

ABSTRACT
Fusaric acid (FA), a mycotoxin contaminant of maize, displays toxicity in plants and animals; however, its epigenetic mechanism is unknown. DNA methylation, an epigenetic modification that regulates gene expression, is mediated by DNA methyltransferases (DNMTs; DNMT1, DNMT3A, and DNMT3B) and demethylases (MBD2). The expression of DNMTs and demethylases are regulated by promoter methylation, microRNAs (miR-29b) and post-translational modifications (ubiquitination). Alterations in these DNA methylation modifying enzymes affect DNA methylation patterns and offer novel mechanisms of FA toxicity. We determined the effect of FA on global DNA methylation as well as a mechanism of FA-induced changes in DNA methylation by transcriptional (promoter methylation), post-transcriptional (miR-29b) and post-translational (ubiquitination) regulation of DNMTs and MBD2 in the human hepatocellular carcinoma (HepG2) cell line. FA induced global DNA hypomethylation (p < 0.0001) in HepG2 cells. FA decreased the mRNA and protein expression of DNMT1 (p < 0.0001), DNMT3A (p < 0.0001), and DNMT3B (p < 0.0001) by upregulating miR-29b (p < 0.0001) and inducing promoter hypermethylation of DNMT1 (p < 0.0001) and DNMT3B (p < 0.0001). FA decreased the ubiquitination of DNMT1 (p = 0.0753), DNMT3A (p = 0.0008), and DNMT3B (p < 0.0001) by decreasing UHRF1 (p < 0.0001) and USP7 (p < 0.0001). FA also induced MBD2 promoter hypomethylation (p < 0.0001) and increased MBD2 expression (p < 0.0001). Together these results indicate that FA induces global DNA hypomethylation by altering DNMT promoter methylation, upregulating miR-29b, and increasing MBD2 in HepG2 cells.
KEYWORDS: Fusaric acid, global DNA hypomethylation, promoter methylation, DNA methyltransferases, miR-29b, DNMT ubiquitination, MBD2
Introduction
Fusaric acid (FA; 5-butylpicolinic acid), a ubiquitous mycotoxin and secondary metabolite produced by pathogenic fungi of the genus Fusarium, contaminates agricultural foods and exhibits low to moderate toxicity [1]. Previously, feed samples were reported to contain an average of 643 µg/kg FA [2] and approximately 2.5–18 µg/kg FA were reported to contaminate commercial foods and feeds [3]. These foods, especially maize, form an essential part of the human and animal diet; and the consumption of FA-contaminated commodities may have serious health implications. Studies evaluating the effects of FA are limited and understanding the molecular and epigenetic effects of FA exposure is important in decreasing FA contamination and lowering the risk of FA-related adverse health outcomes.
FA is phytotoxic to several plants by inhibiting root and leaf cell function [4] and has been implicated in the pathogenesis of wilt diseases [4–7]; it is a highly lipophilic toxin that traverses cellular membranes and induces toxicity by altering various biochemical processes. Known mechanisms of FA toxicity include alterations in membrane permeability [5,7], oxidative stress [8,9], mitochondrial dysfunction [6,10,11], DNA damage [12,13], and apoptosis [10,12,14,15]. It is also immunotoxic to peripheral blood mononuclear cells (PBMCs) and human monocytic (THP-1) cells [14]. FA has tumouristatic and tumouricidal effects in several mammalian tumour cell lines, thereby, displaying anti-cancer activity [13,16]. It has neurochemical effects in mice brain and reduced aggressive behaviour and motor activity [17]. FA also attenuates isoproterenol induced heart failure by preventing the development of cardiac hypertrophy and fibrosis [18].
FA is a chelating agent and the removal of essential divalent cations such as calcium affects bone ossification [19] and blood coagulation [20]; it also chelates copper causing hypotension [21,22] and notochord malformation [23]. The toxicity of FA may also be attributed to synergistic interactions with other co-occurring mycotoxins such as fumonisin B1 (FB1) [24], deoxynivalenol (DON) [25], and 4,15-diacetoxyscirpenol (DAS) [26].
DNA methylation is a common epigenetic modification that regulates gene expression and plays a major role in cell signalling pathways that are essential in the normal growth and development of higher organisms. Dysregulation in the DNA methylation pattern has been observed in several human diseases such as cancer [27] and neurodegeneration [28]. DNA methylation is catalyzed by DNA methyltransferases (DNMTs) such as DNMT1, DNMT3A, and DNMT3B. DNMT1 is a maintenance DNMT that binds specifically to hemi-methylated DNA and is responsible for conserving the methylation pattern from one generation to the next [29]. DNMT3A and DNMT3B are de novo DNMTs that target unmethylated cytosine bases to initiate methylation [29]. DNMTs are the major regulators of DNA methylation and alterations in their expression and activity affects DNA methylation patterns and cellular function. The activity and stability of DNMTs are regulated by promoter methylation, microRNAs, and post-translational modifications (PTMs).
Promoter methylation, methylation of CpG islands within the promoter region of specific genes, is important in regulating gene transcription; promoter hypermethylation prevents binding of transcription factors and inhibits gene transcription, whereas promoter hypomethylation activates gene transcription.
MicroRNAs are small non-coding RNA molecules that post-transcriptionally regulate gene expression by binding to the 3ʹ untranslated region (3ʹUTR) of the target messenger RNA (mRNA) and negatively regulating the processing, stability, and translation of the mRNA [30]. MiR-29 plays a major role in cell proliferation, differentiation, and apoptosis [31,32]. The miR-29 family consists of two clusters: cluster 1, located on chromosome 7q32.3, consists of miR-29a and miR-29b-1; and cluster 2, located on chromosome 1q32.2, consists of miR-29b-2 and miR-29c. MiR-29b-1 and miR-29b-2 have identical mature sequences and are collectively referred to as miR-29b. Several effects of miR-29b have been identified such as activating the tumour suppressor protein, p53 and regulating cell proliferation, and apoptosis by targeting p85α and the cell division cycle 42 (CDC42) [31,32]. It prevents liver fibrosis by targeting the PI3K/AKT signalling pathway [33], and targets AKT2 and AKT3 to regulate the Warburg effect in ovarian cancer cells [34]. MiR-29b can also regulate the DNA methylation status of the cell in a negative feedback loop by directly targeting DNMT3A and DNMT3B [35,36]. Furthermore, the expression of miR-29b is itself epigenetically regulated and thus inversely correlated with the DNA methylation status of the cell.
PTMs also regulate the expression and activity of DNMTs. These modifications occur in the N- and C-terminal regions of the protein and include acetylation and ubiquitination [29]. The acetylation of DNMTs is regulated by the acetyltransferase, Tip60 and the deacetylases, HDAC1 and HDAC2 [29,37,38]. The ubiquitination of DNMTs is triggered by DNMT acetylation and is regulated by the E3 ligase, ubiquitin-like and ring finger domain 1 (UHRF1), and the deubiquitylating enzyme, ubiquitin specific peptidase 7 (USP7) [29,37,38]. The ubiquitination of DNMTs play a major role in inhibiting DNMT stability and promoting proteasomal degradation.
DNA methylation forms a platform for several methyl binding proteins. Methyl-CpG binding domain proteins (MBDs) are a family of nuclear proteins that play an important role in regulating DNA methylation and gene transcription by recruiting chromatin remodelling complexes to regions of methylated DNA. Several MBDs have been identified (MBD1-6); however, MBD2 is the major MBD that binds specifically to methylated CpG islands and acts as a methylation-dependent transcriptional repressor and DNA demethylase [39].
Although several effects of FA have been described, the effect of FA on epigenetic regulation has not been determined. This study aimed to determine an epigenetic effect of FA in the human hepatocellular carcinoma (HepG2) cell line, as a mechanism of FA-induced toxicity. The effect of FA on global DNA methylation as well as the mechanism of FA-induced changes in DNA methylation by transcriptional (promoter methylation), post-transcriptional (miR-29b), and post-translational (ubiquitination) regulation of DNMTs and MBD2 was determined.
Results
Fusaric acid induced global DNA hypomethylation in HepG2 cells
We first determined the effect of FA on global DNA methylation in liver (HepG2) cells. 5-methylcytosine, a common marker of global DNA methylation, was quantified using a commercialized kit (Abcam, ab117128) and 5-aza-2-DC was used as a negative control. The percentage of 5-methylcytosine in the 5-aza-2-DC and FA-treated HepG2 cells were decreased compared to the control (p < 0.0001; Figure 1). This suggested that FA induced a dose-dependent decrease in global DNA methylation in HepG2 cells.
Figure 1.
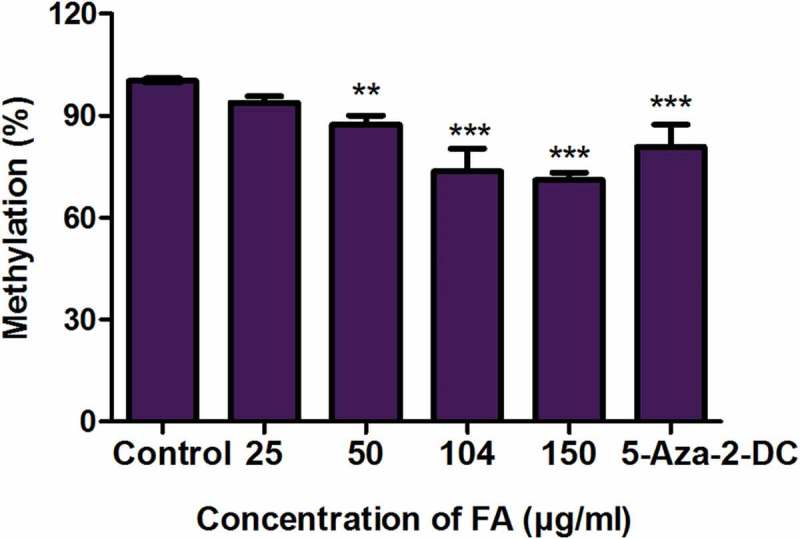
Fusaric acid induced global DNA hypomethylation in HepG2 cells. DNA isolated from control and FA-treated HepG2 cells were assayed for global DNA methylation by quantifying 5-methylcytosine using a Colorimetric Methylated DNA Quantification Kit. Fusaric acid decreased the percentage of 5-methylcytosine in HepG2 cells compared to the control. Results are represented as mean fold-change ± SD (n = 3). Statistical significance was determined by one-way ANOVA with the Bonferroni multiple comparisons test (**p < 0.005, ***p < 0.0001).
Fusaric acid decreased the expression of DNMT1, DNMT3A, and DNMT3B in HepG2 cells
The DNMTs, DNMT1, DNMT3A, and DNMT3B, play a major role in initiating and maintaining DNA methylation patterns. Due to the FA-induced global DNA hypomethylation in the HepG2 cells, we evaluated the mRNA and protein expressions of DNMT1, DNMT3A, and DNMT3B. FA significantly decreased the mRNA expression of DNMT1 (p < 0.0001; Figure 2(a)), DNMT3A (p < 0.0001; Figure 2(a)), and DNMT3B (p < 0.0001; Figure 2(a)) in HepG2 cells compared to the control. The protein expression of DNMT1 (p < 0.0001; Figure 2(b)), DNMT3A (p < 0.0001; Figure 2(b)), and DNMT3B (p < 0.0001; Figure 2(b)) was also significantly decreased in the FA-treated HepG2 cells compared to the control.
Figure 2.
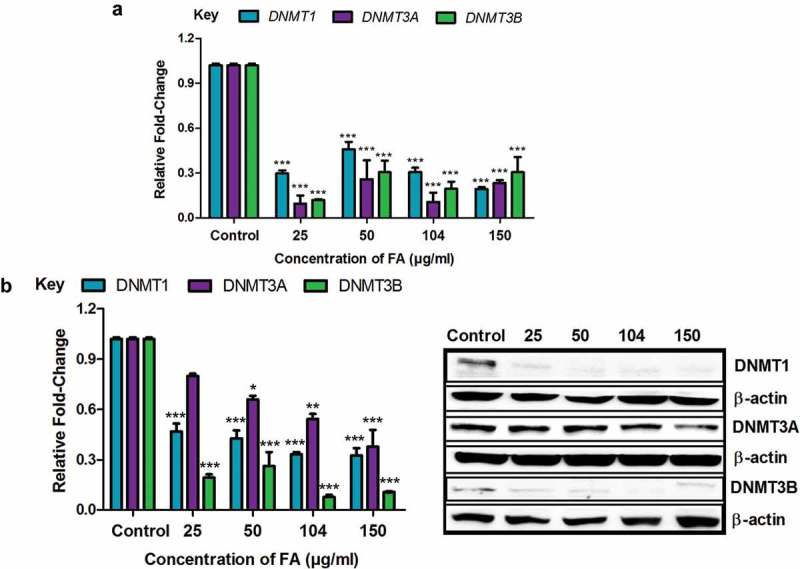
The effect of FA on DNA methyltransferases in HepG2 cells. (a) RNA isolated from control and FA-treated HepG2 cells were reverse transcribed into cDNA and analyzed by qPCR. Fusaric acid significantly decreased the mRNA expression of DNMT1, DNMT3A, and DNMT3B in HepG2 cells. (b) Protein expression of DNMT1, DNMT3A, and DNMT3B were determined by Western blot. Fusaric acid decreased the protein expression of DNMT1, DNMT3A, and DNMT3B in HepG2 cells. Results are represented as mean fold-change ± SD (n = 3). Statistical significance was determined by one-way ANOVA with the Bonferroni multiple comparisons test (*p < 0.05, **p < 0.005, ***p < 0.0001).
Fusaric acid altered DNMT promoter methylation in HepG2 cells
The methylation of gene promoters plays a major role in determining transcriptional activity and gene expression. We determined if the decrease in the mRNA expression of DNMT1, DNMT3A, and DNMT3B observed in the FA-treated HepG2 cells were a result of promoter methylation. FA significantly increased promoter methylation of DNMT1 (p < 0.0001; Figure 3) and DNMT3B (p < 0.0001; Figure 3) in HepG2 cells compared to the control; however, the promoter methylation of DNMT3A was decreased in the lower FA concentrations (25, 50, and 104 µg/ml) and increased in the higher FA concentration (150 µg/ml) (p < 0.0001; Figure 3).
Figure 3.
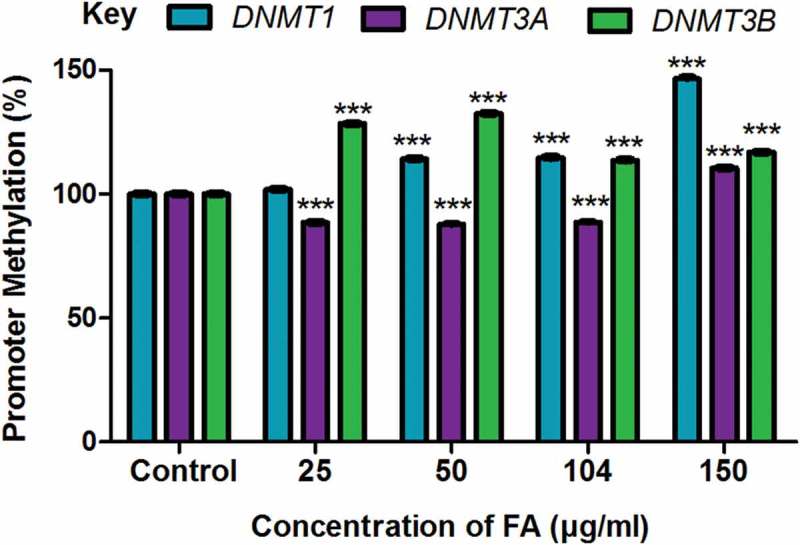
The effect of FA on the promoter methylation of DNMT1, DNMT3A, and DNMT3B in HepG2 cells. DNA isolated from control and FA-treated HepG2 cells were assayed for DNMT promoter methylation using the OneStep qMethyl Kit. Fusaric acid induced promoter hypermethylation of DNMT1 and DNMT3B, and altered promoter methylation of DNMT3A in HepG2 cells. Results are represented as mean fold-change ± SD (n = 3). Statistical significance was determined by one-way ANOVA with the Bonferroni multiple comparisons test (***p < 0.0001).
Fusaric acid decreased miR-29b promoter methylation, upregulated miR-29b, and decreased the expression of Sp1 in HepG2 cells
The expression of miR-29b is regulated by DNA methylation; miR-29b is silenced by DNA hypermethylation whereas DNA hypomethylation is known to upregulate miR-29b [36]. Since FA induced DNA hypomethylation in HepG2 cells, we determined the effect of FA on the promoter methylation and expression of miR-29b. FA significantly decreased the promoter methylation of miR-29b (p < 0.0001; Figure 4(a)) and increased the expression of miR-29b (p < 0.0001; Figure 4(b)) in HepG2 cells compared to the control. The expression of miR-29b was also significantly increased by 5-aza-2-DC (p < 0.0001; Figure 4(b)).
Figure 4.
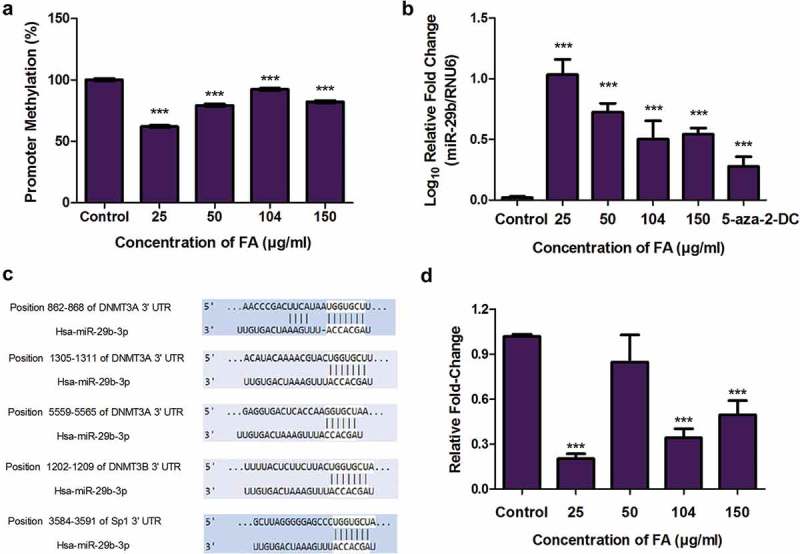
The effect of FA on miR-29b and Sp1 in HepG2 cells. (a) DNA isolated from control and FA-treated HepG2 cells were assayed for miR-29b promoter methylation using the OneStep qMethyl Kit. Fusaric acid induced promoter hypomethylation of miR-29b in HepG2 cells. (b) RNA isolated from control and FA-treated HepG2 cells were reverse transcribed into cDNA and analyzed by qPCR. Fusaric acid significantly increased the expression of miR-29b in HepG2 cells. (c) TargetScan analysis of miR-29b to the 3ʹUTRs of DNMT3A, DNMT3B, and Sp1. (d) RNA isolated from control and FA-treated HepG2 cells were reverse transcribed into cDNA and analyzed for Sp1 expression by qPCR. Fusaric acid decreased the mRNA expression of Sp1 in HepG2 cells. Results are represented as mean fold-change ± SD (n = 3). Statistical significance was determined by one-way ANOVA with the Bonferroni multiple comparisons test (***p < 0.0001).
MiR-29b is also a known regulator of DNMT expression. MiR-29b was previously shown to directly target DNMT3A and DNMT3B and indirectly target DNMT1 via repression of the transcriptional activator, Sp1 [35,36,40]. This was confirmed using the bioinformatics prediction algorithm software, TargetScan version 7.1. MiR-29b was found to have complementary base pairs with DNMT3A at positions 862-868, 1305-1311, and 5559-5565; DNMT3B at position 1202-1209; and Sp1 at position 3584-3591 (Figure 4(c)). DNMT1 was not a direct target of miR-29b. Due to the increase in miR-29b and decrease in DNMT expression by FA, we then determined the effect of FA on the mRNA expression of Sp1. FA significantly decreased the expression of Sp1 (p < 0.0001; Figure 4(d)) in HepG2 cells compared to the control. These data suggest that the decrease in the mRNA expression of DNMT1, DNMT3A, and DNMT3B may be influenced by miR-29b.
Fusaric acid decreased the ubiquitination of DNMT1, DNMT3A, and DNMT3B by decreasing the expression of UHRF1 and USP7 in HepG2 cells
PTMs such as acetylation and ubiquitination regulate the activity and expression of DNMTs. The acetylation of DNMTs triggers the ubiquitination of DNMTs leading to proteasomal degradation. We determined if the decrease in the protein expression of DNMT1, DNMT3A, and DNMT3B in the FA treatments were a result of the ubiquitination and proteasomal degradation of the DNMTs. FA significantly decreased the ubiquitination of DNMT1 (p = 0.0753; Figure 5(a)), DNMT3A (p = 0.0008; Figure 5(a)), and DNMT3B (p < 0.0001; Figure 5(a)) in HepG2 cells compared to the control. However, at 150 µg/ml FA the ubiquitination of DNMT1 and DNMT3B were increased.
Figure 5.
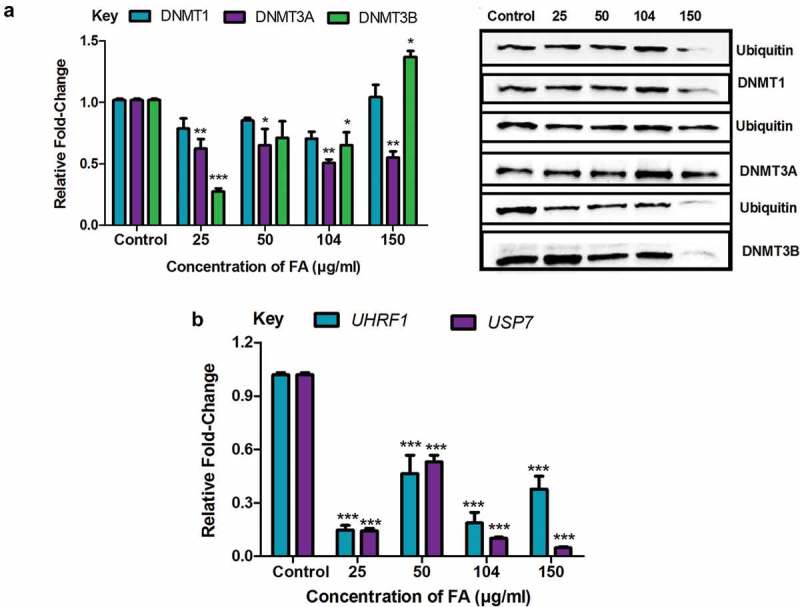
The effect of FA on the ubiquitination of DNMT1, DNMT3A, and DNMT3B in HepG2 cells. (a) The ubiquitination of DNMT1, DNMT3A, and DNMT3B were detected by immuno-precipitation and Western blot. Fusaric acid altered the ubiquitination of DNMT1, DNMT3A, and DNMT3B in HepG2 cells. (b) RNA isolated from control and FA-treated HepG2 cells were reverse transcribed into cDNA and analyzed by qPCR. Fusaric acid significantly decreased the expression of UHRF1 and USP7 in HepG2 cells. Results are represented as mean fold-change ± SD (n = 3). Statistical significance was determined by one-way ANOVA with the Bonferroni multiple comparisons test (*p < 0.05, **p < 0.005, ***p < 0.0001).
The ubiquitination regulators, UHRF1 and USP7, are the major enzymes responsible for ubiquitinating and deubiquitinating DNMTs, respectively. The FA-induced decrease in the ubiquitination of DNMT1, DNMT3A, and DNMT3B led to the assessment of UHRF1 and USP7. FA significantly decreased the mRNA expression of UHRF1 (p < 0.0001; Figure 5(b)) and USP7 (p < 0.0001; Figure 5(b)) in HepG2 cells compared to the control. These results indicate that the decrease in the protein expression of DNMT1, DNMT3A, and DNMT3B observed in the FA-treated cells is not due to the ubiquitination and proteasomal degradation of DNMTs.
Fusaric acid induced MBD2 promoter hypomethylation and increased the expression of MBD2 in HepG2 cells
Methyl CpG binding domain protein 2 (MBD2), a major MBD, promotes global DNA hypomethylation by binding specifically to methylated DNA and functioning as a methylation-dependent transcriptional repressor and DNA demethylase. We determined if the FA-induced decrease in global DNA methylation occurred as a result of MBD2. FA significantly decreased MBD2 promoter methylation (p < 0.0001; Figure 6(a)) and increased the protein expression of MBD2 (p < 0.0001; Figure 6(b)) in HepG2 cells compared to the control. The mRNA expression of MBD2 (p < 0.0001), and other MBDs such as MBD1 (p < 0.0001), MBD3 (p < 0.0001), MBD4 (p < 0.0001), MBD5 (p < 0.0001), and MBD6 (p < 0.0001) were significantly decreased in the FA-treated cells compared to the control (Supplementary Table S1).
Figure 6.
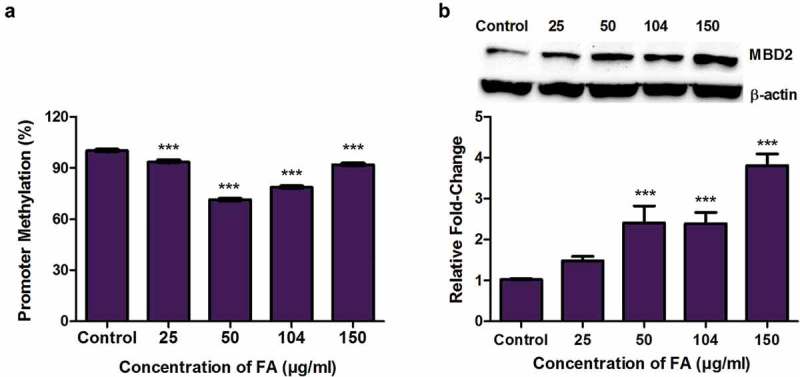
The effect of FA on MBD2 promoter methylation and MBD2 expression in HepG2 cells. (a) DNA isolated from control and FA-treated HepG2 cells were assayed for MBD2 promoter methylation using the OneStep qMethyl Kit. Fusaric acid significantly induced promoter hypomethylation of MBD2 in HepG2 cells. (b) Protein expression of MBD2 was determined by Western blot. Fusaric acid significantly increased the protein expression of MBD2 in HepG2 cells. Results are represented as mean fold-change ± SD (n = 3). Statistical significance was determined by one-way ANOVA with the Bonferroni multiple comparisons test (***p < 0.0001).
Discussion
FA, a neglected mycotoxin found in agricultural foods, alters biological pathways causing toxicity in various plant and animal models. To date several mechanisms of FA toxicity have been described [10,12,14,15,20,21,23]; however, the effect of FA on epigenetic modifications is unknown. DNA methylation is an important epigenetic modification that regulates chromatin structure and alters gene expression and thus may play a crucial role in FA toxicity. In this study, we provide evidence that FA alters global DNA methylation in HepG2 cells by modulating the expression of DNMTs and demethylases in a mechanism that involves alterations in promoter methylation and miR-29b expression, but not the ubiquitination of DNMTs.
FA induced global DNA hypomethylation in HepG2 cells as evidenced by the significant decrease in 5-methylcytosine content (Figure 1); this global DNA hypomethylation is due to a concomitant decrease in the expression of the de novo methyltransferases, DNMT3A and DNMT3B, and the maintenance methyltransferase, DNMT1 (Figure 2(a,b)) as well as an increase in the demethylase, MBD2 (Figure 6(b)). Furthermore, FA altered the mRNA expression of DNMT1 and DNMT3B by inducing promoter hypermethylation (Figure 3). This is in agreement with previous studies in which promoter hypermethylation of DNMT1 and DNMT3B decreased the mRNA expression of DNMT1 and DNMT3B, respectively [41,42]. Although promoter hypomethylation of DNMT3A is associated with an increase in the transcription of DNMT3A, the decrease in DNMT3A mRNA transcript levels observed in the FA-treated HepG2 cells suggests possible regulation at the post-transcriptional level.
MicroRNAs regulate gene expression at the post-transcriptional level. This occurs in a sequence specific manner and leads to either the degradation of the target mRNA or inhibition of translation. MiR-29b, regulated by DNA methylation, was previously shown to repress DNA methylation by directly targeting DNMT3A and DNMT3B, and indirectly targeting DNMT1 by inhibiting the transcriptional activator, Sp1 [35,36]. This was further confirmed using TargetScan version 7.1 (Figure 4(c)). FA significantly upregulated the expression of miR-29b in HepG2 cells (Figure 4(b)) and the expression of miR-29b was inversely correlated with the DNA methylation status in the FA-treated HepG2 cells, as evidenced by the significant decrease in miR-29b promoter methylation (Figure 4(a)). The upregulation of miR-29b also corresponds with the decrease in the mRNA expression of Sp1, DNMT1, DNMT3A, and DNMT3B in the FA-treated cells. This is in agreement with previous studies where overexpression of miR-29b was found to downregulate the expression of DNMT3A and DNMT3B, and induce global DNA hypomethylation in acute myeloid leukaemia (AML) and lung cancer cells [35,36]. Overexpression of miR-29b in AML was also shown to downregulate the expression of Sp1 causing a subsequent decrease in DNMT1 expression and global DNA hypomethylation [35,40]. Therefore, these results indicate that the FA-induced increase in miR-29b expression may be an alternative mechanism for the reduced DNMT3A mRNA expression and an additional mechanism for the reduced DNMT1 and DNMT3B mRNA expressions.
The protein expression of DNMT1, DNMT3A, and DNMT3B was also significantly decreased in the FA-treated HepG2 cells (Figure 2(b)). PTMs such as acetylation and ubiquitination play a major role in influencing the catalytic activity, stability, and protein-protein interactions of DNMTs. The acetylation of DNMTs is mediated by Tip60 and primes DNMTs for UHRF1-mediated ubiquitination and proteasomal degradation [29,37,38]. The DNMTs are deacetylated by HDAC1 and HDAC2, and deubiquitinated by USP7.
The role of acetylation and ubiquitination on the regulation of DNMT1 is well understood. The acetylation of DNMT1 on lysine (K) residues, K1349 and K1415, in the catalytic domain decreases DNMT1 activity whereas the acetylation of K1111, K1113, K1115, and K1117 in the lysine-glycine rich (KG)-repeat increases the transcriptional repressor activity of DNMT1 [43]. The acetylation of lysine residues in the KG-repeat also increases the DNMT1-UHRF1 interaction and impairs the DNMT1-USP7 interaction, thereby, promoting the ubiquitination and degradation of DNMT1 [44,45]. The overexpression of UHRF1 was also shown to increase the ubiquitination of DNMT1 and decrease DNMT1 expression [44]. Previous studies also indicate that UHRF1 physically interacts with DNMT3A and DNMT3B, thereby, inhibiting the activity of both DNMT3A and DNMT3B and promoting proteasomal degradation [46].
The decrease in the protein expression of DNMT1, DNMT3A, and DNMT3B in the FA-treated HepG2 cells suggested that FA may also decrease the protein expression of DNMTs by ubiquitination and proteasomal degradation. In fact, FA actually decreased the ubiquitination of DNMT1, DNMT3A, and DNMT3B in HepG2 cells (Figure 5(a)). The expression of UHRF1 and USP7 was also significantly decreased in the FA-treated cells (Figure 5(b)), suggesting that the decrease in the ubiquitination of DNMT1, DNMT3A, and DNMT3B was a result of UHRF1 and USP7. Thus, the FA-induced decrease in the protein expression of DNMT1, DNMT3A, and DNMT3B was due to the increased DNMT promoter methylation and/or miR-29b expression and a subsequent inhibition of translation, and not the ubiquitination and proteasomal degradation of the DNMT protein.
UHRF1 also contains a methyl DNA-binding domain, SRA (SET and RING associated) domain, that binds preferentially to hemi-methylated DNA and functions to recruit DNMT1 to hemi-methylated CpG islands to facilitate maintenance of DNA methylation [47]. The observed decrease in global DNA methylation in the FA-treated HepG2 cells may also occur as a result of the decrease in UHRF1 and DNMT1 leading to a loss in the maintenance of DNA methylation.
In addition to alterations in the expression of DNMTs and UHRF1, FA may also induce global DNA hypomethylation by targeting the transcriptional repressor and demethylase, MBD2. MBD2 plays an essential role in hypomethylation and was previously shown to activate gene expression by promoting demethylation of several target genes. Our results indicate that FA induced MBD2 promoter hypomethylation (Figure 6(a)) and increased the protein expression of MBD2 (Figure 6(b)) in HepG2 cells. This occurred despite the significant decrease in the mRNA expression of MBD2 (Supplementary Table S1), and suggests that the FA-induced expression of MBD2 may contribute to global DNA hypomethylation. Previous studies indicate MBD2 promoter hypomethylation to be associated with active gene transcription and an increase in MBD2 expression. Although, MBD2 is associated with gene activation, overexpression of MBD2, and global DNA hypomethylation leads to genomic instability in several human cancers [48,49].
Global DNA hypomethylation is considered a hallmark of cancer as it leads to genomic instability and increases the frequency of mutations [50]. Global DNA hypomethylation also inhibits cellular differentiation [51] and induces apoptosis [51–54]. Previously, FB1, a Fusarium-derived mycotoxin often co-produced with FA, was shown to induce global DNA hypomethylation (by modulating the expression of DNMTs and MBD2) and histone demethylation, possibly leading to chromatin instability and liver tumourigenesis [55]. FB1 also alters promoter methylation of tumour suppressor genes (c-myc, p15, p16, and e-cadherin) [56,57], inhibits miR-27b and increases cytochrome P450 1B1 [58] leading to hepatic neoplastic transformation. Zearalenone also induces global DNA hypomethylation and reduces the viability of human bronchial epithelial cells via DNA damage, cell cycle arrest, and apoptosis [59]. In contrary, other Fusarium produced mycotoxins such as deoxynivalenol and T2 toxin induce global DNA hypermethylation and histone demethylation [60,61]. The toxicity of FA has been mainly attributed to oxidative stress, DNA damage, and apoptosis [10,12–15,62], and the FA-induced global DNA hypomethylation may provide an alternative mechanism by which FA induces its genotoxic and cytotoxic effects.
In conclusion, this study provides an alternative mechanism of FA-induced genotoxicity and cytotoxicity at the epigenetic level. The results indicate that FA induces global DNA hypomethylation in HepG2 cells by decreasing the expression of DNMT1, DNMT3A, and DNMT3B and increasing the expression of MBD2 (Figure 7). The results further indicate that FA decreases the expression of DNMT1, DNMT3A, DNMT3B, and MBD2 proteins by increasing promoter methylation and/or by upregulating miR-29b. It has also been shown that miR-29b itself can be regulated by DNA methylation, and that reduced methylation as seen globally following treatment with FA may lead to increased expression of miR-29b. These findings suggest that FA-induced changes in DNA methylation may potentially be used as a biomarker for FA exposure and toxicity. Finally, targeting the DNA methylation pathway via epigenetic modulation of DNMTs and miR-29b may provide a therapeutic intervention against FA toxicity; this is particularly important in poverty stricken areas where maize forms a staple diet and the risk of FA contamination is high.
Figure 7.
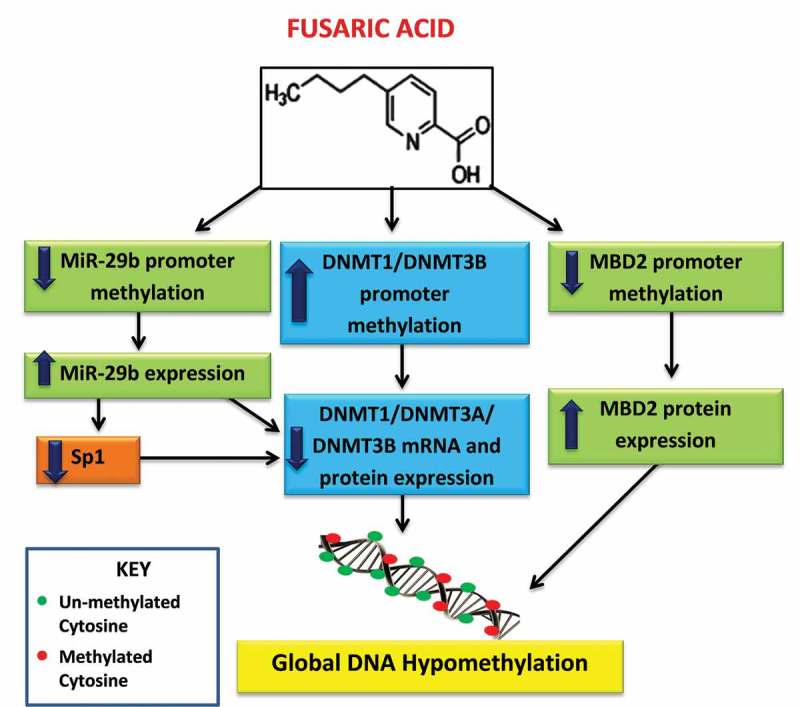
Proposed mechanism of FA-induced global DNA hypomethylation in HepG2 cells. FA induces global DNA hypomethylation by decreasing the mRNA and protein expression of DNMT1, DNMT3A, and DNMT3B. The decrease in DNMTs is caused by promoter hypermethylation of DNMT1 and DNMT3B, and promoter hypomethylation and upregulation of miR-29b. MiR-29b negatively regulates the mRNA expression of DNMT1, DNMT3A, and DNMT3B. In addition, FA may also induce global DNA hypomethylation by causing promoter hypomethylation and upregulation of MBD2.
Materials and methods
Materials
FA (Gibberella fujikuroi, F6513) and the DNA methylation inhibitor, 5-aza-2-deoxycytidine (5-aza-2-DC; A3653) were purchased from Sigma-Aldrich. The HepG2 cell line was purchased from Highveld Biologicals. Cell culture consumables were obtained from Lonza Biotechnology. Western Blot reagents were purchased from Bio-Rad. All other reagents were purchased from Merck.
Cell culture and treatment
HepG2 cells (1.5 × 106) were cultured (37°C, 5% CO2) in complete culture media (CCM; Eagle’s Minimum Essentials Medium (EMEM) containing 10% foetal calf serum, 1% penicillin-streptomycin fungizone, and 1% L-glutamine), until 90% confluent. Stocks of FA (1 mg/ml) were prepared in 0.1 M PBS and the cells were incubated (37°C, 5% CO2, 24 h) with various concentrations of FA (25, 50, 104, and 150 µg/ml). These FA concentrations were obtained from literature [10] and represented 90%, 75%, 50%, and 40% cell viabilities, respectively. The 5-aza-2-DC (50 mM) stock was prepared in 100% DMSO. The concentration of 5-aza-2-DC (10 µM, 24 h) inducing DNA hypomethylation in HepG2 cells was obtained from literature [63] and used as a negative control. An untreated control (CCM only) was also prepared. Cell viability was determined using the trypan blue cell exclusion method. All results were verified by performing two independent experiments in triplicate.
DNA isolation and quantification of DNA methylation
Genomic DNA was isolated from control and FA-treated HepG2 cells. Briefly, HepG2 cells were incubated in cell lysis buffer (600 µl, 15 min, RT; 0.5 M EDTA (pH 8.0), 1 M Tris-Cl (pH 7.6), 0.1% SDS) and potassium acetate buffer (600 µl, 8 min, RT; 5 M potassium acetate, glacial acetic acid) before centrifugation (13,000×g, 5 min, 24°C). The supernatant containing genomic DNA was transferred into fresh 1.5 ml micro-centrifuge tubes and 100% isopropanol (600 µl) was added to precipitate the DNA which was recovered by centrifugation (13,000×g, 5 min, 24°C). The DNA was washed in 100% ethanol (300 µl) and centrifuged (13,000×g, 5 min, 24°C). The DNA pellets were air dried (30 min, RT), resuspended in DNA hydration buffer (40 µl; 10 mM EDTA (pH 8.0), 100 mM Tris-Cl (pH 7.4)), and heated (65°C, 15 min). DNA concentration was determined using the Nanodrop2000 spectrophotometer (Thermo-Fischer Scientific) and standardized to 100 ng/µl. DNA purity was assessed using the A260/A280 absorbance ratios.
The DNA was used to quantify global DNA methylation using the Colorimetric Methylated DNA Quantification Kit (Abcam, ab117128), as per manufacturer’s instructions. The percentage 5-methylcytosine (5-mC) content was calculated using the supplied formula (Supplementary Information) and represented as fold-change relative to the control.
Promoter methylation of miR-29b, DNMTs, and MBD2
Genomic DNA was isolated from control and FA-treated HepG2 cells using the Quick-g-DNA MiniPrep Kit (Zymo Research, D3007), as per manufacturer’s instructions. The isolated DNA was then eluted in nuclease-free water and purified using the DNA Clean and Concentrator™-5 Kit (Zymo Research, D4003), as per manufacturer’s instructions. The DNA was quantified using the Nanodrop2000 spectrophotometer and standardized to 4 ng/µl. The promoter methylation of DNMT1, DNMT3A, DNMT3B, MBD2, and miR-29b was assessed using the OneStep qMethyl Kit (Zymo Research, 5310), as per manufacturer’s instructions. Briefly, 20 ng DNA was subject to a test and reference reaction containing specific primers (Supplementary Table S2). Cycling conditions were as follows: digestion by methyl sensitive restriction enzymes (AccII, HpaII, and HpyCH4IV) (37°C, 2 h), initial denaturation (95°C, 10 min), followed by 45 cycles of denaturation (95°C, 30 s), annealing (Supplementary Table S2, 60 s), extension (72°C, 60 s), final extension (72°C, 60 s), and a hold at 4°C. The percentage methylation was calculated using the supplied formula (Supplementary Information) and represented as fold-change relative to the control.
RNA isolation and quantitative polymerase chain reaction (qPCR)
Total RNA was extracted from control and FA-treated HepG2 cells using Qiazol Reagent (Qiagen, 79306). Briefly, HepG2 cells were rinsed in 0.1 M PBS and incubated (5 min, RT) in 500 µl Qiazol and 500 µl 0.1 M PBS before extraction with a cell scraper. Cellular lysates were incubated overnight (−80°C). Thereafter, chloroform (100 µl) was added and centrifuged (12,000×g, 4°C, 15 min). The aqueous phase containing RNA was transferred to fresh 1.5 ml micro-centrifuge tubes and 100% cold isopropanol (250 µl) was added to each sample before overnight incubation (−80°C). Samples were centrifuged (12,000×g, 4°C, 20 min) and the RNA pellets were washed in 75% cold ethanol (500 µl). Finally, samples were centrifuged (7,400×g, 4°C, 15 min), RNA pellets were air dried (30 min, RT), resuspended in nuclease-free water (15 µl), and incubated (3 min, RT). The RNA was quantified using the Nanodrop2000 spectrophotometer and standardized to 1,000 ng/µl. The A260/A280 absorbance ratio was used to assess RNA purity.
The RNA was used to prepare cDNA using the miScript II RT Kit (Qiagen, 218161), as per manufacturer’s instructions. The expression of miR-29b was analyzed using the miScript SYBR Green PCR Kit (Qiagen, 218073) and specific 10X miScript primer assay [Hs_miR-29b_1, Qiagen, MS00006566], as per manufacturer’s instructions. Human RNU6 (Qiagen, MS000033740) was used as the housekeeping gene to normalize microRNA expression.
For mRNA expression, cDNA was synthesized using the iScript cDNA Synthesis Kit (Bio-Rad, 1708891), as per manufacturer’s instructions. The expression of DNMT1, DNMT3A, DNMT3B, MBD1-MBD6, Sp1, UHRF1, and USP7 was determined using the Sso Advanced™ Universal SYBR Green Supermix (Bio-Rad, 1725270), as per manufacturer’s instructions. GAPDH was used as the housekeeping gene to normalize mRNA expression. Primer sequences and annealing temperatures are listed in Supplementary Table S2. All qPCR experiments were conducted using the CFX96 Real Time PCR System (Bio-Rad) and analyzed using the Bio-Rad CFX Manager™ Software version 3.1. The comparative threshold cycle (Ct) method was used to determine relative changes in expression [64].
Protein isolation and Western blot
The protein expression of DNMT1, DNMT3A, DNMT3B, and MBD2 was determined using Western blot. Briefly, crude protein extracts were isolated from control and FA-treated HepG2 cells using cytobuster reagent (200 µl; Novagen, 71009) supplemented with protease and phosphatase inhibitors (Roche; 05892791001 and 04906837001, respectively). The protein was quantified using the bicinchoninic acid (BCA) assay, standardized to 1 mg/ml and boiled (100°C, 5 min) in a 1:1 dilution with 1X Laemmli buffer [dH2O, 0.5 M Tris-HCl (pH 6.8), glycerol, 10% SDS, 5% β-mercaptoethanol, 1% bromophenol blue]. Thereafter, the proteins were separated using sodium dodecyl sulphate polyacrylamide gel electrophoresis (10% resolving gel, 4% stacking gel; 1 h, 150 V) and transferred onto nitrocellulose membranes using the Bio-Rad Trans-Blot® Turbo Transfer System (20 V, 30 min). Following transfer, the membranes were blocked in 5% BSA in Tris buffered saline with 0.05% Tween 20 [TTBS; 150 mM NaCl, 3 mM KCl, 25 mM Tris, 0.05% Tween 20, dH2O, pH 7.5; 1 h, RT] and probed overnight (4°C) with primary antibody [DNMT1 (Cell Signalling Technology, #5032S; 1:250), DNMT3A (Cell Signalling Technology, #3598S; 1:500), DNMT3B (Santa Cruz, sc-130740; 1:250), and MBD2 (Santa Cruz, sc-271562; 1:500)]. The membranes were rinsed five times with TTBS (10 min, RT) and probed with a horse-radish peroxidase (HRP)-conjugated secondary antibody [goat anti-rabbit (Cell Signalling Technology, #7074S; 1:10,000) and goat anti-mouse (Cell Signalling Technology, #7076P2; 1:5,000); 1 h, RT]. The membranes were rinsed five times in TTBS (10 min, RT). The Clarity™ Western ECL Substrate Kit (Bio-Rad, #170-5060) was used to detect specific protein bands and the images were captured using the ChemiDoc™ XRS+ Molecular Imaging System (Bio-Rad). The membranes were then quenched in hydrogen peroxide (5%, 37°C, 30 min), rinsed once in TTBS (10 min, RT) and probed with the housekeeping protein, anti-β-actin (Sigma-Aldrich, A3854; 1:5,000; 30 min, RT) to normalize protein expression. Densitometric analysis was performed using the Bio-Rad Image Lab Software version 5.1 and the results were represented as a fold-change in band density (RBD) relative to the control.
Immuno-precipitation
Immuno-precipitation was used to determine ubiquitinated DNMT1, DNMT3A, and DNMT3B levels. Briefly, crude protein extracts were isolated from control and FA-treated HepG2 cells using 1X cell lysis buffer [500 µl; 20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 10% glycerol, 1% Triton X-100]. The protein was quantified using the BCA assay and standardized to 1.5 mg/ml. Thereafter, the protein lysates (200 µl) were incubated with primary antibody [DNMT1 (Cell Signalling Technology, #5032S); DNMT3A (Cell Signalling Technology, #3598S); and DNMT3B (Santa Cruz, sc-130740); 1:100] overnight (4°C) and the antigen-antibody complex was precipitated using protein A beads (20 µl 50% bead slurry; Cell Signalling Technology, #9863) for 1–3 h at 4°C. The immuno-precipitates were recovered by centrifugation (14,000×g, 4°C, 30 s), washed five times in 1X cell lysis buffer (500 µl), resuspended in 3X Laemmli buffer (20 µl) and boiled (100°C, 5 min). The samples were then analyzed by Western blotting using the following antibodies: primary antibody [ubiquitin (BD BioSciences, BD550944; 1:1,000), DNMT1 (Cell Signalling Technology, #5032S; 1:1,000), DNMT3A (Cell Signalling Technology, #3598S; 1:1,000), and DNMT3B (Santa Cruz, sc-130740; 1:500)] and secondary antibody [goat anti-rabbit (Cell Signalling Technology, #7074S) and goat anti-mouse (Cell Signalling Technology, #7076P2); 1:5,000]. The protein expression of ubiquitin was divided by the total protein expressed to determine the ratio of ubiquitinated protein.
Statistical analysis
GraphPad Prism version 5.0 (GraphPad Prism Software Inc.) was used to perform all statistical analyses. The one-way analysis of variance (ANOVA) with the Bonferroni multiple comparisons test was used to analyze the data. The results were expressed as the mean fold-change ± standard deviation (SD) (n = 3), unless otherwise indicated. Statistical significance was considered at p < 0.05.
Funding Statement
This work was supported by the National Research Foundation Innovation Doctoral Scholarship (Grant no.: SFH160703175722) and the College of Health Sciences (University of Kwa-Zulu Natal; Grant no.: 570869).
Author contributions
TG, SN, PN, and AC conceptualized and designed the study. TG conducted all laboratory experiments, analyzed the data, and wrote the manuscript. PN assisted in conducting laboratory experiments. SN, PN, and AC revised the manuscript. All authors have read the manuscript prior to submission.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability
All datasets generated in this study are available in Supplementary Information and from the corresponding author on reasonable request.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Bacon C, Porter J, Norred W, et al. Production of fusaric acid by Fusarium species. Appl Environ Microbiol. 1996. November;62:4039–4043. PubMed PMID: 8899996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Streit E, Schwab C, Sulyok M, et al. Multi-mycotoxin screening reveals the occurrence of 139 different secondary metabolites in feed and feed ingredients. Toxins (Basel). 2013. March;5:504–523. PubMed PMID: 23529186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chen Z, Luo Q, Wang M, et al. A rapid method with UPLC for the determination of fusaric acid in fusarium strains and commercial food and feed products. Indian J Microbiol 2016. August;57:68–74. PubMed Central PMCID: PMC5243244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Singh VK, Singh HB, Upadhyay RS.. Role of fusaric acid in the development of Fusarium wilt symptoms in tomato: physiological, biochemical and proteomic perspectives. Plant Physiol Biochem. 2017. September;118:320–332. PubMed PMID: 28683401 [DOI] [PubMed] [Google Scholar]
- [5].D’Alton A, Etherton B.. Effects of fusaric acid on tomato root hair membrane potentials and ATP levels. Plant Physiol. 1984. January;74:39–42. PubMed PMID: 16663382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Diniz S, Oliveira R. Effects of fusaric acid on Zea mays L. seedlings. Phyton Int J Exp Bot. 2009. January;78:155–160. [Google Scholar]
- [7].Pavlovkin J, Mistrik I, Prokop M. Some aspects of the phytotoxic action of fusaric acid on primary Ricinus roots. Plant Soil Environ. 2004. September;50:397–401. [Google Scholar]
- [8].Singh VK, Upadhyay RS. Fusaric acid induced cell death and changes in oxidative metabolism of Solanum lycopersicum L. Bot Stud. 2014. December;55:66–76. PubMed PMID: 28510945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sapko O, Utarbaeva AS, Makulbek S. Effect of fusaric acid on prooxidant and antioxidant properties of the potato cell suspension culture. Russian J Plant Physiol 2011. September;58:828–835. [Google Scholar]
- [10].Abdul NS, Nagiah S, Chuturgoon AA. Fusaric acid induces mitochondrial stress in human hepatocellular carcinoma (HepG2) cells. Toxicon. 2016. September;119:336–344. PubMed PMID: 27390038 [DOI] [PubMed] [Google Scholar]
- [11].Köhler K, Bentrup FW. The effect of fusaric acid upon electrical membrane properties and ATP level in photoautotrophic cell suspension cultures of Chenopodium rubrum L. Z Pflanzenphysiol. 1983. March;109:355–361. [Google Scholar]
- [12].Ghazi T, Nagiah S, Tiloke C, et al. Fusaric acid induces DNA damage and post‐translational modifications of p53 in human hepatocellular carcinoma (HepG2) cells. J Cell Biochem. 2017. November;118:3866–3874. PubMed PMID: 28387973 [DOI] [PubMed] [Google Scholar]
- [13].Stack BC Jr, Hansen JP, Ruda JM, et al. Fusaric acid: a novel agent and mechanism to treat HNSCC. Otolaryngol Head Neck Surg. 2004. July;131:54–60. PubMed PMID: 15243558 [DOI] [PubMed] [Google Scholar]
- [14].Dhani S, Nagiah S, Naidoo DB, et al. Fusaric acid immunotoxicity and MAPK activation in normal peripheral blood mononuclear cells and Thp-1 cells. Sci Rep. 2017. June;7:3051–3060. PubMed PMID: 28596589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ogata S, Inoue K, Iwata K, et al. Apoptosis induced by picolinic acid-related compounds in HL-60 cells. Biosci Biotechnol Biochem. 2001. October;65:2337–2339. PubMed PMID: 11758936 [DOI] [PubMed] [Google Scholar]
- [16].Fernandez-Pol J, Klos D, Hamilton P. Cytotoxic activity of fusaric acid on human adenocarcinoma cells in tissue culture. Anticancer Res. 1993;13(Jan):57–64. PubMed PMID: 8476229 [PubMed] [Google Scholar]
- [17].Diringer MN, Kramarcy NR, Brown JW, et al. Effect of fusaric acid on aggression, motor activity, and brain monoamines in mice. Pharmacol Biochem Behav. 1982. January;16:73–79. PubMed PMID: 6173886 [DOI] [PubMed] [Google Scholar]
- [18].Li X, Zhang Z-L, Wang HF. Fusaric acid (FA) protects heart failure induced by isoproterenol (ISP) in mice through fibrosis prevention via TGF-β1/SMADs and PI3K/AKT signaling pathways. Biomed Pharmacother. 2017. September;93:130–145. PubMed PMID: 28624424 [DOI] [PubMed] [Google Scholar]
- [19].Reddy R, Larson C, Brimer G, et al. Developmental toxic effects of fusaric acid in CD1 mice. Bull Environ Contam Toxicol. 1996. September;57:354–360. PubMed PMID: 8672059 [DOI] [PubMed] [Google Scholar]
- [20].Devaraja S, Girish KS, Santhosh MS, et al. Fusaric acid, a mycotoxin, and its influence on blood coagulation and platelet function. Blood Coagul Fibrinolysis. 2013. June;24:419–423. PubMed PMID: 23343693 [DOI] [PubMed] [Google Scholar]
- [21].Hidaka H, Nagatsu T, Takeya K, et al. Fusaric acid, a hypotensive agent produced by fungi. J Antibiot. 1969. May;22:228–230. PubMed PMID: 5811396 [DOI] [PubMed] [Google Scholar]
- [22].Terasawa F, Kameyama M. The clinical trial of a new hypotensive agent, fusaric acid (5-butylpicolinic acid): the preliminary report. Circ J. 1971. March;35:339–357. PubMed PMID: 4932992 [DOI] [PubMed] [Google Scholar]
- [23].Yin ES, Rakhmankulova M, Kucera K, et al. Fusaric acid induces a notochord malformation in zebrafish via copper chelation. Biometals. 2015. August;28:783–789. PubMed PMID: 25913293 [DOI] [PubMed] [Google Scholar]
- [24].Bacon CW, Porter JK, Norred WP. Toxic interaction of fumonisin B1 and fusaric acid measured by injection into fertile chicken egg. Mycopathologia. 1995;129(Jan):29–35. PubMed PMID: 7617015 [DOI] [PubMed] [Google Scholar]
- [25].Smith T, MacDonald E. Effect of fusaric acid on brain regional neurochemistry and vomiting behavior in swine. J Anim Sci. 1991. May;69:2044–2049. PubMed PMID: 1712354 [DOI] [PubMed] [Google Scholar]
- [26].Fairchild AS, Grimes JL, Porter JK, et al. Effects of diacetoxyscirpenol and fusaric acid on poults: individual and combined effects of dietary diacetoxyscirpenol and fusaric acid on turkey poult performance. Int J Poult Sci. 2005;4(3):350–355. [Google Scholar]
- [27].Gopalakrishnan S, Van Emburgh BO, Robertson KD. DNA methylation in development and human disease. Mutat Res. 2008. December;647:30–38. PubMed PMID: 18778722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Iraola‐Guzmán S, Estivill X, Rabionet R. DNA methylation in neurodegenerative disorders: a missing link between genome and environment? Clin Genet. 2011. July;80:1–14. PubMed PMID: 21542837 [DOI] [PubMed] [Google Scholar]
- [29].R-K L, Y-C W. Dysregulated transcriptional and post-translational control of DNA methyltransferases in cancer. Cell Biosci. 2014. August;4:46–56. PubMed PMID: 25949795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Winter J, Jung S, Keller S, et al. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009. March;11:228–234. PubMed PMID: 19255566 [DOI] [PubMed] [Google Scholar]
- [31].Park SY, Lee JH, Ha M, et al. miR-29 miRNAs activate p53 by targeting p85 alpha and CDC42. Nat Struct Mol Biol. 2009. January;16:23–29. PubMed PMID: 19079265 [DOI] [PubMed] [Google Scholar]
- [32].Wei W, He HB, Zhang WY, et al. miR-29 targets Akt3 to reduce proliferation and facilitate differentiation of myoblasts in skeletal muscle development. Cell Death Dis. 2013. June;4:e668–e678. PubMed PMID: 23764849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wang J, Esh C, H-Y C, et al. microRNA-29b prevents liver fibrosis by attenuating hepatic stellate cell activation and inducing apoptosis through targeting PI3K/AKT pathway. Oncotarget. 2015. March;6:7325–7338. PubMed PMID: 25356754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Teng Y, Zhang Y, Qu K, et al. MicroRNA-29B (mir-29b) regulates the Warburg effect in ovarian cancer by targeting AKT2 and AKT3. Oncotarget. 2015. December;6:40799–40814. PubMed PMID: 26512921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Garzon R, Liu S, Fabbri M, et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009. June;113:6411–6418. PubMed PMID: 19211935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fabbri M, Garzon R, Cimmino A, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci USA. 2007. October;104:15805–15810. PubMed PMID: 17890317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Scott A, Song J, Ewing R, et al. Regulation of protein stability of DNA methyltransferase 1 by post-translational modifications. Acta Biochim Biophys Sin. 2014. March;46:199–203. PubMed PMID: 24389641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Denis H, Ndlovu MN, Fuks F. Regulation of mammalian DNA methyltransferases: a route to new mechanisms. EMBO Rep. 2011. July;12:647–656. PubMed PMID: 21660058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Detich N, Theberge J, Szyf M. Promoter-specific activation and demethylation by MBD2/demethylase. J Biol Chem. 2002. September;277:35791–35794. PubMed PMID: 12177048 [DOI] [PubMed] [Google Scholar]
- [40].Qin H, Zhu X, Liang J, et al. MicroRNA-29b contributes to DNA hypomethylation of CD4+ T cells in systemic lupus erythematosus by indirectly targeting DNA methyltransferase 1. J Dermatol Sci. 2013. January;69:61–67. PubMed PMID: 23142053 [DOI] [PubMed] [Google Scholar]
- [41].Novakovic B, Wong NC, Sibson M, et al. DNA methylation-mediated down-regulation of DNA methylatransferase-1 (DNMT1), is coincident with, but not essential for, global hypomethylation in human placenta. J Biol Chem. 2010. March;285:9583–9593. PubMed PMID: 20071334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Naghitorabi M, Asl JM, Sadeghi HMM, et al. Quantitative evaluation of DNMT3B promoter methylation in breast cancer patients using differential high resolution melting analysis. Res Pharm Sci. 2013. July;8:167–175. PubMed PMID: 24019826 [PMC free article] [PubMed] [Google Scholar]
- [43].Peng L, Yuan Z, Ling H, et al. SIRT1 deacetylates the DNA methyltransferase 1 (DNMT1) protein and alters its activities. Mol Cell Biol. 2011. December;31:4720–4734. PubMed PMID: 21947282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Du Z, Song J, Wang Y, et al. DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Sci Signal. 2010. November;3:ra80–ra101. PubMed PMID: 21045206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Cheng J, Yang H, Fang J, et al. Molecular mechanism for USP7-mediated DNMT1 stabilization by acetylation. Nat Commun. 2015. May;6:7023–7033. PubMed PMID: 25960197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Jia Y, Li P, Fang L, et al. Negative regulation of DNMT3A de novo DNA methylation by frequently overexpressed UHRF family proteins as a mechanism for widespread DNA hypomethylation in cancer. Cell Discov. 2016. April;2:16007–16026. PubMed PMID: 27462454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Bostick M, Kim JK, Estève PO, et al. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007. September;317:1760–1764. PubMed PMID: 17673620 [DOI] [PubMed] [Google Scholar]
- [48].Yuan K, Xie K, Fox J, et al. Decreased levels of miR-224 and the passenger strand of miR-221 increase MBD2, suppressing maspin and promoting colorectal tumor growth and metastasis in mice. Gastroenterology. 2013. October;145:853–864. PubMed PMID: 23770133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Stirzaker C, Song JZ, Ng W, et al. Methyl-CpG-binding protein MBD2 plays a key role in maintenance and spread of DNA methylation at CpG islands and shores in cancer. Oncogene. 2017. March;36:1328–1338. PubMed PMID: 27593931 [DOI] [PubMed] [Google Scholar]
- [50].Fazio C, Covre A, Cutaia O, et al. Immunomodulatory properties of DNA hypomethylating agents: selecting the optimal epigenetic partner for cancer immunotherapy. Front Pharmacol. 2018. December;9:1443–1456. PubMed PMID: 30581389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Jackson M, Krassowska A, Gilbert N, et al. Severe global DNA hypomethylation blocks differentiation and induces histone hyperacetylation in embryonic stem cells. Mol Cell Biol. 2004. October;24:8862–8871. PubMed PMID: 15456861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Rodriguez J, Frigola J, Vendrell E, et al. Chromosomal instability correlates with genome-wide DNA demethylation in human primary colorectal cancers. Cancer Res. 2006. September;66:8462–9468. PubMed PMID: 16951157 [DOI] [PubMed] [Google Scholar]
- [53].Dodge JE, Okano M, Dick F, et al. Inactivation of Dnmt3b in mouse embryonic fibroblasts results in DNA hypomethylation, chromosomal instability, and spontaneous immortalization. J Biol Chem. 2005. May;280:17986–17991. PubMed PMID: 15757890 [DOI] [PubMed] [Google Scholar]
- [54].Jackson-Grusby L, Beard C, Possemato R, et al. Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat Genet. 2001. January;27:31–39. PubMed PMID: 11137995 [DOI] [PubMed] [Google Scholar]
- [55].Chuturgoon A, Phulukdaree A, Moodley D. Fumonisin B1 induces global DNA hypomethylation in HepG2 cells – an alternative mechanism of action. Toxicology. 2014. January;315:65–69. PubMed PMID: 24280379 [DOI] [PubMed] [Google Scholar]
- [56].Demirel G, Alpertunga B, Ozden S. Role of fumonisin B1 on DNA methylation changes in rat kidney and liver cells. Pharm Biol. 2015. April;53:1302–1310. PubMed PMID: 25858139 [DOI] [PubMed] [Google Scholar]
- [57].Sancak D, Ozden S. Global histone modifications in fumonisin B1 exposure in rat kidney epithelial cells. Toxicol In Vitro. 2015. October;29:1809–1815. PubMed PMID: 26208285 [DOI] [PubMed] [Google Scholar]
- [58].Chuturgoon AA, Phulukdaree A, Moodley D. Fumonisin B1 modulates expression of human cytochrome P450 1b1 in human hepatoma (Hepg2) cells by repressing Mir-27b. Toxicol Lett. 2014. May;227:50–55. PubMed PMID: 24614526 [DOI] [PubMed] [Google Scholar]
- [59].So MY, Tian Z, Phoon YS, et al. Gene expression profile and toxic effects in human bronchial epithelial cells exposed to zearalenone. PLoS One. 2014;9(May):e96404–e96422. PubMed PMID: 24788721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lan M, Han J, Pan MH, et al. Melatonin protects against defects induced by deoxynivalenol during mouse oocyte maturation. J Pineal Res. 2018. August;65:e12477–e12487. PubMed PMID: 29453798 [DOI] [PubMed] [Google Scholar]
- [61].Zhu C-C, Zhang Y, Duan X, et al. Toxic effects of HT-2 toxin on mouse oocytes and its possible mechanisms. Arch Toxicol. 2016. June;90:1495–1505. PubMed PMID: 26138683 [DOI] [PubMed] [Google Scholar]
- [62].Mamur S, Ünal F, Yılmaz S, et al. Evaluation of the cytotoxic and genotoxic effects of mycotoxin fusaric acid. Drug Chem Toxicol. 2018. September;11:1–9. PubMed PMID: 30204001. [DOI] [PubMed] [Google Scholar]
- [63].Ahn EY, Kim JS, Kim GJ, et al. RASSF1A-mediated regulation of AREG via the Hippo pathway in hepatocellular carcinoma. Mol Cancer Res. 2013. July;11:748–758. PubMed PMID: 23594797 [DOI] [PubMed] [Google Scholar]
- [64].Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods. 2001. December;25:402–408. PubMed PMID: 11846609 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets generated in this study are available in Supplementary Information and from the corresponding author on reasonable request.