

Abstract
The M2 isoform of pyruvate kinase (PK) is upregulated in most cancers including glioblastoma. Although PKM2 has been reported to use dual kinase activities to regulate cell growth, it also interacts with phosphotyrosine (pY)-containing peptides independently of its kinase activity. The potential for PKM2 to use the binding of pY-containing proteins to control tumor growth has not been fully examined. We here describe a novel mechanism by which PKM2 interacts in the nucleus with the RNA binding protein HuR to regulate HuR sub-cellular localization, p27 levels, cell cycle progression and glioma cell growth. Suppression of PKM2 in U87, T98G and LN319 glioma cells resulted in increased p27 levels, defects in entry into mitosis, increased centrosome number, and decreased cell growth. These effects could be reversed by shRNA targeting p27. The increased levels of p27 in PKM2 knock-down cells were caused by a loss of the nuclear interaction between PKM2 and HuR, and a subsequent cytoplasmic re-distribution of HuR, which in turn led to increased cap-independent p27 mRNA translation. Consistent with these results, the alterations in p27 mRNA translation, cell cycle progression and cell growth caused by PKM2 suppression could be reversed in vitro and in vivo by suppression of HuR or p27 levels, or by introduction of forms of PKM2 that could bind pY, regardless of their kinase activity. These results define a novel mechanism by which PKM2 regulates glioma cell growth, and also define a novel set of potential therapeutic targets along the PKM2-HuR-p27 pathway.
Keywords: pyruvate kinase, HuR, p27, cell cycle, metabolism
Pyruvate kinase (PK) is an enzyme that catalyzes the conversion of phosphoenolpyruvate (PEP) to pyruvate in the final and rate-limiting step of glycolysis.1 PK is expressed in four isoforms, L, R, Ml and M2.2 PKML and PKMR have a limited, tissue-specific pattern of expression, being expressed in liver and erythrocytes, respectively. PKM1 in contrast is expressed in tissues thought to have high energy requirements such as skeletal muscle and brain.3 PKM1 forms homotetramers, which have a high affinity for PEP and generate high levels of pyruvate,2 which in turn is funneled into the citric acid cycle for further production of energy.2 PKM2 in contrast, is expressed in many differentiated tissues as well as in cells with a high rate of nucleic acid synthesis such as stem cells and many (but not all) tumors, including gliomas.4,5 In these tissues PKM2 exists in both tetrameric and monomeric/dimeric forms.6 The tetrameric form of PKM2 is the most active with regard to PK activity.2 In tumors, however, growth-related signaling events lead to phosphorylation of PKM2 at Y105,7 and increased binding of PKM2 to phospho-tyrosine (pY)-containing proteins.8 These events, along with low levels of the PKM2 allosteric activator fructose 1,6 -bisphosphate, limit tetramer formation, limit PK activity, and in turn are associated with increased tumor growth.2,4,7,8 Consistent with this idea, the complete deletion of PKM2 in models of breast cancer results in increased tumor growth.10 In its dimeric form as is found in tumors, however, PKM2 has been reported to display a second distinct kinase activity9 that phosphorylates molecules that directly or indirectly promote cell cycle entry10,11 facilitate chromosomal segregation,12 and promote cellular proliferation.13 In this manner the protein kinase functions of PKM2 have been suggested to contribute to tumor growth independently of the PK activity of the enzyme.8,9,11,12
Although PKM2 uses kinase-dependent mechanisms to alter cell behavior, PKM2 also interacts with a variety of metabolites and pY-containing proteins independently of its kinase activity.2,4,8 Furthermore, although the kinase-dependent activities of PKM2 have been defined in cell lines with extremely high growth factor pathway activation,11,13–15 the potential for PKM2 to use its pY-binding ability to control basal states of tumor growth has not been fully examined. In this report we show that PKM2 uses its pY-binding ability to reduce cytoplasmic levels of HuR, a master regulator of RNA metabolism. Loss of PKM2 limits the binding to HuR and leads to a cytoplasmic re-distribution of HuR. This in turn leads to increased cap-independent p27 mRNA translation, increased levels of p27 and a variety of cell cycle defects associated with high p27 levels. These results define a new HuR- and p27-mediated pathway by which PKM2 controls RNA metabolism and regulates glioma cell growth.
Material and Methods
Cell culture
U87MG, T98G and LN319 human glioma cells were provided by the UCSF Brain Tumor Center Tissue Core and were cultured as described.16
Modulation of PKM2, p27 and HuR expression
Nontargeted, or one of two different lentiviral PKM2 shRNAs were used to transduce U87, T98G and LN319 cells as described.16 Parental and PKM2 knock-down cells were also either transiently transfected (Fugene 6) with one of the two different siRNA targeting p27 or HuR, transduced with shRNAs targeting p27 or HuR, or transduced with lentiviral constructs encoding human HuR or one of four different forms of mouse PKM2 (WT, pY binding-deficient K433E15, kinase-dead K367M17 or R399E10). In each case the appropriate scrambled siRNA, scrambled shRNA or blank vector control was also used.
Cell synchronization and analysis of cell cycle and centrosome number
Cells were synchronized by incubation (48 hr) in serum-free DMEM-H21 media, released into 10% serum-containing media, collected at the indicated time points, fixed and stained with phospho (Ser 28) histone H3.3 antibody (Cell Signaling), and analyzed by FACS. For BrdU incorporation analysis, released cells were incubated with BrdU (10 μM, 30 min), fixed, and stained in PBS containing 20 μg/ml anti-BrdU (Roche). Cell cycle distribution was assessed in fixed, propidium iodide-labeled cells by FACS in combination with Flowjo software (Treestar). DNA content was measured by DAPI staining and quantitative FACS analysis. Bright field microscopy was performed to monitor the cell size. Centrosome number was assessed using a pericentrin-specific antibody (Abcam) followed by a fluorescent secondary antibody (anti rabbit Alexa 488, Invitogen) and 40 × immunofluorescence microscopic analysis of >400 cells per group.
Translation assays
Control and PKM2 knock-down cells were transiently transfected with a full length or a 71 bp deleted 5’ UTR of p27 bicistronic (pCMV–Myc–RL–p27 5 UTR –FL) expression construct. p27 cap-independent translation analysis was performed using a Dual Luciferase kit (Promega). p27 IRES bicistronic mRNA expression levels were determined by quantitative PCR and were used to normalize luciferase activities.
Sucrose density gradient fractionation and RNA analysis
Cells (5 × 107) were incubated with cycloheximide (15 min, 100 mg/ml). Cytoplasmic lysates (500 μl, NE-PER Extraction Kit, Pierce) were subjected to linear sucrose density gradient (10–50%) centrifugation18 and divided into 10 fractions of increasing density. RNA was extracted (Trizol LS, Invitrogen), dissolved in nuclease-free water, and analyzed (Agilent 2100 Bioanalyzer) to identify the polysome fractions with a 28S/18S rRNA ratio equal to 2.19 Relevant fractions were analyzed in triplicate for p27 and β-actin mRNA levels by real time PCR.16 For p27, the primers were 5- CGCTTT GTTTTGTTCGGTTT-3 and 5- TCGCACGTTTGACATC TTTC-3. For β -actin the primers were 5-GATGAGATT GGCATGGCTTT-3 and 5- CACCTTCACCGTTCCAGTTT −3. Thermal cycling conditions consisted of an initial 958C for 3 min followed by 40 cycles at 95°C for 15 sec, 60°C for 15 sec and 72°C for 30 sec (Rotor Gene, Qiagen).
Cell fractionation, protein extraction, cross-linking, immunoprecipitation and western blot analysis
Protein lysates from control, rapamycin (100 nM, 18 hr) or leptomycin B (0 or 5 ng/ml, 16 hr) treated cells or cell sub-fractions (NE-PER Extraction kit, Pierce) were prepared in lysis buffer supplemented with protease and phosphatase inhibitors (Roche). For immunoprecipitation, protein lysates were precleared with protein A/G-agarose beads (Santa Cruz, 3 hr, 4°C), then incubated with primary antibody (16 hr, 4°C). Immune complexes were precipitated (2 hr, 4°C) with protein A/G-agarose beads. In control samples, the primary antibody was substituted with control IgG. Immunoprecipitates were washed four times with RIPA buffer containing 0.5 M NaCl and 2% SDS, three times with PBS, then resuspended in Laemmli buffer. Proteins were separated on 4–20% gradient polyacrylamide gels and transferred onto Immuno-Blot PVDF membranes (Bio-Rad Laboratories). Membranes were then incubated in blocking buffer (1X TBS containing 5% milk and 0.05% Tween-20, 2 hr), probed overnight with antibodies specific for the cytoplasmic marker β-tubulin (1:1000), the nuclear marker histone H3 (1:1000), PKM2 (1:1000), β-actin (1:20,000), pS6 (1:1000), 4E-BP1,(1:1000), cyclin B (1:1000), pY (1:2000), pY15 Cdk1 (1:2000), (all Cell Signaling), PKM1 (Proteintech, 1:1000), p27 (1:500)(Santa-Cruz), phospho (T187) p27 (1:500)(Abcam), HuR (1:500, Santa Cruz) or FLAG (1:1000, Sigma), washed, then incubated with appropriate horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology). Antibody binding was detected by incubation with ECL reagents (Amersham Pharmacia Biotech).
Cyclin B/Cdk1 activity assays
Cyclin B/Cdk1 activity was determined using a cyclinB/Cdk1 activity assay (MBL International Corp.)
Immunofluorescence staining of cells, xenografts and primary human GBM sections
Cultured cells grown overnight in four-well-chambered slides overnight were fixed in paraformaldehyde (4%, 10 min). Formalin-fixed, paraffin-embedded 5 μm sections of intracranial U87 xenografts and primary human GBM (obtained from the UCSF Brain Tumor Center Tissue Core using protocols approved by the UCSF Institutional Review Board and neuropathologically verified as grade IV based on the WHO classification scheme) were mounted on slides, deparafinized, washed and rehydrated in graded alcohol. For antigen unmasking, slides were placed in 1 mM EDTA pH 8 (10 min, 100°C under pressure) followed by 15 min at a sub-boiling temperature. After rinsing twice in PBST, all slides were blocked (3% normal goat serum and 0.2% triton X-100 in PBS, 30 min, room temperature), then incubated with PKM2 (1:200), p27 (1:100), FLAG (1:300) and/or HuR (1:100) primary antibodies in 1% goat serum and 0.2% triton X-100 in PBS (18–20 hr, 4°C). After washing, slides were incubated with fluorescent-tagged secondary antibodies (647, 588, 546, 1:200, 2 hr, Invitrogen) appropriate for the host species of the primary antibody. Following washing (PBS, 3 × 5 min each), sections were incubated with DAPI, washed and mounted. Negative controls for antibody labeling were performed by omitting primary or secondary antibodies.
Image acquisition and analysis
Confocal laser scanning fluorescence microscopy was performed on a Zeiss LSM 510 equipped with violet 405 nm, argon 488 nm, helium-neon 543 nm and helium-neon 633 nm lasers for excitation (DAPI, HuR, PKM2 and p27 imaging, respectively). Images were obtained using a 63 × objective. Random fields (15–18, 4–6 cells per field) were chosen from tissue sections from each of three GBM exhibiting low or high nuclear PKM2 staining. The absolute amount of PKM2 and HuR fluorescence localized to the nucleus or cytoplasm in each cell was calculated using ZEN software (Zeiss), and expressed as a percentage of the total cellular fluorescence. The total p27 cellular fluorescence was calculated and expressed as a percentage of the average p27 fluorescence noted in five positive control, p27-expressing, immune-infiltrated cells in each section.
In vivo studies
Immunodeficient mice (nu/nu; Charles River) (n = 7 in each group) were injected intracranially with 4 × 105 luciferase-expressing U87 cells containing blank lentiviral constructs or constructs encoding p27 or HuR, as well as lentiviral constructs encoding scrambled shRNAs or shRNAs targeting PKM2, p27 or HuR. Tumor growth was monitored weekly by treating mice with D-luciferin (150 mg/kg IP, Gold-Bio-technology) and measuring bioluminescence using a Xenogen IVIS Bioluminescence imaging station (Caliper). Tumor growth was calculated by normalizing luminescence measurements to Day 1 postinjection values. The guidelines of the UCSF Institutional Animal Care and Use Committee were followed for all animal work.
Statistical analysis
The unpaired Student’s t test was applied (p values) for comparing two groups while a one-way ANOVA test with post hoc Turkey-Kramer multiple comparisons test was used for multiple groups. A k-means test was used to define cutpoints of high and low nuclear PKM2, cytoplasmic HuR and p27 expression, while a χ2 goodness of fit test was used to determine if values displayed a random uniform distribution.
Results
PKM2 knock-down causes defects in entry to mitosis
We and others have shown that shRNA-mediated suppression of PKM2 decreases tumor cell proliferation in vitro and in vivo.10,15,16 The growth suppression we noted in PKM2 knock-down glioma cells in vitro, however, was not associated with decreased cyclin D1 protein levels (Fig. 1a, Supporting Information Fig. S1a), decreased cyclin D1 mRNA levels (Supporting Information Fig. S1b), increased PKM1 levels (Fig. 1a, Supporting Information Fig. S1a) or an accumulation of cells with a sub G0, G0/G1 or S phase DNA content (Fig. 1b). Rather, cells accumulated with a greater than 2N DNA content (G2/M, Fig. 1b). Serum-starved control or PKM2 knock-down cells re-entered the cell cycle 4–6 hr after releasing into serum-containing media, and displayed comparable percentages of cells entering S phase (BrdU + cells, Supporting Information Fig. S1c). Significantly fewer PKM2-knock-down cells, however, progressed into mitosis (phospho-histone H3.3+ cells) over the first 12 hr following release compared to control cells (Fig. 1c, Supporting Information Fig. S1d). Strikingly, cells expressing either of two shRNAs targeting PKM2 also exhibited increased DNA content (Supporting Information Fig. S1e), increased cell size (Supporting Information Fig. S1f) and an increased percentage of cells with >2 centrosomes (Fig. 1d). Although the PKM2 knock-down cells did not exhibit increased β-galactosidase staining associated with senescence (not shown), their phenotype resembled that of Skp2-deficient cells, which have high levels of p27 and similar difficulties progressing into mitosis.20 We therefore considered the possibility that PKM2 uses control of p27 to regulate mitotic progression and tumor cell growth.
Figure 1.
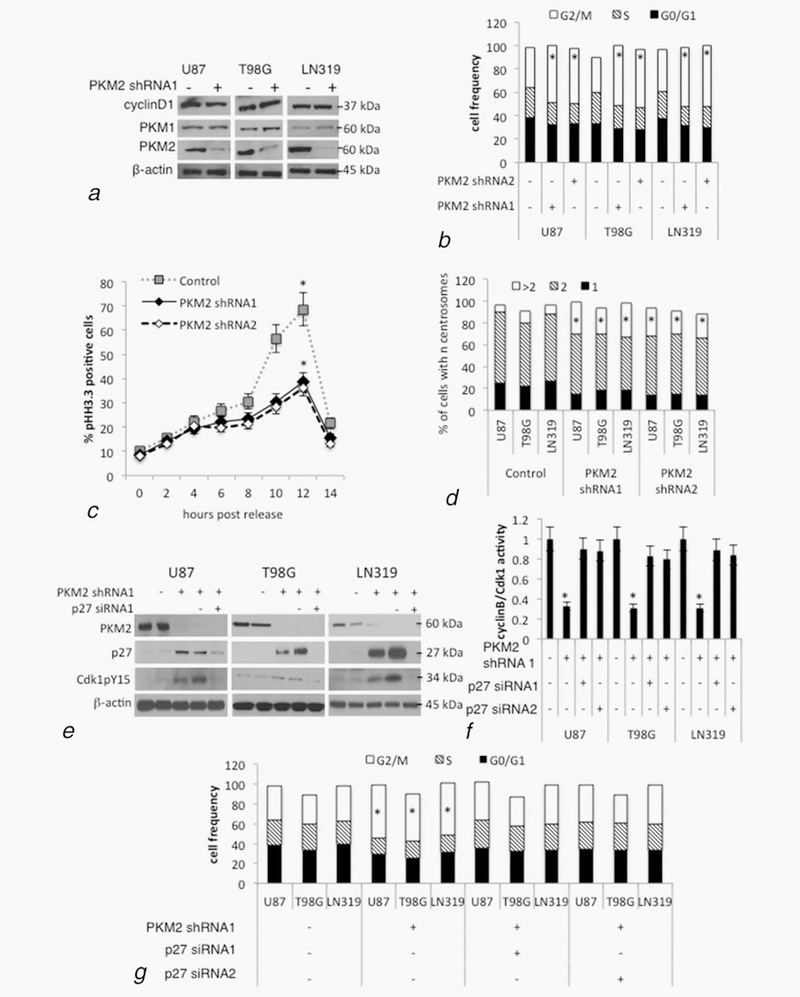
Suppression of PKM2 causes p27-dependent defects in cell cycle progression and mitotic entry. (a, b) U87, T98 or LN319 glioma cells were lentivirally infected with a scrambled shRNA (PKM2 shRNA-, control) or one of two constructs encoding shRNAs targeting human PKM2 (PKM2 shRNA1+ and PKM2 shRNA2+). Following drug selection, polyclonal populations were examined by Western blot for cyclin D1, PKM1, PKM2 and β-actin expression (a), or by FACS for cell cycle distribution (b). See also Supporting Information Figure S1a. *, p < .05, n =3. (c) U87 cells from panel a were serum starved for 48 hr, after which serum was added and the cells were then assessed for the percentage of mitotic pH3.3+ cells by immunohistochemistry and FACS. See also Supporting Information Figures S1b and S1c. *, p < .05, n = 3. (d) Cells from panel a and Supporting Information Figure S1a were fixed and incubated with a centrosome-specific pericentrin antibody, after which the cells were examined for centrosome number. See also Supporting Information Figures S1d and S1e. *, p < .05, n = 3. (e, f) Levels of PKM2, p27, phosphoY15-Cdk1 and β-actin (E), and cyclin B/Cdk1 activity (normalized to control)(f), in U87, T98 and LN319 cells from Figure 1a, or cells additionally containing scramble siRNA (p27 siRNA-) or one of two siRNAs targeting p27 (p27 siRNA1 & p27 siRNA2). See also Supporting Information Figure S1f and S1g. *, p < .05, n = 3. (g) FACS-based cell cycle distribution in U87, T98G and LN319 cells from panel e and f. See also Supporting Information Figures S1h–S1j. *, p < .05, n = 3.
PKM2-mediated effects on growth and cell cycle progression are p27 dependent
p27 is a repressor of cyclin A/Cdk2 complexes and the G1/S transition.21 p27-mediated inhibition of cyclin A/Cdk2, however, also limits the movement of cells through the G2-M transition by indirectly increasing Cdkl pY15 inhibitory phosphorylation and decreasing Cdkl/cyclin B activity.20 Consistent with this observation, cells expressing either of two PKM2 shRNAs had significantly higher levels of p27 and pY15 Cdkl (Figs. 1e, Supporting Information Fig. Slg), significantly less Cdkl activity (Fig. 1f Supporting Information Fig. Slh), and accumulated in the G2/M phase of the cell cycle (Fig. 1g, Supporting Information Fig. Sli) relative to their nontargeted controls. Additional introduction of either of two siRNAs targeting p27 decreased p27 and Cdkl pY15 levels by >90%, and reversed the decrease in cyclinB/Cdkl activity and the accumulation of cells in G2/M, all without changing levels of PKM2 (Figs. 1e–g, Supporting Information Figs. Slg–Sli). Furthermore, siRNA-mediated suppression of p27 reversed the suppressive effects of PKM2 knock-down on colony growth in vitro (Supporting Information Fig. Slj) and restored the ability of cells to retain a normal cell cycle distribution and centrosome number (Fig. 1g, Supporting Information Fig. Slk). The phenotypic changes and growth suppression noted following loss of PKM2 expression were therefore dependent on increased p27 expression.
PKM2 loss increases p27 mRNA cap-dependent translation
To understand how PKM2 influences expression of p27 and cell cycle progression, we first determined how p27 expression was altered in PKM2 knock-down cells. Control and PKM2 knock-down cells had comparable levels of p27 mRNA (Supporting Information Fig. S2a), suggesting that the higher p27 levels noted in the PKM2 knock-down cells were not a result of the ability of PKM2-mediated Stat3 activation to transcriptionally up-regulate p27.9 p27 levels can also, however, be regulated translationally, and the p27 mRNA contains a 5’UTR with an internal ribosome entry site that allows for cap-independent, as well as cap-dependent translation.22 The incubation of control or PKM2 knock-down cells with rapamycin, which blocks mTOR signaling and cap-dependent translation,23 did not, however, block the increase in p27 levels caused by PKM2 knock-down (Supporting Information Fig. S2b). The effects of PKM2 knock-down on cap-independent translation of the p27 mRNA were therefore examined in cells transiently transfected with a bicistronic expression construct that yielded a transcript that could be translated in both a cap-dependent manner to generate both Renilla and Firefly luciferase, and in a cap-independent manner (by virtue of an intervening p27 5’ UTR) to yield additional F. luciferase. Control and PKM2 knock-down cells exhibited similar cap-dependent expression of the R. luciferase (Fig. 2a). PKM2 knock-down cells, however, exhibited significantly increased expression of F. luciferase expression when the intact p27 5’UTR was included in the expression construct (Fig. 2a) but not when cells contained a 71 bp deleted 5’UTR construct (data not shown), indicative of increased cap-independent translation. Polysome analysis also showed that although the amount of β-actin mRNA in monosomal and polysomal fractions was similar in control and PKM2 knock-down cells, significantly more p27 mRNA was in the polysomal fractions of PKM2 knock-down cells (Fig. 2b, Supporting Information Fig. S2c), confirming that PKM2 loss increases p27 mRNA translation.
Figure 2.
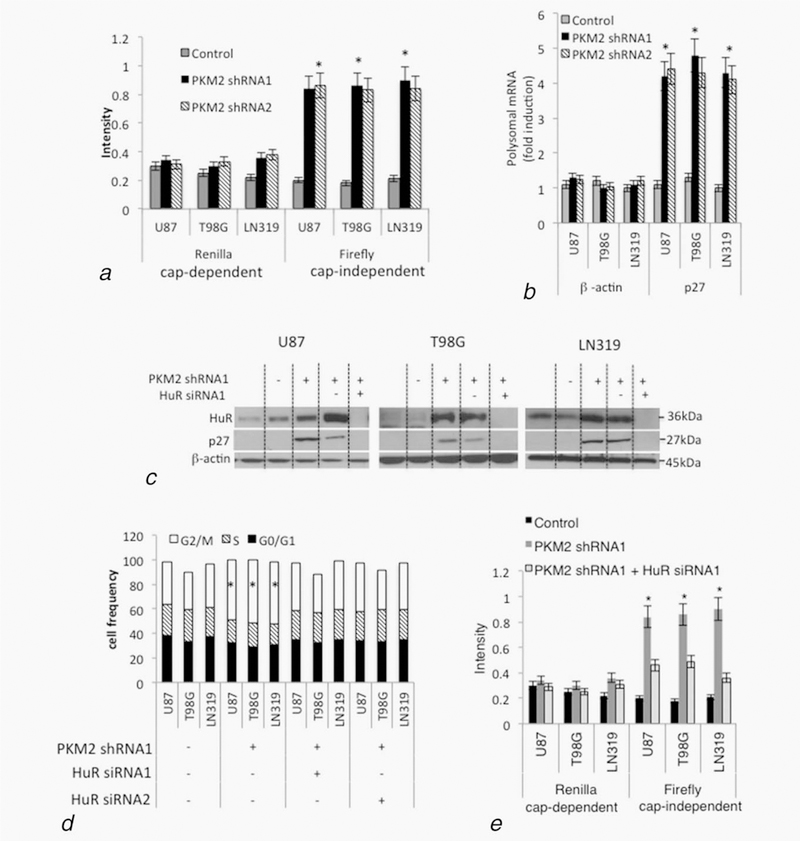
PKM2 suppression increases p27 levels and cell cycle arrest via HuR-dependent increased p27 mRNA translation. (a) Expression of Renilla and Firefly luciferase genes in lysates from cells from Figure 1aand Supporting Information Figure S1a containing a bicistronic reporter construct in which cap-dependent translation of both genes is driven by the CMV promoter while expression of Firefly can additionally be driven in a cap-independent manner from the internal ribosome entry site in an upstream p27 5’LTR. Quantitative PCR was used to normalize luciferase expression to bicistronic transcript levels. See also Supporting Information Figures S2a–S2b. *, p < .05, n = 3. (b) Control or PKM2 knock-down U87 cells from Figure 1a and Supporting Information Figure S1a were lysed and subjected to sucrose density gradient centrifugation, with subsequent RNA from fractions containing unassembled ribosomal subunits (fractions 2–5) or assembled polyribosomes (fractions 6–10) analyzed for p27 and β-actin mRNA content by quantitative PCR. Each bar represents the fraction of β-actin or p27 mRNA contained in the polysomal fractions in control or PKM2 knock-down cells. See also Supporting Information Figure S2c. Error bars indicate standard deviations, *, p < .05, n = 3. (c–e) Levels of HuR, p27 and β-actin (c), cell cycle distribution (d), and cap-dependent and cap-independent translation of p27 mRNA in cells from Figure 1a also expressing nontargeted (HuR siRNA-) or one of two HuR-targeted (HuR siRNA1 and HuR siRNA2) siRNA. See also Supporting Information Figures S2d–S2f. *, p < .05, n = 3.
The effects of PKM2 loss on p27 levels and cell cycle progression are HuR-dependent
To define the mechanism by which PKM2 regulates p27 capindependent translation, we first tested the role of HuR, an RNA-binding protein previously described as a regulator of p27 translation.21,24 The PKM2 knock-down that increased p27 levels and promoted accumulation of cells in G2/M also led to increased levels of HuR (Fig. 2c, Supporting Information Fig. S2d). Both the increased p27 levels and cell cycle arrest caused by PKM2 knock-down could be reversed by siRNA-mediated suppression of HuR levels (Figs.2c–d, Supporting Information Figs. S2d–S2e). Furthermore, the siRNA-mediated suppression of HuR in PKM2 knock-down cells also decreased the cap-independent translation of p27 (Fig. 2e, Supporting Information Fig. S2f). These results show that loss of PKM2 leads to increased levels of HuR. The subsequent stimulation of cap-independent p27 mRNA translation and increases in p27 protein levels is in turn a significant contributor to the deficits in mitotic entry in PKM2 knock-down cells.
PKM2 influences HuR cellular localization
Because HuR is a nuclear protein that shuttles to the cytoplasm to increase p27 mRNA translation,22 we next examined the effect of PKM2 knock-down on HuR localization. HuR was predominantly in the nuclear, histone-containing sub-fraction of control cells (Fig. 3a) and co-localized with DAPI-stained cell nuclei (Fig. 3b). In PKM2 knock-down cells, however, HuR was shifted to the cytoplasm. Incubation of cells with leptomycin, a specific pharmacologic inhibitor of nuclear export25 had no effect on the nuclear localization of HuR in control cells, but blocked the cytoplasmic accumulation of HuR in PKM2 knock-down cells (Fig. 3c). These results therefore show that loss of PKM2 expression allows increased export of HuR to the cytoplasm.
Figure 3.
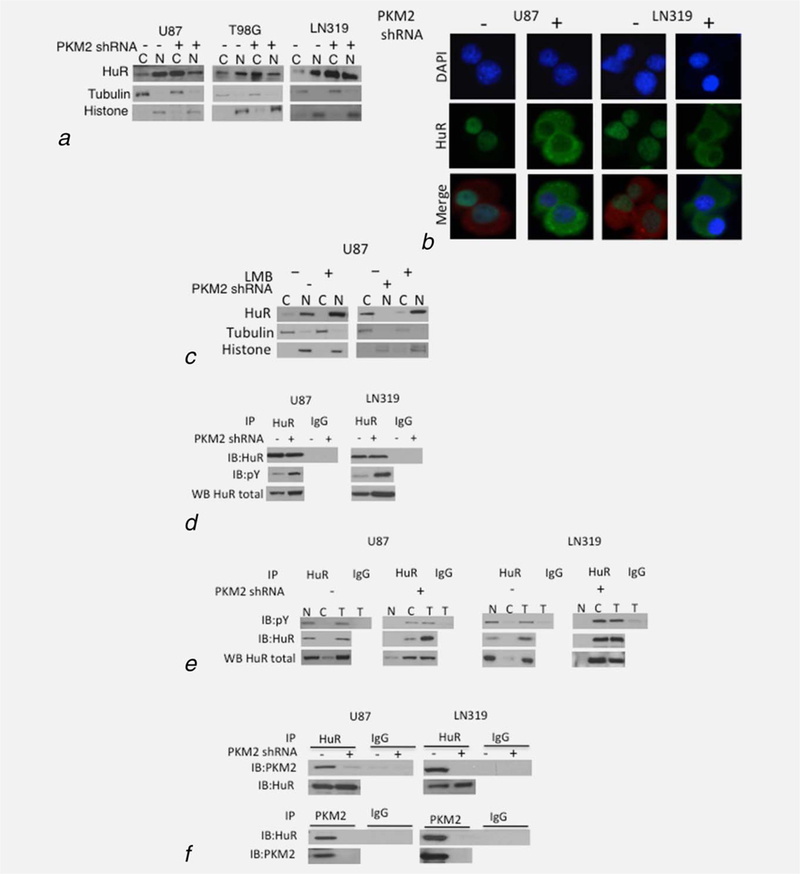
PKM2 controls the sub-cellular localization of HuR. (a, b) Levels of HuR, histone H3 and tubulin in the cytoplasmic (C) and nuclear (N) fractions of cells expressing a scramble or PKM2-targeted shRNA as assessed by Western blot (a) and DAPI(blue)/HuR (green) co-immunofluorescence analysis (b). PKM2 staining (red) delineates the cytoplasm in groups with minimal cytoplasmic HuR. (c) Western blot analysis of HuR, tubulin and histone in nuclear and cytoplasmic fractions of control and shPKM2 expressing U87 cells incubated with 0 or 5ng leptomycin B (LMB, 8 hr). (d) Western blot analysis of HuR and pY proteins in total cellular HuR or IgG immunoprecipitates from control or PKM2-suppressed U87 and LN319 cells. (e) Western blot analysis of HuR and pY proteins in total cellular (T) nuclear (N), or cytoplasmic (C) HuR or IgG immunoprecipitates from control or PKM2-suppressed U87 and LN319 cells. (f) Western blot analysis of PKM2 and HuR levels in HuR immunoprecipitates, and HuR levels in PKM2 immunoprecipitates from U87 and LN319 cells expressing a scramble or PKM2-targeted shRNA.
PKM2 binds and retains pY HuR in the nucleus to regulate p27 and cell cycle progression
Cellular stress alters the phosphorylation of HuR (S202, 221 and 242),26–29 which in turn leads to export of HuR into the cytoplasm and accumulation in stress granules.29 Levels of pS202 HuR in PKM2 knock-down cells, however, were not different from those in control cells, and the nuclear and cytoplasmic HuR were similarly S202 phosphorylated in both groups (Supporting Information Figs. S3a and S3b). There was also no evidence of stress granules (Supporting Information Fig. S3c), activation of AMP kinase (not shown) or of the mTOR inhibition (Supporting Information Fig. S2b) noted in metabolically stressed cells.30 Combined with recent evidence that PKM2 may not function as a protein kinase,31 these data suggest that PKM2 may regulate HuR sub-cellular localization by events unrelated to direct or indirect HuR serine phosphorylation.
HuR also contains seven tyrosines, and the phosphorylation of at least one of these (Y200) plays a role in HuR sub-cellular localization.29 Because PKM2 is reported to be a kinase that can also bind pY-containing proteins, we considered the possibility that PKM2 could control HuR nuclear export by directly or indirectly phosphorylating tyrosine residues of HuR, or by directly interacting with pY HuR. HuR immunoprecipitates from PKM2 knock-down cells contained similar or higher levels of protein recognized by a pY-specific antibody than control cells (Fig. 3d) suggesting that while HuR is tyrosine phosphorylated in GBM cells, PKM2 does not directly or indirectly increase tyrosine phosphorylation of HuR. Furthermore, the pY HuR in control cells was located in the nucleus while that in PKM2 knock-down cells was in the cytoplasm (Fig. 3e), suggesting that the pY status of HuR did not per se control the cytoplasmic redistribution of HuR following PKM2 suppression.
In light of this data we considered the possibility that direct binding of PKM2 to pY HuR contributes to HuR nuclear localization and function. In control cells, PKM2 was found in HuR immunoprecipitates and HuR was found in PKM2 immunoprecipitates, while both these interactions were less apparent in PKM2 knock-down cells (Fig. 1f). Furthermore, when PKM2 knock-down cells were infected with constructs encoding FLAG-tagged WT PKM2 (mM2), or forms of PKM2 that could (R399E, K367M) or could not (K433E) bind pY-containing proteins,8 HuR was associated only with FLAG-tagged immunoprecipitates containing forms of PKM2 that retained pY-binding ability (Fig. 4a). Of note, this association was independent of PKM2 kinase activity as the K367M PKM2 mutant encodes a kinase-dead form of the enzyme (Supporting Information Fig. S4b). Additionally, only the forms of PKM2 that could bind HuR (mM2, K367M, R399E) suppressed the increase in p27 and HuR levels mediated by knock-down of endogenous PKM2 (Fig. 4b, Supporting Information Fig. S4a). These forms of PKM2 were also uniquely able to restore the ability of PKM2 knock-down cells to enter mitosis and to retain a normal DNA content and centrosome number (Supporting Information Figs. S4c–S4e).
Figure 4.
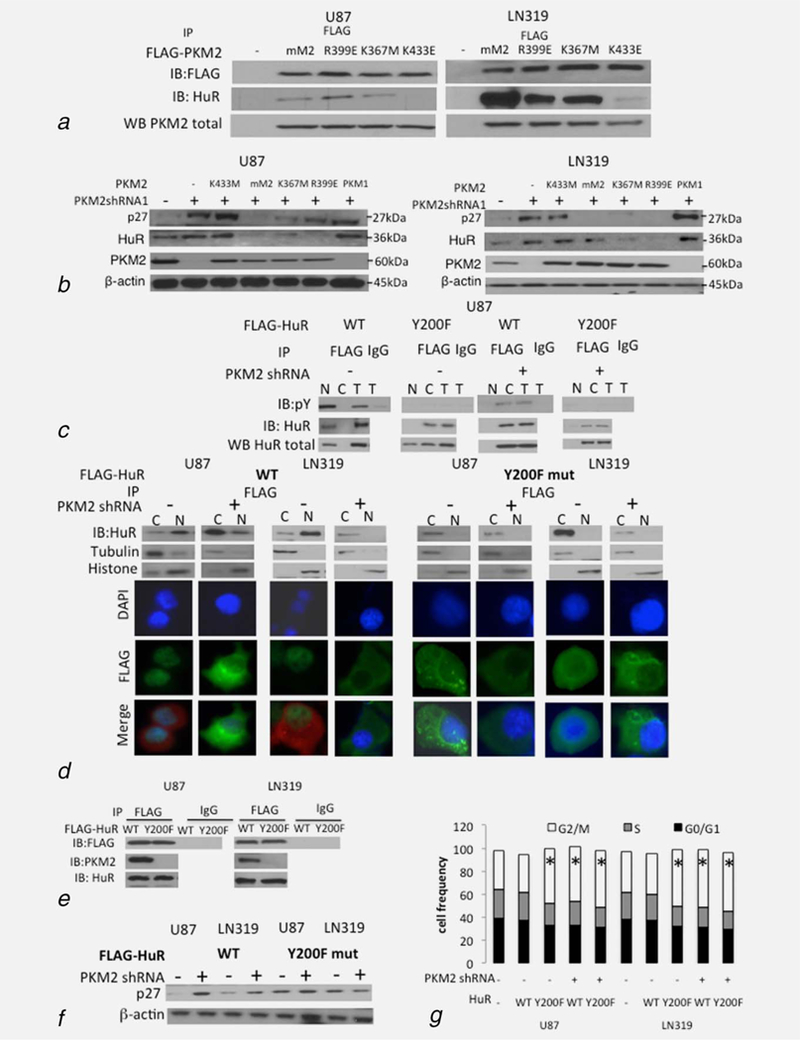
PKM2 interacts with HuR in a pY-dependent manner to control p27 levels and cell cycle progression. (a)Western blot analysis of levels of FLAG and HuR in FLAG immunoprecipitates from control or PKM2 shRNA-expressing U87 and LN319 cells expressing FLAG-tagged mouse WT (mM2) or R399E, K367M or K433E forms of PKM2. Western blot analysis of total PKM2 (bottom of panel) was used to verify equal input. (b) Western blot analysis of levels of p27, HuR, PKM2 and bactin in total cell lysates from control or PKM2 shRNA 1-expressing U87 (left) and LN319 (right) cells expressing WT (mM2) or R399E, K367M, or K433E forms of PKM2, or PKM1. See also Supporting Information Figures S4a–S4e. (c) Western blot analysis of pY and HuR levels in FLAG immunoprecipitates from U87 cells expressing WT Y200F FLAG-tagged HuR and a scramble or PKM2-targeted shRNA. (d)Western blot analysis of HuR in the FLAG immunoprecipitates from the nuclear (N) and cytoplasmic (C) fractions of control or PKM2 shRNA-containing U87 and LN319 cells expressing FLAG-tagged WT (left) or Y200F (right) HuR. DAPI (blue)/FLAG (green) co-immunofluorescence analysis was used to verify the Western blot assignment of sub-cellular fractionation. PKM2 staining (red) delineates the cytoplasm in groups with minimal cytoplasmic HuR. (e) Western blot analysis of FLAG, PKM2 and HuR in the FLAG or IgG immunoprecipitates from U87 and LN319 cells expressing FLAG-tagged WT or Y200F HuR. (f,g)Western blot analysis of p27 and b-actin (f) and cell cycle analysis (g) in control and PKM2 shRNA-expressing U87 and LN319 cells also expressing WT or HuR Y200F-encoding, FLAG-tagged constructs. *,p<.05,n = 3
Because phosphorylation of HuR Y200 has been suggested to be involved in the control of HuR sub-cellular localization, we also performed converse experiments in which control or PKM2-knock-down cells were infected with a construct encoding FLAG-tagged WT HuR or a mutant form of HuR (Y200F) limited in its ability to be tyrosine phosphorylated. As expected, FLAG-tagged WT HuR exhibited tyrosine phosphorylation, accumulated in the nuclear fractions of control cells and co-localized with nuclear DAPI staining (Figs. 4c and 4d), while FLAG-tagged WT HuR was similarly tyrosine phosphorylated but accumulated in the cytoplasmic fractions and cytoplasm of PKM2-knock-down cells (Figs. 4c and 4d). FLAG-tagged Y200F mutant HuR exhibited no detectable tyrosine phosphorylation in either control or PKM2 knock-down cells, perhaps consistent with the observation that tyrosine phosphorylation of HuR occurs in a co-ordinated manner.29 Of note, however, Y200F HuR accumulated not in the nuclear fractions but in the cytoplasmic fractions and nonDAPI-stained regions of both cell groups (Figs. 4c and 4d). Furthermore, PKM2 was found in FLAG-immunoprecipitates from cells expressing FLAG-tagged WT HuR, but was not present in those from cells expressing FLAG-tagged Y200F HuR (Fig. 4e). The introduction of the Y200F HuR into control cells also led to increased levels of p27 and accumulation of cells in G2/M comparable to that induced by PKM2 knock-down, and this effect was not enhanced by combining Y200F HuR expression with PKM2 suppression (Figs. 4f and 4g). These data show that PKM2 uses its pY-binding ability to retain pY-HuR in the nucleus. Accordingly, loss of PKM2 releases HuR for export to the cytoplasm where it increases translation of p27 and limits cell cycle progression.
Levels of nuclear PKM2 correlate inversely with cytoplasmic HuR and p27 expression in vitro and in human GBM samples
To more fully address the relationship between PKM2, HuR and p27 localization and expression, control and PKM2-suppressed U87 cells grown in vitro and as xenografts were examined by triple immunofluorescence. PKM2 was primarily nuclear in control U87 cells grown in culture, and this staining co-incided with nuclear HuR staining and a relative lack of p27 staining (Fig. 5a). In contrast, in PKM2 knock-down cells, HuR exhibited a cytoplasmic localization in association with p27 expression. The same was true in vivo, where U87 tumor cells grown as xenografts expressed nuclear PKM2 and HuR, but little p27, while tumors derived from U87 PKM2 knock-down cells exhibited cytoplasmic PKM2 and HuR expression accompanied by increased expression of p27. To address the relationship in primary human GBM samples, 15 GBM samples were separated into groups of low (0–10% positive), intermediate (10–70% positive) or high (>70% positive) nuclear PKM2 staining. Fifty cells in each of three human GBM in the low and high expressing groups were then examined by triple immune-fluorescence (Supporting Information Table S1). Roughly 75–100% of the cells in each group stained positively for PKM2, and total PKM2 immunofluorescence was comparable between the low and high groups. The nuclear PKM2 immunofluorescence in the low group however contributed <5% of the total PKM2 staining while in the high group it contributed > 20% of the total PKM2 (Fig. 5b). Consistent with this observation, the total intensity of HuR staining was higher in the group that retained more PKM2 in the nucleus, and over 95% of this staining was nuclear, versus a roughly equal distribution of HuR between the nucleus and cytoplasm in the cells that retained little nuclear PKM2. A significantly higher percentage of the cells with low levels of nuclear PKM2 and/or containing high levels of cytoplasmic HuR also were positive for p27 staining relative to cells in other groups (Fig. 5c). Furthermore, using unsupervized k-means clustering to define high/low cut-points, the 300 cells analyzed did not display a random uniform distribution across all eight possible combinations of high/low expression (Supporting Information Fig. S5), but rather separated cleanly into two groups (high nuclear PKM2/high cytoplasmic HuR/low p27 vs. low nuclear PKM2/high cytoplasmic HuR/low p27) (χ2 goodness of fit test, p < 2.2e-16) (Fig. 5c). These results indicate that the associations between PKM2, HuR and p27 noted in cultured GBM cells are also present in GBM cells in vivo and in primary GBM samples.
Figure 5.
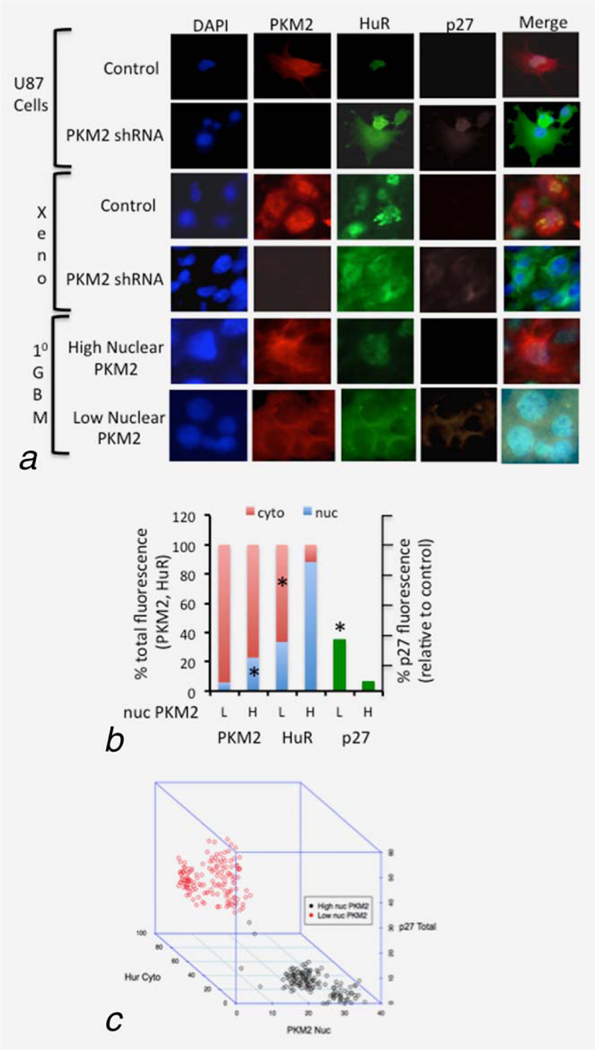
Levels of nuclear PKM2 correlate inversely with cytoplasmic HuR and p27 expression in vitro and in human GBM samples. (a) Representative results from immunofluorescence analysis of PKM2 (red), HuR (green) and p27(violet) nuclear/cytoplasmic expression in control and PKM2 shRNA U87 cells in vitro, the same cells grown as intracranial tumor xenografts (xeno), and of primary human GBM determined by PKM2 immunohistochemistry to have high or low expression of nuclear PKM2. (b) Average sub-cellular distribution of PKM2 and HuR immunofluorescence (left) and of total p27 immunofluorescence (right, normalized to infiltrated immune cells) in 50 cells from each of three primary human GBM determined by PKM2 immunohistochemistry to have low (L) or high (H) expression of nuclear PKM2. *,p<.05, n5150. (c) Three-dimensional plot of cytoplasmic HuR, nuclear PKM2 and total p27 immunofluorescence from 300 individual cells derived from three primary human GBM with low (black circles) or high (red circles) expression of nuclear PKM2, each. See also Supporting Information Figure S4a and Table S1.
The PKM2-HuR-p27 pathway controls glioma growth in vivo
To determine if the events noted in vitro are relevant in vivo, U87 cells modulated with respect to PKM2, p27 and/or HuR (Fig. 6a) were intracranially injected into mice, after which the effect on tumor growth was monitored by bioluminescence imaging. Control tumors grew rapidly over an initial 4 week period (Fig. 6b), necessitating the sacrifice of all animals by Day 40 postimplantation (Fig. 6c). Tumors generated from cells expressing PKM2 shRNA, or constructs encoding HuR or p27 exhibited a significant reduction in growth relative to controls (Fig. 6b), and animals bearing these tumors exhibited a significant survival advantage (Fig. 6c). Furthermore, the growth inhibition and survival advantage noted in PKM2 knock-down cells could be reversed by the introduction of shRNAs targeting either HuR or p27. These results show that the PKM2-HuR-p27 pathway modulates the growth of GBM cells both in vitro and in the xenograft setting.
Figure 6.
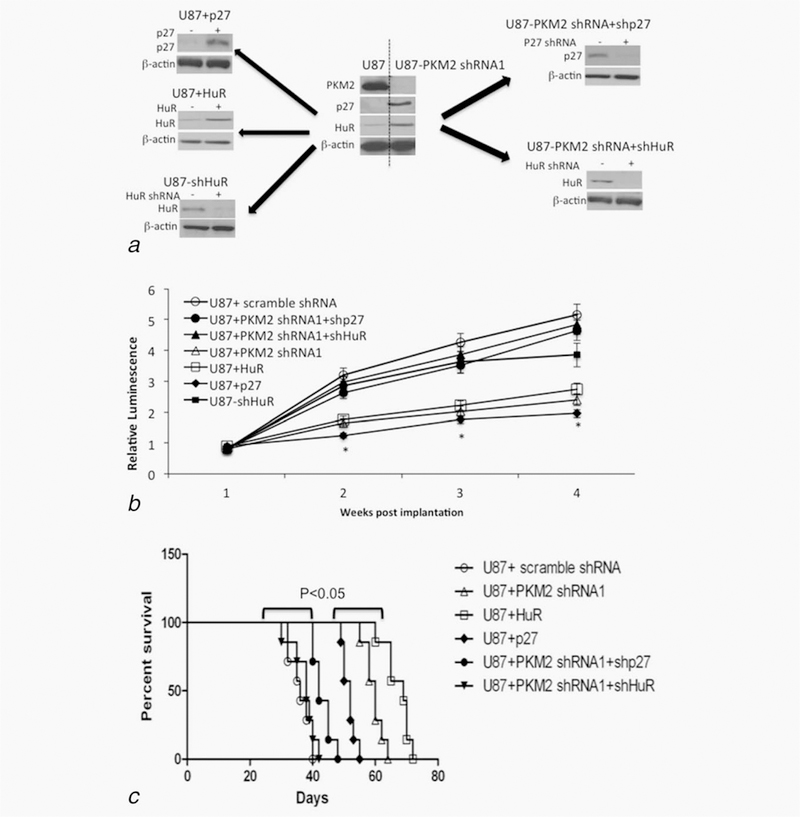
The PKM2-HuR-p27 pathway controls glioma growth in vivo. (a) Western blot validation of levels of PKM2, p27, HuR and β-actin in cells injected intracranially in panel b. (b) In vivo tumor growth curves of bioluminescently labeled control and PKM2, HuR and p27 modified U87 tumors formed following intracranial implantation of cells. *, p < .05, n = 7. (c) Kaplan-Meyer survival curves for animals (N = 7 for each group) intracranially implanted with control U87+ scramble shRNA, U87 + PKM2 shRNA, U87+PKM2 shRNA+shHuR, U87 + PKM2 shRNA+p27 shRNA, U87 + HuR or U87+p27 cells.
Discussion
PKM2 has been suggested to use both PK and protein kinase activities to regulate cell growth. PKM2, however, also binds pY-containing proteins independently of its kinase activity. Although binding of pY-containing peptides regulates PK activity, the potential for PKM2 to use phosphorylated tyrosine residue binding to directly control tumor cell cycle progression and growth has not been fully examined. In this paper we show that PKM2 uses its pY-binding ability to retain HuR in the nucleus, limit cytoplasmic cap-independent p27 mRNA translation, and promote glioma cell growth. These results define a new HuR- and p27-mediated pathway by which PKM2 controls RNA metabolism and regulates glioma cell growth.
Novel links between PKM2, HuR, p27 and cell cycle arrest
The present data provide a novel link between PKM2, HuR, p27 and cell cycle arrest. The cell cycle arrest noted following PKM2 knock-down in the present study occurred in the absence of increased levels of PKM1 and could not be reversed by exogenous expression of PKM1. These results therefore differ from those described in normal mouse fibroblasts in which genetic suppression of PKM2 led to increased PKM1 expression, PKMl-dependent nucleotide depletion and accumulation of cells in S phase.32 While the basis for these differences remain to be defined, the results suggest that tumor cells respond to changes in the PKM2/PKM1 ratio differently than normal nonimmortalized cells. The cell cycle arrest noted following PKM2 knock-down in the present study was also a direct consequence of increased levels of p27 and was independent of the ability of PKM2 to transcriptionally control levels of Stat3 and cyclin D.10,11 Given that the effects of PKM2 knock-down on cyclin D and cell cycle entry were uncovered in tumor cells following EGFR pathway activation,1,13–15 the effects noted here on HuR, p27 and mitotic entry may represent a more basal level of PKM2-mediated growth control. The identification of p27 as a PKM2 effector in this process is also consistent with the observation that p27, in addition to controlling the cyclin A/Cdk2 complex and G1 transition, can also indirectly reduce cyclin B/Cdk1 activity and cause G2/M arrest, especially in tumor cells with a compromized G1/S checkpoint.33 The increase in p27 levels in response to suppression of PKM2 was not associated with increased levels of p27 transcripts, but rather with HuR-mediated increase in p27 cap-independent translation. HuR is a master regulator of RNA metabolism whose sequence-specific RNA binding alters pre-mRNA processing,34,35 increases mRNA stability,36–38 and, through control of the translational repressor lincRNA-p21, increases mRNA translation.39 Although HuR stabilizes mRNAs encoding for pro-growth molecules,40 it also stabilizes and/or increases the translation of mRNAs encoding negative regulators of growth such as TSP-1 41 and other IRES-containing transcripts such as p27.42 Furthermore, HuR over-expression correlates with increased as well as decreased aggressiveness in breast cancer,43,44 and in at least one study impairs the growth of triple negative breast cancers.41 In this regard it appears that the targets and outcomes of HuR expression may be highly dependent on the cellular context in ways that at present are only partially understood.
In addition to linking PKM2 to the control of HuR, p27 and cell cycle arrest, the data presented also define a mechanism by which this is accomplished. HuR is a pY-containing protein,26–29 and its stability and function are known to be controlled by sub-cellular location.45,46 PKM2 in turn is a nuclear protein that binds pY-containing peptides. The data presented here show that in control cells, pY-containing HuR is predominantly a nuclear protein, and that forms of PKM2 that retain pY-binding ability co-immunoprecipitate with HuR as well as rescue cells from defects in cell cycle progression caused by PKM2 knock-down. The accumulation of HuR in the cytoplasm following suppression of PKM2 levels also supports the idea that PKM2 binds and traps HuR, and in particular pY-containing HuR (as well as possibly p27 itself)47 in the nucleus, limiting the ability of HuR to facilitate p27 mRNA translation in the cytoplasm. Under these conditions the trapped nuclear HuR may be able to function as a progrowth regulator of splicing and a stabilizer of nuclear pre-mRNAs involved in stimulating cell growth,38 including PKM2.48,49 Conversely, in cells with reduced levels of PKM2, HuR assumes a cytoplasmic location, much as it does in stressed cells.49 Under these circumstances HuR may regulate a different set of transcripts (including p27), that favor cell cycle arrest, suppress apoptosis and contribute to cell survival rather than cell proliferation.50 The exact identity of the forms of pY HuR critical for PKM2 binding and nuclear retention cannot be identified from the present studies. Although the inability of Y200F HuR to bind PKM2 and be retained in the nucleus suggests that phosphorylation of Y200 may be critical, other HuR tyrosine residues whose phosphorylation is altered by Y200 HuR mutation may also be involved. The abundance and significance of these multiple forms of tyrosine-phosphorylated HuR in GBM cells remains to be determined.
The association between nuclear PKM2 levels, cytoplasmic HuR levels and total p27 levels noted in cultured GBM cells are also present in GBM xenograft cells and more importantly, in primary GBM samples, suggesting that the PKM2-HuR-p27 axis functions not only in vitro but also in primary GBM. Furthermore, the heterogeneity of PKM2 expression and PKM2 sub-cellular localization noted in this and other studies suggests that the PKM2-HuR-p27 axis may play a role in balancing the pro-growth and pro-survival signaling that ultimately regulates tumor progression. The further unraveling of the interconnections between PKM2, HuR and RNA metabolism will likely be of interest in assessing this possibility, as well as in determining the role of this pathway in glioma development and therapy.
Supplementary Material
What’s new?
Pyruvate kinase M2 (PKM2) is overexpressed in many tumors, where it facilitates glycolysis and promotes tumor growth through interactions with phosphotyrosine (pY)-containing peptides. Its exact contributions to tumor cell growth, however, are not fully understood. Here, PKM2 is shown to leverage its pY-binding ability to interact in the nucleus with the RNA-binding protein HuR, thereby promoting glioma cell growth. Disruption of the interaction resulted in cytoplasmic redistribution of HuR, increased cap-independent p27 mRNA translation, and cell cycle arrest. The work defines a novel set of potential therapeutic targets along the PKM2-HuR-p27 pathway.
Acknowledgements
We thank Lewis Cantley and Zhimin Lu for PKM2 constructs, Myriam Gorospe for HuR constructs, and William Weiss and Joe Costello for helpful discussions in the preparation of the manuscript.
References
- 1.Valentini G, Chiarelli L, Fortin R, et al. The allosteric regulation of pyruvate kinase. J Biol Chem 2000;275:18145–52. [DOI] [PubMed] [Google Scholar]
- 2.Mazurek S, Boschek CB, Hugo F, et al. Pyruvate kinase type M2 and its role in tumor growth and spreading. Sem Can Biol 2005;15: 300–8. [DOI] [PubMed] [Google Scholar]
- 3.Bluemlein K, Gruning NM, Feichtinger RG, et al. No evidence for a shift in pyruvate kinase PKM1 to PKM2 expression during tumorigenesis. Oncotarget 2011;2: 393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guminska M, Ignacak J, Kedryna T, et al. Tumor-specific pyruvate kinase isoenzyme M2 involved in biochemical strategy of energy generation in neoplastic cells. Acta Biochem Pol 1997; 44:711–24. [PubMed] [Google Scholar]
- 5.Steinberg P, Klingelhoffer A, Schafer A, et al. Expression of pyruvate kinase M2 in preneoplastic hepatic foci of N-nitrosomorpholine-treated rats. Virchows Arch 1999;434:213–20. [DOI] [PubMed] [Google Scholar]
- 6.Anastasiou D, Yu Y, Israelsen WJ, et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol 2012; 8:839–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hitosugi T, Kang S, Vander Heiden MG, et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal 2009;2:ra73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christofk HR, Vander Heiden MG, Wu N, et al. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 2008;452:181–6. [DOI] [PubMed] [Google Scholar]
- 9.Gao X, Wang H, Yang JJ, et al. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell 2012;45:598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Israelsen WJ, Dayton TL, Davidson SM, et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 2013;155:397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang W, Xia Y, Hawke D, et al. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 2012;150: 685–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang Y, Li X, Yang W, et al. PKM2 regulates chromosome segregation and mitosis progression of tumor cells. Mol Cell 2014;53:75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keller KE, Doctor ZM, Dwyer ZW, et al. SAICAR induces protein kinase activity of PKM2 that is necessary for sustained proliferative signaling of cancer cells. Mol Cell 2014;53:700–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang HS, Nagane M, Klingbeil CK, et al. The enhanced tumorigenic activity of a mutant epi-dermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J Biol Chem 1997; 272:2927–35. [DOI] [PubMed] [Google Scholar]
- 15.Yang W, Xia Y, Ji H, et al. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature 2011;480:118–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mukherjee J, Phillips JJ, Zheng S, et al. Pyruvate kinase M2 expression, but not pyruvate kinase activity, is up-regulated in a grade-specific manner in human glioma. PloS One 2013;8:e57610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Christofk HR, Vander Heiden MG, Harris MH, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008;452:230–3. [DOI] [PubMed] [Google Scholar]
- 18.Lal A, Mazan-Mamczarz K, Kawai T, et al. Con-current versus individual binding of HuR and AUF1 to common labile target mRNAs. Embo J 2004;23:3092–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fromm-Dornieden C, von der Heyde S, Lytovchenko O, et al. Novel polysome messages and changes in translational activity appear after induction of adipogenesis in 3T3-L1 cells. BMC Mol Biol 2012;13:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakayama K, Nagahama H, Minamishima YA, et al. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev Cell 2004;6:661–72. [DOI] [PubMed] [Google Scholar]
- 21.Toyoshima H, Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell 1994;78:67–74. [DOI] [PubMed] [Google Scholar]
- 22.Millard SS, Vidal A, Markus M, et al. A U-rich element in the 5’ untranslated region is necessary for the translation of p27 mRNA. Mol Cell Biol 2000;20:5947–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beretta L, Gingras AC, Svitkin YV, et al. Rapa-mycin blocks the phosphorylation of 4E-BP1 and inhibits cap-dependent initiation of translation. Embo J 1996;15:658–64. [PMC free article] [PubMed] [Google Scholar]
- 24.Kullmann M, Gopfert U, Siewe B, et al. ELAV/ Hu proteins inhibit p27 translation via an IRES element in the p27 5’UTR. Genes Dev 2002;16: 3087–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Komiyama K, Okada K, Tomisaka S, et al. Anti-tumor activity of leptomycin B. J Antibiot 1985; 38:427–9. [DOI] [PubMed] [Google Scholar]
- 26.Kim HH, Abdelmohsen K, Lal A, et al. Nuclear HuR accumulation through phosphorylation by Cdk1. Genes Dev 2008;22:1804–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim HH, Yang X, Kuwano Y, et al. Modification at HuR(S242) alters HuR localization and proliferative influence. Cell Cycle 2008;7:3371–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim HH, Gorospe M. Phosphorylated HuR shut-tles in cycles. Cell Cycle 2008;7:3124–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoon JH, Abdelmohsen K, Srikantan S, et al. Tyrosine phosphorylation of HuR by JAK3 triggers dissociation and degradation of HuR target mRNAs. Nucleic Acids Res 2014;42:1196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hofmann S, Cherkasova V, Bankhead P, et al. Translation suppression promotes stress granule formation and cell survival in response to cold shock. Mol Biol Cell 2012;23:3786–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hosios AM, Fiske BP, Gui DY, et al. Lack of Evidence for PKM2 Protein Kinase Activity. Mol Cell Vol 2015;59:850–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lunt SY, Muralidhar V, Hosios AM, et al. Pyruvate kinase isoform expression alters nucleotide synthesis to impact cell proliferation. Mol Cell 2015;57:95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Enders GH. Gauchos and ochos: A Wee1-Cdk tango regulating mitotic entry. Cell Div 2010;5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caput D, Beutler B, Hartog K, et al. Identification of a common nucleotide sequence in the 3’-untranslated region of mRNA molecules specifying inflammatory mediators. Proc Natl Acad Sci USA 1986;83:1670–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lebedeva S, Jens M, Theil K, et al. Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol Cell 2011;43: 340–52. [DOI] [PubMed] [Google Scholar]
- 36.Abdelmohsen K, Pullmann R Jr., Lal A, et al. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol Cell 2007;25:543–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fan XC, Steitz JA. Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. Embo J 1998;17:3448–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gallouzi IE, Brennan CM, Stenberg MG, et al. HuR binding to cytoplasmic mRNA is perturbed by heat shock. Proc Natl Acad Sci USA 2000;97: 3073–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Legnini I, Morlando M, Mangiavacchi A, et al. A feedforward regulatory loop between HuR and the long noncoding RNA linc-MD1 controls early phases of myogenesis. Mol Cell 2014;53:506–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Filipova N, Yang X, Wang Y, et al. The RNA-binding protein HuR promotes glioma growth and treatment resistance. Mol Cancer Res 2011;9:648–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gubin MM, Calaluce R, Davis JW, et al. Overex-pression of the RNA binding protein HuR impairs tumor growth in triple negative breast cancer associated with deficient angiogenesis. Cell Cycle 2010;9:3337–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoon JH, Abdelmohsen K, Srikantan S, et al. LincRNA-p21 suppresses target mRNA translation. Mol Cell 2012;47:648–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heinonen M, Bono P, Narko K, et al. Cytoplasmic HuR expression is a prognostic factor in invasive ductal breast carcinoma. Cancer Res 2005;65:2157–2161. [DOI] [PubMed] [Google Scholar]
- 44.Heinonen M, Fagerholm R, Aaltonen K, et al. Prognostic role of HuR in hereditary breast cancer. Clin Cancer Res 2007;13:6959–6963. [DOI] [PubMed] [Google Scholar]
- 45.Doller A, Pfeilschifter J, Eberhardt W. Signalling pathways regulating nucleo-cytoplasmic shuttling of the mRNA-binding protein HuR. Cell Signal 2008;20:2165–73. [DOI] [PubMed] [Google Scholar]
- 46.Nabors LB, Gillespie GY, Harkins L, et al. HuR, a RNA stability factor, is expressed in malignant brain tumors and binds to adenine- and uridine-rich elements within the 3’ untranslated regions of cytokine and angiogenic factor mRNAs. Can Res 2001;61:2154–61. [PubMed] [Google Scholar]
- 47.Zhang J, Feng G, Bao G, et al. Nuclear transloca-tion of PKM2 modulates astrocyte proliferation via p27 and -catenin pathway after spinal cord injury. Cell Cycle 2015;14:2609–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mukherjee N, Corcoran DL, Nusbaum JD, et al. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol Cell 2011;43: 327–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang J, Guo Y, Chu H, et al. Multiple functions of the RNA-binding protein HuR in cancer progression, treatment responses and prognosis. Int J Mol Sci 2013;14:10015–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim HH, Abdelmohsen K, Gorospe M. Regual-tion of HuR by DNA Damge Response Kinases. J Nucl Acids 2010;Article ID 981487. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.