

Abstract
Sodium channel Nav1.7, encoded by SCN9A, is expressed in DRG neurons and regulates their excitability. Genetic and functional studies have established a critical contribution of Nav1.7 to human pain disorders. We have now characterized a novel Nav1.7 mutation (R1279P) from a female human subject with distal limb pain, in which depolarized fast inactivation overrides impaired activation to produce hyperexcitability and spontaneous firing in DRG neurons. Whole-cell voltage-clamp recordings in human embryonic kidney (HEK) 293 cells demonstrated that R1279P significantly depolarizes steady-state fast-, slow-, and closed-state inactivation. It accelerates deactivation, decelerates inactivation, and facilitates repriming. The mutation increases ramp currents in response to slow depolarizations. Our voltage-clamp analysis showed that R1279P depolarizes channel activation, a change that was supported by our multistate structural modeling. Because this mutation confers both gain-of-function and loss-of-function attributes on the Nav1.7 channel, we tested the impact of R1279P expression on DRG neuron excitability. Current-clamp studies reveal that R1279P depolarizes resting membrane potential, decreases current threshold, and increases firing frequency of evoked action potentials within small DRG neurons. The populations of spontaneously firing and repetitively firing neurons were increased by expressing R1279P. These observations indicate that the dominant proexcitatory gating changes associated with this mutation, including depolarized steady-state fast-, slow-, and closed-state inactivation, faster repriming, and larger ramp currents, override the depolarizing shift of activation, to produce hyperexcitability and spontaneous firing of nociceptive neurons that underlie pain.
Keywords: DRG neurons, mutation, Nav1.7, pain, sodium channel
Introduction
Voltage-gated sodium channels play an indispensable role in action potential initiation and propagation in neurons (Hodgkin and Huxley, 1952). One of the nine sodium channel subtypes (Nav1.1-Nav1.9), the Nav1.7 channel, encoded by SCN9A, has attracted significant attention as a potential therapeutic target for pain disorders (Dib-Hajj et al., 2009a; Lee et al., 2014). The Nav1.7 channel is preferentially expressed in small DRG neurons, which include nociceptive and sympathetic ganglion neurons (Black et al., 1996, 2012; Toledo-Aral et al., 1997; Djouhri et al., 2003; Rush et al., 2006). Discovery of missense mutations in SCN9A in inherited erythromelalgia (IEM) (Cummins et al., 2004; Yang et al., 2004; Dib-Hajj et al., 2005; Drenth et al., 2005), a hereditary disorder characterized by recurrent attacks of red, warm, and burning pain in distal extremities, has established a causal relationship between gain-of-function mutations of Nav1.7, which enhance activation and IEM. More than a dozen gain-of-function mutations that enhance activation of the Nav1.7 channel have been described in IEM patients over the past decade (Dib-Hajj et al., 2013). Paroxysmal extreme pain disorder (PEPD), a different type of pain disorder, has also been linked to gain-of-function missense mutations in Nav1.7 channel, which impair fast inactivation (Fertleman et al., 2006). Additionally, gain-of-function variants of Nav1.7 have been identified in patients with painful small-fiber neuropathy (Faber et al., 2012). Loss-of-function mutations in SCN9A, in contrast, have been found in individuals with channelopathy-associated insensitivity to pain (Cox et al., 2006). These human genetic studies support a critical role of Nav1.7 in pain pathways.
In this study, we describe a novel SCN9A/Nav1.7 mutation (R1279P) in a patient with distal leg pain. We show that, despite depolarized activation, this mutation produces gain-of-function changes due to depolarized inactivation, which enhances spontaneous and evoked excitability of DRG neurons.
Materials and Methods
Cell culture and plasmid transfection.
The plasmid carrying the human Nav1.7 cDNA was cloned into a mammalian expression vector (Klugbauer et al., 1995) and converted to tetrodotoxin-resistance (TTX-R) by Y362S substitution (Herzog et al., 2003) and to the adult-long splice isoform (Raymond et al., 2004) (hNav1.7R/AL); designated as wild-type (WT). The R1279P missense mutation from a female human subject with distal leg pain (i.e., replacement of arginine by proline residue at position 1279 in the S4 segment of domain III within the α subunit of hNav1.7 channel) was introduced using QuikChangeII XL site-directed mutagenesis (Stratagene) and referred to as R1279P hereinafter. Identity of inserts was confirmed by sequencing at the Howard Hughes Medical Institute/Keck Biotechnology Center at Yale University (New Haven, CT).
Plasmids of hNav1.7R/AL WT or R1279P were cotransfected with EGFP into HEK293 cells using Optifect Reagent (Invitrogen). HEK293 cells were grown under standard culture conditions (5% CO2, 37°C) in DMEM supplemented with 10% FBS.
Isolation and transfection of primary sensory neurons.
Animal studies followed a protocol approved by the Veterans Administration West Haven Hospital Institutional Animal Care and Use Committees. DRGs from 4- to 6-week-old female and male Sprague–Dawley rats were harvested and dissociated as described previously (Dib-Hajj et al., 2009b; Huang et al., 2013, 2014). Briefly, DRG neurons were dissociated with a 20 min incubation in 1.5 mg/ml Collagenase A (Roche) and 0.6 mm EDTA, followed by a 18 min incubation in 1.5 mg/ml Collagenase D (Roche), 0.6 mm EDTA, and 30 U/ml papain (Worthington Biochemical). DRGs were then centrifuged and triturated in 0.5 ml of DRG media containing 1.5 mg/ml BSA (low endotoxin) and 1.5 mg/ml trypsin inhibitor (Sigma). After trituration, 2 ml of DRG media was added to the cell suspension, which was filtered with 70 μm nylon mesh cell strainer (BD Biosciences). The mesh was washed twice with 2 ml of DRG media. The cells were then transfected with EGFP and WT or mutant hNav1.7R/AL constructs using a Nucleofector IIS (Lonza) and Basic Neuron SCN Nucleofector Kit (VSPI-1003; Amaxa). Briefly, the cell suspension was centrifuged (100 × g for 3 min), and the cell pellet was resuspended in 20 μl of Nucleofector solution, mixed with 2 μg of hNav1.7R/AL WT or mutant construct plus 0.2 μg of EGFP, and transfected using Nucleofector IIS and protocol SCN-BNP 6. After transfection, cells were recovered in calcium-free DMEM, fed with DRG media supplemented with nerve growth factor (50 ng/ml) and glial cell line-derived neurotrophic factor (50 ng/ml), and maintained at 37°C in a 95% air/5% CO2 (v/v) incubator for 40–55 h before current-clamp recording.
Whole-cell electrophysiology.
Voltage-clamp recordings were performed on isolated, GFP-labeled HEK293 cells 24–36 h after transfection using an EPC-10 amplifier and the Patchmaster program (v 53; HEKA Elektronik) at room temperature (22 ± 1°C). Fire-polished electrodes were fabricated from 1.6 mm outer diameter borosilicate glass micropipettes (World Precision Instruments) using a Sutter Instruments P-97 puller and had a resistance of 0.7–1.5 MΩ. Pipette potential was adjusted to zero before seal formation. Liquid junction potential was not corrected. To reduce voltage errors, 80%–90% series resistance compensation was applied. Cells were excluded from analysis if the predicted voltage error exceeded 3 mV. Linear leak currents were subtracted out using the P/N method. Sodium current recordings were initiated after a 5 min equilibration period once whole-cell configuration was achieved. Current traces were sampled at 50 kHz and filtered with a low-pass Bessel setting of 10 kHz. The pipette solution contained the following (in mM): 140 CsF, 10 NaCl, 1 EGTA, and 10 HEPES, 10 dextrose, pH 7.30 with CsOH (adjusted to 310 mOsmol/L with sucrose). The extracellular bath solution contained the following (in mM): 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, 10 dextrose, pH 7.30 with NaOH (adjusted to 320 mOsmol/L with sucrose). TTX (300 nM) was included in the bath to block the endogenous sodium currents in HEK293 cells (Cummins et al., 2009; Dib-Hajj et al., 2009b; He and Soderlund, 2010). Transfected HEK293 cells were held at −120 mV for all parameters examined. Current–voltage (I-V) relationships were measured using a series of 100 ms step depolarizations (−80 mV to 60 mV in 5 mV increments at 5 s intervals) from holding potential. Current density was calculated by normalizing maximal peak currents with cell capacitance. Peak inward currents obtained from activation protocols were converted to conductance values using the equation G = I/(Vm − ENa), where G is the conductance, I is the peak inward current, Vm is the membrane potential step used to elicit the response, and ENa is the reversal potential for sodium channel, which is determined for each cell using the x-axis intercept of a linear fit of the peak inward current responses to the last six voltage steps of the activation protocol. Conductance data were normalized by the maximum conductance value and fit with a Boltzmann equation of the form G = Gmax + (Gmin − Gmax)/(1 + exp [(Vm − V1/2)/k)], where V1/2 is the midpoint of activation and k is a slope factor. The kinetics of deactivation were examined by 50 ms pulses at a range of potentials from −40 mV to −120 mV in −5 mV increments following a brief activation of the channels at −10 mV for 0.5 ms. The tail currents were fit with a single-exponential equation of the form I = A × exp (−t/τ) + Ic, where A is the amplitude of the fit, t is time, τ is the time constant of decay, and Ic is the asymptotic minimum to which the tail currents decay. To assess the open-state inactivation, decaying currents after the peak during the activation protocol were fit with the same single-exponential equation as the one measuring deactivation kinetics. Steady-state fast inactivation was assessed with a series of 500 ms prepulses (−140 mV to −10 mV in 10 mV increments); the remaining noninactivated channels were activated by a 40 ms step depolarization to −10 mV. Peak inward currents obtained from steady-state fast inactivation were normalized by the maximum current amplitude and fit with a Boltzmann equation of the form I/Imax = 1/(1 + exp [(Vm − V1/2)/k]), where Vm represents the inactivating prepulse membrane potential and V1/2 represents the midpoint of fast inactivation. Steady-state slow inactivation was assessed by prepulsing the cells with a 30 s stimulus at a range of voltages from −130 mV to 30 mV in 10 mV increments, followed by a 100 ms step to −120 mV to remove fast inactivation and then a 20 ms depolarizing step to −10 mV to elicit a test response, which reflects the remaining currents available for activation. Peak inward currents obtained from steady-state slow inactivation were normalized by the maximum current amplitude and fit with a Boltzmann equation of the form I/Imax = A + (1 − A)/(1 + exp [(Vm − V1/2)/k]), where Vm represents the inactivating prepulse membrane potential, V1/2 represents the midpoint of slow inactivation, and A represents the percentage of noninactivating channels. Recovery of hNav1.7 channels from fast inactivation, repriming, was examined using a two-pulse protocol with interpulse intervals varying from 1 to 513 ms. Recovery rates were measured by normalizing peak current elicited by the test pulse (10 ms depolarization to −10 mV) to that of the prepulse (20 ms at −10 mV) at voltages ranging from −110 mV to −80 mV in 10 mV increments. Closed-state inactivation was examined by stepping from the holding potential of −120 mV to more depolarized potentials ranging from −110 mV to −60 mV in 10 mV increments, followed by a 20 ms test pulse at −10 mV to activate the available channels. The duration of conditioned voltages ranged from 1 to 513 ms. The peak currents obtained at different time intervals were normalized and plotted against the time interval at each voltage potential tested. The normalized current acquired at the time interval of 513 ms was used to calculate the voltage dependence of steady-state closed-state inactivation. The same Boltzmann equation used in fitting steady-state fast inactivation was adopted for closed-state inactivation, where V1/2 represents the midpoint of steady-state closed-state inactivation. Ramp currents were elicited with ramp depolarizations from −120 to 0 mV at multiple rates of 0.2, 0.24, 0.3, 0.4, 0.6, and 1.2 mV/ms. The amplitude of ramp current was presented as a percentage of the maximal peak current that was obtained from the activation protocol.
For current-clamp recordings, the pipette solution contained the following (in mm): 140 KCl, 0.5 EGTA, 5 HEPES, and 3 Mg-ATP, 10 dextrose, pH 7.30 with KOH (adjusted to 310 mOsm with sucrose), and the bath solution contained the following (in mm): 140 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2, 10 HEPES, 10 dextrose, pH 7.30 with NaOH (adjusted to 320 mOsm with sucrose). TTX was not used in the bath solution for current-clamp recordings (Cummins et al., 2009). Transfection of DRG neurons does not permit a direct measurement of relative expression of recombinant Nav1.7 to the endogenous channel. In addition to Nav1.7, there are other endogenous TTX-S channels expressed in adult DRG neurons that constitute ∼30% of the total TTX-S currents (Dib-Hajj et al., 2010; Vasylyev et al., 2014). We compared cultures transfected with WT and R1279P channels that were prepared under identical conditions, so that our experiments provide a firm basis for assessing relative excitability of neurons expressing WT versus mutant channels. Whole-cell configuration was obtained in voltage-clamp mode before proceeding to the current-clamp recording mode. DRG neurons (20–30 μm) with GFP labeling and neurites were selected for analysis. Small DRG neurons with stable (<10% variation) resting membrane potentials (RMPs) more negative than −40 mV and overshooting action potentials (>85 mV RMP to peak) were used for additional data collection to ensure sufficient amount of functional Nav1.8 channels for producing the upstroke of action potentials. Input resistance was determined by the slope of a linear fit to hyperpolarizing responses to current steps from −5 pA to −40 pA in −5 pA increments. Threshold was determined by the first action potential elicited by a series of depolarizing current injections (200 ms) that increased in 5 pA increments. Action potential frequency was determined by quantifying the number of action potentials elicited in response to depolarizing current injections (500 ms). Finally, the expression level of endogenous Nav1.8 channels was examined by holding neurons at −50 mV (Cummins and Waxman, 1997). Cells that expressed small Nav1.8 currents (<1 nA) and unable to generate all-or-none action potentials in response to 200 ms current stimulus were excluded from analysis (Cheng et al., 2011).
Data were analyzed using Fitmaster (HEKA Elektronik) and Origin (Microcal Software) software. Unless otherwise noted, statistical significance was determined using an independent t test. Two-proportion z test was used for comparing populations of spontaneously firing neurons and repetitively firing neurons. Mann–Whitney test was used for comparison of firing frequencies between neurons expressing Nav1.7 WT and R1279P mutant in response to stimuli ranging from 25 to 500 pA. Results are presented as mean ± SEM, and error bars in the figures indicate SEM.
Multistate structural modeling.
Modeling of the Nav1.7 channel was performed based on previously described methods (Yang et al., 2012, 2013b). Briefly, the sequence of voltage-sensing domain (VSD) of Nav1.7 domain III was aligned to Kv1.2/Kv2.1 “paddle chimera” voltage-gated K+ channel (Long et al., 2007) using ClustalW2 and manually refined based on previous studies (Yarov-Yarovoy et al., 2012). Modeling of the S1-S4 VSD in six gating states was performed with SWISS-MODEL (Arnold et al., 2006; Bordoli et al., 2009) based on templates generated in an ultra-long molecular dynamics simulation (Jensen et al., 2012) and refined as described previously (Yang et al., 2011, 2013a). Models were further energy minimized, analyzed, and visualized in Molecular Operating Environment (MOE; Chemical Computing Group). Virtual mutagenesis was performed using MOE.
Results
Clinical phenotype and identification of the SCN9A R1279P mutation
The R1279P mutation in SCN9A was identified in a 49-year-old patient with a history of an irritating sensation in both calves and an irrepressible desire to move her legs, beginning at age 33. Approximately 10 years later, she developed severe tingling pain, which she rated as an 8 in severity on a scale from 0 to 10, which initially began at age 44 in the toes, but progressed to the knees. She also noted that her skin was painful to touch. The patient denied numbness or weakness but described palpitations, intermittent cold and white fingers, hot and cold flushes, and recurrent urinary tract infections. The patient denied sicca symptoms, orthostatic hypotension, and constipation. She had an unremarkable medical history and was not taking any medication. Family history included her mother with rheumatic arthritis and her son, aged 18, with complaints of painful and sometimes red hands and feet, who did not consent to further analysis. Neurological examination was normal except for allodynia from the knees down. Laboratory examinations were unremarkable. EMG and nerve conduction studies showed normal motor and sensory conduction in arms and legs, with no evidence for an axonal polyneuropathy. The patient declined quantitative sensory testing and a skin biopsy for analysis of intraepidermal nerve fibers. The patient met clinical criteria, including length-dependent symptoms consistent with small-fiber damage without large-fiber involvement, together with autonomic signs, for probable small-fiber neuropathy (Faber et al., 2012; Lauria et al., 2012), and was also given a diagnosis of restless legs syndrome (Rios Romenets and Postuma, 2013), which is well established as a concomitant of painful neuropathy (Iannaccone et al., 1995; Polydefkis et al., 2000; Gemignani et al., 2006). Tizanidine and amitriptryptiline did not provide relief, and acetaminophen provided partial relief. Gabapentin was initially discontinued because of side effects. Symptoms were, however, progressive and gabapentin was restarted, providing adequate symptom control for a number of years.
Genetic analysis of SCN9A, the gene encoding Nav1.7, revealed a mutation (c.3836G>C, R1279P). The mutation that was identified (c.3836G>C, R1279P) has not previously been described. Arginine 1279 is located within the voltage sensor (S4) in domain III (Fig. 1). The mutation substitutes arginine 1279, a highly conserved residue among the sodium channel family, by a proline, which changes a positively charged amino acid residue to a secondary amine whose amine nitrogen is bound to two alkyl groups and is unable to form a hydrogen bond with neighboring residues.
Figure 1.

Schematic of a voltage-gated sodium channel α subunit showing the location of the R1279P mutation and the aligned sequences for DIII/S4. R1279P is conserved in all known human voltage-gated sodium channels.
Voltage-clamp characterization of WT and R1279P mutant Nav1.7
To investigate the effect of the R1279P mutation on electrophysiological properties of the Nav1.7 channel, HEK293 cells were transiently transfected with either hNav1.7R/AL WT or R1279P mutant construct followed by voltage-clamp studies 24 h after transfection. Representative whole-cell currents from WT and R1279P mutant channels are presented in Figure 2A and Figure 2B, respectively. The average peak inward current density was not significantly different between WT and R1279P Nav1.7 channels (Table 1).
Figure 2.

The R1279P mutation of Nav1.7 depolarizes activation and accelerates deactivation. A, Inward currents recorded from an HEK293 cell expressing WT hNav1.7R/AL sodium channels. B, Inward currents recorded from an HEK293 cell expressing R1279P mutant. C, Comparison of the voltage dependence of activation for WT (n = 24) and R1279P (n = 32) channels. The midpoint of activation (V1/2, act) was depolarized by 5.9 mV for R1279P mutant currents (Table 1). D, Time constants for tail current deactivation for WT (n = 18) and R1279P (n = 25) hNav1.7 channels. Error bars indicate SEM. *Statistical significance (p < 0.05).
Table 1.
Biophysical properties of WT and mutant Nav1.7 channels
| Nav1.7 | Current density |
Activation (mV) |
Steady-state fast inactivation (mV) |
Steady-state slow inactivation (mV) |
Closed-state inactivation (mV) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pA/pF | n | V1/2,act | k | n | V1/2,fast | k | n | V½,slow | k | A% | n | V½,closed | k | n | |
| WT | −285 ± 41 | 28 | −24.0 ± 1.3 | 8.77 ± 0.40 | 24 | −84.2 ± 0.80 | 6.33 ± 0.15 | 19 | −65.6 ± 2.0 | 11.4 ± 0.68 | 10.7 ± 1.1 | 5 | −87.0 ± 0.70 | 6.26 ± 0.68 | 5 |
| R1279P | −237 ± 30 | 34 | −18.1 ± 0.78*** | 9.19 ± 0.23 | 32 | −74.6 ± 0.79*** | 6.82 ± 0.26 | 30 | −58.0 ± 2.4* | 11.6 ± 0.52 | 10.6 ± 1.6 | 6 | −78.6 ± 1.5*** | 6.99 ± 0.53 | 5 |
*p < 0.05 versus WT channels;
***p < 0.001 versus WT channels.
The voltage dependence of activation for WT and R1279P is shown in Figure 2C. The threshold for activation of R1279P mutant channels was ∼5 mV more depolarized than for WT channels. The midpoint of activation (V1/2, act) was depolarized by 5.9 mV for R1279P compared with WT (Table 1). The slope factor of voltage-dependent activation, which represents the steepness of the Boltzmann fits, remained unaffected by the mutation (Table 1). As shown in Figure 2D, we examined the kinetics of deactivation, which reflect the transition from the open to the closed state. Most likely because of the depolarizing shift of activation threshold, R1279P mutant currents exhibited significantly faster kinetics for deactivation at −40 mV and −45 mV, which appeared to be the activation threshold of hNav1.7 channels.
The kinetics for open-state inactivation, which reflects the transition from the open to the inactivated state, were significantly decelerated in the mutant channel at voltages from −45 to 0 mV (Fig. 3A). The time constant of fast inactivation for R1279P almost doubled at voltages ranging from −45 mV to −25 mV. For example, the time constant was 7.30 ± 0.58 ms (n = 24) for R1279P and 3.50 ± 0.27 ms (n = 21) for WT at −35 mV (p < 0.001). However, the inactivation kinetics were similar between WT and R1279P at more depolarized potentials from 5 to 40 mV.
Figure 3.
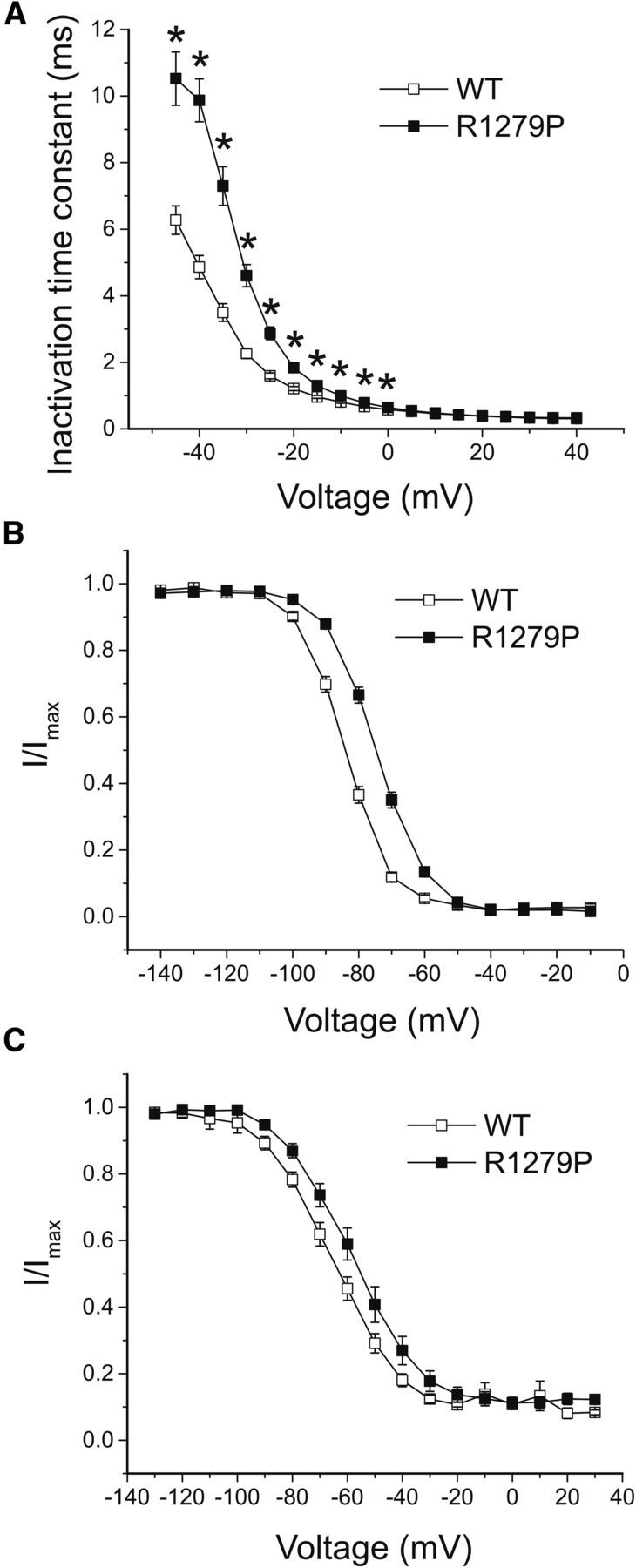
The R1279P mutation depolarizes inactivation. A, The kinetics of open-state inactivation measured as a function of voltage for WT (n = 21) and R1279P (n = 24) hNav1.7 channels. Time constants were obtained by fitting currents elicited as described in Figure 2A, B with single-exponential function. B, Comparison of steady-state fast inactivation for WT and R1279P Nav1.7 channels. The midpoint of fast inactivation (V1/2, fast) for R1279P was depolarized by 9.6 m compared with WT (Table 1). C, Comparison of steady-state slow inactivation for WT and R1279P hNav1.7 channels. The voltage dependence of slow inactivation for R1279P (V1/2, slow) was depolarized by 7.6 mV compared with WT (Table 1). Error bars indicate SEM. *Statistical significance (p < 0.05).
In addition to the significant deceleration of open-state inactivation, we found that the R1279P mutation dramatically depolarized the voltage-dependent fast inactivation curve by 9.6 mV (Fig. 3B; Table 1). We also asked whether slow inactivation was affected by this mutation. As shown in Figure 3C, R1279P shifted the voltage-dependent slow inactivation curve in a depolarizing direction by 7.6 mV (Table 1). There was no statistically significant difference in slope factor between WT and R1279P either for steady-state fast or slow inactivation (Table 1). The fraction of noninactivating currents during slow inactivation (A%) for R1279P did not differ from that for WT either (Table 1).
We tested recovery from fast inactivation (repriming) of WT and R1279P mutant channels using a two-pulse protocol with varied interpulse interval from 1 to 513 ms at voltage potentials of −110 mV (black), −100 mV (blue), −90 mV (magenta), and −80 mV (green), respectively (Fig. 4). The current evoked by the test pulse at −10 mV was normalized to the current elicited by the prepulse at −10 mV, to represent the fraction of recovering currents, and was plotted against the interpulse interval to demonstrate the rate and extent of repriming for WT (n = 6) and R1279P mutant (n = 9) channels (Fig. 4). At hyperpolarized potentials, such as shown for −110 mV, the repriming rate was rapid and similar between WT and R1279P. For interpulse potentials at −100 mV, −90 mV, and −80 mV, however, both the rates and the fractions of recovery for R1279P (filled squares) were larger than those for WT channels (open squares). As shown in Figure 4, Nav1.7 channels reprimed less completely and at a slower rate when the conditioned voltage potential depolarized from −110 mV to −80 mV. However, R1279P mutant channels were able to recover to the same extent of WT with a comparable rate at a voltage potential 10 mV more depolarized than WT. For example, R1279P recovered at a similar rate and to a similar extent at −100 mV (blue, solid squares) compared with WT channels at −110 mV (black, open squares), at −90 mV (magenta, solid squares) compared with WT channels at −100 mV (blue, open squares), and at −80 mV (green, solid squares) compared with WT channels at −90 mV (magenta, open squares) (Fig. 4). These observations are consistent with our findings that R1279P depolarized the voltage dependence of fast inactivation by ∼10 mV (Fig. 3B; Table 1).
Figure 4.

The R1297P mutation facilitates repriming of hNav1.7 channel. Recovery from fast inactivation for WT (n = 6) and R1279P (n = 9) hNav1.7 channels was assessed at recovery potentials ranging from −110 mV to −80 mV that varied in time (1–513 ms). Error bars indicate SEM.
We further examined the effect of the R1279P mutation on closed-state inactivation. The time-dependent development of closed-state inactivation at conditioning potentials ranging from −110 mV to −60 mV, which were below the threshold for activating hNav1.7 channels, is displayed in Figure 5A for WT and Figure 5B for R1279P mutant channels. For steady-state conditions, the R1279P mutation shifted the voltage dependence of closed-state inactivation in a depolarized direction by 8.4 mV (Fig. 5C; Table 1), which is similar to the depolarizing shift in the voltage dependence of steady-state fast inactivation for R1279P (9.6 mV) (Fig. 3B; Table 1).
Figure 5.
The R1279P mutation depolarizes closed-state inactivation of hNav1.7 channels. A, Development of closed-state inactivation of WT hNav1.7 channels. B, Development of closed-state inactivation of R1279P hNav1.7 channels. C, Voltage dependence of steady-state closed-state inactivation. Normalized currents after a recovery period of 513 ms were plotted as a function of the inactivation voltage potential. The R1279P mutation depolarized closed-state inactivation by 8.4 mV (Table 1). Error bars indicate SEM.
Finally, we examined the responses of WT and R1279P channels to slow or fast depolarizations using ramp stimuli at rates of 0.2, 0.24, 0.3, 0.4, 0.6, and 1.2 mV/ms (Table 2). The ramp current was normalized to the peak inward current recorded during the activation protocol for each cell (Fig. 2A,B). Representative ramp current traces in response to slow depolarizations at a rate of 0.2 mV/ms for WT (gray) and R1279P (black) are shown in Figure 6A and demonstrate a pronounced inward current for R1279P near −60 mV. The R1279P mutation resulted in an ∼3-fold increase of the current amplitude in response to small and slow depolarizations (0.2 mV/ms) (Table 2). Compared with WT, the ramp currents of R1279P mutant channels were significantly increased at slow depolarizing rates (0.2, 0.24, 0.3, and 0.4 mV/ms) (p < 0.05), whereas the enhancement in R1279P diminished at fast depolarizing rates (0.6 and 1.2 mV/ms) (Fig. 6B; Table 2). Consistent with a depolarizing shift in voltage-dependent activation by 5.9 mV for R1279P, the voltage at which the peak ramp current of the mutant channel occurred was also significantly depolarized by ∼5 mV at every ramp rate tested (p < 0.05) (Fig. 6C; Table 2).
Table 2.
Ramp currents of WT and mutant Nav1.7 channels at multiple depolarizing rates
| Nav1.7 ramp current | 0.2 mV/ms |
0.24 mV/ms |
0.3 mV/ms |
0.4 mV/ms |
0.6 mV/ms |
1.2 mV/ms |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % of Ipeak | Vpeak (mV) | % of Ipeak | Vpeak (mV) | % of Ipeak | Vpeak (mV) | % of Ipeak | Vpeak (mV) | % of Ipeak | Vpeak (mV) | % of Ipeak | Vpeak (mV) | |
| WT (n = 7) | 0.608 ± 0.15 | −45.3 ± 2.3 | 0.843 ± 0.20 | −44.8 ± 1.8 | 1.22 ± 0.28 | −44.5 ± 1.5 | 1.94 ± 0.45 | −44.0 ± 1.8 | 3.46 ± 0.83 | −41.6 ± 1.8 | 9.20 ± 1.7 | −38.5 ± 1.9 |
| R1279P (n = 25) | 1.61 ± 0.15** | −40.4 ± 0.84* | 1.96 ± 0.20** | −40.0 ± 1.1* | 2.53 ± 0.24** | −39.6 ± 0.96* | 3.55 ± 0.37* | −39.0 ± 0.93* | 5.82 ± 0.57 | −37.5 ± 0.88* | 13.2 ± 1.0 | −33.8 ± 1.1* |
| Δ | 2.65X** | 4.9 mV* | 2.33X** | 4.8 mV* | 2.07X** | 4.9 mV* | 1.83X* | 5.0 mV* | 1.68X | 4.1 mV* | 1.43X | 4.7 mV* |
*p < 0.05 versus WT channels;
**p < 0.01 versus WT channels.
Figure 6.
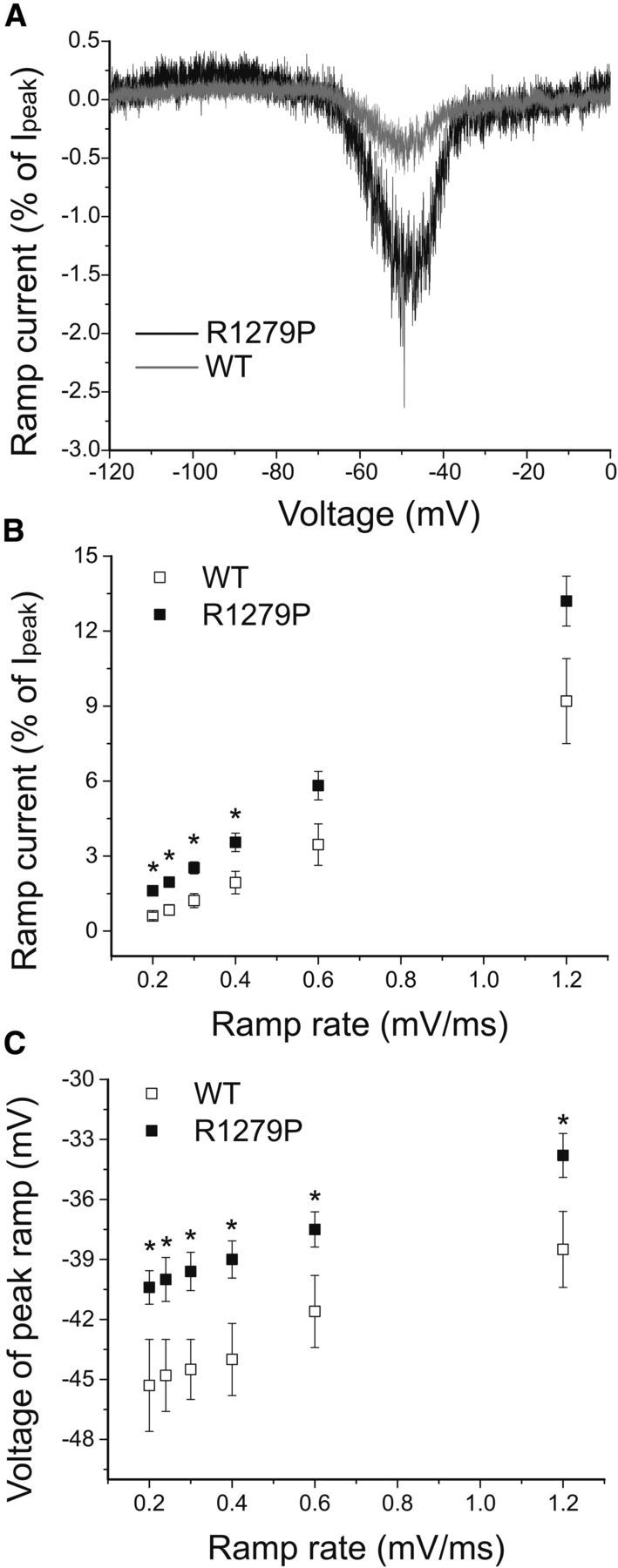
The R1279P alters responses of hNav1.7 channels to ramp stimuli at multiple rates. A, Representative ramp currents in response to slow depolarizations (0.2 mV/ms) were normalized to the peak current acquired during activation protocol and plotted as a function of membrane potential for WT and R1279P hNav1.7 channels. B, Changes in ramp amplitude in WT (n = 7) and R1279P (n = 25) hNav1.7 channels when depolarizing rate increased from 0.2 to 1.2 mV/ms (Table 2). C, Voltages at which the peak responses of ramp occur at depolarizing rates ranging from 0.2 to 1.2 mV/ms for WT (n = 7) and R1279P (n = 25) hNav1.7 channels (Table 2). Error bars indicate SEM. *Statistical significance (p < 0.05).
Current-clamp characterization of DRG neurons expressing WT and R1279P mutant Nav1.7
Because R1279P conferred both gain-of-function and loss-of-function attributes on the Nav1.7 channel, we tested the effect of this mutation on DRG neuron excitability using current-clamp recording. To assess the effects of R1279P mutation on DRG neuronal excitability, current-clamp experiments were performed in two groups of small DRG neurons: electroporated with GFP and either the hNav1.7R/AL WT or R1279P construct. Only one of 27 (3.7%) of DRG neurons expressing WT channels fired action potentials spontaneously, whereas a significantly larger population of cells that express R1279P mutant channels (29%, 10 of 35 cells) produced spontaneous action potentials with no injected current stimulus (Table 3). Thus, there is a close to 8-fold increase in the population of spontaneously firing neurons for R1279P compared with that for WT (p < 0.05; Fig. 7A). Spontaneous action potentials recorded for 30 s continuously in a representative DRG neuron expressing R1279P mutant channels are presented in Figure 7A. The spontaneous firing DRG neurons were excluded from the analysis of RMP, input resistance, action potential amplitude, half-width, voltage threshold, and current threshold for WT and R1279P. There were no significant differences (p > 0.05) in input resistance, voltage threshold at which an action potential took off, action potential amplitude, or half-width of an action potential between the two groups of neurons expressing WT (n = 26) or R1279P mutant (n = 25) channels that fire action potentials in response to injected current stimuli (Table 3). The RMP of neurons expressing R1279P, however, was significantly depolarized by 6.3 mV (Fig. 7B; Table 3). R1279P and WT also differed with regard to current threshold at which an all-or-none action potential is elicited. R1279P reduced the current threshold by 42% compared with WT (Fig. 7C; Table 3). Figure 7D shows a small DRG neuron expressing WT channels, which responded to subthreshold current injections (180–200 pA in 5 pA increments) with graded membrane potential depolarizations. At a threshold of 205 pA, an overshooting action potential was produced in this cell and a further increase in injected current (e.g., 210 pA) generated a similar spike. Representative action potentials recorded from a small DRG neuron expressing R1279P mutant channels are shown in Figure 7E. The cell only produced graded depolarizations when injected current was ≤95 pA, and an action potential did not occur until the current stimulus reached ≥100 pA, which was defined as the current threshold for this cell (Fig. 7E).
Table 3.
Action potential characterization for WT and mutant Nav1.7 channels in small DRG neurons
| Nav1.7 | Spontaneously firing neurons | Evoked firing neurons | Input resistance (GΩ) | RMP (mV) | Action potential amplitude (mV) | Half-width (ms) | Voltage threshold (mV) | Current threshold (pA) | Repetitively firing neurons |
|---|---|---|---|---|---|---|---|---|---|
| WT | 1 of 27, 3.7% | n = 26 | 0.878 ± 0.083 | −53.2 ± 0.98 | 122 ± 1.6 | 4.16 ± 0.19 | −22.6 ± 1.5 | 208 ± 21 | 7 of 26, 27% |
| R1279P | 10 of 35, 29%* | n = 25 | 0.763 ± 0.067 | −46.9 ± 0.96*** | 119 ± 2.1 | 4.73 ± 0.26 | −23.3 ± 1.7 | 121 ± 18** | 18 of 25, 72%** |
*p < 0.05 versus WT channels;
**p < 0.01 versus WT channels;
***p < 0.001 versus WT channels.
Figure 7.

The R1279P mutation depolarizes RMP and reduces current threshold for initiation of single action potentials. A, Representative recording of spontaneous firing in a small DRG neuron expressing R1279P mutant channels. The trace was recorded for 30 s without current injection. Bar graph showing a 7.8-fold increase in the proportion of spontaneous firing cells for DRG neurons expressing R1279P (gray) compared with WT hNav1.7 channels (black); numbers to the right of the bar graph show values for WT (lower value in parentheses) and R1279P (upper value). B, RMP of neurons expressing WT was depolarized by 6.3 mV compared with that of R1279P hNav1.7 channels (Table 3). C, Current threshold was significantly reduced after expressing R1279P channels (Table 3). D, Responses of a current-clamped small DRG neuron transfected with WT hNav1.7 channels to a series of subthreshold (180–200 pA) and suprathreshold depolarizing current steps (205 and 210 pA). Starting at a subthreshold stimulus intensity, the current amplitude was increased in 5 pA increments to an intensity well beyond threshold. The current threshold was 205 pA for this neuron. E, The same threshold protocol was applied to a small DRG neuron transfected with R1279P hNav1.7 channel. The current threshold was 100 pA for this neuron. Arrows with numbers indicate the current amplitude used to elicit the labeled response. Error bars indicate SEM. Statistical significance: *p < 0.05; **p < 0.01; ***p < 0.001.
Finally, we examined the numbers of action potentials evoked by graded (25–500 pA), prolonged current injections (500 ms) in both groups of neurons. As shown in Figure 8B, C, cells expressing WT channels were more likely to generate only one action potential when injected with currents of amplitudes 1.5- or 2-fold the current threshold. In contrast, cells expressing R1279P tended to fire multiple action potentials in response to similar stimuli (Fig. 8E,F). The fraction of repetitively firing neurons was significantly increased with the expression of R1279P mutant channels (18 of 25, 72%) compared with WT (7 of 26, 27%, p < 0.01) (Table 3). The average numbers of action potentials elicited by graded current injections in small DRG neurons expressing WT or R1279P are plotted in Figure 8G. Small DRG neurons expressing R1279P mutant construct fired at a higher-than-normal frequency in response to injected currents ranged from 75 to 500 pA (Fig. 8G).
Figure 8.

The R1279P mutation increases excitability in small DRG neurons. Responses of a small DRG neuron expressing WT (A–C) or R1279P (D–F) hNav1.7 channels to 500 ms depolarizing current steps that are 1-fold (A, D), 1.5-fold (B, E), and 2-fold (C, F) of its current threshold, respectively. G, Summary of firing frequency in response to graded inputs. The total number of action potentials elicited by the indicated depolarizing current step from 25 to 500 pA in 25 pA increments was compared between two groups of small DRG neurons expressing WT (n = 26) or R1279P (n = 25) channels. Error bars indicate SEM. *Statistical significance (p < 0.05).
Multistate structural modeling of Nav1.7 DIII VSD
Multistate modeling provides a high-resolution method for predicting atomic-level changes associated with the gating of a voltage-gated ion channel as it transits from closed to activated and back to closed states (Yang et al., 2013b). Because the IFM inactivation particle is not located within a membrane-spanning segment that can be accurately modeled, channel inactivation is not amenable to analysis by multistate modeling. However, channel activation is regulated in large part by changes in the relative conserved membrane-spanning segments, especially the VSD where R1279 is located. We therefore studied the Nav1.7 DIII VSD using multistate modeling and assessed the effects of the R1279P mutation on channel activation. The VSDs of Nav1.7 DIII containing R1279 or R1279P were analyzed in six gating states: (1) activated state; (2) early deactivation state; (3) late deactivation state; (4) resting/closed state; (5) early activation state; and (6) late activation state (Fig. 9A–F).
Figure 9.
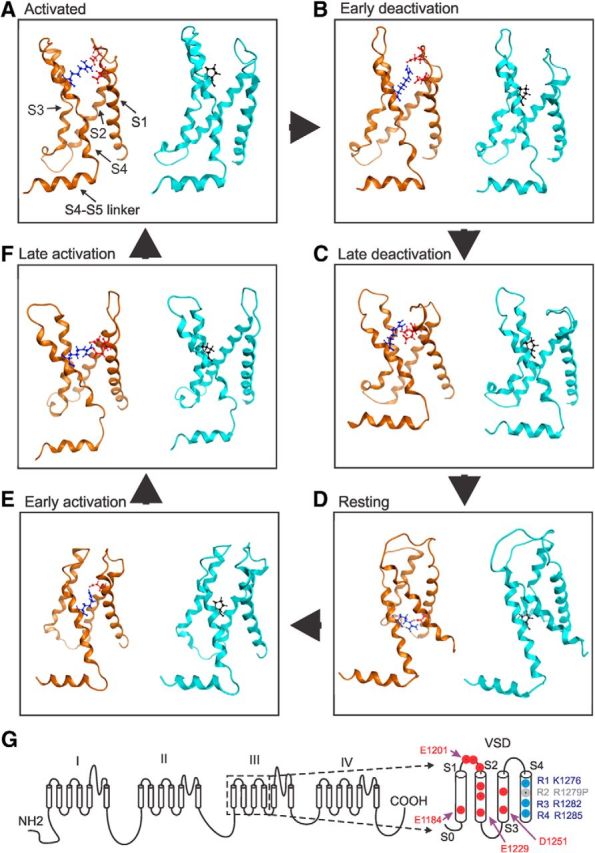
Multistate modeling of VSD of WT and mutant Nav1.7 DIII in six gating states. In each panel, the WT channel with VSD containing R1279 is shown on the left, wheat-colored, and the mutant channel with VSD containing R1279P is shown on the right, in cyan. S1–S4 helices are labeled in the left of A. A, Activated state. R1279 interacts with E1201 and E1206. B, Early deactivation state. R1279 interacts with E1201 and E1206. C, Late deactivation state. R1279 interacts with E1201 and D1202. D, Resting state. R1279 interacts with E1229. E, Early activation state. R1279 interacts with D1219. F, Late activation state. R1279 interacts with E1201 and D1202. R1279 residue is shown as stick and blue. R1279P is shown as stick and black. The interaction partners of R1279 are shown as stick and red. G, Schematic of the human Nav1.7 channel. Boxed area represents the VSD, which is enlarged at the right. Negatively charged residues are labeled red, and positively charged residues are labeled blue. R1279 is labeled gray.
The VSD domain contains negatively charged residues that might be critical interaction partners for R2, including E1184 of S1 helix; E1201, D1202, and E1206 of S1-S2 linker; E1216, D1219, and E1229 of S2 helix; and D1251 and D1256 of S3 helix (Fig. 9G). Using multistate modeling, we observed that positively charged R1279 has strong ionic interactions with negatively charged residues in the channel open states but not in the closed states (Fig. 9 A–F, left, wheat-colored). In particular, R1279 interacts with two negatively charged residues (E1201 and E1206) in the activated state and early deactivation state. In the late deactivation state, R1279 also interacts with two negatively charged residues (E1201 and D1202). However, in the resting state and early activation state, R1279 only interacts with one negatively charged residue (E1229 and D1219, respectively). When the channel enters late activation, R1279 again interacts with two negatively charged residues (E1201 and D1202).
Using R1279 as a “probe,” we calculated the interaction energy between R1279 and negatively charged residues with MOE, and we found a clear trend of interaction energy decrease when the channel transits from open to closed states. Interaction energies between R1279 and negatively charged residues during the different channel gating states are as follows: (1) activated state: −65.8 kcal/mol; (2) early deactivation: −47.2 kcal/mol; (3) late deactivation: −23.7 kcal/mol; (4) resting state: −35.0 kcal/mol; (5) early activation: −25.7 kcal/mol; and (6) late activation: −61.7 kcal/mol. Together, these results suggest that R1279 has a stronger interaction with negatively charged residues during open states, which may help to stabilize the channel open conformations.
On the other hand, mutation R1279P resulted in the loss of ionic interactions in all these gating states (Fig. 9A–F, right, cyan), as proline is not a charged residue. The strong ionic interactions between R1279 and E1201, D1202 and E1206 in the activated state, early deactivation state, and late activation state are abolished by the R1279P mutation (Fig. 10). Moreover, the interaction energy between R1279 and surrounding residues decreases from −65.8 kcal/mol to −2.4 kcal/mol in the activated state, from −47.2 kcal/mol to −2.3 kcal/mol in the early deactivation state, and from −61.7 kcal/mol to −2.7 kcal/mol in the late activation state, due to the R1279P mutation. As strong ionic interactions between R1279 and surrounding residues occur mostly during channel open states, R1279 is likely to stabilize the channel open conformations, which are disrupted by R1279P mutation, making the channel harder to open. This finding is consistent with our observations showing that R1279P depolarizes voltage-dependent activation (Fig. 2C; Table 1).
Figure 10.

Interaction map of R1279 and R1279P mutation with surrounding residues. A, Activated state. R1279 has ionic interactions with E1201 and E1206. B, Early deactivation state. R1279 has ionic interactions with E1201 and E1206. C, Late deactivation state. R1279 has ionic interactions with E1201 and D1202. D, Resting state. R1279 has ionic interactions with E1229. E, Early activation state. R1279 has ionic interactions with D1219. F, Late activation state. R1279 has ionic interactions with E1201 and D1202. Red shadow represents ionic interactions. Fuzzy blue circles represent noninteracting space of R1279 or R1279P. The ionic interactions get weaker when the channel transits into resting states and get stronger when the channel transits into activated state. R1279P mutation abolishes the ionic interactions seen in the WT channel.
Discussion
In this study, we report a novel mutation in the Nav1.7 sodium channel (R1279P) identified in a patient with adult-onset distal leg pain and restless legs. This mutation depolarizes voltage-dependent activation and depolarizes steady-state fast, slow, and closed-state inactivation. Our findings that activation and inactivation show a similar depolarizing shift in the R1279P mutant fits well with the conclusion that activation and inactivation of voltage-gated sodium channels are not independent processes and inactivation derives its voltage dependence from activation (Bezanilla and Armstrong, 1977). In small DRG neurons, expression of R1279P depolarized RMP and produced hyperexcitability, reducing current threshold for single-action potentials, and dramatically increasing the populations of spontaneously firing and repetitively firing neurons as well as the firing frequency in response to graded depolarizing inputs.
Over the past decade, Nav1.7 mutations have been found in individuals with IEM (Yang et al., 2004; Dib-Hajj et al., 2013) that typically hyperpolarize channel activation, resulting in an increased window current, which renders sensory neurons hyperexcitable (Vasylyev et al., 2014). Approximately 10 mutants in Nav1.7 have been associated with a different pain disorder characterized by paroxysmal pain (PEPD), and these generally depolarize steady-state fast inactivation (Fertleman et al., 2006; Dib-Hajj et al., 2013). Gain-of-function variants of Nav1.7 that subtly impair fast and/or slow inactivation have also been linked to painful idiopathic small-fiber neuropathy (Faber et al., 2012; Han et al., 2012). In the current study, a Nav1.7 mutation that increases excitability of DRG neurons was identified in a patient with distal leg pain, who met clinical criteria for small-fiber neuropathy and for restless legs syndrome, which is well established as a concomitant of painful neuropathy (Rutkove et al., 1996; Polydefkis et al., 2000; Gemignani et al., 2007; Bachmann et al., 2010).
The mutation site of R1279P is located within the voltage sensor in domain III of Nav1.7 channel, unlike the majority of IEM-linked SCN9A mutants, which occur in domains I and II (Drenth and Waxman, 2007; Dib-Hajj et al., 2013). In contrast to enhanced activation, the typical gain-of-function attribute in IEM-related Nav1.7 mutations (Dib-Hajj et al., 2010; Waxman, 2013), R1279P impairs activation by a depolarizing shift of 5.9 mV. Notably, steady-state fast inactivation is depolarized by 9.6 mV, which is a typical attribute for gain-of-function Nav1.7 mutants in PEPD (Fertleman et al., 2006). Slow inactivation is depolarized by 7.6 mV in the mutant channel. Mutations of Nav1.7 that impair fast and/or slow inactivation have been described in painful small-fiber neuropathy (Faber et al., 2012; Han et al., 2012).
Precedent for a link between depolarized fast inactivation and distal limb pain in humans is provided by the G616R Nav1.7 mutation (Choi et al., 2010). This mutation depolarizes steady-state fast inactivation without affecting activation of hNav1.7R/AL. As in the present case, clinical onset of pain was delayed. Recently, a novel erythromelalgia mutation A1632T was found in a patient who first presented at the age of 17 with recurrent episodes of burning pain, redness, warmth, and swelling in her feet (Eberhardt et al., 2014). Interestingly, A1632T rendered sensory neurons hyperexcitable via a depolarization of steady-state fast inactivation by 7.3 mV, although it did not influence activation (Eberhardt et al., 2014). In the case of R1279P, the depolarizing shift in fast inactivation was sufficient to override the depolarizing shift in activation, producing hyperexcitability and spontaneous firing of small DRG neurons and, at the clinical level, distal leg pain.
We have previously studied a voltage-gated potassium channel mutation (Kv10.2-R327H) associated with epilepsy/autism (Yang et al., 2013b). R327 is one of the critical positively charged residues (R2) within the S4 helix of the Kv10.2 channel. Together with three additional positively charged residues (commonly known as R1-R4), these residues sense changes in membrane potential and produce a conformational change of the VSD to control channel gating. During the dynamic channel activation process, the VSD undergoes conformational changes, and these positively charged residues appear to form ionic interactions with multiple negatively charged residues of the S1-S4 helices. Using multistate modeling, we showed that R327 has strong ionic interaction with negatively charged residues of the S1-S3 helices in the closed but not open state of the channel. The R327H Kv10.2 mutation weakens these interactions, thus making it easier to open the channel (Yang et al., 2013b).
The present results suggest that the loss of positive charge of R1279 due to the R1279P mutation may affect ionic interactions between R1279 of S4 and negatively charged residues of other helices of the VSD during channel gating. Interestingly, although R1279P is a loss of charge mutation located at the same R2 position as in the R327H Kv10.2 mutation, the R1279P Nav1.7 mutation has an effect opposite to that of R327H (depolarizing vs hyperpolarizing channel activation). Using multistate modeling, we found that negatively charged residues within the Nav1.7 channel are likely to contribute to the effect of the R1279P mutation on channel activation. Indeed, because of the existence of negatively charged residues of the S1-S2 linker and the different location of negatively charged residues of S2 of Nav1.7 DIII, it is predicted that R1279 has strong interactions with negatively charged residues in the open states (rather than closed states) during the gating of Nav1.7 channel. In contrast, the locations and interactions of negatively charged residues of the VSD of the Kv10.2 channel make the R327 residue a stabilizing element for the channel closed state. Together, multistate structural modeling thus suggests a critical role of nonconservative negatively charged residues within the VSD in shaping channel activation of both potassium and sodium channels, and supports our observation of depolarized activation in R1279P mutant.
Our previous study of ionic interactions between R2 and negatively charged residues of VSD in a voltage-gated potassium channel (Kv10.2) (Yang et al., 2013b) permits us to compare the differences between charged residues in Kv10.2 and Nav1.7. Although both Kv10.2 and Nav1.7 contain negatively charged residues distributed across the S1 to S3 helices, the orientation and relative locations of these residues are different. Notably, Nav1.7 has three negatively charged residues (E1201, D1202, and E1206) located at the S1-S2 linker that are key partners with which R1279 establishes ionic interactions in the channel open states as suggested by our current study, whereas these three residues are missing from the Kv10.2 channel. Thus, the ionic interactions between R2 and negatively charged residues in activated states of Kv10.2 are lacking.
The ionic interactions between R1-R4 and acidic residues of VSD of sodium channels have been studied previously, mainly in NaChBac, a fourfold symmetric bacterial Nav channel, in an effort to understand the movements of the VSD during gating. Nav1.7 shares many conserved residues with NaChBac, but there are also clear differences: Among the three negatively charged residues within the Nav1.7 S1-S2 linker discussed above, only one is conserved between NaChBac and Nav1.7 (E1201 of Nav1.7 vs E43 of NaChBac). Other conserved negatively charged residues are as follows: D1219, E1229, and D1215 of Nav1.7 versus D60, E70, D93, of NaChBac, respectively. It has been suggested that R2 of NaChBac may form ion pairs with E43 and D60 in channel-activated states while establishing ionic interactions with D60 in channel resting states (DeCaen et al., 2009, 2011; Yarov-Yarovoy et al., 2012). However, as NaChBac lacks some negatively charged residues present in Nav1.7, we cannot definitively extrapolate results obtained in NaChBac to the Nav1.7 channel.
The Nav1.7 R1279P mutation also causes a significant shift of channel fast inactivation. However, the structural basis for the effect of the R2 mutation on fast inactivation, which mainly involves the interaction of IFM inactivation particle of the loop between domain III and IV and the channel S5 and S6 pore-forming helices (Goldin, 2003; Ulbricht, 2005), is not clear from our current model because it does not include this loop.
We have previously demonstrated that slow closed-state inactivation permits the Nav1.7 channel to produce larger ramp currents compared with other sodium channels, such as Nav1.4 and Nav1.6 (Cummins et al., 1998; Herzog et al., 2003). A study on Nav1.3 found that the early component of ramp current, which depends on closed-stated inactivation, increases with ramp rate (Estacion and Waxman, 2013). Consistent with this observation, we found that averaged ramp currents of WT and R1279P gradually increased when the depolarizing rate increased from 0.2 to 1.2 mV/ms. We also found that, for both WT and R1279P, the ramp current elicited by fast depolarizations occurred at more depolarized potentials compared with those generated by slow depolarizations, possibly because fast depolarizations of transmembrane potential do not permit voltage-gated sodium channels to reach equilibrium.
In addition to linking depolarized fast inactivation of Nav1.7 to pain, the present results show that depolarized inactivation is sufficient to overcome attenuated activation of the Nav1.7 channel, thereby producing hyperexcitability and spontaneous firing in DRG neurons. Our results thus support the idea that agents that enhance inactivation or stabilize the inactivated state of Nav1.7 may provide a novel approach to pain pharmacotherapy.
Footnotes
This work was supported in part by the Rehabilitation Research Service and Medical Research Service, Department of Veterans Affairs, and the Erythermalgia Association. The Center for Neuroscience and Regeneration Research is a Collaboration of the Paralyzed Veterans of America with Yale University. We thank Lynda Tyrrell, Palak Shah, and Fadia Dib-Hajj for technical assistance; and Dr. Mark Estacion, Dr. Chongyang Han, Dr. Xiaoyang Cheng, and Dr. Dmytro Vasylyev for valuable comments.
The authors declare no competing financial interests.
References
- Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- Bachmann CG, Rolke R, Scheidt U, Stadelmann C, Sommer M, Pavlakovic G, Happe S, Treede RD, Paulus W. Thermal hypoaesthesia differentiates secondary restless legs syndrome associated with small fibre neuropathy from primary restless legs syndrome. Brain. 2010;133:762–770. doi: 10.1093/brain/awq026. [DOI] [PubMed] [Google Scholar]
- Bezanilla F, Armstrong CM. Inactivation of the sodium channel: I. Sodium current experiments. J Gen Physiol. 1977;70:549–566. doi: 10.1085/jgp.70.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JA, Dib-Hajj S, McNabola K, Jeste S, Rizzo MA, Kocsis JD, Waxman SG. Spinal sensory neurons express multiple sodium channel alpha-subunit mRNAs. Brain Res Mol Brain Res. 1996;43:117–131. doi: 10.1016/s0169-328x(96)00163-5. [DOI] [PubMed] [Google Scholar]
- Black JA, Frézel N, Dib-Hajj SD, Waxman SG. Expression of Nav1.7 in DRG neurons extends from peripheral terminals in the skin to central preterminal branches and terminals in the dorsal horn. Mol Pain. 2012;8:82. doi: 10.1186/1744-8069-8-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nat Protoc. 2009;4:1–13. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- Cheng X, Dib-Hajj SD, Tyrrell L, Te Morsche RH, Drenth JP, Waxman SG. Deletion mutation of sodium channel Na(V)1.7 in inherited erythromelalgia: enhanced slow inactivation modulates dorsal root ganglion neuron hyperexcitability. Brain. 2011;134:1972–1986. doi: 10.1093/brain/awr143. [DOI] [PubMed] [Google Scholar]
- Choi JS, Cheng X, Foster E, Leffler A, Tyrrell L, Te Morsche RH, Eastman EM, Jansen HJ, Huehne K, Nau C, Dib-Hajj SD, Drenth JP, Waxman SG. Alternative splicing may contribute to time-dependent manifestation of inherited erythromelalgia. Brain. 2010;133:1823–1835. doi: 10.1093/brain/awq114. [DOI] [PubMed] [Google Scholar]
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM, Woods CG. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–898. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Waxman SG. Downregulation of tetrodotoxin-resistant sodium currents and upregulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons after nerve injury. J Neurosci. 1997;17:3503–3514. doi: 10.1523/JNEUROSCI.17-10-03503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Howe JR, Waxman SG. Slow closed-state inactivation: a novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J Neurosci. 1998;18:9607–9619. doi: 10.1523/JNEUROSCI.18-23-09607.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Dib-Hajj SD, Waxman SG. Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. J Neurosci. 2004;24:8232–8236. doi: 10.1523/JNEUROSCI.2695-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Rush AM, Estacion M, Dib-Hajj SD, Waxman SG. Voltage-clamp and current-clamp recordings from mammalian DRG neurons. Nat Protoc. 2009;4:1103–1112. doi: 10.1038/nprot.2009.91. [DOI] [PubMed] [Google Scholar]
- DeCaen PG, Yarov-Yarovoy V, Sharp EM, Scheuer T, Catterall WA. Sequential formation of ion pairs during activation of a sodium channel voltage sensor. Proc Natl Acad Sci U S A. 2009;106:22498–22503. doi: 10.1073/pnas.0912307106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaen PG, Yarov-Yarovoy V, Scheuer T, Catterall WA. Gating charge interactions with the S1 segment during activation of a Na+ channel voltage sensor. Proc Natl Acad Sci U S A. 2011;108:18825–18830. doi: 10.1073/pnas.1116449108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj SD, Rush AM, Cummins TR, Hisama FM, Novella S, Tyrrell L, Marshall L, Waxman SG. Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain. 2005;128:1847–1854. doi: 10.1093/brain/awh514. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Black JA, Waxman SG. Voltage-gated sodium channels: therapeutic targets for pain. Pain Med. 2009a;10:1260–1269. doi: 10.1111/j.1526-4637.2009.00719.x. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Choi JS, Macala LJ, Tyrrell L, Black JA, Cummins TR, Waxman SG. Transfection of rat or mouse neurons by biolistics or electroporation. Nat Protoc. 2009b;4:1118–1126. doi: 10.1038/nprot.2009.90. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. Sodium channels in normal and pathological pain. Annu Rev Neurosci. 2010;33:325–347. doi: 10.1146/annurev-neuro-060909-153234. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Yang Y, Black JA, Waxman SG. The Na(V)1.7 sodium channel: from molecule to man. Nat Rev Neurosci. 2013;14:49–62. doi: 10.1038/nrc3640. [DOI] [PubMed] [Google Scholar]
- Djouhri L, Newton R, Levinson SR, Berry CM, Carruthers B, Lawson SN. Sensory and electrophysiological properties of guinea-pig sensory neurones expressing Nav 1.7 (PN1) Na+ channel alpha subunit protein. J Physiol. 2003;546:565–576. doi: 10.1113/jphysiol.2002.026559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenth JP, Waxman SG. Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J Clin Invest. 2007;117:3603–3609. doi: 10.1172/JCI33297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenth JP, te Morsche RH, Guillet G, Taieb A, Kirby RL, Jansen JB. SCN9A mutations define primary erythermalgia as a neuropathic disorder of voltage gated sodium channels. J Invest Dermatol. 2005;124:1333–1338. doi: 10.1111/j.0022-202X.2005.23737.x. [DOI] [PubMed] [Google Scholar]
- Eberhardt M, Nakajima J, Klinger AB, Neacsu C, Hühne K, O'Reilly AO, Kist AM, Lampe AK, Fischer K, Gibson J, Nau C, Winterpacht A, Lampert A. Inherited pain: sodium channel Nav1.7 A1632T mutation causes erythromelalgia due to a shift of fast inactivation. J Biol Chem. 2014;289:1971–1980. doi: 10.1074/jbc.M113.502211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estacion M, Waxman SG. The response of Na(V)1.3 sodium channels to ramp stimuli: multiple components and mechanisms. J Neurophysiol. 2013;109:306–314. doi: 10.1152/jn.00438.2012. [DOI] [PubMed] [Google Scholar]
- Faber CG, Hoeijmakers JG, Ahn HS, Cheng X, Han C, Choi JS, Estacion M, Lauria G, Vanhoutte EK, Gerrits MM, Dib-Hajj S, Drenth JP, Waxman SG, Merkies IS. Gain of function Nav1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol. 2012;71:26–39. doi: 10.1002/ana.22485. [DOI] [PubMed] [Google Scholar]
- Fertleman CR, Baker MD, Parker KA, Moffatt S, Elmslie FV, Abrahamsen B, Ostman J, Klugbauer N, Wood JN, Gardiner RM, Rees M. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52:767–774. doi: 10.1016/j.neuron.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Gemignani F, Brindani F, Negrotti A, Vitetta F, Alfieri S, Marbini A. Restless legs syndrome and polyneuropathy. Mov Disord. 2006;21:1254–1257. doi: 10.1002/mds.20928. [DOI] [PubMed] [Google Scholar]
- Gemignani F, Brindani F, Vitetta F, Marbini A, Calzetti S. Restless legs syndrome in diabetic neuropathy: a frequent manifestation of small fiber neuropathy. J Peripher Nerv Syst. 2007;12:50–53. doi: 10.1111/j.1529-8027.2007.00116.x. [DOI] [PubMed] [Google Scholar]
- Goldin AL. Mechanisms of sodium channel inactivation. Curr Opin Neurobiol. 2003;13:284–290. doi: 10.1016/S0959-4388(03)00065-5. [DOI] [PubMed] [Google Scholar]
- Han C, Hoeijmakers JG, Ahn HS, Zhao P, Shah P, Lauria G, Gerrits MM, te Morsche RH, Dib-Hajj SD, Drenth JP, Faber CG, Merkies IS, Waxman SG. Nav1.7-related small fiber neuropathy: impaired slow inactivation and DRG neuron hyperexcitability. Neurology. 2012;78:1635–1643. doi: 10.1212/WNL.0b013e3182574f12. [DOI] [PubMed] [Google Scholar]
- He B, Soderlund DM. Human embryonic kidney (HEK293) cells express endogenous voltage-gated sodium currents and Na v 1.7 sodium channels. Neurosci Lett. 2010;469:268–272. doi: 10.1016/j.neulet.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog RI, Cummins TR, Ghassemi F, Dib-Hajj SD, Waxman SG. Distinct repriming and closed-state inactivation kinetics of Nav1.6 and Nav1.7 sodium channels in mouse spinal sensory neurons. J Physiol. 2003;551:741–750. doi: 10.1113/jphysiol.2003.047357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Yang Y, Zhao P, Gerrits MM, Hoeijmakers JG, Bekelaar K, Merkies IS, Faber CG, Dib-Hajj SD, Waxman SG. Small-fiber neuropathy Nav1.8 mutation shifts activation to hyperpolarized potentials and increases excitability of dorsal root ganglion neurons. J Neurosci. 2013;33:14087–14097. doi: 10.1523/JNEUROSCI.2710-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Han C, Estacion M, Vasylyev D, Hoeijmakers JG, Gerrits MM, Tyrrell L, Lauria G, Faber CG, Dib-Hajj SD, Merkies IS, Waxman SG. Gain-of-function mutations in sodium channel NaV1.9 in painful neuropathy. Brain. 2014;137:1627–1642. doi: 10.1093/brain/awu079. [DOI] [PubMed] [Google Scholar]
- Iannaccone S, Zucconi M, Marchettini P, Ferini-Strambi L, Nemni R, Quattrini A, Palazzi S, Lacerenza M, Formaglio F, Smirne S. Evidence of peripheral axonal neuropathy in primary restless legs syndrome. Mov Disord. 1995;10:2–9. doi: 10.1002/mds.870100103. [DOI] [PubMed] [Google Scholar]
- Jensen MØ, Jogini V, Borhani DW, Leffler AE, Dror RO, Shaw DE. Mechanism of voltage gating in potassium channels. Science. 2012;336:229–233. doi: 10.1126/science.1216533. [DOI] [PubMed] [Google Scholar]
- Klugbauer N, Lacinova L, Flockerzi V, Hofmann F. Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. EMBO J. 1995;14:1084–1090. doi: 10.1002/j.1460-2075.1995.tb07091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauria G, Merkies IS, Faber CG. Small fibre neuropathy. Curr Opin Neurol. 2012;25:542–549. doi: 10.1097/WCO.0b013e32835804c5. [DOI] [PubMed] [Google Scholar]
- Lee JH, Park CK, Chen G, Han Q, Xie RG, Liu T, Ji RR, Lee SY. A monoclonal antibody that targets a NaV1.7 channel voltage sensor for pain and itch relief. Cell. 2014;157:1393–1404. doi: 10.1016/j.cell.2014.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Long SB, Tao X, Campbell EB, MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- Polydefkis M, Allen RP, Hauer P, Earley CJ, Griffin JW, McArthur JC. Subclinical sensory neuropathy in late-onset restless legs syndrome. Neurology. 2000;55:1115–1121. doi: 10.1212/WNL.55.8.1115. [DOI] [PubMed] [Google Scholar]
- Raymond CK, Castle J, Garrett-Engele P, Armour CD, Kan Z, Tsinoremas N, Johnson JM. Expression of alternatively spliced sodium channel alpha-subunit genes: unique splicing patterns are observed in dorsal root ganglia. J Biol Chem. 2004;279:46234–46241. doi: 10.1074/jbc.M406387200. [DOI] [PubMed] [Google Scholar]
- Rios Romenets S, Postuma RB. Treatment of restless legs syndrome. Curr Treat Options Neurol. 2013;15:396–409. doi: 10.1007/s11940-013-0241-x. [DOI] [PubMed] [Google Scholar]
- Rush AM, Dib-Hajj SD, Liu S, Cummins TR, Black JA, Waxman SG. A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc Natl Acad Sci U S A. 2006;103:8245–8250. doi: 10.1073/pnas.0602813103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkove SB, Matheson JK, Logigian EL. Restless legs syndrome in patients with polyneuropathy. Muscle Nerve. 1996;19:670–672. doi: 10.1002/(SICI)1097-4598(199605)19:5<670::AID-MUS20>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Toledo-Aral JJ, Moss BL, He ZJ, Koszowski AG, Whisenand T, Levinson SR, Wolf JJ, Silos-Santiago I, Halegoua S, Mandel G. Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc Natl Acad Sci U S A. 1997;94:1527–1532. doi: 10.1073/pnas.94.4.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbricht W. Sodium channel inactivation: molecular determinants and modulation. Physiol Rev. 2005;85:1271–1301. doi: 10.1152/physrev.00024.2004. [DOI] [PubMed] [Google Scholar]
- Vasylyev DV, Han C, Zhao P, Dib-Hajj S, Waxman SG. Dynamic-clamp analysis of wild-type human Nav1.7 and erythromelalgia mutant channel L858H. J Neurophysiol. 2014;111:1429–1443. doi: 10.1152/jn.00763.2013. [DOI] [PubMed] [Google Scholar]
- Waxman SG. Painful Na-channelopathies: an expanding universe. Trends Mol Med. 2013;19:406–409. doi: 10.1016/j.molmed.2013.04.003. [DOI] [PubMed] [Google Scholar]
- Yang Y, Wang Y, Li S, Xu Z, Li H, Ma L, Fan J, Bu D, Liu B, Fan Z, Wu G, Jin J, Ding B, Zhu X, Shen Y. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet. 2004;41:171–174. doi: 10.1136/jmg.2003.012153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Shi W, Chen X, Cui N, Konduru AS, Shi Y, Trower TC, Zhang S, Jiang C. Molecular basis and structural insight of vascular K(ATP) channel gating by S-glutathionylation. J Biol Chem. 2011;286:9298–9307. doi: 10.1074/jbc.M110.195123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Dib-Hajj SD, Zhang J, Zhang Y, Tyrrell L, Estacion M, Waxman SG. Structural modelling and mutant cycle analysis predict pharmacoresponsiveness of a Na(V)1.7 mutant channel. Nat Commun. 2012;3:1186. doi: 10.1038/ncomms2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Estacion M, Dib-Hajj SD, Waxman SG. Molecular architecture of a sodium channel S6 Helix: radial tuning of the voltage-gated sodium channel 1.7 activation gate. J Biol Chem. 2013a;288:13741–13747. doi: 10.1074/jbc.M113.462366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Vasylyev DV, Dib-Hajj F, Veeramah KR, Hammer MF, Dib-Hajj SD, Waxman SG. Multistate structural modeling and voltage-clamp analysis of epilepsy/autism mutation Kv10.2-R327H demonstrate the role of this residue in stabilizing the channel closed state. J Neurosci. 2013b;33:16586–16593. doi: 10.1523/JNEUROSCI.2307-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarov-Yarovoy V, DeCaen PG, Westenbroek RE, Pan CY, Scheuer T, Baker D, Catterall WA. Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc Natl Acad Sci U S A. 2012;109:E93–E102. doi: 10.1073/pnas.1118434109. [DOI] [PMC free article] [PubMed] [Google Scholar]