

Abstract
Porous organic cages have emerged over the last 10 years as a subclass of functional microporous materials. However, among all of the organic cages reported, large multicomponent organic cages with 20 components or more are still rare. Here, we present an [8 + 12] porous organic imine cage, CC20, which has an apparent surface area up to 1752 m2 g–1, depending on the crystallization and activation conditions. The cage is solvatomorphic and displays distinct geometrical cage structures, caused by crystal-packing effects, in its crystal structures. This indicates that larger cages can display a certain range of shape flexibility in the solid state, while remaining shape persistent and porous.
Short abstract
By increasing the bend angle between amine groups in diaminocyclohexane precursors, we have successfully directed the formation of an [8 + 12] porous organic cage. The cuboctahedral-shaped cage is flexible in the solid state, and it can be processed into two different porous crystalline solids, which have contrasting stabilities and sorption properties.
Porous organic cages (POCs) are discrete polymacrocyclic molecules that contain a permanent, guest-accessible intrinsic cavity and that are porous to guests such as gases in the solid state. Since the first reports in 2009,1 the number of porous organic cages (POCs) appearing in the literature has increased dramatically, although in comparison to other classes of porous materials, they are still relatively uncommon.2−6 POCs with different shapes and sizes have now been reported, and promising applications have been demonstrated, such as gas storage,7 molecular separations,8−11 sensing,12,13 catalyst supports,14−17 and porous additives.18 In contrast to extended porous frameworks, such as metal–organic frameworks (MOFs)19 and covalent organic frameworks (COFs),20,21 the permanent porosity of organic cages is mainly attributed to the intrinsic cavities present in these discrete, shape-persistent molecules. These molecular pores can also be solubilized to produce porous liquids.22,23 Previous efforts toward the preparation of new organic cages have tended to focus on the assembly, via reversible reactions, of one24 or two distinct starting materials into cages that comprise fewer than 20 subunits. The synthesis of larger multicomponent organic cages with high surface areas and large pore volume is rare. To date, Warmuth,25,26 Mastalerz,7,27 Gawronski,28 Beuerle,29,30 and our group31 have successfully obtained large organic cages from at least 20 subunits. However, with the exception of Mastalerz’s [8 + 12] boronic ester cages,7,27 none of these large cages exhibits permanent porosity in the solid state. This could be due to collapse of the desolvated cages as a cumulative effect of limited rotation in multiple “rigid” bonds.31 Also, not all studies provide gas sorption data; thus, it is possible that some systems are, in fact, porous but that this was not explored.
For the rational design of large organic cages via a bottom-up strategy, it is important to recognize that small changes in the bond angles between the reactive functionalities in the starting materials can have a profound effect on the outcome of the reaction. For example, we reported the synthesis of two imine-based organic cages, CC5 and CC7, synthesized from tris(4-formylphenyl)amine and homochiral trans-1,2-cyclopentanediamine or trans-1,2-cyclohexanediamine (trans-1,2-CHDA), respectively.31 The addition of a single extra carbon atom into the vicinal diamine-functionalized ring resulted in a minor change to the bond angle between the diamine groups, which increased the size of the cage product from a 10-component [4 + 6] cage to a 20-component [8 + 12] cage. Likewise, Fujita demonstrated that slight changes to bond angles between pyridyl ligand donors significantly affect the structures of metal–organic polyhedra, which he referred to as “emergent behavior”.33 A similar phenomenon was observed by Iwasawa and Beuerle for the synthesis of boroxine and boronate ester cages, respectively.24,30 Fujita, Iwasawa, and Beuerle all found that increasing the angle between the reacting groups led to the formation of larger cages containing more components, while decreasing the bond angle between the reacting groups often led to the formation of smaller cages.
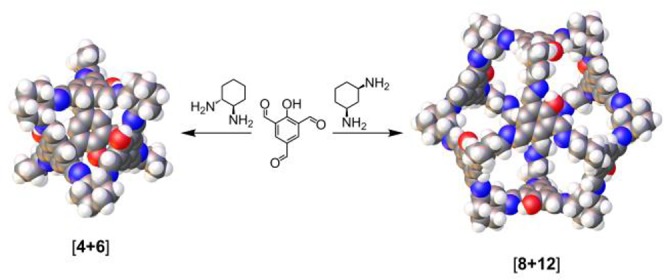
Large cages can be targeted either by increasing the dimensions of the building blocks34 or by keeping the building blocks the same size and increasing the number of units in the cage. As discussed above, the latter may be achieved by increasing the bond angle between the reactive groups in the starting materials. Recently, Petryk et al.35 reported the synthesis of a CC3(1) analogue in which each aromatic ring contains a single hydroxyl group that is disordered throughout the structure (Scheme 1). Here, we report the synthesis of an expanded analogue of this cage, synthesized using cis-1,3-cyclohexanediamine (cis-1,3-CHDA) in place of the homochiral trans-1,2-CHDA that was used in the synthesis of the smaller cage. The increase in the bond angle between the amines in the cis-1,3-CHDA results in a change in cage topology from a 10-component tetrahedral [4 + 6] cage to a 20-component cubic [8 + 12] cage (Scheme 1).
Scheme 1. Synthesis of the [4 + 6] Cage Reported by Petryk et al.35 and the [8 + 12] Cage Reported Here, Both by Imine Condensation.
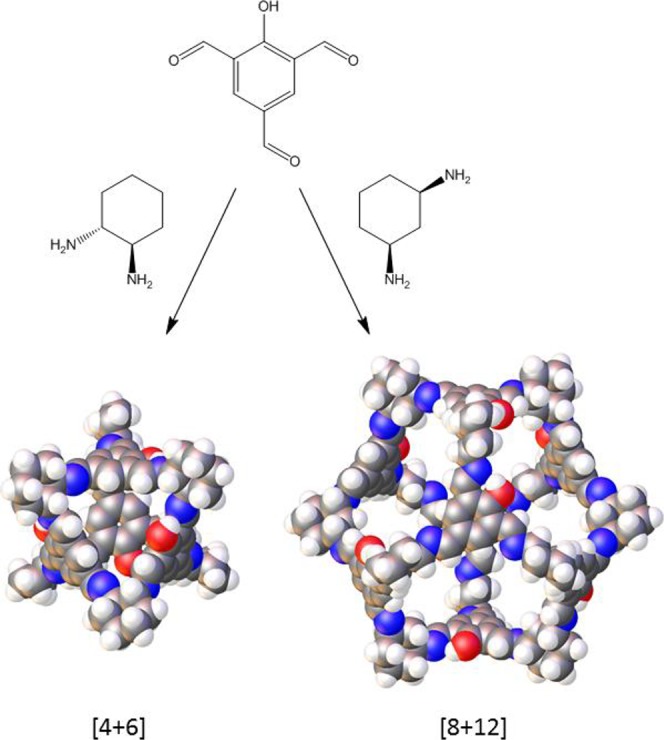
In each cage, all aromatic rings contain a single hydroxyl group that is disordered throughout the structure. Atoms colors are as follows: gray, carbon; white, hydrogen; blue, nitrogen; red, oxygen.
Results and Discussion
CC20 was obtained by the self-assembly of 8 molecules of 2-hydroxy-1,3,5-triformylbenzene (HO-TFB) with 12 molecules of cis-1,3-CHDA. CC20 is similar in composition to the recently reported [4 + 6] cage, synthesized from HO-TFB and homochiral trans-1,2-CHDA.35 Topologically, it can be regarded as a cube where the arene panels map onto the vertices of the cube and the cyclohexanes define the edges. Geometrically, it can be defined as a cuboctahedron with 8 triangular faces (arenes) and 6 square faces (windows). Interestingly, CC20 was observed to form multiple solvatomorphs. A number of different solvates were obtained in which the shape of the cage was distorted from the ideal, symmetrical structure. Several of these solvates were found to be porous to N2 at 77 K after desolvation.
Initial attempts to synthesize a cis-1,3-CHDA-containing cage focused on reacting the diamine with 1,3,5-triformylbenzene (TFB). However, multiple experimental attempts (using different solvents, concentrations, and stirring rates)36 all gave complex product mixtures by 1H NMR, alongside an insoluble white precipitate. It has been reported that the introduction of a suitably positioned hydroxyl group can aid the formation of cage product.37 With this in mind, we switched from using TFB to HO-TFB. HO-TFB was recently reported by Petryk et al. to form a [4 + 6] cage by reaction with chiral trans-1,2-CHDA (Scheme 1). On mixing solutions of cis-1,3-CHDA with HO-TFB in DCM/MeOH, an insoluble yellow precipitate was formed initially. After the reaction mixture stood for 1 week, crystals began to grow on the walls of the flask. During optimization of the experimental conditions, it was found that layering a dilute solution of the cis-1,3-CHDA onto solid HO-TFB, without stirring, avoided the formation of the initial insoluble precipitate; a small amount of TFA was also observed to catalyze the reaction. After 2 weeks, a yellow crystalline product formed on the inner wall of the flask, this time without any insoluble coprecipitate. 1H and 13C NMR, diffusion NMR, and high-resolution mass spectrometry were consistent with the formation of an [8 + 12] cage (Figures S1–S5). The successful outcome of this reaction suggests that the hydroxyl group on the aromatic ring is required to stabilize the geometrical structure of the [8 + 12] cage intermediates by forming salicylic imines with intramolecular hydrogen bonding—in the absence of hydroxyl groups, this [8 + 12] cage did not form.
To better understand the formation of CC20, the reaction was repeated in a d2-DCM/d4-MeOH solution (1/1 by volume) without TFA catalyst and the progress of the reaction was monitored by 1H NMR (Figure S6). From the 1H NMR spectra, the appearance of a broad peak between 8.6 and 8.8 ppm after 15 min indicates the rapid formation of imine bonds, although the broad peak shape suggests the formation of multiple misaligned soluble oligomers and proto-cages. Over time, the aldehyde peaks (9.7 and 10.4 ppm) disappeared, and the two broad peaks (between 8.0 and 8.8 ppm) were resolved into four sharp peaks consistent with the formation of a highly symmetrical imine cage as the initial kinetic products slowly equilibrate to the thermodynamic cage product. During this period, crystals were observed to form on the inner wall of the NMR tube, which accounts for the decrease in the intensity of the cage signals in the 1H NMR.
Four solvated single-crystal structures of CC20 were identified. Directly from the synthesis in CH2Cl2 and MeOH we obtained large solvated crystals (Figure S7) with cubic Pm3 symmetry. In the Pm3 structure, there are two crystallographically distinct CC20 conformers (Figure 1a,b). While both CC20 conformers have open cuboctahedral shapes, comparable to the shape of the [8 + 12] cage reported by Gawronski,28 one of the cage conformers has an elliptical shape, which elongates the cage windows (Figure 1a). CC20 does not have bulky tert-butyl substituents on the vertices like the Gawronski cage. In the CC20Pm3 structure, the cages are packed inefficiently window to window, resulting in the formation of large interconnected extrinsic voids (Figure 1c). These voids are filled with disordered solvent in the crystal structure.
Figure 1.

Single-crystal structure and crystal packing of different CC20 solvates recorded at 100 K: (a, b) two distinct CC20 conformers in Pm3 DCM/MeOH solvate, which pack window to window in the crystal structure; (c, d) window to windows packing in the Im3n-pentane phase; (e) window to vertex packing of CC20 in the CHCl3/MeCN P1 solvate, which reduces the size of extrinsic voids between cages in the crystal structure; (f, g) crystal packing in the n-pentane P1 solvate. Cage conformers with pronounced elliptical shapes are highlighted in green, and disordered OH groups and all H atoms are omitted for clarity.
Gas-phase computational simulations show that both conformations geometry optimize to the same conformation with no other conformer present within 20 kJ mol–1 of this low-energy conformer. Thus, these two conformations of the cage are not distinct minima when the molecule is considered in isolation and their presence is likely due to crystal-packing effects in the solid state. This suggests that there is some flexibility in the cage, although computational modeling supported by experimental porosity measurements also suggests that the cage remains shape-persistent upon desolvation.
To evaluate the stability of the Pm3 structure, solvent exchanges of the reaction solvent were carried out in the crystal pores. After the reaction solvent was exchanged with n-pentane, the Pm3 crystals underwent a single-crystal to single-crystal transformation to a cubic Im3 structure. In the higher symmetry Im3 crystal structure, there is just one crystallographically distinct cage molecule, but the open cuboctahedral cage shape and crystal packing of CC20 were both retained in the n-pentane-solvated Im3 structure (Figure 1c vs Figure 1d). This transformation highlights the influence that solvation effects can have upon the conformation of cage molecules in the solid-state.
Recrystallization of CC20 from a CHCl3 and MeCN solution yielded needle-shaped crystals with triclinic P1 symmetry (Figure 1e,f). In the P1 crystal structure, the drop in crystallographic symmetry in comparison to the cubic structures is related to the crystal packing of the cages, rather than a loss of the open cuboctahedral cage shape, although one of the CC20 conformers in the P1 solvate phase also has an elliptical shape. In the CHCl3/MeCN P1 solvate, the crystallographically distinct cages are packed in a window to vertex fashion (Figure 1e), which reduces the size of extrinsic voids in CC20 (Figure 1c vs Figure 1f). The packing density of cages in this solvated structure is 0.62 g cm–3, in comparison with 0.51 g cm–3 calculated for the cubic solvates. Interestingly, this P1 solvated phase also underwent a single-crystal to single-crystal transformation to a second P1 phase, after exchanging the CHCl3 and MeCN crystallization solvent in the pores with n-pentane. In the n-pentane P1 solvate, the elliptical shape of the two crystallographically distinct cage conformers are less pronounced, but the crystal packings in the two P1 solvate structures are different, and the packing density of cages in this structure is 0.70 g cm–3 (Figure 1f vs Figure 1g). These crystallization experiments have demonstrated that solvent can be used to direct the packing of CC20, which we have used here to prevent the formation of energetically unstable extrinsic voids.
In comparison with the [4 + 6] cage synthesized by Petryk et al., the larger CC20 must be desolvated more carefully before gas sorption. The as-synthesized Pm3 structure is poorly crystalline after the reaction solvent is removed from the pores, and this phase was found to be nonporous to N2 after desolvation under vacuum at 90 °C (Figure 2 and Figures S8–S10). However, careful desolvation of the n-pentane activated Im3 phase under a flow of nitrogen followed by vacuum at ambient temperature afforded a porous phase (Figure 2 and Figures S11 and S12). In contrast, drying the pentane solvate in air for 2 days results in loss of crystallinity (Figure S13). The carefully desolvated sample was found to be porous with an SABET value of 829 m2/g. However, after activation and post gas sorption, PXRD patterns indicate that this material transforms after the n-pentane is removed from the crystal structure (Figure S11).
Figure 2.
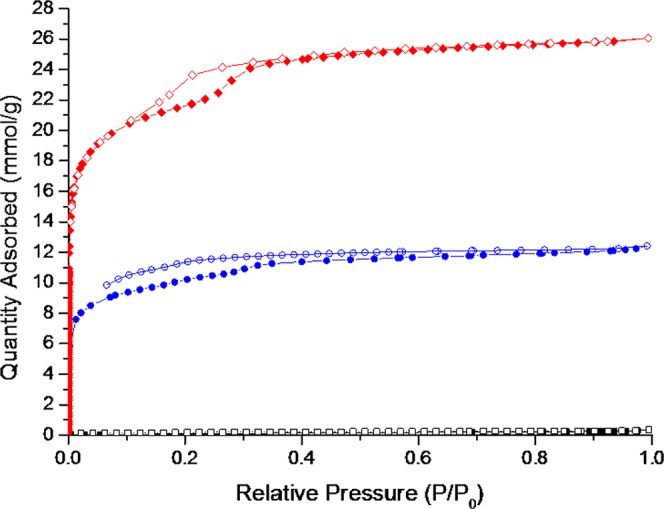
Nitrogen sorption isotherms of CC20 polymorphs at 77.3 K: DCM/MeOH Pm phase activated at 90 °C under vacuum (black squares), n-pentane Im phase activated under vacuum at room temperature (blue circles), and CHCl3/MeCN crystallized P phase activated at 30 °C under vacuum after exchanging the crystallization solvent with n-pentane (red diamonds) (closed symbols for adsorption, open symbols for desorption).
The as-crystallized P1 CHCl3/MeCN phase was carefully activated by initially exchanging the crystallization solvent with n-pentane and then by heating this P1n-pentane solvate at 30 °C under vacuum. After activation using these conditions, we found that this phase performed better in terms of structural stability and it remained crystalline after gas-sorption analysis, according to PXRD measurements (Figure S14). Thermogravimetric analysis (TGA) also indicates that the CC20 material, crystallized and activated using the same conditions, is stable and crystalline up to 300 °C, when it is heated under N2 (Figures S15 and S16). The SABET value for this phase was 1752 m2/g, which is one of the higher values reported so far for POCs (Figure 2 and Figure S17); the current record is 3758 m2/g,7 but the vast majority of POCs have SABET values <1000 m2/g.6 The desolvated P1 phase also shows a large xenon uptake of 3.44 mmol/g at 1 bar and 273 K; by comparison, the smaller [4 + 6] CC3 has a Xe uptake of 2.69 mmol/g at 1 bar and 298 K (Figures S17 and S18), but unlike CC3, CC20 does not appear to be saturated at 1 bar Xe pressure.8
Differences were observed between the simulated PXRD patterns of the Im n-pentane solvate and the P1 solvates, in comparison to the PXRD patterns recorded after desolvation and gas sorption analysis, indicating that the cage structures transform further during these processes (Figures S19 and S20). This is likely due to changes in the shape of CC20 upon desolvation and/or reorientation of the cages in the crystal lattice. We ascribe this to the “shape flexibility” exhibited by CC20 and the spherical nature of this cage. We have previously found that smaller spherical cages can be highly solvatomorphic38 and difficult to desolvate without pronounced structural transformations.
CC20 also showed fluorescent emission (Figures S20 and S21) when it was dissolved in chloroform (1 mg/mL) and irradiated with 357 or 497 nm light. Both excitation wavelengths result in emission at 534 nm (yellow-green light). This could potentially allow the application of this cage for molecular sensing.13
Summary
In conclusion, by increasing the bond angle between the reactive functionality on one of the starting materials, we have successfully synthesized a new [8 + 12] cage, CC20, by imine condensation. CC20 was observed to form several solvatomorphs by SCXRD, which contain a number of distinctly shaped cages due to crystal-packing effects. To the best of our knowledge, this is the first time that a “shape-persistent” POC has displayed flexibility in the solid state. The formation of the large CC20 was found to be more sensitive to the synthesis conditions than its smaller analogue, and the stabilizing effects of hydroxyl groups were required to form the cage. CC20 exhibits a very high SABET value of 1752 m2/g, which is among the highest reported for an organic molecular solid.
Acknowledgments
We thank the Engineering and Physical Sciences Research Council (EP/H000925/1) and the European Research Council under FP7 (RobOT, ERC grant agreement no. 321156). K.E.J. and T.H. thank the Royal Society for a University Research Fellowship. We thank the STFC for access to the Diamond Light Source and the staff at beamlines I19 (MT15777). We also thank Dr. Edward Eden-Rump for running the diffusion NMR experiment. We thank Dr. Rebecca Greenaway and the Microbiorefinery for assistance with HRMS/QTOF measurements.
Glossary
Abbreviations
- CCX
a “covalent cage” in a series published by the Cooper group, X being the number of cages reported in order of publishing
- PXRD
powder X-ray diffraction
- BET
Brunauer–Emmett–Teller
- SCXRD
single-crystal X-ray diffraction
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.cgd.8b01761.
Full synthetic and experimental details and characterization data, crystallographic details, and computational details (PDF)
Accession Codes
CCDC 1881384–1881387 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- Tozawa T.; Jones J. T. A.; Swamy S. I.; Jiang S.; Adams D. J.; Shakespeare S.; Clowes R.; Bradshaw D.; Hasell T.; Chong S. Y.; Tang C.; Thompson S.; Parker J.; Trewin A.; Bacsa J.; Slawin A. M. Z.; Steiner A.; Cooper A. I. Nat. Mater. 2009, 8, 973–978. 10.1038/nmat2545. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Mastalerz M. Chem. Soc. Rev. 2014, 43, 1934–1947. 10.1039/C3CS60358J. [DOI] [PubMed] [Google Scholar]
- Chong S. Y.; Cooper A. I.. Porous Organic Cages, 2nd ed.; Elsevier: 2017. [Google Scholar]
- Beuerle F.; Gole B. Angew. Chem., Int. Ed. 2018, 57, 4850–4878. 10.1002/anie.201710190. [DOI] [PubMed] [Google Scholar]
- Evans J. D.; Sumby C. J.; Doonan C. J. Chem. Lett. 2015, 44, 582–588. 10.1246/cl.150021. [DOI] [Google Scholar]
- Hasell T.; Cooper A. I. Nat. Rev. Mater. 2016, 1, 16053. 10.1038/natrevmats.2016.53. [DOI] [Google Scholar]
- Zhang G.; Presly O.; White F.; Oppel I. M.; Mastalerz M. Angew. Chem., Int. Ed. 2014, 53, 1516–1520. 10.1002/anie.201308924. [DOI] [PubMed] [Google Scholar]
- Chen L.; Reiss P. S.; Chong S. Y.; Holden D.; Jelfs K. E.; Hasell T.; Little M. A.; Kewley A.; Briggs M. E.; Stephenson A.; Thomas K. M.; Armstrong J. A.; Bell J.; Busto J.; Noel R.; Liu J.; Strachan D. M.; Thallapally P. K.; Cooper A. I. Nat. Mater. 2014, 13, 954–960. 10.1038/nmat4035. [DOI] [PubMed] [Google Scholar]
- Miklitz M.; Jiang S.; Clowes R.; Briggs M. E.; Cooper A. I.; Jelfs K. E. J. Phys. Chem. C 2017, 121, 15211–15222. 10.1021/acs.jpcc.7b03848. [DOI] [Google Scholar]
- Hasell T.; Miklitz M.; Stephenson A.; Little M. A.; Chong S. Y.; Clowes R.; Chen L.; Holden D.; Tribello G. A.; Jelfs K. E.; Cooper A. I. J. Am. Chem. Soc. 2016, 138, 1653–1659. 10.1021/jacs.5b11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; Wang Q.; Long H.; Zhang W. J. Am. Chem. Soc. 2011, 133, 20995–21001. 10.1021/ja210418t. [DOI] [PubMed] [Google Scholar]
- Brutschy M.; Schneider M. W.; Mastalerz M.; Waldvogel S. R. Adv. Mater. 2012, 24, 6049–6052. 10.1002/adma.201202786. [DOI] [PubMed] [Google Scholar]
- Zwijnenburg M. A.; Berardo E.; Peveler W. J.; Jelfs K. E. J. Phys. Chem. B 2016, 120, 5063–5072. 10.1021/acs.jpcb.6b03059. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Xiong Y.; Ge J.; Lin R.; Chen C.; Peng Q.; Wang D.; Li Y. Chem. Commun. 2018, 54, 2796–2799. 10.1039/C7CC09918E. [DOI] [PubMed] [Google Scholar]
- Mondal B.; Acharyya K.; Howlader P.; Mukherjee P. S. J. Am. Chem. Soc. 2016, 138, 1709–1716. 10.1021/jacs.5b13307. [DOI] [PubMed] [Google Scholar]
- Qiu L.; McCaffrey R.; Jin Y.; Gong Y.; Hu Y.; Sun H.; Park W.; Zhang W. Chem. Sci. 2018, 9, 676–680. 10.1039/C7SC03148C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffrey R.; Long H.; Jin Y.; Sanders A.; Park W.; Zhang W. J. Am. Chem. Soc. 2014, 136, 1782–1785. 10.1021/ja412606t. [DOI] [PubMed] [Google Scholar]
- Jiang S.; Chen L.; Briggs M. E. E.; Hasell T.; Cooper A. I. I. Chem. Commun. 2016, 52, 6895–6898. 10.1039/C6CC01034B. [DOI] [PubMed] [Google Scholar]
- Zhou H.-C.; Long J. R.; Yaghi O. M. Chem. Rev. 2012, 112, 673–674. 10.1021/cr300014x. [DOI] [PubMed] [Google Scholar]
- Segura J. L.; Mancheño M. J.; Zamora F. Chem. Soc. Rev. 2016, 45, 5635–5671. 10.1039/C5CS00878F. [DOI] [PubMed] [Google Scholar]
- Feng X.; Ding X.; Jiang D. Chem. Soc. Rev. 2012, 41, 6010–6022. 10.1039/c2cs35157a. [DOI] [PubMed] [Google Scholar]
- Giri N.; Del Pópolo M. G.; Melaugh G.; Greenaway R. L.; Rätzke K.; Koschine T.; Pison L.; Gomes M. F. C.; Cooper A. I.; James S. L. Nature 2015, 527, 216. 10.1038/nature16072. [DOI] [PubMed] [Google Scholar]
- Greenaway R. L.; Holden D.; Eden E. G. B.; Stephenson A.; Yong C. W.; Bennison M. J.; Hasell T.; Briggs M. E.; James S. L.; Cooper A. I. Chem. Sci. 2017, 8, 2640–2651. 10.1039/C6SC05196K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono K.; Johmoto K.; Yasuda N.; Uekusa H.; Fujii S.; Kiguchi M.; Iwasawa N. J. Am. Chem. Soc. 2015, 137, 7015–7018. 10.1021/jacs.5b02716. [DOI] [PubMed] [Google Scholar]
- Xu D.; Warmuth R. J. Am. Chem. Soc. 2008, 130, 7520–7521. 10.1021/ja800803c. [DOI] [PubMed] [Google Scholar]
- Liu X.; Warmuth R. J. Am. Chem. Soc. 2006, 128, 14120–14127. 10.1021/ja0644733. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Presly O.; White F.; Oppel I. M.; Mastalerz M. Angew. Chem., Int. Ed. 2014, 53, 1516–1520. 10.1002/anie.201308924. [DOI] [PubMed] [Google Scholar]
- Skowronek P.; Warzajtis B.; Rychlewska U.; Gawroński J. Chem. Commun. 2013, 49, 2524–2526. 10.1039/c3cc39098e. [DOI] [PubMed] [Google Scholar]
- Klotzbach S.; Scherpf T.; Beuerle F. Chem. Commun. (Cambridge, U. K.) 2014, 50, 12454–12457. 10.1039/C4CC01794C. [DOI] [PubMed] [Google Scholar]
- Klotzbach S.; Beuerle F. Angew. Chem., Int. Ed. 2015, 54, 10356–10360. 10.1002/anie.201502983. [DOI] [PubMed] [Google Scholar]
- Jelfs K. E.; Wu X.; Schmidtmann M.; Jones J. T. A.; Warren J. E.; Adams D. J.; Cooper A. I. Angew. Chem., Int. Ed. 2011, 50, 10653–10656. 10.1002/anie.201105104. [DOI] [PubMed] [Google Scholar]
- Sun Q.-F. Q.-F.; Iwasa J.; Ogawa D.; Ishido Y.; Sato S.; Ozeki T.; Sei Y.; Yamaguchi K.; Fujita M. Science 2010, 328, 1144–1147. 10.1126/science.1188605. [DOI] [PubMed] [Google Scholar]
- Ding H.; Yang Y.; Li B.; Pan F.; Zhu G.; Zeller M.; Yuan D.; Wang C. Chem. Commun. 2015, 51, 1976–1979. 10.1039/C4CC08883B. [DOI] [PubMed] [Google Scholar]
- Petryk M.; Szymkowiak J.; Gierczyk B.; Spólnik G.; Popenda Ł.; Janiak A.; Kwit M. Org. Biomol. Chem. 2016, 14, 7495–7499. 10.1039/C6OB01429A. [DOI] [PubMed] [Google Scholar]
- Briggs M. E.; Cooper A. I. Chem. Mater. 2017, 29, 149–157. 10.1021/acs.chemmater.6b02903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M. W.; Oppel I. M.; Griffin A.; Mastalerz M. Angew. Chem., Int. Ed. 2013, 52, 3611–3615. 10.1002/anie.201208156. [DOI] [PubMed] [Google Scholar]
- McMahon D. P.; Stephenson A.; Chong S. Y.; Little M. A.; Jones J. T. A.; Cooper A. I.; Day G. M. Faraday Discuss. 2018, 211, 383–399. 10.1039/C8FD00031J. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.