

Abstract
Corchorusoside C (1), isolated from Streptocaulon juventas collected in Vietnam, was found to be non-toxic in a zebrafish (Danio rerio) model and to induce cytotoxicity in several cancer cell lines with notable selective activity against prostate DU-145 cancer cells (IC50 0.08 μM). Moreover, corchorusoside C induced DU-145 cell shrinkage and cell detachment. In CCD-112CoN colon normal cells, 1 showed significantly reduced cytotoxic activity (IC50 2.3 μM). A preliminary mechanistic study indicated that 1 inhibits activity and protein expression of NF-κB (p50 and p65), IKK (α and β) and ICAM-1 in DU-145 cells. ROS concentrations increased at 5 h post-treatment and MTP decreased in a dose-dependent manner. Moreover, decreased protein expression of Bcl-2 and increased expression of PARP-1 was observed. Furthermore, corchorusoside C increased both the activity and protein levels of caspases 3 and 7. Additionally, 1 induced sub-G1 population increase of DU-145 cells and modulated caspases in zebrafish with non-differential morphological effects. Therefore, corchorusoside C (1) induces apoptosis in DU-145 cells and targets the same pathways both in vitro and in vivo in zebrafish. Thus, the use of zebrafish assays seems worthy of wider application than is currently employed for the evaluation of potential anticancer agents of natural origin.
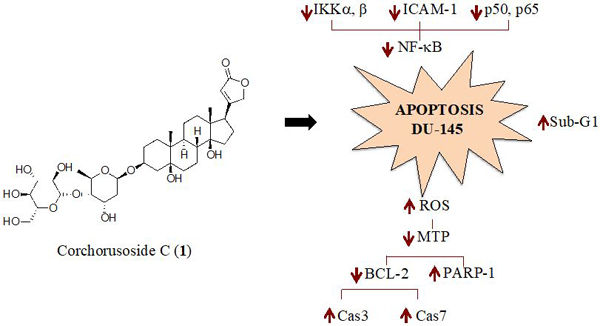
GRAPHICAL ABSTRACT
Cancer is a complex of several diseases, of which the principal characteristic is the uncontrolled proliferation of cells in the body. Currently, cancer is the second leading cause of morbidity and mortality worldwide, and, according to an estimate by the World Health Organization, there were 9.6 million cancer related deaths in 2018.1 Prostate and lung cancer are the most common types of cancer in men. In the United States there were an estimated 164,690 new cases and 29,430 deaths due to prostate cancer in 2018.2 Although there are numerous drugs for cancer treatment available currently, there is undoubtedly a need to discover new safe and effective chemotherapy drugs. Even as the end of the second decade of the 21th century is reached, plants continue to be among the most important sources of natural products with therapeutic potential.3
Streptocaulon juventas (Lour.) Merr. (Apocynaceae) is a perennial herb, native to the Indochina region, and is commonly known as “Ha thu o trang” in Vietnam.4 In folkloric medicine, this plant has been used for the treatment of anemia, malaria, rheumatism, menstrual disorders, neurasthenia, dyspepsia, dysentery, diarrhea, stomachache, fever, chronic nephritis and traumatic injuries.5 It has been reported that the main active secondary metabolites of S. juventas are cardenolides, which display a range of biological activities, including antimicrobial, antiplasmodial, and antioxidant effects. Recently, these compounds have been shown to exhibit in vitro cytotoxic activities in various human cancer cell lines.6 Among the cardenolides isolated from this species, corchorusoside C (1), a steroid with a five-membered lactone ring and two sugar units, has previously displayed cytotoxic activity against the A549, NCI-H460, HT-1080, HeLa and HGC-27 cancer cell lines.7–10
It is well known that the main mechanism of action of cardenolides is Na+/K+ ATPase inhibition, which induces an increase in intracellular Ca2+ concentrations.11 Due to this property, cardenolides have been utilized previously to treat heart failure.11,12 Recently, it has been found that antineoplastic properties of cardenolides may involve activation or inhibition of several cellular signal transduction mechanisms, including those that may or may not be relevant to the suppression mechanism of Na+ / K+ ATPase protein. For instance, periplocin, a cardenolide isolated from Periploca sepium, induces DNA double strand breaks and death-receptor mediated apoptosis.13 Nerigoside, a cardenolide from Nerium oleander, has been demonstrated that induces inhibition of ERK/GSK3β/β-catenin signaling pathway.14 (+)-strebloside, a cardenolide from Streblus asper, induces repression of p53 and inhibits NF-κB in addition of the inhibition of Na+ / K+ ATPase pump.15 Oleandrin, derivated from Nerium oleander, also induces a block of nuclear factor kappa-light-chain B (NF-κB).16
NF-κB is a protein complex that regulates the expression of several genes associated with cell survival by activation of immune and inflammatory responses.17 Furthermore, NF-κB inhibition has been associated with cytotoxic effects through the induction of apoptotic signaling in cancer cells.18 In this way, NF-κB represents a promising form of targeted cancer chemotherapy and may provide insight into therapeutic options for cardenolides.19
In a continuing search for anticancer agents, the potential mechanism of action of the antiproliferative activity of corchorusoside C (1) isolated from S. juventas collected in Vietnam was investigated in both the DU-145 human prostate cancer cell line and zebrafish. The effects shown were found to be associated with the inhibition of the NF-κB complex and compromise of the mitochondrial transmembrane potential.
RESULTS AND DISCUSSION
Phytochemical investigation of S. juventas led to the isolation and identification of corchorusoside C (1),20 which was then prioritized for preliminary mechanistic studies. Recently, the zebrafish (Danio rerio) has been shown suitable for the investigation of in vivo developmental toxicity.21 Thus, to evaluate the potential toxicity of corchorusoside C, zebrafish at 24 hours post fertilization (hpf) were treated for 24 h with 1 and cycloheximide and digoxin as positive controls at 50 μM. As shown in Figure 1, 1 did not produce any visible developmental abnormalities, including those in the brain, vasculature, heart, gut, trunk, and tail in the zebrafish. However, cycloheximide caused developmental delay and digoxin induced a type of lordosis, a spine malformation or curvature disorder (Figure 1). The lack of toxic effects in this regard and the high yield of corchorusoside C (1) from its plant of origin encouraged further studies to determine its antiproliferative effect in cancer cells. The antiproliferative effects of corchorusoside C were evaluated against colon (HT-29), cervical (HeLa), prostate (DU-145 and PC-3) and breast (MCF-7 and MDA-MB-231) cancer cell lines, using the SRB assay. Corchorusoside C displayed cytotoxic effects in a dose-dependent manner with the following IC50 values: 0.12, 0.19, 0.08, 0.2, 0.1, 0.2 μM, respectively (Table 1), with a notable activity against prostate DU-145 cancer cells (0.08 μM). Since their initial use in 1978,22 DU-145 prostate cancer cells have been utilized extensively to find new drug leads against prostate cancer. DU-145 cells are considered androgen non-responsive, making them aggressive and interesting for drug development studies.23 Due to its submicromolar potency against HT-29 colon cancer cells (IC50 0.12 μM), corchorusoside C was tested for cytotoxicity against CCD-112-CoN human normal colon cells, resulting in an IC50 value of 2.7 μM (Table 1), indicating a selectivity index of 22.5 fold toward the cancerous colon cells.
Figure 1.

(A) Structure of corchorusoside C (1). (B) Zebrafish toxicity determination of 1 from Streptocaulon juventas. Zebrafish were treated for 24 h at 50 μM with 1, or cycloheximide or digoxin as the positive controls, with an untreated control also used.
Table 1.
Cytotoxicity of Corchorusoside C (1) Against Human Cancer and Normal Cell Lines.
| compound | HT-29 a | HeLa a | DU-145 a | PC-3 a | MCF-7 a | MDA-MB-231 a | CCD-112CoN a |
|---|---|---|---|---|---|---|---|
| 1 | 0.12 | 0.19 | 0.08 | 0.2 | 0.1 | 0.2 | 2.7 |
| paclitaxelb | 0.002 | 0.005 | 0.004 | 0.17 | 0.1 | 23 | |
| ellipticineb | 0.8 |
IC50 values are the concentration (μM) required for 50% inhibition of cell viability after 72 h incubation for test compound treatment and were calculated using nonlinear regression analysis with measurements performed in triplicate and representative of three independent experiments, where the values generally agreed within 10%.
Positive controls.
The effects of corchorusoside C (1) on DU-145 cell proliferation was studied over periods of 24, 48, and 72 hours. The cell proliferation rate was found to be significantly reduced in a concentration-dependent manner, especially for the first 24 hours, and further reduced at 48 and 72 hours (Figure S3A), with timed IC50 values of 11.2, 1.2 and 0.08 μM, respectively. Phenotypically, in early apoptosis, cell shrinkage and pyknosis are visible by light microscopy as apoptosis is characterized by chromatin condensation, plasma membrane blebbing, DNA fragmentation, and collapse of the cell into small fragments (apoptotic bodies).24 The photomicrographs in Figure S3B show that 1 decreased the number of cells and induced cell shrinkage as well as cell detachment from the monolayer surface, when compared with untreated cells. Also, indicated in Figure S3B is an increase in the number of apoptotic cells corresponding to the length of incubation with this cardenolide. Morphological investigation of cells by microscopy revealed that 1 induced apoptotic cell death in DU-145 cancer cells.
In recent years, numerous cardenolides have shown apoptosis through caspase-dependent apoptosis including (+)-strebloside,15 12,16-dihydroxicalotropin, calotropin, corotoxigenin 3-O-glucopyranoside, desglucouzarin,25 periplocymarin and periplocin.26 Additionally, selected cardenolides have entered phase I and II clinical trials for the treatment of solid tumors with satisfactory safety and efficacy profiles. A retrospective study with digoxin (a cardenolide), for instance, showed that users of digoxin with congestive heart failure exhibited a lower risk of developing prostate cancer versus those who did not take this drug.27 Also, a phase II trial on digoxin as a stand-alone agent showed a rate of positive prostate specific antigen doubling time (PSADT) outcome after six months of treatment.27,28
Concomitantly, most anticancer compounds induce cell cycle arrest and cause apoptotic cell death. Based on selective growth inhibition of DU-145 cells and the results shown above, the effect of corchorusoside C (1) was determined on the cell cycle distribution of prostate DU-145 cells by flow cytometric analysis. After 24 h of treatment, 1 showed a significant decrease in the percentage of cells in the G1 phase and exhibited an increase in cells in the sub-G1 phase when compared to a control (p < 0.05, p < 0.01; Figure 2). Regulation of cell cycle progression in cancer cells is considered a potentially effective strategy for tumor control growth. Previous studies have demonstrated that the accumulation of sub-G1 phase population is prominently associated with apoptosis. The sub-G1 phase is also known as the death phase.29
Figure 2.

(A) Effect of corchorusoside C (1) on the cell cycle distribution in DU-145 prostate cancer cells. Cells were treated for 24 h with 1 (0–5 μM) and then stained with PI. Following flow cytometry, the cellular DNA profile was analyzed using the software WinMDI 2.8. (B) Data represent the percentage of cell counts in each cell cycle phase. The results are expressed as the means ± SEM of two independent experiments. *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001 for significant differences against control treatment.
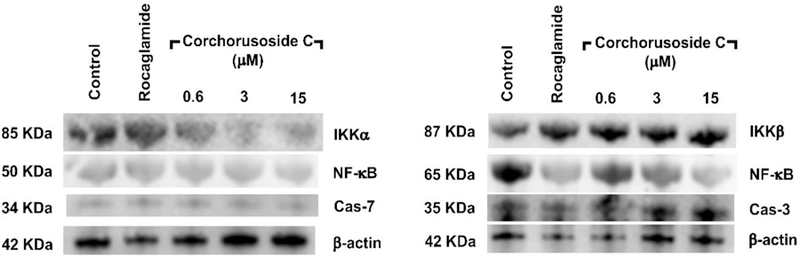
Different mechanisms have been proposed for how cardenolides exert their apoptotic effect. Thus, an approach to determine a potential mechanism of cytotoxic activity of corchorusoside C (1) was pursued to assess its inhibition to nuclear factor kappa B (NF-κB). Inhibition of the NF-κB activity is related to tumor cell proliferation, apoptotic induction, and increasing sensitivity of cells to anticancer drugs.17 Furthermore, tumor necrosis factor alpha (TNFα) is known to activate NF-κB through a kinase pathway. Hence, 1 was examined for its effect on TNFα-induced NF-κB activity. In an enzyme-based ELISA NF-κB assay after 5 h of treatment, corchorusoside C (1) showed NF-κB inhibitory activity with an IC50 value of 4.3 μM (Table 2). To corroborate this result, the effects of 1 on mediators of the NF-κB pathway were analyzed. DU-145 cells were treated for 12 h with corchorusoside C. Protein expression levels of subunits p50 and p65 of NF-κB and the inhibitor-κB kinases α and β (IKKα and IKKβ) were measured by immunoblot techniques. As shown in Figure 3A, 1 inhibited the production of both NF-κB p50 and p65 in a concentration-dependent manner (p < 0.05; Figure 3B). Similarly, the expression of IKKα and β, an upstream mediator in the NF-κB pathway, was found to be significantly down-regulated in a concentration-dependent manner (p < 0.05). The NF-κB molecule exists as a homodimer or heterodimer complex in the cytoplasm and five members of this family have been identified, designated as NF-κB1 (p50/p105), NF-κB2 (p52/p100), REL, RelA (p65/NF-κB3) and RelB.16 In general, degradation of IκB by activation of IKK enzyme induces NF-κB to cross the nuclear membrane and the expression of molecules related to the survival of the cell,30 including FLIP, Bcl-XL, A1/Bfl-1, cellular inhibitor of apoptosis (c-IAP), X chromosome–linked inhibitor of apoptosis (XIAP), TRAF1, and TRAF2.31 NF-κB activation is necessary to trigger inducible expression of intercellular cell adhesion molecule-1 (ICAM-1),32 which mediate adhesion of cells including DU-145 prostate cancer cells.33 It was then examined as to whether 1 can modify ICAM-1 expression in DU-145 prostate cancer cell lines, using Western blot. Figure 4 shows that 12 h of treatment with the cardenolide reduced ICAM-1 protein expression in a concentration-dependent manner when compared to the control (p < 0.05). Taken together, the results described above indicate that the cytotoxic activity of corchorusoside C (1) may be related to apoptosis mediated by inhibition in activity and down-regulation of expression of NF-κB in DU-145 prostate cancer cells.
Table 2.
Target-based Assay Activity of Corchorusoside C (1).
IC50 value (μM) for inhibition of nuclear factor kappa B (NF-κB p65).
IC50 value (μM) for inhibition of mitochondrial transmembrane potential (MTP) in HT-29 cells.
Positive controls.
Figure 3.

Effects of corchorusoside C (1) on the NF-κB signaling pathway in DU-145 prostate cancer cells. (A) Effects of 1 on the protein levels of NF-κB p50 and p65. B) Effects of 1 on protein levels of IKK α and β. Cells were treated with 1 (0–50 μM) for 12 h, and the total protein was prepared for Western blot analysis. Rocaglamide was used as the positive control (3 μM). Data are expressed as the means ± SEM (n = 3) and analyzed by one-way ANOVA using Dunnett’s multiple-comparison test (*p < 0.05, **p < 0.01, ***p < 0.01) of three independent assays.
Figure 4.

Effects of corchorusoside C (1) on protein levels of ICAM in DU-145 prostate cancer cells. Cells were treated with 1 (0–50 μM) for 12 h. Total protein was prepared for Western blot analysis. Rocaglamide was used as the positive control (3 μM). Data are expressed as the means ± SEM (n = 3) and analyzed by one-way ANOVA using Dunnett’s multiple-comparison test (*p < 0.05, **p < 0.01) of three independent assays.
Accumulating evidence indicates that reactive oxygen species (ROS) play a crucial role in apoptosis signaling pathways.34 Thus, it was hypothesized that the antiproliferative activity induced by corchorusoside C (1) might be associated with increased production of intracellular ROS in DU-145 cells. As shown in Figure 5, quantification of fluorescence (due to DCFH-DA conversion) suggested that after 5 h of treatment with 1 a significant increment of ROS production was observed when compared to the control (p < 0.05). Excess cellular levels of ROS cause damage to proteins, nucleic acids, lipids, membranes and organelles such as mitochondria.35 Mitochondrial dysfunction is related to increased ROS. To explore whether 1 induces a loss of mitochondrial transmembrane potential (MTP, ΔΨM), the JC-1 fluorescence indicator assay was performed. The results showed that 1 significantly reduced MTP in a concentration-dependent manner (Figure 6), with an IC50 value of 0.3 μM (Table 2), suggesting that apoptosis induced by corchorusoside C in DU145 cells might be associated with the intrinsic apoptotic pathway. Intrinsic apoptosis is a mitochondrion-centered cell death that is mediated by mitochondrial outer membrane permeabilization (MOMP). Changes in MOMP can induce the release of several pro-apoptotic proteina such as cytochrome c, Smac/Diablo and AIF.36 Cytochrome c (Cyt c) is an essential component of the apoptosome complex by caspase proteases activation.37
Figure 5.
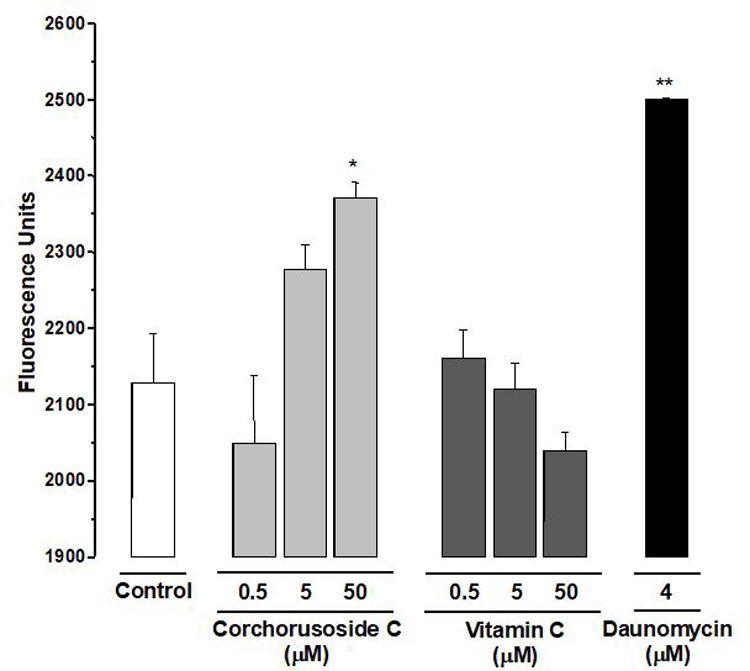
Evaluation of ROS inducing effect of corchorusoside C (1) on DU-145 prostate cancer cells. Daunomycin was used as a positive control. Data are expressed as the means ± SEM (n = 3) and analyzed by one-way ANOVA using Dunnett’s multiple-comparison test compared to a control; *p < 0.05, **p < 0.01.
Figure 6.
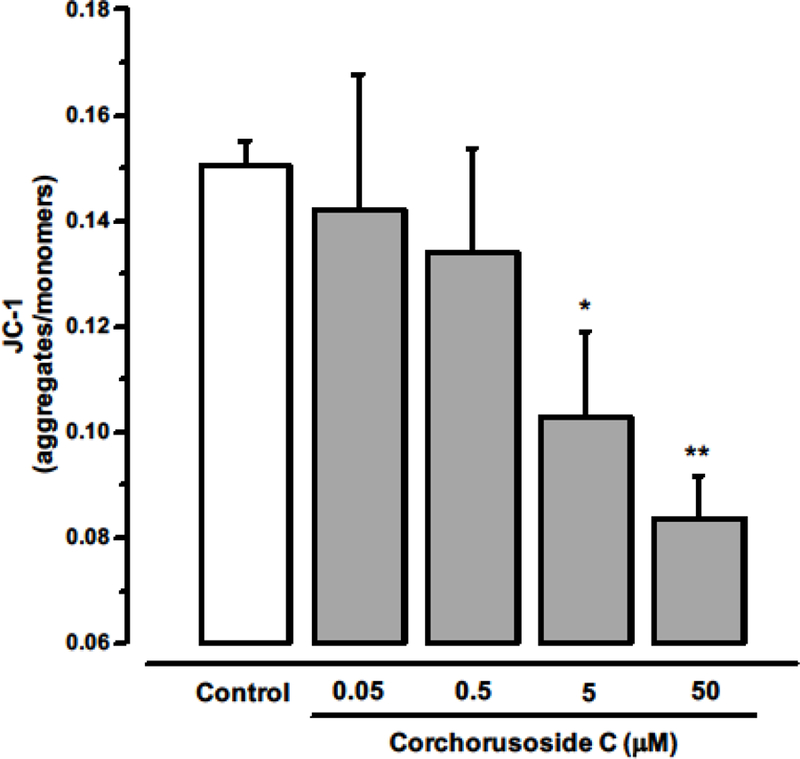
Effect of corchorusoside C (1) on the mitochondrial outer membrane potential (ΔΨM). Cells were treated with 1 (0–50 μM) for 5 h. The lipophilic cationic dye JC-1 was used to stain cells and the ratio of red to green fluorescence (aggregates/monomers) was calculated. Data are expressed as the means ± SEM (n = 3) and analyzed by one-way ANOVA using Dunnett’s multiple-comparison test compared to control; *p < 0.05, **p < 0.01.
The B cell lymphoma-2 (Bcl-2) family proteins regulate MOMP, leading to protein liberation from the space between the mitochondrial inner and outer membranes.38 The Bcl-2 protein family can be divided into antiapoptotic proteins, such as Bcl-2, Bcl-XL, Bcl-w, Mcl-1, A1, Bcl-Rambo, Bcl-L10, and Bcl-G, and proapoptotic proteins, such as Bax, Bak, and Bok.39 Hence, Western blot analysis was used to investigate the effect of corchorusoside C (1) on protein expression of Bcl-2 antiapoptotic protein. The results showed significant reductions of Bcl-2 protein expression at 5 and 50 μM concentrations (p < 0.05, p < 0.01, respectively) of 1, when compared to a control (Figure 7A).
Figure 7.

(A) Effects of corchorusoside C (1) on protein levels of Bcl-2 and (B) PARP-1 in DU-145 prostate cancer cells. Cells were treated with 1 (0–50 μM) for 12 h, and the total protein was prepared for Western blot analysis. Rocaglamide was used as the positive control (3 μM). Data are expressed as the means ± SEM (n = 3) and analyzed by one-way ANOVA using Dunnett’s multiple-comparison test (*p < 0.05, **p < 0.01, ***p < 0.01) of three independent assays.
Recent studies have indicated that Bcl-2 suppress poly(ADP-ribose) polymerases-1 (PARP-1) protein expression, for which the main function is to repair DNA damage.40 Western blot analysis showed that corchorusoside C treatment induced a significant increment in protein expression of PARP-1 in DU-145 cells at 5 and 50 μM (p < 0.05, p < 0.01, respectively; Figure 7B). This outcome is in agreement with the assumption that corchorusoside C induces damage in DU-145 cells by deregulating MOMP. Thus, an inverse correlation is reported between Bcl-2 expression and PARP-1.
In order to obtain evidence of the effects of corchorusoside C (1) on caspase activity, a luminescence assay caspase 3/7®GLO was used. The results in Figure 8A reveal that after 5 h of treatment with this cardenolide, DU-145 cells exhibited caspase enzyme activity in a dose-dependent manner (p < 0.01, p < 0.001). In this context, to better characterize the effects of 1 on the caspase pathway, the expression of caspases-3 and -7 were examined using Western blotting. DU-145 cells were treated 12 h at different concentrations (0–50 μM) of 1. The results showed that this cardenolide significantly increased protein expression of both caspase-3 and -7 (p < 0.05; Figure 8B), with particular emphasis on caspase-3. Caspases-3 and -7 are known as caspase effectors, which are responsible for initiating the hallmarks of the degradation phase of apoptosis, through the cleavage of caspase-activated DNAse, which then translocate to the nucleus and cleave DNA.41
Figure 8.

Effects of corchorusoside C (1) on the caspase pathway in DU-145 prostate cancer cells. (A) Effects of 1 on activity of caspase 3 and 7 after 5 h of treatment with corchorusoside C (0–8 μM). B) Effects of 1 (0–50 μM) on the protein levels of caspase 3 and 7 after 12 h of treatment and the total protein was prepared for Western blot analysis. Rocaglamide was used as positive control (3 μM). Data are expressed as the means ± SEM (n = 3) and analyzed by one-way ANOVA using Dunnett’s multiple-comparison test (*p < 0.05, **p < 0.01, ***p < 0.01) of three independent assays.
Zebrafish has become an invaluable model organism for the study of vertebrate biology,42 particularly to study mechanism of action of drug leads with limited availability. Hence, the effects of corchorusoside C (1) were assessed in an embryo zebrafish model to compare with the results observed in vitro with DU-145 cells. The results in Figure 9 show that treatment with 1 reduced protein expression of NF-κB p65 and IKKα, with observable changes in NF-κB p50 and IKKβ nearly undetected. On caspase expression in vivo, 1 induced a significant increase of caspase-3 protein levels, but not of caspase-7. These results are in agreement with those found in DU-145 cells.
Figure 9.
Effects of corchorusoside C (1) on key zebrafish proteins involved in the apoptosis signaling pathway.
Since corchorusoside C (1) showed significant results both in vitro and in vivo, efforts were made to compare its potential against the drug digoxin, another cardenolide derived from the Digitalis lanata. It is well known that cardenolides are a class of compounds that inhibit the plasma membrane Na+/K+-ATPase pump. This inhibition induces an increase on intracellular Na+ concentrations, which in turn decrease Na+/Ca2+ exchange activity. Reducing Na+/Ca2+ exchange activity would also induce an increment of intracellular Ca2+ ions, and as result more Ca2+ is released by the sarcoplasmic reticulum to affect myocyte contractility and to increase risk of cell calcification which may correlate with the outcome of the overall inotropic response in cells/myocytes.11, 12 This mechanism of action has been correlated with the toxic effect reported for digoxin in human myocytes. To investigate if corchorusoside C (1) elevates intracellular concentrations of Ca2+ in the same manner as digoxin, DU-145 cells were incubated with fura-2 AM and the fluorescence values were registered to correlate with intracellular Ca2+ levels. The results showed that after five hours of treatment of DU-145 cells at 50 μM with either 1 or digoxin, an increase in Ca2+ levels was observed compared to the negative control (p < 0.01 and p < 0.001, respectively). However, the increase in Ca2+ concentration induced by digoxin at 50 μM showed a significant difference compared with the effects of 1 at the same concentration (p < 0.01, Figure 10). Moreover, digoxin is known to effect zebrafish development (Figure 1), which could be related to its effects on the Ca2+. In contrast, corchorusoside C (1) did not affect zebrafish development when compared with digoxin at the same concentration. This suggests that 1 might not exhibit toxicity similar to that of digoxin.
Figure 10.
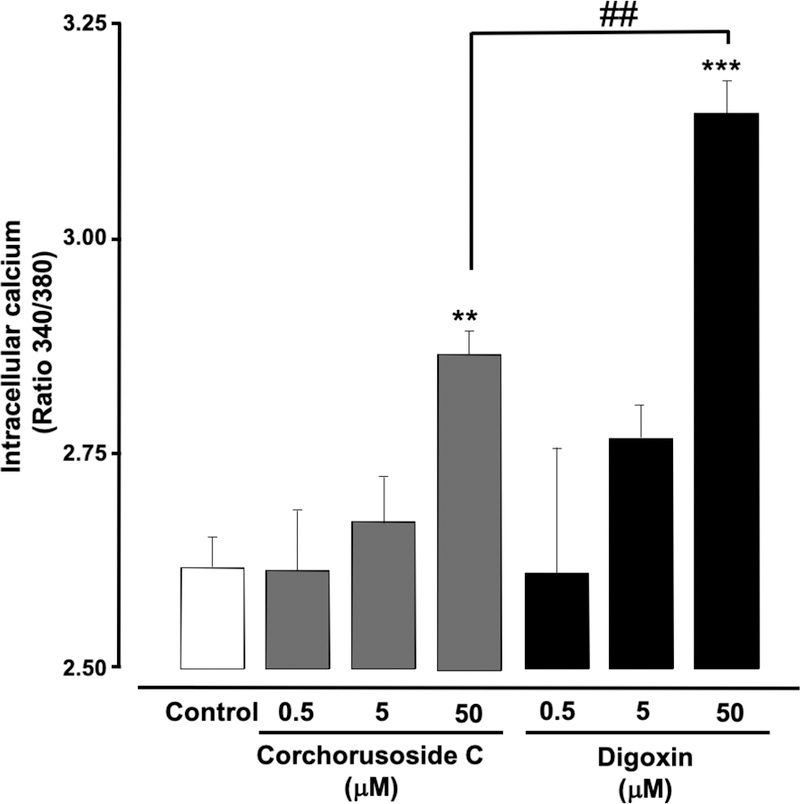
Effects of corchorusoside C (1) on intracellular calcium levels in DU-145 prostate cancer cells. DU-145 cells were treated with 1 (0–50 μM) for 5 h. Fluorescence was determined using fura-2 AM. Data are expressed as the means ± SEM (n = 3) and analyzed by one-way ANOVA using Dunnett’s multiple-comparison test (**p < 0.01, ***p < 0.01) compared to negative control or by a Student’s t-test (## p < 0.01) of two independent experiments.
In conclusion, corchorusoside C (1), a cardenolide isolated from Streptocaulon juventas, induces apoptosis in DU-145 prostate cancer cells by inhibition of the NF-κB pathway and activation of the intrinsic apoptotic pathway. Additionally, the increment in sub-G1 phase of the cell cycle suggests that 1 induces cell death by apoptosis. In the zebrafish, 1 exhibited similar effects on the NF-κB pathway. It is worthy of mention that corchorusoside C exhibited a high selectivity index toward cancer cells and did not show developmental toxicity in zebrafish at 50 μM when compared with the positive controls digoxin (another cardenolide) and cycloheximide. Hence, corchorusoside C (1) seems worthy of further studies in relation to its anticancer potential.
EXPERIMENTAL SECTION
General Experimental Procedures.
The optical rotation for corchorusoside C (1) was acquired on an Anton Paar MCP 150 polarimeter (Anton Paar GmbH, Graz, Austria). 1D (1H, 13C, DEPT-90, DEPT-135) and 2D (COSY, HSQC, HMBC, ROESY) NMR spectra were acquired on Bruker AVANCE II 400 MHz NMR instrument (Bruker, Billerica, MA, USA) at 298 K. Spectra were referenced with respect to solvent residual signals (CD3OD, δ 3.31 ppm for 1H, δ 49.00 ppm for 13C). High-resolution mass spectrometric data were acquired on a LTQ Orbitrap™ (Thermo Fisher Scientific Inc., Waltham, MA, USA) mass spectrometer with electrospray ionization (ESI) as ionization source. The mass spectrum was acquired in the positive mode and sodium iodide was used as an external calibrant. Column chromatography was performed using silica gel (particle size 40 × 63 μm (230 × 400 mesh) or 63–200 µm (65 × 250 mesh)], Sorbent Technologies, Norcross, GA, USA). Analytical TLC was performed on pre-coated silica gel TLC plates (aluminum backed, thickness 200 μm, UV254 fluorescent, Sorbent Technologies). The developed plates were either dipped in H2SO4 or sprayed with Godin’s reagent43 (spray solution A: 1% vanillin in ethanol mixed with an equal portion (1:1) of 3% perchloric acid in water and spray solution B: 10% H2SO4 in ethanol) and heated for chemical visualization. Reversed-phase (C18) HPLC purification was performed on a Waters HPLC system equipped with a 600 controller, a 717 Plus autosampler and a 2487 dual wavelength absorbance detector using a YMC Pack ODS-A semi-preparative column (10 ×250 mm, 5 μm).
Plant Material.
An initial small-scale collection of the stems of Streptocaulon juventas were made in January 2010 from Turtle Conservation Site at Núi Chúa National Park, Vietnam and a voucher specimen (AA06945/ST; voucher no.: Soejarto et al. 14880) was deposited at the Field Museum of Natural History, Chicago, IL, USA, under accession number 2300834. For isolation purposes and further studies, a large-scale re-collection of the same plant parts were made from the same location in July 2011.
Extraction and Isolation.
The dried stems of S. juventas (1.7 kg) were extracted with methanol and dried in vacuo. The dried methanol extract was successively partitioned with hexanes, chloroform (in three portions), ethyl acetate and water. The first and second portions of the chloroform partition were combined and subjected to partition with 1% NaCl to remove plant polyphenols.44 This de-tannified chloroform partition (26.4 g) was then fractioned by silica gel column chromatography using a gradient of CH2Cl2/MeOH (100:0 to 0:100), to produce 33 sub-fractions (F1 through F33), which were pooled according to their TLC similarities. F30 (1.4 g) was subjected to another silica gel (40 × 63 μm particle size, 300 g) column chromatography step (5.5 (i.d.) × 40 cm), eluted with step gradient mixture of CH2Cl2/MeOH (50/0, 50/1, 40/1, 30/1, 20/1, 10/1, 5/1, 3/1, 2/1, 1/1 and 0/1), after which, for the 3:1 solvent system, altogether 187 sub-fractions (each 10 mL) were collected. Then, using TLC, fractions were combined to give ten (F30F1 through F30F10) pooled sub-fractions. Fraction F30F3 (90 mg) was subjected to purification by semi-preparative HPLC [YMC Pack ODS-A column (10 ×250 mm, 5 μm); solvent system CH3CN/H2O, gradient elution: 25% to 33 % CH3CN for 26 min, 33% to 100% CH3CN for 1 min, followed by a 10 min wash with 100% CH3CN; flow rate 2 mL/min] to give corchorusoside C (1, 11.1 mg, tR 26.39 min). This compound [= periplogenin 3-O-β-D-glucopyranosyl-(1→4)-O-β-D-digitoxopyranoside; (c 0.17, MeOH); lit.3 (c 0.2, MeOH)] was identified from its 1D and 2D NMR spectroscopic and HRMS data (see Supporting Information, Figures 1S and 2S), in comparison with literature values.20
Zebrafish Toxicity.
Zebrafish (Danio rerio) were provided by the Department of Neuroscience, The Ohio State University. Experiments were completed under an approved animal protocol (Ohio State University IACUC protocol number 2004A00000006, entitled “Vertebrate Model for Natural Product Drug Discovery”; PI: Esperanza J. Carcache de Blanco). Fish were housed in an automated fish housing system at 28 °C. Embryos were grown to 24 hours post fertilization (hpf), and then exposed to corchorusoside C (1), digoxin, or cycloheximide at 50 μM or vehicle (1% DMSO in water) for 24 h,45 then zebrafish were observed under an Axiovert 40 CFL Zeiss microscope and pictures were taken using a ProgRes C10 plus camera.
Cell Culture.
HT-29 (colon), MDA-MB-231 (breast), MCF-7 (breast), PC3 (prostate), DU-145 (prostate), HeLa (cervical) cancer cells, and CCD-112CoN normal colon cells were obtained from the American Type Culture Collection, Manassas, VA, USA. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) or Roswell Park Memorial Institute medium (RPMI-1640) or Eagle’s Minimum Essential medium (EMEM) containing 10% fetal bovine serum (FBS) and 10% antibiotic-antimycotic from Gibco (Rockville, MD, USA). In the case of EMEM, sodium pyruvate (1mM), essential amino acids solution (0.1mM) and sodium bicarbonate (1.5 g/L) were added. The cells were grown as a monolayer in T75 tissue culture flasks and kept at 37 °C and in an atmosphere with 5% of CO2.
Cytotoxicity Assays.
The antiproliferative activity of corchorusoside C (1) was determined on human cancer and normal cell lines, using a sulforhodamine B (SRB) assay.46 Cells were seeded on a 96-well plate at a density of 5×104 cells/milliliter. Compound 1 was added at different concentrations (30–3×10−4 μM) to the plates. Each cell suspension was incubated at 37 °C in 5% CO2 for 24, 48 or 72 h. After this, cells were fixed with 20% cold trichloroacetic acid and then washed with tap water to be dried at room temperature. Cells were stained with SRB (0.4%), and unbound dye was washed with 1% acetic acid. After drying, bound dye was solubilized with Tris-base (10 mM), and absorbance was read at 515 nm using a FLUOstar Optima plate reader (BMG Labtech Inc., Durham, NC, USA). Paclitaxel or ellipticine were used as positive controls. The inhibitory concentration 50 (IC50) values were calculated by non-linear regression analysis using TableCurve 2Dv4 (System Software Inc., San Jose, CA, USA).
Cell Cycle Analysis.
Cell cycle analysis was carried out in a similar way as previously reported.47 Briefly, DU-145 cells were seeded on 6-cm plates and treated with corchorusoside C (1, 0.005–5 μM) for 24 h. Cells were then washed with PBS and treated with trypsin EDTA, harvested, centrifuged, and re-suspended in cold water. Cells were fixed using ethanol (70%), washed and re-suspended in PBS. Then, RNAse (20 mg/mL) was added to be incubated for 1 h at 37 °C. Cells were stained with propidium iodide (PI, 1.0 mg/mL; Sigma Aldrich) and protected from light until analysis using a BD FACS Calibur cytometer at an excitation wavelength of 488 nm and an emission wavelength of 617 nm.
NF-κB Inhibition Assay.
The NF-κB inhibition assay was carried out according to a previous procedure.47 Briefly, cells were treated with four different concentrations of corchorusoside C (1, 0.05–50 μM) or rocaglamide (positive control) and tumor necrosis factor alpha (TNF-α). Then, nuclear extracts were obtained, which were used to measure the ability of corchorusoside C (1) to interfere with the translocation of NF-κB to the nucleus. This interaction was measured by detecting the absorbance signal in a FLUOstar Optima plate reader (BMG Labtech, Inc.). The IC50 values were obtained using TableCurve 2D v4 (System Software Inc., San Jose, CA, USA).
Reactive Oxygen Species (ROS) Assay.
This assay was performed in a similar way as in a previous reported procedure.47 Prostate DU-145 cancer cells were seeded (5 × 104 per mL) in a 96-well black plate and treated with corchorusoside C (1, 0.5 to 50 μM), vitamin C (0.5 to 50 μM), daunomycin (4 μM), or a control (DMSO 10%). After incubation at 37 °C, 2′,7′-dichlorofluorescein-diacetate (DCFH-DA, 10 μM) was administrated to detect intracellular ROS. Fluorescence was recorded using a Fluostar Optima fluorescence plate reader (BMG Labtech, Inc.), with an excitation wavelength of 485 nm and emission wavelength of 530 nm. All treatments were performed in triplicate and data are representative of two independent experiments.
Mitochondrial Transmembrane Potential (MTP) Assay.
Cancer cells were used to determine the effect of corchorusoside C (1) on MTP following a previously published protocol.48 Briefly, cells were seeded on 96-well black culture plate. After incubation at 37 °C in 5% CO2, cells were treated with 1, staurosporine or a control (DMSO 1 %). To all wells were added 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazoylcarbocyanine iodide (JC-1 staining solution) and incubation was conducted under the same conditions as described above. Finally, 100 μL of PBS buffer were added, and fluorescence analysis was carried out using a FLUOstar Optima plate reader. In healthy cells, JC-1 forms aggregates that cause a strong fluorescent intensity, which was measured at an excitation and emission wavelength of 535 nm and 595 nm, respectively. Unhealthy (apoptotic) cells in JC-1 as monomers were measured with excitation and emission at 485 nm and 535 nm, respectively. The results are presented in μM as the half maximal inhibitory concentration (IC50) in relation to the control, with staurosporine used as the positive control.
Western Blot Analysis.
DU-145 cells in DMEM medium were treated with corchorusoside C (1) at different concentrations (0.05–50 μM) for 12 h. Rocaglamide (8 μM) and DMSO/water were used as controls. Cells were lysed using PhosphoSafe ™ extraction reagent from Novagen (Madison, WI, USA). For zebrafish studies, animals (72 hpf) were treated for three hours at different concentrations with 1, and then were frozen in dry ice and homogenized. Protein concentration was determined using the BCA method for both cells and animal samples. Twenty μg of each lysed sample were separated in Nu-PAGE 10% SDS-PAGE Bis-Tris gel by electrophoresis using SDS-PAGE. Proteins were transferred to a polyvinyldiene fluoride membrane (PVDF) and blocked with 3 % of bovine serum albumin (BSA). The membranes were incubated with the corresponding primary antibodies overnight and with secondary antibodies for 1 h. Proteins were detected using chemiluminescent substrate Supersignal Femto kit (Thermo Fisher Scientific, Waltham, MA, USA) and band densities were analyzed using ImageJ 1.52a.49
Caspase-3/-7 Activity.
Caspase-3 and -7 activities were measured in DU-145 (1×104 cells /well) prostate cancer cells using a Caspase-Glo 3/7® kit. After 5 h of treatment in a white 96 plate with corchorusoside C (1, 0.08–8 μM), paclitaxel (0.01μM) or DMSO/water, Caspase-Glo 3/7® reagent was added to cells, and then were incubated for 2 h.50 Luminescence was measured using a FLUOstar Optima plate reader (BMG Labtech, Inc.).
Intracellular Calcium Assay.
Calcium concentrations were determined using fura-2-acetoxymethyl ester (fura-2 AM) radiometric dye.51 Briefly, DU-145 prostate cancer cells seeded in a black-well 96 plate (1 × 104 cells/well) were treated for 5 h with corchorusoside C (1), digoxin (0–50 μM, positive control) or DMSO/water (negative control). After treatment, the medium was removed and cells were first incubated with 5 μM of fura-2 AM, then incubated with probenecid (2.5 mM). Intracellular calcium was determined by calculation of the fluorescence ratio at 510 nm emission, in response to 340/380 nm excitation wavelength, using a Synergy H1 Hybrid Reader (Biotek®) and Gen5 2.09 software.
Statistical Analysis.
Experimental results are presented as the means ± standard error of the mean (SEM), and all measurements and analyses were carried out in triplicate on two or three independent experiments. TableCurve 2D 4v (System Software Inc., San Jose, CA, USA) was used for statistical evaluations to obtain IC50 values. The statistical significance differences were ascertained by ANOVA and post-hoc Dunett’s test or an unpaired Studentś T-test using GrapPad Prism 5.0 software. A value of *p < 0.05, **p < 0.01, ***p < 0.001 indicated significant differences between groups against to the controls.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Program Project grant P01 CA125066 from the National Cancer Institute, National Institutes of Health. The plant samples were collected under a collaborative arrangement between the University of Illinois at Chicago (USA) and the Institute of Ecology and Biological Resources of the Vietnam Academy of Science and technology, Hanoi (Vietnam). Dr. C. A. McElroy (College of Pharmacy), The Ohio State University, is thanked for access to the NMR instrumentation used in this investigation.
Footnotes
The authors declare no conflict of interest.
REFERENCES
- (1).World Health Organization. Fact Sheet. Cancer 2018. https://www.who.int/cancer/en/ (Accessed December 11, 2018).
- (2).Siegel RL; Miller KD; Jemal A CA: Cancer J. Clin 2018, 68, 7–30. [DOI] [PubMed] [Google Scholar]
- (3).Mann K Nat. Rev. Cancer 2002, 2, 143–148. [DOI] [PubMed] [Google Scholar]
- (4).Zhou JS; Zhang TT; Chen JJ; Wang Q Chin. J. Nat. Med 2009, 7, 108–110. [Google Scholar]
- (5).Vo VC Dictionary of Vietnamese Medicinal Plants; Publishing House Medicine: Ho Chi Minh City, Vietnam, 1997. [Google Scholar]
- (6).Can NMC; Le DT; Kamei K; Dang TPT Wound Rep. Reg 2017, 25, 956–963. [DOI] [PubMed] [Google Scholar]
- (7).Xue R; Han N; Ye C; Wang H-B; Yin J Phytochemistry 2013, 88, 105–111. [DOI] [PubMed] [Google Scholar]
- (8).Xue R; Han N; Ye C; Wang L; Yang J; Wang Y; Yin J Fitoterapia 2014, 98, 228–233. [DOI] [PubMed] [Google Scholar]
- (9).Ueda J-Y; Tezuka Y; Banskota AH; Tran QL; Tran QK; Saiki I; Kodata S Biol. Pharm. Bull 2003, 26, 1431–1435. [DOI] [PubMed] [Google Scholar]
- (10).Zhang X-H; Zhu H-L; Yu Q; Xuan L-J Chem. Biodivers 2007, 4, 998–1002. [DOI] [PubMed] [Google Scholar]
- (11).Schoner W; Scheiner-Bobis G Am. J. Physiol. Cell Physiol 2007, 293, C509–C536. [DOI] [PubMed] [Google Scholar]
- (12).Lin Y; Chen D; Wang L; Ye D In Natural Products; Ramawat K, Mérillon JM, Eds.; Springer: Berlin, Heidelberg, 2013; pp 3743–3755. [Google Scholar]
- (13).Lohberger B; Wagner S; Wohlmuther J; Kaltenegger H; Stuendl N; Leithner A; Rinner B; Kunert O; Bauer R; Kretschmer N Phytomedicine 2018, 51, 162–170. [DOI] [PubMed] [Google Scholar]
- (14).Shi-Yuan W; Yan-Yan C; Chun-Miao D; Cui-Qiong Z; Miao-Miao J; Phytomedicine 2019, 57, 352–363.30831484 [Google Scholar]
- (15).Chen W-L; Ren Y; Ren J; Erxleben C; Johnson ME; Gentile S; Kinghorn AD; Swanson SM; Burdette JE J. Nat. Prod 2017, 80, 659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Manna SK; Sah NK; Newman RA; Aggarwal BB Cancer Res 2000, 60, 3838–3847. [PubMed] [Google Scholar]
- (17).Hoesel B; Schmid JA Mol. Cancer 2013, 12, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Nakanishi C; Toi M Nat. Rev. Cancer 2005, 4, 297–309. [DOI] [PubMed] [Google Scholar]
- (19).Newman RA; Yang P; Pawlus AD; Block KI Mol. Interv 2008, 8, 36–49. [DOI] [PubMed] [Google Scholar]
- (20).Nakamura T; Goda Y; Sakai S; Kondo K; Akiyama H; Toyoda M Phytochemistry 1998, 49, 2097–2101. [DOI] [PubMed] [Google Scholar]
- (21).Sipes NS; Padilla S; Knudsen TB Birth Defects Res. C. Embryo Today 2011, 93, 256–67. [DOI] [PubMed] [Google Scholar]
- (22).Stone KR; Mickey DD; Wunderli H; Mickey GH; Paulson DF Int. J. Cancer 1978, 3, 274–281. [DOI] [PubMed] [Google Scholar]
- (23).Alimirah F; Chen J; Basrawala Z; Xin H; Choubey D FEBS Lett 2006, 580, 2294–2300. [DOI] [PubMed] [Google Scholar]
- (24).Elmore S Toxicol. Pathol 2007, 35, 495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Rascón-Valenzuela LA; Velázquez C; Garibay-Escobar A; Vilegas W; Medina-Juárez LA; Gámez-Meza N; Robles-Zepeda RE J. Ethnopharmacol 2016, 193, 303–311. [DOI] [PubMed] [Google Scholar]
- (26).Bloise E; Braca A; De Tommasi N; Belisario MA Cancer Chemother. Pharmacol 2009, 64, 793–802 [DOI] [PubMed] [Google Scholar]
- (27).Menger L; Vacchelli E; Kepp O; Eggermont A; Tartour E; Zitvogel L; Kroemer G; Galluzzi L Oncoimmunology 2013, 2, e23082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Mekhail T; Kaur H; Ganapathi R; Budd GT; Elson P; Bukowski RM Investig. New Drugs 2006, 24, 423–427. [DOI] [PubMed] [Google Scholar]
- (29).Chuang JY; Tsai YY; Chen SC; Hsieh TJ; Chung JG In Vivo 2005, 19, 683–688. [PubMed] [Google Scholar]
- (30).Oeckinghaus A; Hayden MS; Ghosh S Nat. Immunol 2011, 8, 695–708. [DOI] [PubMed] [Google Scholar]
- (31).Luo JL; Kamata H; Karin MJ Clin. Invest 2005, 115, 2625–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Xia YF; Liu LP; Zhong CP; Geng JG Biochem. Biophys. Res. Commun 2001, 289, 851–856. [DOI] [PubMed] [Google Scholar]
- (33).Ren Z; Kang W; Wang L; Sun B; Ma J; Zheng C; Sun J; Tian Z; Yang X; Xiao W Mol. Cancer 2014, 13, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Simon H-U; Haj-Yehia A; Levi-Schaffer F Apoptosis 2000, 5, 415–418. [DOI] [PubMed] [Google Scholar]
- (35).Redza-Dutordoir M; Averill-Bates DA Biochim. Biophys. Acta 2016, 1863, 2977–2992. [DOI] [PubMed] [Google Scholar]
- (36).Uren RT; Dewson G; Bonzon C; Lithgow T; Newmeyer DD; Kluck RM J. Biol. Chem 2005, 280, 2266–2274. [DOI] [PubMed] [Google Scholar]
- (37).Wang C; Youle RJ Annu. Rev. Genet 2009, 43, 95–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kalkavan H; Green DR Cell Death Differ 2018, 25, 46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kale J; Osterlund EJ; Andrews DW Cell Death Differ 2018, 25, 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Dutta C; Day T; Kopp N; Van Bodegom D; Davids MS; Ryan J; Bird L; Kommajosyula N; Weigert O; Yoda A; Fung H; Brown JR; Shapiro GI; Letai A; Weinstock DM Cancer Res 2012, 72, 4193–4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Fiandalo MV; Kyprianou N Exp. Oncol 2012, 34, 165–175. [PMC free article] [PubMed] [Google Scholar]
- (42).Lieschke GJ; Currie PD Nat. Rev. Genet 2007, 8, 353–67. [DOI] [PubMed] [Google Scholar]
- (43).Marston A; Hostettmann K Saponins; Cambridge University Press: Cambridge, U.K,1995; pp 122–174. [Google Scholar]
- (44).Wall ME; Wani MC; Brown DM; Fullas F; Oswald JB; Josephson FF; Thornton NM; Pezzuto JM; Beecher CWW; Farnsworth NR; Cordell GA; Kinghorn AD Phytomedicine 1996, 3, 281–285. [DOI] [PubMed] [Google Scholar]
- (45).Li Y; Huang W; Huang S; Du J; Huang C Biochem. Biophys. Res. Commun 2012, 422, 85–90. [DOI] [PubMed] [Google Scholar]
- (46).Likhitwitayawuid K; Angerhofer CK; Cordell GA; Pezzuto JM; Ruangrungsi NJ Nat. Prod 1993, 56, 30–38. [DOI] [PubMed] [Google Scholar]
- (47).Kim JA; Lau EK; Pan L; Carcache de Blanco EJ Anticancer Res 2010, 30, 3295–3300. [PMC free article] [PubMed] [Google Scholar]
- (48).Deng Y; Balunas MJ; Kim J-A; Lantvit DD; Chin Y-W; Chai H; Sugiarso S; Kardono LBS; Fong HHS; Pezzuto JM; Swanson SM; Carcache de Blanco EJ; Kinghorn AD J. Nat. Prod 2009, 72, 1165–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Muñoz-Acuña U; Wittwer J; Ayer S; Pearce C; Oberlies NH; Carcache de Blanco EJ Anticancer Res 2012, 32, 2415–2422. [PMC free article] [PubMed] [Google Scholar]
- (50).Acuña UM; Shen Q; Ren Y; Lantvit DD; Wittwer JA; Kinghorn AD; Swanson SM; Carcache de Blanco EJ Int. J. Cancer Res 2013, 9, 36–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Martínez M; Martínez NA; Silva WI BioProtocol 2017, 7, 1–6. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.