

Abstract
Sleep disturbances are common in people with monogenic neurological disorders and they dramatically affect the life of individuals with the disorders and their families. The associated sleep problems are probably caused by multiple factors that have not been elucidated. Study of the underlying molecular cause, behavioral phenotypes, and reciprocal interactions in several single-gene disorders (Angelman Syndrome, Fragile X Syndrome, Rett Syndrome, and Huntington’s Disease) leads to the suggestion that sleep disruption and other symptoms may directly result from abnormal operation of circadian systems due to genetic alteration and/or conflicting environmental cues for clock entrainment. Therefore, because circadian patterns modify the symptoms of neurological disorders, treatments that modulate our daily rhythms may identify heretofore unappreciated therapies for the underlying disorders.
Introduction: single gene neurological disorders
The vast majority of neurological disorders are complex or multifactorial disorders. Those diseases include schizophrenia, depression, autism, epilepsy, Alzheimer’s Disease, Parkinson’s Disease, etc., all of which are influenced by multiple genetic and environmental factors. In contrast, single-gene neurological disorders are caused by variations/mutations in the DNA sequence of a specific gene [1]. For example, the core symptoms of Angelman Syndrome (AS), Fragile X Syndrome (FXS), Rett Syndrome (RTT), and Huntington’s Disease (HD) are caused by single-gene variants [2,3]. Moreover, environmental factors often play important roles in the initiation and development of single gene disorders [4,5]. Although single-gene neurological disorders are not very common, they are extremely valuable to investigate the genetic and pathological causes of disorders because of the simpler genetic underpinings as compared with complex genetic disorders [6●●]. Investigation of monogenic diseases can create a foundation for new preventive treatments and therapeutic drug discovery, as well as providing conceptual and technical bases for studying more complex disorders.
Interplay of sleep and circadian rhythms
Many physiological processes display day–night rhythms, including sleep–wake behavior and metabolism. These daily oscillations are regulated and coordinated by the master circadian clock in the suprachiasmatic nucleus (SCN) of the hypothalamus and by tissue-specific clocks [7]. In mammals, the expression of approximately 50% of genes are circadianly regulated at primarily posttranscriptional levels in at least one tissue [8,9]. Those rhythmic transcripts are translated into proteins and processed by posttranslational modifications, and then act functionally and rhythmically in myriad cellular processes [10,11]. Disruption of these rhythmic gene expression patterns, as in human circadian desynchrony (e.g. from shiftwork, jet-lag, and/or sleep disruption) can have profound effects on mental health [12●,13]. Improper circadian entrainment is associated with the onset of neurological disorders, and circadian disruption may interact with other susceptibility factors to precipitate disease states [14].
Circadian rhythms also help determine our sleep patterns. The body’s master SCN clock controls the production of melatonin and corticosterone, two hormones that are involved in sleep regulation [15,16]. The SCN receives environmental light/dark information via retinal-hypothalamic tracts from specialized ganglion cells in the eyes to the brain. The circadian clock is thereby enabled to anticipate, sense, and respond to light-dark changes so as to create physiological plasticity to predictable alterations in the external environment [4]. For decades, circadian rhythms have been thought to dictate sleep timing. In addition to affecting the timing of sleep, emerging evidence shows that circadian rhythms in the brain and even in peripheral tissues such as muscle [14,17●] are also able to regulate and coordinate the sleep quality and sleep duration by affecting ‘sleep homeostasis.’ Circadian rhythms and sleep therefore interact, balancing each other to fine-tune the daily cycles of behavior, metabolism and physiology in the body [18].
Ube3a imprinting and AS
AS is a neurodevelopmental disorder of imprinting characterized by mental disability, developmental delays, sleep disorders, epileptic seizures, motor difficulties, and speech impairment [19●●,20,21]. There is no specific therapy for AS and treatment for seizures usually becomes necessary. About 70% of AS patients have deletions of the maternal copy of chromosome 15 in the region of 15q11-q13 (Figure 1). The Ube3a gene within this region was identified as the genetic locus for AS [21]. Ube3a encodes a HECT-domain E3 ubiquitin ligase that adds ubiquitin to substrates, thereby targeting them for destruction in the proteasome. AS is an example of genomic imprinting that is caused by the deletion or inactivation of the maternal copy of Ube3a, while the paternal copy is imprinted and therefore silenced. It has been thought that the paternal imprinting involved in AS occurs only in the brain and is not imprinted in non-neural peripheral tissues or in glia [22,23]. However, detailed analyses in a variety of cell types using the Ube3a::YFP mouse as a tool demonstrated spatiotemporal confirmation of allele-specific Ube3a expression in neurons, astrocytes and oligodendrocytes [24–26]. Besides the allele-specific expression of Ube3a in different cell types, the three known isoforms of its transcript may play different and critical roles in the pathology of AS [27,28●●]. Therefore, improving our understanding of the developmental parameters of paternal Ube3a imprinting, including its isoform-specific silencing and cellular basis could facilitate the therapeutic treatment of AS.
Figure 1.
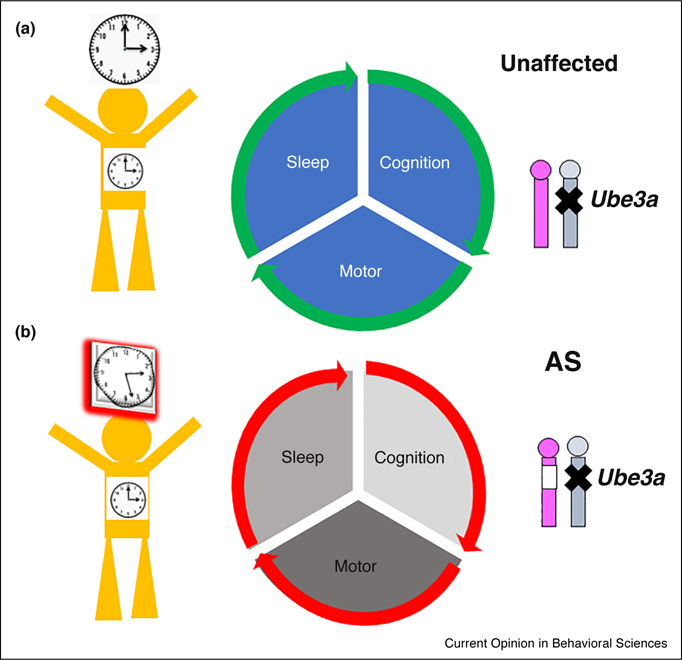
Angelman Syndrome (AS) results from deletion (open box on chromosome 15 in lower right corner of the figure) of the Ube3a gene on maternal chromosome 15 and concomitant imprinting (X) of the Ube3a gene on paternal chromosome 15. In addition to motor and cognition deficits (middle panels), AS has significant effects on sleep and the circadian clock (note desynchrony of brain vs. peripheral clocks in left panels) [19●●].
Sleep/circadian disruption in AS
Sleep syndrome in AS individuals
Sleep problems are very common in AS patients. Up to 75% of subjects with AS suffer from sleep disturbances [29,30], and these sleep disruptions are one of the syndrome’s most stressful manifestations to families with an AS member [31]. Disrupted sleep phenotypes such as short sleep duration, frequent arousal during sleep, and increased sleep onset latency are observed very often in AS patients. In addition, snoring and parasomnias are frequent. These sleep problems in AS are most likely multifactorial, and include epilepsy, medication, anxiety, GI symptoms, etc. as contributing factors [32,33]. The severity of sleep problems varies with the extent of genetic disruption; for example, patients with a deletion of the entire 15q11-q13 region often have more severe sleep traits than patients with point mutations in Ube3a [30]. This observation suggests that other (adjacent) genes contribute to the severity. For instance, abnormal GABA transmission may be a direct effect of AS in the majority of cases in which a large portion of chromosome 15 is deleted so that maternal deletion of GABA receptors (e.g. Gabrb3 in the 15q11-q13 region) could contribute to sleep anomalies in AS [34]. Interestingly, sleep problems are more severe and prevalent during childhood but improve in adolescence and adulthood, indicating developmental modulation [35]. However, AS children might naturally need less sleep than their typically developing counterparts [36,37].
In terms of sleep structure, polysomnography studies showed distinct intermittent rhythmic delta and theta waves, and interictal epileptiform discharges in AS individuals [38,39]. Recently, increased delta rhythmicity is observed as the most common EEG phenotype in AS (~84% of patients) [40●]. Furthermore, children with AS exhibit increased long-range EEG coherence both during wakefulness and in the gamma band during sleep. In addition, sleep spindles (a thalamocortical oscillation in the sigma band that occurs during NREM sleep) are less frequent and briefer in children with AS indicating an impaired function of AS in memory consolidation [41].
Animal models and AS
The human sleep studies of AS subjects are confounded by the variety of methods for sleep assessment, medications used by the subjects, and the large age range of the subject population. In order to gain insight into the mechanisms underlying the AS disorder and to obtain preclinical data to evaluate drug/therapeutic treatment to AS, animal models have been developed [23,42,43]. Because the main cause of single-gene neurological disorders is due to the alteration of one gene, animal models are suitable and relevant tools to study the behavior and physiology of the disorder, as well as paving the way towards therapeutic and/or pharmacological treatments. Mouse, zebrafish, and fruit flies are commonly used animal models by virtue of their genetic malleability, although rats are increasingly the model of choice for disorders that involve cognitive dysfunction and metabolism [44].
Traditional gene targeting and transgene technologies have provided many useful animal models for human diseases. However, some diseases with genetic bases cannot be easily mimicked with those techniques [45]. Thus, new genetic engineering tools like CRISPR (Clustered Regular Interspaced Short Palindromic Repeats), TALEN (Transcription Activator-like Effector Nucleases) and ZFN (Zinc-finger nucleases) to create small in situ lesions, and the analytic capability of sequencing and precision medicine’s ‘big data’ provide unprecedented opportunities to create animal models including nonhuman primate models that better resemble the genetic alteration of these disorders. These tools will enable basic research to be more relevant to the clinic.
Different mouse models have been able to recapitulate some but not all of the human symptoms of AS [23,42,43]. The phenotypic effects of AS models are influenced by multiple factors including knockout strategy, age, gender, diet, laboratory conditions, etc. In particular, there are pronounced differences in the AS phenotypes that depend upon mouse strain (e.g. C57BL/6 {B6}, 129, and mixed C57BL/6 and 129). For example, AS 129 mice performed poorly on contextual fear conditioning and exhibited a lower seizure threshold than do AS C57 mice [46●]. Noteworthy is the observation that gene expression and behavior in genotypically identical AS mice are influenced by whether their mothers are affected (m–/p+) or not (m+/p–) by the gene loss [47]. What’s more, phenotypic analysis of C57BL/6J versus C57BL/6N substrains showed that even these subtle changes at the genomic level could lead to changes of circadian behavior (e.g. for the effects of cocaine on circadian behavior [48].
Sleep phenotypes in AS animal models
Ube3a(m–/p+) mice have a markedly reduced capacity to accumulate sleep pressure both during their active period and in response to forced sleep deprivation [49]. The majority of AS human subjects have a large chromosomal deletion that removes multiple genes, including Atp10a and Gabrb3. Atp10a is not imprinted in mouse, and therefore it is insensitive to the AS imprinting center [50]. However, loss of Gabrb3 in the AS mouse model is sufficient to cause EEG abnormalities and disturbed rest-activity cycles that are similar to the clinical features of AS, indicating that impaired expression of the Gabrb3 gene in humans probably contributes to the sleep phenotypes of AS [51]. In agreement with this idea, an animal model with specifically GABAergic Ube3a deficiency showed an increase in cortical EEG total and delta power [52●].
Circadian disruption in AS animal models
Although it has been known for decades that sleep is disrupted in AS human subjects, only in the last few years have researchers started to investigate whether circadian rhythmicity is affected by Ube3a in AS. In a Drosophila model for AS based on a null mutation of the fly homolog of Ube3a (dube3a), circadian rhythmicity and activity/rest cycles were abnormal [53]. Consistent with that observation, knockdown of Ube3a in cultured mammalian cells lengthens the circadian period [54]. Further studies using two widely used AS mouse models (Ube3a m–/p+ and Ube3a-Gabrb3 m–/p+ [19●●]) reported lengthened behavioral period. In congruence with the previous observations of GABRB3 deficiency influencing circadian/sleep phenotypes, we found that ‘big deletion’ mice (i.e. Ube3a-Gabrb3 m–/p+ mice) appeared to have very significant circadian phenotypes [19●●]. While we also found a significant lengthening of circadian period in the ‘small deletion’ (i.e. Ube3a m–/p+ mice) AS model [19●●], a different research group using the same small deletion AS mouse reported a similar trend of lengthened circadian period, but their results did not achieve statistical significance [49] (they tested different ages and used a slightly different breeding protocol, and these differences might account for the discrepancies between their results and ours with the small deletion AS model).
Paternal imprinting involved in AS occurs mainly in neurons but is either not imprinted in non-neural peripheral tissues or is bi-allelic in non-neural brain cells such as glia [24,26]. A recent publication using small deletion Ube3a m /p+ mice reported that SCN neurons maintain persistent expression of paternal UBE3A protein, which the authors interpreted to mean there is a relaxation of Ube3a imprinting in the SCN that is not typical of most neurons [55]. However, UBE3A still appeared to express in the SCN of Ube3a-null mice (~20%), suggesting non-specific binding of their UBE3A-antibody or leaky expression of Ube3a in the knockout animal. Both observations are common limitations of immunohistological cytochemistry and conventional knockout strategies. In contrast, we found that Ube3a is indeed imprinted in the SCN of both mice and rats (unpublished data). We concluded that Ube3a expression constitutes a directmechanistic connectionbetweensymptoms of AS and the circadian mechanism [19●●], suggesting that chronotherapeutics may be effective for AS sleep disorders [56].
In order to further explore the crosstalk between UBE3A and the core clockwork, we and others pinpointed a central component of the mammalian circadian clock, BMAL1 (ARNTL in humans), as a potential ubiquitinylation target of UBE3A [19●●,54]. These results imply that the BMAL1 protein in AS-modelrodents may be under-ubiquitinylated (due to the deficiency in UBE3A levels) and therefore not degraded efficiently. The prediction, therefore is that BMAL1 abundance is increased in AS mouse neurons. If so, reduction of BMAL1 levels by experimental reduction of Bmal1 genedosagecould rescue AS phenotypes in Ube3a m–/p+ and Ube3a-Gabrb3 m–/p+ mouse models. Indeed, we find that the suppression of wheel-running activity by constant illumination in AS mice [19●●] is rescued when maternal deletion of Ube3a (Ube3a m–/p+ AS model) is combined with lower gene dosage of Bmal1 (accomplished by heterozygosity for Bmal1) (Figure 2). While we presume that this ‘rescue by reduction of Bmal1 gene dosage’ is mediated in the SCN region (since it is the master coordinator of circadianbehavior), we cannotexclude by this level of analysis the possibility that other brain regions may contribute to the behavioral rescue by Bmal1 reduction depicted in Figure 2.
Figure 2.
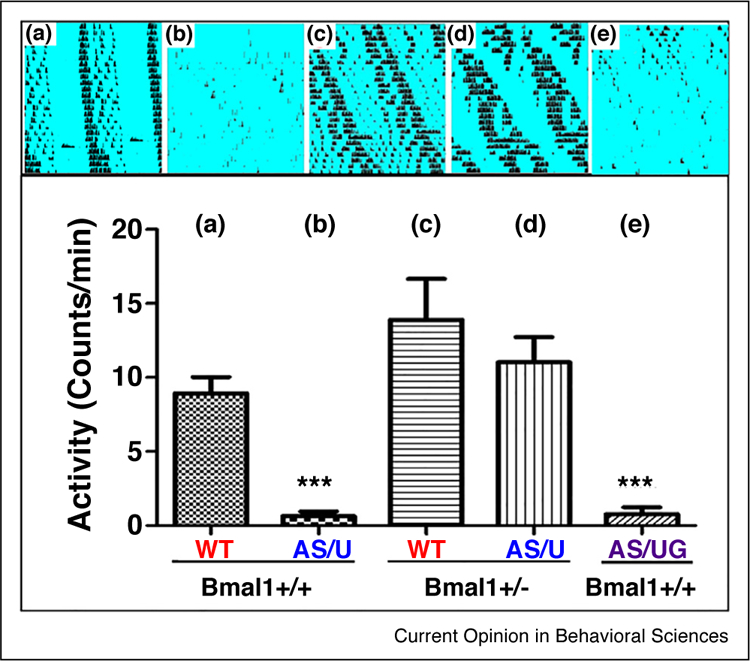
Rescue of light-suppression of wheel-running activity by reducing Bmal1 gene dosage. Constant light suppresses the wheel-running activity of AS mice [19●●], as shown here in the comparison between panel A and panels B and E. Heterozygosity for the Ube3a-target clock gene Bmal1 reduces the gene dosage of Bmal1 and rescues the level of wheel-running activity in constant illumination (blue background) as shown by comparing panel D with panel B. Upper panels are actographs of circadian locomotor rhythms, and lower panels are quantifications of the activity on running wheels. (a) WT-m +/p+, Bmal1+/+; (b) ‘small deletion’ Ube3a-m−/p+, Bmal1+/+; (c) WT-m+/p+, Bmal1+/−; (d) ‘small deletion’ Ube3a-m−/p+, Bmal1+/−; (e) ‘big deletion’ Ube3a-Gabrb3 m−/p+, Bmal1+/+ (unpublished data). WT = wild-type mice.
Sleep/circadian phenotypes in other single-gene neurological disorders
Like AS, most single-gene neurological disorders have significant sleep and circadian phenotypes. FXS is an X-linked disorder caused by a CGG repeat expansion in the 5′UTR of the Fmr1 gene, resulting in gene silencing. Sleep problems are commonly observed, and the abnormalities appeared to be regulated by circuits involved in the dysregulation of melatonin and the circadian system. In animal studies, Fmr1/Fxr2 double knockout (dKO) and Fmr1-KO/Fxr2 heterozygous animals exhibit a loss of rhythmic activity in a light:dark (LD) cycle, whereas Fmr1-KO or Fxr2-KO single knockout mice display a shorter free-running period of locomotor activity in constant darkness (DD) [57]. In addition, total sleep time in adult Fmr1 KO mice are significantly different from wild-type mice and depend on age [58]. Remarkably, TALEN-edited Mecp2 mutant monkeys (diurnal cynomolgus monkey) have more fragmented sleep, which is similar to the most prevalent sleep problem reported as night waking in RTT individuals [59,60●]. In line with those observations in primates, abnormal circadian rhythms were also reported in Mecp2 mutant mice.
The most extensively studied example of a monogenic disorder that causes perturbed sleep and circadian rhythms is Huntington’s disease (HD). HD is a neuro-degenerative disorder caused by a CAG repeat expansion in the HTT gene. Animal model studies demonstrate that changes in the molecular clock contribute greatly to the observed sleep/circadian phenotypes in a sex-dependent manner [61,62], and that when circadian or sleep function is restored pharmacologically, the rate of cognitive decline is reduced [63,64●,65].
Conclusions: application of sleep/circadian phenotypes to drug discovery and biomarker identification
Regarding neurological disorders, circadian and sleep abnormalities have generally been considered to be consequences of the associated pathological changes in the brain [66]. We suggest, however, that the circadian/sleep disruption is the cause of some of the pathologies, not just a consequence (Figure 3). With the development of new technologies to monitor circadian behaviors and sleep noninvasively (even at home in a subject’s everyday environment [67●,68]), the patterns of daily rhythmicity and sleep may provide a more robust biological marker for determining the effects of novel therapeutic strategies relevant to neurological disorders and suggest new diagnostic approaches for these diseases. Emerging data show that sleep disruption may be a direct result of circadian disruption [19●●,57,61,69], and we contend that a disrupted clock network contributes directly to the pathology of many diseases. Consequently, our sleeping and circadian patterns might be a modifiable cause of neurological disorders, and therapies that manipulate our daily rhythms may tap into heretofore unappreciated therapies. Therefore, identification of disrupted circadian networks as novel targets (as in the case of AS [19●●]) may provide mechanistic and clinical insights into how the circadian clock and sleep interacts with pathological pathways that are mediated by genetic disorders.
Figure 3.
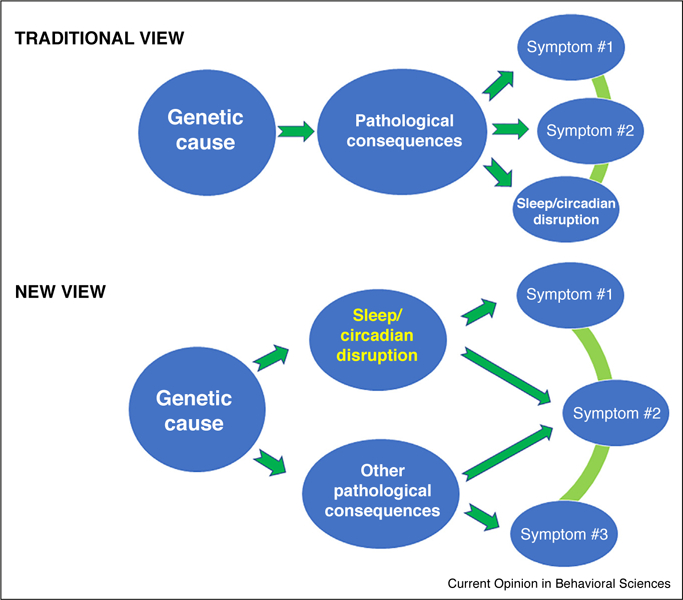
Two views of the contribution of sleep/circadian disruption in genetics-based disorders. The traditional view is that sleep/circadian disruption is an endpoint symptom of the pathological consequences of a genetic disorder. Recent studies support the new view that the sleep/circadian disruption is a direct contributor to the severity of endpoint symptoms.
Acknowledgements
We thank the USA National Institutes of Health for funding (R37 Circadian clock in muscle could have a function in sleep regulation. GM067152 from NIH/NIGMS and R01 NS104497 from NIH/NINDS). We are grateful to Dr Terry Jo Bichell (who has a child with AS) for introducing us to Angelman Syndrome and its potential sleep/circadian phenotypes.
Footnotes
Conflict of interest statement
Nothing declared.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
● of special interest
●● of outstanding interest
- 1.Beaudet AL, Meng L: Gene-targeting pharmaceuticals for single-gene disorders. Hum Mol Genet 2016, 25:R18–R26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Powell WT, LaSalle JM: Epigenetic mechanisms in diurnal cycles of metabolism and neurodevelopment. Hum Mol Genet 2015, 24:R1–R9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Santoro MR, Bray SM, Warren ST: Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu Rev Pathol 2012, 7:219–245. [DOI] [PubMed] [Google Scholar]
- 4.Johnson CH, Elliott JA, Foster R: Entrainment of circadian programs. Chronobiol Int 2003, 20:741–774. [DOI] [PubMed] [Google Scholar]
- 5.Kennedy DP, Adolphs R: The social brain in psychiatric and neurological disorders. Trends Cogn Sci 2012, 16:559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. ●●.Buiting K, Williams C, Horsthemke B: Angelman syndrome — insights into a rare neurogenetic disorder. Nat Rev Neurol 2016:584–593.An important review article regarding the Angelman syndrome and its meaning for rare neurogenetic disorder.
- 7.Mohawk JA, Green CB, Takahashi JS: Central and peripheral circadian clocks in mammals. Annu Rev Neurosci 2012, 35:445–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, Takahashi JS: Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science 2012, 338:349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mure LS, Le HD Benegiamo G, Chang MW, Rios L, Jillani N, Ngotho M, Kariuki T, Dkhissi-Benyahya O, Cooper HM, Panda S: Diurnal transcriptome atlas of a primate across major neural and peripheral tissues. Science 2018, 359. [DOI] [PMC free article] [PubMed]
- 10.Egli M, Johnson CH: A circadian clock nanomachine that runs without transcription or translation. Curr Opin Neurobiol 2013, 23:732–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dunlap JC: Molecular bases for circadian clocks. Cell 1999, 96:271–290. [DOI] [PubMed] [Google Scholar]
- 12. ●.Shi SQ, White MJ, Borsetti HM, Pendergast JS, Hida A, Ciarleglio CM, de Verteuil PA, Cadar AG, Cala C, McMahon DG, Shelton RC, Williams SM, Johnson CH: Molecular analyses of circadian gene variants reveal sex-dependent links between depression and clocks. Transl Psychiatry 2016, 6:e748.Functional analysis of polymorphisms in circadian clock genes in major depressive disorder and sex difference is explored.
- 13.Veatch OJ, Keenan BT, Gehrman PR, Malow BA, Pack AI: Pleiotropic genetic effects influencing sleep and neurological disorders. Lancet Neurol 2017, 16:158–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hastings MH, Goedert M: Circadian clocks and neurodegenerative diseases: time to aggregate? Curr Opin Neurobiol 2013, 23:880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malow BA: Sleep disorders, epilepsy, and autism. Ment Retard Dev Disabil Res Rev 2004, 10:122–125. [DOI] [PubMed] [Google Scholar]
- 16.Scheer FA, Czeisler CA: Melatonin, sleep, and circadian rhythms. Sleep Med Rev 2005, 9:5–9. [DOI] [PubMed] [Google Scholar]
- 17. ●.Ehlen JC, Brager AJ, Baggs J, Pinckney L, Gray CL, DeBruyne JP, Esser KA, Takahashi JS, Paul KN: Bmal1 function in skeletal muscle regulates sleep. Elife 2017, 6:e26557.Circadian clock in muscle could have a function in sleep regulation.
- 18.Hurley JM, Loros JJ, Dunlap JC: Circadian oscillators: around the transcription-translation feedback loop and on to output. Trends Biochem Sci 2016, 41:834–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. ●●.Shi SQ, Bichell TJ, Ihrie RA, Johnson CH: Ube3a imprinting impairs circadian robustness in Angelman syndrome models. Curr Biol 2015, 25:537–545.The conclusions of this review are largely based on the findings from this paper from our laboratory.
- 20.Tan WH, Bird LM, Thibert RL, Williams CA: If not Angelman, what is it? A review of Angelman-like syndromes. Am J Med Genet A 2014, 164A:975–992. [DOI] [PubMed] [Google Scholar]
- 21.Jiang Y, Lev-Lehman E, Bressler J, Tsai TF, Beaudet AL: Genetics of Angelman syndrome. Am J Hum Genet 1999, 65:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Albrecht U, Sutcliffe JS, Cattanach BM, Beechey CV, Armstrong D, Eichele G, Beaudet AL: Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat Genet 1997, 17:75–78. [DOI] [PubMed] [Google Scholar]
- 23.Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, Sweatt JD, Beaudet AL: Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron 1998, 21:799–811. [DOI] [PubMed] [Google Scholar]
- 24.Dindot SV, Antalffy BA, Bhattacharjee MB, Beaudet AL: The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum Mol Genet 2008, 17:111–118. [DOI] [PubMed] [Google Scholar]
- 25.Gustin RM, Bichell TJ, Bubser M, Daily J, Filonova I, Mrelashvili D, Deutch AY, Colbran RJ, Weeber EJ, Haas KF: Tissue-specific variation of Ube3a protein expression in rodents and in a mouse model of Angelman syndrome. Neurobiol Dis 2010, 39:283–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Judson MC, Sosa-Pagan JO, Del Cid WA, Han JE, Philpot BD: Allelic specificity of Ube3a expression in the mouse brain during postnatal development. J Comp Neurol 2014, 522:1874–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamamoto Y, Huibregtse JM, Howley PM: The human E6-AP gene (UBE3A) encodes three potential protein isoforms generated by differential splicing. Genomics 1997, 41:263–266. [DOI] [PubMed] [Google Scholar]
- 28. ●●.Copping NA, Christian SGB, Ritter DJ, Islam MS, Buscher N, Zolkowska D, Pride MC, Berg EL, LaSalle JM, Ellegood J, Lerch JP, Reiter LT, Silverman JL, Dindot SV: Neuronal overexpression of Ube3a isoform 2 causes behavioral impairments and neuroanatomical pathology relevant to 15q.2-q13.3 duplication syndrome. Hum Mol Genet 2017, 26:3995–4010.Isoforms of Ube3a gene play important roles in regulation of neuronal activity and behaviors.
- 29.Smith A, Wiles C, Haan E, McGill J, Wallace G, Dixon J, Selby R, Colley A, Marks R, Trent RJ: Clinical features in 27 patients with Angelman syndrome resulting from DNA deletion. J Med Genet 1996, 33:107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pelc K, Cheron G, Boyd SG, Dan B: Are there distinctive sleep problems in Angelman syndrome? Sleep Med 2008:434–441. [DOI] [PubMed]
- 31.Goldman SE, Richdale AL, Clemons T, Malow BA: Parental sleep concerns in autism spectrum disorders: variations from childhood to adolescence. J Autism Dev Disord 2012, 42:531–538. [DOI] [PubMed] [Google Scholar]
- 32.Allen AM, Coon DW, Uriri-Glover J, Grando V: Factors associated with sleep disturbance among older adults in inpatient rehabilitation facilities. Rehabil Nurs 2013, 38:221–230. [DOI] [PubMed] [Google Scholar]
- 33.Larson AM, Ryther RC, Jennesson M, Geffrey AL, Bruno PL, Anagnos CJ, Shoeb AH, Thibert RL, Thiele EA: Impact of pediatric epilepsy on sleep patterns and behaviors in children and parents. Epilepsia 2012, 53:1162–1169. [DOI] [PubMed] [Google Scholar]
- 34.Lalande M, Minassian BA, DeLorey TM, Olsen RW: Parental imprinting and Angelman syndrome. Adv Neurol 1999, 79:421–429. [PubMed] [Google Scholar]
- 35.Sueri C, Ferlazzo E, Elia M, Bonanni P, Randazzo G, Gasparini S, D’Agostino T, Sapone AR, Ascoli M, Bellavia MA, Cianci V, Gambardella A, Labate A, Aguglia U: Epilepsy and sleep disorders improve in adolescents and adults with Angelman syndrome: a multicenter study on 46 patients. Epilepsy Behav 2017, 75:225–229. [DOI] [PubMed] [Google Scholar]
- 36.Zhdanova IV, Wurtman RJ, Wagstaff J: Effects of a low dose of melatonin on sleep in children with Angelman syndrome. J Pediatr Endocrinol Metab 1999, 12:57–67. [DOI] [PubMed] [Google Scholar]
- 37.Barry RJ, Leitner RP, Clarke AR, Einfeld SL: Behavioral aspects of Angelman syndrome: a case control study. Am J Med Genet A 2005, 132A:8–12. [DOI] [PubMed] [Google Scholar]
- 38.Miano S, Bruni O, Leuzzi V, Elia M, Verrillo E, Ferri R: Sleep polygraphy in Angelman syndrome. Clin Neurophysiol 2004, 115:938–945. [DOI] [PubMed] [Google Scholar]
- 39.Vendrame M, Loddenkemper T, Zarowski M, Gregas M, Shuhaiber H, Sarco DP, Morales A, Nespeca M, Sharpe C, Haas K, Barnes G, Glaze D, Kothare SV: Analysis of EEG patterns and genotypes in patients with Angelman syndrome. Epilepsy Behav 2012, 23:261–265. [DOI] [PubMed] [Google Scholar]
- 40. ●.Sidorov MS, Deck GM, Dolatshahi M, Thibert RL, Bird LM, Chu CJ, Philpot BD: Delta rhythmicity is a reliable EEG biomarker in Angelman syndrome: a parallel mouse and human analysis. J Neurodev Disord 2017, 9:17.Sleep EEG could be used as a biological marker for Angelman syndrome drug discovery.
- 41.den Bakker H, Sidorov MS, Fan Z, Lee DJ, Bird LM, Chu CJ, Philpot BD: Abnormal coherence and sleep composition in children with Angelman syndrome: a retrospective EEG study. Mol Autism 2018, 9:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang YH, Pan Y, Zhu L, Landa L, Yoo J, Spencer C, Lorenzo I, Brilliant M, Noebels J, Beaudet AL: Altered ultrasonic vocalization and impaired learning and memory in Angelman syndrome mouse model with a large maternal deletion from Ube3a to Gabrb3. PLoS One 2010, 5:e12278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Silva-Santos S, van Woerden GM, Bruinsma CF, Mientjes E, Jolfaei MA, Distel B, Kushner SA, Elgersma Y: Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. J Clin Investig 2015, 125:2069–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ellenbroek B, Youn J: Rodent models in neuroscience research: is it a rat race? Dis Model Mech 2016, 9:1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jucker M: The benefits and limitations of animal models for translational research in neurodegenerative diseases. Nat Med 2010, 16:1210–1214. [DOI] [PubMed] [Google Scholar]
- 46. ●.Born HA, Dao AT, Levine AT, Lee WL, Mehta NM, Mehra S, Weeber EJ, Anderson AE: Strain-dependence of the Angelman Syndrome phenotypes in Ube3a maternal deficiency mice. Sci Rep 2017, 7:8451.Different strains of mice have profound effects on behavior in AS animal models.
- 47.Grier MD, Carson RP, Lagrange AH: Of mothers and myelin: aberrant myelination phenotypes in mouse model of Angelman syndrome are dependent on maternal and dietary influences. Behav Brain Res 2015, 291:260–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kumar V, Kim K, Joseph C, Kourrich S, Yoo SH, Huang HC, Vitaterna MH, de Villena FP, Churchill G, Bonci A, Takahashi JS: C57BL/6N mutation in cytoplasmic FMRP interacting protein 2 regulates cocaine response. Science 2013, 342:1508–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ehlen JC, Jones KA, Pinckney L, Gray CL, Burette S, Weinberg RJ, Evans JA, Brager AJ, Zylka MJ, Paul KN, Philpot BD, DeBruyne JP: Maternal Ube3a loss disrupts sleep homeostasis but leaves circadian rhythmicity largely intact. J Neurosci 2015, 35:13587–13598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DuBose AJ, Johnstone KA, Smith EY, Hallett RA, Resnick JL: Atp10a, a gene adjacent to the PWS/AS gene cluster, is not imprinted in mouse and is insensitive to the PWS-IC. Neurogenetics 2010, 11:145–151. [DOI] [PubMed] [Google Scholar]
- 51.DeLorey TM, Handforth A, Anagnostaras SG, Homanics GE, Minassian BA, Asatourian A, Fanselow MS, Delgado-Escueta A, Ellison GD, Olsen RW: Mice lacking the beta3 subunit of the GABAA receptor have the epilepsy phenotype and many of the behavioral characteristics of Angelman syndrome. J Neurosci 1998, 18:8505–8514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. ●.Judson MC, Wallace ML, Sidorov MS, Burette AC, Gu B, van Woerden GM, King IF, Han JE, Zylka MJ, Elgersma Y, Weinberg RJ, Philpot BD: GABAergic neuron-specific loss of Ube3a causes Angelman syndrome-like EEG abnormalities and enhances seizure susceptibility. Neuron 2016, 90:56–69.Ube3a in GABAergic cells are critical for normal EEG and regulation of neuronal circuits.
- 53.Wu Y, Bolduc FV, Bell K, Tully T, Fang Y, Sehgal A, Fischer JA: A Drosophila model for Angelman syndrome. Proc Natl Acad Sci U S A 2008, 105:12399–12404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gossan NC, Zhang F, Guo B, Jin D, Yoshitane H, Yao A, Glossop N, Zhang YQ, Fukada Y, Meng QJ: The E3 ubiquitin ligase UBE3A is an integral component of the molecular circadian clock through regulating the BMAL1 transcription factor. Nucleic Acids Res 2014, 42:5765–5775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones KA, Han JE, DeBruyne JP, Philpot B: Persistent neuronal Ube3a expression in the suprachiasmatic nucleus of Angelman syndrome model mice. Sci Rep 2016, 6:28238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schroeder AM, Colwell CS: How to fix a broken clock. Trends Pharmacol Sci 2013, 34:605–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang J, Fang Z, Jud C, Vansteensel MJ, Kaasik K, Lee CC, Albrecht U, Tamanini F, Meijer JH, Oostra BA, Nelson DL: Fragile X-related proteins regulate mammalian circadian behavioral rhythms. Am J Hum Genet 2008, 83:43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saré RM, Harkless L, Levine M, Torossian A, Sheeler CA, Smith CB: Deficient sleep in mouse models of fragile X syndrome. Front Mol Neurosci 2017, 10:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boban S, Wong K, Epstein A, Anderson B, Murphy N, Downs J, Leonard H: Determinants of sleep disturbances in Rett syndrome: novel findings in relation to genotype. Am J Med Genet A 2016, 170:2292–2300. [DOI] [PubMed] [Google Scholar]
- 60. ●.Chen Y, Yu J, Niu Y, Qin D, Liu H, Li G, Hu Y, Wang J, Lu Y, Kang Y, Jiang Y, Wu K, Li S, Wei J, He J, Wang J, Liu X, Luo Y, Si C, Bai R, Zhang K, Liu J, Huang S, Chen Z, Wang S, Chen X, Bao X, Zhang Q, Li F, Geng R, Liang A, Shen D, Jiang T, Hu X, Ma Y, Ji W, Sun YE: Modeling Rett syndrome using TALEN-edited MECP2 mutant cynomolgus monkeys. Cell 2017, 169:945–955.Abnormal circadian/sleep patterns were observed in MECP2 mutant cynomolgus monkeys (a novel nonhuman primate model for RTT using TALEN gene editing technology).
- 61.Morton AJ, Wood NI, Hastings MH, Hurelbrink C, Barker RA, Maywood ES: Disintegration of the sleep–wake cycle and circadian timing in Huntington’s disease. J Neurosci 2005, 25:157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kuljis DA, Gad L, Loh DH, MacDowell Kaswan Z, Hitchcock ON, Ghiani CA, Colwell CS: Sex differences in circadian dysfunction in the BACHD mouse model of Huntington’s disease. PLOS ONE 2016, 11:e0147583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pallier PN, Morton AJ: Management of sleep/wake cycles improves cognitive function in a transgenic mouse model of Huntington’s disease. Brain Res 2009, 1279:90–98. [DOI] [PubMed] [Google Scholar]
- 64. ●.Wang HB, Loh DH, Whittaker DS, Cutler T, Howland D, Colwell CS: Time-restricted feeding improves circadian dysfunction as well as motor symptoms in the Q175 mouse model of Huntington’s disease. eNeuro 2018, 5.Restore circadian behavior is able to rescue the abnormality of motor syndromes in HD mouse model.
- 65.Maywood ES, Fraenkel E, McAllister CJ, Wood N, Reddy AB, Hastings MH, Morton AJ: Disruption of peripheral circadian timekeeping in a mouse model of Huntington’s disease and its restoration by temporally scheduled feeding. J Neurosci 2010, 30:10199–10204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Trickett J, Heald M, Oliver C, Richards C: A cross-syndrome cohort comparison of sleep disturbance in children with Smith–Magenis syndrome, Angelman syndrome, autism spectrum disorder and tuberous sclerosis complex. J Neurodev Disord 2018, 10:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. ●.Han S, Kim J, Won SM, Ma Y, Kang D, Xie Z, Lee KT, Chung HU, Banks A, Min S, Heo SY, Davies CR, Lee JW, Lee CH, Kim BH, Li K, Zhou Y, Wei C, Feng X, Huang Y, Rogers JA: Battery-free, wireless sensors for full-body pressure and temperature mapping. Sci Transl Med 2018, 10.A new technology is discovered to monitor the circadian rhythms of temperature noninvasively in many different parts of the body simultaneously.
- 68.Jackson CL, Patel SR, Jackson WB 2nd, Lutsey PL, Redline S: Agreement between self-reported and objectively measured sleep duration among white, black, Hispanic, and Chinese adults in the United States: Multi-Ethnic Study of Atherosclerosis. Sleep 2018, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cutler TS, Park S, Loh DH, Jordan MC, Yokota T, Roos KP, Ghiani CA, Colwell CS: Neurocardiovascular deficits in the Q175 mouse model of Huntington’s disease. Physiol Rep 2017, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]