

Abstract
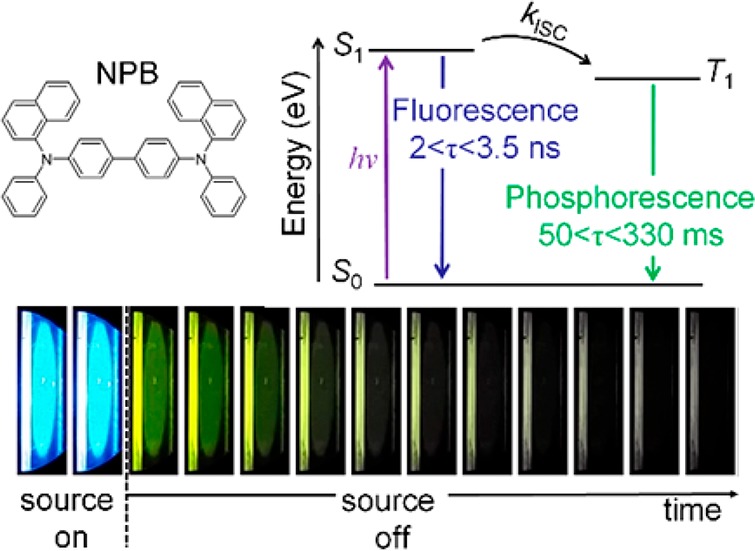
Biluminescent organic emitters show simultaneous fluorescence and phosphorescence at room temperature. So far, the optimization of the room-temperature phosphorescence in these materials has drawn the attention of research. However, the continuous-wave operation of these emitters will consequently turn them into systems with vastly imbalanced singlet and triplet populations, which is due to the respective excited-state lifetimes. This study reports on the exciton dynamics of the biluminophore NPB (N,N′-di(1-naphthyl)-N,N′-diphenyl-(1,1-biphenyl)-4,4-diamine). In the extreme case, the singlet and triplet exciton lifetimes stretch from 3 ns to 300 ms, respectively. Through sample engineering and oxygen quenching experiments, the triplet exciton density can be controlled over several orders of magnitude, allowing us to study exciton interactions between singlet and triplet manifolds. The results show that singlet–triplet annihilation reduces the overall biluminescence efficiency already at moderate excitation levels. Additionally, the presented system represents an illustrative role model to study excitonic effects in organic materials.
Introduction
The electronic properties of organic molecules are characterized by distinct spin manifolds as a consequence of joint effects of highly localized excitations and, compared with inorganic materials, low dielectric constants.1 The results are typical energetic splitting of first singlet and triplet excited states, respectively, on the order of a few hundred millielectronvolts.1 Organic molecules typically only show efficient fluorescence, that is, the emission from the singlet state, as it is an allowed transition.2 On the contrary, the transition strength from the triplet state to ground state under emission of a photon (phosphorescence) is extremely weak because it is quantum mechanically forbidden, requiring the participating electron to undergo a spin flip.2
In the vast of organic molecules, a low rate of radiative transition in the triplet manifold remains due to weak coupling; however, it is outcompeted by nonradiative recombination at room temperature. As a consequence, the triplet state in organic molecules is typically considered a “dark” state. Still, phosphorescence of organic molecules can be achieved by two means (cf. Figure 1a): (i) enhancement of the radiative phosphorescence rate kr,P to outcompete the nonradiative channel knr,P or (ii) suppress knr,P in favor of a dominating kr,P. The first route is followed in a concerted way for the development of phosphorescent emitters for organic light-emitting diodes (OLEDs), owing to the fact that ∼75% of the formed excitons are born as triplets.3 Here heavy metal atoms are incorporated in the core of a molecular structure to enhance kr,P due to strong spin–orbit coupling (SOC).4 A weaker version of the same strategy is the increase in kr,P, making use of SOC induced by a metal atom in the vicinity of the emitter (external heavy atom effect).5 Furthermore, special molecular designs can equally be in favor of a moderately enhanced phosphorescence rate.6−8 Route (ii) is most effectively realized by incorporating the emitter molecules at low concentration in rigid polymer hosts9 or alternative matrices10 in addition to engineering the molecular packing to effectively restrict the intramolecular motions by the intermolecular interactions of the molecules in crystal phase.11−14 Such room-temperature phosphorescence (RTP) from purely organic molecules has attracted much attention recently15−20 because the persistent nature of the phosphorescence is a unique luminescence feature allowing for various novel applications.21,22
Figure 1.

(a) General energy diagram of an organic biluminophore. Above this scheme, a photograph of a spin-coated film on a quartz substrate showing the emission from the singlet state (S1) mainly and the afterglow emission from the triplet state (T1). After photon absorption upon excitation (hv,ex) from the ground state (S0), the S1 can be deactivated radiatively as fluorescence (kr,F) and nonradiatively via internal conversion (knr,F) or intersystem crossing (kISC) to T1. Following energy transfer from S1, the possible deactivation paths of T1 are radiative as phosphorescence (kr,P) and nonradiative (knr,P) via internal conversion and quenching. (b) Photoluminescence (PL) spectra of a typical biluminescent system, under a nitrogen atmosphere at room temperature, displaying the contributions from the singlet (S1) and triplet (T1) excited states. (c) From left to right, chemical structures of the polymer matrix PMMA, the biluminophore NPB, and the small-molecule matrix TCTA. Arrows point out the techniques used to deposit the films as follows: sc for PMMA:NPB (black, 1) and TCTA:NPB (red, 2) as well as thermal evaporation for TCTA:NPB (blue, 3).
In all studies to date, the sole focus has been put on the enhancement of the phosphorescence of organic molecules. However, light is emitted in such systems from both their singlet (fluorescence) and triplet excited states (Figure 1a,b), which is due to a competition of kISC, kr,F, and knr,F (i.e., intersystem crossing, fluorescence radiative, and nonradiative rates). Principally, because the oscillator strength is much larger in the singlet manifold, excitation of the molecules is almost exclusively realized via absorption to singlet states (S0 → S1). This biluminescence, or dual-state emission, can be efficient for any given mixing ratio between fluorescence and phosphorescence if both kr,F ≫ knr,F and kr,P ≫ knr,P. Essentially, the typical lifetimes of fluorescence (nanoseconds) and phosphorescence (milliseconds to seconds) differ greatly with direct influence on the overall population of the molecules. Of course, these novel emitters are of particular interest for potential applications because their luminescence characteristics can be retained at room temperature.
Experimental Section
The studies were carried out on thin films deposited either by spin coating (sc) or thermal evaporation in ultrahigh vacuum, respectively. The biluminescent emitter is NPB (N,N′-di(1-naphthyl)-N,N′-diphenyl-(1,1-biphenyl)-4,4-diamine) (Lumtec), which is dispersed into either the polymer host PMMA (Alfa Aesar) or the small-molecule host TCTA (Lumtec). PMMA was used as received, and TCTA and NPB were purified by vacuum train sublimation twice before use. Solvent polarity and dilution level were optimized for the wet processing of the small-molecule host system (TCTA) to obtain homogeneous solutions and films.23
Here all materials were dissolved in methoxybenzene (Anisol, purity 99.7%) at concentrations of 80, 40, and 10 mg/mL for PMMA, TCTA, and NPB, respectively. Quartz substrates were cleaned based on the following sequence: 15 min treatment in ultrasonic bath (UB15) in DIW and soap; rinsing with DIW; UB15 in ethanol, UB15 in IPA, and finally 10 min of O2 plasma after drying with nitrogen. For the spin-coated films, NPB is embedded at a concentration of 2 wt % onto the host (PMMA or TCTA). Such dilutions are spin-coated from a methoxybenzene solution onto glass substrates (1″ by 1″ by 1 mm) at a spin speed of 2000 rpm (1000 rpm/s ramp) for 60 s.
Thermal evaporated films were formed by coevaporation of TCTA and NPB on an unheated quartz glass substrate using an ultrahigh vacuum deposition system (Kurt J. Lesker Company) at a deposition rate of 0.8 A/s under a pressure below 3 × 10–7 mbar. The target concentration was 2 wt % as well to match the solution-processed samples.
PL measurements were performed under a nitrogen atmosphere (N2 6.0, purity 99.9999%) and ambient conditions (air) at room temperature. For the acquisition of time-resolved phosphorescence and emission spectra data, a customized setup was used, where nitrogen is flowing continuously in a sample box to guarantee an inert atmosphere during measurements. PL spectra were recorded with a spectrometer (CAS 140CTS, Instrument Systems) and the LED (M365L2, λmax = 365 nm, Thorlabs) operating in continuous-wave (cw) mode to obtain the integrated spectrum. The LED was driven by a T-Cube LED driver (LEDD1B, Thorlabs), and a band-pass filter (FB370-10, Thorlabs) was placed in front of the LED to avoid an overlap between excitation and emission spectra. Phosphorescence spectra were collected with the same instruments with the addition of the pulse generator (Keysight (Agilent) Technologies 8114A) to separate phosphorescence from fluorescence in a time-gated scheme. Therefore, a pulse train of 500 ms LED-on and 500 ms LED-off was generated by the pulse generator. The spectrometer was triggered with a delay of 20 ms to the falling edge of the LED-on pulse and integrated for 450 ms, while the excitation source was off, assuring no detection of the next pulse. The spectrometer CAS 140CTS was gated to 20 ms integration windows to resolve the PL during a 500 ms long pump pulse of the UV LED. These windows were started with different delays (0, 50, ..., 450 ms) to the beginning of the pump pulse. The time-resolved PL intensity of a complete duty cycle was recorded with a switchable gain silicon photodetector (PDA100A, Thorlabs).
The time-resolved PL setup to measure the fluorescence and phosphorescence lifetimes consisted of a time-correlated single-photon-counting (TCSPC) system (TimeHarp 260 PICO, Picoquant). The excitation source was a pulsed diode laser PDL 820 with a 374 nm laser head running at 40 MHz repetition rate. A single photon-sensitive PMA hybrid was used for detection. The count rate was adjusted to <1% of the laser rate to prevent pile-up. Data acquisition was set to a resolution of 0.2 ns to record the prompt fluorescence lifetime and 1.3 ms for the phosphorescence lifetime. Phosphorescence decays were recorded following a high repetition (80 MHz) burst excitation composed of many subnanosecond pulses (λex = 374 nm), which, for the long phosphorescence lifetime, functions as a high-intensity excitation pulse.
Photoluminescence quantum yield (PLQY) measurements were performed in a customized setup for measurements under a nitrogen atmosphere at oxygen levels below 0.1%. This setup consists of an antistatic glovebox with transfer chamber (SICCO), 6″ integrating sphere (Labsphere) using an LED (M340L4, λmax = 340 nm, Thorlabs) as excitation source driven by the LEDD1B driver (Thorlabs), and a spectrometer (CAS 140CTS, Instrument Systems), following de Mello method24 for the estimation of the absolute PLQY, and the spectra were corrected for wavelength-dependent instrument sensitivity.
The thin films’ morphology was analyzed by X-ray reflectivity (XRR). The mean film density was determined from a 13-point XRR measurement using a Jordan Valley JVX 5200 XRR thin-film measurement system. The characterization of the vibrational modes was performed using a Nicolet iS 10 FT-IR spectrometer (Thermo Fisher Scientific) in attenuated total reflection (ATR) mode, with a diamond crystal used as window and a 20 nm gold layer underneath the film of interest.
Results and Discussion
In this study, we present an investigation on the interplay between fluorescence and phosphorescence in such organic biluminophores that puts the consequences of the of the more than six orders of magnitude difference between singlet and triplet states into the spotlight. Here we vary the host matrices embedding the archetypical biluminescent emitter NPB (N,N′-di(1-naphthyl)-N,N′-diphenyl-(1,1-biphenyl)-4,4-diamine), which allows for a variation of the triplet population under steady-state conditions as a result of a changed knr,P. Furthermore, we investigate the PL under both nitrogen and ambient (air) environments, where in the latter case, the triplet population is effectively quenched by oxygen.25
For NPB, we have reported that it shows efficient phosphorescence at room temperature (RTP) in addition to conventional fluorescence.9 Furthermore, NPB is widely known and used in the field of OLEDs as hole-transporting material, which is why we use it here as an archetypical biluminescent emitter in this study. It is known that the RTP strongly depends on the host material, as it is the latter that represents the interface to nonradiative deactivation pathways.9,10 Similar to our previous reports, we use PMMA with triplet energy of 3.1 eV26 and glass-transition temperature Tg of 108 °C27 as a reference matrix material based on its ability to form rigid glasses, which is beneficial for observation of phosphorescence at room temperature.9 Additionally, to vary the environment of the emitter NPB and at the same time resemble the high triplet energy requirement and glassy character of the host in a small molecule, we chose the starburst-shaped small-molecule TCTA (4,4′,4″-tris (N-carbazolyl)-triphenylamine) with triplet energy of 2.79 eV28 and Tg of 151 °C29 as an alternative host material. This material choice is further motivated by the possibility to integrate the effect of biluminescence to electroluminescent devices, which has to date only been done using specially designed host materials.30Figure 1c shows the chemical structures of all three molecules of this study and also illustrates which processing schemes are used to fabricate the samples. Because of its physical dimensions, the polymer PMMA-based system is only fabricated by wet-processing techniques, while for TCTA, the mixed thin films are processed by spin-coating (sc) and thermal evaporation. All samples are made with the same NPB concentration of 2 wt % to allow for best comparability. The biluminescence reference system [PMMA:NPB]sc 2 wt % shows oxygen-dependent phosphorescence, with an excited-state lifetime in the range of a few hundred milliseconds (see later discussion for details). The typical fluorescence and phosphorescence emission bands with emission maxima at 425 and 540 nm, respectively, are depicted in the biluminescent spectrum plotted in Figure 1b.
Figure 2a shows the time-resolved fluorescence and phosphorescence of all samples, which were acquired in the respective time window needed. The corresponding fluorescence and phosphorescence emission bands are plotted for all samples in Figure 2b. The phosphorescence band is detected after the excitation pulse is turned off. All decay curves deviate from monoexponential decay. Therefore, the transient intensities I are fitted with a biexponential decay of the following form (cf. Figure S1)
| 1 |
where A1 and A2 are the relative contributions of the individual decays with lifetimes τ1 and τ2, respectively. Abg is a constant offset that describes the instrument background intensity. It is used in fit calculations, if reached within the time window. Deviation from monoexponential decay, as observed here for all systems, has been reported before for similar systems.31 Average weighted lifetimes for the transients are calculated according to the following expression
| 2 |
These lifetimes are used in the following discussion as representative excited-state lifetimes for reasons of simplicity. For all samples, we have made two identical samples and calculated a mean value obtained from the individual samples. The fluorescence lifetimes vary only slightly between the three samples in the range of 2 to 3 ns (cf. Table 1, values obtained in nitrogen). In stark contrast, the phosphorescence lifetime differs significantly. Here [PMMA:NPB]sc shows the longest lifetime of 323 ms, followed by the spin-coated [TCTA:NPB]sc system with a lifetime of 96 ms. Clearly the shortest average weighted lifetime is observed for the thermally evaporated (evap) system [TCTA:NPB]evap with 54 ms. Hence, the phosphorescence lifetimes differ by as much a factor of 6 between the [PMMA:NPB]sc reference and the [TCTA:NPB]evap system.
Figure 2.

Characteristics of biluminescent systems when the emitting molecule (NPB) is diluted at 2 wt % in the host matrix. (a) Normalized dynamic response under nitrogen conditions in the nanosecond range, corresponding to the prompt fluorescence radiative decay rate. (b) Normalized dynamic response under nitrogen conditions in the millisecond range, corresponding to the delayed (phosphorescence) radiative decay rate. (c) Normalized fluorescence spectra (dashed line) of biluminescent films excited at λex = 365 nm and continuous wave source and phosphorescence spectra (solid line) of biluminescent films excited at λex = 365 nm with a pulsed source under a nitrogen atmosphere at room temperature. Residual contributions from delayed fluorescence are cut in the spectra of panel c. For the complete delayed emission spectra, refer to Figure S3.
Table 1. Photophysical Properties of Biluminescent Systems at Room Temperature.
| τ́F (ns)d | τ́P (ms)e | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| systema | Fmax (nm)b | Pmax (nm)c | N2 | air | N2 | air | PLQY (%)f | PLQYF (%)g | PLQYP (%)h | IF,N2/IF,O2 (%)i | ITOTAL,N2/ITOTAL,O2j |
| [PMMA:NPB]sc | 425 | 540 | 2.6 | 2.6 | 322.6 | 10.4 | 26 ± 3 | 23.1 | 2.6 | 77 | 0.81 |
| [TCTA:NPB]sc | 440 | 540 | 3.4 | 3.4 | 95.6 | 10.1 | 31 ± 8 | 30.4 | 0.4 | 96 | 0.96 |
| [TCTA:NPB]evap | 430 | 540 | 2.0 | 2.3 | 53.8 | 7.1 | 32 ± 3 | 31.6 | 0.2 | 98 | 0.99 |
Data for systems in which NPB is diluted at 2 wt % in the host matrix.
Measured fluorescence emission maxima.
Measured phosphorescence emission maxima.
Fitted (biexponential, average weighted) fluorescence lifetimes.
Fitted (biexponential, average weighted) phosphorescence lifetimes.
Photoluminescence quantum yield.
Fluorescence quantum yield.
Phosphorescence quantum yield (cf. Figure S5 for details of PLQY determination).
Nitrogen-to-air fluorescence intensity maxima ratio.
Nitrogen-to-air total integrated intensity ratio.
The experimental phosphorescence lifetime can be expressed as
| 3 |
The change in phosphorescence lifetime cannot be caused by an alteration in the radiative rate kr,P, simply because the different chosen environments cannot introduce a coupling strong enough to influence the phosphorescence transition strength because neither PMMA nor TCTA contains elements such as metals or halogens atoms that can enhance SOC or induce heavy atom effect to the NPB molecules. Therefore, the nonradiative rate knr,P must be responsible for the observed changes of the phosphorescence lifetime. Here, in general, various mechanisms exist that possibly contribute to this rate. While vibrational deactivation kvib is considered to be the primary cause,32 higher order effects like triplet–triplet annihilation (TTA), or even quenching at static impurity sites with a rate kimp can easily add to an effective nonradiative rate. The coefficient rate for TTA (kTTA) depends on the actual interaction partner concentration
| 4 |
While the intermolecular forces for small molecules are the same as for polymers (i.e., van der Waals forces), polymer molecules are large systems, where the magnitude of their intermolecular forces can exceed those between small molecules.33 Therefore, a polymer matrix like PMMA brings higher rigidity to embedded NPB molecules, which, in turn, suppresses nonradiative deactivation.9,10,34 For the difference between the two TCTA-based systems (sc and evap), we can only speculate in this stage. It is possible that the formation through spin-coating, being a highly nonequilibrium process, induces mechanical constraints to the system that similarly introduces rigidity to the film that suppresses nonradiative channels. In contrast, the fabrication via thermal evaporation will allow the molecules to find their energetically most favorable conformation within the mixed film, which leads to a softer packing allowing for more nonradiative modes. We have investigated these systems systematically with X-ray diffraction and Fourier-transform infrared spectroscopy (cf. Figure S2) to possibly identify differences in the constitution of the different samples that may hint to the arguments from above. However, we did not observe significant differences. All samples are amorphous, allowing us to exclude effects of crystal formation to affect our observations, which is an alternative route to enhance RTP.35−37 We have observed delayed fluorescence as a result of TTA. Here the large singlet–triplet energy splitting of NPB (∼0.6 eV38) rules out up-conversion via thermally activation, which can only overcome barriers of <0.37 eV.39 TTA-mediated delayed fluorescence has been previously reported only for organic crystals exhibiting RTP40 as a side effect of the proximity between molecules and formation of agglomerates. In the three biluminescent systems, TTA is strongest for the [TCTA:NPB]evap samples, which is monitored through delayed fluorescence (cf. Figure S3). For the remaining discussion, we consider nonradiative losses as one effective rate knr,P, as given in eq 4, influencing the phosphorescent lifetime τp, knowing that it, strictly seen, is a quantity that decreases during the excited-state decay if TTA is significant.
Possible applications making use of biluminescence such as optical sensing do require operation in quasi-cw regime to allow the phosphorescence intensity to build up properly. Here quasi-cw is understood as a scenario where the pump duration is on the same order of magnitude as the triplet state lifetime τp. For a molecule similar to NPB, namely, (BzP)PB [N,N′-bis(4-benzoyl-phenyl)-N,N′-diphenyl-benzidine], we have already observed a decrease in the integrated luminescence, that is, fluorescence and phosphorescence, during a quasi-cw excitation pulse.31 While appointing this effect to singlet-triplet annihilation (STA) with a coefficient rate kSTA,41 we did not investigate this effect further.31
This important interplay between the two luminescent states will be discussed in the following. The strength of STA is proportional to both singlet nS and triplet exciton densities nT(41)
| 5 |
With the three systems discussed above having distinctively different triplet lifetimes τp (cf. Table 1), the magnitude of STA should also vary between the samples, as the triplet exciton density nT scales with the lifetime τp. To reduce STA to an absolute minimum, we performed additional measurements (transient and quasi-cw) in an ambient environment (air), making use of the fact that oxygen very effectively quenches the triplet states with a rate kq,O2.25,32 Hence, under the presence of oxygen, eq 3 becomes
| 6 |
Figure 3 shows the fluorescence (a) and phosphorescence (b) decays following a pulsed excitation for all three systems. The fluorescence shows no differences between nitrogen and air measurements. This result with the additional information that all three systems show slightly different fluorescence decays (cf. Figure 2a and Table 1) leads to the conclusion that none of the three involved rates, that is, kr,F, knr,F, and kISC, are altered by the presence of oxygen. In contrast, the phosphorescence decays of all samples are much faster in air compared with the measurement in a nitrogen atmosphere as a result of an effective kq. Interestingly, all three transients in air show very similar time constants showing some residual radiative contribution giving rise to τp ≈ 10 ms (cf. Table 1). This suggests that the oxygen quenching is strong but not completely outcompeting kr,P. For all samples, the oxygen quenching effectively reduces the triplet density under steady-state conditions by at least one order of magnitude (cf. Table 1).
Figure 3.

Time resolved luminescence of the different biluminescent systems [PMMA:NPB]sc (black), [TCTA:NPB]sc (red), and [TCTA:NPB]evap (blue) in nitrogen (solid line) and air (dashed line). (a) Fluorescence decays following a single, subns pulse with λex = 374 nm. (b) Phosphorescence decays of the samples following a high repetition (80 MHz) burst excitation composed of many subns pulses (λex = 374 nm). This is to ensure a sufficiently high triplet population to detect the long-lived phosphorescence. The horizontal gray lines indicate the instrument background.
Figure 4a–c show the cw-PL of all samples obtained under exactly the same measurement configuration for both nitrogen and ambient environments. In air, the remaining phosphorescence contribution (cf. Figure 3b) is so weak that it is masked by the low-energy tail of the fluorescence spectrum. The PL spectra in a nitrogen atmosphere show two important differences: (i) the relative phosphorescence intensity scales with the phosphorescence lifetimes, where the reference system [PMMA:NPB]sc shows the highest phosphorescence intensity. (ii) The fluorescence intensity of all samples decreases with respect to the measurement in air. As mentioned above in the discussion of the time-resolved data, none of the outgoing rates from the S1 level can be the reason for this observation. Figure 4d shows the ratio between peak fluorescence under nitrogen and air (also cf. Table 1), showing the strongest reduction for the [PMMA:NPB]sc system (77%) and the weakest one for the [TCTA:NPB]evap (98%), again in agreement with STA scaling with the triplet exciton density nT.
Figure 4.

Comparison of the spectral characteristics of the different samples in nitrogen and air. PL spectra are taken under continuous-wave excitation in both air (dashed line) and nitrogen (solid line) environments without changing the geometry of the setup, allowing for comparability of the respective two measurements. Spectra are normalized with respect to the fluorescence maximum to the luminescence in air. (a) [PMMA:NPB]sc (black), (b) [TCTA:NPB]sc (red), (c) [TCTA:NPB]evap (blue), (d) ratio of the absolute fluorescence peaks between nitrogen and air environment for the three different systems (a–c).
Figure 5a shows
the PL intensity as a function of time for the [PMMA:NPB]sc reference system during 500 ms on, 500 ms off duty cycle quasi-cw
pulse. While the air measurement shows constant intensity during the
on-pulse, the corresponding intensity obtained under a nitrogen atmosphere
gradually decreases, reaching a steady state close to the off-time. Figure 5b shows the intensity
of the fluorescence and phosphorescence as a function of time during
the on-pulse, as obtained from spectrally resolved time-gated measurements.
Here the fluorescence decreases, while at the same time the phosphorescence
increases. For the phosphorescence data points, it is important to
note that the spectral integration window (500–700 nm) accounts
not only for the phosphorescence but also for the low-energy tail
of the fluorescence band (cf. Figure 4a). Therefore, the increase in the phosphorescence
is counteracted by a fraction of the fluorescence, which decreases
with time. A detailed analysis of the phosphorescence during the on-pulse
is given in the Supporting Information (cf.
Figure S6). The increase in phosphorescence and by that the triplet
exciton density, nT, is due to the long
phosphorescent lifetime τp ≈ 330 ms of the
[PMMA:NPB]sc system. In accordance with eq 4, the singlet exciton density nS, and consequently the fluorescence intensity,
will decrease with increasing nT, as excited
singlet states are absorbed by excited triplet states with a coefficient
rate kSTA. This process is an inherent
loss channel for the biluminescent system, as singlet exciton energy
is dissipated in a nonradiative  cycle. This is also reflected in the PLQY
data of the three systems. While the two systems comprising TCTA as
host, which show only minor reductions in fluorescence between air
and nitrogen measurement (cf. Figure 4d), show 31 and 32% PLQY for [TCTA:NPB]sc and [TCTA:NPB]evap, respectively, the corresponding value
for the [PMMA:NPB]sc reference systems is lower (26%).
This difference in PLQY fits well to the reduction of 77% observed
in Figure 4d (cf. Table 1). The same trend
is observed if we compare the total integrated spectrum in nitrogen
and air (cf. Table 1). Note that under these circumstances, the PLQY must be considered
to be a function of the excitation density, because of STA and other
possible nonlinear processes that depend on the density of the involved
excitons.
cycle. This is also reflected in the PLQY
data of the three systems. While the two systems comprising TCTA as
host, which show only minor reductions in fluorescence between air
and nitrogen measurement (cf. Figure 4d), show 31 and 32% PLQY for [TCTA:NPB]sc and [TCTA:NPB]evap, respectively, the corresponding value
for the [PMMA:NPB]sc reference systems is lower (26%).
This difference in PLQY fits well to the reduction of 77% observed
in Figure 4d (cf. Table 1). The same trend
is observed if we compare the total integrated spectrum in nitrogen
and air (cf. Table 1). Note that under these circumstances, the PLQY must be considered
to be a function of the excitation density, because of STA and other
possible nonlinear processes that depend on the density of the involved
excitons.
Figure 5.
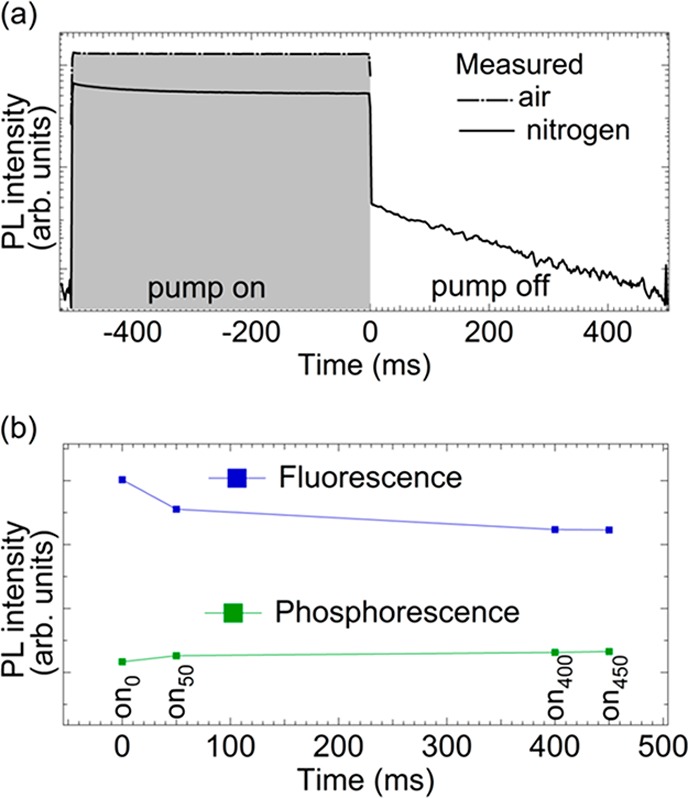
Development of singlet and triplet populations during quasi-cw excitation pulses. (a) PL intensity of the [PMMA:NPB]sc reference system during and after a 500 ms excitation pulse under air (dashed line) and nitrogen (solid line) environments. (b) Intensities of fluorescence (380–500 nm) and phosphorescence (500–700 nm) obtained in 20 ms integration windows with different delays to the pump pulse start. Here on0 = no delay, on50 = 50 ms delay, on400 = 400 ms delay, and on450 = 450 ms delay.
Conclusions
This study discusses the exciton dynamics of the efficient, room-temperature biluminescent emitter NPB. The radiative lifetimes of fluorescence and phosphorescence are in the range of 3 ns and 300 ms, respectively, spanning eight orders of magnitude. This difference in lifetimes puts the biluminescent system in strong imbalance between singlet and triplet exciton densities under cw operation, which can lead to a limitation of the biluminescence efficiency as a result of nonlinear processes. Here STA reduces the fluorescence intensity significantly. By sample engineering, the triplet exciton lifetime can be reduced through an increase in the nonradiative rate knr,P, which leads to an effectively suppressed STA. Hence, depending on the actual pump intensities that will dictate the exciton densities in both spin manifolds, an upper limit for the phosphorescence lifetime can be deduced until which STA is only of minor effect. Moreover, a convenient and simple method to observe biluminescence from purely organic molecules is demonstrated, in which commercially available small molecules and polymer are mixed to form a biluminescent host–guest system, with no need of complicated material synthesis or functionalization. Our report shows that small-molecule matrix materials, which normally diverge significantly in their nanostructure from polymers, are also capable of promoting biluminescence of purely organic molecules at room temperature.
While for some applications the persistent nature of the RTP is a key feature, other areas of use may exclusively call for high-efficiency and broadband, dual-state emission. Future material design for biluminescent or RTP emitters will have a careful optimization of the triplet state’s dynamics to be of central importance.
Acknowledgments
We are grateful for the support of the German Excellence Initiative via the Cluster of Excellence EXC 1056 Center for Advancing Electronics Dresden (cfaed). This work has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 679213). We thank Caroline Walde, Tobias Günther, and Andreas Wendel for film evaporation, Dr. Lutz Wilde from Fraunhofer IPMS for XRR measurements, Dr. Norwid-Rasmus Behrnd and Dr. Ramunas Lygaitis for discussions concerning the morphology of the films, and, finally, Dr. Axel Fischer and Dr. Reinhard Scholz for further discussions on the topic of this manuscript.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcc.7b04529.
Additional data of the fluorescence and phosphorescence transients and curve fittings; FTIR, XRR and GIXRD spectra of the TCTA:NPB system; additional data of the PL spectra showing TTA-based delayed fluorescence in PMMA:NPB and TCTA:NPB systems; calculation of the fluorescence and phosphorescence quantum yield; and experimental determination of the singlet and triplet populations of the PMMA:NPB system. (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Bässler H.; Köhler A. Charge Transport in Organic Semiconductors. Top. Curr. Chem. 2011, 312, 1–66. 10.1007/128_2011_218. [DOI] [PubMed] [Google Scholar]
- Harris D. C.; Bertolucci M. D.. Symmetry and Spectroscopy: An Introduction to Vibrational and Electronic Spectroscopy; Oxford University Press: Oxford, U.K., 1978. [Google Scholar]
- Baldo M. A.; O’Brien D. F.; Thompson M. E.; Forrest S. R. Excitonic Singlet-Triplet Ratio in a Semiconducting Organic Thin Film. Phys. Rev. B: Condens. Matter Mater. Phys. 1999, 60, 14422–14428. 10.1103/PhysRevB.60.14422. [DOI] [Google Scholar]
- Klessinger M.; Michl J.. Excited States and Photochemistry of Organic Molecules; Wiley-VCH: New York, 1995. [Google Scholar]
- Samonina-Kosicka J.; De Rosa C. A.; Morris W. A.; Fan Z.; Fraser C. L. Dual-Emissive Difluoro Boronnaphthyl-Phenyl β-Diketonate Polylactide Materials: Effects of Heavy Atom Placement and Polymer Molecular Weight. Macromolecules 2014, 47, 3736–3746. 10.1021/ma5006606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.; Mao Z.; Zhang X.; Ou D.; Mu Y.; Zhang Y.; Zhao C.; Liu S.; Chi Z.; Xu J.; et al. Intermolecular Electronic Coupling of Organic Units for Efficient Persistent Room-Temperature Phosphorescence. Angew. Chem., Int. Ed. 2016, 55, 2181–2185. 10.1002/anie.201509224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata S.; Totani K.; Watanabe T.; Kaji H.; Vacha M. Relationship Between Room Temperature Phosphorescence and Deuteration Position in a Purely Aromatic Compound. Chem. Phys. Lett. 2014, 591, 119–125. 10.1016/j.cplett.2013.11.019. [DOI] [Google Scholar]
- Zhao W.; He Z.; Lam J. W. Y.; Peng Q.; Ma H.; Shuai Z.; Bai G.; Hao J.; Tang B. Z. Rational Molecular Design for Achieving Persistent and Efficient Pure Organic Room Temperature Phosphorescence. Chem. 2016, 1, 592–602. 10.1016/j.chempr.2016.08.010. [DOI] [Google Scholar]
- Reineke S.; Baldo M. A. Room Temperature Triplet State Spectroscopy of Organic Semiconductors. Sci. Rep. 2015, 4, 3797. 10.1038/srep03797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata S.; Totani K.; Zhang J.; Yamashita T.; Kaji H.; Marder S. R.; Watanabe T.; Adachi C. Efficient Persistent Room Temperature Phosphorescence in Organic Amorphous Materials Under Ambient Conditions. Adv. Funct. Mater. 2013, 23, 3386–3397. 10.1002/adfm.201203706. [DOI] [Google Scholar]
- Xie Y.; Ge Y.; Peng Q.; Li C.; Li Q.; Li Z. How the Molecular Packing Affects the Room Temperature Phosphorescence in Pure Organic Compounds: Ingenious Molecular Design, Detail Crystal Analysis, and Rational Theoretical Calculations. Adv. Mater. 2017, 29, 1606829. 10.1002/adma.201606829. [DOI] [PubMed] [Google Scholar]
- Gong Y.; Zhao L.; Peng Q.; Fan D.; Yuan W. Z.; Zhang Y.; Tang B. Z. Crystallization-Induced Dual Emission from Metal and Heavy Atom-Free Aromatic Acids and Esters. Chem. Sci. 2015, 6, 4438. 10.1039/C5SC00253B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An Z.; Zheng C.; Chen R. C.; Shi H.; Chen T.; Wang Z.; li H.; Deng R.; Liu X.; et al. Stabilizing Triplet Excited States for Ultralong Organic Phosphorescence. Nat. Mater. 2015, 14, 685–690. 10.1038/nmat4259. [DOI] [PubMed] [Google Scholar]
- Hong Y.; Lam J. W. Y.; Tang B. Z. Aggregation-Induced Emission: Phenomenon, Mechanism and Applications. Chem. Commun. 2009, 4332–4352. 10.1039/b904665h. [DOI] [PubMed] [Google Scholar]
- Kwon M. S.; Yu Y.; Coburn C.; Phillips A. W.; Chung K.; Shanker A.; Jung J.; Kim G.; Pipe K.; Forrest S. R.; et al. Suppressing Molecular Motions for Enhanced Room-Temperature Phosphorescence of Metal-Free Organic Materials. Nat. Commun. 2015, 6, 8947. 10.1038/ncomms9947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Xie T.; Cui M.; Yang L.; Sun X.; Jiang J.; Zhang G. General Design Strategy for Aromatic Ketone-Based Single-Component Dual-Emissive Materials. ACS Appl. Mater. Interfaces 2014, 6, 2279–2284. 10.1021/am405209w. [DOI] [PubMed] [Google Scholar]
- Lee D.; Bolton O.; Kim B. C.; Youk J. H.; Takayama; Kim J. Room Temperature Phosphorescence of Metal-Free Organic Materials in Amorphous Polymer Matrices. J. Am. Chem. Soc. 2013, 135, 6325–6329. 10.1021/ja401769g. [DOI] [PubMed] [Google Scholar]
- Gong Y.; Chen G.; Peng Q.; Yuan W. Z.; Xie Y.; Li S.; Zhang Y.; Tang B. Z. Achieving Persistent Room Temperature Phosphorescence and Remarkable Mechanochromism from Pure Organic Luminogens. Adv. Mater. 2015, 27, 6195–6201. 10.1002/adma.201502442. [DOI] [PubMed] [Google Scholar]
- Xu J.; Takai A.; Kobayashi Y.; Takeuchi M. Phosphorescence from a Pure Organic Fluorene Derivative in Solution at Room Temperature. Chem. Commun. 2013, 49, 8447–8449. 10.1039/c3cc44809f. [DOI] [PubMed] [Google Scholar]
- Li C.; Tang X.; Zhang L.; Li C.; Liu Z.; Bo Z.; Dong Y. Q.; Tian Y.-H.; Dong Y.; Tang B. Z. Reversible Luminescence Switching of an Organic Solid: Controllable On–Off Persistent Room Temperature Phosphorescence and Stimulated Multiple Fluorescence Conversion. Adv. Opt. Mater. 2015, 3, 1184–1190. 10.1002/adom.201500115. [DOI] [Google Scholar]
- Hirata S.; Totani K.; Yamashita T.; Adachi C.; Vacha M. Large Reverse Saturable Absorption Under Weak Continuous Incoherent Light. Nat. Mater. 2014, 13, 938–946. 10.1038/nmat4081. [DOI] [PubMed] [Google Scholar]
- Lehner P.; Staudinger C.; Borisov S. M.; Klimant I. Ultra-Sensitive Optical Oxygen Sensors for Characterization of Nearly Anoxic Systems. Nat. Commun. 2014, 5, 4460. 10.1038/ncomms5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan L.; Hou L.; Lee T.-W.; Qiao J.; Zhang D.; Dong G.; Wang L.; Qiu Y. Solution Processable Small Molecules for Organic Light-Emitting Diodes. J. Mater. Chem. 2010, 20, 6392–6407. 10.1039/b926348a. [DOI] [Google Scholar]
- De Mello J. C.; Wittmann H. F.; Friend R. H. An Improved Experimental Determination of External Photoluminescence Quantum Efficiency. Adv. Mater. 1997, 9, 230–232. 10.1002/adma.19970090308. [DOI] [Google Scholar]
- Garner A.; Wilkinson F. Quenching of Triplet States by Molecular Oxygen and the Role of Charge-Transfer Interactions. Chem. Phys. Lett. 1977, 45, 432–435. 10.1016/0009-2614(77)80057-2. [DOI] [Google Scholar]
- Avdeenko A. A.; Dobrovolskaya T. L.; Kultchitsky V. A.; Naboikin Y. V.; Pakulov S. N. Temperature-Dependence of Luminescence Decay Time of Benzyl. J. Lumin. 1976, 11, 331–337. 10.1016/0022-2313(76)90017-X. [DOI] [Google Scholar]
- Porter C. E.; Blum F. D. Thermal Characterization of PMMA Thin Films Using Modulated Differential Scanning Calorimetry. Macromolecules 2000, 33, 7016–7020. 10.1021/ma000302l. [DOI] [Google Scholar]
- Reineke S.; Schwartz G.; Walzer K.; Leo K. Direct Observation of Host-Guest Triplet-Triplet Annihilation in Phosphorescent Solid Mixed Films. Phys. Status Solidi RRL 2009, 3, 67–69. 10.1002/pssr.200802266. [DOI] [Google Scholar]
- Xiao L.; Chen Z.; Qu B.; Luo J.; Kong S.; Gong Q.; Kido J. Recent Progresses on Materials for Electrophosphorescent Organic Light-Emitting Devices. Adv. Mater. 2011, 23, 926–952. 10.1002/adma.201003128. [DOI] [PubMed] [Google Scholar]
- Kabe R.; Notsuka N.; Yoshida K.; Adachi C. Afterglow Organic Light-Emitting Diode. Adv. Mater. 2016, 28, 655–660. 10.1002/adma.201504321. [DOI] [PubMed] [Google Scholar]
- Reineke S.; Seidler N.; Yost S. R.; Prins F.; Tisdale W. A.; Baldo M. A. Highly Efficient, Dual State Emission from an Organic Semiconductor. Appl. Phys. Lett. 2013, 103, 093302. 10.1063/1.4819444. [DOI] [Google Scholar]
- Turro N. J.Modern Molecular Photochemistry; University Science Books: Sausalito, CA, 1991. [Google Scholar]
- Israelachvili J. N.Intermolecular and Surface Forces; Academic Press: San Diego, CA, 2011. [Google Scholar]
- Oster G.; Geacintov N.; Ullah Khan A. Luminescence in Plastics. Nature 1962, 196, 1089–1090. 10.1038/1961089a0. [DOI] [Google Scholar]
- Yuan W. Z.; Shen X. Y.; Zhao H.; Lam J. W. Y.; Tang L.; Lu P.; Wang C.; Liu Y.; Wang Z.; Zheng Q.; et al. Crystallization-Induced Phosphorescence of Pure Organic Luminogens at Room Temperature. J. Phys. Chem. C 2010, 114, 6090–6099. 10.1021/jp909388y. [DOI] [Google Scholar]
- Gong Y.; Zhao L.; Peng Q.; Fan D.; Yuan W. Z.; Zhang Y.; Tang B. Z. Crystallization-Induced Dual Emission from Metal- and Heavy Atom-Free Aromatic Acids and Esters. Chem. Sci. 2015, 6, 4438–4444. 10.1039/C5SC00253B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H.; An Z.; Li P.-Z.; Yin J.; Xing G.; He T.; Chen H.; Wang J.; Sun H.; Huang W.; et al. Enhancing Organic Phosphorescence by Manipulating Heavy-Atom Interaction. Cryst. Growth Des. 2016, 16, 808–813. 10.1021/acs.cgd.5b01400. [DOI] [Google Scholar]
- Jankus V.; Winscom C.; Monkman A. P. Dynamics of Triplet Migration in Films of N,N′-Diphenyl-N, N′-Bis(1-naphthyl)-1, 1′-Biphenyl-4, 4″-Diamine. J. Phys.: Condens. Matter 2010, 22, 185802. 10.1088/0953-8984/22/18/185802. [DOI] [PubMed] [Google Scholar]
- Chen T.; Zheng L.; Yuan J.; An Z.; Chen R.; Tao Y.; Li H.; Xie X.; Huang W. Understanding the Control of Singlet-Triplet Splitting for Organic Exciton Manipulating: a Combined Theoretical and Experimental Approach. Sci. Rep. 2015, 5, 10923. 10.1038/srep10923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno S.; Akeno H.; Ohtani H.; Yuasa H. Visible Room-Temperature Phosphorescence of Pure Organic Crystals Via a Radical-Ion-Pair Mechanism. Phys. Chem. Chem. Phys. 2015, 17, 15989–15995. 10.1039/C5CP01203A. [DOI] [PubMed] [Google Scholar]
- Gärtner C.; Karnutsch C.; Lemmer U.; Pflumm C. The Influence of Annihilation Processes on the Threshold Current Density of Organic Laser Diodes. J. Appl. Phys. 2007, 101, 023107. 10.1063/1.2425003. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.