study. Since MRE11A does not contain any canonical LC8 binding motif (KXTQT or G(I/V)Q), in-depth follow-up studies on LC8-MRE11A complex to investigate how LC8 inhibits MRE11A would help us to understand the genetic and structural interplay between LC8–53BP1 and the resection machinery.
LC8 is a small subunit of cytoplasmic dynein complex which is involved in positioning the organelles in the cells, retrograde cargo transport and cell division regulation (69). However, a large fraction of LC8 is not associated with dynein (70). Besides the function of LC8 as cargo adapters of the dynein motor complex, it is also recognized as a regulatory hub protein and interacts with a large number of proteins involved in diverse biological functions (46,56). LC8 dimerizes and creates two symmetric grooves at the dimer interface. The dimerized surface provides a binding site with the short linear motif located in the intrinsically disordered protein region of its interacting proteins. Given the well-characterized function of LC8 as ‘molecular velcro’ (46), the interaction between LC8 and 53BP1 may potentially promote the 53BP1 dimerization on chromatin.
LC8 is a highly evolutionary conserved protein that is an essential protein in Drosophila and C. elegans (71,72). Surprisingly, LC8 is not required for survival in mammalian cells. LC8 is involved in a variety of biological functions. It will be challenging to separate the DNA repair function of LC8 from the cytoplasmic dynein motor complex by genetic study in vivo. Our data showed that at least a portion of LC8 is tightly associated with chromatin and 53BP1 which implicates its potential role in the DDR pathway. The ATQTIK sequence on 53BP1 also overlapped with the potential ATM-phosphorylated TQ site (73). However, the interaction does not seem to be phosphorylation dependent (30). Further exploration of the LC8-DDR protein complex will provide us a better understanding of 53BP1-LC8 in the context of DNA repair.
LC8 is an estrogen response gene that is overexpressed in a subset of breast cancer cell lines (74,75). Identification of the LC8 depletion phenotype provides insight into the mechanism of how LC8 mutations could potentially cause poor prognosis and PARP inhibitor resistance in BRCA1-mutant cells. On the other hand, ectopic LC8 expression in a subset of breast cancer cells may lead to suppression of wildtype BRCA1-circuitry on damaged replicating chromatin, which confers a BRCA1 mutant phenotype. A detailed analysis of LC8 mutations will be important for deciphering the molecular functions of LC8 in BRCA1 regulation and clinical outcome. In summary, our study uncovered a novel role for LC8 in the DDR pathway, which can potentially provide critical insight into cancer prognosis and treatment in targeting BRCA1-mutant cells.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank the University of Arkansas for Medical Sciences (UAMS) Proteomics Core Facility and Dr Fen Xia's group from the Department of Radiation Oncology for the insightful discussion and comments. We also thank Drs. Jeremy Stark and Maria Jasin for generous sharing of reagents.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The research in the J.W.L. laboratory was supported in part by start-up funds from the University of Arkansas for Medical Sciences and National Institute of Health [P20GM121293] National Institute of Health [K22CA204354] to J.W.L. Funding for open access charge: National Institute of Health [K22CA204354].
Conflict of interest statement. None declared.
REFERENCES
- 1. Jackson S.P., Bartek J.. The DNA-damage response in human biology and disease. Nature. 2009; 461:1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lord C.J., Ashworth A.. The DNA damage response and cancer therapy. Nature. 2012; 481:287–294. [DOI] [PubMed] [Google Scholar]
- 3. Aplan P.D. Causes of oncogenic chromosomal translocation. Trends Genet. 2006; 22:46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Polo S.E., Jackson S.P.. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011; 25:409–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Panier S., Boulton S.J.. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014; 15:7–18. [DOI] [PubMed] [Google Scholar]
- 6. Zimmermann M., de Lange T.. 53BP1: pro choice in DNA repair. Trends Cell Biol. 2014; 24:108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bunting S.F., Callen E., Wong N., Chen H.T., Polato F., Gunn A., Bothmer A., Feldhahn N., Fernandez-Capetillo O., Cao L. et al.. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010; 141:243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li M., Cole F., Patel D.S., Misenko S.M., Her J., Malhowski A., Alhamza A., Zheng H., Baer R., Ludwig T. et al.. 53BP1 ablation rescues genomic instability in mice expressing ‘RING-less’ BRCA1. EMBO Rep. 2016; 17:1532–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bouwman P., Aly A., Escandell J.M., Pieterse M., Bartkova J., van der Gulden H., Hiddingh S., Thanasoula M., Kulkarni A., Yang Q. et al.. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010; 17:688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cao L., Xu X., Bunting S.F., Liu J., Wang R.H., Cao L.L., Wu J.J., Peng T.N., Chen J., Nussenzweig A. et al.. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol. Cell. 2009; 35:534–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Daley J.M., Sung P.. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol. Cell Biol. 2014; 34:1380–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huen M.S., Grant R., Manke I., Minn K., Yu X., Yaffe M.B., Chen J.. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007; 131:901–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mailand N., Bekker-Jensen S., Faustrup H., Melander F., Bartek J., Lukas C., Lukas J.. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007; 131:887–900. [DOI] [PubMed] [Google Scholar]
- 14. Doil C., Mailand N., Bekker-Jensen S., Menard P., Larsen D.H., Pepperkok R., Ellenberg J., Panier S., Durocher D., Bartek J. et al.. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009; 136:435–446. [DOI] [PubMed] [Google Scholar]
- 15. Stewart G.S., Panier S., Townsend K., Al-Hakim A.K., Kolas N.K., Miller E.S., Nakada S., Ylanko J., Olivarius S., Mendez M. et al.. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009; 136:420–434. [DOI] [PubMed] [Google Scholar]
- 16. Bartocci C., Denchi E.L.. Put a RING on it: regulation and inhibition of RNF8 and RNF168 RING finger E3 ligases at DNA damage sites. Front Genet. 2013; 4:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xie A., Hartlerode A., Stucki M., Odate S., Puget N., Kwok A., Nagaraju G., Yan C., Alt F.W., Chen J. et al.. Distinct roles of chromatin-associated proteins MDC1 and 53BP1 in mammalian double-strand break repair. Mol. Cell. 2007; 28:1045–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Scully R., Xie A.. Double strand break repair functions of histone H2AX. Mutat. Res. 2013; 750:5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wilson M.D., Benlekbir S., Fradet-Turcotte A., Sherker A., Julien J.P., McEwan A., Noordermeer S.M., Sicheri F., Rubinstein J.L., Durocher D.. The structural basis of modified nucleosome recognition by 53BP1. Nature. 2016; 536:100–103. [DOI] [PubMed] [Google Scholar]
- 20. Botuyan M.V., Lee J., Ward I.M., Kim J.E., Thompson J.R., Chen J., Mer G.. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006; 127:1361–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dulev S., Tkach J., Lin S., Batada N.N.. SET8 methyltransferase activity during the DNA double-strand break response is required for recruitment of 53BP1. EMBO Rep. 2014; 15:1163–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tuzon C.T., Spektor T., Kong X., Congdon L.M., Wu S., Schotta G., Yokomori K., Rice J.C.. Concerted activities of distinct H4K20 methyltransferases at DNA double-strand breaks regulate 53BP1 nucleation and NHEJ-directed repair. Cell Rep. 2014; 8:430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gatti M., Pinato S., Maspero E., Soffientini P., Polo S., Penengo L.. A novel ubiquitin mark at the N-terminal tail of histone H2As targeted by RNF168 ubiquitin ligase. Cell Cycle. 2012; 11:2538–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fradet-Turcotte A., Canny M.D., Escribano-Diaz C., Orthwein A., Leung C.C., Huang H., Landry M.C., Kitevski-LeBlanc J., Noordermeer S.M., Sicheri F. et al.. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature. 2013; 499:50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zgheib O., Pataky K., Brugger J., Halazonetis T.D.. An oligomerized 53BP1 tudor domain suffices for recognition of DNA double-strand breaks. Mol. Cell Biol. 2009; 29:1050–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mattiroli F., Vissers J.H., van Dijk W.J., Ikpa P., Citterio E., Vermeulen W., Marteijn J.A., Sixma T.K.. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell. 2012; 150:1182–1195. [DOI] [PubMed] [Google Scholar]
- 27. Wang Y., Jia S.. Degrees make all the difference: the multifunctionality of histone H4 lysine 20 methylation. Epigenetics. 2009; 4:273–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jorgensen S., Schotta G., Sorensen C.S.. Histone H4 lysine 20 methylation: key player in epigenetic regulation of genomic integrity. Nucleic Acids Res. 2013; 41:2797–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chapman J.R., Barral P., Vannier J.B., Borel V., Steger M., Tomas-Loba A., Sartori A.A., Adams I.R., Batista F.D., Boulton S.J.. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell. 2013; 49:858–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Di Virgilio M., Callen E., Yamane A., Zhang W., Jankovic M., Gitlin A.D., Resch W., Oliveira T.Y., Chait B.T. et al.. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science. 2013; 339:711–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Escribano-Diaz C., Orthwein A., Fradet-Turcotte A., Xing M., Young J.T., Tkac J., Cook M.A., Rosebrock A.P., Munro M., Canny M.D. et al.. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell. 2013; 49:872–883. [DOI] [PubMed] [Google Scholar]
- 32. Feng L., Fong K.W., Wang J., Wang W., Chen J.. RIF1 counteracts BRCA1-mediated end resection during DNA repair. J. Biol. Chem. 2013; 288:11135–11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zimmermann M., Lottersberger F., Buonomo S.B., Sfeir A., de Lange T.. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science. 2013; 339:700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Manke I.A., Lowery D.M., Nguyen A., Yaffe M.B.. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science. 2003; 302:636–639. [DOI] [PubMed] [Google Scholar]
- 35. Jowsey P.A., Doherty A.J., Rouse J.. Human PTIP facilitates ATM-mediated activation of p53 and promotes cellular resistance to ionizing radiation. J. Biol. Chem. 2004; 279:55562–55569. [DOI] [PubMed] [Google Scholar]
- 36. Boersma V., Moatti N., Segura-Bayona S., Peuscher M.H., van der Torre J., Wevers B.A., Orthwein A., Durocher D., Jacobs J.J.L.. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5′ end resection. Nature. 2015; 521:537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu G., Chapman J.R., Brandsma I., Yuan J., Mistrik M., Bouwman P., Bartkova J., Gogola E., Warmerdam D., Barazas M. et al.. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature. 2015; 521:541–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dev H., Chiang T.W., Lescale C., de Krijger I., Martin A.G., Pilger D., Coates J., Sczaniecka-Clift M., Wei W., Ostermaier M. et al.. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018; 20:954–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ghezraoui H., Oliveira C., Becker J.R., Bilham K., Moralli D., Anzilotti C., Fischer R., Deobagkar-Lele M., Sanchiz-Calvo M., Fueyo-Marcos E. et al.. 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature. 2018; 560:122–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gupta R., Somyajit K., Narita T., Maskey E., Stanlie A., Kremer M., Typas D., Lammers M., Mailand N., Nussenzweig A. et al.. DNA repair network analysis reveals shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell. 2018; 173:972–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mirman Z., Lottersberger F., Takai H., Kibe T., Gong Y., Takai K., Bianchi A., Zimmermann M., Durocher D., de Lange T.. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature. 2018; 560:112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Noordermeer S.M., Adam S., Setiaputra D., Barazas M., Pettitt S.J., Ling A.K., Olivieri M., Alvarez-Quilon A., Moatti N., Zimmermann M. et al.. The shieldin complex mediates 53BP1-dependent DNA repair. Nature. 2018; 560:117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang J., Aroumougame A., Lobrich M., Li Y., Chen D., Chen J., Gong Z.. PTIP associates with Artemis to dictate DNA repair pathway choice. Genes Dev. 2014; 28:2693–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huen M.S., Huang J., Leung J.W., Sy S.M., Leung K.M., Ching Y.P., Tsao S.W., Chen J.. Regulation of chromatin architecture by the PWWP domain-containing DNA damage-responsive factor EXPAND1/MUM1. Mol. Cell. 2010; 37:854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lo K.W., Kan H.M., Chan L.N., Xu W.G., Wang K.P., Wu Z., Sheng M., Zhang M.. The 8-kDa dynein light chain binds to p53-binding protein 1 and mediates DNA damage-induced p53 nuclear accumulation. J. Biol. Chem. 2005; 280:8172–8179. [DOI] [PubMed] [Google Scholar]
- 46. Barbar E. Dynein light chain LC8 is a dimerization hub essential in diverse protein networks. Biochemistry. 2008; 47:503–508. [DOI] [PubMed] [Google Scholar]
- 47. Leung J.W., Makharashvili N., Agarwal P., Chiu L.Y., Pourpre R., Cammarata M.B., Cannon J.R., Sherker A., Durocher D., Brodbelt J.S. et al.. ZMYM3 regulates BRCA1 localization at damaged chromatin to promote DNA repair. Genes Dev. 2017; 31:260–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F.. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013; 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. An L., Dong C., Li J., Chen J., Yuan J., Huang J., Chan K.M., Yu C.H., Huen M.S.Y.. RNF169 limits 53BP1 deposition at DSBs to stimulate single-strand annealing repair. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E8286–E8295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Maya-Mendoza A., Moudry P., Merchut-Maya J.M., Lee M., Strauss R., Bartek J.. High speed of fork progression induces DNA replication stress and genomic instability. Nature. 2018; 559:279–284. [DOI] [PubMed] [Google Scholar]
- 51. Barbar E., Kleinman B., Imhoff D., Li M., Hays T.S., Hare M.. Dimerization and folding of LC8, a highly conserved light chain of cytoplasmic dynein. Biochemistry. 2001; 40:1596–1605. [DOI] [PubMed] [Google Scholar]
- 52. Her J., Ray C., Altshuler J., Zheng H., Bunting S.F.. 53BP1 mediates ATR-Chk1 signaling and protects replication forks under conditions of replication stress. Mol. Cell Biol. 2018; 38:e00472-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Iwabuchi K., Bartel P.L., Li B., Marraccino R., Fields S.. Two cellular proteins that bind to wild-type but not mutant p53. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:6098–6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lo K.W., Naisbitt S., Fan J.S., Sheng M., Zhang M.. The 8-kDa dynein light chain binds to its targets via a conserved (K/R)XTQT motif. J. Biol. Chem. 2001; 276:14059–14066. [DOI] [PubMed] [Google Scholar]
- 55. Rapali P., Szenes A., Radnai L., Bakos A., Pal G., Nyitray L.. DYNLL/LC8: a light chain subunit of the dynein motor complex and beyond. FEBS J. 2011; 278:2980–2996. [DOI] [PubMed] [Google Scholar]
- 56. Rapali P., Radnai L., Suveges D., Harmat V., Tolgyesi F., Wahlgren W.Y., Katona G., Nyitray L., Pal G.. Directed evolution reveals the binding motif preference of the LC8/DYNLL hub protein and predicts large numbers of novel binders in the human proteome. PLoS One. 2011; 6:e18818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Houston S.I., McManus K.J., Adams M.M., Sims J.K., Carpenter P.B., Hendzel M.J., Rice J.C.. Catalytic function of the PR-Set7 histone H4 lysine 20 monomethyltransferase is essential for mitotic entry and genomic stability. J. Biol. Chem. 2008; 283:19478–19488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Oda H., Okamoto I., Murphy N., Chu J., Price S.M., Shen M.M., Torres-Padilla M.E., Heard E., Reinberg D.. Monomethylation of histone H4-lysine 20 is involved in chromosome structure and stability and is essential for mouse development. Mol. Cell Biol. 2009; 29:2278–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Harrison J.C., Haber J.E.. Surviving the breakup: the DNA damage checkpoint. Annu. Rev. Genet. 2006; 40:209–235. [DOI] [PubMed] [Google Scholar]
- 60. Xu Y., Ning S., Wei Z., Xu R., Xu X., Xing M., Guo R., Xu D.. 53BP1 and BRCA1 control pathway choice for stalled replication restart. Elife. 2017; 6:e30523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zou L., Cortez D., Elledge S.J.. Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev. 2002; 16:198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vassin V.M., Anantha R.W., Sokolova E., Kanner S., Borowiec J.A.. Human RPA phosphorylation by ATR stimulates DNA synthesis and prevents ssDNA accumulation during DNA-replication stress. J. Cell Sci. 2009; 122:4070–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. MacDougall C.A., Byun T.S., Van C., Yee M.C., Cimprich K.A.. The structural determinants of checkpoint activation. Genes Dev. 2007; 21:898–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shiotani B., Nguyen H.D., Hakansson P., Marechal A., Tse A., Tahara H., Zou L.. Two distinct modes of ATR activation orchestrated by Rad17 and Nbs1. Cell Rep. 2013; 3:1651–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kim D.H., Lee K.H., Kim J.H., Ryu G.H., Bae S.H., Lee B.C., Moon K.Y., Byun S.M., Koo H.S., Seo Y.S.. Enzymatic properties of the Caenorhabditis elegans Dna2 endonuclease/helicase and a species-specific interaction between RPA and Dna2. Nucleic Acids Res. 2005; 33:1372–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bohgaki M., Bohgaki T., El Ghamrasni S., Srikumar T., Maire G., Panier S., Fradet-Turcotte A., Stewart G.S., Raught B., Hakem A. et al.. RNF168 ubiquitylates 53BP1 and controls its response to DNA double-strand breaks. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:20982–20987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. He Y.J., Meghani K., Caron M.C., Yang C., Ronato D.A., Bian J., Sharma A., Moore J., Niraj J., Detappe A. et al.. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature. 2018; 563:522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Becker J.R., Cuella-Martin R., Barazas M., Liu R., Oliveira C., Oliver A.W., Bilham K., Holt A.B., Blackford A.N., Heierhorst J. et al.. The ASCIZ-DYNLL1 axis promotes 53BP1-dependent non-homologous end joining and PARP inhibitor sensitivity. Nat. Commun. 2018; 9:5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Roberts A.J., Kon T., Knight P.J., Sutoh K., Burgess S.A.. Functions and mechanics of dynein motor proteins. Nat. Rev. Mol. Cell Biol. 2013; 14:713–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. King S.M., Barbarese E., Dillman J.F. 3rd, Patel-King R.S., Carson J.H., Pfister K.K.. Brain cytoplasmic and flagellar outer arm dyneins share a highly conserved Mr 8,000 light chain. J. Biol. Chem. 1996; 271:19358–19366. [DOI] [PubMed] [Google Scholar]
- 71. Kamath R.S., Fraser A.G., Dong Y., Poulin G., Durbin R., Gotta M., Kanapin A., Le Bot N., Moreno S., Sohrmann M. et al.. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003; 421:231–237. [DOI] [PubMed] [Google Scholar]
- 72. Dick T., Ray K., Salz H.K., Chia W.. Cytoplasmic dynein (ddlc1) mutations cause morphogenetic defects and apoptotic cell death in Drosophila melanogaster. Mol. Cell Biol. 1996; 16:1966–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lottersberger F., Bothmer A., Robbiani D.F., Nussenzweig M.C., de Lange T.. Role of 53BP1 oligomerization in regulating double-strand break repair. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:2146–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Vadlamudi R.K., Bagheri-Yarmand R., Yang Z., Balasenthil S., Nguyen D., Sahin A.A., den Hollander P., Kumar R.. Dynein light chain 1, a p21-activated kinase 1-interacting substrate, promotes cancerous phenotypes. Cancer Cell. 2004; 5:575–585. [DOI] [PubMed] [Google Scholar]
- 75. Rayala S.K., den Hollander P., Balasenthil S., Yang Z., Broaddus R.R., Kumar R.. Functional regulation of oestrogen receptor pathway by the dynein light chain 1. EMBO Rep. 2005; 6:538–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.