

Abstract
Overstimulation of NMDA-type glutamate receptors is believed to be responsible for neuronal death of the CNS in various disorders, including cerebral and spinal cord ischemia. However, the intrinsic and physiological mechanisms of modulation of these receptors are essentially unknown. Here we report that cholestane-3β,5α,6β-triol (triol), a major metabolite of cholesterol, is an endogenous neuroprotectant and protects against neuronal injury both in vitro and in vivo via negative modulation of NMDA receptors. Treatment of cultured neurons with triol protects against glutamate-induced neurotoxicity, and administration of triol significantly decreases neuronal injury after spinal cord ischemia in rabbits and transient focal cerebral ischemia in rats. An inducible elevation of triol is associated with ischemic preconditioning and subsequent neuroprotection in the spinal cord of rabbits. This neuroprotection is effectively abolished by preadministration of a specific inhibitor of triol synthesis. Physiological concentrations of triol attenuate [Ca2+]i induced by glutamate and decrease inward NMDA-mediated currents in cultured cortical neurons and HEK-293 cells transiently transfected with NR1/NR2B NMDA receptors. Saturable binding of [3H]triol to cerebellar granule neurons and displacement of [3H]MK-801 binding to NMDA receptors by triol suggest that direct blockade of NMDA receptors may underlie the neuroprotective properties. Our findings suggest that the naturally occurring oxysterol, the major cholesterol metabolite triol, functions as an endogenous neuroprotectant in vivo, which may provide novel insights into understanding and developing potential therapeutics for disorders in the CNS.
Keywords: calcium; cerebral ischemia; cholestane-3β, 5α, 6β-triol; NMDA; spinal cord ischemia; steroid
Introduction
Steroids and oxysterols are ubiquitous lipid components of eukaryotic plasma membranes and play vital roles in regulating the properties of cell membranes in mammalian cells (Mauch et al., 2001; Steck and Lange, 2010). Physiologically, the main metabolic pathways of cholesterol involve the biosynthesis of bile acids and steroid hormones, as well as the oxidation into oxysterols, the most abundant of which is cholestane-3β,5α,6β-triol (triol; Javitt, 1994; Payne and Hales, 2004). According to the ancient Chinese Pharmacopoeia, gallstones from oxen, which mainly contain cholesterol and its metabolites, have been used medicinally to treat various CNS disorders, including epilepsy (Gu, 1955). In traditional Indian medicine, animal gallstones have also been used as drugs for at least 4000 years (Thiyagarajan and Sunderrajan, 1992). More recent clinical epidemiological studies have shown that higher total plasma cholesterol levels correlate with lower mortality and better outcomes in patients who have had a stroke (Iso et al., 1989; Dyker et al., 1997; Olsen et al., 2007). A recent epidemical research with meta-analysis also revealed that low cholesterol is a risk of all-cause mortality in Japan (Kirihara et al., 2008), including stroke.
Ischemic stroke is a leading cause of death and disability worldwide, with few if any effective therapies (Al Hasan and Murugan, 2012). Numerous pathophysiological studies have implicated excessive stimulation of glutamate receptors, changes in the expression and function of these receptors, and/or aberrant glutamatergic neurotransmission in the mechanisms of ischemic neuronal injury and death (Budd, 1998; Mori et al., 2004; Giffard and Swanson, 2005). It is also generally accepted that disturbances in intracellular calcium ([Ca2+]i) homeostasis contribute to ischemia-induced neuronal injury and death (Cross et al., 2010). Overstimulation of NMDA receptors is believed to be responsible for neuronal death in the CNS induced by a variety of ischemic and traumatic insults. NMDA receptor channel-mediated Ca2+ overload, which plays a critical role in neuronal excitotoxicity, has also been implicated in neuronal death after spinal cord ischemia (Kocaeli et al., 2005; Villmann and Becker, 2007). NMDA receptors are modulated by many endogenous and exogenous substances. For example, the amino acid glycine and many neurosteroids, including 24(S)-hydroxycholesterol, facilitate the activation of NMDA receptors by glutamate (Fahey et al., 1995; Paul et al., 2013). To our knowledge, no naturally occurring NMDA receptor antagonists have so far been identified or shown to be active in vivo as neuroprotectants.
In the current study, we demonstrate that an endogenous oxysterol metabolite of cholesterol, triol, is able to protect neurons against glutamate-induced neurotoxicity in vitro and ischemia-induced neuronal injury in vivo. Triol is also a potent NMDA receptor negative modulator that may underlie its observed neuroprotective properties. Our findings suggest that the metabolism of cholesterol to triol may serve to reduce neuronal injury and death after ischemic CNS insults.
Materials and Methods
Reagents.
Triol and 6-ketocholestanol (KC) were provided by Sigma-Aldrich. The purity of triol was >99.0%, determined under 1260 HPLC (Agilent) with an 2000ES ELSD detector (Alltech).
Animals.
Sprague Dawley rats (240–270 g, males) and New Zealand white rabbits (2.0–2.5 kg, males) were used. All animals were provided by the Animal Research Laboratory at Sun Yat-sen University (Guangdong, China). The experimental protocol used was approved by the Ethics Committee for Animal Experimentation.
Human blood donors.
Six healthy volunteers (20–30 years old, three males) provided their blood for triol determination in plasma.
Determination of triol.
All plasma and tissue samples were stored at −80°C and determined as soon as possible. Determination of triol was performed as described previously (Sevanian et al., 1994; Razzazi-Fazeli et al., 2000) with modification. To avoid oxidation during sample preparation, the whole process was carefully kept away from light and oxygen, such as solvent degasification and saturation with nitrogen, solvent evaporation under a stream of nitrogen, and addition of butylated hydroxytoluene and EDTA before sample preparation.
A total of 1.0 g of plasma or 2.0 g of 50% tissue homogenate containing 0.9% NaCl was used for analysis, and 3β,6β-dihydroxyl-cholestane-24-one was used for the internal standard (IS). First, plasma or tissue homogenate was vortex extracted by adding ethyl acetate. After ethyl acetate evaporated, the residue was dissolved in hexane-ethyl acetate (9:1, v/v) and loaded onto the SPE column (Sep-Pak Vac, 3 ml/500 mg; Waters). The column was then washed with 10 ml of hexane-diethyl ether (95:5, v/v), 15 ml of hexane-diethyl ether (90:10, v/v), and 10 ml of hexane-diethyl ether (80:20, v/v) in order. Finally, the column was eluted by 6 ml of acetone. Then the acetone was evaporated, and the residue was dissolved in 100 μl of methanol. An aliquot of the aqueous layer (10 μl) was injected into the liquid chromatography–mass spectrometry (LC–MS) system.
The LC–MS system consists of an Agilent 1200 HPLC with a 6120 quadrupole MS equipped with atmospheric pressure chemical ionization (APCI) source and operated in positive ion mode. Chromatographic separation was performed by a XBridge C18 column (250 × 4.6 mm inner diameter, 5 μm; Waters) at 30°C using a mobile phase of water–methanol–acetonitrile (6:44:50, v/v/v) at a flow rate of 1.0 ml/min. Determination of triol was applied in the selective ion monitoring (SIM) mode with a mass-to-charge ratio (m/z) of 384.7–385.7 and 400.7–401.7 for IS. Linearity was assessed by triol IS peak area ratio versus the concentrations of triol. All calibration curves were corrected by subtracting the ratio of triol IS peak area in biological samples. Assay precisions and recoveries were determined and approved for all tested plasma and tissues.
Cell culture and in vitro experiments.
Primary cultures of spinal cord neurons, cerebellar granule neurons, and cortical neurons of rats were prepared as described previously (Dichter, 1978; Hughes et al., 1993; Berman and Murray, 1996). For glutamate exitotoxicity assay, the neuronal cultures were washed three times with Mg2+-free Locke's buffer and then incubated with 200 μm glutamate and 10 μm glycine in Mg2+-free Locke's buffer for 45 min. After that, the cultures were washed three times and maintained in the normal culture medium (DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin) for 24 h in the incubator. HEK-293 cells were transfected with the plasmids containing NR1/NR2B subunits using Lipofectamine 2000 as described previously (Qiu et al., 2005). The survival rate of cells exposed to various concentrations of triol was measured by MTT assay as described previously (Sun et al., 2013).
In vivo animal model of spinal cord ischemia.
The animal model of spinal cord ischemia in rabbits was performed as described previously (Johnson et al., 1993; Celik et al., 2002). Rabbits were randomly assigned into four groups (n = 12) as follows: (1) the ischemia group (ISC), in which the infrarenal aorta was occluded for 20 min to produce spinal cord ischemic injury; (2) the triol group, in which an intravenous infusion of 8 mg/kg triol [10% of dimethylsulfoxide (DMSO) as vehicle] was administered 30 min before spinal cord ischemia; (3) the vehicle group, in which an intravenous infusion of the same volume 10% of DMSO was administered; and (4) the sham group, in which only the aorta was exposed. During the surgical operation, a 22-gauge catheter was inserted into the ear artery to measure proximal blood pressure, and another catheter was inserted into the left femoral artery to measure distal blood pressure. Blood pressure was monitored continuously by using a calibrated pressure transducer connected to an invasive pressure monitor (Spacelabs Healthcare). Rectal temperature was maintained between 38°C and 39°C by a temperature-controlled heating pad during the experiments. Arterial blood was sampled at the preischemic state, 10 min after ischemia, and 10 min after reperfusion for the determination of the partial pressure of oxygen in arterial blood (PaO2) and partial pressure of carbon dioxide in arterial blood (PaCO2). Arterial blood gases were measured by means of the OMNI Modular System (AVL List).
Both the neurological and histopathological evaluations were assessed at 48 h after reperfusion. A modified Tarlov criteria (Johnson et al., 1993) was used: score 0, no voluntary hindlimb function; score 1, perceptible joint movement; score 2, active movement but no ability to stand; score 3, to be able to stand but not to walk; and score 4, complete normal hindlimb motor function. For the histopathological analysis, the lumbar spinal cord was removed, and coronal sections of the spinal cord (L5 segment) were cut at a thickness of 6 μm and stained with hematoxylin and eosin (H&E).
Middle cerebral artery occlusion-induced brain injury in rats.
Male Sprague Dawley rats were randomly divided into seven groups (n = 10): a saline group, a 20% hydroxypropyl-β-cyclodextrin (HP-β-CD) group (vehicle group), and five different therapeutic time windows of triol groups, respectively. The middle cerebral artery occlusion (MCAO) model and physiological monitoring were performed as described previously (Longa et al., 1989). Briefly, the right common carotid artery and the right external carotid artery were exposed through a ventral midline neck incision and were ligated. A 3-0 nylon monofilament suture (Ethicon nylon suture) with a blunt tip was inserted into the internal carotid artery to a point ∼17–18 mm distal to the carotid bifurcation until a mild resistance was felt, thereby occluding the origins of the anterior cerebral and middle cerebral arteries. Reperfusion was accomplished by withdrawing the suture after 120 min of ischemia. During the surgical operation, the right femoral artery was cannulated for continuous monitoring of arterial blood pressure and for sampling blood to measure PaO2 and PaCO2, and rectal temperature was monitored and kept between 37.0°C and 37.5°C by a temperature-controlled heating pad. The five triol groups were administered intravenously with 12 mg/kg triol at 0.5, 1, 2, 3, and 4 h after the onset of MCAO. The saline and vehicle groups were administered intravenously with the same volume of saline and vehicle as the triol groups, respectively, at 2 h after onset of MCAO.
After 24 h of reperfusion, each animal was assigned a neurological score (Longa et al., 1989): 0, no neurologic deficit; 1, failure to extend the left forepaw fully; 2, circling to the left; 3, falling to left; 4, no spontaneous walking with a depressed level of consciousness; and 5, dead.
For observation of the persistent neuroprotection of triol, one group of animals administered triol intravenously at 1 h after onset of MCAO were evaluated at day 7 after reperfusion.
After the neurological assessment, the rats were anesthetized with 10% chloral hydrate and decapitated. Brains were quickly removed and coronally sectioned with a tissue chopper at 2 mm interval, beginning 1 mm posterior to the anterior pole, incubated in 1.0% 2,3,5-triphenyltetrazolium chloride (TTC) at 37°C for 30 min, and fixed in 4.0% paraformaldehyde solution. Each brain slice was photographed and quantified using an image analysis system (Adobe Photoshop CS5). The infarct volume of each rat was corrected for swelling of the ischemic hemisphere by applying a derived formula (Swanson et al., 1990): the corrected infarct volume percentage = 100 × (contralateral hemisphere volume − non-infarct ipsilateral hemisphere volume)/contralateral hemisphere volume.
Ischemic preconditioning.
Rabbits were randomly assigned into six groups (n = 10) as follows: (1) the ISC group, in which the infrarenal aorta was occluded for 20 min to produce spinal cord ischemic injury; (2) the ischemic preconditioning (IPC) group, in which 6 min of infrarenal aorta occlusion below the threshold of the ischemic injury was performed alone without the second ischemia; (3) the IPC + ISC group, in which 6 min of infrarenal aorta occlusion was followed by 30 min of reperfusion before the 20 min of infrarenal aorta occlusion; (4) the KC + IPC + ISC group, in which KC, a specific inhibitor of triol biosynthesis, was preadministered intravenously with 8 mg/kg 1 h before the IPC, followed by 30 min of reperfusion before the 20 min of infrarenal aorta occlusion; (5) the vehicle + IPC + ISC group, in which the same volume of vehicle used to dissolve KC was preadministered 1 h before the IPC, followed by 30 min of reperfusion before the 20 min of infrarenal aorta occlusion; and (6) the sham group, in which the infrarenal aorta was exposed but not occluded.
Additionally, to exclude the possibility that KC could abolish the neuroprotection of IPC through exacerbating spinal cord ischemic injury by some other mechanism rather than inhibition of triol biosynthesis, 32 rabbits were randomly assigned into four groups (n = 8) as follows: (1 and 2) the IPC group and the ISC group were performed the same as described above; (3) the KC + IPC group, in which KC was preadministered intravenously with 8 mg/kg 1 h before the IPC; and (4) the KC + ISC group, in which KC was preadministered intravenously with 8 mg/kg 96 min before the ISC.
During the experiments, rectal temperature was maintained between 38°C and 39°C by a temperature-controlled heating pad, blood pressure was monitored continuously, and arterial blood was sampled at the preischemic state, 10 min after ischemia, and 10 min after reperfusion for the determination of PaO2 and PaCO2. Both the neurological and histopathological evaluations were assessed at 48 h after reperfusion.
Measurement of [Ca2+]i and patch-clamp recording.
Cells were prepared and loaded with the fluorescence dye Fluo-3/AM, as described previously (Földes-Papp et al., 2003). Briefly, the cells were grown in culture medium on six-well tissue plates at 37°C. Cells were incubated for 30 min in the dark at room temperature with 5 μm membrane-permeant Fluo-3/AM and 0.02% pluronic F-127 in HBSS (in mm: 125 NaCl, 0.62 KCl, 1.8 CaCl2, 20 HEPES, and 6 glucose, adjusted to pH 7.4 with NaOH). The fluorescence signal was monitored and recorded by a laser scanning confocal imaging system (Qiu et al., 2005) (TCS SP2; Leica). The data analysis was processed with Origin 6.1. The change in fluorescence intensity after drug treatment as indicated was normalized with the initial intensity.
Whole-cell patch-clamp recordings were made under voltage-clamp mode at room temperature (22–25°C). Recording was performed using a Multiclamp700A amplifier and Digidata 1322 series interface (AXON Instrument). Signals were filtered at 10 kHz. The pClamp 9.0 software system (Molecular Devices) was used for data recording and analysis. Patch pipettes were pulled from 1.2 mm outside diameter, 0.5 mm inner diameter glass pipettes (local product) using a P-97 horizontal puller (Sutter Instruments). The pipettes were filled with internal solution containing the following (in mm): 140 CsF, 1 CaCl2, 10 HEPES, 11 EGTA, 2 tetraethylammonium, and 4 MgCl2, pH 7.3, adjusted with CsOH (290–300 mOsm). The bath solution contained the following (in mm): 140 NaCl, 5.4 KCl, 20 HEPES, 10 glucose, and 2 CaCl2, pH 7.4, adjusted with NaOH and HCl (320–330 mOsm). Unless stated otherwise, inward NMDA-mediated currents (INMDA) were induced by local application of 200 μm NMDA with 3 μm glycine, 10 μm bicuculline, 10 μm 6-cyano-7-nitroquinoxaline-2,3-dione, 1 μm TTX, and 1 μm strychnine in bath solution. The maximal inward current value was measured as the peak current. The effect of triol was not tested until three stable NMDA-induced currents were achieved, after the formation of whole-cell configuration.
Binding assay.
Saturation experiments were performed with cultured cerebellar granule neurons and HEK-293 cells transfected with or without NR1/NR2B receptors. Briefly, the cultured cerebellar granule neurons and HEK-293 cells were washed three times, and then different concentrations of [3H]triol were added to the cultures to incubate for 20 min, after which the cultures were washed three times with ice-cold incubation buffer containing the following (in mm): 10 glucose, 124 NaCl, 5 KCl, 1.2 CaCl2, 1.2 MgCl2, and 15.6 Tris, pH 7.4, at an interval of 10 min. Finally, 300 μl of 10% SDS was added to lyse the neurons and HEK-293 cells, and the radioactivity of the lysate was measured by liquid scintillation 1 h later. Nonspecific binding was defined in the presence of 1 mm triol, which is 2000 times the highest concentration of [3H]triol used in the saturation analysis.
For the competition analysis, 90 nm [3H]MK-801, a standard radioactive ligand for NMDA receptor binding assays, was added to the cultured cerebellar granule neurons to incubate for 20 min. Then different concentrations of triol were added to cultures to incubate for 30 min to displace the [3H]MK-801 binding. After that, the cultures were washed three times with ice-cold incubation buffer at an interval of 10 min. Finally, 300 μl of 10% SDS was added to lyses the neurons, and the radioactivity of the lysate was measured by liquid scintillation 1 h later.
Statistic analysis.
The scores of hindlimb motor function and the numbers of normal neurons were analyzed using a nonparametric method (Kruskal–Wallis test), followed by the Mann–Whitney U test. The other variables were expressed as mean ± SD and were analyzed using one-factor ANOVA, followed by a post hoc least significant difference test. The correlation between the neurological function Tarlov score and the number of normal neurons in the anterior spinal cord was analyzed using Spearman's rank correlation. p < 0.05 was considered to be statistically significant.
Results
Tissue distribution of triol
In preliminary experiments, we found that rabbits with hypercholesterolemia had reduced neuronal injury and neurological impairment after spinal cord ischemia compared with rabbits with normal cholesterol (our unpublished data). However, previous studies, including our own, failed to demonstrate neuroprotective effects of cholesterol itself (Olsen et al., 2007; our unpublished observations). In many tissues, cholesterol is rapidly oxidized to cholesterol-5,6-epoxide, followed by hydration to triol by cholesterol 5,6-epoxide hydrolase (ChEH; Nashed et al., 1985; Fig. 1A). Triol has been demonstrated to be a major metabolite of cholesterol in humans (Sevanian et al., 1994; Schroepfer, 2000), and triol plasma levels are elevated in hypercholesterolemic rabbits and humans (Schroepfer, 2000; Leonarduzzi et al., 2005). We next determined triol concentrations in human and rabbit plasma by LC–MS and found plasma levels of 0.37 and 0.12 μm in humans and rabbits, respectively (Fig. 1B). We then systematically measured the distribution of triol in various rat tissues. As shown in Figure 1D, triol levels in various rat tissues ranged from 1.07 to 0.32 μm (spinal cord > liver > brain ≥ kidney > plasma).
Figure 1.

Physiological distribution of triol. A, The fundamental metabolic pathway of cholesterol. Cholesterol is transformed into cholesterol-5,6-epoxide (5,6-epoxide) via oxidation and then is hydrated into triol by ChEH. B, The plasma concentrations of triol in rabbits and humans. All values represent mean ± SD of six individuals. C, LC–MS spectra of triol in the SIM mode with m/z of 384.7–385.7 in rat blood, brain, spinal cord (S.C.), liver, and kidney. IS indicates the internal standard (3β,6β-dihydroxyl-cholestane-24-one) in the SIM with m/z of 400.7–401.7 in rat liver. The APCI source and the positive ion mode were involved in the measurement. D, The distribution of triol in rats. All values represent mean ± SD of six rats.
Neuroprotective effects of triol in vitro
Exposure of cultured spinal cord and cortical and cerebellar granule neurons to high neurotoxic concentrations of glutamate results in a significant reduction in the number of surviving neurons (Fig. 2A). Pretreatment of neurons with triol (5–15 μm) for 30 min before glutamate exposure increases the survival of spinal cord motoneurons, cortical neurons, and cerebellar granule neurons (Fig. 2B–D) in a concentration-dependent manner. In contrast, cholesterol itself had no neuroprotective effects at these same concentrations, and neither compound had an effect on neuronal survival in untreated neurons (data not shown).
Figure 2.

Neuroprotective effects of triol on glutamate-induced excitotoxicity in cultured neurons in vitro. A, Representative photomicrographs of the neuroprotective effects of triol against glutamate (Glu)-induced neurotoxicity in cultured spinal motoneurons. The control group (Control), neurons without any treatment; the glutamate group (Glu), neurons treated with 200 μm glutamate; the MK801 + Glu and Triol + Glu groups, neurons pretreated with 10 μm triol or 10 μm MK-801 for 30 min before the addition of glutamate. Motoneurons were observed after being incubated with glutamate for 24 h. Scale bars, 30 μm. B–D, Survival rates of spinal cord motoneurons (B), cortical neurons (C), and cerebellar granule neurons (D) with or without triol. The survival rates were calculated by comparison with the corresponding control cultures. All values represent mean ± SD. *p < 0.05 and **p < 0.01 compared with the glutamate group, respectively.
Neuroprotective effects of triol in the ischemia-induced spinal cord injury model in rabbits
We next studied the possible neuroprotective effect of triol in a model of spinal cord ischemia in rabbits (Johnson et al., 1993; Celik et al., 2002). After spinal cord ischemia, we observed a good correlation between neurological scores and the number of normal motoneurons in the anterior spinal cord 48 h after reperfusion (r = 0.827, p < 0.01; Fig. 3A). Neurological evaluation indicated that rabbits in the triol-treated group (8 mg/kg) exhibited higher neurological scores than those in the ISC (control) group (average score of 2.50 vs 0.83, p < 0.05; Fig. 3B). Three rabbits in the triol-treated group showed completely normal motor function after spinal cord ischemia (neurological score of 4; Fig. 3B). In contrast, no rabbit in the control (or vehicle-treated) group showed normal motor function (all neurological scores ≤2). There was no difference in neurological scores between the untreated control group and the DMSO vehicle-treated group (average score of 0.83 vs 0.92, p > 0.05; Fig. 3B). In both the untreated control and vehicle-treated group, rabbits rated with neurological scores of approximately ≤1 showed vacuolation of gray matter and disappearance of most normal motoneurons (Fig. 3C). In contrast, in the triol-treated group, rabbits rated with a neurological score of 3 showed mostly normal motoneurons (Fig. 3C, arrows), with some eosinophilic neurons (Fig. 3C, arrowheads). The number of surviving anterior spinal cord neurons in the triol-treated group was significantly higher than those of both the control and vehicle-treated groups (p < 0.01; Fig. 3D). In addition, physiologic variables, including blood pressure, blood gases, and rectal temperature, among these surgical operation groups had no significant differences.
Figure 3.

Neuroprotective effects of triol on spinal cord ischemic injury in rabbit. A, Correlation between the neurological function score and the number of normal neurons in anterior spinal cord 48 h after reperfusion. ▴, ○, ●, and ▵ represent animals in the ischemia, DMSO, triol, and sham groups, respectively. B, Neurological scores of rabbits at 48 h after reperfusion. The ischemia group (ISC), in which the infrarenal aorta was occluded for 20 min to produce spinal cord ischemia injury; the vehicle DMSO group (Vehicle), received intravenous infusion of the same volume vehicle of DMSO; the triol group (Triol), received intravenous infusion of triol with 8 mg/kg 30 min before spinal cord ischemia; the sham group (Sham), underwent exposure of the aorta only. The ISC, vehicle, triol, and sham groups had an average neurological score of 0.83, 0.92, 2.50, and 4.00, respectively. C, Representative results of lumbar spinal cord sections (L5) stained with H&E. Slices were chosen from rabbits rated as having an average score. Note that the darkly stained cytoplasm of dead neurons (arrowheads) compared with the fine granular cytoplasm and Nissl substance of the viable cells (arrows). Scale bars, 50 μm. D, The number of normal neurons of the anterior spinal cord in each animal 48 h after reperfusion. *p < 0.05 and **p < 0.01 compared with the ischemia group.
Therapeutic time window of triol after transient cerebral ischemia in rats
We used the transient MCAO model in rats to test whether triol is an effective neuroprotectant and the therapeutic time window after cerebral ischemia. As shown in Figure 4, transient MCAO in rats leads to a large infarct volume in the ipsilateral hemisphere at 24 h after reperfusion, which affects a significant portion of the frontal and parietal cortices, caudate–putamen, and diencephalon (Fig. 4A). The infarct volume induced by MCAO was 31.53 ± 1.69% for the saline group and 29.87 ± 2.52% for the vehicle-treated group (20% HP-β-CD), respectively (Fig. 4B). To explore a possible therapeutic time window of triol treatment, 12 mg/kg triol was administrated to rats at 0.5, 1.0, 2.0, 3.0, and 4.0 h after onset of MCAO, respectively. Triol treatment markedly reduced the extent of the infarct volume compared with saline- or vehicle-treated animals (Fig. 4A,B) at the all time points. The infarct volumes were reduced to 7.17 ± 1.43, 9.70 ± 1.20, 10.86 ± 0.84, 14.76 ± 1.25, and 16.98 ± 2.12%, respectively (vs 29.87 ± 2.52% in vehicle-treated rats). Triol treatment also improved functional neurological outcome as demonstrated by better neurologic scores in the triol-treated animals compared with those in the vehicle-treated ones (p < 0.05; Fig. 4C). Together, these morphological and behavioral data demonstrate that neuronal injury induced by MCAO can be markedly reduced by administration of triol even if the treatment takes place 4 h after ischemic injury. Furthermore, the neuroprotection of triol is persistent rather than temporal, simply delaying cell death, as evidenced by smaller infarct volumes and better neurologic scores at 7 d after reperfusion compared with those of vehicle-treated rats (p < 0.01; Fig. 4D–F). However, the physiological parameters, such as blood pressure, blood gases, and rectal temperature, among the surgical group animals had no significant changes.
Figure 4.
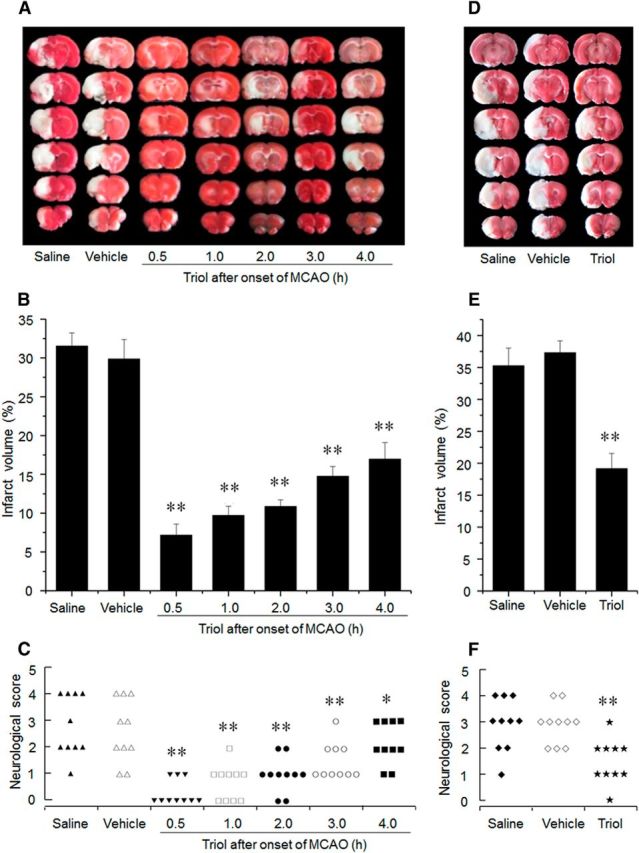
The neuroprotective effects of triol treatment on brain injury induced by MCAO at 24 h and 7 d after reperfusion. A, Representative TTC-stained brain sections at 24 h after reperfusion. Infarction area (pale region) is significantly reduced by intravenous administration of triol (12 mg/kg) at five different therapeutic time windows, respectively, compared with that of the saline or vehicle group. B, Corresponding percentage of infarction volume to the contralateral hemisphere volume at 24 h after reperfusion. The therapeutic time window of triol was used at 0.5, 1, 2, 3, and 4 h after onset of MCAO. C, Corresponding neurological outcome of rats at 24 h after reperfusion. D, Representative TTC-stained brain sections at 7 d after reperfusion. Infarction area (pale region) is significantly reduced by intravenous administration of triol (12 mg/kg) at 1 h after onset of MCAO compared with that of the saline or vehicle group. E, Corresponding percentage of infarction volume to the contralateral hemisphere volume at day 7 after reperfusion. F, Corresponding neurological outcome of rats at 7 d after reperfusion. *p < 0.01 and **p < 0.01 compared with the saline group.
Triol mediates neuroprotection after ischemic preconditioning
We next used a rabbit IPC model to explore the relationship between neuroprotection and the spinal cord concentrations of endogenous triol. The rabbits in the IPC + ISC group exhibit significantly higher neurological scores than those in the ISC group (average score of 3.1 vs 0.8, respectively, p < 0.01; Fig. 5A). As shown in Figure 5, B and C, the histopathological analysis further confirmed the marked neuroprotection observed after IPC. Coincidentally, the tissue levels of triol were significantly increased at 2 h after the IPC, were approximately fourfold greater than control levels at 4 h, and remained significantly elevated at 6 h after the IPC (Fig. 5G). Furthermore, after intravenous administration of triol (8 mg/kg), the concentration of triol in the spinal cord was ∼1.0 μm between 2 and 8 h (Fig. 5H), which is approximately fourfold of the basal concentration (0.25 μm) and equivalent to the highest endogenous triol concentration induced by IPC (1 μm, 4 h after IPC treatment).
Figure 5.
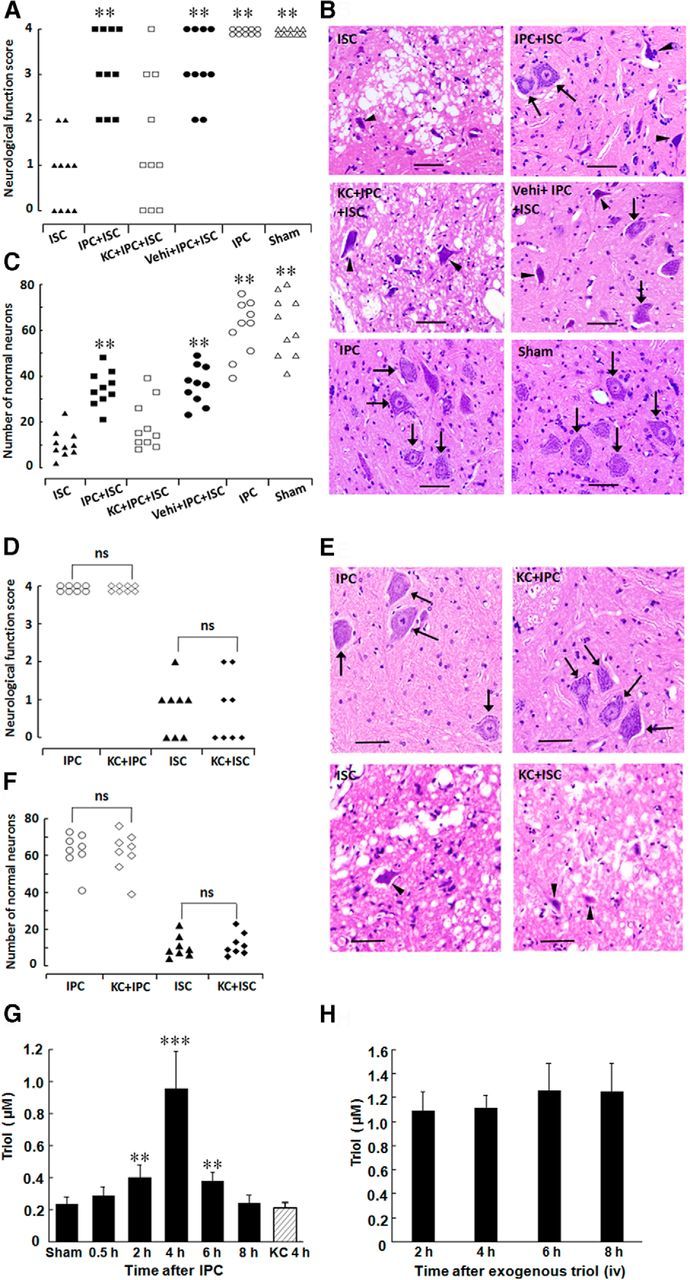
Neuroprotective effects of spinal cord IPC on spinal cord injury and triol elevation in the spinal cord in rabbits. A, Neurological scores of animals at 48 h after reperfusion in the ISC, IPC + ISC, KC + IPC + ISC, Vehi + IPC + ISC, IPC, and Sham groups, which had an average neurological score of 0.8, 3.1, 1.5, 3.2, 4.0, and 4.0, respectively. ISC, Ischemic injury; IPC, ischemic preconditioning; Vehi, vehicle; KC, 6-ketocholestanol (a specific ChEH inhibitor of triol biosynthesis). B, Representative results of lumbar spinal cord sections (L5) stained with H&E. Slices were chosen from rabbits rated as having an average score. Note the darkly stained cytoplasm of dead neurons (arrowheads) compared with the fine granular cytoplasm and Nissl substance of the viable cells (arrows). Scale bars, 50 μm. C, The number of normal neurons of the anterior spinal cord at 48 h after reperfusion in the ISC, IPC + ISC, KC + IPC + ISC, Vehi + IPC + ISC, IPC, and Sham groups. *p < 0.05 and **p < 0.01 compared with the ISC group, respectively. D, Neurological scores of animal at 48 h after reperfusion in the IPC, KC + IPC, ISC, and KC + ISC groups. The rabbits in the IPC group and KC + IPC group all displayed normal neurological function. Also, the neurological scores in the rabbits of the ISC group were not different from those of the KC + ISC group. ns, No significance. E, Representative results of lumbar spinal cord sections (L5) stained with H&E. Slices were chosen from rabbits rated as having an average score. Note the darkly stained cytoplasm of dead neurons (arrowheads) compared with the fine granular cytoplasm and Nissl substance of the viable cells (arrows). Scale bars, 50 μm. F, The number of normal neurons of the anterior spinal cord at 48 h after reperfusion in the IPC, KC + IPC, ISC, and KC + ISC groups. The rabbits in the IPC group and KC + IPC group all displayed almost intact neurons. Also, the number of normal neurons in the rabbits of ISC group was not different from those of the KC + ISC group. G, Triol levels in the rabbit spinal cord were significantly elevated 2 h after IPC, peaked at 4 h, and kept significantly high at 6 h. However, when KC, an inhibitor of ChEH, was preadministered intravenously 1 h before IPC, triol in the spinal cord 4 h after IPC was at a relatively low level and showed no difference from the sham group. **p < 0.01 and ***p < 0.001 compared with the sham group. H, Triol levels in spinal cord after intravenous administration.
Pretreatment of rabbits with a specific ChEH inhibitor of triol biosynthesis (Sevanian and McLeod, 1986), KC (8 mg/kg), 60 min before IPC abolished both the neuroprotection (Fig. 5A–C) and triol increase (Fig. 5G) induced by IPC. However, as shown in Figure 5D–F, pretreatment with KC did not exacerbate the relatively severe ischemic injury (20 min of spinal cord ischemia, ISC) as well as the mild ischemia (6 min of spinal cord ischemia, IPC). Together with the observed neuroprotective effects of administered triol in the rabbit spinal cord ischemia and rat MCAO models, these data suggest that the increase of endogenous triol after IPC and the neuroprotection induced by IPC are casually related to one another.
Triol blocks [Ca2+]i increase induced by glutamate and INMDA in cortical neurons
Using laser scanning confocal calcium imaging (Földes-Papp et al., 2003), we found that triol reduced the [Ca2+]i increase triggered by glutamate in a concentration-dependent manner (Fig. 6A,B). We further examined the effects of triol on NMDA currents in primary cultured cortical neurons. The formal recording was not started until three stable current traces were obtained. No significant NMDA current reduction was observed after recording for 7 min, in the absence of triol (Fig. 6C). In contrast, NMDA currents were time and concentration dependently decreased by ∼25% and ∼60% at 7 min in the presence of 1 μm (Fig. 6D,G) and 10 μm (Fig. 6E,G) triol, respectively. The “time-dependent” decrease of NMDA currents by triol is reminiscent of a use-dependent inhibition. To provide evidence supporting this hypothesis, we did not activate NMDA receptors between 2 and 7 min (5 min interval, no use) and found, under such a paradigm, that NMDA current was only decreased by 13.94% at 7 min in the presence of 10 μm triol (Fig. 6F,G), which was much less than that of the ∼60% decrease observed in Figure 6, E and G. Obviously, this inhibition is use dependent: less NMDA receptor use produces less NMDA current inhibition.
Figure 6.
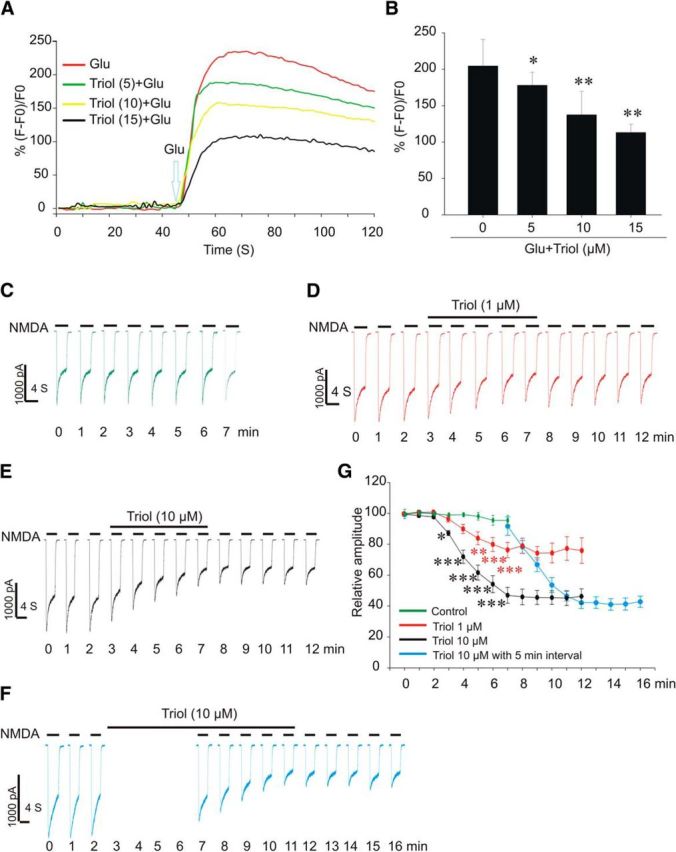
Inhibitory effect of triol on the [Ca2+]i increase induced by glutamate (Glu) and INMDA in cortical neurons. A, Representative results of live fluorescence intensity from cortical neurons exposed to 200 μm glutamate, pretreated with triol (numbers in parentheses indicate concentration, in micromolar). B, Summary of the inhibitory effects of triol on [Ca2+]i induced by glutamate. The data were expressed as (F − F0)/F0 (F and F0 indicate the fluorescence value after and before exposure to glutamate). Data represent mean ± SD (n = 20). *p < 0.05 and **p < 0.01 compared with glutamate alone. C, D, Green and red current traces showing time-dependent changes of the NMDA-gated currents in the absence or presence of 1 μm triol, respectively. NMDA (200 μm) was used to induce NMDA currents. E, F, Black and blue traces showing the inhibition of NMDA currents by 10 μm triol without or with 5 min interval, respectively. G, Summary data showing the concentration-dependent and use-dependent inhibition of NMDA-gated currents by triol (two-way ANOVA, followed by Bonferroni's post hoc tests, *p < 0.05, **p < 0.01, ***p < 0.001).
Inhibitory effects of triol on [Ca2+]i induced by NMDA in HEK-293 cells transfected with NMDA receptors
We next transfected HEK-293 cells with NR1/NR2B NMDA receptors and examined the effects of triol on [Ca2+]i induced by NMDA. As shown in Figure 7A–C, application of 200 μm NMDA fails to increase [Ca2+]i in wild-type HEK-293 cells (Fig. 7A), but a prominent elevation in [Ca2+]i was observed in HEK-293 cells transfected with NR1/NR2B NMDA receptors (Fig. 7B). Triol significantly reduces [Ca2+]i responses in a concentration-dependent manner (Fig. 7B,C). MK-801 (10 μm), used as a positive control, also reduces NMDA-induced [Ca2+]i elevation. In contrast, cholesterol (10 μm) has no effect on NMDA-induced [Ca2+]i increases in these same cells.
Figure 7.
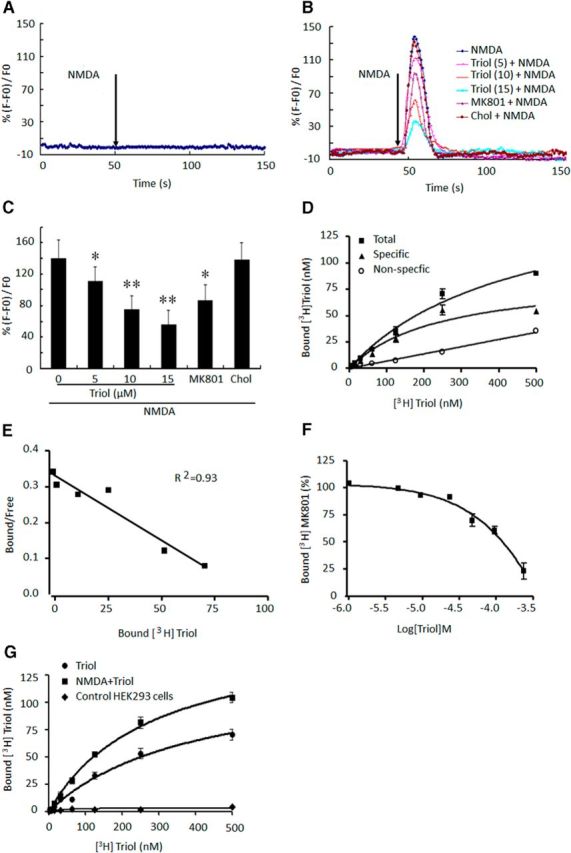
Inhibitory effects of triol on calcium influx induced by NMDA in HEK-293 cells transfected with NMDA receptor and binding assays of [3H]triol and [3H]MK-801. A, NMDA (200 μm) did not induce an increase in [Ca2+]i in wild-type HEK-293 cells. B, Representative recordings of NMDA-evoked Ca2+ influx after addition of triol, cholesterol (Chol, 10 μm), and MK-801 (10 μm) for 15 min in HEK-293 cells transiently transfected with NR1/NR2B receptors (numbers in parentheses are concentration, in micromolar). C, Summary data indicate that triol dose dependently prevents NMDA-induced increase of [Ca2+]i in HEK-293 cells transfected with NR1/NR2B receptors. All values are presented as mean ± SD (n = 20). *p < 0.05 and **p < 0.01 compared with the NMDA group. D, Saturation binding assay of [3H]triol to primary cultured rat cerebellar granule neurons. The experiment repeated three times show similar results. E, Scatchard analysis for [3H]triol binding to primary cultured rat cerebellar granule neurons. F, Competitive effect of triol on [3H]MK-801 binding to NMDA receptors. G, Saturation binding assay of [3H]triol to HEK-293 cells transfected with NMDA receptors. All values are presented as mean ± SD of three separate experiments.
Binding of [3H]triol to primary cultured cerebellar granule neurons
To further investigate the action of triol on NMDA receptors, we studied the binding of [3H]triol to primary cultured cerebellar granule neurons. The results demonstrate specific and saturable binding of [3H]triol with an apparent Kd of 279.21 ± 43.88 nm, whereas nonspecific binding was linear with increased [3H]triol concentrations (Fig. 7D,E). Finally, we studied the effects of triol on the specific binding of [3H]MK-801 to NMDA receptors expressed on cultured cerebellar granule neurons. We found that triol readily displaced [3H]MK-801 binding (Fig. 7F) in a concentration-dependent manner. Furthermore, we found that 100 μm NMDA could significantly increase the maximum binding of [3H]triol in HEK-293 cells transfected with NR1/NR2B receptors, but [3H]triol did not bind to the wild-type HEK-293 cells (Fig. 7G).
Discussion
In the present study, we found that triol, a major metabolite of cholesterol that is present in many tissues, including the brain and spinal cord, has neuroprotective properties when studied in vitro and in vivo. Although triol was once considered a toxic metabolite of cholesterol, after prolonged exposure of endothelial cells to high concentrations (Matthias et al., 1987), we found no evidence that lower physiologic concentrations were toxic to cultured neurons. In fact, we observed neuroprotective properties of triol in primary cultured neurons, including cortical neurons, cerebellar granular neurons, and neurons from the anterior spinal cord, when these cells were subsequently exposed to excitotoxic concentrations of glutamate. We also tested triol in a spinal cord ischemia model in rabbits in which ischemic injury induced by infrarenal aortic occlusion results in a highly reproducible deficit in neurological function attributable to loss of spinal cord motoneurons. Parenteral administration of triol before spinal cord ischemic injury markedly inhibited both the behavioral deficits and loss of motoneurons observed after ischemic injury. Although there exists a rather limited “time window” for neuroprotective agents after ischemic injury, we observed that triol can significantly protect against ischemic injury when administered as long as 4 h after MCAO. We also found that the neuroprotection of triol is persistent rather than simply delaying cell death, as evidenced by a potent neuroprotective effect of triol on MACO animals 7 d after reperfusion.
Does triol play a physiological role as an endogenous neuroprotectant? The presence in the CNS of abundant sources of cholesterol and the presence of its auto-oxidation product cholesterol-5,6-epoxide, as well as a biosynthetic pathway for additional hydration via CheH (Nashed et al., 1985) and relatively high tissue levels of triol present in a variety of species support a potential physiological or pathophysiological role.
Ischemic preconditioning has been used to elucidate endogenous neuroprotective mechanisms (Dirnagl and Meisel, 2008). In this experimental paradigm, subtoxic ischemic insult results in subsequent neuroprotection to an otherwise toxic ischemic injury. A number of cellular mechanisms have been proposed to mediate the neuroprotective state after the processes of preconditioning (Clarkson, 2007; Zhao et al., 2013; Stetler et al., 2014). However, the actual functional effecter molecules are still essentially unknown. In the present study, markedly improved neurological and histopathological outcomes were observed after ischemic preconditioning in the rabbit spinal cord as reported by a number of laboratories (Yu et al., 2006; Huang et al., 2007; Yang et al., 2008; Fang et al., 2013). Using this experimental neuroprotective paradigm, we observed increased triol concentrations in the spinal cord up to 4 h after preconditioning. This inducible increase of spinal cord triol levels was blocked by pretreatment with a specific inhibitor of triol synthesis, which also blocked the neuroprotection induced by preconditioning. These observations, combined with our in vitro and in vivo data, strongly suggest that triol may function as an endogenous neuroprotectant, possibly induced by subtoxic ischemic injury or preconditioning.
However, we found a dose difference of triol between physiological concentrations and the ones used in experiments in vitro and in vivo. From the point of view of the current methodology, the dose differences may be associated with the determination of endogenous triol. The brain or spinal cord concentrations of triol tested in our study may only represent the average ones of triol in a tissue homogenate, which may not truly reflect the concentrations in a specific region or a specific subcellular localization, such as the nerve terminal, in which triol may accumulate to reach a higher concentration to execute its neuroprotection.
Another potential factor responsible for the dose differences may also be related to the high concentrations of excitoxins used. In the in vitro study, 200 μm glutamate was used to ensure the induction of a massive neuronal death. Under this condition, a relatively high dose of triol should be needed to reach its notable neuroprotective effect. A similar phenomenon has been reported in studies of other endogenous neuroprotective agents in the literature. For example, taurine, a well established endogenous modulator, exerts its neuroprotection in rat primary cultured neurons in a high concentration of 25 mm for antagonizing 250 μm glutamate (Chen et al., 2001; Wu et al., 2005), whereas its measured endogenous concentrations are ∼2–9 mm in rat brain (Porcellati, 1963; Palkovits et al., 1986). The main consideration for using relatively high doses (8 mg/kg) of triol in in vivo experiments is aimed to obtain a maximal neuoroprotection effect under the severe pathological condition of spinal cord ischemia, because only one single dose was administrated to the animals. In addition, triol binds with plasma proteins as high as >90% of the amount administrated, which may limit the concentrations of the drug in the action sites. Conversely, it is notable that the concentrations of triol in spinal cord after exogenous administration with 8 mg/kg of triol at multiple time points are comparable with those induced by IPC. In both situations, triol displays definite neuroprotections. However, we believe that the exact reasons for the dose differences deserve additional investigation.
Intracellular calcium overload is generally accepted to be the main mechanism underlying neuronal toxicity (Szydlowska and Tymianski, 2010; Piccolini et al., 2013). Elevation of [Ca2+]i may activate Ca2+-dependent proteases, break down critical proteins or enzymes, and lead to neuronal death (Xiong et al., 2004). Molecular neuroscience has further revealed that glutamate/NMDA receptors consist of ligand-gated ion channels, by which they control Ca2+ influx (Evans et al., 2012; Guo et al., 2013). Glutamate-induced [Ca2+]i elevation is mainly through NMDA receptor channels (Murphy and Miller, 1988). In the current study, we demonstrate that triol markedly attenuates glutamate-induced [Ca2+]i increases in a concentration-dependent manner, which may account for the observed neuroprotective effects of triol in both in vitro and in vivo models that implicate NMDA receptor-mediated [Ca2+]i overload. Furthermore, we observe a significant use-dependent inhibition of NMDA current by triol starting from 1 μm, which is a concentration equivalent to the basal triol level (1.07 μm) in spinal cord in rats and an elevated triol level (0.96 μm) at 4 h after IPC in rabbits.
The findings from radioactive triol binding assays demonstrate a specific binding site for triol in neurons. Furthermore, we provide evidence of direct interplay between triol and NMDA receptors with radioactive MK-801. MK-801 is widely used as a use-dependent or open channel blocker for NMDA receptors. When an NMDA receptor channel is in an open status (in use), MK-801 enters into and binds at the inside of the channels, resulting in an inhibition (Huettner and Bean, 1988; Rodríguez-Moreno et al., 2011). The displacement of MK-801 binding by triol suggests modulation on the open channel site in which MK-801 binds, implying that triol may function as an endogenous NMDA receptor open channel blocker, which is consistent with the patch-clamp findings that triol use dependently inhibit NMDA current. This observation was further supported by the fact that the [3H]triol maximum binding with NMDA receptors is significantly stimulated in the presence of 100 μm NMDA. The recombinant NMDA receptor expressed in HEK-293 cells is a more stringent molecular model and may provide a useful transgenic cellular model to clarify the effects of any potential modulators on NMDA receptors. It was reported that the functional NMDA receptor is a heteromeric protein and requires coassembly of the NR1 (which binds glycine) subunit with at least one NR2 (which binds glutamate) subunit (Paoletti and Neyton, 2007). Lines of evidence show the predominant extrasynaptic NMDA subunit NR2B acts as a central mediator for stroke injury (Lai et al., 2011) and may represent a promising therapeutic target for stoke intervention. In our experiment, we investigate the effects of triol on NMDA receptors with a focus on NR1/NR2B subtype. These data further confirm the role of triol as a negative modulator of NMDA receptors. Additionally, it would be interesting to investigate the selectivity of triol on other NMDA subunits in the future.
Footnotes
This work was supported by National Natural Science Foundation of China Grant 81173045. We thank Dr. Tianming Gao for assistances in patch-clamp recording and Dr. Jianhong Luo for providing plasmid NR1/NR2B.
The authors are named inventors on Patent CN200810198703 held by Sun Yat-sen University for the use of cholestane-3β, 5α, 6β-triol as treatments of neuronal damage.
References
- Al Hasan M, Murugan R. Stenting versus aggressive medical therapy for intracranial arterial stenosis: more harm than good. Crit Care. 2012;16:310. doi: 10.1186/cc11326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman FW, Murray TF. Characterization of glutamate toxicity in cultured rat cerebellar granule neurons at reduced temperature. J Biochem Toxicol. 1996;11:111–119. doi: 10.1002/(SICI)1522-7146(1996)11:3<111::AID-JBT2>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Budd SL. Mechanisms of neuronal damage in brain hypoxia/ischemia: focus on the role of mitochondrial calcium accumulation. Pharmacol Ther. 1998;80:203–229. doi: 10.1016/S0163-7258(98)00029-1. [DOI] [PubMed] [Google Scholar]
- Celik M, Gökmen N, Erbayraktar S, Akhisaroglu M, Konakc S, Ulukus C, Genc S, Genc K, Sagiroglu E, Cerami A, Brines M. Erythropoietin prevents motor neuron apoptosis and neurologic disability in experimental spinal cord ischemic injury. Proc Natl Acad Sci U S A. 2002;99:2258–2263. doi: 10.1073/pnas.042693799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WQ, Jin H, Nguyen M, Carr J, Lee YJ, Hsu CC, Faiman MD, Schloss JV, Wu JY. Role of taurine in regulation of intracellular calcium level and neuroprotective function in cultured neurons. J Neurosci Res. 2001;66:612–619. doi: 10.1002/jnr.10027. [DOI] [PubMed] [Google Scholar]
- Clarkson AN. Anesthetic-mediated protection/preconditioning during cerebral ischemia. Life Sci. 2007;80:1157–1175. doi: 10.1016/j.lfs.2006.12.022. [DOI] [PubMed] [Google Scholar]
- Cross JL, Meloni BP, Bakker AJ, Lee S, Knuckey NW. Modes of neuronal calcium entry and homeostasis following cerebral ischemia. Stroke Res Treat. 2010;2010:316862. doi: 10.4061/2010/316862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichter MA. Rat cortical neurons in cell culture: culture methods, cell morphology, electrophysiology, and synapse formation. Brain Res. 1978;149:279–293. doi: 10.1016/0006-8993(78)90476-6. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Meisel A. Endogenous neuroprotection: mitochondria as gateways to cerebral preconditioning? Neuropharmacology. 2008;55:334–344. doi: 10.1016/j.neuropharm.2008.02.017. [DOI] [PubMed] [Google Scholar]
- Dyker AG, Weir CJ, Lees KR. Influence of cholesterol on survival after stroke: retrospective study. BMJ. 1997;314:1584–1588. doi: 10.1136/bmj.314.7094.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RC, Morera-Herreras T, Cui Y, Du K, Sheehan T, Kotaleski JH, Venance L, Blackwell KT. The effects of NMDA subunit composition on calcium influx and spike timing-dependent plasticity in striatal medium spiny neurons. PLoS Comput Biol. 2012;8:e1002493. doi: 10.1371/journal.pcbi.1002493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahey JM, Lindquist DG, Pritchard GA, Miller LG. Pregnenolone sulfate potentiation of NMDA-mediated increases in intracellular calcium in cultured chick cortical neurons. Brain Res. 1995;669:183–188. doi: 10.1016/0006-8993(94)01223-5. [DOI] [PubMed] [Google Scholar]
- Fang B, Li XM, Sun XJ, Bao NR, Ren XY, Lv HW, Ma H. Ischemic preconditioning protects against spinal cord ischemia-reperfusion injury in rabbits by attenuating blood spinal cord barrier disruption. Int J Mol Sci. 2013;14:10343–10354. doi: 10.3390/ijms140510343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Földes-Papp Z, Demel U, Tilz GP. Laser scanning confocal fluorescence microscopy: an overview. Int Immunopharmacol. 2003;3:1715–1729. doi: 10.1016/S1567-5769(03)00140-1. [DOI] [PubMed] [Google Scholar]
- Giffard RG, Swanson RA. Ischemia-induced programmed cell death in astrocytes. Glia. 2005;50:299–306. doi: 10.1002/glia.20167. [DOI] [PubMed] [Google Scholar]
- Gu G. Shen Nong's herbal classic. Peking, China: People's Medical Publishing House; 1955. [Google Scholar]
- Guo ZY, Li CZ, Li XJ, Wang YL, Mattson MP, Lu CB. The developmental regulation of glutamate receptor-mediated calcium signaling in primary cultured rat hippocampal neurons. Neuroreport. 2013;24:492–497. doi: 10.1097/WNR.0b013e32836206b5. [DOI] [PubMed] [Google Scholar]
- Huang H, Zhang L, Wang Y, Yao J, Weng H, Wu H, Chen Z, Liu J. Effect of ischemic post-conditioning on spinal cord ischemic-reperfusion injury in rabbits. Can J Anaesth. 2007;54:42–48. doi: 10.1007/BF03021898. [DOI] [PubMed] [Google Scholar]
- Huettner JE, Bean BP. Block of N-methyl-d-aspartate-activated current by the anticonvulsant MK-801: selective binding to open channels. Proc Natl Acad Sci U S A. 1988;85:1307–1311. doi: 10.1073/pnas.85.4.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes RA, Sendtner M, Thoenen H. Members of several gene families influence survival of rat motoneurons in vitro and in vivo. J Neurosci Res. 1993;36:663–671. doi: 10.1002/jnr.490360607. [DOI] [PubMed] [Google Scholar]
- Iso H, Jacobs DR, Jr, Wentworth D, Neaton JD, Cohen JD. Serum cholesterol levels and six-year mortality from stroke in 350,977 men screened for the multiple risk factor intervention trial. N Engl J Med. 1989;320:904–910. doi: 10.1056/NEJM198904063201405. [DOI] [PubMed] [Google Scholar]
- Javitt NB. Bile acid synthesis from cholesterol: regulatory and auxiliary pathways. FASEB J. 1994;8:1308–1311. doi: 10.1096/fasebj.8.15.8001744. [DOI] [PubMed] [Google Scholar]
- Johnson SH, Kraimer JM, Graeber GM. Effects of flunarizine on neurological recovery and spinal cord blood flow in experimental spinal cord ischemia in rabbits. Stroke. 1993;24:1547–1553. doi: 10.1161/01.STR.24.10.1547. [DOI] [PubMed] [Google Scholar]
- Kirihara Y, Hamazaki K, Hamazaki T, Ogushi Y, Tsuji H, Shirasaki S. The relationship between total blood cholesterol levels and all-cause mortality in Fukui city, and meta-analysis of this relationship in Japan. J Lipid Nutr. 2008;17:67–78. [Google Scholar]
- Kocaeli H, Korfali E, Oztürk H, Kahveci N, Yilmazlar S. MK-801 improves neurological and histological outcomes after spinal cord ischemia induced by transient aortic cross-clipping in rats. Surg Neurol. 2005;64(Suppl 2):S22–S26. doi: 10.1016/j.surneu.2005.07.034. discussion S27. [DOI] [PubMed] [Google Scholar]
- Lai TW, Shyu WC, Wang YT. Stroke intervention pathways: NMDA receptors and beyond. Trends Mol Med. 2011;17:266–275. doi: 10.1016/j.molmed.2010.12.008. [DOI] [PubMed] [Google Scholar]
- Leonarduzzi G, Gamba P, Sottero B, Kadl A, Robbesyn F, Calogero RA, Biasi F, Chiarpotto E, Leitinger N, Sevanian A, Poli G. Oxysterol-induced up-regulation of MCP-1 expression and synthesis in macrophage cells. Free Radical Bio Med. 2005;39:1152–1161. doi: 10.1016/j.freeradbiomed.2005.06.024. [DOI] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.STR.20.1.84. [DOI] [PubMed] [Google Scholar]
- Matthias D, Becker CH, Gödicke W, Schmidt R, Ponsold K. Action of cholestane-3 beta,5 alpha,6 beta-triol on rats with particular reference to the aorta. Atherosclerosis. 1987;63:115–124. doi: 10.1016/0021-9150(87)90111-0. [DOI] [PubMed] [Google Scholar]
- Mauch DH, Nägler K, Schumacher S, Göritz C, Müller EC, Otto A, Pfrieger FW. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- Mori T, Tateishi N, Kagamiishi Y, Shimoda T, Satoh S, Ono S, Katsube N, Asano T. Attenuation of a delayed increase in the extracellular glutamate level in the peri-infarct area following focal cerebral ischemia by a novel agent ONO-2506. Neurochem Int. 2004;45:381–387. doi: 10.1016/j.neuint.2003.06.001. [DOI] [PubMed] [Google Scholar]
- Murphy SN, Miller RJ. A glutamate receptor regulates Ca2+ mobilization in hippocampal neurons. Proc Natl Acad Sci U S A. 1988;85:8737–8741. doi: 10.1073/pnas.85.22.8737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashed NT, Michaud DP, Levin W, Jerina DM. Properties of liver microsomal cholesterol 5,6-oxide hydrolase. Arch Biochem Biophys. 1985;241:149–162. doi: 10.1016/0003-9861(85)90371-6. [DOI] [PubMed] [Google Scholar]
- Olsen TS, Christensen RH, Kammersgaard LP, Andersen KK. Higher total serum cholesterol levels are associated with less severe strokes and lower all-cause mortality: ten-year follow-up of ischemic strokes in the Copenhagen Stroke Study. Stroke. 2007;38:2646–2651. doi: 10.1161/STROKEAHA.107.490292. [DOI] [PubMed] [Google Scholar]
- Palkovits M, Elekes I, Láng T, Patthy A. Taurine levels in discrete brain nuclei of rats. J Neurochem. 1986;47:1333–1335. doi: 10.1111/j.1471-4159.1986.tb00761.x. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol. 2007;7:39–47. doi: 10.1016/j.coph.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Paul SM, Doherty JJ, Robichaud AJ, Belfort GM, Chow BY, Hammond RS, Crawford DC, Linsenbardt AJ, Shu HJ, Izumi Y, Mennerick SJ, Zorumski CF. The major brain cholesterol metabolite 24(S)-hydroxycholesterol is a potent allosteric modulator of N-methyl-d-aspartate receptors. J Neurosci. 2013;33:17290–17300. doi: 10.1523/JNEUROSCI.2619-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne AH, Hales DB. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr Rev. 2004;25:947–970. doi: 10.1210/er.2003-0030. [DOI] [PubMed] [Google Scholar]
- Piccolini VM, Bottone MG, Bottiroli G, De Pascali SA, Fanizzi FP, Bernocchi G. Platinum drugs and neurotoxicity: effects on intracellular calcium homeostasis. Cell Biol Toxicol. 2013;29:339–353. doi: 10.1007/s10565-013-9252-3. [DOI] [PubMed] [Google Scholar]
- Porcellati G. On the occurence and distribution of free phospholipid phosphoric esters and some amino acid compounds in the nervous tissues of some animal species. Riv Biol. 1963;56:209–226. [PubMed] [Google Scholar]
- Qiu S, Hua YL, Yang F, Chen YZ, Luo JH. Subunit assembly of N-methyl-d-aspartate receptors analyzed by fluorescence resonance energy transfer. J Biol Chem. 2005;280:24923–24930. doi: 10.1074/jbc.M413915200. [DOI] [PubMed] [Google Scholar]
- Razzazi-Fazeli E, Kleineisen S, Luf W. Determination of cholesterol oxides in processed food using high performance liquid chromatography–mass spectrometry with atmospheric pressure chemical ionization. J Chromatog A. 2000;896:321–334. doi: 10.1016/s0021-9673(00)00719-6. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Moreno A, Kohl MM, Reeve JE, Eaton TR, Collins HA, Anderson HL, Paulsen O. Presynaptic induction and expression of timing-dependent long-term depression demonstrated by compartment-specific photorelease of a use-dependent NMDA receptor antagonist. J Neurosci. 2011;31:8564–8569. doi: 10.1523/JNEUROSCI.0274-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroepfer GJ., Jr Oxysterols: modulators of cholesterol metabolism and other processes. Physiol Rev. 2000;80:361–554. doi: 10.1152/physrev.2000.80.1.361. [DOI] [PubMed] [Google Scholar]
- Sevanian A, McLeod LL. Catalytic properties and inhibition of hepatic cholesterol-epoxide hydrolase. J Biol Chem. 1986;261:54–59. [PubMed] [Google Scholar]
- Sevanian A, Seraglia R, Traldi P, Rossato P, Ursini F, Hodis H. Analysis of plasma cholesterol oxidation products using gas- and high-performance liquid chromatography/mass spectrometry. Free Radical Bio Med. 1994;17:397–409. doi: 10.1016/0891-5849(94)90166-X. [DOI] [PubMed] [Google Scholar]
- Steck TL, Lange Y. Cell cholesterol homeostasis: mediation by active cholesterol. Trends Cell Biol. 2010;20:680–687. doi: 10.1016/j.tcb.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetler RA, Leak RK, Gan Y, Li P, Zhang F, Hu X, Jing Z, Chen J, Zigmond MJ, Gao Y. Preconditioning provides neuroprotection in models of CNS disease: paradigms and clinical significance. Prog Neurobiol. 2014;114:58–83. doi: 10.1016/j.pneurobio.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Zheng X, Zhou Y, Zhu W, Ou Y, Shu M, Gao X, Leng T, Qiu P, Yan G. Alphaxalone inhibits growth, migration and invasion of rat C6 malignant glioma cells. Steroids. 2013;78:1041–1045. doi: 10.1016/j.steroids.2013.06.008. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. Semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium. 2010;47:122–129. doi: 10.1016/j.ceca.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Thiyagarajan R, Sunderrajan A. Gunapadam thatu jeeva vagupp. Chennai, India: Directorate of Indian Medicine and Homeopathy; 1992. Gunapadam thatu jeeva vagupp; pp. 474–478. [Google Scholar]
- Villmann C, Becker CM. On the hypes and falls in neuroprotection: targeting the NMDA receptor. Neuroscientist. 2007;13:594–615. doi: 10.1177/1073858406296259. [DOI] [PubMed] [Google Scholar]
- Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ, Simon RP. Neuroprotection in ischemia: Blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–698. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- Wu H, Jin Y, Wei J, Jin H, Sha D, Wu JY. Mode of action of taurine as a neuroprotector. Brain Res. 2005;1038:123–131. doi: 10.1016/j.brainres.2005.01.058. [DOI] [PubMed] [Google Scholar]
- Yang C, Ren Y, Liu F, Cai W, Zhang N, Nagel DJ, Yin G. Ischemic preconditioning suppresses apoptosis of rabbit spinal neurocytes by inhibiting ASK1-14-3-3 dissociation. Neurosci Lett. 2008;441:267–271. doi: 10.1016/j.neulet.2008.06.037. [DOI] [PubMed] [Google Scholar]
- Yu QJ, Wang YL, Zhou QS, Huang HB, Tian SF, Duan DM. Effect of repetitive ischemic preconditioning on spinal cord ischemia in a rabbit model. Life Sci. 2006;79:1479–1483. doi: 10.1016/j.lfs.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Zhao L, Liu X, Liang J, Han S, Wang Y, Yin Y, Luo Y, Li J. Phosphorylation of p38 MAPK mediates hypoxic preconditioning-induced neuroprotection against cerebral ischemic injury via mitochondria translocation of Bcl-xL in mice. Brain Res. 2013;1503:78–88. doi: 10.1016/j.brainres.2013.01.051. [DOI] [PubMed] [Google Scholar]