

Abstract
Transition state theory teaches that chemically stable mimics of enzymatic transition states will bind tightly to their cognate enzymes. Kinetic isotope effects combined with computational quantum chemistry provides enzymatic transition state information with sufficient fidelity to design transition state analogues. Examples are selected from various stages of drug development to demonstrate the application of transition state theory, inhibitor design, physicochemical characterization of transition state analogues, and their progress in drug development.
Graphical Abstract
1. INTRODUCTION
Linus Pauling proposed over 70 years ago that “…the only reasonable picture of the catalytic activity of enzymes is that which involves an active region of the surface of the enzyme which is closely complementary in structure not to the substrate molecule itself, in its normal configuration, but rather to the substrate molecule in a strained configuration, corresponding to the “activated complex” for the reaction catalyzed by the enzyme: the substrate molecule is attracted to the enzyme, and caused by the forces of attraction to assume the strained state which favors the chemical reaction…”. And “…an enzyme complementary to a strained substrate molecule would attract more strongly to itself a molecule resembling the strained substrate molecule than it would the substrate molecule.”1 “The attraction of the enzyme molecule for the activated complex would thus lead to a decrease in its energy, and hence to a decrease in the energy of activation of the reaction, and to an increase in the rate of the reaction.”2 It was this recognition of the activated complex that Richard Wolfenden used to explain the tight binding of oxalate to lactate dehydrogenase, unsaturated analogues of substrates to 3-keto-steroid isomerase and proline racemace, the potent inhibition of cytidine deaminase by tetrahydrouridine and triose phosphate isomerase inhibition by 2-phosphoglycolate.3 Wolfenden formalized the Pauling principles by considering a thermodynamic box comparing solution (uncatalyzed) formation of product to that from the enzyme and enzyme affinity to substrate relative to that of the transition state (Figure 1). By doing so, Wolfenden concluded that ‘The attractive force between the enzyme and transition state for nonenzymatic reaction (if it could be measured) should be characterized by a binding constant exceeding by the factor F (enzyme rate acceleration) the binding constant of the substrate in the Michaelis complex… An ideal inhibitor, which perfectly resembled in its binding properties the substrate part of ETX (the transition state complex), should thus be bound as much more tightly to the enzyme than the substrate (the “binding ratio”) as the rate of the enzymatic reaction exceeds that of it nonenzymatic counterpart (the “rate ratio”). This conclusion applies whatever the nature of the binding forces (covalent, noncovalent, or both) and regardless of whether the enzyme itself is much distorted during binding and catalysis.”3 This description suggested an upper limit to the strong interactions between enzymes and putative transition state analogues.4 A sustained quest for the factor F in experimentally accessible reactions has placed upper limits near 1020 with many common enzymatic reactions in the region from 1012 to 1016.5–9 As enzymatic Km values are typically in the 10−3−10−6 M range, perfect transition states are predicted to bind with dissociation constants of 10−15−10−22 M according to the Pauling–Wolfenden proposal. These considerations place an upper limit for the affinity of transition state analogues, which by virtue of their chemical stability necessarily differ from the properties of the unstable and ethereal transition states. In practice, few if any inhibitors have reached these affinities. Most commonly, compounds considered transition state analogues bind with dissociation constants in the nanomolar to femtomolar range (10−9 −10−15 M).10
Figure 1.
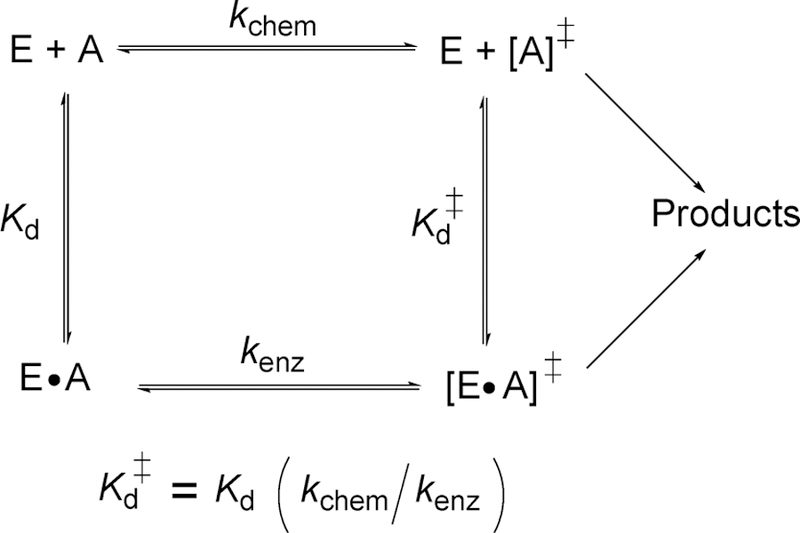
Thermodynamic box for describing the equilibrium binding constant of the transition state as described by Pauling and formalized by Wolfenden. E and A are enzyme and reactant, kchem and kenz are the rates of transition state formation without and with enzyme, and Kd and Kd‡ are dissociation constants for the Michaelis and transition state complexes, respectively.
2. PROPERTIES OF THE TRANSITION STATE
Understanding that mimics of the transition state will provide powerful inhibitors for the cognate enzymes, the search for transition state information was underway. Initially, identification of powerful inhibitors suggested transition state features. Thus, the tight binding of 2-phosphoglycolate to triose phosphate isomerase supported the formation of an ene–diol intermediate, and the tight binding of tetrahydrouridine to cytidine deaminase suggested a Meisenheimer intermediate for the proposed nucleophilic aromatic substitution reaction for cytidine deamination.11,12 Intermediates are not transition states but are expected to lie at higher energy levels than reactants, and mimics of intermediates would also be consistent with the suggestions of the tight-binding hypothesis.13 The transition states of chemical and enzymatic reactions correspond to bond loss and bond formation. Bond vibrational modes in biological molecules have time constants on the femtosecond time scale; thus, the transition state lifetimes, the loss of a restoring mode, are very much shorter than the catalytic turnover numbers of enzymes, typically 1–103 s−1. The brief transition state lifetimes are experimentally problematic in providing spectroscopic characterization, and it is necessary to use deductive measures of kinetic isotope effects and computational chemistry to provide well-defined geometric and electrostatic views of enzymatic transition states.14,15
2.1. Kinetic Isotope Effect (KIE) Approach to Enzymatic Transition States
The development of KIEs applied to chemical reactions and later extended to enzyme-catalyzed reactions provided the chemical detail needed for the essential features of the transition state, namely, geometry (bond angles and lengths) and electrostatic potentials. These descriptors are conveniently provided from the wave function of the transition state, derived from experimental KIEs, and treated as a fixed structure for the purpose of building a computational model of the transition state as a guide to design transition state analogues. This approach has been detailed in readily available methods and review articles.16–19 Here, the steps in this approach will be summarized and the methods for enzymatic synthesis of the isotopically labeled substrates will be outlined. A more complete guide to “how to do it” literature is provided in section 15.1.
The approach to understand the nature of enzymatic transition states begins with a primary data set of kinetic isotope effects, refines these to intrinsic values, and compares them to quantum-chemically defined potential transition states for experimental and theoretical agreement. Experimental determination of enzymatic transition states begins with (1) synthesis of the substrates with isotope labels at individual positions involved in the reaction coordinate and remote reporter groups. (2) Intrinsic isotope effects are required for valid analysis of the chemical step and can be established from kinetic and commitment measurements. (3) Gaussian (or similar) quantum mechanical (QM) approaches are used to iterate systematically through possible transition states to find a transition state with the nearest match of the intrinsic kinetic isotope effects. (4) Electrostatic potential surfaces are calculated from the wave functions of the reactant and transition state, treating the transition state as a stationary structure. (5) Stable chemical mimics of the transition state are designed based on a geometric and molecular electrostatic match of the transition state. (6) Chemical synthesis of transition state analogues is followed by kinetic analysis against the target enzyme. (7) Inhibitory analogues are evaluated for biological efficacy by testing in cellular and animal models.20
2.2. Synthesis of Isotopically Labeled Reactants
For the N-ribosyltransferases, a focus of this review, the experimental range of KIEs requires isotopic labels at the anomeric carbon of ribose (the reaction center) and the nitrogen of the leaving group, both of which give primary isotope effects to define the extent of leaving group bond breaking and the extent of bond-forming participation of the incipient nucleophiles (water for hydrolases and phosphate for phosphorylases). The α-secondary KIE from the hydrogen at the anomeric carbon provides information on the rehybridization of the reaction center at the transition state. The β-secondary KIE from the hydrogen at C2′ of ribose provides ribose pucker information, as this isotope effect is strongly affected by hyperconjugation to the breaking leaving group bond and the dihedral angle between C2′–H2′. These four isotope effects define the major properties of the transition state. Every additional measurement contributes additional details. For example, the α-O4′ isotope effect from the ribosyl ring provides a second estimate of the rehybridization of the anomeric carbon at the transition state. Leaving group effects, e.g., the N7 KIE in purine leaving groups, indicate the extent of leaving group protonation at the transition state. In competitive isotope label experiments, the atoms mentioned above are made heavy with 18O, 15N, 14C, 13C, 3H, or 2H and remote labels report on the discrimination between the chemically influenced KIE and the remote, chemically uninfluenced isotope label. Examples for the synthesis of isotopically labeled ATP, AMP, 5-phosphoribosyl 5-pyrophosphate, inosine, and adenosine are provided (Figure 2).
Figure 2.

Enzymatic synthesis of radiolabeled reactants PRPP from specifically labeled glucose or ribose. Product of a single-pot coupled synthesis yields ATP. Subsequent conversions to AMP, PRPP, inosine, and adenosine are shown. Isotopic labels in any of the reactants can be used to label the desired position in products. Other transferases can replace APRTase to generate other nucleotides as intermediates or products.21–23
2.3. Binding Isotope Effects and Remote Labels
Remote labels at isotopically KIE-”silent” positions are required for the competitive radioisotope method. Chemical studies that break the N-riboside bond of AMP to form adenine and ribose 5-phosphate provide an example. The [5′–14C]AMP molecule provides an isotope label remote from the site of chemistry. A mixture of [5′–14C]AMP and [1′–3H]AMP provides ribose 5-phosphate products with a ratio of 14C to 3H representing the 1′–3H KIE. Likewise, a mixture of [5′–3H]AMP and [1′–14C]AMP provides ribose 5-phosphate products with a ratio of 14C to 3H representing the 1′–14C KIE. Double remote labels can be used to measure isotope effects from chemically stable isotopes like 2H, 13C, 15N, and 18O. A mixture of [5′–14C; 9-15N]AMP and [5′–3H]AMP provides ribose 5-phosphate products with a ratio of 14C to 3H representing the [9-15N]AMP KIE. Remote labels require extra caution as remote binding isotope effects are common with 3H.24 On formation of the Michaelis complex, 3H–C bonds can be influenced by the catalytic site environment. For each degree of distortion from the solution sp3 geometry, an approximate 1% 3H binding isotope effect will be observed from 3H–C bonds.25 These values are normal or inverse (the 3H-labeled molecule binding weaker or stronger) if the 3H–C modes are less or more constrained in the bound complexes, respectively. For this reason it is always essential to measure the BIE and/or KIE of remote 3H–C labels. This is experimentally direct by either binding or kinetic experiments with mixtures of [5′–3H]AMP and [5′–14C]AMP. Remote labels with 14C are preferred, as there are no significant binding isotope effects for remote 14C labels, in contrast to those for 3H.26,27 We will see some examples of these isotope effects in the transition state analyses described herein. Binding isotope effects can be a significant contribution to experimentally measured KIEs. For example, in binding isotope effect studies for glucose to human brain hexokinase, binding isotope effects from 6.5% normal to −7.3% inverse were observed in individually 3H-labeled glucose molecules.28 Experimentally, these effects have been considered a boon by providing bond distortional information for understanding ground state stabilization or destabilization and a bane, by requiring binding isotope effect corrections when measuring KIEs for transition state analysis.29,30
3. AMP NUCLEOSIDASE
3.1. AMP Nucleosidase Kinetic Isotope Effects
AMP nucleosidase catalyzes the hydrolysis of AMP to adenine and ribose 5-phosphate with allosteric activation by MgATP2– and inhibition by inorganic phosphate.31,32 The enzyme is found only in bacteria and has been proposed to regulate the relative ratio of ATP to less-phosphorylated nucleotides by controlled hydrolysis of AMP to products that are easily recycled to the adenylate pool.33 As AMP nucleosidase is not found in mammals, it has been proposed as a potential antibacterial target.34 The crystal structure of the Escherichia coli enzyme is homohexameric with each of the six catalytic sites composed of contacts from two subunits (Figure 3).35 A test of its suitability as a target came from genetic inactivation of the enzyme in E. coli. Loss of the catalytic activity had the effect of increasing intracellular ATP concentrations, resulting in cellular cryoprotection.36 Thus, AMP nucleosidase is an unlikely drug target. Nevertheless, the enzyme played an important role in the early application of kinetic isotope effects to understand transition state structure. Kinetic isotope effect studies compared KIE profiles for AMP nucleosidases from different sources with the nonenzymatic chemical solvolysis of AMP.21,22 This catalytic activity was first characterized in the enzyme from Azotobacter vinelandii, a hexameric enzyme that has been well characterized kinetically.31,32,37 The crystal structure of the A. vinelandii enzyme has not been reported.
Figure 3.

Crystal structure of E. coli AMP nucleosidase hexamer with formycin 5′-phosphate at the catalytic sites (A), and detailed contacts between the enzyme and formycin 5′-phosphate at the catalytic sites (B). Catalytic site contacts are from the parental and adjacent (*) subunit contacts. From PDB structure 1T8S. Adapted with permission from ref 35. Copyright 2004 Elsevier.
3.2. Comparing Chemical and Enzymatic KIEs
The transition states of AMP nucleosidases (AMPNs) have been interrogated by KIE analysis using three distinct forms of the enzyme. The A. vinelandii enzyme requires MgATP2− as an essential kcat activatior with the unactivated enzyme being 200-fold less active than the activated form (Av – ATP and Av MgATP in Figure 4). A mutant form of the A. vinelandii enzyme was developed with a 50-fold decreased kcat but unchanged binding of the allosteric regulators, MgATP2− or inorganic phosphate (mAv – ATP and mAv MgATP in Figure 4).38 The E. coli enzyme differs from the A. vinelandii homologue by a mechanism of allosteric activation with MgATP2− causing decreased Km values by several orders of magnitude (Ec – ATP and Ec MgATP in Figure 4).39 This early application of isotope effects provided an experimental approach toward understanding the sensitivity of the transition state structure in response to remote regulators. It also probed changes in transition states in enzymes with mutationally altered kinetic values. A direct chemical comparison was possible for this reaction.
Figure 4.
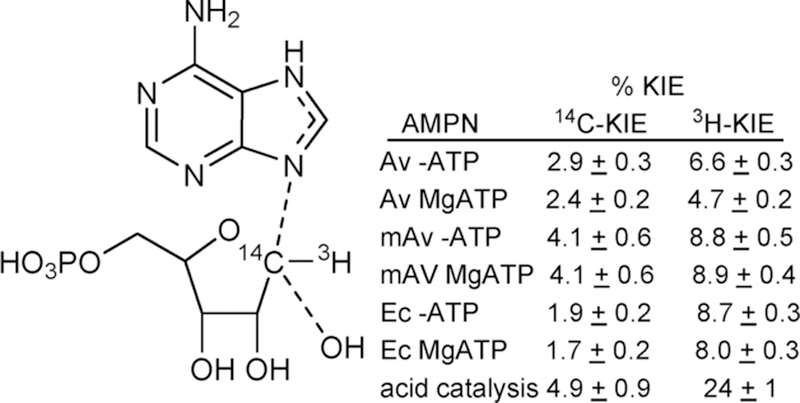
Kinetic isotope effects for [1′–14C]AMP primary effect and [1′–3H]AMP α-secondary effect with different enzyme conditions and compared to the acid-catalyzed solvolysis of AMP in 0.1 M HCl at 50 °C.21,22 % KIE = 100% (KIE – 1.000).
3.3. Qualitative Analysis of AMP Nucleosidase KIEs
Nucleophilic displacements at carbon generate 14C primary KIE values of up to 14% for transition states with symmetric (SN2) attack of the nucleophile and departure of the leaving group.40 Atomic crowding around the reaction center restrains the out-of-plane bending modes for the 1′–3H atom to be similar to the reactant state, giving KIE values near unity. Conversely, classic SN1 reactions give near unity 1′–14C primary KIE values but large 1′–3H values (above 30%) as the transition state ribocation has a fully developed sp2 geometry where increased out-of-plane modes for the C–3H bond contribute to large seconday 3H-KIE values.40,41 On the basis of similar KIE values for the enzymatic reactions with altered catalytic rates of over 3 orders of magnitude, intrinsic isotope effects were assumed. Small 14C and modest 3H KIE values were interpreted as transition states dominated by SN1 character, with significant participation of the attacking water (or hydroxyl ion) nucleophile with catalytic site crowding to prevent out-of-plane freedom for the 3H atom, resulting in small 3H KIE values on the enzyme but large ones in solution. Dissociated transition states with ribocation character generate an anionic leaving group that requires neutralization for efficient departure from the transition state. Leaving group neutralization was proposed by N7 protonation. Thus, an elevated pKa and protonation at N7 described part of the transition state for the AMP nucleosidases, suggesting formycin 5′-phosphate as an inhibitor (Figure 5).
Figure 5.

Formycin 5′-phosphate differs from AMP by a chemically stable C–C ribosidic bond and elevated pKa at N7 to mimic protonation of this group at the transition state.
3.4. Formycin 5′-Phosphate as a Transition State Analogue
Formycin 5′-phosphate was proposed as a transition state analogue for AMP nucleosidase based on kinetic, KIE, and substrate specificity studies.42 pKa profiles suggested an ionizable enzyme group (R1, pKa 6.2) to be the proton donor for N7 at the transition state.37 Formycin (7-amino-3-(β-D-ribofuranosyl)pyrazolo-(4,3-d)-pyrimidine) is a natural product isolated from culture media of Nocardia interforma.43 It is phosphorylated in mammalian cells and can be readily converted to the 5′-phosphate by chemical phosphorylation.44 Formycin 5′-phosphate gave a dissociation constant of 43 nM for the A. vinelandii AMP nucleosidase compared to a Km of 120 μM for the AMP substrate, a factor of 2800 tighter binding for the analogue (Figure 5).44 The crystal structure of the E. coli AMP nucleosidase with formycin 5′-phosphate at the catalytic sites revealed Asp428 to be responsible for protonation of N7 in a bidentate interaction between the N6 amino group and N7 (Figure 3). The reaction is completed by water addition to the ribocationic transition state; however, no catalytic water was observed in the crystal structure nor is there a nearby catalytic base to assist water ionization toward an attacking hydroxide ion.35 Formycin analogues have provided useful mechanistic tools but have not found use as antibiotics or antimetabolites.
4. ADENOSINE DEAMINASE
4.1. Function and Deficiency
Adenosine deaminases catalyze the hydrolytic conversion of adenosine (and other 6-substituted purine ribosides) and their 2′-deoxy-counterparts to the 6-oxypurine ribosides. Genetic deficiency of human adenosine deaminase causes severe combined immune deficiency leading to infections and death in the first years of life.45–47 Lack of adenosine deaminase (less than 1% of normal) causes accumulation of 2′-deoxyadenosine in the blood, enzymatic conversion 2′-deoxyATP in rapidly dividing cells of the immune system, errors in DNA replication, and active repair processes leading to p53 activation and apoptotic cell death.48 The preferred treatment is compatible bone marrow (stem cell) transplant, and a secondary treatment can be accomplished by enzyme replacement therapy with stabilized adenosine deaminase constructs to remove 2′-deoxyadenosine from the circulation.49 Transition state analogue inhibitors of human adenosine deaminase have been used as anticancer agents because of the high specificity of adenosine deaminase deficiency for rapidly dividing cells of the immune system. In addition, many 6-amino purine nucleoside antimetabolites used as anticancer agents are susceptible to inactivation by the action of adenosine deaminase, and their use in combination with 2′-deoxycoformycin prevents inactivation by deamination.
4.2. Mechanism and Structure
The catalytic mechanism of adenosine deaminase was first proposed to be a two-step C6 hydrolytic mechanism with the discovery that 6-amino, 6-Cl, and several other 6-substituents could be hydrolyzed from purine ribosides with a common rate-limiting step (Figure 6).50,51 The mechanism was confirmed by the NMR finding that an equilibrium existed between adenosine deaminase and [2-13C]-and [6-13C]purine ribosides where the hybridization of C6 changed from sp2 to sp3 on the enzyme as a result of a new bond to oxygen or sulfur.52 Thus, the enzyme is capable of reversibly adding to the C6 of the purine base nucleoside, also supported by the solvent exchange of H218O into the O6 of inosine.53 Earlier kinetic isotope effect studies demonstrated an unusual, large inverse solvent isotope effect for the deamination of adenosine, and this was interpreted as protonation of N1 by a Cys group to form a thiol anion at an early step in the mechanism, in coordination with addition of the hydroxyl group at C6, part of the sp2 to sp3 rehybridization.54 At the time the involvement of Zn2+ at the catalytic site was not known, making the thiol active site contact a logical hypothesis. The first crystal structure of an adenosine deaminase (mouse) in complex with 6-hydroxyl-1,6-dihydropurine ribonucleoside, a transition state analogue, revealed that the catalytic site contained a tightly bound zinc cofactor also chelated to the 6R-hydroxyl group isomer of the transition state analogue (Figure 7).55 The structure explained the inverse solvent isotope effect reported earlier, as metal-based ionization of water prior to nucleophilic addition also generates an inverse solvent isotope effect.56 No Cys groups are near the catalytic site, including the chelation sphere of the Zn2+. Three His nitrogens, a carboxylate oxygen, and the 6-hydroxyl group of the bound intermediate complete the coordination complex.
Figure 6.

Substrate 2′-deoxyadenosine (2′-dAdo) enzyme-stabilized intermediate at the catalytic site and the natural product transition state analogue inhibitor, deoxycoformycin. The Zn2+-activated water nucleophile is shown. In clinical use, the analogue is known as pentostatin.
Figure 7.
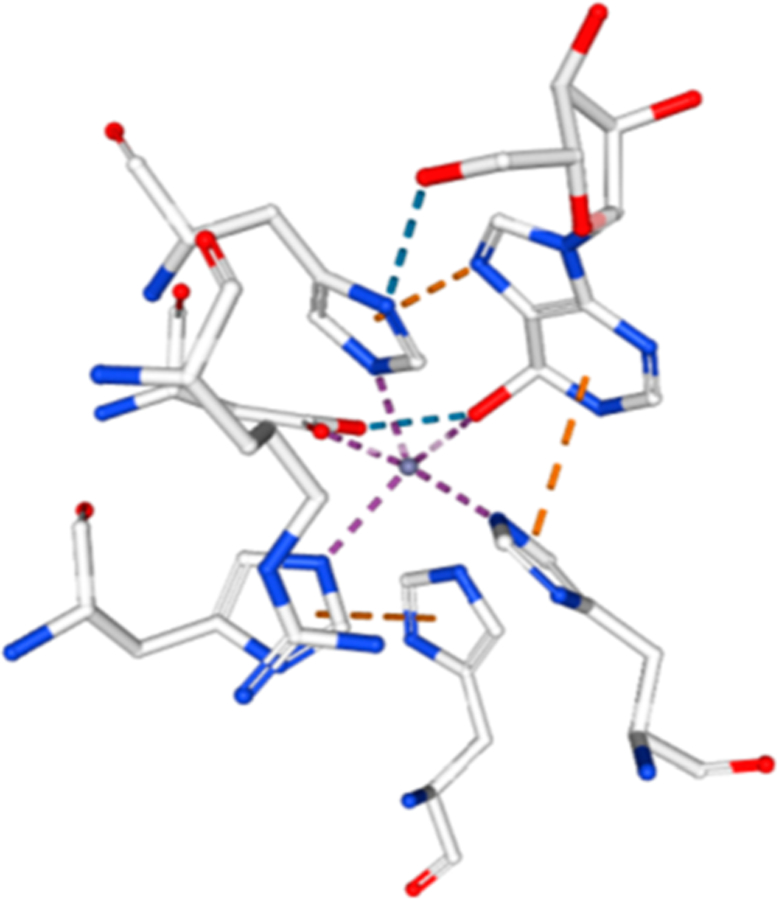
Zn2+ cation at the catalytic site of bovine adenosine deaminase from PDB 1FKX, D296A mutant. Zn2+ ion is the central sphere in the figure and is in contact with the protein ligands and the 6-hydroxyl group of 6-hydroxydihydroadenosine.
4.3. Coformycins
Two natural product transition state analogues for adenosine deaminase, coformycin (1) and 2′-deoxycoformycin (2), have been isolated from culture media of Streptomyces antibioticus, N. interforma, and Streptomyces kaniharaensis SF-557 (Figure 8).57–59 The same cultures that produce formycin also produce coformycin, hence the name. Their structural determination, characterization as inhibitors of adenosine deaminase, and chemical synthesis were completed within a few years of the initial discovery. Dissociation constants for human adenosine deaminase with 2′-deoxycoformycin (pentostatin) and coformycin have been reported to be 2.5 and 10 pM, respectively, matching the 4-fold difference in affinities for adenosine and 2′-deoxyadenosine.60–62 On the basis of the original observations that adenosine deaminase deficiency causes severe combined immunodeficiency disease (both B and T cells), pentostatin was entered into clinical trials for a variety of lymphoid malignancies.63–66 Strong responses have been observed in chronic lymphocytic leukemia (CLL), but even more impressive responses are observed with the relatively rare hairy cell leukemia (HCL). Treatment with pentostatin causes adenosine and 2′-deoxyadenosine to accumulate in the blood with specific accumulation of dAMP and dATP in lymphocytes, where high expression of deoxycytidine kinase mistakenly converts 2′-deoxyadenosine to dAMP which accumulates as dATP because of low expression of 5′-nucleotidase activity in these rapidly dividing cells.67 The expression of enzymes causing dATP accumulation provides the cancer cell specificity.68
Figure 8.

Dissociation constants (Kd) for inhibiton of ADAs from bovine (Bt), human (Hs), and Plasmodium falciparum (Pf) sources. Reproduced from ref 434. Copyright 2007 American Chemical Society.
The picomolar binding affinity of the coformycins with adenosine deaminase permits atomic dissection of the energy of binding by inhibitor analogues. Changing the stereochemistry of the 8-R hydroxyl center to the 8-S configuration reduces the binding affinity by 9.8 kcal/mol.69 Deletion of the HN CH=N group from the 7-membered ring decreases binding by 7 kcal/ mol, and deletion of the 2′-deoxyribose group decreases binding by 8.5 kcal/mol.70 Wolfenden has interpreted these large energy changes as “Cooperativity is so great that every substituent must be in exactly the right position, or the consequent losses in binding affinity may be catastrophic”. In other words, binding of the transition state analogue must reflect faithfully all of the interactions involved in the actual transition state. PfADA, but not human ADA, accepts 5′-substituents, providing species specificity for these inhibitors (3–6; Figure 8).
Coformycin and 2′-deoxycoformycin are equipotent for bovine, human, and P. falciparum adenosine deaminases. Both compounds are slow-onset, tight-binding inhibitors with dissociation constants of 60–110 pM (Figure 8). The similar inhibitory properties suggest similar transition states for these enzymes, verified by the isotope effect studies detailed below.
4.4. Adenosine Deaminase Transition State Structures
Transition state analysis for the adenosine deaminases compared enzymes from human, bovine, and P. falciparum sources. Kinetic isotope effects were measured for the reaction center at C6, N1, and the exocyclic N6 amino group. All of the enzymes catalyze nucleophilic aromatic substitutions (SNAr transition states) where the essential N1 protonation is partial and the bond order to the attacking Zn2+-activated hydroxyl group is nearly complete. A defining difference for these transition states is the extent to which N1 protonation is complete.71 Transition states for P. falciparum, human, and bovine enzymes have N1–H bond lengths of 1.915, 1.550, and 1.275, respectively, at their transition states. Molecular electrostatic potential maps indicate that all of these transition states are similar in geometry and charge to the coformycins (Figure 9).
Figure 9.
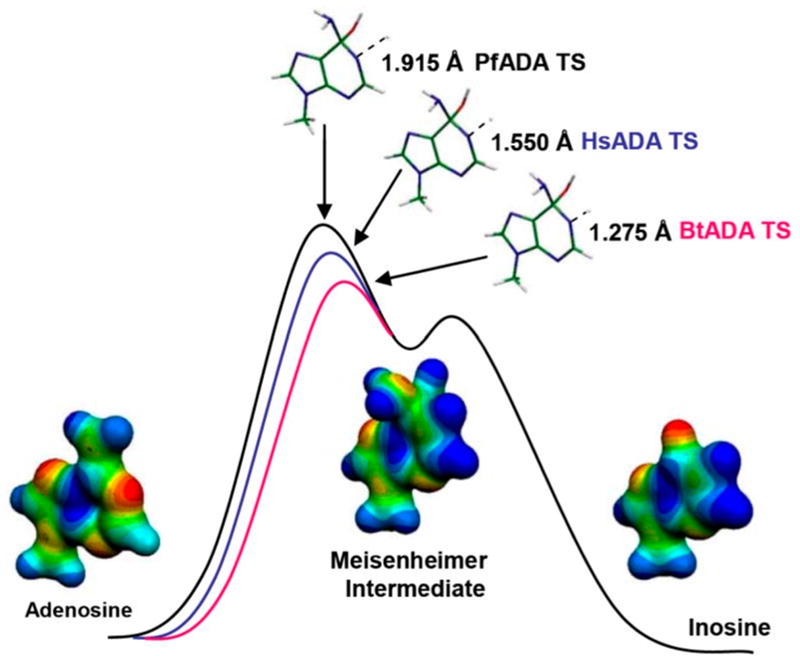
Reaction coordinate and transition state structures for the P. falciparum (PfADA), human (HsADA), and bovine (BrADA) transition states with molecular electrostatic potential maps of reactant, transition state, and product. Reproduced from ref 71. Copyright 2007 American Chemical Society.
5. NUCLEOSIDE HYDROLASES
5.1. Biological Function
The nucleoside hydrolases are a broadly distributed family of purine and pyrimidine N-ribosyl hydrolases found in single-cell organisms and plants but not in mammals.72–74 They form free nucleobases and ribose as products. The nucleosidases are found in protozoan parasites that cause human disease including Trypanosoma, Leishmania, and Giardia. They are thought to play a metabolic role by converting host nucleosides into nucleobases for salvage into the metabolic pools of the parasite.75–77 Protozoan parasites are purine auxotrophs, and purine salvage is essential to growth. Nucleoside hydrolases have been identified for all purine ribosides and have been proposed as antiparasitic targets. Targeting purine salvage is complicated by the presence of other purine salvage enzymes that may bypass nucleoside hydrolases.
The nucleoside hydrolases also act as antigens. Circulating antibodies to a Leishmania nucleoside hydrolase (called NH36) is a biomarker for infection. Immunization using this enzyme or its C-terminal fragment as antigens is protective in animal infections.78,79 Transition state analogue inhibitors of the nucleoside hydrolases also show some efficacy against visceral leishmaniasis (L. chagasi) in animal models but with limited knowledge of the mechanism of action.80 Mechanistic information and transition state analogue design for the nucleoside hydrolases has used the enzyme from a nonparasitic protozoan, Crithidia fasciculata, a biological model for Trypanosoma brucei (African trypanosomiasis) and Leishmania species (Leishmaniasis).81–83 The normal host for C. fasciculate is plant-feeding mosquitoes with transmission thought to be through plant-to-insect contacts.84 The nucleoside hydrolase from C. fasciculate (CfNH or IU-nucleoside hydrolase) hydrolyzes all naturally occurring purine and pyrimidine ribonucleosides with similar catalytic efficiencies. Others, for example the protozoan GI-nucleoside hydrolase, strongly prefer 6-oxypurine leaving groups, and the IAG-nucleoside hydrolase from T. brucei prefers these purine leaving groups. In contrast, AMP nucleosidase is specific for adenine, and purine nucleoside phosphorylase is specific for 6-oxypurine leaving groups.85
5.2. Transition State Analogue Design Features
Inhibitor design for the nucleoside hydrolases was initiated from the family of kinetic isotope effects obtained with specific labeled inosine molecules (Figure 10).86 Nucleoside hydrolases permitted extensive measurement of isotope effects and revealed some surprising features of the transition state. The N-ribosidic bond has a Pauling bond order of 0.2 at the transition state as indicated by the 15N9 and 14C1′ KIE values. The water nucleophile has very weak participation at 3 Å and defines an early transition state on an SN1 reaction coordinate. The relatively large α-KIE for 1′–3H confirmed the rehybridized C1′ toward sp2, and an increased out-of-plane mode gives rise to the large isotope effect. Surprisingly, the β-secondary 2′–3H KIE is as large as the α-effect, indicating dihedral orbital overlap (hyperconjugation) between the C2′–3H2 bond and the nearly vacant orbitals of the C1′–N9 bond.41 The remote 5′–3H KIE is a surprising 5.1%, a binding isotope effect associated with the formation of the transition state. The effects are directional such that the collective modes of the C5–3H bonds exhibit more freedom of motion when bound than when in solution. This distortion also affects the nearby C4′–3H bond to give a small inverse isotope effect of −0.8%, indicating a more constrained bond vibrational environment for this atom. A catalytic site hydrogen bond to the 5′-hydroxyl group was proposed to be responsible for the binding isotope effect, a prediction later confirmed by crystallography (see below).
Figure 10.
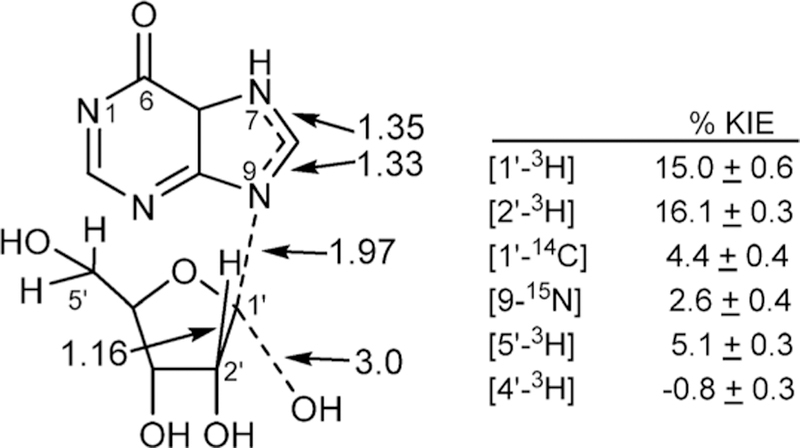
Kinetic isotope effects and bond lengths(Angstroms)at the transition state of CfNH. The N-ribosidic bond is nearly cleaved, with weak nucleophilic participation to form a partially developed ribocation.
Nucleoside analogues with substituted purine or ribosyl rings are poor inhibitors of CfNH (Ki values of 2–44 mM compared to a Km of 0.38 mM).87 Transition state analogues were designed from the geometry and electrostatic potential maps of the transition state (Figure 11).88 As individual features of the transition state were designed into analogues of the potential inhibitors, Ki values decreased from 380 μM for the substrate inosine by 4-fold for sp2 geometry in ribolactone, 38-fold for the ribocation effect, 120-fold for the ribocation and mimicry of the imidazole, 10 000-fold for the ribocation and a hydrophobic group as a purine analogue, 50 000-fold for the chemically stable immucillin-H incorporating the protonated deazapurine leaving group and the ribocation, and 150 000-fold for the combined sp2 reaction center, ribocation, hydrophobic group, and proton acceptor in the p-nitro group of p-nitrophenyl riboamidrazone. An isotope-edited resonance Raman study of CfNH in complex with the p-nitrophenyl riboamidrazone revealed enzyme stabilization of one resonance state. The p-nitrophenyl group is in the quinonoid form, and the exoribosyl nitrogen bonded to the C1′ of the ribosyl group is protonated, while that bound to the phenyl group is not (Figure 11).89 The zwitterionic tautomer has a distributed cationic charge centered at C1′. The molecular electrostatic potential of p-nitrophenyl riboamidrazone bound to the catalytic site closely resembles that of the transition state.89
Figure 11.

Chemical features of the transition state for CfNH incorporated into candidates as transition state analogues.88
5.3. Nucleosidase Mechanistic Probe Substrate
The transition state of CfNH demonstrated that N7 protonation of the purine leaving group occurs prior to transition state formation.86 The pKa of this group increases as the C1′–N9 ribosidic bond is lost, and transition state analysis does not indicate if N7 protonation is an early or late event with respect to formation of the transition state or if protonation is from a catalytic site proton donor. Substrate specificity studies were designed to test the effects of leaving group protonation on CfNH compared to five additional purine/pyrimidine N-ribosyltransferases (Table 1). Synthesis of p-nitrophenyl β-D-riboside and p-nitrophenyl β-D-riboside 5′-phosphate provided the test substrates.91 They permit analysis of leaving group activation at the enzyme catalytic sites. If the test enzyme achieves the transition state through protonation of the leaving group, p-nitrophenyl β-D-riboside is predicted to be a poor substrate, as the leaving group cannot be additionally activated by protonation. If the test enzyme achieves the transition state through formation of the ribocation or by enforced participation of the nucleophile, p-nitrophenyl β-D-riboside will be an excellent substrate, as the p-nitrophenyl leaving group is highly activated. N-Ribosyltransferases use three mechanisms to achieve the transition state: leaving group activation (e.g., protonation of the N7 of purines), formation of the ribocation transition state, and activation of the attacking nucleophile (water in CfNH and phosphate in phosphorolysis reactions). In this analysis the broad-specificity CfNH and LmNH the only excellent catalysts for p-nitrophenyl β-D-riboside, indicating its transition state formation occurs by ribocation activation. With inosine as substrate for CfNH, one proton donor and one proton acceptor (pKa values 9.1 and 7.1) are required for catalysis. Hydrolysis of p-nitrophenyl β-D-riboside shows no required proton donors or acceptors in the pH profiles, appropriate with the activated leaving group. The transition state structure showed weak participation of the water nucleophile, establishing ribocation formation as the primary force for transition state formation in CfNH and LmNH but not for the other enzymes.
Table 1.
Chemical Test of Leaving Group Activation for N-Ribosyltransferases*
| Enzyme | [kcat p-NO2-φ-riboside]/[kcat substrate] | |
|---|---|---|
| CfNH (IU-nucleoside hydrolase)a | 8.5 |  |
| LmNH (IU-nucleoside hydrolase)b | 1.8 | |
| IAG-nucleoside hydrolasec | 0.024 | |
| GI-nucleoside hydrolased | 0.0003 | |
| Purine nucleoside phosphorylasee | 0.000017 | |
| AMP nucleosidasef | 0.000015 | |
| Reactions compared p-nitrophenyl β-D-riboside as substrate to a,b,cinosine, dguanosine, and einosine. fp-Nitrophenyl β-D-riboside 5′-phosphate and AMP were compared as substrates for AMP nucleosidase. bLmNH is the enzyme from Leishmania major. Results from reference 89, 90. | ||
5.4. Structural Analysis with a Transition State Analogue
The catalytic sites of NHs have been characterized crystallo-graphically to determine the mechanism of water activation and leaving group proton donor and to explore enzymatic contacts that could be involved in stabilization of the ribocationic transition state.92–95 p-Amino-phenyl-iminoribitol (pAPIR) was used as the catalytic site ligand for CfNH. The transition state analogue is held in proximity to the catalytic water nucleophile. This water is in contact with a tightly bound catalytic site Ca2+ ion (Figure 12). The Ca2+ ion is coordinated to the 2′-and 3′-vicinal hydroxyl groups of the iminoribitol, oxygen atoms from Thr126, Asp242, and Asp10, and both side chain oxygens of Asp15 to complete an octahedral coordination with all contacts at 2.6 Å or less. The Ca2+ copurifies with the enzyme and is sufficiently tightly bound that catalytic assays in the presence of 10 mM EGTA are not inhibitory, although the Ca2+ can be removed by more extensive treatments and can be replaced.96 The water molecule is in favorable geometry for catalysis, 3.2 Å from the carbon analogous to the C1′ anaomeric carbon of normal substrates (Figure 12). Protonation of the leaving group was not an essential feature of the catalytic site, but transition state analysis indicated N7 protonation does occur at the transition state, implying protonation from solvent. The crystal structure shows no candidate ionizable groups near the leaving group pocket, consistent with the leaving group analysis. The KIE analysis of CfNH indicated binding distortion at C5′, an interaction that may be involved in formation of the ribocation. A 2.5 Å strong hydrogen bond between Glu166 and the 5′-hydroxyl group places the 5′-oxygen 2.7 Å from the position corresponding to the O4′-ring oxygen of ribose. Forcing two electron-rich oxygens close together destabilizes the ribosyl group toward the ribocation. This interaction requires anchoring of the ribosyl group, clearly provided by the Ca2+ interaction with both ribosyl vicinal hydroxyl groups together with hydrogen bonds from Asp14, Asp242, and Thr126 (Figures 12 and 13).
Figure 12.

Stereoviews of a transition state analogue bound to the catalytic site of CfNH. Upper panel shows the contacts to the catalytic site Ca2+, and lower panel indicates the contacts to the pAPIR transition state analogue. Reproduced from ref 96. Copyright 1996 American Chemical Society.
Figure 13.

Distance map for the catalytic site of CfNH with contacts to the catalytic site and pAPIR as a catalytic site ligand. His82 is 3.6 Å from the leaving group and has been considered a potential leaving group proton donor. Note the 2.7 Å neighboring group interaction between O5′ and O4′. Reproduced from ref 96. Copyright 1996 American Chemical Society.
Transition state analysis for CfNH is summarized by formation of the ribocation by neighboring group interaction between the O5′ and the O4′ oxygens of a tightly tethered ribosyl, no significant leaving group assistance, and ionization of the attacking water nucleophile by a 2.4 Å contact with the Ca2+ ion. The nearby Asp10 acts as a proton acceptor from the water. The reaction is formally a nucleophilic displacement, but the nucleophilic attack lags behind N-ribosidic bond breaking, as the transition state analysis has the water nucleophile at 3 Å in agreement with its crystallographic position (Figures 12 and 13).
5.5. Transition State Specificity for Isozymes
CfNH and LmNH are outliers in the leaving group activation scale (Table 1). In the IAG-nucleoside hydrolase from Trypanosoma vivax (TvNH), leaving group protonation is implicated.97 The catalytic site Ca2+ binding is similar to CfNH, but instead of the weakly interacting groups at the purine/ pyrimidine leaving group site, TvNH has the purine leaving group in a hydrophobic stack with a pair of tryptophan residues, Trp83 and Trp260 (Figures 13 and 14). This interaction is proposed to favor electron donation into the leaving group, raising the pKa to permit protonation by solvent. On the basis of the relative reactivity of p-nitrophenyl β-D-riboside with the nucleoside hydrolases, it is clear that the Trp pair plays a more important role in leaving group activation for TvNH than in CfNH.
Figure 14.
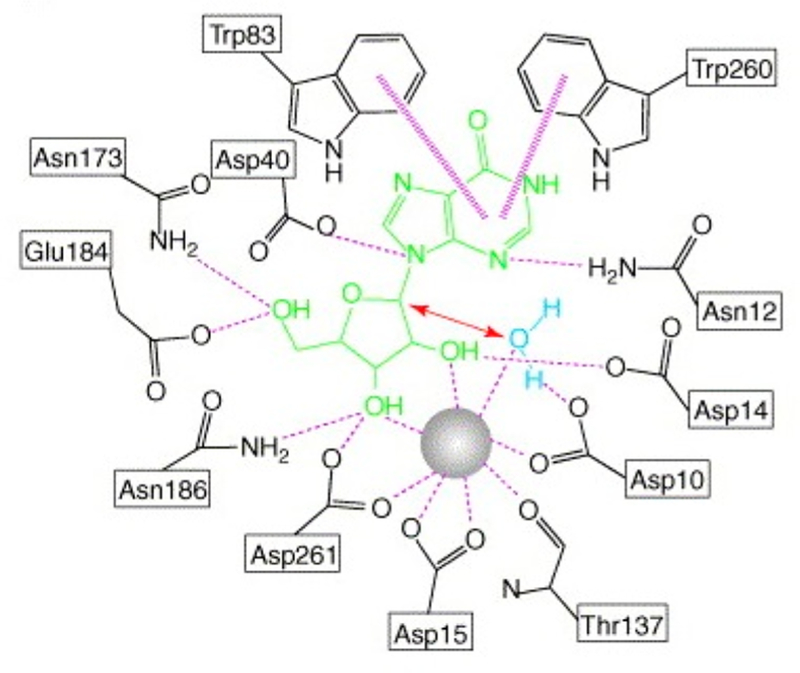
Interaction map for the catalytic site of TvNH with contacts to the catalytic site Ca2+ and inosine as a catalytic site ligand. Asp10Ala mutant prevented hydrolysis of the inosine. Trp83 and Trp260 are stacked with the leaving group. Reproduced with permission from ref 94. Copyright 2006 Elsevier.
Leaving group interactions are also reflected in transition state analogue specificity. Transition state analogues with a hydrogen-bond pattern of the appropriate purine leaving groups are good inhibitiors of CfNH and TvNH.98 Both CfNH (IU-NH in Figure 15) and IAG-NH bind tightly to ImmH, as both enzymes accept inosine as substrates (hypoxanthine leaving group), to give Kd values of 42 and 24 nM. Loss of the 2′-OH is more detrimental to CfNH (IU-NH) than to IAG-NH, where more of the transition state interaction comes from the leaving group activation. Thus, 2′-deoxy-ImmH binding decreases affinity by >1000-fold for IU-NH but only 27-fold for IAG-NH. In contrast, the leaving group interactions for IAG-NH make binding of the 4-amino-1-naphthalenyl-iminoribitol >2100-fold weaker for IAG-NH but only 10-fold weaker for IU-NH.
Figure 15.
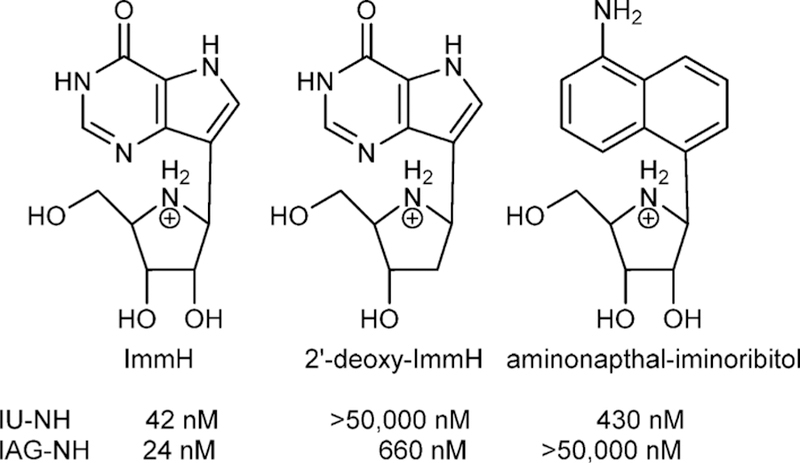
Analogues of the transition states for IU-NH (CfNH) and IAG-NH (TbbNH) demonstrating ribocation and leaving group interaction differences.97,98
5.6. Nucleoside Hydrolases as Drug Targets
Studies using a family of transition state analogues against the bloodstream form of T. brucei brucei indicated that nucleoside hydrolases are not essential.99 RNA interference to inhibit expression of IAG-NH, IG-NH, and methylthioadenosine phosphorylase did not induce purine starvation. Nucleoside hydrolases have provided a thorough understanding of reaction mechanism, transition state structure, reaction coordinate motion, and design of transition state analogues but are not suitable drug targets in the Trypanosoma. Parallel studies with a broader range of protozoan parasites have yet to be completed, but the essential nature of purine salvage appears to have forced the evolution of multiple salvage pathways, making the targeting of a single enzyme a difficult drug design strategy. Each organism has distinct pathways, and in some important parasites, including P. falciparum, there are metabolic steps related to purine salvage that can be used as suitable drug targets (see below).100–102
6. BACTERIAL ADP-RIBOSYLATING TOXINS AND THEIR TRANSITION STATES
6.1. Introduction
Bacterial toxins are responsible for disease symptoms when invading bacteria are induced to produce the damaging toxins.103–105 Several bacterial toxins work by a common chemical mechanism, the ADP-ribosylation of regulatory G-proteins using NAD+ as an ADP-ribosyl donor. Covalent modification of G-proteins causes loss of function, leading to damage and/or death of affected cells. Cholera, diphtheria, and pertussis toxins are produced by Vibrio cholerae, Bordetella pertussis, and Corynebacterium diphtheria to cause intestinal cholera, whooping cough, and diphtheria.106–108 The bacterial exotoxins disrupt G-protein signaling to cause cell death. The common toxic mechanism for these agents is mono-ADP-ribosylation of specific amino acids in Gsα, eEF-2, and Giα proteins, respectively, by the catalytic A chains of the toxins once they have entered the target cells.109 In the absence of acceptor proteins, these toxins also act as NAD+ N-ribosyl hydrolases. The same NAD+ hydrolytic reaction can be accomplished by nonenzymatic chemical solvolysis providing the opportunity to compare transition state structures of solution chemistry with those formed by enzymes in hydrolase or transferase reactions, Figure 16.110–112
Figure 16.

ADP ribosylation of eukaryotic elongation factor 2 (eEF2), G-stimulatory protein α, and G-inhibitory protein α by diphtheria, pertussis, and cholera toxins, respectively. Attacking nucleophile atoms are designated by the electrons. Note the inversion of configuration at C1 of the ribosyl group.
6.2. Synthesis of Labeled NAD+ Molecules
All ADP-ribosylation reactions involve the transfer of the ADP-ribosyl group from NAD+ to yield the ADP-ribosylated receptor protein and nicotinamide. Understanding the transition state structures of the ADP-ribosyltransferases requires kinetic isotope effect analysis from the NAD+ reactant. Isotopic labels are needed in the same positions as indicated for other N-ribosyltranserases. Synthesis of isotopically labeled NAD+ molecules is a parallel approach to the synthesis of labeled ATP and its derivatives (Figure 2). Synthesis of NAD+ proved more challenging because of its relative instability. With care, yields of NAD+ with the desired labels can exceed 90% from the limiting starting isotopic label (Figure 17A).113
Figure 17.

(A) Synthesis of isotopically labeled [1′–14C]NAD+ by coupled enzymatic reactions. Enzymes are (1) hexokinase, (2) glucose 6-phosphate dehydrogenase, (3) 6-phosphogluconate dehydrogenase, (4) 5-phosphoriboisomerase, (5) 5-phosphoribosyl 1-pyrophosphate synthatase, (6) nicotinate phosphoribosyltransferase, (7) NAD+ pyrophosphorylase, (8) pyruvate kinase, (9) glutamate dehydrogenase, and (10) adenylate kinase. Single-pot incubation (steps 1–10) converts glucose to NaAd+. Reaction is stopped at step 11; NaAD+ is purified and converted to NAD+ by NAD+ synthetase (12).113 Different labels in the starting glucose or the nicotinic acid added in step 6 provide NAD+ with any desired label in the NMN+ portion of the molecule. From the method of Figure 2, label can be placed at any position in ATP and incorporated into the AMP portion of NAD+ by incorporation at step (B) Reaction coordinate distances for ADP-ribosylating cholera, pertussis, and diphtheria toxins at their transition states as determined by kinetic isotope effect analysis. All distances are in Angstroms. Hydrolysis refers to the toxin-catalyzed solvolysis of NAD+ in the absence of the protein nucleophile ADP-ribosylation acceptor. All of these toxins catalyze NAD+ solvolysis. ADP-ribosyl transferase activity to Gαi3 peptide for pertussis toxin is to the C20-terminal peptide in which transfer occurs to Cys at amino acid 4 of the peptide. ADP-ribosyl transferase to eEF-2 used full-length eEF2 isolated from baker’s yeast.
6.3. Cholera Toxin
The slow hydrolysis of NAD+ (kcat= 8 min–1) catalyzed by cholera toxin in the absence of the Gαi protein has been studied to characterize the hydrolytic transition state.114 Kinetic isotope effects support an asymmetric, dissociative transition state with the bond to the leaving group nicotinamide nearly broken (2.16 Å) and even weaker participation of the attacking water nucleophile at >3.0 Å (Figure 17B). The ribocationic transition state generated a large, normal KIE of 18.6% from [1′–3H]NAD+, indicating increased out-of-plane modes. Small KIEs are anticipated from [1′–14C]NAD+ in a dissociative mechanism, and the value of 3% is consistent with low remaining bond order to the leaving group. Water is not the preferred nucleophile for this reaction, and it is not surprising that its participation is not enforced with a distance of >3.0 Å at the transition state. Despite the remote location of the water nucleophile, it is a specific interaction, as methanolysis does not occur with the reaction enforced by cholera toxin.
6.4. Pertussis Toxin
Isotope effect experiments with pertussis toxin were designed to compare the nature of the transition states formed by the enzyme in the absence (hydrolytic reaction) and presence of the thioate nucleophile from the Gαi3 protein.115,116 The toxin is active with the N-terminal 20 amino acids of the G-protein, where the nucleophilic thioate anion is 4 amino acids from the N-terminal. The KIE values and the transition state for the hydrolysis reaction catalyzed by pertussis toxin were similar to that for cholera toxin (Figure 17). When the nucleophile from the G-protein peptide is present, the KIE values are altered. Thus, the KIE for [1′–14C]NAD+ changed from 2% for hydrolysis to 5% for ADP-ribosyl transfer to the Cys nucleophile. Transition state analysis indicated an earlier transition state with the C1′ to N1 leaving group decreasing from 2.14 to 2.07 Å and the attacking nucleophile also demonstrating increased participation at the transition state, increasing from >3.0 Å for hydrolysis to 2.47 Å for the attack of the thioate nucleophile (Figure 17). Contacts at the enzyme catalytic site in the ternary complex are more optimized, with improved activation of the leaving group and increased participation of the peptide nucleophile.
6.5. Diphtheria Toxin
The nucleophile targeted for ADP-ribosylation by diphtheria toxin is a post-translationally modified histidine in eukaryotic elongation factor 2 (eEF-2) called diphthamide (Figure 15).117 The transition states for hydrolysis of NAD+ and ADP-ribosylation of eEF-2 by diphtheria toxin are similar to those for cholera and pertussis toxins by being dissociative with strong ribocation character at the transition state and by becoming earlier in the presence of the protein nucleophile.118,119 In the case of diphtheria toxin, the transition states were determined for the hydrolytic reaction and full-length eEF-2 isolated from bakers yeast.120 Increased nucleophile participation at the transition state is apparent in the primary KIE values. For the hydrolytic reaction, the [1′–14C]NAD+ KIE was 3.4% and increased to 5.5% for ADP-ribosylation of eEF-2. Analysis of these and other isotope effects indicated a weak bond of 2.64 Å to the leaving group at the hydrolysis transition state, changing to 1.99 Å in the presence of eEF-2. The main effect is on leaving group activation, as the water and diphthamide nucleophiles are at similar distances of 2.45 and 2.58 Å, respectively, at the transition states. This difference has been speculated to be caused by activation of the nicotinamide group from a decreased dielectric of the leaving group pocket.
6.6. Nucleophilic Displacement by Electrophile Migration
Transition state analysis from KIEs yields a two-state comparison between reactant and transition state, with no information about intervening or following chemical steps.121 However, it is of interest that in many N-ribosyltransferases the transition states are similar with low Pauling bond order to the leaving group and even lower bond order to the attacking nucleophile. In these mechanisms, the leaving group and the nucleophile are relatively fixed by the enzymatic architecture during the reaction coordinate. Enzymatic interactions generate the ribocation and the C1′ anomeric carbon migrates 1.8–2.1 Å between the leaving group and the nucleophile.122–125 Crystallographic evidence from reactants, transition state analogues, and products at the catalytic sites of these enzymes also supports the anomeric carbon migration. The mechanism has been termed “nucleophilic displacement by electrophile migration” based on the chemical formality of nucleophilic displacement evidenced by the inversion of configuration, with the electrophile migration referring to the excursion of the ribocation between the leaving group and the nucleophile. An interesting point for the electrophile migration mechanism in the context of the ADP-ribosylating toxins is that it permits a wide variation of pKa values between the leaving group and the nucleophile. These asymmetric transition states permit formation of highly reactive ribocations that will react with nucleophiles of different chemical reactivity. For the NAD+-based ADP ribosyltransferases discussed here, the nucleophile diversity includes water, Arg, Cys, and diphthamide. In other enzymes it includes the N1 of adenosine in CD38, the N1 of ATP in the ATP-phosphoribosyltransferase, and carbonyl oxygens in the Sirtuins.126–128 This concept will be revisited in the phosphorylase reactions of section 12.
6.7. Transition State Design for ADP-Ribosylating Toxins
The transition states of ADP-ribosylating toxins share common features of a nicotinamide leaving group approaching neutral charge as the N-ribosidic bond breaks, a ribocation, and a long bond between the ribocation and the leaving group. In all cases, the participation of the nucleophile is weak. It was hypothesized that transition state analogues might be synthesized with similar characteristics to mimic these transition states (Figure 18.).129
Figure 18.

Design of transition state analogues for ADP-ribosylating cholera, pertussis, and diphtheria toxins. Features of the ribocation are provided by the hydroxypyrrolidine, a long bond to the leaving group is provided by the methylene bridge, and recognition elements of the carboxamide group are retained.129
6.7.1. Inhibitor Synthesis.
Potential inhibitors were synthesized using the strategy of Figure 19. Although ADP-ribosylating toxins, phosphorylases, and nucleoside hydrolases all proceed through ribocation transition states, transition state analogue affinities are proportional to the enzyme-enforced rate enhancements according to the Wolfenden proposal.3,4 Transition state analogue design may be challenging for the ADP-ribosylating toxins because of the relatively small k /k ratio and the relatively weak binding of the NAD substrate. In the nucleoside hydrolases, rate enhancements are estimated to be around 1012, while for NAD+ hydrolysis and ADP-ribosylation, enhancements are 3–6 orders of magnitude smaller at 106–109. These considerations raised concern that even though transition state analysis provides novel insight to catalysis poor catalysts may resist efforts to design tight-binding transition state analogues.
Figure 19.

Chemical synthetic scheme for 3-hydroxypyrrolidine transition state analogues for ADP-ribosylating cholera, pertussis, and diphtheria toxins.129
6.7.2. Cholera Toxin Inhibitors.
Inhibitors 1 and 2 (Figure were tested as potential transition state analogues against cholera, pertussis, and diphtheria toxins for both the NAD+ hydrolytic reaction and the ADP-ribosylation reactions.129 Cholera toxin binds NAD+ with a Km value of 10.8 mM for the hydrolytic reaction. Inhibitors 1 and 2 gave Ki values of 17.4 and 32.7 μM, respectively. Therefore, they give Km/Ki values of 620 and 330 relative to NAD+. Although these are impressive ratios, suggesting capture of transition state features, the relatively high Km values for NAD+ make the absolute inhibition constants rather modest compared to the nanomolar inhibitors described above and the picomolar to femtomolar constants to be discussed below. In the presence of O-methyl-arginine as an ADP-ribosyl group acceptor, the Km value for NAD+ decreased to 4.7 mM and inhibitors 1 and 2 also bound tighter at 10.9 and 23.6 μM, respectively, to give Km/Ki values of 431 and 200 relative to NAD+. The ribocationic character of the 3-hydroxypyrrolidine analogues apparently generates binding affinity because of their similarity to the transition state. However, the catalytic activity of cholera toxin for hydrolysis and transfer to O-methyl-arginine is slow, at 8.7 and 20.0 min–1, providing a limited kcat/kchem target for tight binding of transition state analogues. The cholera toxin system deserves additional study with these inhibitors, as the Km values for NAD+ in this in vitro system are unlikely to be functional in vivo, where other activating factors are known to interact with cholera toxin for catalytic activation.130–132 Increased catalytic function is likely to enhance the interaction of transition state analogues.
6.7.3. Pertussis Toxin Inhibitors.
Pertussis toxin binds NAD+ with a Km value of 19 μM for the hydrolytic reaction.115,129 Inhibitors 1 and 2 gave K values of 24.4 and 39.7 μM, respectively. Therefore, they give Km/Ki values of 0.78 and 0.48 relative to NAD+. The relatively low Km value for NAD+ makes these micromolar inhibition constants near equivalent when compared to the binding of substrate. Additional studies are needed to design effective transition state analogues for pertussis toxin, as there are no reports of inhibitiors for the ADP-ribosylation of G-protein inhibitory peptides or for reaction with the intact Gαi3 protein. Surprisingly, the ribocationic character of the 3-hydroxypyrrolidine analogues does not contribute to binding as transition state analogues in the case of in vitro action of pertussis toxin. Synthesis and testing of the iminoribitol analogues of 1 and 2 (Figure 19) would add information relative to the 2-hydroxyl group and placement of the ribocation mimic.
6.7.4. Diphtheria Toxin Inhibitors.
Diphtheria toxin gives a Km value of 85 μM for the hydrolysis of NAD+ and a slow reaction of 0.11 min−1.133 Diphtheria toxin is much more active for diphthamide ADP-ribosylation of yeast eEF-2, increasing the reaction rate 1650-fold to 182 min−1.134 Inhibitors 1 and 2 (Figure 19) gave Ki values of 48.2 and 32.9 μM, respectively, for the hydrolysis of NAD+, slightly better inhibitors than NAD+ is a substrate. They give Km/Ki values of 1.8 and 2.6 relative to NAD+. The Km value for NAD+ decreases to 6 μM in the presence of eEF-2, but the Ki values for inhibitors 1 and 2 do not decrease in proportion with Ki values of 30.5 and 19.1 μM, respectively. Therefore, the Km/Ki values are 0.2 and 0.3 for the inhibitors relative to NAD+. Transition state analogues are anticipated to exhibit increased binding as the reaction rate increases, a property not seen with diphtheria toxin. No other transition state analogues have been explored for this toxin, although there are reports of inhibition of uptake by amine compounds and semicarbazoles.135–137
Because catalytic rate enhancements by the bacterial exotoxins are small, transition state analogues do not capture large energies of activation.129 Although Km/Ki values are large for cholera toxin, the weak binding of NAD+ results in rather weak (micromolar) affinities for the putative transition state analogues. In the cases of diphtheria and pertussis toxins, the transition state analogues bind with approximately the same affinity as substrates, suggesting modest transition state stabilization. Additionally, diphtheria and pertussis toxins may catalyze ADP-ribosyl transferase reactions from ground state destabilization by increasing the activation of the already activated nicotinamide leaving group while making the ADP-ribosyl receiving nucleophile the nearest reactive atom to capture the ribocation formed by nicotinamide loss. Although the original goals of transition state analogue design to block toxin action have not been accomplished, the transition state analysis of three ADP-ribosylating toxins establishes the mechanistic features of their transition states.
7. RIBOSOME-INACTIVATING PROTEINS
7.1. Ricin A-Chain
Among the most toxic of all natural products are the plant-derived ribosome-inactivating proteins, typified by ricin.138–140 The notoriety of ricin as a toxin was enhanced when the KGB used it to assassinate the Bulgarian dissident and defector Georgi Markov at a bus stop in London in 1976.141–143 Ricin is composed of a membrane-penetrating B-chain and a catalytically active A-chain. The subunits are linked by disulfide bonds that are reduced as the AB-toxin complex travels from the membrane by retrograde transport to the golgi and the cytosol where the reduction of disulfide bonds releases the catalytically active ricin A-chain.144 Its physiological substrate is functional eukaryotic ribosomes where it hydrolyzes a single adenine by base excision from the ricin–sarcin loop of 28 S rRNA (position 4324 in rat rRNA).145,146 Adenine depurination from the 5′-GAGA-3′tetraloop modifies the binding site for eEF-2 and destroys the catalytic function of the ribosome. The A-chain of ricin has been linked to antibodies that recognize specific cancer cell types.147–150 Delivery kills the target cancer cells, but the efficacy of this therapy is limited by toxic side effects of ricin A-chain that remains in the circulation and/or is released from cancer cells undergoing toxin-induced cell death.151–153 Transition state analogue, inhibitors of ricin A-chain could be used as rescue agents to prevent circulating ricin A-chain from causing cell lysis in the vascular bed.
7.1.1. Ricin A-Chain Transition State Structure.
Kinetic isotope effects on the hydrolysis of a small 10mer stem– tetraloop oligonucleotide substrate with isotopic labels incorporated specifically into the target adenine (bold) of the 5′-CGCGAGAGCG-3′ stem loop established the mechanism of the reaction as DN*AN, with a ribocationic, highly dissociative transition state (Figure 20).154 Small stem–loop DNA structures are also substrates, and KIE measurements with the same sequence of DNA altered the KIE values, but the reaction stayed within the general description of the DN*AN mechanism.155
Figure 20.
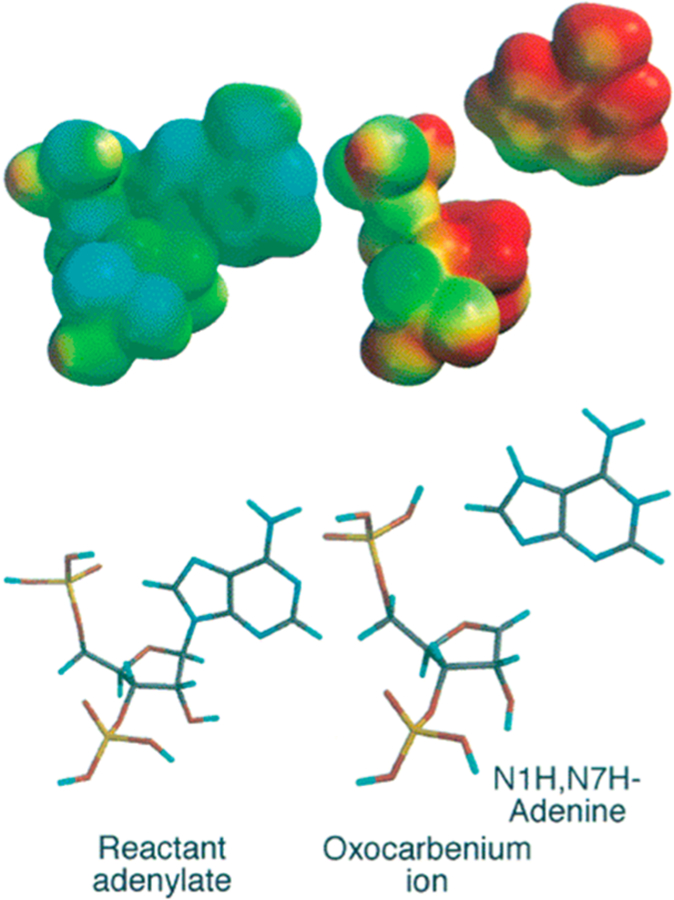
Transition state structure of ricin A-chain acting on stem– loop RNA at pH 4.0. (Upper left) Reactant adenylate electrostatic potential at the van der Waals surface. (Upper right) Transition state electrostatic potential showing fully dissociated adenine. Reproduced from ref 154. Copyright 2000 American Chemical Society.
7.1.2. Inhibitor Design and Synthesis for Ricin A-Chain.
Inhibitors were designed and synthesized to mimic the stem–loop substrate. Alterations toward the transition state included replacing the substrate adenylate with a hydroxypyrrolidine mimic of the ribocation in an N1′-methylene bridge to 9-deazaadenine placed at the depurination site in tetraloops of varied length (Figures 21 and 22).156,157 An abasic, 14mer construct inhibited ricin A-chain with a Kd of 480 nM (1N-14, Figure 22). Filling the catalytic site with adenine improved binding of IN-14 to 12 nM, demonstrating the benefit of the adenine in combination with the transition state ribocation mimic.158 A stem–tetraloop inhibitor was synthesized that incorporates a methylene-bridged hydroxypyrrolidine (1-a zasugar mimic, namely, (3 S,4 R )-3-hydroxy-4-(hydroxymethyl)pyrrolidine) to a 9-deazaadenyl group to mimic the N7-protonated leaving group at the transition state (Figure 21). The 240 nM inhibition constant for the 9-deazaadenyl 14mer (DA-14) was improved to 26 nM in a construct with a phenyl-substituted inhibitor with a 2′-deoxyguanosine-nucleoside placed 5′ to the depurination site (Figure 22). This molecule is the tightest binding inhibitor reported for ricin A-chain. Important features of these molecules are their ribocation mimics and the increased pKa at N7 of the adenine leaving group, both properties of the transition state. The phenyl-substituted molecule combines the ribocation mimic with a planar hydrophobic substituent replacing the adenine leaving group, suggesting a complimentary hydrophobic feature at the leaving group pocket.
Figure 21.

Synthetic route to hydroxypyrrolidine transition state analogues of ribosome-inactivating proteins: (i) DMTrCl (1.5 equiv), DMAP (catalytic), (iPr)2NEt (2 equiv), pyridine, room temperature, 5 h; (ii) 2-cyanoethyl N,N-diisopropylchlorophosphoramidite (2.5 equiv), 2,4,6-collidine (2.5 equiv), 1-methylimidazole (1 equiv), methylene chloride, 0 °C, 30 min; (iii) Expedite DNA/RNA synthesis system. Reproduced from ref 156. Copyright 2004 American Chemical Society.
Figure 22.
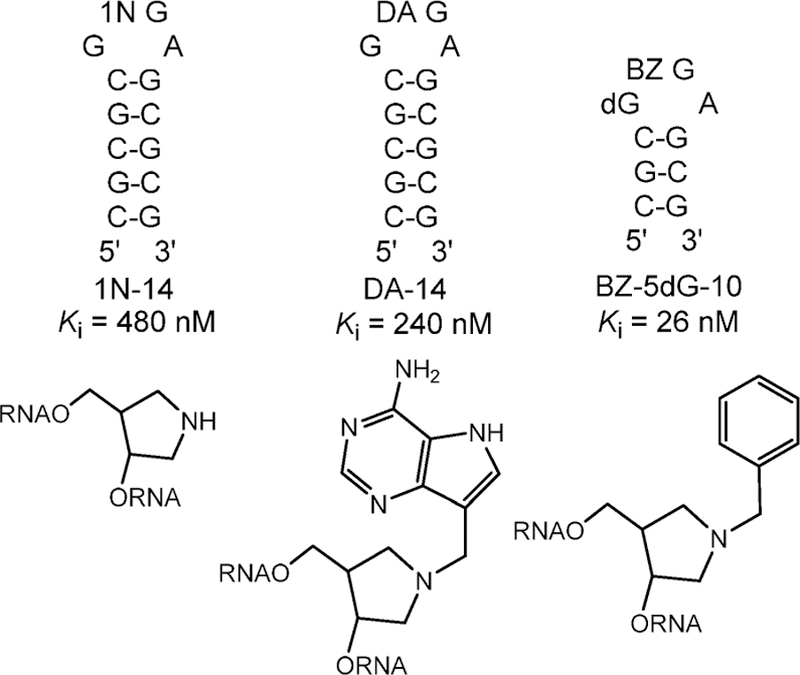
Transition state analogue inhibitors of ricin A-chain. 1N, DA, BZ, and deoxyG (dG) inserts into the stem–loop structures replace adenosine at the depurination site for the reaction and are shown below the respective stem loops.159
7.1.3. Structure of Ricin A-Chain with a Transition State Analogue.
The value of hydrophobic interactions at the catalytic site only became apparent when the crystal structure of ricin A-chain was solved with a covalently circularized stem– loop transition state analogue at the catalytic site (Figure 23).160,161 Ricin A-chain structures with fragments or single nucleotides at the catalytic site did not induce the full architecture revealed by the transition state analogues.162–166 The adenine leaving group is sandwiched between two tryosine groups. The nearby Arg has the effect of favoring protonation of the adenine leaving group. The results of these mechanistic, transition state, inhibitor design, and structural studies made ricin A-chain the best-understood ribosome inactivation protein. In vitro, the ricin A-chain has an unusual pH optimum of pH 4.0 and is nearly inactive at pH 7. The KIEs and transition state analysis were conducted at pH 4.0. Inhibitors functioned well as stem–loop RNA competitors at low pH but did not bind to the enzyme at physiological pH values. The inhibitors did not protect rabbit reticulocyte ribosomes from ricin A-chain inactivation under physiological conditions evaluated by protein translation assays.
Figure 23.

Catalytic site contact maps of ricin A-chain (A) and saporin L3 (B) with a cyclic transition state analogue inhibitor bound to the active sites. Purines and catalytic site groups involved in π-stacking are in orange. Water molecules are drawn as red dots. Hydrogen bonds are shown as dashed lines (green). Hydrogen bonds are in Angstroms. Reproduced with permission from ref 161. Copyright 2009 Proceedings of the National Academy of Sciences.
7.2. Saporin L3
A screen of other ribosome-inactivating proteins that might be susceptible to inhibitors designed for ricin A-chain led to the development of saporin L3, a poorly understood leaf form of a ribosomal-inactivating protein from Soapwort plants and earlier known as saporin L1.167–169 Other isozymes of saporin, especially saporin-6, have been used as a toxic agent in antibody-targeted toxin therapy. Saporin L3 was found to be a superior agent in its catalytic action on mammalian rRNA and as a target for the development of transition state analogues as rescue agents.169–172 Characterization of saporin L3 demonstrated it to be a promiscuos RNA adenine depurinating enzyme.173,174 In addition to depurinating ribosomes and rRNA, saporin L3 catalyzed the depurination of adenines in the GAGA tetraloop of short sarcin–ricin stem–loops and multiple adenines within eukaryotic rRNA, tRNAs, and mRNAs. The transition state structure of saporin L3 was solved in experiments similar to those used for ricin A-chain, except that the stem–loop RNA substrate (5′-GGGAGGGCCC-3′) contained only a single A to prevent multiple depurinations in kinetic isotope effect experiments.175
7.2.1. Saporin L3 Transition State.
Despite catalysis of the same reactions and homology in ricin A-chain and saporin (30% identical), the transition states differ. Intrinsic kinetic isotope effects are a direct reflection of the environment at the transition states. Thus, the intrinsic 1′–14C KIE for the ricin A-chain-catalyzed depurination of RNA was 0.993, indicating a completely dissociated adenine leaving group with no significant participation of the attacking water nucleophile.154 The intrinsic primary 1′–14C KIE for saporin L3 depurination is 1.052, less than expected for a fully developed SN2 mechanism, where the 1′–14C KIE value can be as large as 1.14.175–177 The smaller α-secondary hydrogen KIEs of 1.045 for saporin L3 compared to 1.163 for ricin A-chain also indicates a transition state with less dissociative character. Where does the bond order reside in the saporin L3 transition state? The 9–15N KIEs report on the degree of N9-ribosyl bond breaking. The KIE values indicate a fully broken C–N bond at both TSs. Therefore, the attacking water nucleophile contributes to the bond structure of the transition state more in saporin L3 than in ricin A-chain. It is located 1.95 Å from the anomeric carbon of the adenine depurination site (Figure 24). The nucleophilic water is well defined in the crystal structure of saporin L3 with a transition state analogue.161 The crystallographic water nucleophile is held in position by two 2.5 Å hydrogen bonds to a carboxylate oxygen of Glu174 and the carbonyl oxygen of Glu206 and is positioned 3.1 Å from the reaction center.
Figure 24.
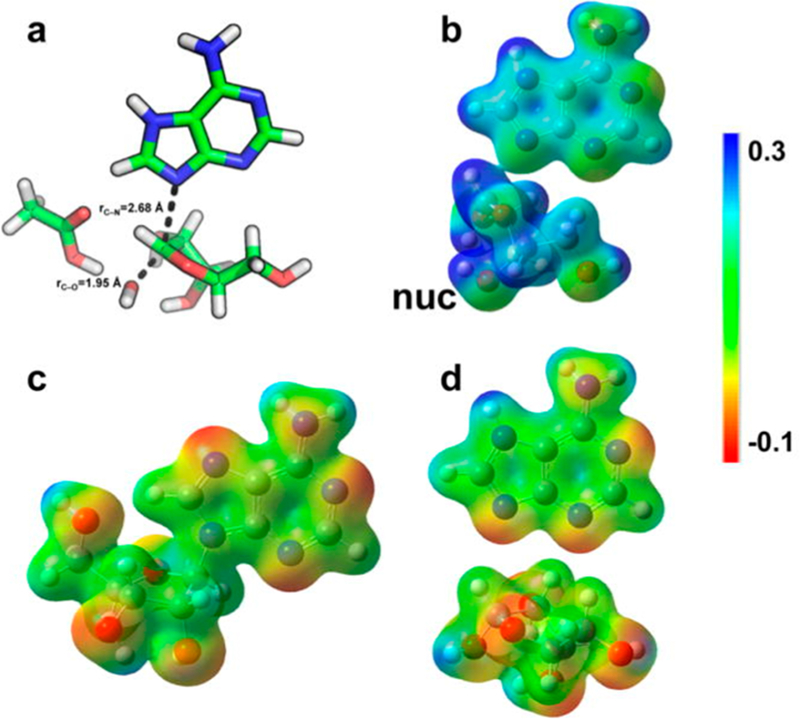
Transition state geometry, electrostatic potential surface (EPS), and NBO charges for the reaction of saporin L3. Transition state geometry (a), EPS values for transition state (b), reactant (c), and products (d) are shown. nuc is the nucleophilic water. Reproduced from ref 175. Copyright 2016 American Chemical Society.
7.2.2. Saporin L3 Inhibitor Design.
Transition state analogues developed to mimic the ricin A-chain transition state were used as a starting point for design of inhibitors for saporin L3. Although the transition states differ in detail, they share ribocation character, leaving group protonation, and specificity for the RNA scaffold. The inhibitor cocrystallized with ricin A-chain and saporin L3 was a four-base cyclic, covalently closed RNA construct. Instead of the RNA stem, the RNA tetraloop mimic was closed with a cyclic oxime linker and contained a transition state analogue at the adenylate depurination site.160 Both DNA and O2′-methylated RNA versions were synthesized. With dissociation constants at 2.3 and 3.9 nM, these are the highest affinity inhibitors reported for saporin L3 (Figure 25). The inhibitors act on purified saporin L3 at physiological Ph and also protect rabbit reticulocyte ribosomes against inactivation by saporin L3 at concentrations consistent with their nanomolar dissociation constants (see below).178
Figure 25.
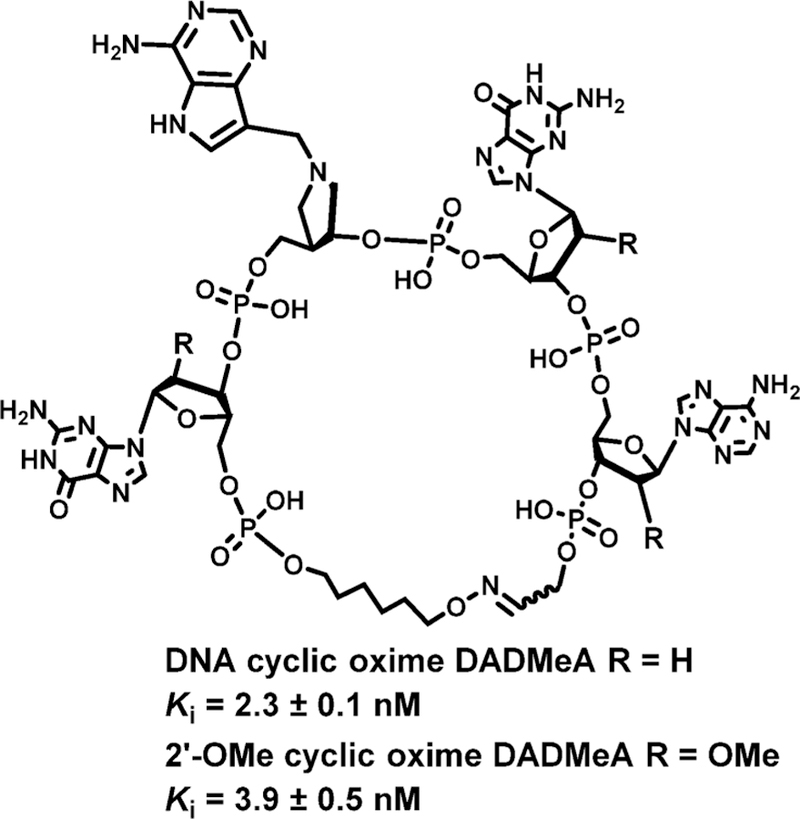
Transition state analogues for saporin L3. Structure is covalently closed and O2′ protected from RNases. It provided a scaffold for A of both ricin A-chain and saporin L3 (Figure 23).178
It is of interest to compare the electrostatic potential surfaces for the saporin L3 transition state to that of a small, linear transition state analogue construct of two backbone links with a transition state analogue linked to a protected guanosine group, a 3.3 nM inhibitor (Figure 26). This construct lacks a stabilized tetraloop group and does not bind to ricin A-chain. The electrostatic potential surface reveals the similarity between the transition state structure and the inhibitor.175
Figure 26.

Comparison of electrostatic potentials for the transition state (a) of saporin L3 and the transition state mimic (b) of a truncated 2-base transition state mimic (c). This truncated inhibitor is a 3.3 nM TS analogue. Reproduced from ref 175. Copyright 2016 American Chemical Society.
7.2.3. Saporin L3 Inhibitors Protect Ribosomes.
Inhibitors of saporin L3 were tested with an in vitro protein translation system using ribosomes from rabbit reticulocyte lysates. The 2-base saporin L3 inhibitor construct (Figure 26) was used. Saporin L3 at a concentration of 300 pM caused ∼90% inhibition of cell-free translation.178 Under the same conditions, varied concentrations of the inhibitor (Figure 26) were added to rescue protein synthesis by protection of the ribosomes from saporin L3. The inhibitor rescued protein synthesis with an EC50 of 36 ± 2 nM, consistent with protection by blocking the saporin L3 catalytic site. This inhibitor also inhibited adenine release from purified 80S rabbit ribosomes with an IC50 of 7.8 ± 1.1 nM. The transition state inhibitors of saporin L3 are effective in preventing ribosome depurination and depurination of small nucleic acid substrates. No studies have yet been reported on the use of saporin L3 inhibitors in protecting cells or animals from the side effects caused by saporin L3.
8. PURINE PHOSPHORIBOSYLTRANSFERASES
8.1. Discovery and Biological Function
Kornberg, Lieberman, and Simms discovered and characterized 5-phosphoribosyl-α-D-1-pyrophosphate (PRPP) in 1954 as the required ribosyl phosphate donor in the synthesis of pyrimidine nucleotides from orotic acid.179 The importance of purine salvage in humans via hypoxanthine-guanine phosphoribosyl-transferase became apparent with the discovery of its genetic deficiency as the cause of Lesch–Nyhan syndrome.180–182 Studies with Plasmodium species indicated an ability to synthesize pyrimidine nucleotides by de novo pathways but not purines, making exogenous purines essential nutrients.183 The pathway for purine salvage in P. falciparum was deduced by direct assay of the enzymes from extracts of cultured parasites, where the high activities of adenosine deaminase, purine nucleoside phosphorylase, and hypoxanthine-guanine-xanthine phosphoribosyltransferase (HGXPRT) supported a robust pathway for salvage of hypoxanthine.184 More recent studies have concluded that purine phosphoribosyltransferases provide an essential step for purine salvage in most purine auxotrophic protozoan parasites.185–187 On the basis of the nature of the transition states for the nucleoside hydrolases and purine nucleoside phosphorylase, transition state analogue candidates were synthesized and found to be powerful inhibitors of the enzymes from both P. falciparum and human sources.188,189 Crystal structures indicated catalytic site contacts essential for catalysis, including the catalytic site magnesium atoms required for catalysis in the HG(X)PRTs (Figures 27 and 28).190–192
Figure 27.

Catalytic site contact map for human HGPRT in complex with ImmGP.190 Light green circles represent crystallographic water oxygens. O2A is the nearest to the reaction center and is proposed to be the nucleophilic oxygen.
Figure 28.

Catalytic site contact map for P. falciparum HGXPRT in complex with ImmHP. Similar to the structure of the human enzyme, O2A is the nearest to the reaction center and is proposed to be the nucleophilic oxygen and the O5′–N4′ distance is 2.7 Å. Reproduced from ref 191. Copyright 1999 American Chemical Society.
8.2. Transition State Structures
The transition states for N-ribosyltransferases exhibit ribocation character with purine leaving groups activated by protonation at N7 of the purine ring.193 As the reaction of HGXPRT is freely reversible (hypoxanthine + PRPP ↔ IMP + PPi), the transition state can be reached from either direction. Analogues of IMP were synthesized with ribocation features of the transition state, similar to the targets described above. Immucillin-G 5′-phosphate (ImmGP) was a 4.6 nM inhibitor of the HGPRT from human sources, binding 14 000 times tighter than the dissociation constant for IMP.188 Considering the full catalytic site ensemble of two Mg2+ ions, PPi, and ImmGP finds 35 interactions at 2.8 Å or less. Thus, the inhibitor complex is highly immobilized, and dilution experiments show slow release of the inhibitor from the inhibited complex.188 Of specific interest for the mechanisms of N-ribosyltransferases is the interaction of Asp137 stabilizing the hydrogen bond to N7, as protonation of the leaving group is a common feature of these enzymes. Another significant interaction is the O5′ with the N4′ iminoribitol nitrogen at 2.7 Å. When nucleotides are at the catalytic site, the close O5′–O4′ distance constitutes a neighboring group interaction assisting formation of the ribocationic transition state.194
8.3. Iminoribitol Transition State Analogues
Immucillin-H 5′-phosphate and Immucillin-G 5′-phosphate were analyzed for P. falciparum HGXPRT and found to be 1 and 14 nM inhibitors, respectively. In the crystal structure of the P. falciparum HGPRT, Asp148 plays the role of N7 proton donor as does Asp137 in the human structure (Figure 28). In both human and P. falciparum enzymes, two Mg2+ ions form bidentate interactions with both phosphoryl groups of the PPi and bridge it to the ribosyl group by another bidentate interaction to the vicinal 2′,3′-hydroxyl groups of the nucleotide. In both enzymes, the transition state analogues are proposed to capture part of the tight interactions at the transition state. An unusually strong hydrogen-bond interaction is observed by the NMR chemical shift of the N7-Asp hydrogen bond. The chemical shift for this proton is detected in the enzyme–immucillinHP–Mg2+– pyrophosphate complexes for both human and P. falciparum enzymes. Downfield 1H signals at 13.9 (human) and 14.3 ppm (P. falciparum) have been assigned to the N7 protons of ImmHP hydrogen bonded to Asp148 (Asp137 for the human enzyme) by 15N-edited proton NMR spectra using [7-15N]ImmHP.188,191 Isotope-edited Raman and FTIR studies of the complex with enzyme–immucillinHP–Mg2+–pyrophosphate found the 5′-phosphate to be dianionic and the pyrophosphate to be the fully ionized tetraanion.195 Mutagenic studies indicate that the Asp is important for catalysis but does not contribute significantly to substrate binding.196,197 Despite the tight binding of the ImmHP and ImmGP for their target enzymes, phosphate esters are not suitable drugs, as anions are membrane impermeable and phosphoesters are susceptible to hydrolysis.
8.4. Asymmetric Acyclic Nucleoside Bisphosphonate Inhibitors
The multiple phosphate binding sites in the catalytic sites of purine phosphoribosyltransferases have led to the synthesis of families of symmetric or asymmetric acyclic nucleoside bisphosphonates (ANbP) as inhibitors of human HGPRT and P. falciparum HGXPRT (Table 2).198–201 Compounds 1a and 20 are 30 and 6 nM inhibitors for the P. falciparum HGXPRT. When converted to the phosphoramidate prodrugs to permit cellular entry, the IC50 values for inhibition of P. falciparum growth in cultured erythrocytes were between 1.4 and 9.7 μM and showed low toxicity for human cell lines.200
Table 2.
Symmetric and Asymmetric Acyclic Nucleoside Bisphosphonate Ki Value with Human and P. falciparum Purine PRTs198–20,1
| No. | Base | Acyclic moiety | Human HGXPRT Ki[μM] | P. f. HGXPRT Ki[μM] |
|---|---|---|---|---|
| 1a | G | 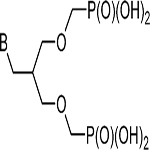 |
0.03 | 0.07 |
| 1b | Hx | 1 | 5 | |
| 2a | G | 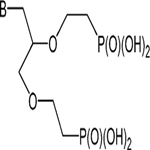 |
0.6 | 0.5 |
| 2b | Hx | 0.7 | NIa | |
| 27 | 8-Br-G | 0.4 | 0.9 | |
| 20 | G | 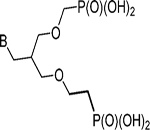 |
0.006 | 0.07 |
| 21 | Hx | 1.8 | 3 | |
| 22 | 8-Br-G | 0.1 | 2 | |
| 23 | 8-Br-Hr | 2.5 | NI | |
| 24 | 7-deaza-G | 0.1 | 4 | |
| 25 | 7-deaza-Hx | 4.6 | 6 |
No inhibition obseved. Base abbreviations are guanine (G), hypoxanthine (Hx), and 8-bromo (8-Br).
What do these constants suggest for potential druggability of the compounds in Table 2? Inhibition of human HGPRT is not a major concern in the treatment of malaria, as only long-term genetic deficiency of the enzyme leads to symptoms in humans. However, two other concerns are dissociation constants above 70 nM, making them weaker by factors of up to 100 than transition state analogues (e.g., 2, Figure 29) for the same target. The phosphonates of Table 2 and Figure 29 suffer from the likely barrier to oral uptake from anionic compounds. Both will need prodrug chemistry to move them toward useful agents.
Figure 29.
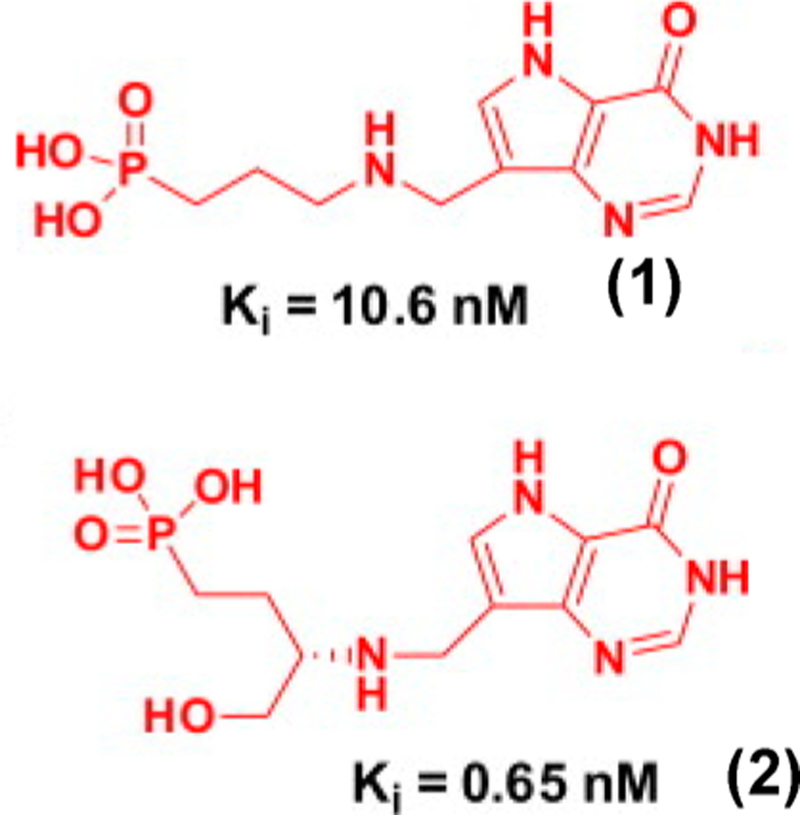
Acyclic aza-C-nucleoside phosphonate inhibitors of P. falciparum HGXPRT.
8.5. Acyclic Aza-C-nucleoside Phosphonate-Substituted 9-Deazahypoxanthine Inhibitors
To maintain the cationic features of the transition state while eliminating the hydrolytic liability of the phosphate monoester, a family of acyclic aza-C-nucleoside phosphonate-substituted 9-deazahypoxanthine inhibitors was synthesized. Five compounds had enzymatic Ki values at 10.6 nM or below, two of which are shown (Figure 29).189,202 A general synthetic scheme was devised to permit variation in the side chain length between the aza and the phosphonate groups, variation in the deazapurine and aza groups, and variation in substituents at asymmetric branching carbons between the aza and phosphonate groups (Figure 30). A remarkable feature of the compounds exemplified in Figure 29 is their specificity for the enzyme from P. falciparum relative to human HGPRT. Thus, of the five compounds with Ki values at 10.6 nM or below, two showed no inhibition of the human enzyme at 10 000 nM and the other three showed Ki human/Ki P. falciparum values of 350, 462, and 585.189
Figure 30.

Synthetic strategy for one family of the acyclic aza-C-nucleoside phosphonate inhibitors: (a) triethyl phosphite, 120 °C; (b) H2NNH2, EtOH; (c) NaBH4, EtOH; (d) 35% aq HCl, 60 °C; (e) 48% HBr 90 °C; (f) 2-picoline borane, MeOH. Reproduced with permission from ref 202. Copyright 2013 Elsevier.
8.6. Prodrugs of Acyclic Aza-C-nucleoside Phosphonate-Substituted 9-Deazahypoxanthine
A prodrug approach to the acyclic aza-C-nucleosides as antimalarials has the goal of delivery to the parasite inside of host erythrocytes. Protecting groups should render the compounds membrane permeable with groups that are cleaved only when the compound enters the P. falciparum compartment. The approach was to protect with groups susceptible to the broad-specificity phospholipase C of P. falciparum while avoiding the narrow specificity of lipases A and D of the erythrocyte (Figure 31). A similar approach has been developed to enhance oral antiviral activity and reduce toxicity of alkoxyalkyl prodrugs of acyclic nucleoside phosphonate antivirals.203,204 Phosphonate groups are anionic at physiologic pH and are poorly adsorbed.205 The phosphonate inhibitors alone (Figure 29) showed no activity against cultured P. falciparum.
Figure 31.

Lysophospholipid prodrug approaches to antimalarials. Prodrug approach summarized in the text was adapted to the AIPs. (A) Phospholipase specificity pattern. (B) Synthetic approach to prevent phospholipase A and D action. Table provides the specific substituents in B and the IC50 values for growth of P. falciparum parasites in human erythrocytes. Reproduced with permission from ref 189. Copyright 2012 Elsevier.
A prodrug strategy that has been used to increase the efficacy of the nucleoside phosphonates cidofovir and tenofovir was tested.206 The lysophospholipid addition allows for transfer through cellular membranes. Inside target cells, phospholipase C (PLC) activity cleaves the prodrug releasing the acyclic phosphonate inhibitor. This system is advantageous for targeting P. falciparum since the Ca2+-dependent PLC activity in erythrocytes displays a narrow substrate specificity for phosphorylated phosphoinositides, and a PLC with broad substrate specificity has been identified in P. falciparum.207–211 To protect the linear acyclic aza-C-nucleoside phosphonate inhibitor (Figure 29) using the lysophosphatidylcholine scaffold, phosphocholine is replaced by the acyclic aza-C-nucleoside phosphonate inhibitor, the sn2 hydroxyl is replaced with a hydrogen, and the sn3 ester linkage is replaced with an ether. These prodrugs demonstrated antiparasite activity against P. falciparum cultured in human erythrocytes.189 Metabolic studies demonstrated that treatment with these prodrugs prevented the incorporation of isotopically labeled hypoxanthine into parasites, establishing HGXPRT as the target.189,212
8.7. Biological Efficacy
Activation of compounds 5–8 (Figure 31) in cultured parasites generates anions that are trapped in the parasites. Incubation of treated parasite cultures with labeled hypoxanthine after the inhibitors were removed prevented metabolic labeling. IC50 values for several of the prodrug constructs on three different strains of P. falciparum, including drug-resistant strains, gave similar IC50 values (∼3 μM). These values are well above the Ki of 10 nM for the parent compounds, suggesting additional optimization of the prodrug scaffold might be useful. These compounds have not yet been tested in animal models of malaria.
8.8. Structure of Pf HGXPRT with an Acyclic Aza-C Transition State Analogue
The crystal structure of Pf HGXPRT in complex with Mg2+, PPi, and an acyclic amino alcohol phosphonate showed how the acyclic transition state analogue could be tightly bound in the active site of Pf HGXPRT.189 The cationic amino group is near the position occupied by the ribocation in the proposed transition state. The cation is close to the pyrophosphate (2.8 Å; all distances are the average value from four monomers in the tetramer) to create a favorable ion pair. The methylene bridge linking the amino cation and 9-deazahypoxanthine allows the deazapurine base to tilt 11° toward Phe197, a residue involved in aromatic ring stacking (Figure 32). The side chains of Asp148 (2.8 Å) and Lys176 (3.0 Å), the amide and carbonyl of Val198 (3.1 and 2.7 Å), and a structural water (2.6 Å) coordinated by magnesium are in hydrogen bonds with N7, O6, O6, N1, and N3 of the deazapurine,. The atomic spacing between the amino cation and the phosphonate group permits favorable and simultaneous inhibitor interactions with the magnesium pyrophosphate and the 5′-phosphate binding sites. The 5′-phosphonate interacts with neighboring residues similar to the phosphate of ImmHP bound to Pf HGXPRT (PDB ID 1CJB). The side-chain residues of Tyr116 (2.6 Å), Thr149 (3.0 Å), and Thr152 (2.6 Å) and amides of Asp148 (3.1 Å), Thr149 (3.1 Å), Gly150 (2.6 Å), and Thr152 (2.9 Å) are in hydrogen-bond distance with the 5′-phosphonate, while the 3′-hydroxyl group forms a hydrogen bond with the side chain of Asp145 (2.8 Å). As the inhibitor has only one hydroxyl group compared to two in the reaction or in complex with the Immucillin-H 5′-phosphate transition state analogue, a water molecule replaces the second hydroxyl group and forms a hydrogen-bond network with the O2 and O4 of pyrophosphate (2.6 and 2.7 Å), the 3′-hydroxyl group (2.9 Å), and the cationic amine (2.9 Å). The single magnesium ion is chelated by Asp204, three water molecules, and pyrophosphate. In complexes crystallized with ImmHP, Mg2+, and PPi (Figure 28) two Mg2+ ions form bidentate ionic interactions chelated to PPi. Here, the single Mg2+ forms a bidentate interaction with PPi, a bidentate interaction with Asp204 and is chelated with 3 water oxygens. Major binding interactions are formed with the phosphonate with 2.6 Å hydrogen bonds to each of the three phosphonate oxygens.
Figure 32.

Active site of Pf HGXPRT bound to the 0.65 nM inhibitor of Figure 29 and MgPPi. Hydrogen bonds are represented as dashed lines. All distances are in Angstroms. Two-dimensional representation of the active site where the ionic bond between pyrophosphate and the ribocation mimic is represented as a thick dashed line. Water molecules are represented as red dots, and the hydrophobic residues interacting with the purine ring are drawn in orange. Reproduced with permission from ref 189. Copyright 2012 Elsevier.
8.9. Summary
Transition state analogues of purine phosphoribosyltransferases support the formation of a ribocationic transition state, also indicated from KIE studies. A drug design challenge comes from the two anionic centers on the required 5′-phosphate (a dianion) and the nucleophilic pyrophosphate (a tetraanion). Monophosphate or monophosphonate prodrugs of transition state analogues show promise but are not yet sufficiently effective to be considerd as drug candidates.
9. PYRIMIDINE PHOSPHORIBOSYLTRANSFERASES
9.1. Function of Human and P. falciparum Orotate Phosphoribosyltransferases
Although P. falciparum requires exogenous purines for salvage, the parasite encodes all enzymes for de novo synthesis of pyrimidines but none of the enzymes for pyrimidine base or nucleoside salvage.213–215 An essential step in pyrimidine synthesis is addition of ribose 5-phosphate (from PRPP) to orotic acid by orotate phosphoribosyltransferase (OPRT; Figure 33). Previous OPRT inhibition studies reported that pyrazofurin, a C-riboside antibiotic, inhibited P. falciparum OPRT (Pf OPRT) activity and demonstrated antimalarial activity with in vitro cultures to give submicromolar IC50 values.216 5-Substituted orotate analogues bind to Pf OPRT and block the growth of parasites with IC50 values at micromolar levels.217–219 5-Selenated uridine derivatives also exhibit submicromolar inhibition against Pf OPRT and Homo sapiens OPRT (HsOPRT).220 None of these compounds can be classified as transition state analogues, and some act by incorporation into cellular nucleic acids with mutagenic potential as drug candidates.
Figure 33.

Reaction, transition state, and proposed transition state analogue inhibitors for P. falciparum and human OPRT-catalyzed OMP pyrophosphorolysis. OPRT transition states are characterized by fully dissociated dianionic orotate, a ribocation, and weak nucleophile (PPi) participation. Iminoribitol, pyrrolidine, or acyclic ribocation groups as mimics of the ribocation were proposed. Methylene or ethylene linkers resemble the extended bond distance to approximate the transition state. PRPP, 5-phospho-α-D-ribosyl 1-pyrophosphate. Reproduced with permission from ref 222. Copyright 2013 American Society for Biochemistry and Molecular Biology.
9.2. Transition State Structures
Transition state analogues for the P. falciparum OPRT with discrimination against the human enzyme could provide new antimalarials. Transition state structures were solved for both human and P. falciparum OPRTs. On the basis of the isotope effects and the analysis of the transition states, both enzymes had similar transition state structures (Figure 33). On the basis of the ribocationic transition states, transition state analogues were designed based on precedent from other N-ribosyltransferase programs, especially that of human purine nucleoside phosphorylase (see below).221,222
9.3. Transition State Analogues
Transition state analogue candidates were synthesized to resemble the geometrical and electrostatic characteristics of the transition states and tested for inhibition with both the human and the P. falciparum OPRTs (Figure 34). For the 20 compounds of Figure 34, most compounds bind better to the human than the P. falciparum enzyme, but the differences are small, no more than 5-fold higher affinity for the human enzyme. The affinity reflects the similar transition states for the OPRTs but where the human OPRT is intrinsically more catalytically active.223 The OPRTs are unusual phosphoribosyltransferases in having a relatively weak affinity for the 5′-phosphate of the nucleotide. A comparison of inhibitor pairs with and without the 5′-phosphate (1 and 2, 3 and 4, 5 and 6, 7 and 8, and 14 and 15) shows less than 5-fold increased affinity when the 5′-hydroxyl groups are replaced with the 5′-phosphate groups.
Figure 34.

Inhibition constants (nM) for potential transition state analogues of P. falciparum (blue) and human (red) OPRT enzymes. Reproduced with permission from ref 222. Copyright 2013 American Society for Biochemistry and Molecular Biology.
Three fundamental contributions toward formation of the transition state are (1) activation of the leaving group, (2) formation of the ribocation, and (3) activation and enforced participation of the nucleophile. Transition state analysis showed full loss of the pyrimidine leaving group at the transition state and weak participation of the nucleophile, consistent with the leaving group activation as a dominant catalytic force. Enforced nucleophilic participation is unlikely. Compounds 1 and 2 of Figure 34 are O-ribosides and test the ability of OPRT to form a ribocation to reach the transition state. The p-nitrophenol groups of 1 and 2 are expected to be substrates of enzymes acting on nucleosides or nucleotides where the major enzymatic contribution to catalysis is formation of the ribocation (see Table 1). They are not substrates for the OPRT enzymes, even though they bind to the catalytic sites with nanomolar affinity. Remarkably, 2 (Figure 34) is the highest affinity inhibitor for both the P. falciparum and the human enzymes. In the absence of a crystal structure for the P. falciparum OPRT, the 40 nM inhibitor 2 was docked into the catalytic site of human OPRT (PDB 2WNS). The binding affinity of the p-nitrophenol group is predicted to be similar in bonding energy to the orotate of OMP. The binding free energy of –10.4 kcal/mol, a predicted binding affinity of 22 nM, is in good agreement with the observed Ki of 40 nM. Compounds 3–20 of Figure 34 test different cationic mimics of the ribocation feature of the OPRT transition states. Iminoribitols (3–10, 12, 16), hydroxypyrro-lidines (11, 14, 15, 17, 18), and dihydroxy propyl amines (19, did not significantly improve the affinity of the simple p-nitophenol ribosides, 1 and 2. The conclusion is that ribocation formation is not the most important contribution to forming the transition state. Transition state analysis demonstrated a lack of nucleophilic enforcement; therefore, the major contribution to the formation of the transition state is proposed to be leaving group activation. Variants of the leaving groups as nucleotides were tested in 2, 4, 6, 8, 12, and 15. None were an improvement from the simple p-nitophenol groups of 1 and 2. Additional structural advances with OPRTs are needed to map leaving group interactions for improved analogue design.
9.4. Inhibitors of OPRT Are Ineffective against Cultured P. falciparum
Compounds 1–3, 5–7, 9–14, and 17–20 (in Figure 34) were tested against the growth of P. falciparum parasites cultured in human erythrocytes. No growth inhibition was found at concentrations to 100 μM. These compounds do not gain access to the OPRT compartment in cultured parasites, do not block pyrimidine synthesis, or are bypassed by pyrimidine sources from erythrocytes in this complex culture medium, although it contained no added pyrimidine source.222,224 The lack of cell growth inhibition may reflect permeability barriers. Detailed transport and metabolic studies will be needed to distinguish these mechanisms. With several of the inhibitors demonstrating dissociation constants below 200 nM, an inhibitor concentration of 100 μM would be expected to cause >99% inhibition of OPRT if freely available to the parasite. Growth of cultured mammalian cell lines was unaffected by these inhibitors up to 10 μM. Transition state analogues for OPRTs provide initial insights for the design of antimalarials and anticancer agents. However, problems of permeability, target access, and species specificity will need to be addressed for these nanomolar inhibitors.
9.5. Amidrazone Inhibitor of Yeast OPRT
A slow-onset, irreversible amidrazone transition state analogue has been reported for Saccharomyces cerevisiae OPRT. Compound 10 (Figure 35) at 2 mM, incubated with PPi, Mg2+, and yeast OPRT, was reported to cause usually slow inactivation of catalytic activity with a t ½ of approximately 200 h (kobs = 0.004 h–1). No recovery of activity could be detected, indicating extreme tight binding or irreversible inactivation of the enzyme.225 Compounds 12–14 showed no inhibition under similar conditions. No other characterization of 10 is apparent from the literature, despite subsequent crystal structures with other inhibitors reported for the S. cerevisiae enzyme from the same group.226
Figure 35.
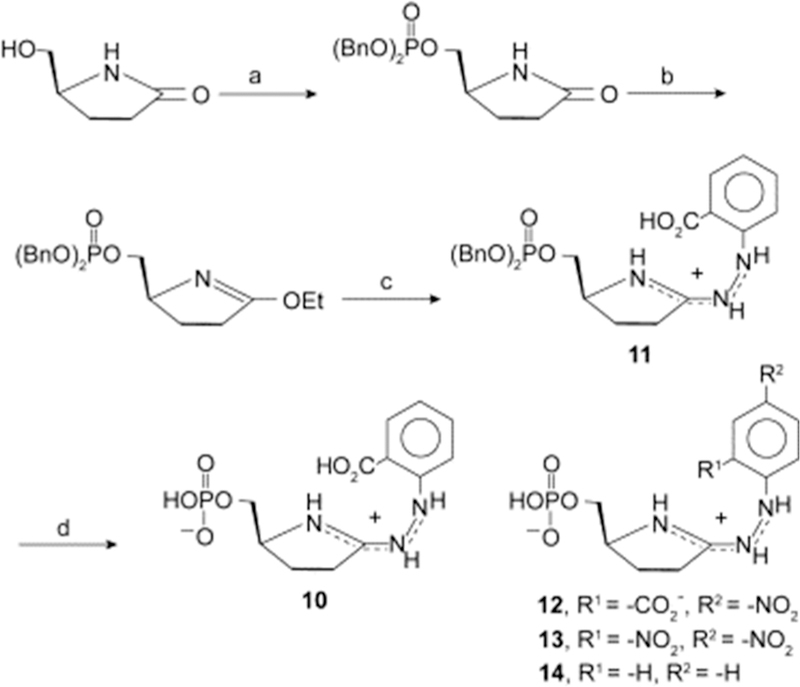
Synthesis of the amidrazone 10, reported to be an inhibitor of yeast OPRT. (a) (BnO)2PO2H, DEAD, THF, 2 h, 25 °C; (b) Et3O+PF6–, CH2Cl2, 2 h, 25 °C; (c) o-hydrazinoPhCO2H, i-Pr2Net, CH2Cl2, 16 h, 25 °C; dil aq HCl; (d) 8 M HCl, 16 h, 25 °C. Reproduced with permission from ref 225. Copyright 2006 Elsevier.
10. HUMAN 5′-METHYLTHIOADENOSINE PHOSPHORYLASE (MTAP)
10.1. Biological Function of Human MTAP
Polyamine synthesis in nucleated mammalian cells produces spermine as the most abundant polyamine, found at millimolar concentrations in rapidly dividing and transformed cells.227,228 Polyamine synthesis involves donation of two propylamino groups to putresine from two molecules of decarboxy-S-adenosylmethionine (SAM) to form a single spermine molecule. Therefore, two SAM molecules are used, and two 5′-methylthioadenosine (MTA) molecules are formed for each spermine molecule. Polyamine synthesis is the only metabolic pathway to generate MTA in humans (although smaller amounts are formed in diphthamide formation229) and is a major use of SAM, a molecule in demand to provide methyl groups for metabolic syntheses and for regulatory control of proteins and nucleic acids.230–232 SAM converted to MTA in the polyamine pathway is not lost to metabolism. MTAP is the only enzyme in humans to metabolize MTA, converting it by phosphorolysis to 5-methylthio-α-D-ribose 1-phosphate and adenine.233 These products are readily converted to SAM precursors, as adenine is converted to AMP by adenine phosphoribosyltransferase and 5-methylthio-α-D-ribose 1-phosphate is converted to methionine. In this way, MTAP provides a metabolic salvage pathway for conversion of MTA to SAM.234 The sole de novo pathway for SAM production involves the reaction of methionine and ATP to form SAM by the human methionine adenosyl transferases (MATs). MAT1 is the liver isoform, and MAT2A is distributed to other human tissues, including cancer cells.235–237
10.2. Transition State and Transition State Analogues
MTAP was targeted for inhibition by transition state analogues to cause cellular accumulation of MTA as a product inhibitor of enzymes of polyamine synthesis.238 Human MTAP was used to measure intrinsic KIE values from isotopically labeled MTA molecules and the transition state structure used to design transition state analogues (Figures 36 and 37). An example of an early transition state analogue (short distance from the ribocation to the leaving group) is MT-ImmA, a 1 nM inhibitor (Figure 37).238 Late transition state analogues, with increased distance between the ribocation mimic and the leaving group, are better mimics of the transition state and include PhT-DADMe-ImmA, MT-DADMe-Immucillin-A (MTDIA), and pClPhT-DADMe-ImmA.239 The pM transition state analogues are slow-onset, tight-binding inhibitors. The affinity of MT-DADMe-ImmA (MTDIA) when compared to the 5 μM Km for MTA as a substrate gives a Km/Ki* ratio of 58 000. MTDIA was tested in cultured mammalian cells to determine the biological effects. Several cancer cell lines responded by undergoing apoptotic cell death when MTDIA was used in combination with MTA.240 Most human cancer cell lines had no response to MTDIA alone, supporting a hypothesis of MTA acting as the active agent. When human cancer cell lines sensitive to MTDIA plus MTA in culture were tested in mouse xenografts, strong responses were observed with human head and neck cell lines FaDu and Cal27 and in human lung cancer lines A549 and H358.240,241 No added MTA is required in mouse models, as systemic inhibition of whole animal MTAP caused the accumulation of MTA in the blood and tissues and excretion into the urine. The original hypothesis of action at the level of polyamine synthesis was disproven by attempts to reverse the effects of MTDIA plus MTA in cultured cells by addition of exogenous polyamines, methionine, or adenine. Likewise, cells and tissues treated with MTDIA had only a modest reduction in cellular polyamine levels.241 Despite the success in mouse models of human cancers, MTAP inhibitors have yot yet advanced to human clinical trials.
Figure 36.

Arsenolysis reaction catalyzed by MTAP, transition state structure, and intrinsic KIE values used to determine the transition state structure.238
Figure 37.

Transition state analogues synthesized to resemble the transition state structure of human MTAP and their dissociation constants.238,239
10.3. Genetic Susceptibility Caused by MTAP Deletion
MTAP is also of interest by being one of the common genomic deletions found in human cancers. Approximately 15% of all human cancers reveal a genetic deletion in a region of chromosome 9p21 that encodes the MTAP gene. A common codeletion is the nearby p16 (CDKN2A, cyclin-dependent kinase inhibitor 2A, multiple tumor suppressor 1) at 9p21.3, a known tumor suppressor gene.242–244 In extensive synthetic-lethal genetic experiments (where the combination of two nonlethal mutations create a lethal one), human cancer cell lines deleted in MTAP (or not, in controls) were scanned for singlegene knockouts that caused cell death in the MTAP-deleted strains but not in controls. Common synthetic lethal interactions were reported to be PRMT5, a histone protein methyl transferase, MAT2A, and RIOK1. These intriguing reports suggest that causing MTAP inhibition by the use of MTDIA would render MTAP-competent cancer cells susceptible to agents to block PRMT5 or MAT2A.245–248 PRMT5 agents are now in clinical development.249–251
The primary hypothesis for the action of MTDIA is currently focused on the role of SAM in the epigenetic progression of cancers. As cancers develop, gene expression patterns are altered to fit the development stage and specific tissue environment. This makes cancers more susceptible to inhibitors of epigenetic change (e.g., PRMT5). As protein and DNA methylation are two sources of epigenetic control, altered SAM salvage caused by inhibition of MTA recycling could have a downstream effect on epigenetic regulation. These links are shown schematically (Figure 38). MTA is formed from decarboxylated SAM (AdoMet) in the synthesis of spermine. The addition of MTDIA causes accumulation of MTA in blood and tissues and prevents MTA conversion to ATP and methionine, substrates for MAT2A to form SAM (step A). Loss of the MTA recycling causes decreased SAM concentration and impairs regulatory methylations of proteins and DNA (step B). Tests of these hypotheses have not yet reached proof of concept in published human clinical trials.
Figure 38.

Hypothetical mechanism of action for MTDIA, a transition state analogue inhibitor of MTAP. MeTR1P is 5-methylthioribose 1-phosphate, and CpG-DNA refers to the DNA methylation sites at CpG islands. AdoHcy is adenosyl homocysteine, Hcy is homocysteine, Ado is adenosine, and Met is methionine.
10.4. Crystal Structure of MTAP with Transition State Analogue
The crystal structure of human MTAP has been solved with MT-ImmA and phosphate to form a complex resembling the transition state and also with MTA and inorganic sulfate at the catalytic sites to resemble the Michaelis complex (Figure 39).238,252 A comparison provides a structural basis to explain the tight binding of transition state analogues. Human MTAP is a homotrimer with catalytic sites at each of the three subunits located near the trimer interfaces. Crystals have a monomer (Figure 39A) in the asymmetric unit with extensive subunit interfaces burying 2500 Å2 from each monomer. The subunit interface is primarily hydrophobic interactions but also includes 12 hydrogen bonds and 2 ion pairs. Each catalytic site is formed mainly from a single subunit contacts but also includes participation from the neighboring subunit, with His137 and Leu279 interactions (Figure 40). The catalytic sites are fully occupied by MT-Imm-A and phosphate, where the nearest phosphate oxygen is 3.0 Å from the cationic imino nitrogen and 3.3 Å from the C1′, the site of the developing electrophilic center in the reaction.
Figure 39.

Human MTAP stereoviews of monomer (A) with bound MT-ImmA and phosphate (red) and trimer (B), and electron density omit map for bound MT-ImmA, phosphate, and two ordered water molecules (C). Reproduced from ref 238. Copyright 2004 American Chemical Society.
Figure 40.

Contacts for MTAP·MTA·SO4 (A) compared to MTAP·MT-ImmA·PO4 (B). Significant changes are in bold for the MTAP·MT-ImmA· PO4 structure. L279B and H137B are from the neighbor subunit and participate in van der Waals interactions. Reproduced from ref 238. Copyright 2004 American Chemical Society.
Transition state analogues, including MT-ImmA, bind at enzymatic catalytic sites and are assumed to stabilize a geometry advanced from the Michaelis complex toward the transition state. Enzyme–inhibitor contacts leading to the transition state stabilize more favorable protein conformational geometries. Conformational changes by transition state analogues bring groups into position to form the transition state but in a stable rather than transient interaction.253 A comparison of MT-ImmA and phosphate with MTA and sulfate supports this idea, as there is only 0.3 Å RMSD difference between the structures at the Cα atoms. However, the close contacts comparing the MT-ImmA and MTA complexes are significantly different (Figure 40). Most important, the distance between the ribocation mimic and the attacking nucleophilic oxygen is 3.3 Å in the transition state complex but 4.2 Å in the Michaelis complex. The cation of the iminoribitol forms an ion pair with phosphate, pulling the MT-ImmA deeper into the catalytic site, where phosphate is held tightly by eight ionic or hydrogen bonds of 2.9 Å or less. Sulfate in the Michaelis complex is held less tightly by six ionic or hydrogen bonds of 2.9 Å or less, and this complex is lacking the ion pair interaction, as MTA is uncharged. Despite being pulled deeper into the catalytic site, presumably by the phosphate-iminoribitol ion pair, the leaving group contacts to D220 and D222 are more favorable for the transition state complex than the Michaelis complex. Thus, contacts improve in multiple dimensions surrounding the transition state complex.
The ion pair with phosphate is most favorable when pClPhT-DADMe-ImmA is at the catalytic site (Figure 41). The phosphate oxygen forms a 2.8 Å ion pair with the 1′-aza group of the riboction mimic and is likely to be a primary force for binding this 10 pM inhibitor.254
Figure 41.

Stereoview of the MTAP catalytic site in complex with pClPhT-DADMe-ImmA and phosphate. Active site residues and those from the adjacent subunit are colored in yellow and cyan, respectively. p-Cl-PhT-DADMe-ImmA and phosphate are colored in gray and orange/red, respectively. Hydrogen bonds (<3.1 Å) are indicated as dashed lines. Reproduced from ref 254. Copyright 2011 American Chemical Society.
10.5. Thermodynamics of MTAP Transition State Analogues
Thermodynamic forces driving ligand binding to the catalytic sites of human MTAP in the presence of phosphate have been explored by direct isothermal titrations.254,255 The subunits demonstrated equivalent and independent ligand binding. Despite the multiple hydrogen bonds and the ion pair at the catalytic sites (Figure 40) for transition state analogues, thermodynamic analysis revealed wholly entropy-driven interactions and even demonstrated small enthalpic binding penalties for the most tightly bound analogues (Figure 42). The product adenine was also compared as a ligand in this analysis because human MTAP copurifies with bound adenine. Crystal structures of apo MTAP and MTAP in complex with pClPhT-DADMe-ImmA revealed inhibitor-induced tightening of the catalytic site, reorganization at the trimer interfaces, release of water from the active sites and subunit interfaces, and compression of the trimeric structure.254 These structural changes drive the entropy-favored binding of transition state analogues. Entro-py-driven inhibitor binding is usually dominated by hydro-phobic inhibitor groups interacting with like areas on the target protein. This is not the case for human MTAP, where only the 5′-thiol substituent group can be characterized as a hydrophobic interaction. Transition state analogue binding reflects local and global protein architecture. Enthalpic interactions at the catalytic site are dominated by entropic reorganization of the homotrimer.
Figure 42.
Thermodynamic signatures for the binding of adenine and transition state analogues to human MTAP·PO4. Reproduced from ref 254. Copyright 2011 American Chemical Society.
These unusual entropic interactions were examined by protein dynamics and heat capacity changes (ΔCp) for binding of transition state analogues and a kinetic analysis of transition state formation (Figure 43).255 Temperature dependence of inhibitor binding and transition state formation by single-turnover stopped-flow kinetics permitted comparison of the heat capacity for the chemical step with various transition state analogues. In this way, the transition state analogue most closely related to the actual transition state can be correlated. The ΔCp of inhibitor binding was negative, variable, and contrary to that expected from the hydrophobic effect.256–258 Thus, inhibitors with increasing hydrophobicity bound with increasing values of ΔCp. Crystal structures of MTAP with different transition state analogues or a substrate analogue, MT-tubercidin, revealed similar overall protein structural parameters, despite large differences in ΔCp for binding. ΔCp values were not correlated with Kd values. Presteady state, stopped flow kinetics revealed the chemical step for the MTAP reaction to also have a negative heat capacity for transition state formation (ΔCp‡ = −558 cal/ mol/K). This value compared most closely to the ΔCp values of −510 for MT-DADMe-ImmA and −580 for MT-ImmA, the compounds that most closely resemble the transition state for the phosphorolysis of MTA. Molecular dynamics simulations of MTAP with MT-DADMe-ImmA showed an increased dynamic motion in the inhibited complex. MTAP-inhibitor complexes demonstrate unusual protein thermal stability for a multimeric protein with a Tm = 99 °C in complex with MT-DADMe-ImmA and phosphate.
Figure 43.
Heat capacity changes and optimal binding temperatures for MTAP. (A) ΔCp for inhibitor binding. DADMe-ImmH binding to PNP (9 pM) is included as a control. (B) Free energy of inhibitor binding to MTAP as a function of temperature. Optimal binding temperature is the minima. DADMe-ImmH binding to PNP is added for comparison. Reproduced from ref 255. Copyright 2016 American Chemical Society.
The structural studies described above indicated the phosphate-ribocation mimic plays a significant role in stabilizing transition state analogues at the catalytic site. A combination of isothermal titrations with and without phosphate permits construction of a thermodynamic box to quantitate this interaction (Figure 44).254 Phosphate binding is improved 425-fold, from 17 μM to 40 nM, by the binding of MT-DADMe-ImmA. Likewise, the dissociation constant for the transition state analogue is improved by a factor of 440-fold by the binding of phosphate. In the simplest interpretation, the presence of the phosphate anionic anchor for the cationic transition state analogue improves binding by 3.6 kcal/mol. In vivo, the physiologic phosphate concentration is millimolar and is expected to maintain the catalytic sites near saturation at all times.259 Thus, the biologically effective dissociation constant for the action of compounds like MTDIA will be the picomolar dissociation constant for the ternary complex.
Figure 44.
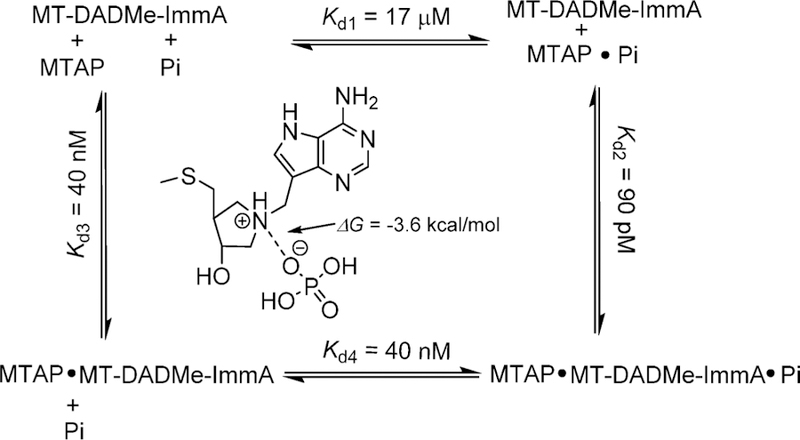
Thermodynamic box for the cooperative binding of phosphate and MT-DADMe-ImmA to human MTAP.254
Despite our deep knowledge of the catalytic mechanism, transition state structure, and inhibitor design for human MTAP, this knowledge has not yet been translated into clinical trials. Animal models of human lung and head and neck cancers support a unique but not yet completely understood mechanism of anticancer action for MTAP inhibitors.240,241 Genetic deletion of MTAP in human cancers causes increased sensitivity to other anticancer agents. Therefore, the combined use of MTAP inhibitors together with inhibitors against targets causing synthetic lethal interactions may provide a new opportunity for multidrug therapy.244–251
11. 5′-METHYLTHIOADENOSINE NUCLEOSIDASES—MTANS
11.1. Biological Roles for Bacterial MTANs
The 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidases (MTANs) are bacterial enzymes with biological roles in (1) the production of quorum-sensing molecules, (2) recycling the MTA from polyamine synthesis to S-adenosylmethionine (SAM), (3) salvaging the adenine and methionine from 5′-deoxyadenosine formed in radical SAM reactions, and (4) forming menaquinone in the few species of bacteria using the futalosine pathway of menaquinone synthesis (Figure 45).260–267 Even with these multiple roles, the scope of MTAN reactions is likely to be underrepresented. Some of the biological functions mentioned above have been revealed only in the past decade. Thus, other functions may remain to be discovered. The chemical reaction is to hydrolytically cleave the N-ribosidic bond between adenine and various 5′-substituted adenosine nucleosides. In bacteria, the MTANs replace the functions of the similar reactions catalyzed by MTAP in mammals. Phosphorolysis reactions by MTAP provide 5-methylthio-α-D-ribose 1-phosphate as a precursor for further conversions, while bacteria express a kinase for 1-phosphoylation of 5-methylthio-α-D-ribose to permit metabolic conversion to methionine.268
Figure 45.

Functions of the bacterial MTANs. Abbreviations: S-adenosylhomocysteine (SAH), 5′-deoxyadenosine (5′-DOA), S-ribosyl homocysteine (SRH), and 5-methylthioribose (MTR). Autoinducer-2 molecules are formed from 4,5-dihydroxy-2,3-pentadione.
Genetic deletion of the MTAN in E. coli (pfs gene) creates strains that are deficient in biotin synthetase and lipoic acid synthase (LipA), reportedly because of accumulation of 5′-deoxyadenosine, a product inhibitior of these reactions.269–271 MTAN deletion also prevents autoinducer-2 (AI-2) synthesis as indicated by AI-2-specific luminescence in the quorum-sensing Vibrio harveyi reporter strain BB170.272 There are reports that inhibition of one or more of the three MTANs expressed in Borrelia burgdorferi (causative agent of Lyme disease) exhibits antibotic action, although the mechanism is not yet established.273,274 Bacterial AI-2 production occurs in Gramnegative and Gram-positive bacteria and has potential as a novel antibacterial target.275–277 Biofilms and toxin production by pathogenic bacteria are under control of autoinducers. As autoinducer pathways are cell-to-cell signaling mechanisms, they control bacterial gene expression but are not essential for growth.278–280 The nonessential target property leads to the possibility that inhibitors of quorum-sensing pathways might provide a decrease in bacterial pathogenecity without inducing resistance, since no selective pressure is placed on the organisms by blocking quorum sensing.
11.2. Early and Late Transition States in the Bacterial MTANs
In the search for inhibitors of quorum sensing, transition state structures were solved for the MTANs from E. coli, Neisseria meningitides, S. pneumoniae, and Mycobacterium tuberculosis.281–284 These transition states provided information for design and synthesis of transition state analogues. Conversely, the specificity of transition state analogues for distinct transition states permitted screening of MTANs from other pathogenic bacteria to deduce transition states and predict additional transition state analogue inhibitors.285 This approach, in addition to the experimental determination of several key kinetic isotope effects for each enzyme, permitted extension of the transition state information to the MTANs from Helicobacter pylori, Staphylococcus aureus, and Klebsiella pneumoniae. Two transition state classes were found. Early transition states, with significant N-ribosyl bond order remaining at the transition states, were found for N. meningitides and H. pylori, while the remaining enzymes had late transition states with fully dissociated leaving groups and a fully developed ribocationic transition state, as exemplified by the transition states for the N. meningitides and E. coli enzymes (Figure 46). Using MT-ImmA as an index for early transition states and MT-DADMe-ImmA as an index for late transition states, other MTANs were classified.285,286 The ratio of Kd values for the two inhibitors is required, as individual enzymes have characteristic affinity for these inhibitors. In every case, MT-DADMe-ImmA binds more tightly than MT-ImmA because of the improved catalytic site contacts mentioned above. However, the ratio of Kd values is diagnostic for MTAN transition state structure (Figure 46).
Figure 46.

(A) Molecular electrostatic potentials of early and late dissociative MTAN transition states compared to MT-ImmA, MT-DADMe-ImmA, early and late transition state analogues. Arrows indicate partial positive charge around C1′ of the E. coli transition state, mimicked at the 1′-pyrolidine nitrogen of MT-DADMe-ImmA and 4′-iminoribitol nitrogen of MT-ImmA. (B) Transition state analogue specificity for early and late transition states. Reproduced from refs 283 and 286. Copyright 2007 American Chemical Society.
11.3. Femtomolar Transition State Analogues for E. coli MTAN
Transition state analogues show exceptional binding affinity to E. coli MTAN (Figures 47 and 48).272,285,287 The DADMe-ImmA inhibitor family requires a 2′-deoxy structure for chemical stability to accommodate the adjacent pyrrolidine nitrogen. Lack of the 2′-hydroxyl group combined with the methylene bridge of the DADMe-Immucillin family permits a closer interaction between the inhibitor cation and the polarized water nucleophile at the catalytic site. Transition state analogues designed for MTANs bind most tightly to E. coli MTAN. For exapmle, the MT-DADMe-ImmA molecule (also an inhibitor of human MTAP; Figures 44 and 46), dissociation constants of 2, 13, 89, 140, 784, 1400, and 24 000 pM for E. coli, V. cholerae, H. pylori, N. meningitides, K. pneumoniae, A. aureus, and S. pneumoniae MTANs, respectively.272,285 This range of interactions is remarkable given the similarity of the catalytic sites of these enzymes.
Figure 47.

Transition state analogues for E. coli MTAN. These are all slow-onset tight-binding inhibitors as indicated by the Ki* designation. Values are for the equilibrium binding constant following slow-onset inhibition. These 5′-substituted-DADMe-ImmA molecules are late transition state analogues to mimic the late, dissociative transition state of E. coli MTAN (Figure 46).287 Selected Ki values are compared for V. cholerae and H. pylori isozymes in Figure 49.
Figure 48.

Molecular electrostatic potential surfaces (MEPs) for (a) the transition state of E. coli MTAN, (b) MT-ImmA, (c) MT-DADMe-ImmA, and (d) pClPhT-DADMe-ImmA. MEPs were calculated in Gaussian98/cube for the optimized geometry and visualized with Molekel 4.0 at a density of 0.008 electron/b. Stick models have the same geometry as the MEP surfaces. Values of Kd are dissociation constants for the inhibitors following slow-onset inhibition or Ki* in slow-onset analysis. Reproduced from ref 281. Copyright 2005 American Chemical Society.
The dissociation constant of 47 fM is the best slow-onset, tight-binding inhibitor for this enzyme and is among the tightest binding for any enzyme–inhibitor interaction (Figure 49). For example, the assembly of click chemistry inhibitory fragments by catalytic site-directed assembly of tarcine and phenanthridinium adducts at the active site of Electophorus acetylcholinesterase gave an azide–alkyne click chemistry adduct with a Kd of 77 fM for T. californica acetylcholinesterase and weaker for acetylcho-linesterases from other species.288,289 Despite the high affinity of catalytic site-directed chemistry for acetylcholinesterases, these compounds bind by virture of large van der Waals interactions in deep catalytic site channels rather than by incorporating transition state features. Even though the MTA substrate for E. coli MTAN has a high apparent affinity, with a Km value of 0.43 μM, the Km/Ki* ratio for pClPhT-DADMe-ImmA is 9.1 × 106. The predicted Km/Ki* ratio for a perfect analogue of purine N-riboside hydrolases is approximately 1012. Therefore, the best inhibitor remains 5 orders of magnitude short of perfection.37
Figure 49.

Examples of transition state analogue affinity for E. coli, V. cholera, and H. pylori MTANs. These are slow-onset tight-binding inhibitors. Values are for the equilibrium binding dissociation constants following slow-onset inhibition.285 nd = not reported.
11.4. MTAN Isozyme Specificity for Transition State Analogues
Crystal stuctures of three inhibitors of different affinity for the E. coli MTAN are instructive.290–292 In all structures the water nucleophile is in position beneath the ribosyl ring in approximate position for nucleophilic addition once the ribocation has been formed. With 5′-methylthiotubercidin, a substrate analogue (0.75 μM Kd), the water nucleophile is 3.5–3.7 Å from the 4′-O or the ribosyl ring.291 With MT-ImmA (Figure.37), a 77 pM transition state analogue, the water nucleophile is 3.0–3.2 Å from the 4′-O, and with MT-DADMe-ImmA (2 pM Kd) the distance to the 1′-N cation of the pyrrolidine ring is 2.5–2.7 Å. This difference in the structures is sufficient to explain the relative inhibitor affinity on the E. coli MTAN. Comparison of the molecular electrostatic potential surfaces for the transition state and three transition state analogues spanning dissociation constants from 77 pM (MT-ImmA) to 47 fM (pClPhT-DADMe-ImmA) does not provide an obvious explanation for the 1600-fold difference in affinity (Figure 48). However, thermodynamic analysis for the binding of transition state analogue inhibitors to the E. coli and other bacterial MTANs reveals transition state analogues bind to the bacterial MTANs with highly favorable enthalpic and, in most cases, also favorable entropic interactions (Figure 50).292 Additional studies were designed to explore protein dynamic interactions as possible contributors to the distince entropic signatures for bacterial MTANs.
Figure 50.

Thermodynamic profiles of ΔH, –TΔS, and ΔG for the binding of first-and second-generation inhibitors to the first active site of E. coli MTAN, S. enterica MTAN, and V. cholera MTAN. Reproduced from ref 292. Copyright 2012 American Chemical Society.
11.5. MTAN Protein Dynamics and Inhibitor Binding
The difference in inhibitor binding affinity for BuT-DADMe-ImmA by the E. coli and V. cholerae enzymes has been investigated by computational protein dynamics.293 High-resolution crystal structures of the MTAN–transition state analogues were used as the starting points for dynamic analysis. Intuitively, tight-binding inhibitors are assumed to cause a more condensed state of the enzyme–inhibitor complex, and indeed, such complexes have been reported for several enzymes.294–299 In most cases, inhibitors bind with some degree of enthalpy– entropy compensation, where multiple hydrogen and/or ionic bonds at the catalytic sites cause strong favorable enthalpic contributions. However, these more ordered, compressed complexes pay an entropic penalty. This is not the case for the bacterial MTANs, where entropy and enthalpy are both favorable for most of these interactions, resulting in large, negative ΔG values of binding (Figure 50). Unexpectedly, a comparison of the RMSF for atoms of BuT-DADMe-ImmA bound at the catalytic sites of E. coli and V. cholerae indicate that the more tightly bound inhibitor at the catalytic site of the E. coli MTAN has more dynamic flexibility than that of the V. cholerae MTAN, where entropic factors are greater for V. cholerae inhibitor binding (Figure 50). Increased dynamic motion is apparent for Glu174, the carboxyl group that activates the nucleophilic water molecule. In E. coli MTAN, Glu174 is 18% more flexible than its equivalent (Glu175) in V. cholerae MTAN. Increased dynamic motion in inhibitor binding is most prominent in the hydrophobic 5′-butylthio group, and this is reflected in increased flexibility of the enzyme surrounding these inhibitor atoms (Figure 51). Thus, inhibitor atoms show more flexibility when bound to E. coli than to V. cholerae MTANs. All atoms of BuT-DADMe-ImmA are more flexible in the EcMTAN active site, providing a dynamic explanation for both entropic and enthalpic contributions to tighter inhibitor binding to the E. coli enzyme.
Figure 51.
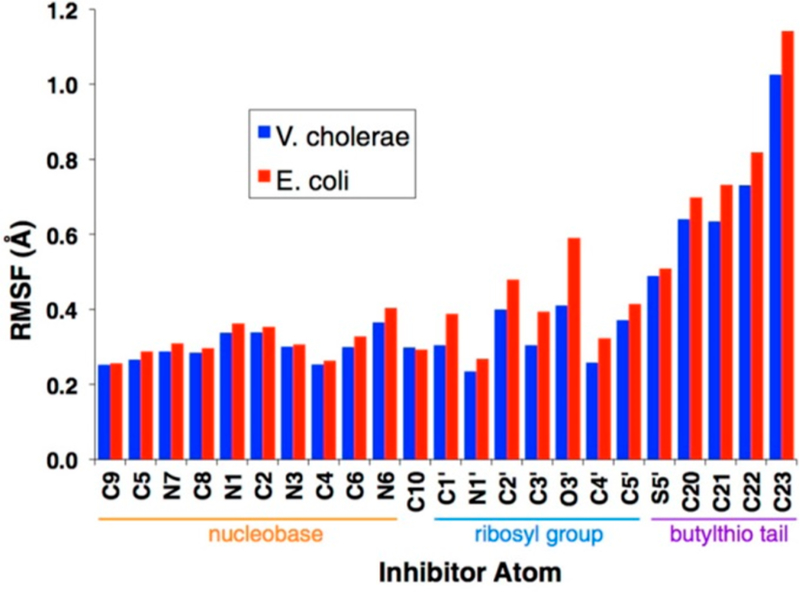
Root-mean-square fluctuation (RMSF) of individual BuT-DADMe-ImmA heavy atoms when occupying the E. coli MTAN active site (red) compared with the V. cholerae MTAN active site (blue). Reproduced from ref 293. Copyright 2013 American Chemical Society.
11.6. Crystal Structure Contacts for MTAN Transition State Analogues
The dynamic explanation for the tight binding of transition state analogues adds insight beyond the structural explanations derived from X-ray crystallography. In 10 distinct enzyme–inhibitor distances for the E. coli and V. cholerae complexes bound to DADMe inhibitors, none vary by more than 0.2 Å (Figure 52). For these MTANs, the purine leaving group interactions include four hydrogen bonds to N1, N6, and N7 from Asp197 or 198 and from backbone atoms of Ile152/ Val153. The nucleophilic water is remarkably stabilized, with five hydrogen-bond interactions with Glu174/175, Glu12 (both MTANs), Arg193/194, and the pyrrolidine nitrogen of the inhibitor. As there is no significant contact difference to the inhibitor and only 0.4 Å RMSD for the Cα of amino acids surrounding the catalytic site, the difference in inhibitor binding affinity appears to have its origin in the dynamic protein components discussed above.
Figure 52.

Inhibitor–enzyme contacts for V. cholerae and E. coli MTANs with DADMe-Immucillin transition state analogues. Data are taken from the Protein Data Bank (PDB) entries 3DP9 and 1Y6Q, respectively. All distances (Angstroms) are between heavy (non-hydrogen) atoms. Backbone RMSD of these active site amino acids from crystal structures is 0.4 Å. Distances in black are from structure of E. coli MTAN in complex with MT-DADMe-ImmA. Those in blue are from the structure of V. cholerae MTAN in complex with BuT-DADMe-ImmA.293
The enzyme-stabilized nucleophilic water in E. coli methyl-thioadenosine nucleosidase is observed both in transition state analysis and in crystallography. Mass spectrometry (MS) experiments were used to determine if the water molecule survives the vacuum of MS analysis, i.e., if it is a stable part of the enzyme–transition state analogue complex. MS analysis of the transition state complex of E. coli MTAN·BuT-DADMe-ImmA (Figures 49 and 52) gave the expected mass + 18, indicating a single trapped water molecule flies as part of the transition state ensemble.300 This result resembles Wolfenden’s earlier experiments demonstrating trapped waters with transition state analogues of cytidine deaminase.301,302 The nucleophilic water is a stable part of hydrolytic transition state complexes.
11.7. MTAN Inhibitors Block AI-2 Quorum Sensing
Powerful inhibitors do not necessarily translate into effective biological function. The cellular barrier for drug uptake into Gram negative bacteria prevents many inhibitors from acting.303–305 MTAN inhibitors were developed as agents to block quorum sensing without placing selective pressure on the parent organisms. In this capacity MTAN inhibitors could reduce gene expression in pathways for biofilm and pathogenicity factors and not induce antibiotic resistance.272,306–308 These theories were tested in cultures of V. cholerae in the presence of BuT-DADMe-ImmA (Figure 53). Overnight growth of pathogenic V. cholerae N16961 with increasing concentrations of BuT-DADMe-ImmA inhibited the AI-2 signal to give an IC50 of 1.4 nM.272 The MTAN activity in actively growing bacterial cells treated the same way was inhibited with an IC50 of 6 nM. Inhibitors at a concentration of 1000 nM did not influence the growth of bacteria in overnight cultures. Similar results were obtained with BuT-DADMe-ImmA, MT-DADMe-ImmA, and EtT-DADMe-ImmA (Figure 47). AI-2 production was also inhibited in similar experiments with pathogenic E. coli O157:H7 to give an IC50 of 125 nM with BuT-DADMe-ImmA. Neither the E. coli nor the V. cholerae strains became resistant to the inhibition of AI-2 by MTAN inhibitors, even after 26 generations of growth at high concentrations (1 μM) of BuT-DADMe-ImmA. Biofilm production was reduced 71% in V. cholerae cultures and 18% in E. coli cultures at 1 μM BuT-DADMe-ImmA. Quorum-sensing pathways remain of interest as a drug target, and several candidates as quorum-sensing inhibitors have been reported to be in clinical trials. Some of these agents also act as bacterial antibiotics, but no transition state analogue acting by inhibition on a quorum-sensing pathway has been approved.309–314
Figure 53.

Inhibition of AI-2 quorum sensing in overnight cultures of V. cholerae by increasing concentrations of BuT-DADMe-ImmA. No growth inhibition was observed. Reproduced with permission from ref 272. Copyright 2009 Nature Publishing.
11.8. MTAN in Menaquinone Synthesis
As transition state analogues for MTAN were being developed for quorum-sensing inhibition, a new pathway was discovered for the bacterial synthesis of menaquinones, essential electron transfer agents in many bacterial species (Figure 54).315–319 In the canonical menaquinone synthetic pathway in E. coli and most other bacteria, the MenA–MenG-encoded enzymes convert chorismate to menaquinone via isochorismate.320,321 In Streptomyces coelicolor, H. pylori, and Campylobacter jejuni, chorismate is first converted to aminofutalosine as an intermediate using the enzymes expressed by the Mqn genes. The adenine ring of aminofutalosine is hydrolyzed from aminofutalosine by MqnB for subsequent conversions of the apurinic intermediated toward menaquinone. Genomic analysis of the Mqn pathway annotated MqnB as an MTAN in these species. Unlike the nonessential role of MTANs in quorum sensing and 5′-deoxyadenosine recycling in enteric organisms, the use of an MTAN in menaquinone synthesis in H. pylori predicted it to be essential in this organism. Human gastric ulcers are caused by H. pylori, and the potential essential function of MqnB here but not in enteric organisms suggested the development of an antibiotic specific for H. pylori by targeting MqnB.
Figure 54.

Synthesis of menaquinone (MK) in E. coli and S. coelicolor. SEPHCHC, 2-succinyl-5-enolpyruvyl-6-hydroxy-3-cyclohexene-1-carboxylate; SHCHC, (1R,6R)-2-succinyl-6-hydroxy-2,4-cyclohexadiene-1-carboxylate; DHNA, 1,4-dihydroxy-2-naphthoate. Pathway enzymes: E. coli Men proteins: MenF, isochorismate synthase; MenD, SEPHCHC synthase; MenH, SHCHC synthase; MenC, o-succinylbenzoate (OSB) synthase; MenE, OSB-CoA synthase; MenB, DHNA-CoA synthase; Ydil, DHNA-CoA thioesterase; MenA, DHNA polyprenyltransferase; MenG, demethylmena-quinone/demethylphylloquinone methyltransferase; MqnA (SCO4506), chorismate dehydratase; MqnE (SCO4494), aminofutalosine synthase; MqnB (SCO4327), aminofutalosine hydrolase; MqnC (SCO4550), DHFL cyclase; MqnD (SCO4326), DHNA synthase; MqnX, amino-deoxyfutalosine deaminase. Compounds are depicted in their quinol forms. Conversion of menaquinol to MK is believed to be nonenzymatic. Reproduced with permission from Biocatalytic Potential of Enzymes Involved in the Biosynthesis of Isoprenoid Quinones. Chemcatchem 2018, 9, 124–135. Copyright 2018 Wiley Online Library.
11.9. MTAN Transition State Analogues Are H. pylori-Specific Antibiotics
Expression of the MqnB protein from H. pylori demonstrated aminofutalosine in addition to the MTA and S-adenosyl-homocysteine as substrates.322,323 Transition state analogues of MTANs from other species (described above) revealed BuT-DADMe-ImmA as a 36 pM, slow-onset tight binding inhibitor of H. pylori MTAN, giving a Km/Kd ratio of 22 000.323 BuT-DADMe-ImmA prevented H. pylori growth on 5% horse blood agar to give an IC90 value of approximately 8 ng/mL, corresponding to a chemical concentration of 23 nM. As the Kd is 36 pM for HpMTAN, there may be a barrier to cell entry or it is possible that the inhibitor is acting on a different target. Additional genetic analysis is needed to establish the mechanism of action. Despite these uncertainties, inhibition of cell growth occurs at inhibitor concentrations 1–3 orders of magnitude lower than approved antibiotics for H. pylori infections, consistent with inhibition of HpMTAN as an essential step.323
The catalytic site of the H. pylori enzyme is similar to other MTANs (Figures 52 and 55).323 The adenine product is a common feature of the MTANs, which are capable of accepting a variety of 5′-substituents on adenosine nucleosides. A flexible hydrophobic pocket at the catalytic site accommodates hydrophobic 5′-substituents. The hydrophobic region in H. pylori MTAN is bordered by Leu104, Phe107, Phe208, Met10, and Ile52, where the Phe107 is a group donated from the adjacent subunit. The flexible hydrophobic region with conserved contacts to 9-deazaadenine led to a synthetic chemistry program to improve binding to H. pylori MTAN and generate favorable IC90 values. Alternative chemical scaffolds were explored.324 Two chemical scaffolds provided tight-binding inhibition, and 14 transition state analogues with IC90 values at or below 16 ng/mL were identified (Figure 56). Of these, the dissociation constants for inhibition of H. pylori MTAN varied from 4 to 170 pM. There was not a linear relationship between Kd value and IC90, indicating that cell entry is a significant parameter for these compounds. The highest affinity compounds generally show the most potent inhibition of H. pylori growth. For example, the four best inhibitors have Kd values of 4, 5, 6, and 7 pM. These same four compounds show IC90 values of 8, 6, 8, and 10 ng/mL values, respectively, for H. pylori growth inhibition. Conversely, the four weakest inhibitors listed in Figure 56, with Kd values of 110, 110, 120, and 170 pM, also gave low IC90 values of 9, 16, 16, and 11 ng/mL, suggesting that these may have superior penetration into the bacteria. Cations are more favorable for uptake than neutral or anionic compounds, and the purine base provides a possible transport recognition element.303–305 Distinct chemical scaffolds effective at the inhibition of H. pylori MTAN and inhibition of growth improve chances of in vivo efficacy.
Figure 55.

Catalytic site contacts for H. pylori MTAN in complex with BuT-DADMe-ImmA. Reproduced from ref 323. Copyright 2012 American Chemical Society.
Figure 56.

Transition state analogue inhibitors selected for low IC90 values for growth of H. pylori. Directed chemical library of transition state analogue inhibitors was synthesized and tested for inhibition of H. pylori MTAN and for bacterial growth inhibition. Reproduced from ref 101 with permission. Copyright 2018 American Chemical Society.
11.10. MTAN Interactions with Acyclic Ribocation Mimics
How do the 1-substituted 2-aminopropanol adducts of 9-deazaadenine interact at the catalytic site to be effective at the inhibition of H. pylori MTAN and inhibition of H. pylori growth? The DADMe inhibitors, for example, BuT-DADMe-ImmA (Figure 55), are stabilized by a hydrogen bond between the nucleophilic water and the 3′-hydroxy group, which is also hydrogen bonded to Glu175. In contrast, the hydroxymethyl group in the acyclic inhibitor (where R = CH3(CH2)3; Figure 56) is hydrogen bonded only to Glu175. The MTAN in complex with an acyclic inhibitor has more conformational freedom and consequently is situated differently in the active site (Figure 57). The torsion angle between the 9-deazaadenine and the CH2-NH substituent is −115° in the acyclic inhibitor but −75° in the 3-hydroxypyrrolidine inhibitors.324 The active site entrance (also the 5′-binding site) is hydrophobic (Met10, Ile52, Leu104, Phe107, Pro115, Phe153, Phe208) and spacious to accommodate MTA, SAH, aminofutalosine, and the 5′-substituents found in inhibitors (Figure 56). The hydrophobic nature of the 5′-substituent region favors van der Waals interactions with 5′-alkylthio substituents that reach beyond the cavity to the exterior of the protein, a channel open to the solvent (Figure 57). Substituents like the R = hydroxybutyl group in the 3-hydroxypyrrolidine inhibitors are extended sufficiently to hydrogen bond to an imidazole nitrogen atom of His109 at the protein surface (N···O = 2.69 Å).
Figure 57.

MTAN from H. pylori bound to R = butyl for the 1-substituted 2-aminopropanol adducts of 9-deazaadenine. (Left) Electron density of 1RMSD for the bound ligand. For maps calculated with 2Fobs – Fcalc coefficients. (Right) Surface representation of the MTAN protein surface showing the buried 9-deazaadenine and the hydrophobic channel leading to the solvent. PDB entry 4YO8. Reproduced from ref 324. Copyright 2015 American Chemical Society.
11.11. MTAN Transition State Analogues Compared to Current Antibiotics
How do the transition state analogues of Figure 56 compare to the current therapy for H. pylori infections associated with human ulcers? Current therapy uses a combination of two or three antibiotics, most commonly metronidazole, amoxicillin, clarithromycin, levofloxacin, and tetracycline, in addition to a proton pump inhibitor (Figure 58).325–327 The rapid spread of antibiotic resistance has also led to sequential therapy initiated with dual therapy containing amoxicillin and a proton pump inhibitor followed by a second, triple therapy including clarithromycin, metronidazole, and a proton pump inhibitor.328–331 A recently recommended first-line therapy is a quadruple therapy including clarithromycin, amoxicillin, and metronidazole and including a proton pump inhibitor for 14 days.332
Figure 58.
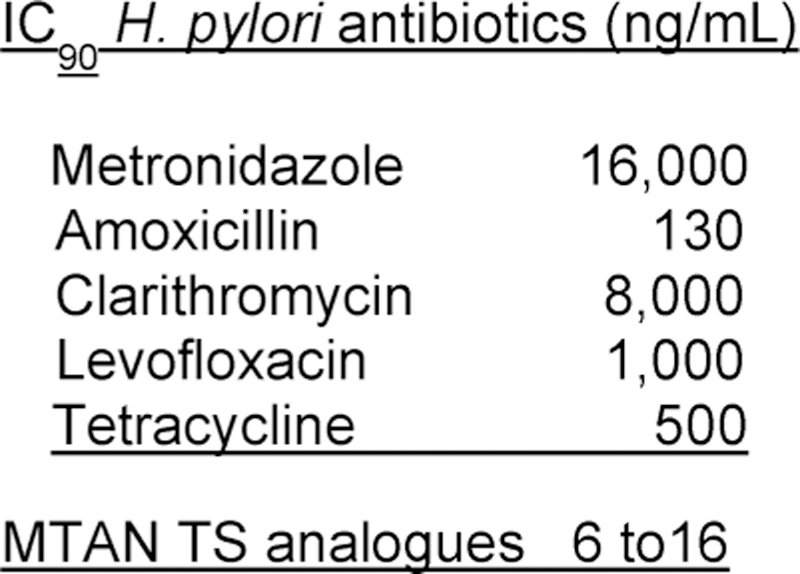
Comparing transition state analogue inhibitors for growth of H. pylori to current antibiotic therapy agents.
A complication of extensive antibiotic therapy is unfavorable alteration of the normal gut flora, leading to repopulation with undesirable organisms, including Clostridium dif f icile. Infections with C. dif f icile are causing an estimated half-million infections per year with an estimated 29 000 deaths in a 2014 report of the CDC.333–335 C. dif f icile is now the most common pathogen causing hospital and care facility infections.336,337 The menaquinone pathway of H. pylori is not present in most organisms of the gut microbiome.315–319 The bacterial species specificity of MTAN inhibitors was tested on E. coli, S. aureus, K. pneumoniae, Shigella flexneri, Slamonella enterica, Psuedomonas aeruginosa, and V. cholerae. None of these organisms showed growth sensitivity to MTAP inhibitors at concentrations causing complete inhibition of H. pylori growth.323 Therefore, the MTAN inhibitors could be useful as species-specific antibiotics, clearing H. pylori infections without causing depletion of the gut microbiome.
12. MAMMALIAN PURINE NUCLEOSIDE PHOSPHORYLASES (PNPS)
12.1. Human PNP Genetic Deficiency and Drug Potential
Interest in purine nucleoside phosphorylase increased following the 1975 report by Eloise Giblett that humans genetically deficient in purine nucleoside phosphorylase (PNP) develop a T-cell immunodeficiency usually apparent by age 2.338–342 Affected infants are normal at birth but gradually develop a loss of T cells, neurological manifestations, and succumb to viral infections. Curative treatment occurs only with hematopoietic stem cell transplantation.343–345 The mechanism of neurological damage is not known, but the loss of T cells is linked to a defect in 2′-deoxyguanosine metabolism.341,342 PNP catalyzes the phosphorolysis of 6-oxypurine nucleosides and 2′-deoxynucleo-sides and is the only enzyme in humans capable of removing the ribosyl group from these compounds to form purine bases. Hypoxanthine and guanine bases can be salvaged to IMP or GMP by hypoxanthine-guanine phosphoribosyltransferase or be further metabolized to uric acid for urinary excretion.346 Genetic deficiency of PNP prevents these reactions, leading to 6-oxynucleosides, including 2′-deoxyguanosine, to accumulate in the blood and to cause a decrease in uric acid levels.340 Activated T-cell clones have active pathways for the transport and phosphorylation of 2′-deoxyguanosine by an induced deoxy-cytidine kinase, normally used to increase deoxynucleoside salvage to support nucleic acid synthesis in T-cell clonal expansion. When excess 2′-deoxyguanosine is present, dGTP accumulates in activated T cells causing apoptotic depletion of these cells with the attendant immune deficiency. Soon after this discovery, PNP became a drug target for the development of new immunosuppressive agents. Over 20 patents from 7 major pharmaceutical efforts were filed or awarded on drug design for human PNP by 1998. None of the agents were successful in recapitulating the T-cell changes observed with genetic deficiency of PNP.347,348 Mutations in human PNP retaining as little as 2% of native PNP activity had reduced disease symptoms.349 Inhibitors capable of greater than 98% continuous inhibition would be needed to regulate T-cell proliferation via PNP inhibition.
12.2. PNP Isozymes Have Distinct KIE Patterns
Transition state analysis for PNP was initiated with the commercially available bovine enzyme, 87% identical in amino acid sequence to the human enzyme and 100% identical in contacts to nucleosides bound at the catalytic site.350–352 There is one amino acid difference in the phosphate binding site.353,354 The intrinsic kinetic isotope effects for bovine PNP were consistent with an early dissociative transition state with partial C1′–N9 bond order remaining at the transition state and significant ribocation character at the ribosyl group.350 There is no significant participation of the nucleophile at the transition state. A large 3H2′ KIE of 1.152 indicated strong hyper-conjugation between the C2′–H and the partial vacant orbital to the leaving group (Figure 59). Using the same experimental protocol, intrinsic KIE analysis for the human PNP gave substantially different isotope effects.355 A diagnostic KIE for this reaction is the value for 14C1′ at the reaction center. Recall that symmetric SN2 reactions have an upper limit of 1.14 for a 14C KIE, while a fully formed ribocation intermediate is predicted to give a KIE of 1.00.356–358 The 1.026 KIE for bovine PNP results from residual bond order between the developing ribocation and the purine leaving group at the transition state. The value of 1.002 for the human enzyme indicates a fully formed ribocation at the transition state in an SN1 reaction. The small 3H2′ β-secondary KIE of 1.031 for human PNP indicated weak hyperconjugation between the C2′–H bond and the partially vacant orbital to the leaving group at the transition state. As this value is large (1.152) in the bovine PNP, the geometry of the ribocations differs at these transition states. Different transition state structures for closely related isozymes with conserved catalytic sites had not been documented before this observation. Additional validation of this observation came from the design of transition state analogues that reflect the differences in these transition states.
Figure 59.
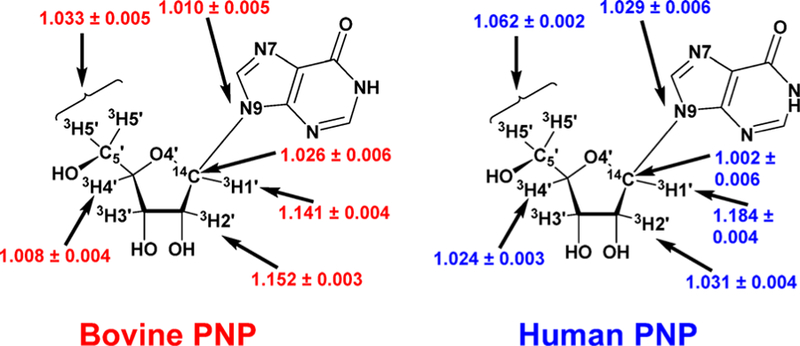
Intrinsic KIE values for the arsenolysis of inosine by bovine and human PNPs. Each KIE shown here was measured in independent experiments with an inosine molecule isotopically labeled in the indicated positions.350,351
12.3. PNP Isozyme-Specific Transition States
The bovine and human PNP transition states corresponding to the intrinsic KIE values (Figure 59) provide information on the bond lengths and electrostatic potential maps (Figure 60). Bovine PNP has approximately 0.3 Pauling bond order remaining in the N-ribosidic bond at the transition state, increasing the bond length to 1.8 Å. The partial loss of the N-ribosidic bond causes protonation at N7 and generates partial cationic character on the ribose. The electrostatic potential maps of the inosine reactant state compared to the bovine PNP transition state reveal the partial positive charge (blue) distributed across the N7-protonated leaving group and the ribocation.359 The nucleophile (arsenate is used to make the reaction irreversible)360 has no significant bond order to C1′ at the transition state, is not included in the electrostatic maps, and is therefore not an essential component in the design of the transition state analogues. The compound proposed to match these features and to confer chemical stability as a transition state analogue was Immucillin-H (Figure 60). The C-nucleoside confers chemical stability and increases the pKa at N7 to favor protonation at neutral pH. Replacement of O4′ with nitrogen gives an iminoribitol with a nitrogen pKa of 6.9, also favoring protonation and generating cationic character at physiological pH values.361 The electrostatic potential match between the bovine PNP transition state and Immucillin-H suggests both geometric and electrostatic similarity, the two features need to convert the catalytic ΔG energy into thermodynamic ΔG energy of transition state analogue binding. ImmH was synthesized and found to be a 23 pM transition state analogue for the bovine PNP and at 56 pM is also a tight-binding inhibitor for human PNP.359 The electrostatic potential map of ImmH assumes it is protonated when bound to PNP, although the pKa of 6.9 does not ensure protonation. Solution and solid-state magic-angle NMR were used to characterize the chemical shifts of [1′–13C,4′–15N]ImmH to compare the ionization states on and off the catalytic sites of human PNP.361 The 4′–15N of ImmH is protonated when bound to the enzyme consistent with a transition state mimic. Also, the sp3 geometry of the 1′–13C of ImmH is distorted toward sp2 geometry when bound to the enzyme. The chemical shift for 1′–1H of ImmH in solution is 4.30 ppm for the neutral and 4.95 ppm for the cationic state. An additional downfield shift to 6.3 ppm occurs in the PNP· [1′–13C,4′–15N]ImmH·Pi complex. Distortion at the anomeric carbon for bound ImmH toward the sp2 geometry of the transition state is evidenced by the corresponding 6 ppm downfield chemical shift observed in [1′–13C]ImmH when bound to the enzyme in solution studies. This result is duplicated in the solid-state NMR spectra of human PNP· [1′–13C,4′–15N]ImmH·Pi. Thus, enforcing full ribocation character and distortion of bound ImmH toward ribocation geometry are features of the transition state deduced both from KIE and from NMR studies.
Figure 60.

PNP reactants, transition state structures, and transition state analogues: (A) Bovine PNP; (B) human PNP.355,359
12.4. Human PNP Transition State Analogues
A chemically stable mimic of the human transition state was designed by using the nitrogen cationic center to replace the C1′ ribocation formed in the fully developed transition state.362 The 3-hydroxypyrrolidine mimic of the ribocation requires the 2′-deoxy structure for chemical stability. The leaving group hypoxanthine is 3.0 Å from the ribocation at the transition state, and this distance was incorporated into the analogue design by a methylene bridge between the 9-deazaadenine leaving group and hydroxypyrrolidine. The electrostatic match between the human PNP transition state and DADMe-ImmH is strong in the ribocationic group, as is the geometry with the hypoxanthine leaving group. An advantage of the DADMe-ImmH structure is in the simplicity of the chemical synthetic approach, where the Mannich reaction can be used under mild conditions, to insert the methylene bridge from formaldehyde between the unprotected 9-deazapurine and the substituted pyrrolidine (Figure 61).362 DADMe-ImmH is a 9 pM transition state analogue of human PNP and a 110 pM inhibitor for the bovine PNP. On the basis of the affinity of these inhibitors for their cognate isozymes, detailed knowledge of the transition state structures provides sufficient information to design inhibitor specificity into the Immucillin transition state analogues.
Figure 61.
Synthesis of the DADMe-Immucillins by the Mannich reaction.362 Conditions: (a) 30% aqueous formaldehyde, NaOAc, H2O, 95 °C. R1 = OH, SBn, SPhpCl, or OAc. R2 = OH or OAc. R3 = OH, NH2, Cl, or N3. R4 = H or NH2.
12.5. Bovine PNP Structures with Transition State Analogues
Crystal structures explored the interaction of substrate analogues (sulfate and inosine) and transition state analogues ImmH and ImmG with phosphate at the catalytic sites of PNPs. How is it that the modest atomic substitutions between inosine and Imm-H increase binding affinity by a factor of nearly 106? The structures compared catalytic site contacts for Michaelis and transition state interactions to explain the nature of the tight-binding interactions (Figure 62). Inosine and guanosine Michaelis complexes with PNP in the presence of PO4 gave Km values of 17 and 13 μM, respectively. ImmH and ImmG are the transition state analogues for each of these substrates (Figure 63). ImmH and ImmG gave equilibrium dissociation constants (Ki*) of 23 and 30 pM to yield Km/Ki* ratios of 7.4 × 105 and 4.3 × 105, respectively, corresponding to increases of –8.1 and −7.8 kcal/mol in binding energy. The structural basis for the increased binding affinity of the Immucillins involves seven or more new hydrogen or ionic bonds forming in the complexes with ImmH and ImmG when compared to the Michaelis complex (Figure 62).363 Specific examples include a leaving group interaction where the Asn243 to inosine N7 decreases from 3.3 Å in the Michaelis complex to 2.8 Å with ImmH. A new ion pair is formed between the cationic iminoribitol and the SO4 or PO4 nucleophiles, where the nucleophile to (neutral) O4′ distance is 3.6 Å with inosine and the nucleophile to N4′ cation is 2.8 Å with the Immucillins. Increased transition state analogue binding energy can be fully explained by an energetic binding gain of only 1.1–1.2 kcal/mol per new ion pair or H-bond interaction. The energetic contributions can be tested for ImmH by systematic alteration of groups on the Immucillin molecules involved in these interactions (Figure 63).
Figure 62.

Catalytic site contacts in the complexes of bovine PNP–inosine–SO4, PNP–ImmH–PO4, and PNP–ImmG-PO4. Distances (in Angstroms) in panels a, b, and c are from Protein Data Base files 1A9S, 1B8O, and 1B8N, respectively. Interactions shown in b and c that are 0.2 Å or more closer than those in panel a are shown in red, and those that are 0.2 Å or more distant in panels b and c than in panel a are shown in blue. All contacts of 3.0 Å or closer are shown together with selected contacts > 3.0 Å for important interactions. Reproduced from ref 122. Copyright 2001 American Chemical Society.
Figure 63.

Transition state analogue variants to test the effect of catalytic site contacts in bovine PNP as indicated in Figure 62. Note the difference in units from pM to μM. Compound numbers 1 and 2 are ImmH and ImmG, respectively. Energetic contributions from important interactions are summarized in Figure 64. Reproduced from ref 364. Copyright 2002 American Chemical Society.
12.6. Bovine PNP Energetics of Transition State Analogue Binding
The remarkable specificity required to capture transition state binding energy is exemplified by comparing 1 (ImmH) and 24 (Figure 63). The difference between 23 pM and 42 μM (1.8 million-fold) is caused by inversion of N3 and C4, causing a decrease of affinity by a factory of 1.8 × 107 in (ΔG = 10.1 kcal/ mol). There are no catalytic site contacts to N3 in the crystal structures with these analogues; therefore, the affinity difference is generated by the altered protonation state at N7. Weak binding of 24 (Figure 63) is also a reminder that the ribocation mimic is a necessary but not sufficient condition for tight binding of transition state analogues to PNP. The ribocation mimic and N7 protonation are both essential features of the transition state. Trivially, from the 1 and 24 comparison (Figure 63), one could say that the total binding energy of ImmH can be accounted for by the N243 hydrogen bond to N7 of the transition state analogue. More accurately, every contact involved in forming the transition state provides a cooperative binding interaction. Loss of any one of the transition state-forming features causes large losses in binding energy. For example, compound 17 (Figure 63) retains the protonated N7 feature but is missing the N4′ cation. It is a 2 μM inhibitor, a loss of 6.8 kcal/mol compared to 1. Each of the catalytic site contacts to ImmH or ImmG that define transition state interactions can be energetically mapped onto the ImmH structure (Figure 64).364 Multiple improved interactions between PNP–substrate and PNP–transition state analogue reflect evolved protein architecture to find the transition state geometry as a preferred protein conformation.
Figure 64.

Energetic differences caused by atomic subustitutions in transition state analogues of bovine PNP. Values are energentic differences expressed in ΔG, kcal/mol, between 1 (ImmH) and the other compounds from Figure 63, as expressed in the subscripts. Reproduced from ref 364. Copyright 2002 American Chemical Society.
12.7. PNP Reaction Coordinate Motion
The transition state structure of bovine PNP together with crystal structures of substrate, transition state analogues, and products bound at the catalytic sites of bovine PNP led to the conclusion that the reaction coordinate motion is dominated by the migration of the anomeric C1′ of the ribosyl group.365 The hypoxanthine ring, the 5′-hydroxyl group, and the phosphate anion remain relatively fixed at the catalytic site, while the anomeric carbon departs the purine leaving group and migrates 1.8 Å to form the sugar phosphate. First recognized in bovine PNP, this mechanism is called nucleophilic displacement by electrophile migration and has also been recognized in many other electrophilic enzymatic transferase reactions.366–370 In the ribosyltransfer reactions catalyzed by three hypoxanthine-guanine phosphoribosyl transferases and an adenine phosphor-ibosyltransferase the C1′ migration is 2.1 Å, while migration in hen egg-white lysozyme is 1.6–1.8 Å (Figure 65).
Figure 65.
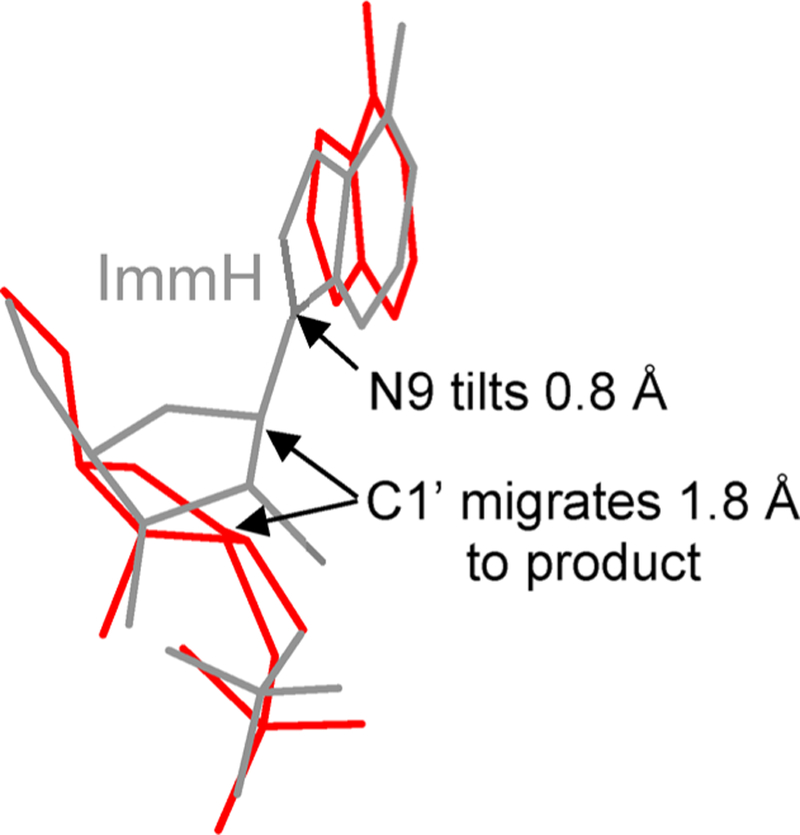
Reaction coordinate motion in bovine PNP based on the crystal structures with ImmH and phosphate bound (gray) compared to enzyme with hypoxanthine and ribose 1-phosphate bound (red).365
12.8. Immucillin-H in Clinical Trials
Immucillin-H was the first molecule to be synthesized as a transition state mimic for bovine PNP but is also an inhibitor of human PNP with a dissociation constant of 56 pM, approximately 3 orders of magnitude more effective as a PNP inhibitor than any of the molecules that had previously entered clinical trials.371–373 A proof-of-principle pharmacokinetic, pharmacodynamic, and clinical study in T-cell malignancies with ImmH (also called BCX-1777 and forodesine) treated patients with an intravenous drug (40 mg/m2) once or twice a day over 5 days. The median drug level (5.4 μM) caused an increased plasma 2′-deoxyguanosine (dGuo) in all patients. Intracellular deoxyguanosine triphosphate (dGTP) increased from 2-to 40-fold in patients and correlated with antileukemia activity. This was the first published clinical study demonstrating the effectiveness of the PNP inhibitor. In this 5-day study there were no objective responses, since leukemia cells in the blood and marrow reappeared after the 5-day therapy.374 Subsequent studies revealed that ImmH has adequate oral availability to permit oral dosing. A phase I study in 13 patients with relapsed or refractory peripheral T/natural killer-cell malignancies and once daily oral ImmH (100, 200, and 300 mg) indicated no dose-limiting toxicities.375 Approximately one-half of the patients showed disease control, and one patient achieved a complete response. Two patients with cutaneous T-cell lymphoma (CTCL) achieved partial responses. This study provided evidence that relapsed or refractory peripheral T/ natural killer-cell malignancies are responsive to PNP inhibition. As inhibition of PNP is a T-cell target, B-cell cancers are less responsive. In a small trial, 8 patients with chronic B-cell lymphocytic leukemia were treated orally with 200 mg/day ImmH. Two had transient responses, and five had disease progression. Cancer cells from these patients showed an insufficient increase in intracellular dGTP (from 6 μM pretreatment to 10 μM post-treatment) relative to dGTP increases in T-cell therapy, which can reach 60-fold.376 A larger study involved 144 heavily pretreated (median 4 prior therapies) cutaneous T-cell lymphoma patients at 200 mg/day oral administration. One-half of the patients had stable disease, 11% achieved partial remission, but there were no complete remissions.377 After a total of 19 clinical trials in various T-and B-cell disorders, ImmH was approved in 2017 in Japan for resistant or relapsed peripheral T-cell lymphoma (PTCL) under the trade name Mundesine.378,379 Oral dosing and low toxicity are features that are expected to provide a convenient alternative therapy for this indication.
12.9. Human PNP Structure with Transition State Analogues
The success of Immucillin-H in human disease, even though it was designed from the transition state of the bovine enzyme, prompted renewed examination of the transition state for the human enzyme. At the time, a difference in transition state structures for isozymes with 87% sequence identity was considered remote as described above (Figures 59 and 60). DADMe-Immucillin-H (DADMe-ImmH) was the first transition state analogue chemically synthesized specifically for the human enzyme. Dissociation constants of 9 pM for human PNP and 110 pM for the bovine enzyme demonstrated the utility of transition state information for the design of transition state analogues. Crystal structures of human PNP with ImmH and DADMe-ImmH, both in complex with SO4 as the nucleophile analogue, revealed that the hydroxypyrrolidine ribocation mimic of DADMe-ImmH is deeper in the catalytic site, permitting it to make a stronger ion pair between the tightly bound SO4 anion and the N1′ cation (Figure 66).380 In the complex with ImmH, the distance is 3.5 Å from the SO4 oxygen to N4′ cation and 3.7 Å to the C1′ position of the anomeric carbon. The 3.7 Å to the C1′ position is the reaction coordinate for the PNP reaction. With DADMe-ImmH bound, the SO4 oxygen to N1′ cation is 3.0 Å, a more powerful ion pair, and consistent with the position of phosphate at the transition state established by KIE analysis. The insertion of the methylene bridge between the 9-deazahypoxanthine and the hydroxypyrrolidine of DADMe-ImmH “elongates” the molecule, allowing more favorable contacts to the nucleophile and the leaving group. The missing 2′-hydroxyl group in DADMe-ImmH, necessary for chemical stability, is replaced by a crystallographic water with contacts similar to that of the 2′-hydroxyl group of ImmH. With DADMe-ImmH, leaving group interactions are stronger to both Asn243 and to Glu201. In general, ImmH is too small in the dimension of the reaction coordinate to make optimal interactions both with the leaving group and with the nucleophile, while the DADMe-ImmH molecule is “just right” in Goldilock terms to fit the catalytic site. These binding interactions reflect the transition state analysis of bovine and human PNPs, where the bovine PNP has a reaction coordinate of 4.8 Å. Human PNP has approximately 6 Å in its reaction coordinate. Insertion of the methylene bridge neatly elongates DADMe-ImmH to span this distance. These procrustean interactions can be explored by solid-state NMR.
Figure 66.

Human PNP catalytic site distances for ImmH and DADMe-ImmH bound at the catalytic sites with SO4 as the nucleophile analogue. Red dashed line highlighted in yellow represents the reaction coordinate distances in the normal reaction. From PDB structures 1RSZ and 3BGS.380
12.10. Distortional Binding of Transition State Analogues by Human PNP
The approach is to synthesize ImmH and DADMe-ImmH with 13C and 15N isotopic labels to provide NMR signals for analysis of the inhibitors free and in complexes with human PNP.361,381,382,385 Distortional forces imposed by the enzyme should be revealed as altered chemical shifts and nuclear interactions on the enzyme. Both molecules carry 13C at the C9 position of the 9-deazahypoxanthine, 13C or 15N at the 1′-position corresponding to the anomeric carbon of inosine, and 15N at the position providing a mimic of the ribocation charge (Figure 67). Rotational echo double resonance (REDOR) experiments are then used for hereronuclear distance measurements, and rotational resonance (R2) experiments were used for homonuclear distance measurements.383,384
Figure 67.
Isotope labels in ImmH (left) and DADMe-ImmH (right) to compare bond distortions at these bonds for inhibitors in solution or bound at the catalytic sites of human PNP. Blue arrows indicate atomic interactions for REDOR distance measurements, and blue brackets are for R2 homonuclear distance measurements. Reproduced with permission from ref 381. Copyright 2013 National Academy of Sciences.
Crystals were made of the labeled ImmH and DADMe-ImmH for analysis without enzyme or were cocrystallized with the enzyme and the crystals subject to magic angle spinning solid-state NMR. Crystallography suggests DADMe-ImmH captures more transition state binding energy by being a closer geometric match to the human PNP transition state than ImmH. These NMR experiments determined if the active site of PNP exerts greater distortional forces on ImmH than on DADMe-ImmH to “achieve” its procrustean transition state geometry. Spectra from the magic angle spinning solid-state NMR of isotope-labeled ImmH and DADMe-ImmH permit precise determinations of internuclear distances. The primary chemical shift data indicated more distortion of ImmH than of DADMe-ImmH at the catalytic sites (Figures 68 and 69). Recoupling techniques, rotational echo double resonance, and rotational resonance established accurate atomistic insight into the geometric changes that occurred upon binding of the Immucillin transition state analogues. Human PNP stretches the C9–C1′ C-nucleoside covalent bond by 0.10 ± 0.05 Å, from 1.47 Å for unbound ImmH to 1.57 Å in the bound complex. This remarkable distortion is approximately 10 kcal/mol distortion energy from the enzymatic interaction. Conversely, the more flexible DADMe-ImmH molecule showed a slight compression of flexible bonds at the catalytic site of human PNP. The C9-methylene bridge carbon was not significantly compressed (0.01 ± 0.1 Å). The distance between the C9 and the N1′ of the pyrrolidine decreased by 0.18 ± 0.06 Å, an indication that no stretching distortion was needed to accommodate DADMe-ImmH into the catalytic site. In solution, the inhibitor cationic nitrogen to C9 distances differ (ImmH = 2.72 Å, DADMe-ImmH = 2.87 Å) but are approximately the same in the PNP complexes (ImmH = 2.71 Å, DADMe-ImmH = 2.69 Å), suggesting an enzymatically favored geometry to resemble the electrostatic nature of the transition state. The energetic distortion of ImmH molecules binding to human PNP by approximately 10 kcal/mol implies that the intrinsic binding force is the observed (from Kd) plus the energy of distortion. With an observed ΔG of binding of −14.1 kcal/mol and ImmH distortional energy of approximately 10 kcal/mol, the intrinsic binding energy is on the order of −24 kcal/mol, a theoretical dissociation constant of 3 × 10−18 M, the same value obtained from the Wolfenden-predicted binding of the transition state for PNP.359 The difference between observed and predicted binding energies causes the mutual distortions of the enzyme–inhibitor complex.
Figure 68.

Magic angle spinning solid-state NMR of labeled ImmH (upper panel) and DADMe-ImmH (lower panel) to compare bond distortions. Peaks at 108.6 and 104 ppm are the 9-13C in bound and free ImmH. Peaks at 62.9 and 54.0 are the 1′–13C label in bound and free ImmH. Peaks at 103.0 ppm and 102.3 ppm are the 9-13C label in bound and free DADMe-ImmH. Peaks at 47.4 and 45.8 ppm are the 13CH2 in diastereomers of free protonated DADMe-ImmH. Pak at 45.0 ppm corresponds to the 13CH2 label in the sole diastereomer present in PNP-bound protonated DADMe-ImmH. Reproduced with permission from ref 381. Copyright 2013 National Academy of Sciences.
Figure 69.
Internuclear distance changes for ImmH (left) and DADMe-ImmH (right) comparing bonds for inhibitors in solution or bound at the catalytic sites of human PNP. Reproduced with permission from ref 381. Copyright 2013 National Academy of Sciences.
12.11. Human PNP Energetics of Transition State Analogue Binding
DADMe-ImmH was the first molecule to be synthesized to mimic the transition state of human PNP. As indicated below, it was also the molecule used in clinical trials for psoriasis and gout. Subsequent syntheses explored chemical specificity and provided mapping of transition state energies (Figures 70 and 71). Energetic properties of DADMe-Immucillin binding to human PNP were compared to the parent compound DADMe-ImmH using a protocol similar to that for bovine PNP (Figures 62 and 63). The 9-deaza guanine derivative DADMe-ImmG binds slightly better than DADMe-ImmH, hence the ΔΔG of −0.5 (Figure 70). All other substitutions caused a reduced binding affinity.385–387 The largest energy loss came from the methylene bridge attachment changed from the C9 to the C8 position of the 9-deazahypoxanthine ring, a change of 7.8 kcal/ mol. Replacing the hydroxypyrrolidine ring with the iminoribitol ring of ImmH (C1′,N4′,2′-OH) misplaces the cation for a loss of 3.1 kcal/mol binding energy. Providing added flexibility in the ribocation mimic by elimination of the 3′–4′ covalent bond of the 3′-hydroxypyrrolidine group decreased binding by 5.4 kcal/ mol. However, other variants of the 3′-hydroxypyrrolidine group, including open ring structures attached to the 9-deazahypoxanthine scaffold, were favorable inhibitors for human PNP (Figure 71). ImmH (56 pM), ImmG (42 pM), DADMe-ImmH (8.5 pM), and DADMe-ImmG (7.0 pM) served as index inhibitions for the synthesis and characterization of inhibitors 1–38 with human PNP (Figure 71). Compounds 8–11 served as slow-onset, tight-binding transition state analogues with dissociation constants of 2.1–8.9 pM, making them comparable to the best of the index inhibitors. Instead of incorporating the iminoribitol and hydroxypyrrolidine mimics of the ribocation, an open-chain nitrogen cationic mimic of the transition state is separated from the 9-deazapurine by one carbon, a distance optimized in both ImmH and DADMe-ImmH. The acyclic Immucillins are characterized by 9-deazahypoxanthine (dHx) or 9-deazaguanine (dG) mimics of the 6-oxypurine leaving group. SerMe-ImmG (8) and SerMe-ImmH (10) are achiral diols formed from the eponymous serinol, while DATMe-ImmG (9) and DATMe-ImmH (11) are chiral triols.387 These compounds include the most powerful inhibitors known for human PNP. Ease of chemical synthesis coupled with low picomolar dissociation constants make these exceptional candidates for eventual clinical applications. Structural and computational studies were used to understand the interactions of acyclic Immucillins at the catalytic sites of human PNP.
Figure 70.

Energetic differences caused by atomic subustitutions in transition state analogues of human PNP. Values are energentic differences, expressed in ΔΔG, kcal/mol, between DADMe-ImmH and related compounds with the atomic substutions indicated by the arrows.
Figure 71.

Dissociation constants for human PNP comparing cyclic with acyclic Immucillins. Compounds marked with an asterisk exhibited slow-onset inhibition kinetics. Values are equilibrium dissociation constants in pM. Source of Ki values. Reproduced from ref 387. Copyright 2009 American Chemical Society.
12.12. Human PNP Structures with Acyclic Ribocation Transition State Analogues
Cocrystallization of the acyclic Immucillins DATMe-ImmH and SerMe-ImmH (11 and 10; Figure 71) with human PNP provided catalytic site contacts to be compared with the structures with ImmH and DADMe-ImmH (1 and 2; Figure 71; see Figure 66). The leaving group interactions are the same as for ImmH and DADMe-ImmH and include bidentate hydrogen bonding from the carbonyl oxygen of Asn243 to NH7 and the imino group with the purine O6 at distances of 2.7–3.1 Å (Figure 72).380 Ribocation formation in the PNP transition state is assisted by hydrogen bonding of His 257 to the 5′-hydroxyl group. This interaction is present for DATMe-ImmH at 2.9 Å but not with SerMe-ImmH where this distance is 3.5 Å. Ion pair formation between the NH2+ ribocation mimic and the nucleophilic oxygen is present at 3.0–3.1 Å, closer than the ion pair in ImmH and equal to that found in DADMe-ImmH. The three ribosyl hydroxyls of ImmH are all hydrogen bonded at the catalytic site. With both DATMe-ImmH and SerMe-ImmH only one or two interact. For both inhibitors, the hydroxyl groups nearest the nucleophile form favorable hydrogen bonds to the oxygens (2.6–2.8 Å). These acyclic Immucillins show remarkable binding affinity considering (1) the His257 interaction is missing in SerMe-ImmH, (2) the third hydroxyl group interactions found in ImmH are missing in both compounds, and (3) that the intrinsic flexibility of these compounds caused by additional rotatable bonds requires an increased entropic penalty on binding. These features were explored in thermodynamic analyses using isothermal calorimetry and computational dynamics.
Figure 72.

Catalytic site contacts between human PNP, DATMe-ImmH, and SerMe-ImmH transition state analogue inhibitors with sulfate or phosphate. Relative distance between inhibitors and the surrounding catalytic site residues is shown in Angstroms. Adapted with permission from ref 380. Copyright 2010 National Academy of Sciences.
12.13. Thermodynamics of Transition State Analogue Binding to Human PNP
The 56 pM binding of ImmH is driven by a large ΔH = –21.2 kcal/mol with a substantial entropic penalty of –TΔS= 7.1 kcal/ mol. DATMe-ImmH binds more tightly at 8.6 pM but with a smaller enthalpic component of only ΔH = −17.5 kcal/mol. The increased binding affinity comes from a large reduction in entropic penalty to −TΔS = 2.3 kcal/mol. The same pattern is seen with SerMe-ImmH, where Kd = 5.2 pM, with ΔH = −20.2 kcal/mol and −TΔS = 4.7 kcal/mol. Chemical flexibility of the acyclic Immucillins requires larger enropic loss on binding than for ImmH, a more rigid compound, but the opposite was observed for the PNP-inhibitor system. Therefore, the altered flexibility is required to come from increased protein dynamic flexibility with these inhibitors bound, thereby imposing a smaller penalty on system entropy.388
Direct evidence for this entropy–enthalpy compensation based on PNP dynamics is provided by H/D exchange experiments and from sedimentation velocity analysis of PNP complexes. The time-dependent peptide backbone amide proton exchange with deuterium was measured for PNP·PO4, the catalytically active, chemically equilibrating Michaelis complexes (PNP·PO4·inosine ↔ PNP·Hx·ribose-1-PO4), and inhibitor complexes PNP·PO4·ImmH and PNP·PO4·DATMe-ImmH. The number of H/D-exchanged peptide bonds and their peptide location was identified by mass spectrometry. Compared to the resting PNP·PO4 complex, 9 peptide bonds were protected from exchange in the equilibrating Michaelis complexes, 13 were protected in PNP·PO4·DATMe-ImmH, and 15 sites per subunit were protected in PNP·PO4·ImmH. The complex undergoing the most tightened, clenched conformation is PNP·PO4·ImmH. The complex with PNP·PO4·DATMe-ImmH permits greater solvent access to peptide bonds but is still tighter than the equilibrating Michaelis complexes under conditions where no free PNP exists. These differences in solvent exchange are reflected in the ultracentrifugation sedimentation velocity of the same complexes. Compared to PNP·PO4, the equilibrating Michaelis complexes, PNP·PO4· DATMe-ImmH and PNP·PO4·ImmH had sedimentation rates 0.68%, 0.95%, and 2.21% faster, respectively. This remarkable hydrodynamic compaction of PNP·PO4·ImmH reflects the large protein dynamic entropic penalty ImmH pays on binding.389 This same property of the PNP·PO4·ImmH complex was seen in the magic angle spinning solid-state NMR studies of the PNP· PO4·ImmH complex, where the enzyme places approximately 10 kcal/mol distortion force on ImmH which also involves reorganization of the protein.381
12.14. Dynamics of Human PNP with Transition State Analogues
Molecular dynamic motions of PNP in complex with transition state analogues were analyzed with the CHARMM27/CMAP force field starting from crystal structures with complexes of various transition state analogues. Complexes with transition state analogues and reactants were compared using 10 ns molecular dynamics simulations with explicit solvent. Comparing the molecular dynamics simulations of the cyclic-carbocation mimic ImmH with acyclic SerMe-ImmH revealed the PNP protein complex of SerMe-Imm-H to be highly flexible. The chemically flexible and rotatable bonds of SerMe-Imm-H allow the inhibitor to flex in concert with the protein environment and to maintain hydrogen bonding during motions of the active sites.390 Tight binding of the picomolar acyclic inhibitors results from increased dynamic motions for interactions within the active sites. Altered numbers of water molecules organized within the different catalytic site complexes may also contribute to the thermodynamic binding properties.
Thermodynamics of PNP interactions with chemically distinct transition state analogue complexes, the hydrogen–deuterium exchange rates, altered sedimentation rates, and molecular dynamic results for human PNP can be interpreted in terms of motions related to the transition state. The PNP geometry needed to form the short-lived transition state is formed only rarely by realizing coincident and optimized distances between catalytic site elements to the leaving group, the ribose, and the phosphate nucleophile.391 When these interactions are simultaneously optimized, the transition state forms. Computational transition path sampling for PNP indicates that the precise geometry required to form the transition state results from local conformational dynamics lasting for a picosecond or less.392–394 Experimental and computational results suggest that the transition state arises from a loose dynamic state rather than a compressed, thermally equilibrated conformation that exists for a significant part of the 5 ms catalytic cycle. Transition path sampling of the PNP reaction indicates a transition state of approximately 10 fs with a reaction coordinate life of approximately 70 fs, occupying less than 10–9 of the catalytic cycle.392
Binding of transition state analogues to human PNP converts the transient dynamic interactions that form the transition state to stable, thermodynamic interactions. The complexes create a stable protein geometry reflecting the transition state. The protein structure selected by catalytic evolution to occur with relatively high probability (among all possible protein conformations) is that promoting transition state formation. The transition state analogue thus becomes favorably bound to a preferred geometry of the protein relative to reactant states. Protein flexibility provides the dynamic catalytic site motions required to search for the transition state. With bound transition state analogues, the enzyme forms longer-lasting hydrogen bonds and ionic interactions that convert the short-lived dynamic excursions of the transition state into long-lived thermodynamic interactions. These structures approximate the transition state. Protein evolution favors formation of the geometric arrangement that promotes barrier crossing. Transition state analogues capture this dynamic state, stabilize it into a thermodynamic state, improve peptide backbone packing within the enzyme, and provide large binding energy.
12.15. Biomedical Significance of PNP Inhibitors
Human PNP deficiency causes metabolic accumulation of 2′-deoxyguanosine (dGuo) in the blood (section 12.8). Activated T cells are the most affected cells because of their ability to accumulate dGuo as excess dGTP. Disruption of DNA synthesis by excess dGTP induces apoptotic cell death specifically in activated T cells. Cellular studies examined a combination of ImmH and dGuo to determine if ImmH recapitulated the human disorder at the cellular level. The combination of ImmH and dGuo was not toxic for unstimulated peripheral T cells, BL-2 or SKW 4.2 B cell leukemia cell lines, or GEO colon carcinoma cell line.395 Activated human T cells, CCRF-CEM, and Molt-4 human T-cell leukemia lines were all sensitive to the combination of ImmH and dGuo. Cell analysis indicated apoptotic cell death. The Jurkat human T-cell leukemia cell line was not sensitive to ImmH and dGuo, attributed to its low level of expression of deoxycytidine kinase (dCK in Figure 73), the enzyme responsible for the conversion of dGuo to dGMP. In the CCRF-CEM susceptible cell line, the dGTP accumulated to approximately 10-fold greater than cellular GTP. A CCRF-CEM AraC-8D cell line deficient in membrane transporters to dGuo is resistant to ImmH + dGuo treatment. In a CEM-SS cell line (T-cell acute lymphoblastic leukemia, T-ALL), incubation with ImmH and dGuo increased the dGTP pool 154-fold, while in nonactivated human lymphocytes the same treatment increased dGTP by 15-fold.396
Figure 73.
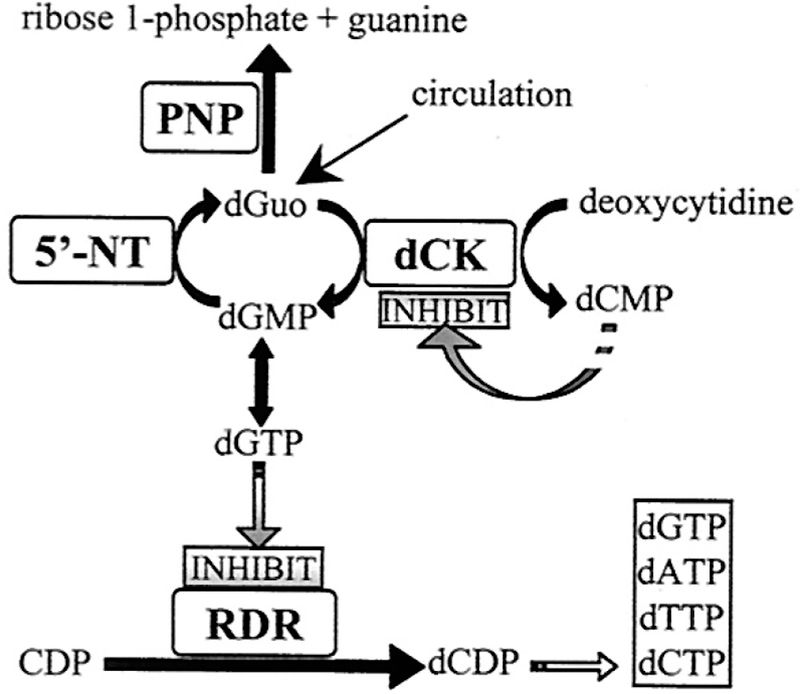
Role of PNP deficiency in dGuo accumulation and T-cell toxicity. DNA recycling generates dGuo which accumulates in the absence of PNP activity. dGuo is phosphorylated in activated T cells by their elevated dCK activity. High levels of dGTP inhibit ribonucleotide diphosphate reductase (RDR). Activated T cells induce dCK and repress 5′-nucleotidases (5′-NT). Reproduced with permission from ref 395. Copyright 2001 National Academy of Sciences.
Pharmacokinetic studies of ImmH (BCX-1777 in animal studies) indicated high oral availability (63%) in mice. In Cynomolgus monkeys, BCX-1777 was absorbed slowly to produce a sustained, low blood concentration of the drug, causing an extended increase in plasma dGuo. Comparing oral and intravenous administration, oral administration of 8.8 mg/ kg was equivalent to 4.4 mg/kg intravenous administration in causing the accumulation of dGuo in the blood. Single oral doses caused a more sustained increase in dGuo, with ∼ 80% of peak dGuo remaining in the blood at 24 h.397 These preclinical studies led to an extended series of human clinical trials, eventually leading to clinical approval for peripheral T-cell lymphoma in Japan as Mundesine (section 12.8). Additional studies examined effects on autoimmune responses and gout as summarized below.379
12.15.1. ImmH in Autoimmune Indications.
Auto-immune disorders involving activated T cells provide another indication for PNP inhibitors.398,399 Self-antigens can be recognized by errant T-cell clones, and their response is directed against human tissues, causing an array of over 70 disorders.400–402 As these are specific T-cell clonal disorders and PNP inhibitors target rapidly dividing, activated T-cell clones, inhibitors of PNP are of interest as T-cell-selective immunosuppressive agents. PNP inhibitors have the potential to remove the activated autoantigenic clone without affecting the unstimulated peripheral T-cell populations.395 ImmH inhibited human lymphocyte proliferation activated by interleukin-2, mixed lymphocyte reaction and phytohemagglutinin. As a PNP inhibitor, ImmH was up to 100-fold more potent than previous PNP inhibitors, Figure 74.395,403 An autoimmune model in mouse is the human peripheral blood lymphocyte test. Human T cells are injected, are activated by the nonhuman environment, and “reject” the SCID-mouse. ImmH prolonged the life span 2-fold or more, equivalent to cyclosporine, an approved immune suppressant.404 The result suggested use of PNP inhibitors as a novel class of selective immunosuppressive agents. Patient reports supporting the use of ImmH (Forodesine in this clinical application) come from treatment of two patients with post-transplant graft-versus-host disease as a consequence of allogeneic hematopoietic stem cell transplantation. Forodesine induced complete remission in both patients. A proposal from this patient observation was that Forodesine contributed to primary antileukemic effects and a secondary immunologic effect, allowing development of ongoing graft-versus-leukemia effect.405,406 This observation is the object of US patent US20170209447A1 “Use of PNP inhibitor to treat relapse of malignancy after hematopoietic stem cell transplant”.407
Figure 74.
SCID mouse–human T-cell autoimmune rejection model. In this trial, ImmH is designated BCX-1777. Reproduced with permission from ref 403. Copyright 2001 Elsevier.
12.15.2. DADMe-ImmH in Psoriasis.
DADMe-ImmH, the first analogue synthesized to mimic the transition state of human PNP, is 6-fold more powerful than ImmH for human PNP; DADMe-ImmH (BCX-4208/R3421 in trials) was entered into a phase 2 study by Hoffmann-La Roche and BioCryst as an oral agent at 20 and 120 mg/day for patients with moderate to severe plaque psoriasis in 2007. This multicenter, randomized, double-blinded, placebo-controlled study ( NCT00504270) was completed in 2009 with no report or additional follow-up.408 No other current development of PNP inhibitors as agents against autoimmune disorders is evident in the NCT database or in the clinical literature.
12.15.3. PNP Inhibitors in Gout.
DADMe-ImmH was repurposed to lower blood uric acid as a gout agent under the names BCX4208 and ulodesine. DADMe-ImmH was found to lower blood urate as a single agent or in combination with allopurinol, a xanthine oxidase inhibitor. The rationale is clear, as PNP is the only enzyme in humans to remove the purine bases from 6-oxypurine nucleosides or 2′-deoxynucleosides. PNP action is required to form purine bases for oxidation to uric acid by xanthine oxidase. Inhibition of PNP causes the increase of urate precursors inosine, 2′-deoxyinosine, guanosine, and 2′-deoxyguanosine in the blood and in the urine to replace urate.409–411 As these nucleosides have higher solubility and do not crystallize under physiological conditions, their presence is preferred to uric acid in gout patients. Once-a-day oral doses of 40, 80, and 120 mg in 60 gout patients reduced serum uric acid in all groups. Inhibition of PNP also increased the circulating dGuo, causing lymphocyte subsets to be decreased by 30–70% depending on ulodesine doses.
Concern about the decreased lymphocytes in the sole-agent study led to a 12-week phase 2 oral study comparing ulodesine doses of 5, 10, 20, and 40 mg/day in combination with allopurinol at 300 mg/day (Figure 75). Compared to allopurinol alone, the combination gave a significantly larger fraction of patients reaching the target urate level below 6 mg/dL serum.412 In a 24 week extension with 160 patients, one-half of the patients receiving ulodesine plus allopurinol achieved urate less than 6 mg/dL compared to 25% of patients in the allopurinol–alone group.413 Placebo and drug adverse effects were similar. With ulodesine doses of 20 and 40 mg/day 15 patient withdrawals occurred with CD4+ < 350 cells/μL. At the end of the test period, the immune response was tested with vaccine standards. Responses were statistically equivalent for ulodesine-treated and placebo subjects. Although four clinical trials have been completed (ClinicalTrials.gov Identifiers: NCT01265264, NCT01407874, NCT01129648, and NCT00985127), there is no literature to indicate that a phase 3 program is underway.414–418
Figure 75.
DADMe-ImmH (called Ulodestin in this trial) lowers serum urate in combination with allopurinol in gout patients. Percent of patients achieving serum urate less than 6.0 mg/dL for Ulodesine and Allopurinol dosing groups.. Reproduced with permission from ref 411. Copyright 2014 Elsevier.
12.15.4. DADMe-ImmH in Vitro vs Human Pharmacokinetics.
DADMe-ImmH binds to PNP with a 9 pM dissociation constant.419 Single-dose oral administration to mice indicated full inhibition of erythrocyte PNP for the life of the cells. Human trial DADMe-ImmH dose-ranging studies also found full inhibition of erythrocyte PNP with a single oral dose, followed by a slow recovery equal to the rate of red cell replacement (120 days).420 Although DADMe-ImmH binding gives kinetic properties consistent with irreversible covalent inactivation in these trials, the inhibitor remains in relatively rapid exchange with PNP in erythrocytes. The mechanism of the target residence time was solved by comparing functional off rates in vitro and in vivo by activity regain and by [14C]DADMe-ImmH exchange rates on the enzyme, in erythrocytes, and in human clinical trials (see Figure 76). The in vitro PNP-DADMe-ImmH dissociation rate (t½) is 8.3 min at 37 °C by catalytic activity regain experiments. Loss of [14C]DADMe-ImmH from erythrocytes during multiple washes is slow and biphasic, a consequence of inhibitor release and rebinding to PNP catalytic sites. The slow phase gave a t½ of 84 h. Loss of [14C]DADMe-ImmH from erythrocytes in the presence of excess unlabeled DADMe-ImmH increased to a t½ of 1.6 h by preventing rebinding. In incubated human erythrocytes, rebinding of DADMe-ImmH is 50-fold more likely than diffusional loss from erythrocytes.
Figure 76.
Single oral administration of DADMe-ImmH (Ulodesine in this trial) rapidly inhibited erythrocyte PNP activity in normal human volunteers. Reproduced with permission from ref 420. Copyright 2017 American Society of Biochemistry and Molecular Biology.
Humans treated with a single oral dose of DADMe-ImmH in phase 1 clinical trials exhibited regain of PNP activity with a t½ of 59 days, corresponding to the erythropoiesis rate in humans. Comparing the intrinsic dissociation rate (t½ = 8.3 min) with the human in vivo regain of PNP activity (t½ = 59 days) requires that each molecule of DADMe-ImmH rebinds > 10 000 times. Catalytic site recapture of DADMe-ImmH by PNP is highly favored in vivo, giving extraordinary biological efficacy. This study quantitated the erythrocyte PNP concentration to be above 1 μM in the erythrocyte. At 37 °C the DADMe-ImmH Kd is 22 pM. Thus, the enzyme concentration exceeds the Kd by a factor of over 45 000. Diffusional escape across the erythrocyte plasma membrane cannot compete with rebinding to the PNP target, even when less than 1% of PNP binding sites are free. Equilibration of the Immucillins across the plasma membrane is known to be rapid, as both equilibrative and ion-linked purine transporters transport the Immucillins.421 Inhibitors with picomolar dissociation constants exhibit long lifetimes on their targets in vivo because the probability of the target enzyme recapturing inhibitor molecules is greater than diffusional loss to the extracellular space (Figure 77).
Figure 77.
Cartoon showing the rebinding (kon) of DADMe-ImmH inside human erythrocytes. As the intrinsic koff (t½) rate is 8.3 min, the on rate is required to be >10 000 more rapid, >20 s−1. Reproduced with permission from ref 420. Copyright 2017 American Society of Biochemistry and Molecular Biology.
13. PURINE SALVAGE IN PROTOZOAN PARASITES
13.1. Structure of Plasmodium PNP
P. falciparum is the protozoan parasite responsible for the most severe forms of human malaria.422,423 Similar to other protozoan parasites, P. falciparum is a purine auxotroph, relying primarily on hypoxanthine as the source for all purine needs of the parasite. In contrast, human cells obtain purines from de novo synthesis and use purine salvage pathways to conserve the need for de novo synthesis. During most of the human infective cycle, P. falciparum lives in erythrocytes where hypoxanthine can be produced from inosine phosphorolysis, in turn produced by dephosphorylation of ATP and adenosine deamination. Human PNP is the only enzyme to form hypoxanthine in erythrocytes. However, P. falciparum also expresses an abundant PNP as a second path for hypoxanthine production. The purine base and nucleoside transporter PfENT1 provides the link between erythrocyte and parasite purine metabolism.424–426 P. falciparum PNP (Pf PNP) differs from the human enzyme. It is a homohexamer with six independent catalytic sites.427–429 In addition to inosine, Pf PNP also uses 5′-methylthioinosine (MTI) as a substrate, formed in P. falciparum by deamination of 5′-methylthioadenosine (MTA). MTI is unknown in normal human metabolism, as human MTAP uses MTA directly and human adenosine deaminase is inactive on MTA.429,430 ImmH is a 56 pM inhibitior of human PNP and a 0.86 nM inhibitor of Pf PNP. ImmH was used to test the theory that human and parasite PNPs are essential in providing the only path to hypoxanthine production in P. falciparum cultured in human erythrocytes. Inosine but not hypoxanthine incorporation into nucleic acids was blocked by ImmH. ImmH prevented P. falciparum growth with an IC50 of 35 nM and was reversed by hypoxanthine but not inosine, establishing the block at the host and parasite PNPs.431 The discovery that MTI is a substrate led to the synthesis and testing of 5′-methylthio-Immucillin-H (MT-ImmH), a 2.7 nM inhibitor for the Pf PNP but a weaker 303 nM inhibitor for human PNP. Structures for ImmH and MT-ImmH with Pf PNP were compared to that from mammalian PNP to demonstrate the differences in mammalian and parasite enzymes (Figure 78).427,429
Figure 78.

Catalytic site contacts for ImmH and SO4 at the catalytic sites of Pf PNP (PDB ID 1NW4) (a) compared with MT-ImmH and SO4 in PfPNP (PDB ID 1Q1G) (b) and ImmH and PO4 in bovine PNP (PDB ID 1B80) (c). Amino acid residues labeled a in panels a and b are from the parent subunit, and those labeled b are from the neighbor subunit across the dimeric interface. Distances are given in Angstroms. Reproduced with permission from ref 427. Copyright 2004 American Society of Biochemistry and Molecular Biology.
13.2. Structure of Plasmodium Adenosine Deaminase
Discovery of MTI as a substrate for PNP in P. falciparum suggested the parasite expression of an MTA deaminase. Adenosine deaminases (ADAs) have been isolated and compared from six Plasmodium species, including P. falciparum.427,432,433 P. falciparum ADAs deaminate adenosine and MTA with approximately equal efficiency, unlike mammalian ADAs which do not use MTA. Coformycins are transition state analogues of the mammalian ADAs and also inhibited P. falciparum ADA with a slow-onset tight-binding Kd of 260 pM. 5′-Methylthiocoformycin (MT-coformycin) was proposed as a way to develop a Plasmodium-specific transition state analogue for P. falciparum ADA.434 The P. falciparum ADA was inhibited by MT-coformycin with a slow-onset tight-binding Kd of 250 pM. In contrast, human ADA showed no significant inhibition by MT-coformycin at 10 μM, a discrimination of greater than 400 000 for the parasite deaminase. The structural basis for MTA and MT-coformycin specificity was solved with the crystal structure of Plasmodium vivax ADA (PvADA).432 The complex with MT-coformycin revealed the expected Zn2+ coordination to be present with both 2′-deoxycoformycin and MT-coformycin but also revealed an unprecedented binding geometry for the 5′-methylthioribosyl group. Compared to Plasmodium ADA complexes with adenosine or 2′-deoxycoformycin, the 5′-methylthioribosyl group of MT-coformycin is rotated 130° with respect to the 5′-ribosyl (Figure 79). The unusual geometry of MT-coformycin is stabilized by a hydrogen-bond network between Asp172, the 3′-hydroxyl, and a structural water molecule that occupies the 5′-hydroxyl binding site when deoxycoformycin is bound. The water found in the MT-coformycin structure is displaced with deoxycoformycin binding. The essential role of Asp172 was demonstrated by site-directed changes deleting Asp172 or mutating Asp172 to Ala or Glu. No catalytic activity is observed in any of these mutants. As anticipated for a transition state analogue, the mutation also destroys MT-coformycin binding.432 Cultured P. falciparum parasites treated with coformycin or MT-coformycin are growth inhibited when MTA is the sole purine source. However, the addition of inosine, hypoxanthine, or MTI restores growth, demonstrating that these compounds are accessible to the parasites and that this MTA deamination pathway is not as essential as the formation of hypoxanthine via PNP, as described above.
Figure 79.

Catalytic site contacts for MT-coformycin (upper panel) and deoxycoformycin (lower panel) at the catalytic sites of PvADA. Relative position of MT-coformycin (PDB entry 3EWC) compared to deoxycoformycin (PDB entry 2PGR) and the active site residues of PvADA. The water molecule is drawn as a dot in the upper panel. Hydrogen bonds and zinc ion interactions are depicted as dashed lines. Distances are given in Angstroms.432
13.3. PNP Inhibitors Are Effective in Primate Infections of P. falciparum
Metabolic production of hypoxanthine in erythrocytes infected with P. falciparum relies on salvage from the purine pool of the host (Figure 80).431,436 Creating a condition of purine starvation for the parasite requires inhibition of human and parasite PNPs. An analysis of DADMe-Immucillin specificity for human and parasite PNPs led to the selection of DADMe-Immucillin-G (DADMe-ImmG; 4, Figure 71), a transition state analogue with Kd values of 7 and 890 pM for human and parasite PNPs, respectively. At near-physiological hypoxanthine concentrations (<10 μM), DADMe-ImmG inhibited growth of drug-sensitive and drug-resistant P. falciparum strains with IC50 values near 150 nM for all strains.435 The efficacy of DADMe-ImmG in blocking hypoxanthine production was demonstrated by direct analysis. Intracellular hypoxanthine in infected erythrocytes was 21 μM without treatment and was below detectable levels (<4 nM) with DADMe-ImmG treatment.
Figure 80.

Purine salvage pathways for formation and utilization of hypoxanthine in human erythrocytes and in P. falciparum parasites. Horizontal arrows indicate transport from red blood cells (RBCs) to the parasite. ADA, PNP, and HGXPRT are adenosine deaminase, purine nucleoside phosphorylase, and hypoxanthine-guanine-xanthine phosphoribosyltransferase. Structures of the human and Plasmodium PNPs are indicated.435
The efficacy of DADMe-ImmG in P. falciparum cultures led to testing in active infections in Aotus lemurinus lemurinus monkeys. Only primates are susceptible to P. falciparum infections, and P. falciparum (FVO strain) is lethal in Aotus if untreated. Oral treatment with DADMe-ImmG, 50 mg kg−1, twice a day for 7 days cleared active P. falciparum infections between the fourth and the seventh day of treatment (Figure 81).435 Aotus blood remained parasite negative for up to 9 days post-treatment. However, the treated monkeys showed regrowth of parasites after treatment was terminated but at a lower rate of growth. DADMe-ImmG showed no drug-associated toxicity in Aotus. Aotus provides a challenging model for inducing hypoxanthine starvation. Humans have normal plasma hypoxanthine of 2.7 μM, but in Aotus, the normal erythrocyte hypoxanthine and inosine concentrations are 64 and 3 μM, respectively.435,437,438 The administration of DADMe-ImmG as a PNP inhibitor
Figure 81.
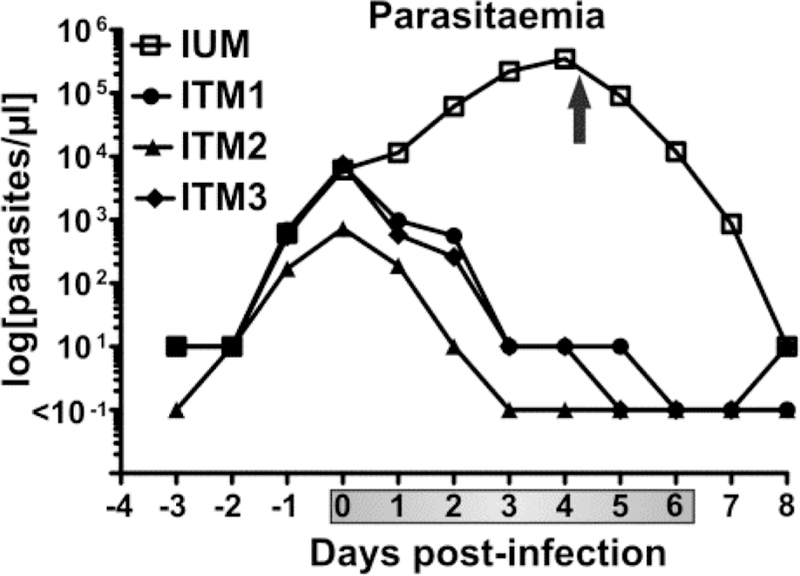
Blood P. falciparum counts in infected control Aotus (IUM, n = 1) or infected treated monkeys (ITM, n = 3). Arrow indicates mefloquine cure of the Aotus control. Shaded bar on the abscissa indicates DADMe-ImmG treatment days. Reproduced with permission from ref 435. Creative Commons 2011 Attribution License. during Aotus infections caused the erythrocyte hypoxanthine to decrease to undetectable levels (<0.3 μM). Inhibition at PNP caused the blood inosine to increase to 50 μM, demonstrating the metabolic block at PNP. It is anticipated that hypoxanthine starvation therapy would be more successful in humans, but to date, there are no reports of this therapy in clinical trials.
13.4. P. falciparum Are Slow to Develop Resistance to DADMe-ImmG
Transition state analogues function by converting the rapidly fluctuating dynamic excursions normally involved in efficiently forming the transition state into stable thermodynamic interactions.253 Mutations against transition state analogue binding are thus mutations away from catalytic function. Transition state analogues used as antibiotics are thus expected to be slower in developing target mutations. When P. falciparum was cultured under incremental DADMe-ImmG drug pressure, drug resistance developed slowly.439 Initial phases of drug resistance were caused by an increase in Pf PNP gene copy number (3–4-fold) and corresponding PNP protein expression. Increased drug pressure caused additional Pf PNP gene copies (up to 14-fold gene copy number), some of which contained point mutations Met183Leu (M183L) or Val181Asp (V181D) at catalytic site residues (Figure 82). Mutant Pf PNPs from resistant clones demonstrated reduced affinity for DADMe-ImmG but also had reduced catalytic efficiency. In native Pf PNP, Glu184 forms bidentate hydrogen bonds with the 2′-and 3′-hydroxyls of the substrate or transition state iminoribitol transition state analogues (Figure 78). Crystal structures of the M183L and V181D mutants demonstrated disrupted catalytic site interactions with the hydroxypyrrolidine group of DADMe-ImmG for V181D. The catalytic defects for V181D (4-fold decrease in kcat/Km; 42 fold increased inhibitor Kd) are overcome by the Pf PNP gene amplification. In contrast, the clone with M183L was nearly inactive (6 × 10–5 of native kcat/ Km), far below viable activity. The M183L clone also had gene amplification with expression of a combination of native and M183L PNPs. It was proposed that subunit hybrids of native and M183L PNPs provide sufficient catalytic activity while decreasing the effect of inhibitor. No test of that hypothesis has been reported. Resistance developed slowly over 136 generations (2136 clonal selection). Thus, transition state analogue inhibitors against Pf PNP are slow to induce resistance. The World Health Organization treatment guidelines recommend multiple drug therapy for treatment of malaria using agents with distinct mechanisms of action.440 No approved antimalarials function by inhibition of purine salvage; thus, PNP inhibitors are anticipated to have favorable characteristics for use in drug combinations.
Figure 82.

Characteristics of P. falciparum resistant to DADMe-ImmG. (A) IC50 values for P. falciparum growth for clones with 5.5–7.2-fold increased IC50 values. (B) IC50 values for P. falciparum growth for clones with 260–980-fold increased IC50 values. (C) Western blot intensity for PfPNP protein in drug-resistant clones. (D) Western blot intensity from highly resistant clones. (E) P. falciparum chromosomes (innermost circle). Peak height is relative to genomic reads of 10 kb contiguous regions. Three control clones (inner tracks) show no amplified regions. Three outer tracks correspond to 2 μM DADMe-ImmG-resistant isolates. Amplified Pf PNP regions are seen in all resistant strains in chr5. (F) Gene amplification in chr5 for three highly–resistant clones. Clones have distinct boundaries for gene amplification, each containing the coding region for the PfPNP gene. Reproduces with permission from ref 439. Copyright 2018 National Academy of Sciences.
13.5. Transition State Analogue for Trichomonas vaginalis PNP
Trichomonas vaginalis is a common human protozoan parasite and the causative agent of trichomoniasis, the most common curable sexually transmitted disease in the Unites States.441–443 Trichomonas vaginalis is unusual among the protozoan parasites in expressing enzymes for adenine and adenosine-based purine salvage pathways.444 Adenine salvage has been proposed to involve a broad-specificity PNP acting to form adenosine coupled to an adenosine kinase to form intracellular AMP. T. vaginalis PNP (TvPNP) is proposed to function in the reverse direction relative to the PNP of the human host as well as an altered purine base specificity. With adenine/adenosine specificity, Immucillin-A (ImmA; Figure 63, 18) and DADMe-Immucillin-A (DADMe-ImmA) were tested as transition state mimics (Figures 83 and 84). They resemble adenosine at early and late transition states with TvPNP. As human PNP does not accept 6-amino purines, ImmA and DADMe-ImmA showed specificity for the T. vaginalis PNP. ImmA was a slow-onset tight-binding inhibitor of TvPNP with an equilibrium dissociation constant of 87 pM.445 TvPNP·ImmA·PO4 inhibitor residence time has a half-life of 17.2 min and a Km/Kd ratio of 70 100. DADMe-ImmA (6-amino analogue of DADMe-ImmH; Figure 71, 2) mimics a late ribocation transition state for TvPNP. It has a dissociation constant of 30 pM, an inhibitor release half-time of 64 min, and a Km/Kd ratio of 203 300. The slightly tighter binding of DADMe-ImmA indicates a late SN1-like transition state. The 6-amino-9-deazapurine group of ImmA and DADMe-ImmA makes them insignificant inhibitors of human PNP. Crystal structures revealed a 2.7 Å ionic interaction between the nucleophilic PO4 oxygen anion and the N1′ cation of the hydroxypyrrolidine. This interaction is weaker in the TvPNP·ImmA·PO4 structure at 3.5 Å (Figure 84). A more favorable interaction in the TvPNP·ImmA·PO4 structure includes hydrogen bonds between the 2′-hydroxyl and the protein that are not present in TvPNP·DADMe-ImmA·PO4 (Figure 83). However, the tighter ion pair is energetically dominant to explain the tighter binding of DADMe-ImmA (Figure 84). Structures with TvPNP reflect the more intimate interactions permitted by the methylene bridge of the DADMe-Immucillins. Subsequent biological experiments with cultured T. vaginalis organisms indicated that TvPNP is not an essential target, possibly due to the direct conversion of adenosine to AMP without the participation of TvPNP. These findings support the earlier work implicating the purine nucleoside kinase (aka adenosine kinase) as a critical enzyme in purine salvage in this organism.444
Figure 83.

Crystal structure of TvPNP with ImmA or DADMe-ImmA and PO4 bound in the active site. The asymmetric unit was used to generate the hexamer by applying 2-fold crystallographic symmetry (upper panel). Monomer pairs are represented with similar colors. (Lower panel) Superposition of the Immucillins at the active sites of the two TvPNP structures. The complex with ImmA is in pink, and the complex with DADMe-ImmA is in cyan. H4* is a catalytic site interaction from the adjacent subunit. Reproduced from ref 445. Copyright 2006 American Chemical Society.
Figure 84.
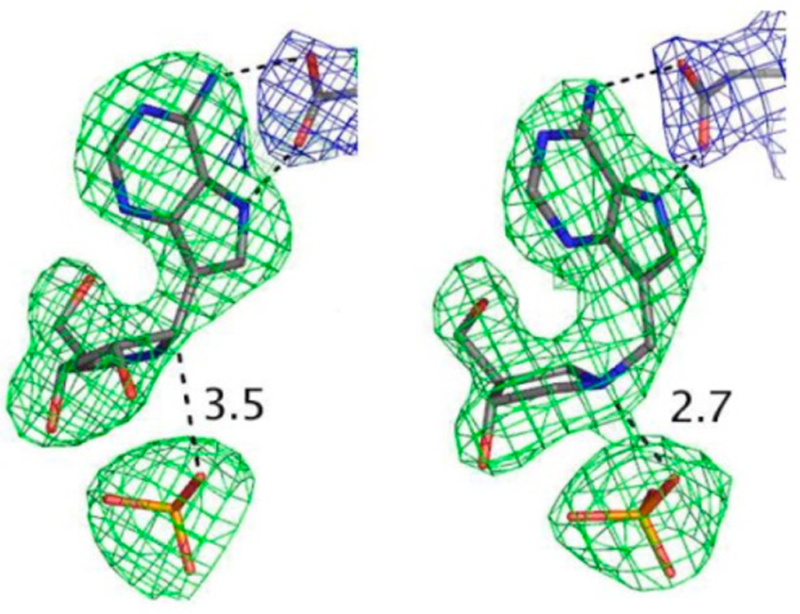
Omit maps for ImmA, DADMe-ImmA, and phosphate (3σ) around the inhibitors (green) and the 2FoFc map contoured at 1σ around the Asp204 (in blue). Distances between the C1′ of ImmA (left) and the N1′ of DADMe-ImmA (right) and the phosphate groups are shown in Angstroms. Reproduced from ref 445. Copyright 2006 American Chemical Society.
13.15.1. Immucillin-A Becomes an Antiviral.
ImmA was included in a small-molecule search for antivirals.446 The compound exhibited antiviral activity in cultured cells against Filoviridae (Marburg, Ebola), Togaviridae (Eastern Equine Encephalitis, Western Equine Encephalitis), Bunyaviridae (Rift Valley Fever, La Crosse), Arenaviridae (Lassa, Junin), Paramyxoviridae (Respiratory Syncytial, Measles), Orthomyxoviridae (Influenze), Picornaviridae (Human rhinovirus 2), and Flavivir-idae (Yellow fever, Japanese encephalitis, Dengue virus) with effective concentration values (EC50) of 3.4–44 μM. The most potent viral inhibition was with the Filoviridae where the EC50 values were 3–12 μM. More recent studies found EC50 values of 1–3 μM for West Nile and Tick-borne encephalitis viruses and 4–12 μM for various strains of Zika virus.447,448 In biosafety level 4 containment animal studies at the United States Army Medical Research Institute of Infectious Diseases (USAM-RIID), postexposure intramuscular administration of ImmA (BCX4430 in preclinical trials) protected against Ebola virus and Marburg virus disease in rodent models.446,449 In nonhuman primate preclinical trials, BCX4430 completely protected cynomolgus macaques from Marburg virus infection when administered as late as 48 h after infection (Figure 85). There were no signs of systemic toxicity or adverse local reactions, supporting additional development as an antiviral.
Figure 85.
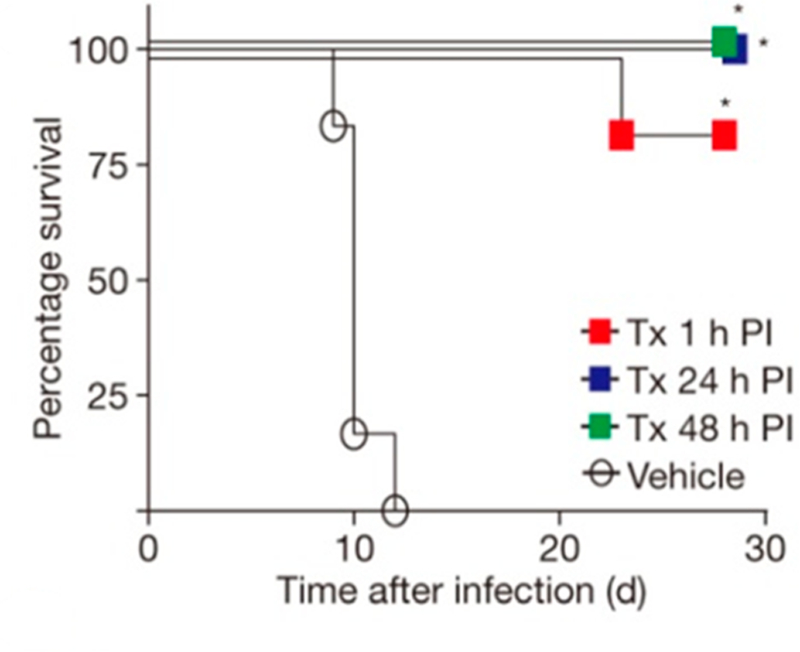
Marburg virus infections in cynomolgus macaques treated with BCX4430 (ImmA; Tx). Animals (n = 6) in each group were infected with virus on day 0. BCX4430 (15 mg/kg) was given twice a day by i.m. injection starting at 1, 24, or 48 h after exposure to the virus. Vehicle indicates no BCX4430 was given. Reproduced with permission from ref 446. Copyright 2014 Nature Publishing.
Although ImmA is a transition state analogue with respect to TvPNP, its antiviral mechanism of action is reported to be as a prodrug for an unusual nucleotide triphosphate chain termination of viral RNA polymerase.446 Human cells phosphorylate ImmA to ImmA 5′-triphosphate with similar efficiency to the conversion of extracellular adenosine to ATP. The ImmA 5′-triphosphate is incorporated into the viral RNA but not the human host cell RNAs. ImmA has both 3′-and 5′-hydroxyl groups and thus would not be considered a traditional chain terminator. Experimental studies with HCV NS5B polymerase demonstrated ImmA incorporation followed by two additional nucleotide additions to the growing RNA with subsequent chain termination. Although the chemical mechanism of this unusual termination has not been reported, it is possible that the cation of ImmA forms an ion pair with one of the adjacent phosphodiester anions to stabilize an RNA conformation that is unproductive for the addition of additional nucleotides.
13.5.2. Phase 1 Clinical Trial for Immucillin-A.
A Phase 1 clinical study to evaluate the safety, tolerability, and pharmacokinetics of BCX4430 (Galidesivir in trials; Clinical-Trials.gov, Identifier: NCT02319772) was initiated in December 2014 by BioCryst Pharmaceuticals with the National Institute of Allergy and Infectious Diseases as a collaborator.450 A total of 94 healthy participants were evaluated for the safety, tolerability, and pharmacokinetics of single, ascending doses of BCX4430 versus placebo in healthy subjects. The latest literature report on the Phase 1 trial was in 2016 and indicated “for single doses of 0.3–10.0 mg/kg, plasma exposure to BCX4430 was dose-related and linear. The first multi-day cohort was evaluated at a dose of 2.5 mg/kg QD for 7 days, and the dose escalation is continuing. To date, BCX4430 injections have been safe and well tolerated.”451 As 5′-phosphorylation of BCX4430 is required to provide the active agent, chemical synthetic efforts have been reported to produce a phosphonate prodrug to improve the delivery to target cells, but these await evaluation for biological efficacy.452
14. HIV-1 PROTEASE TRANSITION STATE MIMICS AS DRUGS
14.1. Brief Background of HIV-1 Protease as an Antiaids Target
A review exemplifying transition state analogues would be incomplete without considering the biological research, inhibitor design, and clinical success story for the application of HIV-1 protease inhibitors. An important chapter of surviving HIV-1 infection is the development of the aspartic protease inhibitors against the essential, viral-encoded HIV-1 protease. This essential enzyme has been called “The triple threat” target for HIV infections because of the multiple steps (at least three) impacted in maturation of the virus when the protease is inhibited.453 Fully functional HIV-1 protease is a homodimer creating a single catalytic site with two essential Asp residues, one from each subunit, whose carboxylate groups are involved in catalysis. Paradoxically, each Gag-Pol poly protein contains a catalytically inactive single half of the protease required for function. Initiation of proteolysis is thought to occur by the interaction of two intact, uncleaved, poly proteins to form active protease in the context of an intact poly protein dimer.454 Immature virus particles containing intact Gag-Pol poly proteins are then processed by the dimer at the nine proteolytic substrate sites to produce fully functional proteins including the reverse transcriptase, RNase H, integrase, and the most active form of the protease. Blocking the protease, either in the Gag-Pol dimeric form or in its fully released form, interferes with all infective processes associated with these proteins, which can only create fully infective viruses after Gag-Pol peptide hydrolysis. Research efforts have led to high-affinity inhibitors with most analogues containing one feature of the transition state, an sp3 center where a substrate carbonyl reaction center has been reduced to the secondary alcohol positioned to occupy the catalytic site where the susceptible peptide bond sp2 carbonyl group is converted at the transition state to the sp3 secondary alcohol in the gem-diol intermediate and in the transition state leading to peptide bond loss (Figure 86).
Figure 86.

Chemical mechanism for HIV-1 protease. Structures are (1) enzyme–substrate complex, (2) water attack TS, (3) tetrahedral gem-diol intermediate, (4) proline N-protonation TS, (5) protonated amide intermediate, (6) cleavage of scissile C–N bond TS, and (7) enzyme–product complex. For transition structure 2, the r(C–O) bond distance is the distance between the oxygen of the attacking water and the carbonyl carbon of the peptide. For transition structure 4, r(N–H) is the bond distance between the nitrogen on the proline and the proton on the catalytic aspartate and r(H–O) is defined as the bond distance between the oxygen and the proton on the catalytic aspartate. Finally, in transition structure 6, r(C–N) is the bond distance of the scissile bond of the peptide. Reproduced with permission from ref 464. Copyright 2012 National Academy of Sciences.
14.2. Clinically Approved HIV-1 Protease Inhibitors
Aspartic proteases constitute a significant drug target group, including HIV-1 protease, renin, BACE1, and an Alzheimer disease target, γ-secretase.455 Humans express 15 aspartic proteases; therefore, drug design approaches for the specific HIV-1 protease target must include specificity motifs at the adjacent amino acid binding sites to direct the inhibitor action toward the target enzyme. The success of the program for development of HIV-1 protease inhibitors has led to the inclusion of HIV-1 inhibitors in every formulation of highly active antiretroviral therapy (HAART). Also included are inhibitors against reverse-transcriptase and/or integrase targets.456 Although the list of approved agents for HIV-1 protease is long and resistance is slow to develop when protease inhibitors are used in combination with agents acting on other targets in HAART therapy, resistance to most HIV-1 protease inhibitors has occurred.457,458 Continued drug design efforts search for substituent groups at N-or C-terminal amino acid side chain binding sites. The current FDA-approved drugs (Figure 87) include an sp3 secondary alcohol at the putative cleavage site surrounded by substituents to occupy adjacent binding sites for amino acids in the normal function of the Gag-Pol peptide.459,460
Figure 87.

Clinically approved inhibitors for HIV-1 protease. Kd values and year of approval are indicated. The transition state center is highlighted. Adapted with permission from ref 459. Copyright 2017 Elsevier Paris.
14.3. Thermodynamics of the HIV-1 Protease Inhibitors
Five HIV-1 protease inhibitors were approved prior to the year 2000 with dissociation constants in the range of 15–2000 pM (Figure 87). Subsequently, four additional drugs were approved with dissociation constants from 1.3 to 16 pM. All of the inhibitors contain the transition state mimic center, namely, the sp3 secondary alcohol at the putative cleavage site, with the exception of Tipranavir, where the alcohol is at an sp2 center. All inhibitors include relatively large hydrophobic surface areas and multiple sites capable of accepting or donating hydrogen bonds. For example, Lopinavir, a 1.3 pM inhibitor, has three freely rotating phenyl rings for hydrophobic interactions, eight sites as potential hydrogen-bond acceptors, and three hydrogen-bond donor groups. Binding thermodynamics indicate that the earlier inhibitors were less driven by enthalpy and more by the entropic hydrophobic interactions than the more recent and more tightly binding analogues.461 Recent information suggests that even more powerful inhibitors can be made to the HIV-1 protease by substituents remote from the site of peptide bond hydrolysis. For example, inhibitors similar to those already approved (Figure 87) but modified to include a boronic acid group have been shown to improve binding affinity to the HIV-1 protease by at least an order of magnitude.462
14.4. Are the HIV-1 Protease Inhibitors Transition State Analogues?
Transition state theory predicts that mutations that decrease the binding of transition state analogues will also reduce catalytic efficiency by a similar degree. The theory has been tested by comparing catalytic site mutants or distinct substrate and transition state analogue pairs for similar changes in chemical potential and binding of transition state analogues.463 This hypothesis was tested by transition state analysis to compare the transition state structures of native and multidrug-resistant HIV-proteases.464 Kinetic isotope effects were measured and interpreted for the chemically rate-controlling step of proton transfer from the active site Asp125 with partial hydrogen-bond formation to the accepting nitrogen and partial bond loss from the carbonyl carbon to the amide leaving group. Near-identical KIEs were observed for native and Ile84Val multidrug-resistant HIV-1 proteases, indicating highly similar transition states. The Ile84Val mutation provides resistance to most approved HIV-1 protease inhibitors. Computational analysis of the cause of the Ile84Val resistance to amprenavir indicated a decreased van der Waals interaction to the hydrophobic ring at the R2 position, not by altered transition state interactions.465,466 The observation that resistance mutations have smaller effects on the reaction rate (transition state formation) than on inhibitor binding indicates that transition state forces may not be the dominant elements in the interactions of HIV-1 protease inhibitors.
15. CONCEPTS AND CONCLUSIONS
15.1. Technology and Theory for Transition State Analysis
In a review covering transition states, mechanism, transition state analogue design, and potential applications, expansion to cover the detailed methods of isotope effect determination and theory would prove (even more) unwieldly. Readers are directed to earlier work that provides comprehensive reviews of methods, theory, practice, and applications to other enzymes. The Sixth Annual Steenbock Symposium in 1976 is recognized as a major event in the application of kinetic isotope effects to enzymatic reactions. The meeting resulted in a thin but valuable monograph combining theory and practice. In addition, it provides a link to the historic development of isotope effect theory.467 W. W. (Mo) Cleland, in addition to his part in organizing the Steenbock Symposium, was an early advocate of using isotope effects to examine enzymatic transition states and to solve mechanistic enzyme problems.468 Practical approaches to measuring small KIEs by competitive methods and statistical analysis of uncertainties and methods to unravel intrinsic KIE values from experimentally observed ones have been reviewed.16–19 The application of KIE principles to glycoside hydrolysis and base-excision repair enzymes overlap with some of the combined experimental and computational work presented here and has also been reviewed.125,469,470 Computational approaches to provide the convergence between KIE measurements and transition state structure is the subject of a recent book chapter and the development of a new suite of computational programs to coordinate isotope effects and bond vibrational properties.471,472
15.2. Transition State Dynamic Concepts
The notion of tight-binding transition state analogues was a well-understood concept, primarily from the work of Wolfenden, long before it developed into a practical approach for the logical design and development of drug candidates. Predictions of Linus Pauling that enzymes evolved to match features of the transition state were prescient, although his proposal that enzymes select out the activated states of reactants from solution did not anticipate the dynamic protein rearrangements needed to bring the Michaelis complex to the transition state. Wolfenden correctly pointed out that enzymes operating near the diffusion limit must, by necessity, function only on ground state reactant molecules. The nature of enzymes in complexes with substrates and transition state analogues is now abundantly informed by structural, spectroscopic, and computational studies comparing Michaelis complexes to those of transition state analogues. Spectral dynamic studies revealed bound reactant molecules exist in multiple, rapidly interchanging energetic states with respect to the bonds in the reaction coordinate.473 The most energetic are converted to the transition state, a protein conformational state with all contacts between enzyme and reactants optimized for the few femtoseconds needed for transition state formation. Because femtosecond motions forming transition states are dictated by local bond vibrational modes, they are mass sensitive, as demonstrated with heavy [2H, 13C, 15N]enzymes.474–478 In the normal reaction coordinate, chemical changes induce release of the products. Transition state analogues take advantage of the protein architecture, evolved to favor the transition state geometry. Transition state analogues convert the favored dynamic conformation of the transition state into a thermodynamically stable state in complex with the analogue. These interactions cause the protein to condense around the transition state analogue, a process well documented by structural approaches.389 Instead of the millisecond reactant and product exchange rates in normal catalysis, transition state analogues are released on the minute to hour time scale, therefore freezing enzyme function to provide extended biological inhibition.420
15.3. Binding of Ribocation Transition State Analogues
Many of the enzymes described here have ribocationic transition states, and a cation defines part of the chemical mimicry of their transition state analogues. In the PNP, MTAP, and HGPRT families, the ribocation mimic is stabilized by an ion pair formed with the anion catalytic site nucleophiles. Without that stabilization, much of the binding energy is lost (e.g., Figure 44). Other ribocation-forming enzymes, for example, the MTANs, ribosome-inactivating proteins, and nucleoside hydro-lases do not have anion nucleophiles to ion pair with transition state analogue cations but contain water molecules as incipient nucleophiles. One might expect that anionic groups from the enzyme would stabilize the ribocation in these enzymes, but that is not the case. Instead, the nearest group to the cation of these transition state analogues is usually the nucleophilic water molecule, often within hydrogen-bond distance of the ribocation. These enzymes often receive assistance in ribocation formation by neighboring-group participation with close O4′– O5′ interactions and hydrogen bonding to the O2′ and O3′ hydroxyls from enzymatic carboxyl groups. Leaving group interactions play a major role in some of the hydrolases, while others receive more activation from formation of the ribosyl cation, exemplified in section 5.3 and Table 1.
15.4. Distinct Transition States from High-Homology Catalytic Sites
Why is it that closely related enzymes form distinct transition states? Human and bovine PNPs are 87% identical overall and are crystallographically indistinguishable in their catalytic site contacts to the purine. One amino acid difference occurs in the contacts to phosphate, but phosphate has no significant bonding to the ribocation at the transition state. Transition state differences were established from intrinsic KIE values and were sufficiently distinct to permit design of species-specific transition state analogues (Figure 60). The ability of near-identical catalytic sites to form distinct transition states has been interpreted in terms of different dynamic excursions at the catalytic sites to form transition state interactions. Differences arise from the altered dynamic architectures of sequences remote from the catalytic sites. Support for this hypothesis came from transition state analysis with a chimeric PNP. Non-conserved amino acids from the bovine enzyme, remote from the catalytic site, were substituted into the human enzyme. The transition state and kinetic properties for the chimeric enzyme differed from either parent.479,480
Another example of distinct catalytic site function from remote interactions comes from the MTANs. E. coli and S. enterica MTANs are 95% identical in amino acid sequence, crystallographically indistinguishable in contacts at the catalytic sites, and catalyze the same reactions. Yet the inhibition with the transition state analogue pClPhT-DADMe-ImmA is 0.04 pM for the E. coli enzyme and 2.7 pM for the S. enterica enzyme, a factor of 67.272 Larger differences occur in other bacterial MTANs, attributed to differences in the dynamic architecture, even in enzymes with near-identical catalytic sites. Large differences in affinity for transition state analogues for closely related MTANs can be attributed to distinct enthalpic contributions of inhibitor binding because of altered dynamic states with distinct but related transition states.293 Creating structurally equivalent catalytic sites from slightly different ones does not create equivalence for transition state interactions. For example, catalytic site contacts to substrates differ by two amino acids between the E. coli and the V. cholerae MTANs, enzymes with 60% identity.481,482 Transition state analogues favor the E. coli enzyme by up to 3 orders of magnitude. Mutation of the catalytic site amino acids (Ala113Pro and Val152Ile) in VcMTAN to match those of EcMTAN resulted in transition state analogue binding even more unlike those of EcMTAN, another indication of the dominant role of remote protein dynamics influencing catalytic site function.
15.5. Applying Transition State Analysis to Drug Design
Every enzyme has one or more transition states, and all enzymes can be explored by KIEs to provide mechanistic and transition state information. However, can we expect to apply transition state drug design to every enzyme? No. Dehydrogenase transition states are defined by the degree to which the hydride (and proton) transfers have occurred between donors and acceptors at their transition states. The role of the enzyme is to align donor–acceptor pairs appropriately and for sufficient time (femtosecond) to support transfer. Understanding these details does not inform design of transition state analogues, as the transition state is defined by the transfer of single atoms. There is not yet a suitable mimic to reflect these single-atom transfers as transition state analogues. Also, although there are inhibitors of dehydrogenases (e.g., oxalate for lactate dehydrogenase) they are not faithful mimics of the transition state.
Phosphotransferases, especially the protein kinases, are a large family of highly desirable drug targets. Their transition states present a challenge similar to the dehydrogenases. Phospho-transferase transition states are defined by the bond orders to the attacking nucleophile, the leaving group, and the extent of distortion in the transferring phosphoryl (PO –) group toward a planar intermediate (SN1, loose transition state) or a compressed SN2 (tight transition state) species.483,483 Transition state analogues are well known for phosphoryl transferases, including AlFx,, BeFx, MgFx, and VOx species.484 While some of these are powerful inhibitors they are nonspecific, toxic, and nondruggable molecules. Presently, we have not been sufficiently clever to incorporate such mimics into stable, druggable molecules, but we have only scratched the surface of the periodic table in transition state analogue design. It is important to note the lack of transition state analogues does not mean the lack of drugs. The pharmaceutical industry is robustly designing and exploiting powerful and occasionally specific inhibitors to target the protein kinases in molecules that capture binding site energy but do not incorporate transition state features.
Astute readers will note that all of the powerful transition state analogues described here arise from enzyme chemistries classified as substitutions at carbon. In the ribosyltransferases, the reaction center is a neutral sp3 carbon in the reactant state and becomes an sp2 planar, cationic reaction center at or near the transition state. The transition state differs from the reactant in both geometry and charge, providing two design principles for stable molecules that mimic the transition state. In the case of the adenosine deaminase family reaction geometries are the opposite, but the same principles apply. The reaction center is sp2 in the reactants, and the enzyme-enforced addition of water creates an sp3 Meisenheimer-like transition state (or possibly an intermediate) with distinct geometry and purine ring ionization states, permitting stable mimics of the transition state to be synthesized. Finally, HIV-1 protease is also substituted at carbon, with the first transition state involving the conversion of the targeted sp2 carbonyl to the sp3 diol from the addition of the aspartic acid-activated water nucleophile. The powerful inhibitors of HIV-1 protease all contain some geometric mimic of this sp3 mimic, although adjacent interactions are also critical in tight-binding interactions.
15.6. Future
Review of a subject as broad as transition states, mechanisms, and analogues requires consideration of a manageable research area, considered retrospectively. Here, the focus has been on targets and transition state analogues informed by experimental and computational isotope effects. The geometric and electrostatic information from transition state analysis has provided design elements as chemical blueprints for synthesis of transition state analogue candidates. Through the ever-evolving power of computational chemistry, at some time in the near future, it is likely that computational analysis alone might be sufficient to provide accurate models of enzymatic transition states. When that happens it will permit a more direct path to the design of these powerful inhibitors.
ACKNOWLEDGMENTS
Richard H. Furneaux, Peter C. Tyler, and Gary B. Evans at the Ferrier Research Institute, Victoria University of Wellington, New Zealand, have been directly involved in the inspired chemical synthesis of the immucillin family described here. Only their long-standing collaboration has brought the design of transition state analogues to the reality of compounds. Generations of Ph.D. students and postdoctoral fellows have contributed to the enzymology, transition state analysis, and inhibitor design. A long-standing collaboration with Steven D. Schwartz, Department of Chemistry, University of Arizona, has provided continued insights into the computational aspects of enzyme–inhibitor interactions. These and other collaborators are frequently cited in the references. The continuing research on transition state analogues and their interactions with proteins has been generously supported by research grants GM041916, CA072444, CA135405, and AI127807 and program project GM068036 from the National Institutes of Health. Generous research support from the Michael Price Family Foundation, Ruth Merns, and the Medicines for Malaria Venture MMV 06/ 0026 is also gratefully acknowledged. Support from Contract DAMD 17–93-C-3051 from the U.S. Army was important for research related to ribosome-inactivating proteins.
Biography
Vern L. Schramm is Professor and Ruth Merns endowed Chair in the Department of Biochemistry at the Albert Einstein College of Medicine. He studied enzymatic mechanisms for his Ph.D. research at the Australian National University with John F. Morrison. He began research in enzymatic reaction mechanisms and transition state analysis at the Department of Biochemistry, Temple University School of Medicine, followed by appointment as Chair, Department of Biochemistry at the Albert Einstein College of Medicine, until 2015. The Schramm laboratory pioneered an approach to use intrinsic kinetic isotope effects combined with computational chemistry to understand the details of enzymatic transition states. Knowledge of enzymatic transition state structure permits the design of stable molecules as transition state mimics. Together with a team of chemistry collaborators in New Zealand, the Schramm laboratory designed and characterized the transition state analogues for many enzyme targets. Three of the inhibitors designed by this approach have entered clinical trials, and others are in earlier development. One analogue was recently approved for use in relapsed or resistant peripheral T-cell lymphoma in Japan. He has received the Rudi Lemberg Award from the Australian Academy of Science, the Repligen Award and the Gordon Hammes Lectureship from the Biological Chemistry Division of the American Chemical Society, and the A. I. Scott Medal from the Texas A&M-ACS Section. He has served as an Associate Editor of the Journal of the American Chemical Society, as a member of Council, General Medical Sciences of the NIH, and as Chair of the Division of Biological Chemistry of the ACS. He was elected to membership in the National Academy of Sciences in 2007.
Footnotes
The author declares the following competing financial interest(s): The author serves as a consultant or board member to several biotech companies involved in development of the immucillin family of transition state analogues, some of them described here. The intellectual property developed around these inhibitors is the joint property of the Albert Einstein College of Medicine and Victoria University of Wellington, New Zealand. The author and collaborators receive royalties through dividend plans of these institutions.
REFERENCES
- (1).Pauling L Molecular Architecture and Biological Reactions. Chem. Eng. News 1946, 24, 1375–1377. [Google Scholar]
- (2).Pauling L Nature of Forces Between Large Molecules of Biological Interest. Nature 1948, 161, 707–709. [DOI] [PubMed] [Google Scholar]
- (3).Wolfenden R Transition State Analogues for Enzyme Catalysis. Nature 1969, 223, 704–705. [DOI] [PubMed] [Google Scholar]
- (4).Wolfenden R Transition State Analog Inhibitors and Enzyme Catalysis. Annu. Rev. Biophys. Bioeng 1976, 5, 271–306. [DOI] [PubMed] [Google Scholar]
- (5).Wolfenden R Thermodynamic and Extrathermodynamic Requirements of Enzyme Catalysis. Biophys. Chem 2003, 105, 559–572. [DOI] [PubMed] [Google Scholar]
- (6).Miller BG; Wolfenden R Catalytic Proficiency: The Unusual Case of OMP Decarboxylase. Annu. Rev. Biochem 2002, 71, 847–885. [DOI] [PubMed] [Google Scholar]
- (7).Wolfenden R; Snider MJ The Depth of Chemical Time and the Power of Enzymes as Catalysts. Acc. Chem. Res 2001, 34, 938–945. [DOI] [PubMed] [Google Scholar]
- (8).Wolfenden R Conformational Aspects of Inhibitor Design: Enzyme-substrate Interactions in the Transition State. Bioorg. Med. Chem 1999, 7, 647–652. [DOI] [PubMed] [Google Scholar]
- (9).Wolfenden R Are There Limits to Enzyme-inhibitor Binding Discrimination? Inferences from the Behavior of Nucleoside Deaminases. Pharmacol. Ther 1993, 60, 235–244. [DOI] [PubMed] [Google Scholar]
- (10).Morrison JF; Walsh CT The Behavior and Significance of Slow-binding Enzyme Inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol 2006, 61, 201–301. [DOI] [PubMed] [Google Scholar]
- (11).Johnson LN; Wolfenden R Changes in Absorption Spectrum and Crystal Structure of Tiose Phosphate Isomerase Brought About by 2-Phosphoglycollate, a Potential Transition State Analogue. J. Mol. Biol 1970, 47, 93–100. [DOI] [PubMed] [Google Scholar]
- (12).Wentworth DF; Wolfenden R On the Interaction of 3,4,5,6-Tetrahydrouridine with Human Liver Cytidine Deaminase. Biochemistry 1975, 14, 5099–5105. [DOI] [PubMed] [Google Scholar]
- (13).Zhang X; Houk KN Why Enzymes are Proficient Catalysts: Beyond the Pauling Paradigm. Acc. Chem. Res 2005, 38, 379–385. [DOI] [PubMed] [Google Scholar]
- (14).Cleland WW Isotope Effects: Determination of Enzyme Transition State Structure. Methods Enzymol 1995, 249, 341–373. [DOI] [PubMed] [Google Scholar]
- (15).Hegazi MF; Borchardt RT; Schowen RL SN2-like Transition State for Methyl Transfer Catalyzed by Catechol-O-methyl-transferase. J. Am. Chem. Soc 1976, 98, 3048–3049. [DOI] [PubMed] [Google Scholar]
- (16).Schramm VL Enzymatic Transition-state Analysis and Transition-state Analogs. Methods Enzymol 1999, 308, 301–355. [DOI] [PubMed] [Google Scholar]
- (17).Singh P; Islam Z; Kohen A Examinations of the Chemical Step in Enzyme Catalysis. Methods Enzymol 2016, 577, 287–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Parkin DW Methods for the Determination of Competitive and Noncompetitive Kinetic Isotope Effects. In Enzyme mechanism from isotope effects; CRC Press, 1991; pp 269–290. [Google Scholar]
- (19).Berti PJ Determining Transition States from Kinetic Isotope Effects. Methods Enzymol 1999, 308, 355–397. [DOI] [PubMed] [Google Scholar]
- (20).Schramm VL Enzymatic Transition States and Transition State Analog Design. Annu. Rev. Biochem 1998, 67, 693–720. [DOI] [PubMed] [Google Scholar]
- (21).Parkin DW; Schramm VL Effects of Allosteric Activation on the Primary and Secondary Kinetic Isotope Effects for Three AMP Nucleosidases. J. Biol. Chem 1984, 259, 9418–9425. [PubMed] [Google Scholar]
- (22).Parkin DW; Leung HB; Schramm VL Synthesis of Nucleotides with Specific Radiolabels in Ribose. Primary 14C and Secondary 3H Kinetic Isotope Effects on Acid-catalyzed Glycosidic Bond Hydrolysis of AMP, dAMP, and Inosine. J. Biol. Chem 1984, 259, 9411–9417. [PubMed] [Google Scholar]
- (23).Markham GD; Parkin DW; Mentch F; Schramm VL A Kinetic Isotope Effect Study and Transition State Analysis of the S-Adenosylmethionine Synthetase Reaction. J. Biol. Chem 1987, 262, 5609–5615. [PubMed] [Google Scholar]
- (24).Horenstein BA; Schramm VL Electronic Nature of the Transition State for Nucleoside Hydrolase. A Blueprint for Inhibitor Design. Biochemistry 1993, 32, 7089–7097. [DOI] [PubMed] [Google Scholar]
- (25).Horenstein BA; Parkin DW; Estupiñán B; Schramm VL Transition-state Analysis of Nucleoside Hydrolase from Crithidia fasciculata. Biochemistry 1991, 30, 10788–10795. [DOI] [PubMed] [Google Scholar]
- (26).Lewis BE; Schramm VL Binding Equilibrium Isotope Effects for Glucose at the Catalytic Domain of Human Brain Hexokinase. J. Am. Chem. Soc 2003, 125, 4785–4798. [DOI] [PubMed] [Google Scholar]
- (27).Lewis BE; Schramm VL Enzymatic Binding Isotope Effects and the Interaction of Glucose with Hexokinase. Isotope effects in Chemistry and Biology; CRC Press, 2006; pp 1019–1053. [Google Scholar]
- (28).Lewis BE; Schramm VL Glucose Binding Isotope Effects in the Ternary Complex of Brain Hexokinase Demonstrate Partial Relief of Ground-state Destabilization. J. Am. Chem. Soc 2003, 125, 4672–4673. [DOI] [PubMed] [Google Scholar]
- (29).Schramm VL Binding Isotope Effects: Boon and Bane. Curr. Opin. Chem. Biol 2007, 11, 529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Stratton CF; Poulin MB; Schramm VL Binding Isotope Effects for Interrogating Enzyme-Substrate Interactions. Methods Enzymol 2017, 596, 1–21. [DOI] [PubMed] [Google Scholar]
- (31).Schramm VL; Leung H Regulation of Adenosine Mono-phosphate Levels as a Function of Adenosine Triphosphate and Inorganic Phosphate. A Proposed Metabolic Role for Adenosine Monophosphate Nucleosidase from Azotobacter vinelandii. J. Biol. Chem 1973, 248, 8313–8315. [PubMed] [Google Scholar]
- (32).Schramm VL Kinetic Properties of Allosteric Adenosine Monophosphate Nucleosidase from Azotobacter vinelandii. J. Biol. Chem 1974, 249, 1729–1736. [PubMed] [Google Scholar]
- (33).Schramm VL; Lazorik FC The Pathway of Adenylate Catabolism in Azotobacter vinelandii. Evidence for Adenosine Monophosphate Nucleosidase as the Regulatory Enzyme. J. Biol. Chem 1975, 250, 1801–1808. [PubMed] [Google Scholar]
- (34).Morrison BA; Shain DH An AMP Nucleosidase Gene Knockout in Escherichia coli Elevates Intracellular ATP Levels and Increases Cold Tolerance. Biol. Lett 2008, 4, 53–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Zhang Y; Cottet SE; Ealick SE Structure of Escherichia coli AMP Nucleosidase Reveals Similarity to Nucleoside Phosphorylases. Structure 2004, 12, 1383–1394. [DOI] [PubMed] [Google Scholar]
- (36).Parry BR; Shain DH Manipulations of AMP Metabolic Genes Increase Growth Rate and Cold Tolerance in Escherichia coli: Implications for Psychrophilic Evolution. Mol. Biol. Evol 2011, 28, 2139–2145. [DOI] [PubMed] [Google Scholar]
- (37).DeWolf WE Jr.; Fullin FA; Schramm VL The Catalytic Site of AMP Nucleosidase. Substrate Specificity and pH Effects with AMP and Formycin 5′-PO4. J. Biol. Chem 1979, 254, 10868–10875. [PubMed] [Google Scholar]
- (38).Parkin DW; Mentch F; Banks GA; Horenstein BA; Schramm VL Transition-state Analysis of a Vmax Mutant of AMP Nucleosidase by the Application of Heavy-atom Kinetic Isotope Effects. Biochemistry 1991, 30, 4586–4594. [DOI] [PubMed] [Google Scholar]
- (39).Leung HB; Schramm VL Adenylate Degradation in Escherichia coli. The Role of AMP Nucleosidase and Properties of the Purified Enzyme. J. Biol. Chem 1980, 255, 10867–10874. [PubMed] [Google Scholar]
- (40).Westaway KC; Fang YR; MacMillar S; Matsson O; Poirier RA; Islam SM Determining the Transition-state Structure for Different SN2 Reactions Using Experimental Nucleophile Carbon and Secondary Alpha-deuterium Kinetic Isotope Effects and Theory. J. Phys. Chem. A 2008, 112, 10264–10273. [DOI] [PubMed] [Google Scholar]
- (41).Adcock W; Trout NA; Vercoe D; Taylor DK; Shiner VJ Jr.; Sorensen TS Solvolysis of (Z)-5-Trimethylstannyl 2-Adamantyl p-Bromobenzenesulfonate: Mechanistic Implications of a Record-breaking Secondary Alpha-deuterium kinetic Isotope Effect for an SN1 Substrate. J. Org. Chem 2003, 68, 5399–5402. [DOI] [PubMed] [Google Scholar]
- (42).Ehrlich JI; Schramm VL Electrostatic Potential Surface Analysis of the Transition State for AMP Nucleosidase and for Formycin 5′-phosphate, a Transition-state Inhibitor. Biochemistry 1994, 33, 8890–8896. [DOI] [PubMed] [Google Scholar]
- (43).Sawa T; Fukagawa Y; Homma I; Wakashiro T; Takeuchi T Metabolic Conversion of Formycin B to Formycin A and to Oxoformycin B in Nocardia interforma. J. Antibiot 1968, 21, 334–339. [DOI] [PubMed] [Google Scholar]
- (44).Rossomando EF; Jahngen J; Eccleston JF A Fluorometric-high-performance Liquid Chromatographic Assay Procedure for Several Enzymatic Activities Using Formycin Analogs of Adenosine 5′-Mono, 5′-Tri, and Cyclic 3′,5′-Monophosphate as Substrates. Anal. Biochem 1981, 116, 80–88. [DOI] [PubMed] [Google Scholar]
- (45).Giblett ER; Anderson JE; Cohen F; Pollara B; Meuwissen HJ Adenosine-deaminase Deficiency in Two Patients with Severely Impaired Cellular Immunity. Lancet 1972, 300, 1067–1069. [DOI] [PubMed] [Google Scholar]
- (46).Mills GC; Schmalstieg FC; Trimmer KB; Goldman AS; Goldblum RM Purine Metabolism in Adenosine Deaminase Deficiency. Proc. Natl. Acad. Sci. U. S. A 1976, 73, 2867–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Cohen A; Hirschhorn R; Horowitz SD; Rubinstein A; Polmar SH; Hong R; Martin DW Jr. Deoxyadenosine Triphosphate as a Potentially Toxic Metabolite in Adenosine Deaminase Deficiency. Proc. Natl. Acad. Sci. U. S. A 1978, 75, 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Chen SH; Ochs HD; Scott CR; Giblett ER; Tingle AJ Adenosine Deaminase Deficiency: Disappearance of Adenine Deoxy-nucleotides from a Patient’s Erythrocytes after Successful Marrow Transplantation. J. Clin. Invest 1978, 62, 1386–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Kohn DB; Gaspar HB How We Manage Adenosine Deaminase-deficient Severe Combined Immune Deficiency (ADA SCID). J. Clin. Immunol 2017, 37, 351–356. [DOI] [PubMed] [Google Scholar]
- (50).Bar̈ HP; Drummond GI On the Mechanism of Adenosine Deaminase Action. Biochem. Biophys. Res. Commun 1966, 24, 584–587. [DOI] [PubMed] [Google Scholar]
- (51).Carter CW Jr. The Nucleoside Deaminases For Cytidine And Adenosine: Structure, Transition State Stabilization, Mechanism, and Evolution. Biochimie 1995, 77, 92–98. [DOI] [PubMed] [Google Scholar]
- (52).Kurz LC; Frieden C Adenosine Deaminase Converts Purine Riboside into an Analogue of a Reactive Intermediate: a 13C NMR and Kinetic Study. Biochemistry 1987, 26, 8450–8057. [DOI] [PubMed] [Google Scholar]
- (53).Evans B; Wolfenden R Hydratase Activity of a Hydrolase. Adenosine Deaminase. J. Am. Chem. Soc 1972, 94, 5902–5903. [DOI] [PubMed] [Google Scholar]
- (54).Weiss PM; Cook PF; Hermes JD; Cleland WW Evidence from Nitrogen-15 and Solvent Deuterium Isotope Effects on the Chemical Mechanism of Adenosine Deaminase. Biochemistry 1987, 26, 7378–7384. [DOI] [PubMed] [Google Scholar]
- (55).Wilson DK; Rudolph FB; Quiocho FA Atomic Structure of Adenosine Deaminase Complexed with a Transition-state Analog: Understanding Catalysis and Immunodeficiency Mutations. Science 1991, 252, 1278–1284. [DOI] [PubMed] [Google Scholar]
- (56).Merkler DJ; Kline PC; Weiss P; Schramm VL Transition-state Analysis of AMP Deaminase. Biochemistry 1993, 32, 12993–13001. [DOI] [PubMed] [Google Scholar]
- (57).Nakamura H; Koyama G; Iitaka Y; Ono M; Yagiawa N Structure of Coformycin, an Unusual Nucleoside of Microbial Origin. J. Am. Chem. Soc 1974, 96, 4327–4328. [DOI] [PubMed] [Google Scholar]
- (58).Ohno M; Yagisawa N; Shibahara S; Kondo S; Maeda K Synthesis of Coformycin. J. Am. Chem. Soc 1974, 96, 4326–4327. [DOI] [PubMed] [Google Scholar]
- (59).Woo PWK; Dion HW; Lange SM; Dahl LF; Durham LJ A Novel Adenosine and Ara-A Deaminase Inhibitor, R-3-(2-Deoxy-β-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]-diazepin-8-ol. J. Heterocycl. Chem 1974, 11, 641–643. [Google Scholar]
- (60).Rogler-Brown T; Agarwal RP; Parks RE Jr. Tight Binding Inhibitors–VI. Interactions of Deoxycoformycin and Adenosine Deaminase in Intact Human Erythrocytes and Sarcoma 180 Cells. Biochem. Pharmacol 1978, 27, 2289–2296. [DOI] [PubMed] [Google Scholar]
- (61).Frieden C; Gilbert HR; Miller WH; Miller RL Adenylate Deaminase: Potent Inhibition by 2’-Deoxycoformycin 5′-Phosphate. Biochem. Biophys. Res. Commun 1979, 91, 278–283. [DOI] [PubMed] [Google Scholar]
- (62).Frieden C; Kurz LC; Gilbert HR Adenosine Deaminase and Adenylate Deaminase: Comparative Kinetic Studies with Transition State and Ground State Analog Inhibitors. Biochemistry 1980, 19, 5303–5309. [DOI] [PubMed] [Google Scholar]
- (63).Poster DS; Penta JS; Bruno S; Macdonald JS 2’-Deoxycoformycin. A New Anticancer Agent. Cancer Clin. Trials 1981, 4, 209–213. [PubMed] [Google Scholar]
- (64).Robak P; Robak T Older and New Purine Nucleoside Analogs for Patients with Acute Leukemias. Cancer Treat. Rev 2013, 39, 851–861. [DOI] [PubMed] [Google Scholar]
- (65).Sarvaria A; Topp Z; Saven A Current Therapy and New Directions in the Treatment of Hairy Cell Leukemia: A review. JAMA Oncol 2016, 2, 123–129. [DOI] [PubMed] [Google Scholar]
- (66).Tedeschi A; Rossi D; Motta M; Quaresmini G; Rossi M; Coscia M; Anastasia A; Rossini F; Cortelezzi A; Nador G; Workgroup CLL; et al. A Phase II Multi-center Trial of Pentostatin Plus Cyclophosphamide with Ofatumumab in Older Previously Untreated Chronic Lymphocytic Leukemia Patients. Haematologica 2015, 100, e501–e504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Kawasaki H; Carrera CJ; Piro LD; Saven A; Kipps TJ; Carson DA Relationship of Deoxycytidine Kinase and Cytoplasmic 5′-Nucleotidase to the Chemotherapeutic Efficacy of 2-Chlorodeox-yadenosine. Blood 1993, 81, 597–601. [PubMed] [Google Scholar]
- (68).Johnston JB Mechanism of Action of Pentostatin and Cladribine in Hairy Cell Leukemia. Leuk. Lymphoma 2011, 52, 43–45. [DOI] [PubMed] [Google Scholar]
- (69).Schramm VL; Baker DC Spontaneous Epimerization of (S)-Deoxycoformycin and Interaction of (R)-Deoxycoformycin, (S)-Deoxycoformycin, and 8-Ketodeoxycoformycin with Adenosine Deaminase. Biochemistry 1985, 24, 641–646. [DOI] [PubMed] [Google Scholar]
- (70).Kati WM; Acheson SA; Wolfenden R A Transition State in Pieces: Major Contributions of Entropic Effects to Ligand Binding by Adenosine Deaminase. Biochemistry 1992, 31, 7356–7366. [DOI] [PubMed] [Google Scholar]
- (71).Luo M; Singh V; Taylor EA; Schramm VL Transition-state Variation in Human, Bovine, and Plasmodium falciparum Adenosine Deaminases. J. Am. Chem. Soc 2007, 129, 8008–8017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Schmidt G; Walter RD; Königk EA Purine Nucleoside Hydrolase from Trypanosoma gambiense, Purification and Properties. Tropenmed. Parasitol 1975, 26, 19–26. [PubMed] [Google Scholar]
- (73).Ribeiro JM; Valenzuela JG The Salivary Purine Nucleosidase of the Mosquito, Aedes aegypti. Insect Biochem. Mol. Biol 2003, 33, 13–22. [DOI] [PubMed] [Google Scholar]
- (74).Bates C; Kendrick Z; McDonald N; Kline PC Transition State Analysis of Adenosine Nucleosidase from Yellow Lupin (Lupinus luteus). Phytochemistry 2006, 67, 5–12. [DOI] [PubMed] [Google Scholar]
- (75).Ashihara H; Stasolla C; Fujimura T; Crozier A Purine Salvage in Plants. Phytochemistry 2018, 147, 89–124. [DOI] [PubMed] [Google Scholar]
- (76).Sańchez-Sancho F; Campillo NE; Paéz JA Chagas Disease: Progress and New Perspectives. Curr. Med. Chem 2010, 17, 423–452. [DOI] [PubMed] [Google Scholar]
- (77).Datta AK; Datta R; Sen B Antiparasitic Chemotherapy: Tinkering with the Purine Salvage Pathway. Adv. Exp. Med. Biol 2008, 625, 116–132. [DOI] [PubMed] [Google Scholar]
- (78).Nico D; Gomes DC; Palatnik-de-Sousa I; Morrot A; Palatnik M; Palatnik-de-Sousa CB Leishmania donovani Nucleoside Hydrolase Terminal Domains in Cross-protective Immunotherapy Against Leishmania amazonensis Murine Infection. Front. Immunol 2014, 5, 273 (eCollection). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Carrillo E; Fernandez L; Ibarra-Meneses AV; Santos MLB; Nico D; de Luca PM; Correa CB; de Almeida RP; Moreno J; Palatnik-de-Sousa CB F1 Domain of the Leishmania (Leishmania) donovani Nucleoside Hydrolase Promotes a Th1 Response in Leishmania (Leishmania) infantum Cured Patients and in Asymptomatic Individuals Living in an Endemic Area of Leishmaniasis. Front. Immunol 2017, 8, 750 (eCollection). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Freitas EO; Nico D; Alves-Silva MV; Morrot A; Clinch K; Evans GB; Tyler PC; Schramm VL; Palatnik-de-Sousa CB Immucillins ImmA and ImmH are Effective and Non-toxic in the Treatment of Experimental Visceral Leishmaniasis. PLoS Neglected Trop. Dis 2015, 9 (12), e0004297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Kipandula W; Smith TK; MacNeill SA Tandem Affinity Purification of Exosome and Replication Factor C Complexes from the Non-human Infectious Kinetoplastid Parasite Crithidia fasciculata. Mol. Biochem. Parasitol 2017, 217, 19–22. [DOI] [PubMed] [Google Scholar]
- (82).Liu W; Arendt CS; Gessford SK; Ntaba D; Carter NS; Ullman B Identification and Characterization of Purine Nucleoside Transporters from Crithidia fasciculata. Mol. Biochem. Parasitol 2005, 140, 1–12. [DOI] [PubMed] [Google Scholar]
- (83).Tetaud E; Lecuix I; Sheldrake T; Baltz T; Fairlamb AH A New Expression Vector for Crithidia fasciculata and Leishmania. Mol. Biochem. Parasitol 2002, 120, 195–204. [DOI] [PubMed] [Google Scholar]
- (84).Schmid-Hempel P; Aebi M; Barribeau S; Kitajima T; du Plessis L; Schmid-Hempel R; Zoller S The Genomes of Crithidia bombi and C. expoeki, Common Parasites of Bumblebees. PLoS One 2018, 13 (1), e0189738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Singh RK; Steyaert J; Verseés W Structural and Biochemical Characterization of the Nucleoside Hydrolase from C. elegans Reveals the Role of Two Active Site Cysteine Residues in Catalysis. Protein Sci 2017, 26, 985–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Horenstein BA; Schramm VL Correlation of the Molecular Electrostatic Potential Surface of an Enzymatic Transition State with Novel Transition-state Inhibitors. Biochemistry 1993, 32, 9917–9925. [DOI] [PubMed] [Google Scholar]
- (87).Boutellier M; Horenstein BA; Semenyaka A; Schramm VL; Ganem B Amidrazone Analogues of D-Ribofuranose as Transition-state Inhibitors of Nucleoside Hydrolase. Biochemistry 1994, 33, 3994–4000. [DOI] [PubMed] [Google Scholar]
- (88).Schramm VL; Horenstein BA; Kline PC Transition State Analysis and Inhibitor Design for Enzymatic Reactions. J. Biol. Chem 1994, 269, 18259–18262. [PubMed] [Google Scholar]
- (89).Deng H; Chan AW; Bagdassarian CK; Estupiñán B; Ganem B; Callender RH; Schramm VL Trypanosomal Nucleoside Hydrolase. Resonance Raman Spectroscopy of a Transition-state Inhibitor Complex. Biochemistry 1996, 35, 6037–6047. [DOI] [PubMed] [Google Scholar]
- (90).Parkin DW; Horenstein BA; Abdulah DR; Estupiñán B; Schramm VL Nucleoside Hydrolase from Crithidia fasciculata. Metabolic Role, Purification, Specificity, and Kinetic Mechanism. J. Biol. Chem 1991, 266, 20658–20665. [PubMed] [Google Scholar]
- (91).Mazzella LJ; Parkin DW; Tyler PC; Furneaux RH; Schramm VL Mechanistic Diagnoses of N-Ribohydrolases and Purine Nucleoside Phosphorylase. J. Am. Chem. Soc 1996, 118, 2111–2112. [Google Scholar]
- (92).Shi W; Schramm VL; Almo SC Nucleoside Hydrolase from Leishmania major. Cloning, Expression, Catalytic Properties, Transition State Inhibitors, and the 2.5-Å Crystal Structure. J. Biol. Chem 1999, 274, 21114–21120. [DOI] [PubMed] [Google Scholar]
- (93).Vandemeulebroucke A; Minici C; Bruno I; Muzzolini L; Tornaghi P; Parkin DW; Verseés W; Steyaert J; Degano M Structure and Mechanism of the 6-Oxopurine Nucleosidase from Trypanosoma brucei brucei. Biochemistry 2010, 49, 8999–9010. [DOI] [PubMed] [Google Scholar]
- (94).Verseés W; Barlow J; Steyaert J Transition-state Complex of the Purine-specific Nucleoside Hydrolase of T. vivax: Enzyme Conformational Changes and Implications for Catalysis. J. Mol. Biol 2006, 359, 331–346. [DOI] [PubMed] [Google Scholar]
- (95).Muzzolini L; Verseés W; Tornaghi P; Van Holsbeke E; Steyaert J; Degano M New Insights into the Mechanism of Nucleoside Hydrolases from the Crystal Structure of the Escherichia coli YbeK Protein Bound to the Reaction Product. Biochemistry 2006, 45, 773–782. [DOI] [PubMed] [Google Scholar]
- (96).Degano M; Gopaul DN; Scapin G; Schramm VL; Sacchettini JC Three-dimensional Structure of the Inosine-uridine Nucleoside N-Ribohydrolase from Crithidia fasciculata. Biochemistry 1996, 35, 5971–5981. [DOI] [PubMed] [Google Scholar]
- (97).Alam R; Barbarovich AT; Caravan W; Ismail M; Barskaya A; Parkin DW; Stockman BJ Druggability of the Guanosine/ Adenosine/Cytidine Nucleoside Hydrolase from Trichomonas vaginalis. Chem. Biol. Drug Des 2018, 92, 1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Furneaux RH; Schramm VL; Tyler PC Transition State Analogue Inhibitors Of Protozoan Nucleoside Hydrolases. Bioorg. Med. Chem 1999, 7, 2599–2606. [DOI] [PubMed] [Google Scholar]
- (99).Figueroa-Villar JD; Sales EM The Importance of Nucleoside Hydrolase Enzyme (NH) in Studies to Treatment of Leishmania: A Review. Chem.-Biol. Interact 2017, 263, 18–27. [DOI] [PubMed] [Google Scholar]
- (100).Ducati RG; Namanja-Magliano HA; Schramm VL Transition-state Inhibitors of Purine Salvage and Other Prospective Enzyme Targets in Malaria. Future Med. Chem 2013, 5, 1341–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Evans GB; Tyler PC; Schramm VL Immucillins in Infectious Diseases. ACS Infect. Dis 2018, 4, 107–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Frame IJ; Deniskin R; Arora A; Akabas MH Purine Import into Malaria Parasites as a Target for Antimalarial Drug Development. Ann. N. Y. Acad. Sci 2015, 1342, 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Harms A; Brodersen DE; Mitarai N; Gerdes K Toxins, Targets, and Triggers: An Overview of Toxin-antitoxin Biology. Mol. Cell 2018, 70, 768–784. [DOI] [PubMed] [Google Scholar]
- (104).Reboud E; Basso P; Maillard AP; Huber P; Attreé I Exolysin Shapes the Virulence of Pseudomonas aeruginosa Clonal Outliers. Toxins 2017, 9, 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Chandrasekaran R; Lacy DB The Role of Toxins in Clostridium difficile infection. FEMS Microbiol. Rev 2017, 41, 723–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Tsuge H; Tsurumura T; Toda A; Murata H; Toniti W; Yoshida T Comparative Studies of Actin-and Rho-specific ADP-Ribosylating Toxins: Insight from Structural Biology. Curr. Top. Microbiol. Immunol 2016, 399, 69–86. [DOI] [PubMed] [Google Scholar]
- (107).Simon NC; Aktories K; Barbieri JT Novel Bacterial ADP-Ribosylating Toxins: Structure and Function. Nat. Rev. Microbiol 2014, 12, 599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Deng Q; Barbieri JT Molecular Mechanisms of the Cytotoxicity of ADP-Ribosylating Toxins. Annu. Rev. Microbiol 2008, 62, 271–288. [DOI] [PubMed] [Google Scholar]
- (109).Popoff MR Bacterial Factors Exploit Eukaryotic Rho GTPase Signaling Cascades to Promote Invasion and Proliferation Within their Host. Small GTPases 2014, 5, e983863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Ferraz JP; Bull HG; Cordes EH Kinetic Alpha-Deuterium Isotope Effects for Enzymatic and Nonenzymatic Hydrolysis of Nicotinamide-beta-riboside. Arch. Biochem. Biophys 1978, 191, 431–436. [DOI] [PubMed] [Google Scholar]
- (111).Bull HG; Ferraz JP; Cordes EH; Ribbi A; Apitz-Castro R Concerning the Mechanism of the Enzymatic and Nonenzymatic Hydrolysis of Nicotinamide Nucleotide Coenzymes. J. Biol. Chem 1978, 253, 5186–5192. [PubMed] [Google Scholar]
- (112).Kuhn I; Kellenberger E; Cakir-Kiefer C; Muller-Steffner H; Schuber F Probing the Catalytic Mechanism of Bovine CD38/NAD+ Glycohydrolase by Site Directed Mutagenesis of Key Active Site Residues. Biochim. Biophys. Acta, Proteins Proteomics 2014, 1844, 1317–1331. [DOI] [PubMed] [Google Scholar]
- (113).Rising KA; Schramm VL Enzymic Synthesis of NAD+ with the Specific Incorporation of Atomic Labels. J. Am. Chem. Soc 1994, 116, 6531–6536. [Google Scholar]
- (114).Rising KA; Schramm VL Transition State Analysis of NAD + Hydrolysis by the Cholera Toxin Catalytic Subunit. J. Am. Chem. Soc 1997, 119, 27–37. [Google Scholar]
- (115).Scheuring J; Schramm VL Kinetic Isotope Effect Characterization of the Transition State for Oxidized Nicotinamide Adenine Dinucleotide Hydrolysis by Pertussis Toxin. Biochemistry 1997, 36, 4526–4534. [DOI] [PubMed] [Google Scholar]
- (116).Scheuring J; Schramm VL Pertussis Toxin: Transition State Analysis for ADP-Ribosylation of G-Protein Peptide αi3C20. Biochemistry 1997, 36, 8215–8223. [DOI] [PubMed] [Google Scholar]
- (117).Abdel-Fattah W; Scheidt V; Uthman S; Stark MJ; Schaffrath R Insights into Diphthamide, Key Diphtheria Toxin Effector. Toxins 2013, 5, 958–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Parikh SL; Schramm VL Transition State Structure for ADP-ribosylation of Eukaryotic Elongation Factor 2 Catalyzed by Diphtheria Toxin. Biochemistry 2004, 43, 1204–1212. [DOI] [PubMed] [Google Scholar]
- (119).Berti PJ; Blanke SR; Schramm VL Transition State Structure for the Hydrolysis of NAD+ Catalyzed by Diphtheria Toxin. J. Am. Chem. Soc 1997, 119, 12079–12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Andersen GR; Nissen P; Nyborg J Elongation Factors in Protein Biosynthesis. Trends Biochem. Sci 2003, 28, 434–441. [DOI] [PubMed] [Google Scholar]
- (121).Hogg JL; Rodgers J; Kovach I; Schowen RL Kinetic Isotope-effect Probes of Transition-state Structure. Vibrational Analysis of Model Transition States for Carbonyl Addition. J. Am. Chem. Soc 1980, 102, 79–85. [Google Scholar]
- (122).Fedorov A; Shi W; Kicska G; Fedorov E; Tyler PC; Furneaux RH; Hanson JC; Gainsford GJ; Larese JZ; Schramm VL; Almo SC Transition State Structure of Purine Nucleoside Phosphorylase and Principles of Atomic Motion in Enzymatic Catalysis. Biochemistry 2001, 40, 853–860. [DOI] [PubMed] [Google Scholar]
- (123).Schramm VL; Grubmeyer C Phosphoribosyltransferase Mechanisms and Roles in Nucleic Acid Metabolism. Prog. Nucleic Acid Res. Mol. Biol 2004, 78, 261–304. [DOI] [PubMed] [Google Scholar]
- (124).Jørgensen R; Wang Y; Visschedyk D; Merrill AR The Nature and Character of the Transition State for the ADP-Ribosyltransferase Reaction. EMBO Rep 2008, 9, 802–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Kuan TC; Wu HR; Adak AK; Li BY; Liang CF; Hung JT; Chiou SP; Yu AL; Hwu JR; Lin CC Synthesis of an S-Linked α (2→ 8) GD3 Antigen and Evaluation of the Immunogenicity of its Glycoconjugate. Chem. - Eur. J 2017, 23, 6876–6887. [DOI] [PubMed] [Google Scholar]
- (126).Sauve AA; Deng H-T; Angeletti RH; Schramm VL A Covalent Intermediate in CD38 is Responsible for ADP-Ribosylation and Cyclization Reactions. J. Am. Chem. Soc 2000, 122, 7855–7859. [Google Scholar]
- (127).Sauve AA; Celic I; Avalos J; Deng H; Boeke JD; Schramm VL Chemistry of Gene Silencing: The Mechanism of NAD+-Dependent Deacetylation Reactions. Biochemistry 2001, 40, 15456–15463. [DOI] [PubMed] [Google Scholar]
- (128).Moggre G-J; Poulin MB; Tyler PC; Schramm VL; Parker EJ Transition State Analysis of Adenosine Triphosphate Phosphoribosyltransferase. ACS Chem. Biol 2017, 12, 2662–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Zhou GC; Parikh SL; Tyler PC; Evans GB; Furneaux RH; Zubkova OV; Benjes PA; Schramm VL Inhibitors of ADP-ribosylating Bacterial Toxins Based on Oxacarbenium Ion Character at their Transition States. J. Biol. Chem 2004, 126, 5690–5698. [DOI] [PubMed] [Google Scholar]
- (130).Moss J; Vaughan M Structure and Function of ARF Proteins: Activators of Cholera Toxin and Critical Components of Intracellular Vesicular Transport Processes. J. Biol. Chem 1995, 270, 12327–12330. [DOI] [PubMed] [Google Scholar]
- (131).O’neal CJ; Jobling MG; Holmes RK; Hol WG Structural Basis for the Activation of Cholera Toxin by Human ARF6-GTP. Science 2005, 309, 1093–1096. [DOI] [PubMed] [Google Scholar]
- (132).Kahn RA; Gilman AG Purification of a Protein Cofactor Required for ADP-Ribosylation of the Stimulatory Regulatory Component of Adenylate Cyclase by Cholera Toxin. J. Biol. Chem 1984, 259, 6228–6234. [PubMed] [Google Scholar]
- (133).Chung DW; Collier RJ The Mechanism of ADP-ribosylation of Elongation Factor 2 Catalyzed by Fragment A from Diphtheria Toxin. Biochim. Biophys. Acta (BBA)-Enzymology 1977, 483, 248–257. [DOI] [PubMed] [Google Scholar]
- (134).Wilson BA; Reich KA; Weinstein BR; Collier RJ Active-site Mutations of Diphtheria Toxin: Effects of Replacing Glutamic Acid-148 with Aspartic Acid, Glutamine, or Serine. Biochemistry 1990, 29, 8643–8651. [DOI] [PubMed] [Google Scholar]
- (135).Schnell L; Mittler AK; Mattarei A; Tehran D; Montecucco C; Barth H Semicarbazone EGA Inhibits Uptake of Diphtheria Toxin into Human Cells and Protects Cells from Intoxication. Toxins 2016, 8, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Kim K; Groman NB In vitro Inhibition of Diphtheria Toxin Action by Ammonium Salts and Amines. J. Bacteriol 1965, 90, 1552–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Mekada E; Uchida T; Okada Y Methylamine Stimulates the action of Ricin Toxin but Inhibits that of Diphtheria Toxin. J. Biol. Chem 1981, 256, 1225–1228. [PubMed] [Google Scholar]
- (138).Olsnes S The history of ricin, abrin and related toxins. Toxicon 2004, 44, 361–370. [DOI] [PubMed] [Google Scholar]
- (139).Doan LG Ricin: Mechanism of Toxicity, Clinical Manifestations, and Vaccine Development. A Review. J. Toxicol., Clin. Toxicol 2004, 42, 201–208. [DOI] [PubMed] [Google Scholar]
- (140).Olsnes S; Refsnes K; Pihl A Mechanism of Action of the Toxic Lectins Abrin and Ricin. Nature 1974, 249, 627–631. [DOI] [PubMed] [Google Scholar]
- (141).Doan LG Ricin: Mechanism of Toxicity, Clinical Manifestations, and Vaccine Development. A review. J. Toxicol., Clin. Toxicol 2004, 42, 201–208. [DOI] [PubMed] [Google Scholar]
- (142).Crompton R; Gall D Georgi Markov Death in a Pellet. Med.-Leg. J 1980, 48, 51–62. [DOI] [PubMed] [Google Scholar]
- (143).Maman M; Yehezkelli Y Ricin: A Possible, Noninfectious Biological Weapon. Bioterrorism and Infectious Agents: A New Dilemma for the 21st Century; Springer: New York, 2009; pp 205–216. [Google Scholar]
- (144).Van Deurs B; Tønnessen TI; Petersen OW; Sandvig K; Olsnes S Routing of Internalized Ricin and Ricin Conjugates to the Golgi Complex. J. Cell Biol 1986, 102, 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Szewczak AA; Moore PB; Chang YL; Wool IG The Conformation of the Sarcin/Ricin Loop from 28S Ribosomal RNA. Proc. Natl. Acad. Sci. U. S. A 1993, 90, 9581–9585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Endo Y; Glü A; Wool IG Ribosomal RNA Identity Elements for Ricin A-chain Recognition and Catalysis. J. Mol. Biol 1991, 221, 193–207. [DOI] [PubMed] [Google Scholar]
- (147).FitzGerald D; Pastan I Targeted Toxin Therapy for the Treatment of Cancer. JNCI: J. Natl. Cancer Inst 1989, 81, 1455–1463. [DOI] [PubMed] [Google Scholar]
- (148).Li C; Yan R; Yang Z; Wang H; Zhang R; Chen H; Wang J BCMab1-Ra, a Novel Immunotoxin that BCMab1 Antibody Coupled to Ricin A Chain, can Eliminate Bladder Tumor. Oncotarget 2017, 8, 46704–46705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Madhumathi J; Devilakshmi S; Sridevi S; Verma RS Immunotoxin Therapy for Hematologic Malignancies: Where are we Heading? Drug Discovery Today 2016, 21, 325–332. [DOI] [PubMed] [Google Scholar]
- (150).Winer LM; O’Dwyer J; Kitson J; Comis RL; Frankel AE; Bauer RJ; Konrad MS; Groves ES Phase I Evaluation of an Anti-breast Carcinoma Monoclonal Antibody 260F9-Recombinant Ricin A Chain Immunoconjugate. Cancer Res 1989, 49, 4062–4067. [PubMed] [Google Scholar]
- (151).Raso V; Ritz J; Basala M; Schlossman SF Monoclonal Antibody-Ricin A Chain Conjugate Selectively Cytotoxic for Cells Bearing the Common Acute Lymphoblastic Leukemia Antigen. Cancer Res 1982, 42, 457–464. [PubMed] [Google Scholar]
- (152).Pietersz GA; McKenzie IF Antibody Conjugates for the Treatment of Cancer. Immunol. Rev 1992, 129, 57–80. [DOI] [PubMed] [Google Scholar]
- (153).Teicher BA; Chari RV Antibody Conjugate Therapeutics: Challenges and Potential. Clin. Cancer Res 2011, 17, 6389–6397. [DOI] [PubMed] [Google Scholar]
- (154).Chen XY; Berti PJ; Schramm VL Ricin A-chain: Kinetic Isotope Effects and Transition State Structure with Stem-loop RNA. J. Am. Chem. Soc 2000, 122, 1609–1617. [Google Scholar]
- (155).Chen XY; Berti PJ; Schramm VL Transition-state Analysis for Depurination of DNA by Ricin A-chain. J. Am. Chem. Soc 2000, 122, 6527–6534. [Google Scholar]
- (156).Roday S; Amukele T; Evans GB; Tyler PC; Furneaux RH; Schramm VL Inhibition of Ricin A-chain with Pyrrolidine Mimics of the Oxacarbenium Ion Transition State. Biochemistry 2004, 43, 4923–4933. [DOI] [PubMed] [Google Scholar]
- (157).Roday S; Saen-oon S; Schramm VL Vinyldeoxyadenosine in a Sarcin-ricin RNA Loop and its Binding to Ricin Toxin A-chain. Biochemistry 2007, 46, 6169–6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Sturm MB; Schramm VL Detecting Ricin: Sensitive Luminescent Assay for Ricin A-chain Ribosome Depurination Kinetics. Anal. Chem 2009, 81, 2847–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Chen XY; Link TM; Schramm VL Ricin A-chain: Kinetics, Mechanism, and RNA Stem– loop Inhibitors. Biochemistry 1998, 37, 11605–11613. [DOI] [PubMed] [Google Scholar]
- (160).Sturm MB; Roday S; Schramm VL Circular DNA and DNA/RNA Hybrid Molecules as Scaffolds for Ricin Inhibitor Design. J. Am. Chem. Soc 2007, 129, 5544–5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Ho MC; Sturm MB; Almo SC; Schramm VL Transition State Analogues in Structures of Ricin and Saporin Ribosome-Inactivating Proteins. Proc. Natl. Acad. Sci. U. S. A 2009, 106 (48), 20276–20281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Montfort W; Villafranca JE; Monzingo AF; Ernst SR; Katzin B; Rutenber E; Xuong NH; Hamlin R; Robertus JD The Three-dimensional Structure of Ricin at 2.8 A. J. Biol. Chem 1987, 262, 5398–5403. [PubMed] [Google Scholar]
- (163).Shi WW; Tang YS; Sze SY; Zhu ZN; Wong KB; Shaw PC Crystal Structure of Ribosome-inactivating Protein Ricin A Chain in Complex with the C-Terminal Peptide of the Ribosomal Stalk Protein P2. Toxins 2016, 8, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Rudolph MJ; Vance DJ; Cheung J; Franklin MC; Burshteyn F; Cassidy MS; Gary EN; Herrera C; Shoemaker CB; Mantis NJ Crystal Structures of Ricin Toxin’s Enzymatic Subunit (RTA) in Complex with Neutralizing and Non-neutralizing Single-chain Antibodies. J. Mol. Biol 2014, 426, 3057–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Monzingo AF; Robertus JD X-ray Analysis of Substrate Analogs in the Ricin A-Chain Active Site. J. Mol. Biol 1992, 227, 1136–1145. [DOI] [PubMed] [Google Scholar]
- (166).Link T; Chen XY; Niu LH; Schramm VL A Hypothesis to Explain the Substrate Reactivity of Ribosomal and Stem-loop RNA with Ricin A-chain. Toxicon 1996, 34, 1317–1324. [DOI] [PubMed] [Google Scholar]
- (167).Tartarini A; Pittaluga E; Marcozzi G; Testone G; Rodrigues-Pousada RA; Giannino D; Spanò L Differential Expression of Saporin Genes Upon Wounding, ABA Treatment and Leaf Development. Physiol. Plant 2010, 140, 141–152. [DOI] [PubMed] [Google Scholar]
- (168).Fabbrini MS; Rappocciolo E; Carpani D; Solinas M; Valsasina B; Breme U; Cavallaro U; Nykjaer A; Rovida E; Legname G; Soria MR Characterization of a Saporin Isoform with Lower Ribosome-inhibiting Activity. Biochem. J 1997, 322, 719–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Ferreras J; Barbieri L; Girbeś T; Battelli MG; Rojo MA; Arias FJ; Rocher MA; Soriano F; Mendeź E; Stirpe F Distribution and Properties of Major Ribosome-inactivating Proteins (28 S rRNA N-Glycosidases) of the Plant Saponaria of f icinalis L. (Caryophyllaceae). Biochim. Biophys. Acta, Gene Struct. Expression 1993, 1216, 31–42. [DOI] [PubMed] [Google Scholar]
- (170).Benatti L; Saccardo MB; Dani M; Nitti G; Sassano M; Lorenzetti R; Lappi DA; Soria M Nucleotide Sequence of cDNA Coding for Saporin-6, a Type-1 Ribosome-inactivating Protein from Saponaria of f icinalis. Eur. J. Biochem 1989, 183, 465–470. [DOI] [PubMed] [Google Scholar]
- (171).Bagga S; Seth D; Batra JK The Cytotoxic Activity of Ribosome-inactivating Protein Saporin-6 is Attributed to its rRNA N-Glycosidase and Internucleosomal DNA Fragmentation Activities. J. Biol. Chem 2003, 278, 4813–4820. [DOI] [PubMed] [Google Scholar]
- (172).Stirpe F; Barbieri L Ribosome-inactivating Proteins Up to Date. FEBS Lett 1986, 195, 1–8. [DOI] [PubMed] [Google Scholar]
- (173).Yuan H; Du Q; Sturm MB; Schramm VL Soapwort Saporin L3 Expression in Yeast, Mutagenesis, and RNA Substrate Specificity. Biochemistry 2015, 54, 4565–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Mason JM; Yuan H; Evans GB; Tyler PC; Du Q; Schramm VL Oligonucleotide Transition State Analogues of Saporin L3. Eur. J. Med. Chem 2017, 127, 793–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Yuan H; Stratton CF; Schramm VL Transition State Structure of RNA Depurination by Saporin L3. ACS Chem. Biol 2016, 11, 1383–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Westaway KC; Waszczylo Z Isotope Effects in Nucleophilic Substitution Reactions. IV. The Effect of Changing a Substituent at the α Carbon on the Structure of SN2 Transition States. Can. J. Chem 1982, 60, 2500–2520. [Google Scholar]
- (177).Matsson O; Dybala-Defratyka A; Rostkowski M; Paneth P; Westaway KC A Theoretical Investigation of α-Carbon Kinetic Isotope Effects and Their Relationship to the Transition-state Structure of SN2 Reactions. J. Org. Chem 2005, 70, 4022–4027. [DOI] [PubMed] [Google Scholar]
- (178).Sturm MB; Tyler PC; Evans GB; Schramm VL Transition State Analogues Rescue Ribosomes from Saporin-L1 Ribosome Inactivating Protein. Biochemistry 2009, 48, 9941–9948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (179).Kornberg A; Lieberman I; Simms ES Enzymatic Synthesis and Properties of 5-Phosphoribosylpyrophosphate. J. Biol. Chem 1955, 215, 389–402. [PubMed] [Google Scholar]
- (180).Nyhan WL The Recognition of Lesch-Nyhan Syndrome as an Inborn Error of Purine Metabolism. J. Inherited Metab. Dis 1997, 20, 171–178. [DOI] [PubMed] [Google Scholar]
- (181).Torres RJ; Puig JG Lesch-Nyhan Syndrome. Drugs Future 2010, 35, 421–427. [Google Scholar]
- (182).McDonald JA; Kelley WN Lesch-Nyhan Syndrome: Altered Kinetic Properties of Mutant Enzyme. Science 1971, 171, 689–691. [DOI] [PubMed] [Google Scholar]
- (183).Sherman IW Biochemistry of Plasmodium (malarial parasites). Microbiol. Rev 1979, 43, 453–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Reyes P; Rathod PK; Sanchez DJ; Mrema JE; Rieckmann KH; Heidrich HG Enzymes of Purine and Pyrimidine Metabolism from the Human Malaria Parasite, Plasmodium falciparum. Mol. Biochem. Parasitol 1982, 5, 275–290. [DOI] [PubMed] [Google Scholar]
- (185).De Koning HP; Bridges DJ; Burchmore RJ Purine and Pyrimidine Transport in Pathogenic Protozoa: from Biology to Therapy. FEMS Microbiol. Rev 2005, 29, 987–1020. [DOI] [PubMed] [Google Scholar]
- (186).Chaudhary K; Roos DS Protozoan Genomics for Drug Discovery. Nat. Biotechnol 2005, 23, 1089–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Ullman B; Carter D Hypoxanthine-guanine Phosphoribosyl-transferase as a Therapeutic Target in Protozoal Infections. Infect. Agents Dis 1995, 4, 29–40. [PubMed] [Google Scholar]
- (188).Li CM; Tyler PC; Furneaux RH; Kicska G; Xu Y; Grubmeyer C; Girvin ME; Schramm VL Transition-state Analogs as Inhibitors of Human and Malarial Hypoxanthine-guanine Phosphoribosyltransferases. Nat. Struct. Biol 1999, 6, 582–587. [DOI] [PubMed] [Google Scholar]
- (189).Hazleton KZ; Ho MC; Cassera MB; Clinch K; Crump DR; Rosario I Jr; Merino EF; Almo SC; Tyler PC; Schramm VL Acyclic Immucillin Phosphonates: Second-generation Inhibitors of Plasmodium falciparum Hypoxanthine-guanine-xanthine Phosphor-ibosyltransferase. Chem. Biol 2012, 19, 721–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (190).Shi W; Li CM; Tyler PC; Furneaux RH; Grubmeyer C; Schramm VL; Almo SC The 2.0 Å Structure of Human Hypoxanthine-guanine Phosphoribosyltransferase in Complex with a Transition-state Analog Inhibitor. Nat. Struct. Biol 1999, 6, 588–593. [DOI] [PubMed] [Google Scholar]
- (191).Shi W; Li CM; Tyler PC; Furneaux RH; Cahill SM; Girvin ME; Grubmeyer C; Schramm VL; Almo SC The 2.0 Å Structure of Malarial Purine Phosphoribosyltransferase in Complex with a Transition-state Analogue Inhibitor. Biochemistry 1999, 38, 9872–9880. [DOI] [PubMed] [Google Scholar]
- (192).Keough DT; Brereton IM; De Jersey J; Guddat LW The Crystal Structure of Free Human Hypoxanthine-guanine Phosphor-ibosyltransferase Reveals Extensive Conformational Plasticity Throughout the Catalytic Cycle. J. Mol. Biol 2005, 351, 170–181. [DOI] [PubMed] [Google Scholar]
- (193).Ducati RG; Firestone RS; Schramm VL Kinetic Isotope Effects and Transition State Structure for Hypoxanthine-guanine-xanthine Phosphoribosyltransferase from Plasmodium falciparum. Biochemistry 2017, 56, 6368–6376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Murkin AS; Birck MR; Rinaldo-Matthis A; Shi W; Taylor DA; Schramm VL Neighboring Group Participation in the Transition State of Human Purine Nucleoside Phosphorylase. Biochemistry 2007, 46, 5038–5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Deng H; Callender R; Schramm VL; Grubmeyer C Pyrophosphate Activation in Hypoxanthine–guanine Phosphoribosyl-transferase with Transition State Analogue. Biochemistry 2010, 49, 2705–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (196).Karnawat V; Gogia S; Balaram H; Puranik M Differential Distortion of Purine Substrates by Human and Plasmodium falciparum Hypoxanthine-guanine phosphoribosyltransferase to Catalyse the Formation of Mononucleotides. ChemPhysChem 2015, 16, 2172–2181. [DOI] [PubMed] [Google Scholar]
- (197).Xu Y; Grubmeyer C Catalysis in Human Hypoxanthine-guanine phosphoribosyltransferase: Asp 137 Acts as a General Acid/ Base. Biochemistry 1998, 37, 4114–4124. [DOI] [PubMed] [Google Scholar]
- (198).Hocková D; Holý A; Masojídková M; Keough DT; de Jersey J; Guddat LW Synthesis of Branched 9-[2-(2-phosphonoe-thoxy) ethyl] Purines as a New Class of Acyclic Nucleoside Phosphonates Which Inhibit Plasmodium falciparum Hypoxanthine– guanine–xanthine Phosphoribosyltransferase. Bioorg. Med. Chem 2009, 17, 6218–6232. [DOI] [PubMed] [Google Scholar]
- (199).Hocková D; Keough DT; Janeba Z; Wang TH; de Jersey J; Guddat LW Synthesis of Novel N-branched Acyclic Nucleoside Phosphonates as Potent and Selective Inhibitors of Human, Plasmodium falciparum and Plasmodium vivax 6-Oxopurine Phosphor-ibosyltransferases. J. Med. Chem 2012, 55, 6209–6223. [DOI] [PubMed] [Google Scholar]
- (200).Špaček P; Keough DT; Chavchich M; Drací̌nsky M; Janeba Z; Naesens L; Edstein MD; Guddat LW; Hockova D Synthesis and Evaluation of Symmetric Acyclic Nucleoside Bisphosphonates as Inhibitors of the Plasmodium falciparum, Plasmodium vivax and Human 6-Oxopurine Phosphoribosyltransferases and the Antimalarial Activity of their Prodrugs. Bioorg. Med. Chem 2017, 25, 4008–4030. [DOI] [PubMed] [Google Scholar]
- (201).Keough DT; Rejman D; Pohl R; Zborníková E; Hocková D; Croll T; Edstein MD; Birrell GW; Chavchich M; Naesens LM; Pierens GK Design of Plasmodium vivax Hypoxanthine-guanine phosphoribosyltransferase Inhibitors as Potential Antimalarial Therapeutics. ACS Chem. Biol 2018, 13, 82–90. [DOI] [PubMed] [Google Scholar]
- (202).Clinch K; Crump DR; Evans GB; Hazleton KZ; Mason JM; Schramm VL; Tyler PC Acyclic Phosph(on)ate Inhibitors of Plasmodium falciparum Hypoxanthine-guanine-xanthine Phosphoribosyltransferase. Bioorg. Med. Chem 2013, 21, 5629–5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Sinokrot H; Smerat T; Najjar A; Karaman R Advanced Prodrug Strategies in Nucleoside and Non-nucleoside Antiviral Agents: A Review of the Recent Five Years. Molecules 2017, 22, 1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).De Clercq E; Field HJ Antiviral Prodrugs–the Development of Successful Prodrug Strategies for Antiviral Chemotherapy. Br. J. Pharmacol 2006, 147, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Spillman NJ; Kirk K The Malaria Parasite Cation ATPase PfATP4 and Its Role in the Mechanism of Action of a New Arsenal of Antimalarial Drugs. Int. J. Parasitol.: Drugs Drug Resist 2015, 5, 149–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (206).Giesler KE; Liotta DC Next-generation Reduction Sensitive Lipid Conjugates of Tenofovir: Antiviral Activity and Mechanism of Release. J. Med. Chem 2016, 59, 10244–10252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Allan D; Michell RH A Calcium-activated Polyphosphoi-nositide Phosphodiesterase in the Plasma Membrane of Human and Rabbit Erythrocytes. Biochim. Biophys. Acta, Biomembr 1978, 508, 277–286. [DOI] [PubMed] [Google Scholar]
- (208).Hostetler KY Alkoxyalkyl Prodrugs of Acyclic Nucleoside Phosphonates Enhance Oral Antiviral Activity and Reduce Toxicity: Current State of the Art. Antiviral Res 2009, 82, A84–A98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Ferrell JE; Huestis WH Phosphoinositide Metabolism and the Morphology of Human Erythrocytes. J. Cell Biol 1984, 98, 1992–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (210).Hanada K; Palacpac NMQ; Magistrado PA; Kurokawa K; Rai G; Sakata D; Hara T; Horii T; Nishijima M; Mitamura T Plasmodium falciparum Phospholipase C Hydrolyzing Sphingomyelin and Lysocholinephospholipids is a Possible Target for Malaria Chemotherapy. J. Exp. Med 2002, 195, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (211).Hanada K; Mitamura T; Fukasawa M; Magistrado PA; Horii T; Nishijima M Neutral Sphingomyelinase Activity Dependent on Mg2+ and Anionic Phospholipids in the Intraerythrocytic Malaria Parasite Plasmodium falciparum. Biochem. J 2000, 346, 671–677. [PMC free article] [PubMed] [Google Scholar]
- (212).Downie MJ; Kirk K; Mamoun CB Purine Salvage Pathways in the Intraerythrocytic Malaria Parasite Plasmodium falciparum. Eukaryotic Cell 2008, 7, 1231–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (213).Gero AM; Brown GV; O’Sullivan WJ Pyrimidine De Novo Synthesis During the Life Cycle of the Intraerythrocytic Stage of Plasmodium falciparum. J. Parasitol 1984, 70, 536–541. [PubMed] [Google Scholar]
- (214).Seymour KK; Lyons SD; Phillips L; Rieckmann KH; Christopherson RI Cytotoxic Effects of Inhibitors of De Novo Pyrimidine Biosynthesis Upon Plasmodium falciparum. Biochemistry 1994, 33, 5268–5274. [DOI] [PubMed] [Google Scholar]
- (215).Painter HJ; Morrisey JM; Mather MW; Vaidya AB Specific Role of Mitochondrial Electron Transport in Blood-stage Plasmodium falciparum. Nature 2007, 446, 88–91. [DOI] [PubMed] [Google Scholar]
- (216).Queen SA; Jagt DL; Reyes P In Vitro Susceptibilities of Plasmodium falciparum to Compounds which Inhibit Nucleotide Metabolism. Antimicrob. Agents Chemother 1990, 34, 1393–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (217).Krungkrai J; Krungkrai SR; Phakanont K Antimalarial Activity of Orotate Analogs that Inhibit Dihydroorotase and Dihydroorotate Dehydrogenase. Biochem. Pharmacol 1992, 43, 1295–1301. [DOI] [PubMed] [Google Scholar]
- (218).Pavadai E; El Mazouni F; Wittlin S; de Kock C; Phillips MA; Chibale K Identification of New Human Malaria Parasite Plasmodium falciparum Dihydroorotate Dehydrogenase Inhibitors by Pharmacophore and Structure-based Virtual Screening. J. Chem. Inf. Model 2016, 56, 548–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (219).Zhu J; Han L; Diao Y; Ren X; Xu M; Xu L; Li S; Li Q; Dong D; Huang J; et al. Design, Synthesis, X-ray Crystallographic Analysis, and Biological Evaluation of Thiazole Derivatives as Potent and Selective Inhibitors of Human Dihydroorotate Dehydrogenase. J. Med. Chem 2015, 58, 1123–1139. [DOI] [PubMed] [Google Scholar]
- (220).Abdo M; Zhang Y; Schramm VL; Knapp S Electrophilic Aromatic Selenylation: New OPRT Inhibitors. Org. Lett 2010, 12, 2982–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (221).Zhang Y; Luo M; Schramm VL Transition States of Plasmodium falciparum and Human Orotate Phosphoribosyltransferases. J. Am. Chem. Soc 2009, 131, 4685–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (222).Zhang Y; Evans GB; Clinch K; Crump DR; Harris LD; Fröhlich RF; Tyler PC; Hazleton KZ; Cassera MB; Schramm VL Transition State Analogues of Plasmodium falciparum and Human Orotate Phosphoribosyltransferases. J. Biol. Chem 2013, 288, 34746–34754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (223).Zhang Y; Deng H; Schramm VL Leaving Group Activation and Pyrophosphate Ionic State at the Catalytic Site of Plasmodium falciparum Orotate Phosphoribosyltransferase. J. Am. Chem. Soc 2010, 132, 17023–17031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (224).Lunev S; Batista FA; Bosch SS; Wrenger C; Groves MR Identification and Validation of Novel Drug Targets for the Treatment of Plasmodium falciparum Malaria: New Insights. Current Topics in Malaria; InTechOpen, 2016; Chapter 12. [Google Scholar]
- (225).Witte JF; Bray KE; Thornburg CK; McClard RW ‘Irreversible’ Slow-onset Inhibition of Orotate Phosphoribosyltransferase by an Amidrazone Phosphate Transition-state Mimic. Bioorg. Med. Chem. Lett 2006, 16, 6112–6115. [DOI] [PubMed] [Google Scholar]
- (226).Gonzaĺez-Segura L; Witte JF; McClard RW; Hurley TD Ternary Complex Formation and Induced Asymmetry in Orotate Phosphoribosyltransferase. Biochemistry 2007, 46, 14075–14086. [DOI] [PubMed] [Google Scholar]
- (227).Tabor CW; Tabor H Polyamines. Annu. Rev. Biochem 1984, 53, 749–790. [DOI] [PubMed] [Google Scholar]
- (228).Michael AJ Polyamines in Eukaryotes, Bacteria, and Archaea. J. Biol. Chem 2016, 291, 14896–14903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).Dong M; Kathiresan V; Fenwick MK; Torelli AT; Zhang Y; Caranto JD; Dzikovski B; Sharma A; Lancaster KM; Freed J; et al. Organometallic and Radical Intermediates Reveal Mechanism of Diphthamide Biosynthesis. Science 2018, 359, 1247–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Mato JM; Martínez-Chantar ML; Lu SC S-Adenosylmethionine Metabolism and Liver Disease. Ann. Hepatol 2015, 12, 183–189. [PMC free article] [PubMed] [Google Scholar]
- (231).Broderick JB; Duffus BR; Duschene KS; Shepard EM Radical S-Adenosylmethionine Enzymes. Chem. Rev 2014, 114, 4229–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (232).Laurino P; Tawfik DS Spontaneous Emergence of S-Adenosylmethionine and the Evolution of Methylation. Angew. Chem., Int. Ed 2017, 56, 343–345. [DOI] [PubMed] [Google Scholar]
- (233).Nobori T; Takabayashi K; Tran P; Orvis L; Batova A; Yu AL; Carson DA Genomic Cloning of Methylthioadenosine Phosphorylase: A Purine Metabolic Enzyme Deficient in Multiple Different Cancers. Proc. Natl. Acad. Sci. U. S. A 1996, 93, 6203–6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (234).Sauter M; Moffatt B; Saechao MC; Hell R; Wirtz M Methionine Salvage and S-Adenosylmethionine: Essential Links Between Sulfur, Ethylene and Polyamine Biosynthesis. Biochem. J 2013, 451, 145–154. [DOI] [PubMed] [Google Scholar]
- (235).Igarashi K; Katoh Y Metabolic Aspects of Epigenome: Coupling of S-Adenosylmethionine Synthesis and Gene Regulation on Chromatin by SAMIT Module. Subcell. Biochem 2013, 61, 105–118. [DOI] [PubMed] [Google Scholar]
- (236).Quinlan CL; Kaiser SE; Bolaños B; Nowlin D; Grantner R; Karlicek-Bryant S; Feng JL; Jenkinson S; Freeman-Cook K; Dann SG; et al. Targeting S-Adenosylmethionine Biosynthesis with a Novel Allosteric Inhibitor of Mat2A. Nat. Chem. Biol 2017, 13, 785–792. [DOI] [PubMed] [Google Scholar]
- (237).Halim AB; LeGros L; Chamberlin ME; Geller A; Kotb M Regulation of the Human MAT2A Gene Encoding the Catalytic α2 Subunit of Methionine Adenosyltransferase, MAT II Gene Organization, Promoter Characterization, and Identification of a Site in the Proximal Promoter that is Essential for its Activity. J. Biol. Chem 2001, 276, 9784–9791. [DOI] [PubMed] [Google Scholar]
- (238).Singh V; Shi W; Evans GB; Tyler PC; Furneaux RH; Almo SC; Schramm VL Picomolar Transition State Analogue Inhibitors of Human 5′-Methylthioadenosine Phosphorylase and X-ray structure with MT-Immucillin-A. Biochemistry 2004, 43, 9–18. [DOI] [PubMed] [Google Scholar]
- (239).Evans GB; Furneaux RH; Lenz DH; Painter GF; Schramm VL; Singh V; Tyler PC Second Generation Transition State Analogue Inhibitors of Human 5′-Methylthioadenosine Phosphorylase. J. Med. Chem 2005, 48, 4679–4689. [DOI] [PubMed] [Google Scholar]
- (240).Basu I; Cordovano G; Das I; Belbin TJ; Guha C; Schramm VL A Transition State Analogue of 5′-Methylthioadenosine Phosphorylase Induces Apoptosis in Head and Neck Cancers. J. Biol. Chem 2007, 282, 21477–21486. [DOI] [PubMed] [Google Scholar]
- (241).Basu I; Locker J; Cassera MB; Belbin TJ; Merino EF; Dong X; Hemeon I; Evans GB; Guha C; Schramm VL Growth and Metastases of Human Lung Cancer Are Inhibited in Mouse Xenografts by a Transition State Analogue of 5′-Methylthioadenosine Phosphorylase. J. Biol. Chem 2011, 286, 4902–4911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Oliveira S. F. V. d.; Ganzinelli M; Chila R; Serino L; Maciel ME; Urban C. d. A.; Lima R. S. d.; Cavalli IJ; Generali D; Broggini M; et al. Characterization of MTAP Gene Expression in Breast Cancer Patients and Cell Lines. PLoS One 2016, 11 (1), e0145647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (243).Xio S; Li D; Vijg J; Sugarbaker DJ; Corson JM; Fletcher JA Codeletion of p15 and p16 in Primary Malignant Mesothelioma. Oncogene 1995, 11, 511–515. [PubMed] [Google Scholar]
- (244).Chen ZH; Zhang H; Savarese TM Gene Deletion Chemoselectivity: Codeletion of the Genes for p16INK4, Methyl-thioadenosine Phosphorylase, and the α-and β-Interferons in Human Pancreatic Cell Carcinoma Lines and its Implications for Chemo-therapy. Cancer Res 1996, 5, 1083–1090. [PubMed] [Google Scholar]
- (245).Marjon K; Cameron MJ; Quang P; Clasquin MF; Mandley E; Kunii K; McVay M; Choe S; Kernytsky A; Gross S; et al. MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Rep 2016, 15, 574–587. [DOI] [PubMed] [Google Scholar]
- (246).Mavrakis KJ; McDonald ER 3rd.; Schlabach MR; Billy E; Hoffman GR; deWeck A; Ruddy DA; Venkatesan K; Yu J; McAllister G; et al. Disordered Methionine Metabolism in MTAP/ CDKN2A-Deleted Cancers Leads to Dependence on PRMT5. Science 2016, 351, 1208–1213. [DOI] [PubMed] [Google Scholar]
- (247).Kryukov GV; Wilson FH; Ruth JR; Paulk J; Tsherniak A; Marlow SE; Vazquez F; Weir BA; Fitzgerald ME; Tanaka M; Bielski CM MTAP Deletion Confers Enhanced Dependency on the PRMT5 Arginine Methyltransferase in Cancer Cells. Science 2016, 351, 1214–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (248).Pfister SX; Ashworth A Marked for Death: Targeting Epigenetic Changes in Cancer. Nat. Rev. Drug Discovery 2017, 16, 241–263. [DOI] [PubMed] [Google Scholar]
- (249).Duncan KW; Chesworth R; Boriack-Sjodin PA; Munchhof MJ; Jin L (Epizyme Inc.) Prmt5 Inhibitors Containing a Dihydro-or Tetrahydroisoquinoline and Uses Thereof. U.S. Patent US 9,745,291, August 29, 2017.
- (250).Duncan KW; Chesworth R; Boriack-Sjodin PA; Munchhof M; Jin L; Penebre E; Barbash OI; inventors (GlaxoSmithKline intellectual property development Ltd., Epizyme Inc.) Prmt5 Inhibitors and Uses Thereof. U.S. Patent Application US 15/501,550, August 10, 2017.
- (251).Penebre E; Kuplast KG; Majer CR; Johnston LD; Rioux N; Munchhof M; Jin L; Boriak-Sjodin A; Wigle TJ; Jacques SL; et al. Identification of a First-in-class PRMT5 Inhibitor with Potent In Vitro and In Vivo Activity in Preclinical Models of Mantle Cell Lymphoma. Blood 2014, 124, 438. [Google Scholar]
- (252).Appleby TC; Erion MD; Ealick SE The Structure of Human 5′-Deoxy-5′-Methylthioadenosine Phosphorylase at 1.7 Å Resolution Provides Insights into Substrate Binding and Catalysis. Structure 1999, 7, 629–641. [DOI] [PubMed] [Google Scholar]
- (253).Schramm VL Enzymatic Transition States: Thermodynamics, Dynamics and Analogue Design. Arch. Biochem. Biophys 2005, 433, 13–26. [DOI] [PubMed] [Google Scholar]
- (254).Guan R; Ho MC; Brenowitz M; Tyler PC; Evans GB; Almo SC; Schramm VL Entropy-driven Binding of Picomolar Transition State Analogue Inhibitors to Human 5′-Methylthioadenosine Phosphorylase. Biochemistry 2011, 50, 10408–10417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (255).Firestone RS; Cameron SA; Karp JM; Arcus VL; Schramm VL Heat Capacity Changes for Transition-State Analogue Binding and Catalysis with Human 5′-Methylthioadenosine Phosphor-ylase. ACS Chem. Biol 2017, 12, 464–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (256).Eftink MR; Anusiem AC; Biltonen RL Enthalpy-entropy Compensation and Heat Capacity Changes for Protein-Ligand Interactions: General Thermodynamic Models and Data for the Binding of Nucleotides to Ribonuclease. Biochemistry 1983, 22, 3884–3896. [DOI] [PubMed] [Google Scholar]
- (257).Hobbs JK; Jiao W; Easter AD; Parker EJ; Schipper LA; Arcus VL Change in Heat Capacity for Enzyme Catalysis Determines Temperature Dependence of Enzyme Catalyzed Rates. ACS Chem. Biol 2013, 8, 2388–2393. [DOI] [PubMed] [Google Scholar]
- (258).Arcus VL; Prentice EJ; Hobbs JK; Mulholland AJ; Van der Kamp MW; Pudney CR; Parker EJ; Schipper LA On the Temperature Dependence of Enzyme-Catalyzed Rates. Biochemistry 2016, 55, 1681–1688. [DOI] [PubMed] [Google Scholar]
- (259).Bergwitz C; Jüppner H Phosphate Sensing. Adv. Chronic Kidney Dis 2011, 18, 132–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (260).Ferro AJ; Barrett A; Shapiro SK Kinetic Properties and the Effect of Substrate Analogues on 5′-Methylthioadenosine Nucleosidase from Escherichia coli. Biochim. Biophys. Acta 1976, 438, 487–494. [DOI] [PubMed] [Google Scholar]
- (261).Della Ragione F; Porcelli M; Cartenì-Farina M; Zappia V; Pegg AE Escherichia coli S-Adenosylhomocysteine/5′-Methylthioa-denosine Nucleosidase. Purification, Substrate Specificity and Mechanism of Action. Biochem. J 1985, 232, 335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (262).Li X; Apel D; Gaynor EC; Tanner ME 5′-Methylthioadenosine Nucleosidase is Implicated in Playing a Key Role in a Modified Futalosine Pathway for Menaquinone Biosynthesis in Campylobacter jejuni. J. Biol. Chem 2011, 286, 19392–19398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (263).Barta ML; Thomas K; Yuan H; Lovell S; Battaile KP; Schramm VL; Hefty PS Structural and Biochemical Characterization of Chlamydia trachomatis Hypothetical Protein CT263 Supports that Menaquinone Synthesis Occurs Through the Futalosine Pathway. J. Biol. Chem 2014, 289, 32214–32229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (264).Park EY; Oh SI; Nam MJ; Shin JS; Kim KN; Song HK Crystal Structure of 5′-Methylthioadenosine Nucleosidase from Arabidopsis thaliana at 1.5-Å Resolution. Proteins: Struct., Funct., Genet 2006, 65, 519–523. [DOI] [PubMed] [Google Scholar]
- (265).Parveen N; Cornell KA Methylthioadenosine/S-Adeno-sylhomocysteine Nucleosidase, a Critical Enzyme for Bacterial Metabolism. Mol. Microbiol 2011, 79, 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (266).Lee JE; Cornell KA; Riscoe MK; Howell PL Structure of E. coli 5′-Methylthioadenosine/S-Adenosylhomocysteine Nucleosi-dase Reveals Similarity to The Purine Nucleoside Phosphorylases. Structure 2001, 9, 941–953. [DOI] [PubMed] [Google Scholar]
- (267).Sedlmayer F; Hell D; Müller M; Auslan̈der D; Fussenegger M Designer Cells Programming Quorum-Sensing Interference with Microbes. Nat. Commun 2018, 9, 1822 DOI: 10.1038/s41467-018-04223-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (268).Sauter M; Moffatt B; Saechao MC; Hell R; Wirtz M Methionine Salvage and S-Adenosylmethionine: Essential Links Between Sulfur, Ethylene and Polyamine Biosynthesis. Biochem. J 2013, 451, 145–154. [DOI] [PubMed] [Google Scholar]
- (269).Sekowska A; Danchin A Identification of yrrU as the Methylthioadenosine Nucleosidase Gene in Bacillus subtilis. DNA Res 1999, 6, 255–264. [DOI] [PubMed] [Google Scholar]
- (270).Sekowska A; Deńervaud V; Ashida H; Michoud K; Haas D; Yokota A; Danchin A Bacterial Variations on the Methionine Salvage Pathway. BMC Microbiol 2004, 4, 9 (open access). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (271).Bao Y; Li Y; Jiang Q; Zhao L; Xue T; Hu B; Sun B Methylthioadenosine/S-Adenosylhomocysteine Nucleosidase (Pfs) of Staphylococcus aureus is Essential for the Virulence Independent of LuxS/AI-2 System. Int. J. Med. Microbiol 2013, 303, 190–200. [DOI] [PubMed] [Google Scholar]
- (272).Gutierrez JA; Crowder T; Rinaldo-Matthis A; Ho MC; Almo SC; Schramm VL Transition State Analogs of 5′-Methylthioadenosine Nucleosidase Disrupt Quorum Sensing. Nat. Chem. Biol 2009, 5, 251–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (273).Parveen N; Cornell KA; Bono JL; Chamberland C; Rosa P; Leong JM Bgp, a Secreted Glycosaminoglycan-Binding Protein of Borrelia burgdorferi Strain N40, Displays Nucleosidase Activity and is Not Essential for Infection of Immunodeficient Mice. Infect. Immun 2006, 74, 3016–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).Cornell KA; Primus S; Martinez JA; Parveen N Assessment of Methylthioadenosine/S-Adenosylhomocysteine Nucle-osidases of Borrelia burgdorferi as Targets for Novel Antimicrobials Using a Novel High-Throughput Method. J. Antimicrob. Chemother 2009, 63, 1163–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Chen X; Schauder S; Potier N; Van Dorsselaer A; Pelczer I; Bassler BL; Hughson FM Structural Identification of a Bacterial Quorum-Sensing Signal Containing Boron. Nature 2002, 415, 545–549. [DOI] [PubMed] [Google Scholar]
- (276).Roy V; Adams BL; Bentley WE Developing Next Generation Antimicrobials by Intercepting AI-2 Mediated Quorum Sensing. Enzyme Microb. Technol 2011, 49, 113–123. [DOI] [PubMed] [Google Scholar]
- (277).Schauder S; Shokat K; Surette MG; Bassler BL The LuxS Family of Bacterial Autoinducers: Biosynthesis of a Novel Quorum-Sensing Signal Molecule. Mol. Microbiol 2001, 41, 463–476. [DOI] [PubMed] [Google Scholar]
- (278).Miller MB; Bassler BL Quorum Sensing in Bacteria. Annu. Rev. Microbiol 2001, 55, 165–199. [DOI] [PubMed] [Google Scholar]
- (279).Papenfort K; Bassler BL Quorum Sensing Signal–Response Systems in Gram-Negative Bacteria. Nat. Rev. Microbiol 2016, 14, 576–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (280).Whiteley M; Diggle SP; Greenberg EP Progress in and Promise of Bacterial Quorum Sensing Research. Nature 2017, 551, 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (281).Singh V; Lee JE; Nuń̃ez S; Howell PL; Schramm VL Transition State Structure of 5′-Methylthioadenosine/S-Adenosylho-mocysteine Nucleosidase from Escherichia coli and its Similarity to Transition State Analogues. Biochemistry 2005, 44, 11647–11659. [DOI] [PubMed] [Google Scholar]
- (282).Singh V; Schramm VL Transition-State Analysis Of S. Pneumoniae 5′-Methylthioadenosine Nucleosidase. J. Am. Chem. Soc 2007, 129, 2783–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (283).Singh V; Luo M; Brown RL; Norris GE; Schramm VL Transition-State Structure of Neisseria meningitides 5′-Methylthioa-denosine/S-Adenosylhomocysteine Nucleosidase. J. Am. Chem. Soc 2007, 129, 13831–13833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (284).Namanja-Magliano HA; Stratton CF; Schramm VL Transition State Structure and Inhibition of Rv0091, a 5′-Deoxyadenosine/5′-Methylthioadenosine Nucleosidase from Myco-bacterium tuberculosis. ACS Chem. Biol 2016, 11, 1669–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (285).Gutierrez JA; Luo M; Singh V; Li L; Brown RL; Norris GE; Evans GB; Furneaux RH; Tyler PC; Painter GF; et al. Picomolar Inhibitors as Transition-State Probes of 5′-Methylthioade-nosine Nucleosidases. ACS Chem. Biol 2007, 2, 725–734. [DOI] [PubMed] [Google Scholar]
- (286).Amyes TL; Richard JP Rational Design of Transition-state Analogues as Potent Enzyme Inhibitors with Therapeutic Applications. ACS Chem. Biol 2007, 2, 711–714. [DOI] [PubMed] [Google Scholar]
- (287).Singh V; Evans GB; Lenz DH; Mason JM; Clinch K; Mee S; Painter GF; Tyler PC; Furneaux RH; Lee JE; et al. Femtomolar Transition State Analogue Inhibitors of 5′-Methylthioa-denosine/S-Adenosylhomocysteine Nucleosidase from Escherichia coli. J. Biol. Chem 2005, 280, 18265–18273. [DOI] [PubMed] [Google Scholar]
- (288).Lewis WG; Green LG; Grynszpan F; Radić Z; Carlier PR; Taylor P; Finn MG; Sharpless KB Click Chemistry in Situ: Acetylcholinesterase as a Reaction Vessel for the Selective Assembly of a Femtomolar Inhibitor from an Array of Building Blocks. Angew. Chem., Int. Ed 2002, 41, 1053–1057. [DOI] [PubMed] [Google Scholar]
- (289).Krasinśki A; Radic Z; Manetsch R; Raushel J; Taylor P; Sharpless KB; Kolb HC In Situ Selection of Lead Compounds by Click Chemistry: Target-Guided Optimization of Acetylcholinesterase Inhibitors. J. Am. Chem. Soc 2005, 127, 6686–6692. [DOI] [PubMed] [Google Scholar]
- (290).Lee JE; Luong W; Huang DJ; Cornell KA; Riscoe MK; Howell PL Mutational Analysis of a Nucleosidase Involved in Quorum-Sensing Autoinducer-2 Biosynthesis. Biochemistry 2005, 44, 11049–11057. [DOI] [PubMed] [Google Scholar]
- (291).Lee JE; Smith GD; Horvatin C; Huang DJ; Cornell KA; Riscoe MK; Howell PL Structural Snapshots of MTA/Adohcy Nucleosidase Along the Reaction Coordinate Provide Insights into Enzyme and Nucleoside Flexibility During Catalysis. J. Mol. Biol 2005, 352, 559–574. [DOI] [PubMed] [Google Scholar]
- (292).Thomas K; Haapalainen AM; Burgos ES; Evans GB; Tyler PC; Gulab S; Guan R; Schramm VL Femtomolar Inhibitors Bind to 5′-Methylthioadenosine Nucleosidases With Favorable Enthalpy and Entropy. Biochemistry 2012, 51, 7541–7550. [DOI] [PubMed] [Google Scholar]
- (293).Motley MW; Schramm VL; Schwartz SD Conformational Freedom in Tight Binding Enzymatic Transition-State Analogues. J. Phys. Chem. B 2013, 117, 9591–9597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (294).Mader MM; Bartlett PA Binding Energy and Catalysis: The Implications for Transition-State Analogs and Catalytic Antibodies. Chem. Rev 1997, 97, 1281–1302. [DOI] [PubMed] [Google Scholar]
- (295).Harel M; Quinn DM; Nair HK; Silman I; Sussman JL The X-ray Structure of a Transition State Analog Complex Reveals the Molecular Origins of the Catalytic Power and Substrate Specificity of Acetylcholinesterase. J. Am. Chem. Soc 1996, 118, 2340–2346. [Google Scholar]
- (296).Eletsky A; Kienhöfer A; Hilvert D; Pervushin K Investigation of Ligand Binding and Protein Dynamics in Bacillus subtilis Chorismate Mutase by Transverse Relaxation Optimized Spectroscopy-Nuclear Magnetic Resonance. Biochemistry 2005, 44, 6788–6799. [DOI] [PubMed] [Google Scholar]
- (297).Wang F; Shi W; Nieves E; Angeletti RH; Schramm VL; Grubmeyer C A Transition-state Analogue Reduces Protein Dynamics in Hypoxanthine-Guanine Phosphoribosyltransferase. Biochemistry 2001, 40, 8043–8054. [DOI] [PubMed] [Google Scholar]
- (298).Finkelstein IJ; Ishikawa H; Kim S; Massari AM; Fayer MD Substrate Binding and Protein Conformational Dynamics Measured by 2D-IR Vibrational Echo Spectroscopy. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 2637–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (299).Seckler JM; Leioatts N; Miao H; Grossfield A The Interplay of Structure and Dynamics: Insights from a Survey of HIV-1 Reverse Transcriptase Crystal Structures. Proteins: Struct., Funct., Genet 2013, 81, 1792–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (300).Wang S; Lim J; Thomas K; Yan F; Angeletti RH; Schramm VL A Complex of Methylthioadenosine/S-Adenosylho-mocysteine Nucleosidase, Transition State Analogue, and Nucleophilic Water Identified by Mass Spectrometry. J. Am. Chem. Soc 2012, 134, 1468–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (301).Schroeder GK; Zhou L; Snider MJ; Chen X; Wolfenden R Flight of a Cytidine Deaminase Complex with an Imperfect Transition State Analogue Inhibitor: Mass Spectrometric Evidence for the Presence of a Trapped Water Molecule. Biochemistry 2012, 51, 6476–6486. [DOI] [PubMed] [Google Scholar]
- (302).Borchers CH; Marquez VE; Schroeder GK; Short SA; Snider MJ; Speir JP; Wolfenden R Fourier Transform Ion Cyclotron Resonance MS Reveals the Presence of a Water Molecule in an Enzyme Transition-State Analogue Complex. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 15341–15345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (303).Masi M; Ref́regiers M; Pos KM; Pages̀ JM Mechanisms of Envelope Permeability and Antibiotic Influx and Efflux in Gram-Negative Bacteria. Nature Microbiol 2017, 2, 17001. [DOI] [PubMed] [Google Scholar]
- (304).Zgurskaya HI; Loṕez CA; Gnanakaran S Permeability Barrier of Gram-Negative Cell Envelopes and Approaches to Bypass it. ACS Infect. Dis 2015, 1, 512–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (305).Miller SI; Salama NR The Gram-Negative Bacterial Periplasm: Size Matters. PLoS Biol 2018, 16, e2004935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (306).Hultqvist LD; Alhede M; Jakobsen TH; Givskov M; Bjarnsholt T Imaging N-Acyl Homoserine Lactone Quorum Sensing In Vivo. Quorum Sensing; Humana Press: New York, 2018; pp 203–212. [DOI] [PubMed] [Google Scholar]
- (307).Ramanathan S; Ravindran D; Arunachalam K; Arumugam VR Inhibition of Quorum Sensing-Dependent Biofilm and Virulence Genes Expression in Environmental Pathogen Serratia marcescens by Petroselinic Acid. Antonie van Leeuwenhoek 2018, 111, 501–515. [DOI] [PubMed] [Google Scholar]
- (308).Kim MK; Zhao A; Wang A; Brown ZZ; Muir TW; Stone HA; Bassler BL Surface-Attached Molecules Control Staphylococcus aureus Quorum Sensing and Biofilm Development. Nature Micorbiol 2017, 2, 17080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (309).Chan KG; Liu YC; Chang CY Inhibiting N-Acyl-Homoserine Lactone Synthesis and Quenching Pseudomonas Quinolone Quorum Sensing to Attenuate Virulence. Front. Microbiol 2015, 6, 1173 (eCollection). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (310).Singh RP; Desouky SE; Nakayama J Quorum Quenching Strategy Targeting Gram-Positive Pathogenic Bacteria. Adv. Exp. Med. Biol 2016, 901, 109–130. [DOI] [PubMed] [Google Scholar]
- (311).Pletzer D; Coleman SR; Hancock RE Anti-Biofilm Peptides as a New Weapon in Antimicrobial Warfare. Curr. Opin. Microbiol 2016, 33, 35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (312).Khan BA; Yeh AJ; Cheung GY; Otto M Investigational Therapies Targeting Quorum-Sensing for the Treatment of Staph-ylococcus aureus Infections. Expert Opin. Invest. Drugs 2015, 24, 689–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (313).Yousif A; Jamal MA; Raad I Biofilm-Based Central Line-Associated Bloodstream Infections. Adv. Exp. Med. Biol 2015, 830, 157–179. [DOI] [PubMed] [Google Scholar]
- (314).Sherrard LJ; Tunney MM; Elborn JS Antimicrobial Resistance in the Respiratory Microbiota of People with Cystic Fibrosis. Lancet 2014, 384, 703–713. [DOI] [PubMed] [Google Scholar]
- (315).Paudel A; Hamamoto H; Panthee S; Sekimizu K Menaquinone as a Potential Target of Antibacterial Agents. Drug Discoveries Ther 2016, 10, 123–128. [DOI] [PubMed] [Google Scholar]
- (316).Ravcheev DA; Thiele I Genomic Analysis of the Human Gut Microbiome Suggests Novel Enzymes Involved in Quinone Biosyn-thesis. Front. Microbiol 2016, 7, 128 (eCollection). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (317).Arakawa C; Kuratsu M; Furihata K; Hiratsuka T; Itoh N; Seto H; Dairi T Diversity of the Early Step of the Futalosine Pathway. Antimicrob. Agents Chemother 2011, 55, 913–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (318).Dairi T Menaquinone Biosyntheses in Microorganisms. Methods Enzymol 2012, 515, 107–122. [DOI] [PubMed] [Google Scholar]
- (319).Cooper LE; Fedoseyenko D; Abdelwahed SH; Kim SH; Dairi T; Begley TP In Vitro Reconstitution of the Radical S-Adenosylmethionine Enzyme Mqnc Involved in the Biosynthesis of Futalosine-Derived Menaquinone. Biochemistry 2013, 52, 4592–4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (320).Cook GM; Greening C; Hards K; Berney M Energetics of Pathogenic Bacteria and Opportunities for Drug Development. Adv. Microb. Physiol 2014, 65, 1–62. [DOI] [PubMed] [Google Scholar]
- (321).Hong W; Chen L; Xie J Molecular Basis Underlying Mycobacterium tuberculosis D-Cycloserine Resistance. Is There a Role for Ubiquinone and Menaquinone Metabolic Pathways? Expert Opin. Ther. Targets 2014, 18, 691–701. [DOI] [PubMed] [Google Scholar]
- (322).Ronning DR; Iacopelli NM; Mishra V Enzyme-Ligand Interactions That Drive Active Site Rearrangements in the Helicobacter Pylori 5′-Methylthioadenosine/S-Adenosylhomocysteine Nucleosi-dase. Protein Sci 2010, 19, 2498–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (323).Wang S; Haapalainen AM; Yan F; Du Q; Tyler PC; Evans GB; Rinaldo-Matthis A; Brown RL; Norris GE; Almo SC; et al. A Picomolar Transition State Analogue Inhibitor of MTAN as a Specific Antibiotic for Helicobacter pylori. Biochemistry 2012, 51, 6892–6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (324).Wang S; Cameron SA; Clinch K; Evans GB; Wu Z; Schramm VL; Tyler PC New Antibiotic Candidates Against Helicobacter pylori. J. Am. Chem. Soc 2015, 137, 14275–14280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (325).Smith S; Boyle B; Brennan D; Buckley M; Crotty P; Doyle M; Farrell R; Hussey M; Kevans D; Malfertheiner P; et al. The Irish Helicobacter pylori Working Group Consensus for the Diagnosis and Treatment of H. Pylori Infection in Adult Patients in Ireland. Eur. J. Gastroenterol. Hepatol 2017, 29, 552–559. [DOI] [PubMed] [Google Scholar]
- (326).Siddique O; Ovalle A; Siddique AS; Moss SF Helicobacter pylori Infection: An Update for the Internist in the Age of Increasing Global Antibiotic Resistance. Am. J. Med 2018, 131, 473–479. [DOI] [PubMed] [Google Scholar]
- (327).Zagari RM; Rabitti S; Eusebi LH; Bazzoli F Treatment of Helicobacter pylori Infection: A Clinical Practice Update. Eur. J. Clin. Invest 2018, 48, e12857. [DOI] [PubMed] [Google Scholar]
- (328).Talebi Bezmin Abadi A; Yamaoka Y Helicobacter pylori Therapy and Clinical Perspective. J. Glob. Antimicrob. Resist 2018, 14, 11. [DOI] [PubMed] [Google Scholar]
- (329).Wang YH; Li Z; Wang L; Zhu-Ge LY; Zhao RL; Wu S; Wang Y; An Y; Xie Y A Systematic Review and Meta-Analysis of Genotypic Methods for Detecting Antibiotic Resistance in Helico-bacter pylori. Helicobacter 2018, 23, e12467. [DOI] [PubMed] [Google Scholar]
- (330).Suzuki H; Mori H World Trends for H. Pylori Eradication Therapy and Gastric Cancer Prevention Strategy by H. pylori Test-and-Treat. J. Gastroenterol 2018, 53, 354–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (331).Mascellino MT; Porowska B; De Angelis M; Oliva A Antibiotic Susceptibility, Heteroresistance, and Updated Treatment Strategies in Helicobacter pylori Infection. Drug Des., Dev. Ther 2017, 11, 2209–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (332).Zagari RM; Rabitti S; Eusebi LH; Bazzoli F Treatment of Helicobacter pylori Infection: A Clinical Practice Update. Eur. J. Clin. Invest 2018, 48, e12857. [DOI] [PubMed] [Google Scholar]
- (333).Luo R; Barlam TF Ten-year Review of Clostridium Difficile Infection in Acute Care Hospitals in the USA, 2005–2014. J. Hosp. Infect 2018, 98, 40–43. [DOI] [PubMed] [Google Scholar]
- (334).McDonald LC; Kutty PK; Kaplan SL Clostridium dif f icile Infection: Prevention and Control UpToDate 2017, 324, 1–12. [Google Scholar]
- (335).McDonald LC; Gerding DN; Johnson S; Bakken JS; Carroll KC; Coffin SE; Dubberke ER; Garey KW; Gould CV; Kelly C; Loo V Clinical Practice Guidelines for Clostridium dif f icile Infection in Adults snd Children: 2017 Update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin. Infect. Dis 2018, 66, e1–e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (336).Forster AJ; Daneman N; van Walraven C Influence of Antibiotics and Case Exposure on Hospital-Acquired Clostridium dif f icile Infection Independent of Illness Severity. J. Hosp. Infect 2017, 95, 400–409. [DOI] [PubMed] [Google Scholar]
- (337).Anderson DJ; Chen LF; Weber DJ; Moehring RW; Lewis SS; Triplett PF; Blocker M; Becherer P; Schwab JC; Knelson LP; et al. Enhanced Terminal Room Disinfection and Acquisition and Infection Caused by Multidrug-Resistant Organisms and Clostridium dif f icile (The Benefits of Enhanced Terminal Room Disinfection Study): A Cluster-Randomised, Multicentre, Crossover Study. Lancet 2017, 389, 805–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (338).Giblett E; Ammann A; Sandman R; Wara D; Diamond L Nucleoside-phosphorylase Deficiency in a Child with Severely Defective T-Cell Immunity and Normal B-Cell Immunity. Lancet 1975, 305, 1010–1013. [DOI] [PubMed] [Google Scholar]
- (339).Giblett ER ADA and PNP Deficiencies: How it All Began. Ann. N. Y. Acad. Sci 1985, 451, 1–8. [DOI] [PubMed] [Google Scholar]
- (340).Markert ML Purine Nucleoside Phosphorylase Deficiency. Immunodef. Rev 1991, 3, 45–81. [PubMed] [Google Scholar]
- (341).Cohen A; Gudas LJ; Ammann AJ; Staal GE; Martin DV Deoxyguanosine Triphosphate as a Possible Toxic Metabolite in the Immunodeficiency Associated with Purine Nucleoside Phosphorylase Deficiency. J. Clin. Invest 1978, 61, 1405–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (342).Roifman CM Primary T-cell Immunodeficiencies. Clinical Immunology, 5th ed.; Elsevier, 2019; pp 489–508. [Google Scholar]
- (343).Yeates L; Slatter MA; Gennery AR Infusion of Sibling Marrow in a Patient with Purine Nucleoside Phosphorylase Deficiency Leads to Split Mixed Donor Chimerism and Normal Immunity. Front. Pediatr 2017, 5, 00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (344).Barbaro M; Ohlsson A; Borte S; Jonsson S; Zetterström RH; King J; Winiarski J; von Döbeln U; Hammarström L Newborn Screening for Severe Primary Immunodeficiency Diseases in Sweden A 2-Year Pilot TREC and KREC Screening Study. J. Clin. Immunol 2017, 37, 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (345).Booth C; Silva J; Veys P Stem Cell Transplantation for the Treatment of Immunodeficiency in Children: Current Status and Hopes for the Future. Expert Rev. Clin. Immunol 2016, 12, 713–723. [DOI] [PubMed] [Google Scholar]
- (346).Mateos FA; Puig JG; Jimenez ML; Fox IH Hereditary Xanthinuria. Evidence for Enhanced Hypoxanthine Salvage. J. Clin. Invest 1987, 79, 847–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (347).Morris PE Jr; Montgomery JA Inhibitors of the Enzyme Purine Nucleoside Phosphorylase. Expert Opin. Ther. Pat 1998, 8, 283–299. [Google Scholar]
- (348).Montgomery JA Purine Nucleoside Phosphorylase: A Target for Drug Design. Med. Res. Rev 1993, 13, 209–228. [DOI] [PubMed] [Google Scholar]
- (349).Fox IH Metabolic Basis for Disorders of Purine Nucleotide Degradation. Metab., Clin. Exp 1981, 30, 616–634. [DOI] [PubMed] [Google Scholar]
- (350).Kline PC; Schramm VL Purine Nucleoside Phosphorylase. Catalytic Mechanism and Transition-State Analysis of the Arsenolysis Reaction. Biochemistry 1993, 32, 13212–13219. [DOI] [PubMed] [Google Scholar]
- (351).Kline PC; Schramm VL Purine Nucleoside Phosphorylase. Inosine Hydrolysis, Tight Binding of the Hypoxanthine Intermediate, and Third-the-Sites Reactivity. Biochemistry 1992, 31, 5964–5973. [DOI] [PubMed] [Google Scholar]
- (352).Erion MD; Takabayashi K; Smith HB; Kessi J; Wagner S; Hönger S; Shames SL; Ealick SE Purine Nucleoside Phosphorylase. 1. Structure– Function Studies. Biochemistry 1997, 36, 11725–11734. [DOI] [PubMed] [Google Scholar]
- (353).Mao C; Cook WJ; Zhou M; Federov AA; Almo SC; Ealick SE Calf Spleen Purine Nucleoside Phosphorylase Complexed with Substrates and Substrate Analogues. Biochemistry 1998, 37, 7135–7146. [DOI] [PubMed] [Google Scholar]
- (354).Ealick SE; Rule SA; Carter DC; Greenhough TJ; Babu YS; Cook WJ; Habash J; Helliwell JR; Stoeckler JD; Parks RE Three-dimensional Structure of Human Erythrocytic Purine Nucleoside Phosphorylase at 3.2 Å Resolution. J. Biol. Chem 1990, 265, 1812–1820. [DOI] [PubMed] [Google Scholar]
- (355).Lewandowicz A; Schramm VL Transition State Analysis for Human and Plasmodium falciparum Purine Nucleoside Phosphorylases. Biochemistry 2004, 43, 1458–1468. [DOI] [PubMed] [Google Scholar]
- (356).Northrop DB Steady-state Analysis of Kinetic Isotope Effects in Enzymic Reactions. Biochemistry 1975, 14, 2644–2651. [DOI] [PubMed] [Google Scholar]
- (357).Melander LC; Saunders WH Reaction Rates of Isotopic Molecules; John Wiley & Sons, 1980. [Google Scholar]
- (358).Bennet AJ; Sinnott ML Complete Kinetic Isotope Effect Description of Transition States for Acid-Catalyzed Hydrolyses of Methyl. Alpha-and. Beta-Glucopyranosides. J. Am. Chem. Soc 1986, 108, 7287–7294. [Google Scholar]
- (359).Miles RW; Tyler PC; Furneaux RH; Bagdassarian CK; Schramm VL One-third-the-sites Transition-State Inhibitors for Purine Nucleoside Phosphorylase. Biochemistry 1998, 37, 8615–8621. [DOI] [PubMed] [Google Scholar]
- (360).Parks RE Jr.; Agarwal RP Purine Nucleoside Phosphorylase. The Enzymes; Academic Press, 1972; Vol. 7, pp 483–514. [Google Scholar]
- (361).Sauve AA; Cahill SM; Zech SG; Basso LA; Lewandowicz A; Santos DS; Grubmeyer C; Evans GB; Furneaux RH; Tyler PC; et al. Ionic States of Substrates and Transition State Analogues at the Catalytic Sites of N-Ribosyltransferases. Biochemistry 2003, 42, 5694–5705. [DOI] [PubMed] [Google Scholar]
- (362).Evans GB; Furneaux RH; Tyler PC; Schramm VL Synthesis of a Transition State Analogue Inhibitor of Purine Nucleoside Phosphorylase via the Mannich Reaction. Org. Lett 2003, 5, 3639–3640. [DOI] [PubMed] [Google Scholar]
- (363).Fedorov A; Shi W; Kicska G; Fedorov E; Tyler PC; Furneaux RH; Hanson JC; Gainsford GJ; Larese JZ; Schramm VL; et al. Transition State Structure of Purine Nucleoside Phosphorylase and Principles of Atomic Motion in Enzymatic Catalysis. Biochemistry 2001, 40, 853–860. [DOI] [PubMed] [Google Scholar]
- (364).Kicska GA; Tyler PC; Evans GB; Furneaux RH; Shi W; Fedorov A; Lewandowicz A; Cahill SM; Almo SC; Schramm VL Atomic Dissection of the Hydrogen Bond Network for Transition-State Analogue Binding to Purine Nucleoside Phosphorylase. Biochemistry 2002, 41, 14489–14498. [DOI] [PubMed] [Google Scholar]
- (365).Schramm VL; Shi W Atomic Motion in Enzymatic Reaction Coordinates. Curr. Opin. Struct. Biol 2001, 11, 657–665. [DOI] [PubMed] [Google Scholar]
- (366).Vocadlo DJ; Davies GJ; Laine R; Withers SG Catalysis by Hen Egg-White Lysozyme Proceeds via a Covalent Intermediate. Nature 2001, 412, 835–838. [DOI] [PubMed] [Google Scholar]
- (367).Bianchet MA; Seiple LA; Jiang YL; Ichikawa Y; Amzel LM; Stivers JT Electrostatic Guidance of Glycosyl Cation Migration Along the Reaction Coordinate of Uracil DNA Glycosylase. Biochemistry 2003, 42, 12455–12460. [DOI] [PubMed] [Google Scholar]
- (368).Lovering AL; De Castro LH; Lim D; Strynadka NC Structural Insight into the Transglycosylation Step of Bacterial Cell-Wall Biosynthesis. Science 2007, 315, 1402–1405. [DOI] [PubMed] [Google Scholar]
- (369).Jin Y; Petricevic M; John A; Raich L; Jenkins H; Portela de Souza L; Cuskin F; Gilbert HJ; Rovira C; Goddard-Borger ED; et al. A β-Mannannase with a Lysozyme Fold and a Novel Molecular Catalytic Mechanism. ACS Cent. Sci 2016, 2, 896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (370).Drohat AC; Maiti A Mechanisms for Enzymatic Cleavage of the N-Glycosidic Bond in DNA. Org. Biomol. Chem 2014, 12, 8367–8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (371).Morris PE Jr.; Elliott AJ; Walton SP; Williams CH; Montgomery JA Synthesis and Biological Activity of A Novel Class Of Purine Nucleoside Phosphorylase Inhibitors. Nucleosides, Nucleo-tides Nucleic Acids 2000, 19, 379–404. [DOI] [PubMed] [Google Scholar]
- (372).Conry RM; Bantia S; Turner HS; Barlow DL; Allen KO; LoBuglio AF; Montgomery JA; Walsh GM Effects of a Novel Purine Nucleoside Phosphorylase Inhibitor, BCX-34, on Activation and Proliferation of Normal Human Lymphoid Cells. Immunopharmacology 1998, 40, 1–9. [DOI] [PubMed] [Google Scholar]
- (373).Duvic M; Olsen EA; Omura GA; Maize JC; Vonderheid EC; Elmets CA; Shupack JL; Demierre MF; Kuzel TM; Sanders DY A Phase III, Randomized, Double-Blind, Placebo-Controlled Study of Peldesine (BCX-34) Cream as Topical Therapy for Cutaneous T-Cell Lymphoma. J. Am. Acad. Dermatol 2001, 44, 940–947. [DOI] [PubMed] [Google Scholar]
- (374).Furman RR; Gore L; Ravandi F; Hoelzer D Forodesine IV (Bcx-1777) Is Clinically Active in Relapsed/Refractory T-Cell Leukemia: Results of a Phase II Study (Interim Report). Blood 2006, 108, 1851. [Google Scholar]
- (375).Duvic M; Forero-Torres A; Foss F; Elsie A; Olsen EA; Kim Y Oral Forodesine (BCX-1777) is Clinically Active in Refractory Cutaneous T-Cell Lymphoma: Results of s Phase I/II Study. Blood 2006, 108, 2467. [Google Scholar]
- (376).Furman RR; Hoelzer D Purine Nucleoside Phosphorylase Inhibition as a Novel Therapeutic Approach for B-Cell Lymphoid Malignancies. Semin. Oncol 2007, 34, S29–S34. [DOI] [PubMed] [Google Scholar]
- (377).Makita S; Maeshima AM; Maruyama D; Izutsu K; Tobinai K Forodesine in the Treatment of Relapsed/Refractory Peripheral T-Cell Lymphoma: An Evidence-Based Review. OncoTargets Ther 2018, 11, 2287–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (378). http://www.mundipharma.com.sg/2017/03/30/mundipharma-wins-approval-antineoplastic-agent-mundesine-treatment-relapsedrefractory-peripheral-t-cell-lymphoma-japan/
- (379).Eryılmaz E; Canpolat C Novel Agents for the Treatment of Childhood Leukemia: An Update. OncoTargets Ther 2017, 10, 3299–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (380).Ho MC; Shi W; Rinaldo-Matthis A; Tyler PC; Evans GB; Clinch K; Almo SC; Schramm VL Four Generations of Transition-State Analogues for Human Purine Nucleoside Phosphor-ylase. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 4805–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (381).Vetticatt MJ; Itin B; Evans GB; Schramm VL Distortional Binding of Transition State Analogs to Human Purine Nucleoside Phosphorylase Probed by Magic Angle Spinning Solid-State NMR. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 15991–15996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (382).Brown SP Applications of High-Resolution 1H Solid-state NMR. Solid State Nucl. Magn. Reson 2012, 41, 1–27. [DOI] [PubMed] [Google Scholar]
- (383).Gullion T; Schaefer J Rotational-echo Double-resonance NMR. J. Magn. Reson 1989, 81, 196–200. [DOI] [PubMed] [Google Scholar]
- (384).Raleigh DP; Levitt MH; Griffin RG Rotational Resonance in Solid State NMR. Chem. Phys. Lett 1988, 146, 71–76. [Google Scholar]
- (385).Lewandowicz A; Ringia EAT; Ting LM; Kim K; Tyler PC; Evans GB; Zubkova OV; Mee S; Painter GF; Lenz DH; et al. Energetic Mapping of Transition State Analogue Interactions with Human and Plasmodium falciparum Purine Nucleoside Phosphorylases. J. Biol. Chem 2005, 280, 30320–30328. [DOI] [PubMed] [Google Scholar]
- (386).Evans GB; Furneaux RH; Lewandowicz A; Schramm VL; Tyler PC Exploring Structure– Activity Relationships of Transition State Analogues of Human Purine Nucleoside Phosphorylase. J. Med. Chem 2003, 46, 3412–3423. [DOI] [PubMed] [Google Scholar]
- (387).Clinch K; Evans GB; Fröhlich RF; Furneaux RH; Kelly PM; Legentil L; Murkin AS; Li L; Schramm VL; Tyler PC; et al. Third-generation Immucillins: Syntheses and Bioactivities of Acyclic Immucillin Inhibitors of Human Purine Nucleoside Phosphor-ylase. J. Med. Chem 2009, 52, 1126–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (388).Edwards AA; Mason JM; Clinch K; Tyler PC; Evans GB; Schramm VL Altered Enthalpy– entropy Compensation in Picomolar Transition State Analogues of Human Purine Nucleoside Phosphorylase. Biochemistry 2009, 48, 5226–5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (389).Edwards AA; Tipton JD; Brenowitz MD; Emmett MR; Marshall AG; Evans GB; Tyler PC; Schramm VL Conformational States of Human Purine Nucleoside Phosphorylase at Rest, at Work, and with Transition State Analogues. Biochemistry 2010, 49, 2058–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (390).Hirschi JS; Arora K; Brooks CL III; Schramm VL Conformational Dynamics in Human Purine Nucleoside Phosphorylase with Reactants and Transition-State Analogues. J. Phys. Chem. B 2010, 114, 16263–16272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (391).Saen-oon S; Quaytman-Machleder S; Schramm VL; Schwartz SD Atomic Detail of Chemical Transformation at the Transition State of an Enzymatic Reaction. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 16543–16548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (392).Schwartz SD; Schramm VL Enzymatic Transition States and Dynamic Motion in Barrier Crossing. Nat. Chem. Biol 2009, 5, 551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (393).Saen-oon S; Schramm VL; Schwartz SD Transition Path Sampling Study of the Reaction Catalyzed by Purine Nucleoside Phosphorylase. Z. Phys. Chem. (Muenchen, Ger.) 2008, 222, 1359–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (394).Silva RG; Murkin AS; Schramm VL Femtosecond Dynamics Coupled to Chemical Barrier Crossing in s Born-Oppenheimer Enzyme. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 18661–18665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (395).Kicska GA; Long L; Hörig H; Fairchild C; Tyler PC; Furneaux RH; Schramm VL; Kaufman HL Immucillin H, a Powerful Transition-state Analog Inhibitor of Purine Nucleoside Phosphorylase, Selectively Inhibits Human T Lymphocytes. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 4593–4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (396).Bantia S; Ananth SL; Parker CD; Horn LL; Upshaw R Mechanism of Inhibition of T-Acute Lymphoblastic Leukemia Cells by PNP Inhibitor BCX-1777. Int. Immunopharmacol 2003, 3, 879–887. [DOI] [PubMed] [Google Scholar]
- (397).Kilpatrick JM; Morris PE; Serota DG Jr; Phillips D; Moore DR II; Bennett JC; Babu YS Intravenous and Oral Pharmacokinetic Study of BCX-1777, A Novel Purine Nucleoside Phosphorylase Transition-State Inhibitor. In Vivo Effects on Blood 2′-Deoxyguanosine in Primates. Int. Immunopharmacol 2003, 3, 541–548. [DOI] [PubMed] [Google Scholar]
- (398).Bantia S; Parker C; Upshaw R; Cunningham A; Kotian P; Kilpatrick JM; Morris P; Chand P; Babu YS Potent Orally Bioavailable Purine Nucleoside Phosphorylase Inhibitor BCX-4208 Induces Apoptosis in B-and T-Lymphocytes A Novel Treatment Approach for Autoimmune Diseases, Organ Transplantation and Hematologic Malignancies. Int. Immunopharmacol 2010, 10, 784–790. [DOI] [PubMed] [Google Scholar]
- (399).Yin L; Dai S; Clayton G; Gao W; Wang Y; Kappler J; Marrack P Recognition of Self and Altered Self by T Cells in Autoimmunity and Allergy. Protein Cell 2013, 4, 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (400).Theofilopoulos AN; Kono DH; Baccala R The Multiple Pathways to Autoimmunity. Nat. Immunol 2017, 18, 716–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (401).Azizi G; Abolhassani H; Asgardoon MH; Alinia T; Yazdani R; Mohammadi J; Rezaei N; Ochs HD; Aghamohammadi A Autoimmunity in Common Variable Immuno-deficiency: Epidemiology, Pathophysiology and Management. Expert Rev. Clin. Immunol 2017, 13, 101–115. [DOI] [PubMed] [Google Scholar]
- (402).Navegantes KC; de Souza Gomes R; Pereira PAT; Czaikoski PG; Azevedo CHM; Monteiro MC Immune Modulation of Some Autoimmune Diseases: The Critical Role of Macrophages and Neutrophils in the Innate and Adaptive Immunity. J. Transl. Med 2017, 15, 36 (Open access). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (403).Bantia S; Miller PJ; Parker CD; Ananth SL; Horn LL; Kilpatrick JM; Morris PE; Hutchison TL; Montgomery JA; Sandhu JS Purine Nucleoside Phosphorylase Inhibitor BCX-1777 (Immucillin-H) A Novel Potent and Orally Active Immunosuppres-sive Agent. Int. Immunopharmacol 2001, 1, 1199–1210. [DOI] [PubMed] [Google Scholar]
- (404).Bantia S; Miller PJ; Parker CD; Ananth SL; Horn LL; Babu YS; Sandhu JS Comparison of In Vivo Efficacy of BCX-1777 and Cyclosporin in Xenogeneic Graft-vs.-Host Disease: The Role of dGTP in Antiproliferative Action of BCX-1777. Int. Immunopharmacol 2002, 2, 913–923. [DOI] [PubMed] [Google Scholar]
- (405).Gandhi V; Kilpatrick JM; Plunkett W; Ayres M; Harman L; Du M; Bantia S; Davisson J; Wierda WG; Faderl S; et al. A Proof-of-principle Pharmacokinetic, Pharmacodynamic, and Clinical Study with Purine Nucleoside Phosphorylase Inhibitor Immucillin-H (BCX-1777, forodesine). Blood 2005, 106, 4253–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (406).Gore L; Stelljes M; Quinones R Forodesine Treatment and Post-Transplant Graft-Versus-Host Disease in Two Patients with Acute Leukemia: Facilitation of Graft-Versus-Leukemia Effect? Semin. Oncol 2007, 34, S35–S39. [DOI] [PubMed] [Google Scholar]
- (407).Bantia S (Nitor Therapeutics) Use of PNP Inhibitor to Treat Relapse of Malignancy After Hematopoietic Stem Cell Transplant U.S. Patent Application 15/329,269, 2017.
- (408).Lu PD; Mazza JM Research Pipeline II: Oral Therapeutics. Advances in Psoriasis; Springer: London, 2014; pp 227–242. [Google Scholar]
- (409).Hollister AS; Maetzel A; Becker MA; Terkeltaub R; Fitz-Patrick D; Smith V; Sheridan WP Ulodesine (BCX4208) Long-Term Safety When Added to Allopurinol in the Chronic Management of Gout: A Phase 2 24-Week Blinded Safety Extension and Vaccine Challenge Study. Arthritis Rheum 2012, 64, S65–S66. [Google Scholar]
- (410).Bantia S; Harman L; Parker C; Papac D; Maetzel A; Taubenheim B; Hollister AS Ulodesine (BCX4208) Add-On Therapy to Allopurinol 300 mg Lowers Hypoxanthine and Xanthine Plasma Levels in a Dose-Dependent Fashion: Results from a 12-Week Randomized Controlled Trial in Patients with Gout. Arthritis Rheum 2012, 64, S698. [Google Scholar]
- (411).Edwards NL; So A Emerging Therapies for Gout. Rheumat. Dis. Clin 2014, 40, 375–387. [DOI] [PubMed] [Google Scholar]
- (412).Chohan S; Becker MA Emerging Therapeutics for Acute and Chronic Gout; Future Med. Ltd., 2013; pp 226–237. [Google Scholar]
- (413).Bantia S; Parker C; Harman L; Papac D; Hollister A Effect of BCX4208 Add-On Therapy to Allopurinol 300 mg on Plasma Hypoxanthine and Xanthine Concentrations in Gout Patients. Ann. Rheum. Dis 2013, 71, 89. [Google Scholar]
- (414).A Randomized, Double-Blind, Dose-Response Study of the Safety and Uric Acid Effects of Oral Ulodesine Added to Allopurinol in Subjects With Gout and Concomitant Moderate Renal Insufficiency NCT01407874; ClinicalTrials.gov, 2011.
- (415).Study to Evaluate sUA Lowering Activity, Safety and Efficacy of Oral Ulodesine Added to Allopurinol NCT01265264; ClinicalTrials.gov, 2010.
- (416).Study to Evaluate the Urate-Lowering Activity and Safety of Oral BCX4208 Administered in Subjects with Gout NCT00985127; ClinicalTrials.gov, 2009.
- (417).Study to Evaluate sUA-Lowering Activity, Safety & PK Interaction of Oral BCX4208 & Allopurinol Administration in Subjects with Gout NCT01129648; ClinicalTrials.gov, 2010.
- (418).Lavan BE; Saha GC; Roberts BK; McWherter CA (CymaBay Therapeutics Inc.) Methods for Treating Gout Flares U.S. Patent Application US 14/930,488, March 1, 2018.
- (419).Lewandowicz A; Tyler PC; Evans GB; Furneaux RH; Schramm VL Achieving the Ultimate Physiological Goal in Transition State Analogue Inhibitors for Purine Nucleoside Phosphorylase. J. Biol. Chem 2003, 278, 31465–31468. [DOI] [PubMed] [Google Scholar]
- (420).Gebre ST; Cameron SA; Li L; Babu YS; Schramm VL Intracellular Rebinding of Transition-state Analogues Provides Extended In Vivo Inhibition Lifetimes on Human Purine Nucleoside Phosphorylase. J. Biol. Chem 2017, 292, 15907–15915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (421).Huang M; Wang Y; Gu J; Yang J; Noel K; Mitchell BS; Schramm VL; Graves LM Determinants of Sensitivity of Human T-Cell Leukemia CCRF-CEM Cells to Immucillin-H. Leuk. Res 2008, 32, 1268–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (422).Cullen KA; Arguin PM Malaria Surveillance United States, 2012. MMWR Surveill. Summ 2014, 63, 1–22. [PubMed] [Google Scholar]
- (423).Fischer PR Decreasing Malaria Mortality in Africa. Infectious Disease Alert, 2017, 36, 5. [Google Scholar]
- (424).Deniskin R; Frame IJ; Sosa Y; Akabas MH Targeting the Plasmodium vivax Equilibrative Nucleoside Transporter 1 (PvENT1) for Antimalarial Drug Development. Int. J. Parasitol.: Drugs Drug Resist 2016, 6, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (425).Frame IJ; Deniskin R; Arora A; Akabas MH Purine Import into Malaria Parasites as a Target for Antimalarial Drug Development. Ann. N. Y. Acad. Sci 2015, 1342, 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (426).Staines HM; Moore CM; Slavic K; Krishna S Transmembrane Solute Transport in the Apicomplexan Parasite Plasmodium. Emer. Topics Life Sci 2017, 1, 553–561. [DOI] [PubMed] [Google Scholar]
- (427).Shi W; Ting LM; Kicska GA; Lewandowicz A; Tyler PC; Evans GB; Furneaux RH; Kim K; Almo SC; Schramm VL Plasmodium falciparum Purine Nucleoside Phosphorylase Crystal Structures, Immucillin Inhibitors, and Dual Catalytic Function. J. Biol. Chem 2004, 279, 18103–18106. [DOI] [PubMed] [Google Scholar]
- (428).Schnick C; Robien MA; Brzozowski AM; Dodson EJ; Murshudov GN; Anderson L; Luft JR; Mehlin C; Hol WG; Brannigan JA; et al. Structures of Plasmodium falciparum Purine Nucleoside Phosphorylase Complexed With Sulfate and its Natural Substrate Inosine. Acta Crystallogr., Sect. D: Biol. Crystallogr 2005, 61, 1245–1254. [DOI] [PubMed] [Google Scholar]
- (429).Ting LM; Shi W; Lewandowicz A; Singh V; Mwakingwe A; Birck MR; Ringia EAT; Bench G; Madrid DC; Tyler PC; et al. Targeting a Novel Plasmodium falciparum Purine Recycling Pathway with Specific Immucillins. J. Biol. Chem 2005, 280, 9547–9554. [DOI] [PubMed] [Google Scholar]
- (430).Zappia V; Oliva A; Cacciapuoti G; Galletti P; Mignucci G; Cartení-Farina M Substrate Specificity of 5′-Methylthioadenosine Phosphorylase from Human Prostate. Biochem. J 1978, 175, 1043–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (431).Kicska GA; Tyler PC; Evans GB; Furneaux RH; Schramm VL; Kim K Purine-less Death in Plasmodium falciparum Induced by Immucillin-H, a Transition State Analogue of Purine Nucleoside Phosphorylase. J. Biol. Chem 2002, 277, 3226–3231. [DOI] [PubMed] [Google Scholar]
- (432).Ho MC; Cassera MB; Madrid DC; Ting LM; Tyler PC; Kim K; Almo SC; Schramm VL Structural and Metabolic Specificity of Methylthiocoformycin for Malarial Adenosine Deaminases. Biochemistry 2009, 48, 9618–9626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (433).Larson ET; Deng W; Krumm BE; Napuli A; Mueller N; Van Voorhis WC; Buckner FS; Fan E; Lauricella A; DeTitta G; et al. Structures of Substrate and Inhibitor-bound Adenosine Deaminase from a Human Malaria Parasite Show a Dramatic Conformational Change and Shed Light on Drug Selectivity. J. Mol. Biol 2008, 381, 975–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (434).Tyler PC; Taylor EA; Fröhlich RF; Schramm VL Synthesis of 5′-Methylthio Coformycins: Specific Inhibitors for Malarial Adenosine Deaminase. J. Am. Chem. Soc 2007, 129, 6872–6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (435).Cassera MB; Hazleton KZ; Merino EF; Obaldia N III; Ho MC; Murkin AS; DePinto R; Gutierrez JA; Almo SC; Evans GB; et al. Plasmodium falciparum Parasites are Killed by a Transition State Analogue of Purine Nucleoside Phosphorylase in a Primate Animal Model. PLoS One 2011, 6, e26916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (436).Madrid DC; Ting LM; Waller KL; Schramm VL; Kim K Plasmodium falciparum Purine Nucleoside Phosphorylase is Critical for Viability of Malaria Parasites. J. Biol. Chem 2008, 283, 35899–35907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (437).Harkness RA; Simmonds RJ; Coade SB Purine Transport and Metabolism in Man: The Effect of Exercise on Concentrations of Purine Bases, Nucleosides and Nucleotides In Plasma, Urine, Leucocytes and Erythrocytes. Clin. Sci 1983, 64, 333–340. [DOI] [PubMed] [Google Scholar]
- (438).Ontyd J; Schrader J Measurement of Adenosine, Inosine, and Hypoxanthine in Human Plasma. J. Chromatogr., Biomed. Appl 1984, 307, 404–409. [DOI] [PubMed] [Google Scholar]
- (439).Ducati RG; Namanja-Magliano HA; Harijan RK; Fajardo JE; Fiser A; Daily JP; Schramm VL Genetic Resistance to Purine Nucleoside Phosphorylase Inhibition in Plasmodium falciparum. Proc. Natl. Acad. Sci. U. S. A 2018, 115, 2114–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (440). http://www.who.int/malaria/areas/drug_resistance/en/
- (441).Alcaide ML; Feaster DJ; Duan R; Cohen S; Diaz C; Castro JG; Golden MR; Henn S; Colfax GN; Metsch LR The Incidence of Trichomonas vaginalis Infection in Women Attending Nine Sexually Transmitted Diseases Clinics in the USA. Sex Transm. Infect 2016, 92, 58–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (442).Fernández-Romero JA; Deal C; Herold BC; Schiller J; Patton D; Zydowsky T; Romano J; Petro CD; Narasimhan M Multipurpose Prevention Technologies: The Future of HIV and STI Protection. Trends Microbiol 2015, 23, 429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (443).Kirkcaldy RD; Augostini P; Asbel LE; Bernstein KT; Kerani RP; Mettenbrink CJ; Pathela P; Schwebke JR; Secor WE; Workowski KA; et al. Trichomonas vaginalis Antimicrobial Drug Resistance in 6 US Cities, STD Surveillance Network, 2009–2010. Emerging Infect. Dis 2012, 18, 939–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (444).Munagala NR; Wang CC Adenosine is the Primary Precursor of All Purine Nucleotides in Trichomonas vaginalis. Mol. Biochem. Parasitol 2003, 127, 143–149. [DOI] [PubMed] [Google Scholar]
- (445).Rinaldo-Matthis A; Wing C; Ghanem M; Deng H; Wu P; Gupta A; Tyler PC; Evans GB; Furneaux RH; Almo SC; et al. Inhibition and Structure of Trichomonas vaginalis Purine Nucleoside Phosphorylase with Picomolar Transition State Analogues. Biochemistry 2007, 46, 659–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (446).Warren TK; Wells J; Panchal RG; Stuthman KS; Garza NL; Van Tongeren SA; Dong L; Retterer CJ; Eaton BP; Pegoraro G; et al. Protection Against Filovirus Diseases by a Novel Broad-Spectrum Nucleoside Analogue BCX4430. Nature 2014, 508, 402–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (447).Taylor R; Kotian P; Warren T; Panchal R; Bavari S; Julander J; Dobo S; Rose A; El-Kattan Y; Taubenheim B; et al. BCX4430–A Broad-Spectrum Antiviral Adenosine Nucleoside Analog Under Development for the Treatment of Ebola Virus Disease. J. Infect. Pub. Health 2016, 9, 220–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (448).Eyer L; Nencka R; De Clercq E; Seley-Radtke K; Růžek D Nucleoside Analogs as a Rich Source of Antiviral Agents Active Against Arthropod-Borne Flaviviruses. Antivir. Chem. Chemother 2018, 26, 204020661876129 (open access). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (449).Cross RW; Mire CE; Feldmann H; Geisbert TW Post-Exposure Treatments for Ebola and Marburg Virus Infections. Nat. Rev. Drug Discovery 2018, 17, 413–434. [DOI] [PubMed] [Google Scholar]
- (450).A Phase 1 Study to Evaluate the Safety, Tolerability and Pharmacokinetics of BCX4430 NCT02319772; ClinicalTrials.gov, 2014.
- (451).Ianevski A; Zusinaite E; Kuivanen S; Strand M; Lysvand H; Teppor M; Kakkola L; Paavilainen H; Laajala M; Kallio-Kokko H; et al. Novel Activities of Safe-in-Human Broad-Spectrum Antiviral Agents. Antiviral Res 2018, 154, 174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (452).Nie P; Groaz E; De Jonghe S; Lescrinier E; Herdewijn P Synthesis of a C-Nucleoside Phosphonate by Base-Promoted Epimerization. Org. Lett 2018, 20, 1203–1206. [DOI] [PubMed] [Google Scholar]
- (453).Potempa M; Lee S-K; Wolfenden R; Swanstrom R The Triple Threat of HIV-1 Protease Inhibitors. Curr. Top. Microbiol. Immunol 2015, 389, 203–241. [DOI] [PubMed] [Google Scholar]
- (454).Freed EO HIV-1 Assembly, Release and Maturation. Nat. Rev. Microbiol 2015, 13, 484–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (455).Eder J; Hommel U; Cumin F; Martoglio B; Gerhartz B Aspartic Proteases in Drug Discovery. Curr. Pharm. Des 2007, 13, 271–285. [DOI] [PubMed] [Google Scholar]
- (456).Volberding PA HIV Treatment and Prevention: An Overview of Recommendations from the 2016 IAS–USA Antiretroviral Guide-lines Panel. Top. Antiviral Med 2017, 25, 17–24. [PMC free article] [PubMed] [Google Scholar]
- (457).Wensing AM; Calvez V; Günthard HF; Johnson VA; Paredes R; Pillay D; Shafer RW; Richman DD Update of the Drug Resistance Mutations in HIV-1. Top. Antiviral Med 2017, 24, 132–133. [PMC free article] [PubMed] [Google Scholar]
- (458).Yilmaz NK; Schiffer CA Drug Resistance to HIV-1 Protease Inhibitors: Molecular Mechanisms and Substrate Coevolution. Antimicrobial Drug Resistance; Springer, 2017; pp. 535–544. [Google Scholar]
- (459).Subbaiah MA; Meanwell NA; Kadow JF Design Strategies in the Prodrugs of HIV-1 Protease Inhibitors to Improve the Pharmaceutical Properties. Eur. J. Med. Chem 2017, 139, 865–883. [DOI] [PubMed] [Google Scholar]
- (460).Mitsuya H; Maeda K; Das D; Ghosh AK Development of Protease Inhibitors and the Fight with Drug-Resistant HIV-1 Variants. Adv. Pharmacol 2008, 56, 169–197. [DOI] [PubMed] [Google Scholar]
- (461).Ohtaka H; Muzammil S; Schön A; Velazquez-Campoy A; Vega S; Freire E Thermodynamic Rules for the Design of High Affinity HIV-1 Protease Inhibitors with Adaptability to Mutations and High Selectivity Towards Unwanted Targets. Int. J. Biochem. Cell Biol 2004, 36, 1787–1799. [DOI] [PubMed] [Google Scholar]
- (462).Raines RT; Windsor I; Palte M; Lukesh J Boronic Acid Inhibitors of HIV Protease U.S. Patent 9,738,664, 2017. (Wisconsin Alumni Research Foundation).
- (463).Hiller NJ; Silva NAAE; Faria RX; Souza ALA; Resende JALC; Borges Farias A; Correia Romeiro N; de Luna Martins D Synthesis and Evaluation of the Anticancer and Trypanocidal Activities of Boronic Tyrphostins. ChemMedChem 2018, 13, 1395–1404. [DOI] [PubMed] [Google Scholar]
- (464).Kipp DR; Hirschi JS; Wakata A; Goldstein H; Schramm VL Transition States of Native and Drug-Resistant HIV-1 Protease are the Same. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 6543–6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (465).Chen J; Wang X; Zhu T; Zhang Q; Zhang JZ A Comparative Insight into Amprenavir Resistance of Mutations V32I, G48V, I50V, I54V, and I84V in HIV-1 Protease Based on Thermodynamic Integration and MM-PBSA Methods. J. Chem. Inf. Model 2015, 55, 1903–1913. [DOI] [PubMed] [Google Scholar]
- (466).Appadurai R; Senapati S How Mutations Can Resist Drug Binding Yet Keep HIV-1 Protease Functional. Biochemistry 2017, 56, 2907–2920. [DOI] [PubMed] [Google Scholar]
- (467).Cleland WW; O’Leary MH; Northrop DB Isotope Effects on Enzyme-catalyzed Reactions. 6th Harry Steenbock Symposium; University Park Press, 1977. [Google Scholar]
- (468).Cleland WW Use of Isotope Effects to Elucidate Enzyme Mechanisms. CRC Critical Rev. Biochem 1982, 13, 385–428. [DOI] [PubMed] [Google Scholar]
- (469).Berti PJ; Tanaka KSE Transition State Analysis Using Multiple Kinetic Isotope Effects: Mechanisms of Enzymatic and Non-Enzymatic Glycoside Hydrolysis and Transfer. Adv. Phys. Org. Chem 2002, 37, 239–314. [Google Scholar]
- (470).Berti PJ; McCann JA Toward a Detailed Understanding of Base Excision Repair Enzymes: Transition State and Mechanistic Analyses of N-Glycoside Hydrolysis and N-Glycoside Transfer. Chem. Rev 2006, 106, 506–555. [DOI] [PubMed] [Google Scholar]
- (471).Williams IH; Wilson PB Kinetic Isotope Effects. Simulating Enzyme Reactivity: Computational Methods in Enzyme Catalysis; RSC Theoretical and Computational Chemistry Series No. 9; RSC, 2017; pp 150–184. [Google Scholar]
- (472).Williams IH; Wilson PB SULISO: The Bath Suite of Vibrational Characterization and Isotope Effect Calculation Software. SoftwareX 2017, 6, 1–6. [Google Scholar]
- (473).Callender R; Dyer RB The Dynamical Nature of Enzymatic Catalysis. Acc. Chem. Res 2015, 48, 407–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (474).Silva RG; Murkin AS; Schramm VL Femtosecond Dynamics Coupled to Chemical Barrier Crossing in s Born-Oppenheimer Enzyme. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 18661–18665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (475).Antoniou D; Ge X; Schramm VL; Schwartz SD Mass Modulation of Protein Dynamics Associated with Barrier Crossing in Purine Nucleoside Phosphorylase. J. Phys. Chem. Lett 2012, 3, 3538–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (476).Kipp DR; Silva RG; Schramm VL Mass-dependent Bond Vibrational Dynamics Influence Catalysis by HIV-1 Protease. J. Am. Chem. Soc 2011, 133, 19358–19361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (477).Świderek K; Ruiz-Pernía JJ; Moliner V; Tuñón I Heavy Enzymes—experimental and Computational Insights in Enzyme Dynamics. Curr. Opin. Chem. Biol 2014, 21, 11–18. [DOI] [PubMed] [Google Scholar]
- (478).Kholodar SA; Ghosh AK; Kohen A Measurement of Enzyme Isotope Effects. Methods Enzymol 2017, 596, 43–83. [DOI] [PubMed] [Google Scholar]
- (479).Luo M; Li L; Schramm VL Remote Mutations Alter Transition-State Structure of Human Purine Nucleoside Phosphorylase. Biochemistry 2008, 47, 2565–2576. [DOI] [PubMed] [Google Scholar]
- (480).Li L; Luo M; Ghanem M; Taylor EA; Schramm VL Second-sphere Amino Acids Contribute to Transition-State Structure in Bovine Purine Nucleoside Phosphorylase. Biochemistry 2008, 47, 2577–2583. [DOI] [PubMed] [Google Scholar]
- (481).Thomas K; Cameron SA; Almo SC; Burgos ES; Gulab SA; Schramm VL Active Site and Remote Contributions to Catalysis in Methylthioadenosine Nucleosidases. Biochemistry 2015, 54, 2520–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (482).Lassila JK; Zalatan JG; Herschlag D Biological Phosphoryl-Transfer Reactions: Understanding Mechanism and Catalysis. Annu. Rev. Biochem 2011, 80, 669–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (483).Cleland WW; Hengge AC Enzymatic Mechanisms of Phosphate and Sulfate Transfer. Chem. Rev 2006, 106, 3252–3278. [DOI] [PubMed] [Google Scholar]
- (484).Jin Y; Richards NG; Waltho JP; Blackburn GM Metal Fluorides as Analogues for Studies on Phosphoryl Transfer Enzymes. Angew. Chem., Int. Ed 2017, 56, 4110–4128. [DOI] [PubMed] [Google Scholar]