

Abstract
Objective:
The opioid epidemic in the United States has led to unprecedented increases in morbidity and mortality, posing a serious public health crisis. Although twin and family studies, as well as genome-wide association studies (GWAS), all identify significant genetic factors contributing to opioid dependence, no studies to date have estimated marker-based heritability estimates of opioid dependence. The goal of the current study was to use a large, genetically imputed, case/control sample of 4,064 participants (after quality control and imputation) with genome-wide data to estimate the unbiased heritability tagged by single nucleotide polymorphisms (SNPs).
Method:
Study data were part of the Genome-wide Study of Heroin Dependence obtained via the Database for Genotypes and Phenotypes (dbGaP). Genomic-Relatedness-Matrix Restricted Maximum Likelihood with adjustment for minor allele frequency (MAF) and linkage disequilibrium (LD; GREML-LDMS) was used to determine the variation in opioid dependence attributable to common SNPs from imputed data. Mixed linear models were used in an exploratory GWAS to assess effects of single SNPs.
Results:
At least 45% of the variance in opioid dependence according to the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, was attributable to common SNPs, after stratifying to account for differences in MAF and LD across the genome. Most of the genetic variance was tagged by SNPs in the 1%–9% MAF range and in low LD with other SNPs in the region. Two markers in LOC101927293 survived multiple-testing correction (i.e., q value < .05).
Conclusions:
Nearly half of the variation in opioid dependence can be attributed to common SNPs. Most of this variation is due to rare variants in low LD with other markers in the region.
The unprecedented increases in morbidity and mortality associated with the opioid epidemic are indicative of the worst drug epidemic in U.S. history (Kolodny et al., 2015). According to the Department of Health and Human Services, in 2015, 33,091 people died from overdosing on opioids (Rudd et al., 2016). This staggering mortality rate reflects more than a 4-fold increase since 1999 (Centers for Disease Control and Prevention, 2016). In 2015, 12.5 million people misused prescription opioids (2.1 million people for the first time), and 828,000 people used heroin (135,000 of these for the first time; Center for Behavioral Health Statistics and Quality [2015 National Survey on Drug Use and Health: Detailed Tables, 2016]). Opioid use cuts across all age, economic, and racial groups (Manchikanti et al., 2012), underscoring the need for studies that can aid in identifying individuals who are at risk for developing opioid dependence.
Twin and family studies provide evidence for the genetic liability of various opioid use–related phenotypes. Studies have estimated a heritability (i.e., the extent to which genetic variation accounts for phenotypic variation) of 43% for lifetime risk of opioid dependence in males (Tsuang et al., 1996, 2001), approximately 50% for lifetime opioid use (Karkowski et al., 2000; Kendler et al., 1999, 2000), and 49%–69% for various opioid use subtypes (Sun et al., 2012). In one study, consisting of all males, opioid abuse had a large contribution of genetic factors not shared by other substances including cannabis, stimulants, sedatives, and psychedelics, sharing only half of its total variance with the common vulnerability across all substances (Tsuang et al., 1998).
Contrary to legalized substances (i.e., alcohol, tobacco, and cannabis [in some states]), there have been fewer genome-wide and candidate-gene association studies for opioid dependence phenotypes. A review of published literature using a combination of search terms —for example, “(alcohol OR tobacco) OR (cannabis OR marijuana)) AND (GWAS [genome-wide association studies] or candidate gene)” for legalized substances; “(opioid or heroin) AND (GWAS or candidate gene)” for opioid/heroin—in PubMed (search completed in July 2018) revealed 3,154 studies of alcohol, tobacco, or cannabis/marijuana versus 321 for heroin/opioid phenotypes. As reviewed by Jensen (2016), only three GWAS identified markers that achieved genome-wide significance (e.g., p < 5 × 10-8) for opioid dependence (Gelernter et al., 2014; Li et al., 2015; Nelson et al., 2016). The results of several studies indicate genetic susceptibility for opioid dependence in dopaminergic, glutamatergic, and opioid receptors, neurotrophins, as well as potassium and calcium transport and signaling mechanisms (Gelernter et al., 2014; Mistry et al., 2014; Nelson et al., 2016; Saxon et al., 2005). Recently, a GWAS on a homogeneous sample of Han Chinese found evidence for an association of opioid dependence with several variants in previously unidentified genes; however, none reached significance at the genome-wide level (Kalsi et al., 2016).
Furthermore, despite the moderate heritability estimates of drug use in twin studies, genetic variants identified in molecular studies of substance use phenotypes explain only a small proportion of the phenotypic variance in substance use behaviors (Ducci & Goldman, 2012), leaving a great deal of genetic variation unaccounted for by GWAS to date. Termed “missing heritability,” this unaccounted-for variation represents the gap between the observed heritability of a trait from family and twin studies and the observed amount of genetic variance explained by genetic markers (Manolio et al., 2009). One potential reason for missing heritability may be due to variants with small effects that do not reach significance at the genome-wide level (i.e., p < 5 × 10-8). However, genetic variance estimates derived from common single nucleotide polymorphisms (SNPs) have consistently been found to be smaller than genetic variance estimated from family-based studies, especially for psychiatric and substance use disorders (Yang et al., 2015). Other potential reasons for missing heritability include insufficient sample sizes to detect significant effects, inflated family-based estimates, and poor tagging of rare causal variants by commonly used genotyping arrays (Evans et al., 2018).
With the growing number of GWAS in psychiatric research, the rarity of large effect findings, and the advancement of methodology and technology to analyze molecular array data, it has become evident that genetic association studies require very large samples (e.g., thousands of cases/controls) to achieve adequate power to detect significant effects (Sullivan, 2010). This is especially true for phenotypes, such as opioid dependence, that have relatively low prevalence rates in the population. Therefore, consortia and data repositories for which large sets of genetic data must be pooled to increase sample size have become essential for the advancement of psychiatric genetic research. Statistical methods that aggregate genetic effects within or across large, genome-wide molecular studies can begin to parse sources of genetic variation, which may help to address the issue of missing heritability (Vinkhuyzen et al., 2013). Such approaches may transcend the limitations of GWAS and candidate gene studies, which typically ignore the polygenic architecture of additive behaviors.
For example, a recent meta-analysis of the OPRM1 variant rs1799971 (A118G) with drug dependence and nonspecific drug dependence suggested that there are protective effects of the G allele on nonspecific drug dependence (Schwantes-An et al., 2016). Unfortunately, the results are obfuscated by the fact that the meta-analytic approach did not account for the effects of other loci, making it difficult to conclude whether these effects are biased because of polygenicity.
A plausible solution to deriving less biased SNP/gene effects is the use of mixed linear models such as Genomic-Relatedness-Matrix Restricted Maximum Likelihood (GREML), which forgoes identification of individual variants for estimating the phenotypic variance that is accounted for by genome-wide SNPs by using an SNP-derived genetic relationship matrix to estimate heritability (Lee et al., 2011). Furthermore, to gain even more insight into the genetic variation underlying complex traits, SNP array data can be genetically imputed using reference panels that can contribute additional variants not captured by the SNPs on commonly used arrays. Yang et al. (2015) found that large portions of genetic variation in human height and body mass index (BMI) can be captured via genetic imputation using the 1000 Genomes (1KG) Reference Panel (Genomes Project et al., 2010) of SNP-array–based genotype data, suggesting that missing heritability for height and BMI is negligible. These findings suggest that 1KG imputation can account for a large amount of variation explained by common and rare genetic variants beyond what might be captured on SNP arrays, leading to more accurate estimates of heritability and minimizing missing heritability that stems from SNP arrays that do not tag causal variants.
However, SNP-based estimates of heritability (h2SNP) can be biased if the linkage disequilibrium (LD; the nonrandom association of alleles at multiple loci) between causal variants and other variants differs, and this bias can be mediated by minor allele frequency (MAF; Yang et al., 2017). For example, GREML has most commonly been used to estimate a single genetic component associated with variation in a trait (i.e., GREML-SC), but the value will be overestimated if causal variants are more common than markers used in the analyses and underestimated if causal variants are rarer than those markers used in the analyses (Yang et al., 2015). One solution to correct for this type of bias is to stratify SNP-based heritability estimates based on LD and MAF in a joint model (i.e., GREML-LDMS), which has been shown to be unbiased by the MAF and LD properties of causal variants, performs better than alternative methods of SNP-heritability estimation, and is recommended for use on whole genome or imputed data (Evans et al., 2018; Yang et al., 2015).
Given the paucity of molecular research examining opioid dependence, the current study aimed to use the 1KG reference panel to genetically impute case/control repository data obtained via the National Institutes of Health Database of Genotypes and Phenotypes in order to (a) derive unbiased estimates of the additive genetic influences on opioid dependence and (b) investigate the extent to which differences in MAF and LD impact the heritability estimate obtained from genetically imputed data. Based on observed patterns with other substance use phenotypes (Palmer et al., 2015a, 2015b), we hypothesized that the SNP-based heritability would account for at least one half of the genetic variance previously observed in twin studies and that variants with lower MAF in regions of low LD would explain the largest proportions of variance.
Method
Sample
The sample consisted of secondary data from 6,487 participants between ages 18 and 78 (M = 40, SD = 11) assessed for substance use and included in the Genome-Wide Association Study of Heroin Dependence as part of the National Human Genome Research Institute’s Gene Environment Association Study Initiative (Database for Genotypes and Phenotypes [dbGaP]; study accession phs000277.v1.p1). Cases and controls from several studies were pooled and made available for secondary analysis as part of a collaborative effort to investigate the genetics of opioid dependence. See Supplemental Materials for a description of each study and Supplemental Table 1 for a summary of cases/controls across each study. (Supplemental material appears as an online-only addendum to the article on the journal’s website.) The present study involves secondary data analysis using data from this archived repository. Consented data access was granted via a Data Use Agreement with the National Institutes of Health and the database of Genotypes and Phenotypes (dbGaP) after receiving study approval from the institutional review board of Emory University.
Assessments
Opioid dependence was determined according to criteria from the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV; American Psychiatric Association, 1994), using the Semi-Structured Assessment for Drug Dependence and Alcoholism (SSADDA), the adapted SSADDA-OZ, or the Semi-Structured Assessment for Genetics of Alcoholism (SSAGA; Bucholz et al., 1994; Hesselbrock et al., 1999; Pierucci-Lagha et al., 2005). The SSADDA and SSAGA yield valid and reliable diagnoses for substance dependence (Bucholz et al., 1994, 1995; Hesselbrock et al., 1999; Malison et al., 2011; Pierucci-Lagha et al., 2005, 2007). All cases were limited to individuals who met lifetime DSM-IV criteria for opioid dependence, and controls were limited to assessed individuals who did not meet DSM-IV criteria for opioid dependence. From the original pooled sample, 1,341 population controls were removed who had not been assessed for DSM-IV opioid dependence.
Genotyping, quality control, and genetic imputation
Data management was conducted using PLINK version 1.9 (Purcell et al., 2007), the GCTA software tool [version 1.25.3], FlashPCA (Abraham et al., 2017), an imputation preparation and checking tool implemented in perl (Rayner et al., 2016), and R version 3.5.0 (R Core Team, 2018). Genomic data were obtained through dbGAP via the National Center for Biotechnology Information (NCBI). Samples were genotyped on three separate platforms (601,273 markers on Illumina Human610 Quad v1; 592,839 on Illumina Human660W Quad v1; and 373,339 on HumanCNV370 Quad v3.0).
Each study sample was prepared and imputed separately, ignoring the presence/absence of phenotypic data. See Supplemental Materials for a detailed description of quality control and data processing. In brief, individuals of European ancestry (EA) were identified using principle components analysis (PCA) with the 1000 Genomes Project (1KG) Phase III (Version 5) reference panel (Auton et al., 2015; Supplemental Figure 1). A total of 5,471 individuals of EA with genetic data were identified, and markers with call rate < .95, MAF < .01 were screened. Genetic imputation was conducted using the 1KG European reference panel and ShapeIT phasing with Minimac3 via the Michigan Imputation Server (https://imputationserver.sph.umich.edu/index.html#!pages/home).
Following imputation, markers with low imputation quality scores (r2 < .70) were removed to ensure that the highest quality markers from each sample were used to reduce potential batch effects across platforms and samples. Next, markers that were not biallelic SNPs were removed, and samples were merged for subsequent analytic quality control. Markers in the final, merged data set that had a call rate < 99%, MAF < 1%, or failed an HWE test (p < .0001) were removed, and individuals with <90% genotyping rate were removed. This resulted in a total of 6,200,495 SNPs. To control for cryptic relatedness, which could artificially inflate h2SNP estimates, a genetic relationship matrix (GRM) was computed using the GCTA software tool, which maximally selects one of any pair of individuals who were more related than second cousins (Yang et al., 2011a). This resulted in 4,064 unrelated opioid dependence cases/controls with genetic and phenotypic data. See Supplemental Tables 2 and 3 for a summary of markers removed at each step of quality control and imputation.
Estimation of additive genetic variance explained by single-nucleotide polymorphisms
GREML, as implemented in the GCTA software tool, was used to decompose phenotypic variance in DSM-IV opioid dependence into additive effects of SNPs (Yang et al., 2013). To explore whether h2SNP estimates vary as a function of MAF and LD, we used GCTA to calculate an individual SNP LD-score for each variant (e.g., representing the sum of LD r2 between a variant and all the variants in a 200Kb region) and then constructed a series of separate GRMs, including (a) a single GRM to estimate the single component h2SNP (GREML-SC), (b) a set of five GRMs partitioned based on the MAF of markers (e.g., bins containing MAF 1%–9%, 10%–19%, 20%–29%, 30%–39%, and 40%–49%) to estimate MAF stratified h2SNP (GREML-MS), (c) a set of four GRMs partitioned based on markers in each LD-score quantile to estimate LD stratified h2SNP (GREML-LD), and (d) a set of 20 GRMs stratified by both MAF and LD-score quantile (GREML-LDMS). Last, to explore whether h2SNP varied by chromosome, we constructed separate GRMs for each chromosome. Models containing multiple GRMs were simultaneously fit in a joint model with multiple genetic components (Yang et al., 2011b).
A series of models were tested to (a) determine the total h2SNP for opioid dependence using a single genetic component, (b) determine whether h2SNP varied as a function of MAF/LD, and (c) determine whether h2SNP differed by chromosome. Each model used a scaling factor to adjust for the prevalence of opioid dependence in the population (i.e., 0.37% for the population prevalence [Compton et al., 2007]). Last, a mixed linear model association using the GCTA software was implemented to identify individual loci associated with opioid dependence (Yang et al., 2014). Multiple comparisons were controlled for using a false discovery rate (FDR) of q < .05 via the qvalue package in R. All models controlled for age, sex, and other drug dependence (including DSM-IV cannabis, alcohol, stimulant, sedative, and cocaine dependence).
Results
Prevalence of opioid dependence
Of the total analytic sample (N = 4,064; 58.98% male; ages 18–78; M = 39.46, SD = 10.59), approximately half (52.17%, n = 2,120) were dependent on opioids. Among cases and controls, 60.47% and 11.78%, respectively, endorsed lifetime dependence on another substance. Nearly half (48.40%) of cases endorsed all seven DSM-IV criteria for opioid dependence. Opioid dependence was related to being male (odds ratio [OR] = 1.26, 95% CI [1.09, 1.46]), being younger (OR = 0.96, 95% CI [0.95, 0.96]), and having used other drugs (OR = 10.25, 95% CI [8.70, 12.11]). See Table 1 for a summary of endorsement by symptom for all cases.
Table 1.
Prevalence of symptom endorsement among individuals with DSM-IV opioid dependence (n = 2,120 cases)

| DSM-IV symptom | n | % |
| Tolerance | 2,031 | 95.80% |
| Withdrawal | 2,095 | 99.34% |
| Longer than intended | 1,315 | 89.21% |
| Attempt to quit | 2,067 | 97.50% |
| Time spent | 1,406 | 95.39% |
| Giving up activities | 1,310 | 88.87% |
| Continued use | 1,312 | 89.01% |
Note: DSM-IV = Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition.
Additive effects of autosomal SNPs on opioid dependence
After we controlled for age and sex, the total single component SNP-based heritability (h2SNP-SC) for opioid dependence was 38% (SE = 3%, p < .001). After we accounted for other drug dependence in addition to age/sex, the total h2SNP-SC was 27% (SE = 3%, p < .001), suggesting that a large portion of genetic variation in opioid dependence exists even after other drug dependence is controlled for. Total heritability estimates for models that stratified across MAF, LD, and MAF/LD varied from h2SNP-MS = 28% (SE = 3%) to h2SNP-LDMS = 45% (SE = 4%), demonstrating the tendency for bias in SNP-heritability estimates of GREML when MAF and LD properties are not accounted for; see Table 2 for a summary of all results. Stratified analyses revealed that the largest portion of the genetic variance in opioid dependence is tagged by common SNPs that were in the lowest MAF range and in the lowest LD-score quartile (h2SNP-LDMS =17%, SE = 3%, p < .001). A linear regression revealed that bins that contained more SNPs were associated with higher estimates of SNP heritability, F(1, 20) = 12.13, p = .003, Adjusted R2 = .37.
Table 2.
GREML-LDMS results: SNP-heritability (h2SNP) partitioned by minor allele frequency (MAF) and linkage disequilibrium (LD) score

| MAF range | 1st quartile LD score | 2nd quartile LD score | 3rd quartile LD score | 4th quartile LD score | Total |
| 1%–9% | 0.17 (0.03)* | 0.01 (0.02) | 0.00 (0.01) | 0.00 (0.01) | 0.08 (0.03)* |
| 10%–19% | 0.05 (0.03)* | 0.04 (0.02)* | 0.00 (0.01) | 0.01 (0.01) | 0.09 (0.03)* |
| 20%–29% | 0.05 (0.02)* | 0.02 (0.02) | 0.02 (0.01) | 0.00 (0.01) | 0.06 (0.03)* |
| 30%–39% | 0.02 (0.02) | 0.01 (0.02) | 0.02 (0.01) | 0.00 (0.01) | 0.05 (0.02)* |
| 40%–49% | 0.00 (0.02) | 0.01 (0.02) | 0.00 (0.01) | 0.00 (0.01) | 0.00 (0.02) |
| Total | 0.30 (0.04)* | 0.11 (0.03)* | 0.03 (0.02) | 0.01 (0.01) | h2SNP-SC = 0.27 (0.03) h2SNP-MS = 0.28 (0.03) h2SNP-LD = 0.45 (0.04) h2SNP-LDMS = 0.45 (0.04) |
Notes: GREML = Genomic-Relatedness-Matrix Restricted Maximum Likelihood; LDMS = LD and MAF stratified. Table displays main effects for analyses with genetic variance partitioned based on MAF and LD (see “Total” column/row) and joint effects with genetic variance partitioned on both MAF and LD (see inner columns/rows). Analyses for joint effect of both MAF and LD comprised 20 genetic components, analyses for main effects of MAF were comprised of 5 genetic components, and analyses for main effects of LD were comprised of 4 genetic components. Total h2SNP for each GREML approach are presented: SC = single component; MS = minor allele frequency stratified.
Indicates that the component contributes a significant portion of genetic variance to the model based on likelihood ratio test (LRT) p < .05.
Figure 1 presents a scatter plot of h2SNP estimates against the number of SNPs in each bin (panel a) as well as h2SNP estimates partitioned by bin (panel b). To test for enrichment and provide a comparison of the observed estimates of h2SNP for each LD/MAF bin relative to what would be expected if the distribution of SNP effects were uniform across all bins, we calculated expected h2SNP estimate as follows: .
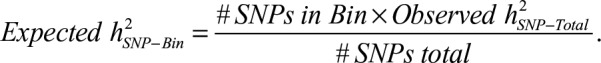 |
Figure 1.

Estimate of the regression of h2SNP for opioid dependence by number of markers in each LD/MAF bin (a) and by LD/MAF bin (b). In panel a, numbers in circles represent bin number. In panel b, dashed lines represent 95% confidence intervals, and dots represent the expected value of h2SNP for a given bin.
Thus, the expected h2SNP for a given bin is based on a weighting of the total h2SNP by the proportion of SNPs in each bin. If the 95% CI of the observed h2SNP excludes the expected h2SNP, then the null hypothesis that the two are equal is rejected at α < .05. Effects of SNPs in the lowest MAF range and in the lowest LD-score quartile (i.e., bin #1) exceeded what would be expected under a uniform distribution, suggesting that this region may be enriched for SNP effects.
When the genetic variance was partitioned across chromosomes, a linear regression revealed that longer chromosomes were associated with higher estimates of SNP heritability, F(1, 20) = 6.59, p = .018, adjusted R2 = .21. Because of the large number of variance components, these analyses did not partition based on MAF or LD. Figure 2 presents a scatter plot of h2SNP estimates against chromosome length and h2SNP estimates partitioned by chromosome for opioid dependence, including expected h2SNP estimates based on a weighting of the total h2SNP by the proportional length of each chromosome
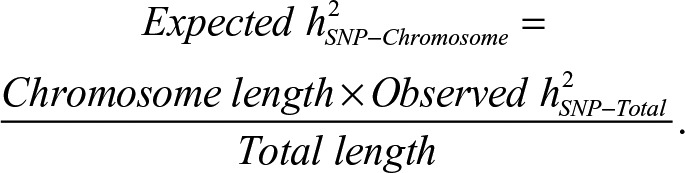 |
Figure 2.

Estimate of the regression h2SNP heritability for opioid dependence against chromosome length (a) and by chromosome (b). In panel a, numbers in circles represent chromosome number. In panel b, dashed lines represent 95% confidence intervals, and dots represent the expected value of h2SNP for a given chromosome.
Several chromosomes contributed to a significant portion of the observed total genetic variance (p < .05), including chromosomes 1, 2, 3, 6, 7, 8, 11, 12, 16, and 18, with effects of chromosome 7 exceeding what would be expected under a uniform distribution of effects, suggesting that this region may be enriched for SNP effects.
Exploratory GWAS of opioid dependence
Figure 3 presents the Q-Q and Manhattan plots of post hoc GWAS results from the mixed linear model association. Two markers (rs1970606, rs1557219) were found significant at the genome-wide level (p < 5 × 10-8), and both passed the FDR correction, p < .05. Each was located on chromosome 6 (LOC101927293) but has not been previously identified in association with opioid dependence or substance use. Five additional markers had p < 5 × 10-7, but none passed FDR. Three of these five markers (rs59675243, rs75449825, rs58356853) were located in the transforming acidic coiled-coil containing protein 2 (TACC2) gene located on chromosome 10. TACC2 has not been previously identified in relation to opioid dependence or use, but it is associated with the centrosome-spindle apparatus during cell cycling and has been implicated, with mixed results, in tumor progression and cancer (Cheng et al., 2010; Lauffart et al., 2003; Onodera et al., 2016; Schuendeln et al., 2004; Shakya et al., 2018) as well as substance use (Drgon et al., 2011; Johnson et al., 2008; Uhl et al., 2008). See Table 3 for GWA summary results for top markers and Supplemental Materials for regional association plots (Pruim et al., 2010). Full summary statistics are available at https://scholarblogs.emory.edu/bgalab/research/paper-supplements.
Figure 3.

Q-Q plot (a) and Manhattan plot (b) of GWAS p values for DSM-IV opioid dependence. Horizontal line represents genome-wide significance.
Table 3.
GWAS summary statistics for top markers

| SNP | Chromosome | Base-pair coordinate | Allele | MAF | β | SE | Odds ratio | p | q | Gene |
| rs1970606 | 6 | 58368572 | G | .29 | .06 | .01 | 1.06 | 7.80E-09 | .029 | LOC101927293 |
| rs1557219 | 6 | 58370925 | C | .29 | .06 | .01 | 1.06 | 9.38E-09 | .029 | LOC101927293 |
| rs59675243 | 10 | 123770622 | A | .04 | .13 | .02 | 1.14 | 1.25E-07 | .242 | TACC2 |
| rs75449825 | 10 | 123769889 | G | .04 | .13 | .02 | 1.14 | 1.56E-07 | .242 | TACC2 |
| rs17300532 | 5 | 71380355 | T | .24 | .06 | .01 | 1.06 | 2.10E-07 | .261 | LOC105379028 |
| rs58356853 | 10 | 123771132 | T | .04 | .12 | .02 | 1.13 | 3.08E-07 | .318 | TACC2 |
| rs4300379 | 11 | 51504993 | G | .38 | -.06 | .01 | 0.94 | 8.87E-07 | .786 | N/a |
Note: GWAS = genome-wide association studies; MAF = minor allele frequency; n/a = not applicable.
Discussion
This study was the first to characterize the SNP-based evidence for heritability of opioid dependence. At least 45% of the variance in DSM-IV opioid dependence was attributable to common SNPs after we stratified to account for differences in MAF and LD across the genome. Although there are no other SNP-based studies with which to compare, the heritability estimates obtained herein are large but consistent with estimates from twin studies that range from 43% (in a male-only sample) to 69% (for opioid use subtypes; Karkowski et al., 2000; Kendler et al., 1999, 2000; Sun et al., 2012; Tsuang et al., 1996, 2001). The differences between GREML estimates of heritability and twin-based estimates for most phenotypes have been attributed to the possibility that the SNPs on the arrays may not be in complete LD with causal variants or that other sources of genetic variance were not included in the model (e.g., extremely rare variants, structural and copy number polymorphisms), as well as epigenetic effects and non-additive genetic effects (e.g., gene-gene interactions; Yang et al., 2017). However, given the similarity between our estimates and those of prior twin studies, it is possible that many of the 1KG-imputed SNPs that passed quality control in the current analysis are in high LD with putative causal variants of mechanisms involved in opioid dependence.
Furthermore, in our analyses, we partitioned genetic variance according to MAF and LD, revealing that most (17%) of the genetic variance in opioid dependence is tagged by SNPs in the low MAF and low LD range. Given that we found that most of the markers were in the low MAF and low LD bins and that SNP-heritability estimates tend to increase as the number of markers increases, the additional coverage of low MAF markers in low LD afforded by the 1KG imputation likely contributes greatly to the large total SNP-heritability estimate.
Last, it is noted that there a few twin and family studies dedicated to estimating the unique heritability of opioid dependence. Therefore, another possible explanation could be that the twin study estimates, of which there are few assessing opioid dependence, may be inaccurate or biased because of differences in the environment and populations from which they were drawn.
Despite the lack of a replication sample, the GWAS revealed two previously unidentified intron variants on chromosome 6 that passed FDR and three markers in the TACC2 gene on chromosome 10 that did not pass genome-wide significance or FDR, but have been previously implicated in substance use research. It is possible that three markers in TACC2 may be associated with opioid dependence either directly or indirectly by serving as a proxy for a causal variant in LD with these variants, but confirmation in an independent sample is needed, as these analyses were exploratory. TACC2 has been associated with tumor research and cancer (Chen et al., 2000; Cheng et al., 2010; Lauffart et al., 2003; Mendrzyk et al., 2006; Onodera et al., 2016; Takayama et al., 2012), although results are mixed (Schuendeln et al., 2004). Less frequent evidence has implicated TACC2 with neuroticism (Eszlari et al., 2017) and bipolar disorder (Johnson et al., 2009). Of particular interest, however, TACC2 has been identified in studies examining the use of several substances, including dependence on illegal substances (Drgon et al., 2011), methamphetamine (Uhl et al., 2008), and any substance (Johnson et al., 2008). Therefore, it is interesting to observe an association between TACC2 and opioid dependence after accounting for involvement with other substances, although these findings are in need of independent replication and these results should be interpreted with caution.
Chromosomal analyses revealed that chromosome 7 accounted for the largest amount of variance in opioid dependence (h2SNP-chr7 = 4%, SE = 1%), and results indicated that longer chromosomes accounted for more variance. Future work could expand on these chromosomal analyses by estimating heritability of more specific genomic regions, such as candidate SNPs based on chromosomal or gene-based regions of interest, in order to isolate regions contributing the most variance to the heritability estimate for opioid dependence. However, as evidenced here, significantly larger sample sizes will be required to partition such small effects.
Several limitations to this work should be noted. First, it is possible that h2SNP values estimated in the present study are biased because of spurious effects based on systematic differences in sample characteristics and ascertainment across the different studies. Although we used stringent quality control to reduce bias from batch effects and other possible confounding and controlled for effects due to age and sex, this work should be replicated in large, independent samples. Second, increasing age is associated with a decrease in the prevalence for opioid dependence. Therefore, the ascertainment-corrected transformation made to the SNP-heritability estimate (and its standard error) may be slightly biased. In the current study, participants ranged in age from 18 to 70, but a constant prevalence for opioid dependence in adults ages 18 and over in the United States was used for all analyses. Therefore, model estimates from this study do not reflect the variation in prevalence across the life span (i.e., the prevalence estimated for adults ages 18 and over is estimated in epidemiological studies as used as a constant value—however, it is possible that the true value is dynamic and varies across age). Similarly, genetic and environmental influences on substance use disorders change across development (Kendler et al., 2008). Because of the wide age range in the present sample and the loss of power when stratifying individuals based on age, we were not able to examine whether heritability estimates were consistent across ages. In addition, the present results incorporated only case and control individuals of European descent; therefore, how these results would generalize to other ancestral populations is unknown.
Last, a majority of the individuals in the control group (73%) had never used opiates in their lifetime, leaving too few individuals to separately compare those who had been exposed to opioids but did not develop dependence to those who had developed dependence. Future research should strive to examine SNP heritability of opioid dependence in a sample of individuals exposed to opioids who differ in their severity of opioid-related problems. Recent evidence has shown that the use of diagnostic measures leads to an underestimation of additive genetic effects for problematic drug outcomes (Palmer et al., 2015).
In sum, we provide the first SNP-based heritability estimates for opioid dependence using a large, case/control study of dependence among individuals of EA. These results contribute to our understanding of the etiology of opioid dependence by providing an avenue for future research aimed at genetic loci contributing to opioid dependence. Given that most of the genetic variance in opioid dependence was attributed to markers in the low MAF and low LD range, future studies should place emphasis on rare variants that are in low LD with other markers to detect specific causal variants implicated in the etiology of opioid dependence. Whole genome sequencing and genetic imputation can be used to overcome the limitations of relying on SNP arrays that only tag a small percentage of common variants. Novel and robust approaches, such as GREML-LDMS, which take into account the complex relationships between MAF and LD, can help to shed light on how rare variants contribute to the genetic liability of opioid dependence as well as other complex traits. However, the development of new research methods is greatly needed to integrate multiple “-omics” evidence (genomic, methylomic, transcriptomic, etc.) as a means of prioritizing and testing sets of genes and associated regulatory elements using statistical methods that account for remaining polygenicity in the genome (Boyle et al., 2017).
Conflict of Interest Statement
All of the listed authors declare that they have no conflicts of interest.
Acknowledgments
The authors acknowledge the contribution of data from GWAS of Heroin Dependence supported by National Institute on Drug Abuse Grant R01DA17305 and accessed through dbGAP to the analysis presented in this communication—that is, publication, presentation, or grant application.
Footnotes
This research was supported by grants from the National Institute on Alcohol Abuse and Alcoholism (T32MH019927 [to Anthony Spirito] and K01AA021113 [to Rohan H. C. Palmer]) and the National Institute of Drug Abuse (T32DA016184 [to Damaris Rohsenow] and DP1DA042103 [Rohan H. C. Palmer].
References
- Abraham G., Qiu Y., Inouye M. FlashPCA2: Principal component analysis of Biobank-scale genotype datasets. Bioinformatics. 2017;33:2776–2778. doi: 10.1093/bioinformatics/btx299. doi:10.1093/bioinformatics/btx299. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Washington, DC: Author; 1994. Diagnostic and statistical manual of mental disorders (4th ed.) [Google Scholar]
- Auton A., Abecasis G. R., Altshuler D. M., Durbin R. M., Bentley D. R., Chakravarti A., Wilson R. K. the 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. doi:10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle E. A., Li Y. I., Pritchard J. K. An expanded view of complex traits: From polygenic to omnigenic. Cell. 2017;169:1177–1186. doi: 10.1016/j.cell.2017.05.038. doi:10.1016/j.cell.2017.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucholz K. K., Cadoret R., Cloninger C. R., Dinwiddie S. H., Hesselbrock V. M., Nurnberger J. I., Jr., Schuckit M. A. A new, semi-structured psychiatric interview for use in genetic linkage studies: A report on the reliability of the SSAGA. Journal of Studies on Alcohol. 1994;55:149–158. doi: 10.15288/jsa.1994.55.149. doi:10.15288/jsa.1994.55.149. [DOI] [PubMed] [Google Scholar]
- Bucholz K. K., Hesselbrock V. M., Shayka J. J., Nurnberger J. I., Jr., Schuckit M. A., Schmidt I., Reich T. Reliability of individual diagnostic criterion items for psychoactive substance dependence and the impact on diagnosis. Journal of Studies on Alcohol. 1995;56:500–505. doi: 10.15288/jsa.1995.56.500. doi:10.15288/jsa.1995.56.500. [DOI] [PubMed] [Google Scholar]
- Center for Behavioral Health Statistics and Quality. 2016 National Survey on Drug Use and Health: Detailed tables. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2017. [Google Scholar]
- Centers for Disease Control and Prevention. Wide-ranging online data for epidemiologic research (WONDER) Atlanta, GA: Author; 2016. Retrieved from https://www.healthdata.gov/dataset/wide-ranging-online-data-epidemiologic-research-wonder. [Google Scholar]
- Chen H. M., Schmeichel K. L., Mian I. S., Lelièvre S., Petersen O. W., Bissell M. J. AZU-1: A candidate breast tumor suppressor and biomarker for tumor progression. Molecular Biology of the Cell. 2000;11:1357–1367. doi: 10.1091/mbc.11.4.1357. doi:10.1091/mbc.11.4.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S., Douglas-Jones A., Yang X., Mansel R. E., Jiang W. G. Transforming acidic coiled-coil-containing protein 2 (TACC2) in human breast cancer, expression pattern and clinical/prognostic relevance. Cancer Genomics & Proteomics. 2010;7:67–73. [PubMed] [Google Scholar]
- Compton W. M., Thomas Y. F., Stinson F. S., Grant B. F. Prevalence, correlates, disability, and comorbidity of DSM-IV drug abuse and dependence in the United States: Results from the National Epidemiologic Survey on Alcohol and Related Conditions. Archives of General Psychiatry. 2007;64:566–576. doi: 10.1001/archpsyc.64.5.566. doi:10.1001/archpsyc.64.5.566. [DOI] [PubMed] [Google Scholar]
- Drgon T., Johnson C. A., Nino M., Drgonova J., Walther D. M., Uhl G. R. “Replicated” genome wide association for dependence on illegal substances: Genomic regions identified by overlapping clusters of nominally positive SNPs. American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics. 2011;156:125–138. doi: 10.1002/ajmg.b.31143. doi:10.1002/ajmg.b.31143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducci F., Goldman D. The genetic basis of addictive disorders. Psychiatric Clinics of North America. 2012;35:495–519. doi: 10.1016/j.psc.2012.03.010. doi:10.1016/j.psc.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eszlari N., Millinghoffer A., Petschner P., Gonda X., Baksa D., Pulay A., Juhász G. Genome-wide gene-based tests replicate the association of the SORCS3 gene with neuroticism. Poster presented at the 30th ECNP Congress; Paris, France: 2017. [Google Scholar]
- Evans L. M., Tahmasbi R., Vrieze S. I., Abecasis G. R., Das S., Gazal S., Keller M. C.& the Haplotype Reference Consortium 2018Comparison of methods that use whole genome data to estimate the heritability and genetic architecture of complex traits Nature Genetics 50737–745.doi:10.1038/s41588-018-0108-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelernter J., Kranzler H. R., Sherva R., Koesterer R., Almasy L., Zhao H., Farrer L. A. Genome-wide association study of opioid dependence: Multiple associations mapped to calcium and potassium pathways. Biological Psychiatry. 2014;76:66–74. doi: 10.1016/j.biopsych.2013.08.034. doi:10.1016/j.biopsych.2013.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselbrock M., Easton C., Bucholz K. K., Schuckit M., Hesselbrock V. A validity study of the SSAGA—a comparison with the SCAN. Addiction. 1999;94:1361–1370. doi: 10.1046/j.1360-0443.1999.94913618.x. doi:10.1046/j.1360-0443.1999.94913618.x. [DOI] [PubMed] [Google Scholar]
- Jensen K. P. A review of genome-wide association studies of stimulant and opioid use disorders. Molecular Neuropsychiatry. 2016;2:37–45. doi: 10.1159/000444755. doi:10.1159/000444755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C., Drgon T., Liu Q.-R., Zhang P.-W., Walther D., Li C.-Y., Uhl G. R. Genome wide association for substance dependence: Convergent results from epidemiologic and research volunteer samples. BMC Medical Genetics. 2008;9:113. doi: 10.1186/1471-2350-9-113. doi:10.1186/1471-2350-9-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C., Drgon T., McMahon F. J., Uhl G. R. Convergent genome wide association results for bipolar disorder and substance dependence. American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics. 2009;150B:182–190. doi: 10.1002/ajmg.b.30900. doi:10.1002/ajmg.b.30900. [DOI] [PubMed] [Google Scholar]
- Kalsi G., Euesden J., Coleman J. R., Ducci F., Aliev F., Newhouse S. J., Breen G. Genome-wide association of heroin dependence in Han Chinese. PLoS One. 2016;11(12):e0167388. doi: 10.1371/journal.pone.0167388. doi:10.1371/journal.pone.0167388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkowski L. M., Prescott C. A., Kendler K. S. Multivariate assessment of factors influencing illicit substance use in twins from female-female pairs. American Journal of Medical Genetics. 2000;96:665–670. doi:10.1002/1096-8628(20001009)96:5<665::AID-AJMG13>3.0.CO;2-O. [PubMed] [Google Scholar]
- Kendler K. S., Karkowski L., Prescott C. A. Hallucinogen, opiate, sedative and stimulant use and abuse in a population-based sample of female twins. Acta Psychiatrica Scandinavica. 1999;99:368–376. doi: 10.1111/j.1600-0447.1999.tb07243.x. doi:10.1111/j.1600-0447.1999.tb07243.x. [DOI] [PubMed] [Google Scholar]
- Kendler K. S., Karkowski L. M., Neale M. C., Prescott C. A. Illicit psychoactive substance use, heavy use, abuse, and dependence in a US population-based sample of male twins. Archives of General Psychiatry. 2000;57:261–269. doi: 10.1001/archpsyc.57.3.261. doi:10.1001/archpsyc.57.3.261. [DOI] [PubMed] [Google Scholar]
- Kendler K. S., Schmitt E., Aggen S. H., Prescott C. A. Genetic and environmental influences on alcohol, caffeine, cannabis, and nicotine use from early adolescence to middle adulthood. Archives of General Psychiatry. 2008;65:674–682. doi: 10.1001/archpsyc.65.6.674. doi:10.1001/archpsyc.65.6.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodny A., Courtwright D. T., Hwang C. S., Kreiner P., Eadie J. L., Clark T. W., Alexander G. C. The prescription opioid and heroin crisis: A public health approach to an epidemic of addiction. Annual Review of Public Health. 2015;36:559–574. doi: 10.1146/annurev-publhealth-031914-122957. doi:10.1146/annurev-publhealth-031914-122957. [DOI] [PubMed] [Google Scholar]
- Lauffart B., Gangisetty O., Still I. H. Molecular cloning, genomic structure and interactions of the putative breast tumor suppressor TACC2. Genomics. 2003;81:192–201. doi: 10.1016/s0888-7543(02)00039-3. [DOI] [PubMed] [Google Scholar]
- Lee S. H., Wray N. R., Goddard M. E., Visscher P. M. Estimating missing heritability for disease from genome-wide association studies. American Journal of Human Genetics. 2011;88:294–305. doi: 10.1016/j.ajhg.2011.02.002. doi:10.1016/j.ajhg.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D., Zhao H., Kranzler H. R., Li M. D., Jensen K. P., Zayats T., Gelernter J. Genome-wide association study of copy number variations (CNVs) with opioid dependence. Neuropsychopharmacology. 2015;40:1016–1026. doi: 10.1038/npp.2014.290. doi:10.1038/npp.2014.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malison R. T., Kalayasiri R., Sanichwankul K., Sughondhabirom A., Mutirangura A., Pittman B., Gelernter J. Inter-rater reliability and concurrent validity of DSM-IV opioid dependence in a Hmong isolate using the Thai version of the Semi-Structured Assessment for Drug Dependence and Alcoholism (SSADDA) Addictive Behaviors. 2011;36:156–160. doi: 10.1016/j.addbeh.2010.08.031. doi:10.1016/j.addbeh.2010.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manchikanti L., Helm S., II, Fellows B., Janata J. W., Pampati V., Grider J. S., Boswell M. V. Opioid epidemic in the United States. Pain Physician. 2012;15(Supplement):ES9–ES38. [PubMed] [Google Scholar]
- Manolio T. A., Collins F. S., Cox N. J., Goldstein D. B., Hindorff L. A., Hunter D. J., Visscher P. M. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. doi:10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendrzyk F., Korshunov A., Benner A., Toedt G., Pfister S., Radlwimmer B., Lichter P. Identification of gains on 1q and epidermal growth factor receptor overexpression as independent prognostic markers in intracranial ependymoma. Clinical Cancer Research. 2006;12:2070–2079. doi: 10.1158/1078-0432.CCR-05-2363. doi:10.1158/1078-0432.CCR-05-2363. [DOI] [PubMed] [Google Scholar]
- Mistry C. J., Bawor M., Desai D., Marsh D. C., Samaan Z. Genetics of opioid dependence: A review of the genetic contribution to opioid dependence. Current Psychiatry Reviews. 2014;10:156–167. doi: 10.2174/1573400510666140320000928. doi:10.2 174/1573400510666140320000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson E. C., Agrawal A., Heath A. C., Bogdan R., Sherva R., Zhang B., Montgomery G. W. Evidence of CNIH3 involvement in opioid dependence. Molecular Psychiatry. 2016;21:608–614. doi: 10.1038/mp.2015.102. doi:10.1038/mp.2015.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onodera Y., Takagi K., Miki Y., Takayama K., Shibahara Y., Watanabe M., Suzuki T. TACC2 (transforming acidic coiled-coil protein 2) in breast carcinoma as a potent prognostic predictor associated with cell proliferation. Cancer Medicine. 2016;5:1973–1982. doi: 10.1002/cam4.736. doi:10.1002/cam4.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer R. H., Brick L., Nugent N. R., Bidwell L. C., McGeary J. E., Knopik V. S., Keller M. C. Examining the role of common genetic variants on alcohol, tobacco, cannabis and illicit drug dependence: Genetics of vulnerability to drug dependence. Addiction. 2015a;110:530–537. doi: 10.1111/add.12815. doi:10.1111/add.12815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer R. H., McGeary J. E., Heath A. C., Keller M. C., Brick L. A., Knopik V. S. Shared additive genetic influences on DSM-IV criteria for alcohol dependence in subjects of European ancestry. Addiction. 2015b;110:1922–1931. doi: 10.1111/add.13070. doi:10.1111/add.13070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierucci-Lagha A., Gelernter J., Chan G., Arias A., Cubells J. F., Farrer L., Kranzler H. R. Reliability of DSM-IV diagnostic criteria using the semi-structured assessment for drug dependence and alcoholism (SSADDA) Drug and Alcohol Dependence. 2007;91:85–90. doi: 10.1016/j.drugalcdep.2007.04.014. doi:10.1016/j.drugalcdep.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierucci-Lagha A., Gelernter J., Feinn R., Cubells J. F., Pearson D., Pollastri A., Kranzler H. R. Diagnostic reliability of the Semi-structured Assessment for Drug Dependence and Alcoholism (SSADDA) Drug and Alcohol Dependence. 2005;80:303–312. doi: 10.1016/j.drugalcdep.2005.04.005. doi:10.1016/j.drugalcdep.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Pruim R. J., Welch R. P., Sanna S., Teslovich T. M., Chines P. S., Gliedt T. P., Willer C. J. LocusZoom: Regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–2337. doi: 10.1093/bioinformatics/btq419. doi:10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M. A., Bender D., Sham P. C. PLINK: A tool set for whole-genome association and population-based linkage analyses. American Journal of Human Genetics. 2007;81:559–575. doi: 10.1086/519795. doi:10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. The R Project for Statistical Computing. 2018. Retrieved from http://www.R-project.org. [Google Scholar]
- Rayner N. W., Robertson N. R., Mahajan A., McCarthy M. I. A suite of programs for pre- and post-imputation data checking. Paper presented at the 66th Annual Meeting of the American Society of Human Genetics; Vancouver, British Columbia: 2016. [Google Scholar]
- Rudd R. A., Seth P., David F., Scholl L. Increases in drug and opioid-involved overdose deaths—United States, 2010–2015. Morbidity and Mortality Weekly Report. 2016;65:1445–1452. doi: 10.15585/mmwr.mm655051e1. doi:10.15585/mmwr.mm655051e1. [DOI] [PubMed] [Google Scholar]
- Saxon A. J., Oreskovich M. R., Brkanac Z. Genetic determinants of addiction to opioids and cocaine. Harvard Review of Psychiatry. 2005;13:218–232. doi: 10.1080/10673220500243364. doi:10.1080/10673220500243364. [DOI] [PubMed] [Google Scholar]
- Schwantes-An T. H., Zhang J., Chen L. S., Hartz S. M., Culverhouse R. C., Chen X., Saccone N. L. Association of the OPRM1 Variant rs1799971 (A118G) with non-specific liability to substance dependence in a collaborative de novo meta-analysis of European-ancestry cohorts. Behavior Genetics. 2016;46:151–169. doi: 10.1007/s10519-015-9737-3. doi:10.1007/s10519-015-9737-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuendeln M. M., Piekorz R. P., Wichmann C., Lee Y., McKinnon P. J., Boyd K., Ihle J. N. The centrosomal, putative tumor suppressor protein TACC2 is dispensable for normal development, and deficiency does not lead to cancer. Molecular and Cellular Biology. 2004;24:6403–6409. doi: 10.1128/MCB.24.14.6403-6409.2004. doi:10.1128/MCB.24.14.6403-6409.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakya M., Zhou A., Dai D., Zhong Q., Zhou Z., Zhang Y., Chen M. High expression of TACC2 in hepatocellular carcinoma is associated with poor prognosis. Cancer Biomarkers. 2018;22:611–619. doi: 10.3233/CBM-170091. doi:10.3233/CBM-170091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan P. F. The psychiatric GWAS consortium: Big science comes to psychiatry. Neuron. 2010;68:182–186. doi: 10.1016/j.neuron.2010.10.003. doi:10.1016/j.neuron.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J., Bi J., Chan G., Oslin D., Farrer L., Gelernter J., Kranzler H. R. Improved methods to identify stable, highly heritable subtypes of opioid use and related behaviors. Addictive Behaviors. 2012;37:1138–1144. doi: 10.1016/j.addbeh.2012.05.010. doi:10.1016/j.addbeh.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama K., Horie-Inoue K., Suzuki T., Urano T., Ikeda K., Fujimura T., Inoue S. TACC2 is an androgen-responsive cell cycle regulator promoting androgen-mediated and castration-resistant growth of prostate cancer. Molecular Endocrinology. 2012;26:748–761. doi: 10.1210/me.2011-1242. doi:10.1210/me.2011-1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. doi:10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuang M. T., Bar J. L., Harley R. M., Lyons M. J. The Harvard Twin Study of Substance Abuse: What we have learned. Harvard Review of Psychiatry. 2001;9:267–279. doi:10.1080/10673220127912. [PubMed] [Google Scholar]
- Tsuang M. T., Lyons M. J., Eisen S. A., Goldberg J., True W., Lin N., Eaves L. Genetic influences on DSM-IIIR drug abuse and dependence: A study of 3,372 twin pairs. American Journal of Medical Genetics. 1996;67:473–477. doi: 10.1002/(SICI)1096-8628(19960920)67:5<473::AID-AJMG6>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Tsuang M. T., Lyons M. J., Meyer J. M., Doyle T., Eisen S. A., Goldberg J., Eaves L. Co-occurrence of abuse of different drugs in men: The role of drug-specific and shared vulnerabilities. Archives of General Psychiatry. 1998;55:967–972. doi: 10.1001/archpsyc.55.11.967. doi:10.1001/archpsyc.55.11.967. [DOI] [PubMed] [Google Scholar]
- Uhl G. R., Drgon T., Liu Q. R., Johnson C., Walther D., Komiyama T., Lin S.-K. Genome-wide association for methamphetamine dependence: Convergent results from 2 samples. Archives of General Psychiatry. 2008;65:345–355. doi: 10.1001/archpsyc.65.3.345. doi:10.1001/archpsyc.65.3.345. [DOI] [PubMed] [Google Scholar]
- Vinkhuyzen A. A., Wray N. R., Yang J., Goddard M. E., Visscher P. M. Estimation and partition of heritability in human populations using whole-genome analysis methods. Annual Review of Genetics. 2013;47:75–95. doi: 10.1146/annurev-genet-111212-133258. doi:10.1146/annurev-genet-111212-133258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Bakshi A., Zhu Z., Hemani G., Vinkhuyzen A. A., Lee S. H., Visscher P. M.& the LifeLines Cohort Study 2015Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index Nature Genetics 471114–1120.doi:10.1038/ng.3390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Lee S. H., Goddard M. E., Visscher P. M. GCTA: A tool for genome-wide complex trait analysis. American Journal of Human Genetics. 2011a;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. doi:10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Lee S. H., Goddard M. E., Visscher P. M. Genomewide complex trait analysis (GCTA): Methods, data analyses, and interpretations. Methods in Molecular Biology. 2013;1019:215–236. doi: 10.1007/978-1-62703-447-0_9. doi:10.1007/978-1-62703-447-0_9. [DOI] [PubMed] [Google Scholar]
- Yang J., Manolio T. A., Pasquale L. R., Boerwinkle E., Caporaso N., Cunningham J. M., Visscher P. M. Genome partitioning of genetic variation for complex traits using common SNPs. Nature Genetics. 2011b;43:519–525. doi: 10.1038/ng.823. doi:10.1038/ng.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Zaitlen N. A., Goddard M. E., Visscher P. M., Price A. L. Advantages and pitfalls in the application of mixed-model association methods. Nature Genetics. 2014;46:100–106. doi: 10.1038/ng.2876. doi:10.1038/ng.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Zeng J., Goddard M. E., Wray N. R., Visscher P. M. Concepts, estimation and interpretation of SNP-based heritability. Nature Genetics. 2017;49:1304–1310. doi: 10.1038/ng.3941. doi:10.1038/ng.3941. [DOI] [PubMed] [Google Scholar]