

Abstract
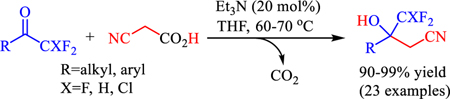
An efficient organocatalytic method for the synthesis of difluoromethyl and trifluoromethyl substituted β-hydroxynitriles is introduced. The decarboxylative cyanomethylation of fluorinated ketones with readily available cyanoacetic acid gives a variety of tertiary alcohols in high yields and without concomitant water elimination. The reaction occurs in the presence of catalytic amounts of triethylamine, can be upscaled and applied to chlorofluoromethyl ketones and difluoromethyl ketimines.
Keywords: Cyanomethylation, Organocatalysis, Organofluorines, β-hydroxynitriles, Cyanoacetic acid
Graphical Abstract
In stark contrast to the wealth of reports on the Henry reaction and other variations of the aldol reaction, relatively few methods that accomplish efficient cyanomethylation of carbonyl substrates exist. The formation of the synthetically versatile β-hydroxynitrile motif often requires large excess of acetonitrile and is in some cases accompanied by the loss of the hydroxy group due to uncontrolled elimination toward unsaturated nitrile derivatives. Substantial progress has been made with regard to the cyanomethylation of aldehydes and imines by the introduction of Me3SiCH2CN or other α-cyano carbananion precursors[1] and through the development of mild transition metal catalyzed procedures that address the challenging low acidity of acetonitrile (pKa 31.3 in DMSO).[2] In addition, decarboxylative cyanomethylation and superbase catalyzed acetonitrile additions have emerged as a practical alternative.[3,4]
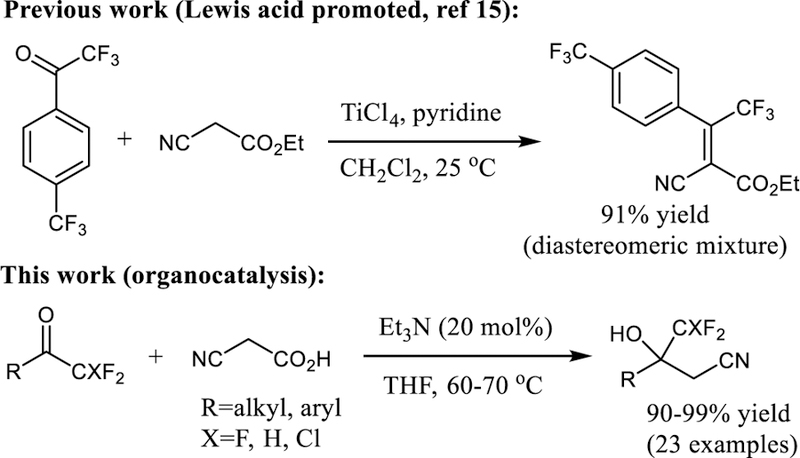
The unique chemical and pharmacological properties of fluorinated compounds continues to receive considerable attention from synthetic and medicinal chemists.[5] The vital role of the C-F moiety in today’s drug design and discovery programs has motivated the introduction of a large variety of organofluorines that either show therapeutic potential in clinical trials or are already in use.[6] Synthetic methods that accomplish C-C bond formation with trifluoromethyl or difluoromethyl ketones or derivatives thereof have become particularly important. The trifluoromethyl group is a key structural motif in several anticonvulsants,[7] metalloproteinase inhibitors,[8] CJ-17,493,[9] a Neurokinin 1 receptor antagonist, ZK (+)-216348,[10] a potent glucocorticoid receptor agonist, and HIV drugs such as Efavirenz.[11] Examples of therapeutics exhibiting a difluoromethylene or difluoromethyl group are the HIV-1 protease inhibitor A-79285[12] and Eflornithine,[13] an inhibitor of ornithine decarboxylase used for the treatment of facial hirsutism and African sleeping sickness. In recent years, we have developed an interest in catalytic nucleophilic addition reactions with trifluoromethyl ketone substrates.[14] We now report the first organocatalytic decarboxylative cyanomethylation of trifluoromethyl and difluoromethyl ketones. This method is applicable to aromatic and aliphatic substrates, operationally simple and it avoids the previously prevailing problem with concurrent water elimination (Scheme 1).[15]
Scheme 1.
Decarboxylative cyanomethylation of halogenated ketones.
We began our search for a practical method for the synthesis of trifluoromethylated β-hydroxynitriles by screening the reaction of 2,2,2-trifluoroacetophenone, 1a, and cyanoacetic acid, 2, in the presence of various organic and inorganic bases. We found that 1a is converted to the desired 4,4,4-trifluoro-3-hydroxy-3-phenylbutanenitrile, 3a, in 49% yield at room temperature after 42 hours when three equivalents of 2 and 50 mol% of DBU are employed in THF (Table 1, entry 1). Importantly, we did not observe formation of by-products under these conditions. The prospect of mild cyanomethylation of 1a without subsequent dehydration encouraged us to test several amines, DMAP, 2-tert-butyl-1,1,3,3-tetramethylguanidine (Barton’s base), K2CO3 and Cs2CO3. The results show that similar yields can be obtained with triethylamine and Barton’s base (Table 1, entries 2–9). When the reaction was carried out in the absence of base no product was formed (Table 1, entry 10).
Table 1.
Optimization of the cyanomethylation of 2,2,2-trifluoroacetophenone.[a]
 | |||||
|---|---|---|---|---|---|
| Entry | Base (mol%) | Solvent | Time (h) | Temp (°C) | Yield (%)[b] |
| 1 | DBU (50) | THF | 42 | 25 | 49 |
| 2 | Et3N (50) | THF | 42 | 25 | 45 |
| 3 | DIPEA (50) | THF | 42 | 25 | 29 |
| 4 | DABCO (50) | THF | 42 | 25 | 13 |
| 5 | TMEDA (50) | THF | 42 | 25 | 23 |
| 6 | DMAP (50) | THF | 42 | 25 | 36 |
| 7 | Barton’s base (50) | THF | 42 | 25 | 42 |
| 8 | K2CO3 (50) | THF | 42 | 25 | <5 |
| 9 | Cs2CO3 (50) | THF | 42 | 25 | 9 |
| 10 | None | THF | 42 | 25 | nr |
| 11 | Et3N (50) | CH2Cl2 | 48 | 25 | <5 |
| 12 | Et3N (50) | 1,4-dioxane | 48 | 25 | 27 |
| 13 | Et3N (50) | toluene | 48 | 25 | <5 |
| 14 | Et3N (50) | CH3CN | 48 | 25 | 8 |
| 15 | Et3N (50) | CH3OH | 48 | 25 | <5 |
| 16 | Et3N (50) | H2O | 48 | 25 | nr |
| 17 | Et3N (10) | neat, MW | 0.75 | 100 | 89 |
| 18 | Et3N (50) | THF | 16 | 60 | 99 |
| 19 | Et3N (20) | THF | 21 | 60 | 98[d] |
| 20[c] | Et3N (20) | THF | 24 | 60 | 98[d] |
| 21[c] | Et3N (10) | THF | 36 | 60 | 91 |
Conditions: 0.3 mmol of 1a, 0.9 mmol of 2, 1 mL of solvent.
By NMR analysis.
Two equivalents of 2 were used. The reaction is slow and takes 3 days with 1.5 equivalents of 2.
Isolated yields.
We then continued with the optimization of solvent, temperature and loading of triethylamine which was chosen because it is commonly present in synthetic laboratories and less expensive than DBU and Barton’s base (see Table 1 and SI). The use of dichloromethane, dioxane, toluene, acetonitrile, methanol and water as solvent did not improve the results. High yields of 3a, albeit with concomitant formation of by-products, were achieved under solvent-free microwave conditions using 10 mol% of Et3N at 100 °C (entry 17). Because milder conditions were considered more promising with regard to a procedure having a wide functional group tolerance we decided to further investigate ambient temperatures using THF as solvent. Finally, we were able to produce 3a in 98% yield after 21 hours using two equivalents of 2 and 20 mol% of Et3N in THF (entry 20).
With the optimized reaction conditions in hand, we continued with exploring the substrate scope and organocatalytic decarboxylative cyanomethylations were carried out with a series of trifluoromethyl ketones (Scheme 2). The reactions with aromatic trifluoromethyl ketones 1a–1c proceeded quantitatively and gave the benzyl alcohols 3a–3c in 97–98% yield. Halogen substituents in ortho or para positions of the trifluoroacetophenones 1d–1g were well tolerated and we isolated 3d–3g in 93–99% yield. The reaction is also applicable to the trifluoroacetophenones 1h–1l carrying a wide range of electron-withdrawing or electron-donating groups. The corresponding tertiary alcohols 3h–3l were obtained in 93–99% yield and side reactions, for example at the ester group, were not observed. Importantly, heteroaryl and aliphatic trifluoromethyl ketones can be used. 4,4,4-Trifluoro-3-hydroxy-3-(thiophen-2-yl)butanenitrile, 3m, was produced with excellent yield and the enolizable trifluoromethyl ketone 1n underwent decarboxylative cyanomethylation to the aliphatic tertiary alcohol 3n in 92% yield. To further demonstrate the general utility of the triethylamine catalyzed decarboxylative cyanomethylation we performed the reaction between 1a and 2 on a gram scale using otherwise essentially the same reaction conditions as described in Scheme 2. In this case, the β-hydroxynitrile 3a was obtained in 93% yield after 24 hours.
Scheme 2.
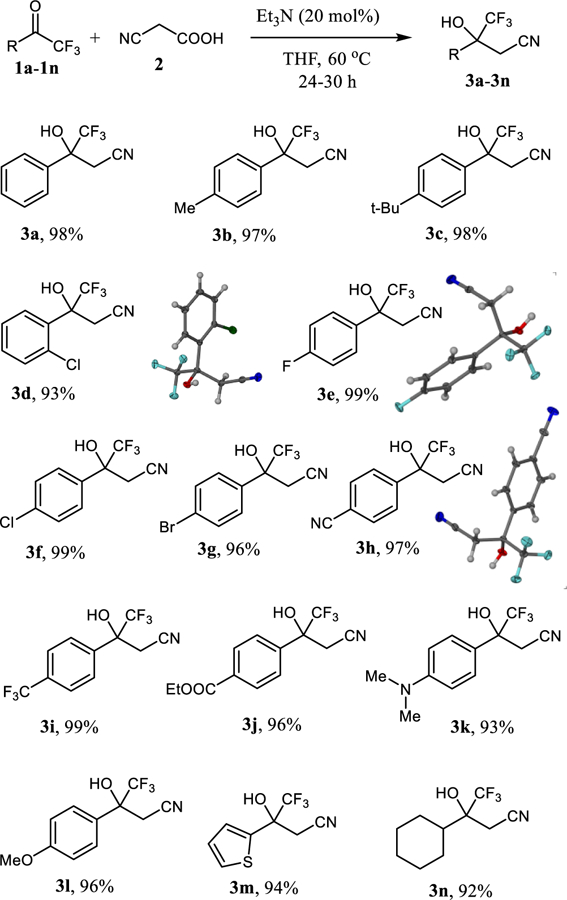
Substrate scope of the cyanomethylation of trifluorome-thyl ketones. Reaction conditions: All reactions were performed with trifluoromethyl ketone 1 (0.3 mmol), cyanoacetic acid 2 (0.6 mmol) and triethylamine (0.06 mmol) in 1 mL of THF at 60 °C. See SI for details.
We then extended our decarboxylative cyanomethylation method to difluormethyl substrates. First, a series of difluoromethyl ketones was synthesized by following previously reported literature protocols.[16] After minor changes of the reaction procedure described above, we were able to prepare the β-difluoromethyl-β-hydroxynitriles 5a–5h (Scheme 3). The cyanomethylation of the phenyl, tolyl, and 2-napthyl ketones 4a–4c gave 5a–5c in 92–99% yield. Again, other functional groups are well tolerated and we obtained the 2-nitro, 4-fluoro and 6-chloro derivatives 5d–5f in 92–99% yield. Excellent results were also achieved with heteroaromatic difluoromethyl ketones and 5g and 5h carrying a 2-furyl and 2-thienyl ring, respectively, were formed in 90–94% yield.
Scheme 3.
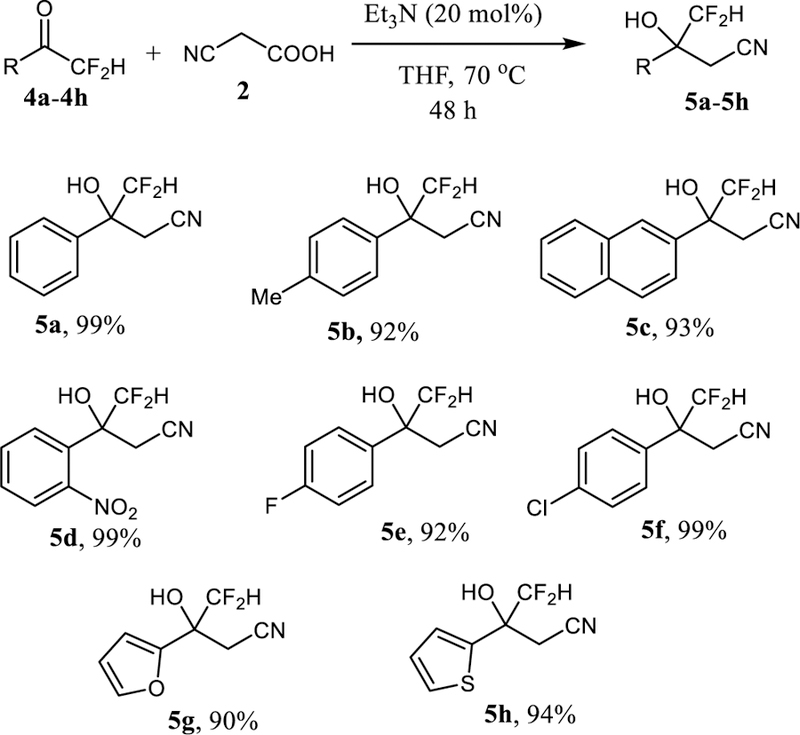
Decarboxylative cyanomethylation of difluoromethyl ketones.
Finally, we employed chlorodifluoromethyl ketone 6 in our reaction protocol (Scheme 4). As expected, the cyanomethylated alcohol 7 was produced in high yield, which proves that other halogens on the alkyl side chain are tolerated. Alternatively, we attempted to prepare 7 by careful addition of one equivalent of LiCH2CN, which was prepared by deprotonation of acetonitrile with n-BuLi at −78 °C, to ketone 6. Under these conditions, 7 was obtained in only 74% yield due to formation of byproducts.[17] We were pleased to find that our protocol is compatible with the use of activated imines. The diastereoselective Mannich reaction with enantiomerically pure tert-butylsulfinyl imide 8 gave 9 with 82% yield and 89:11 diastereoselectivity.[18] If desired, ethyl malonic acid, 10, can be employed in the same protocol to produce 11 in high yields.
Scheme 4.
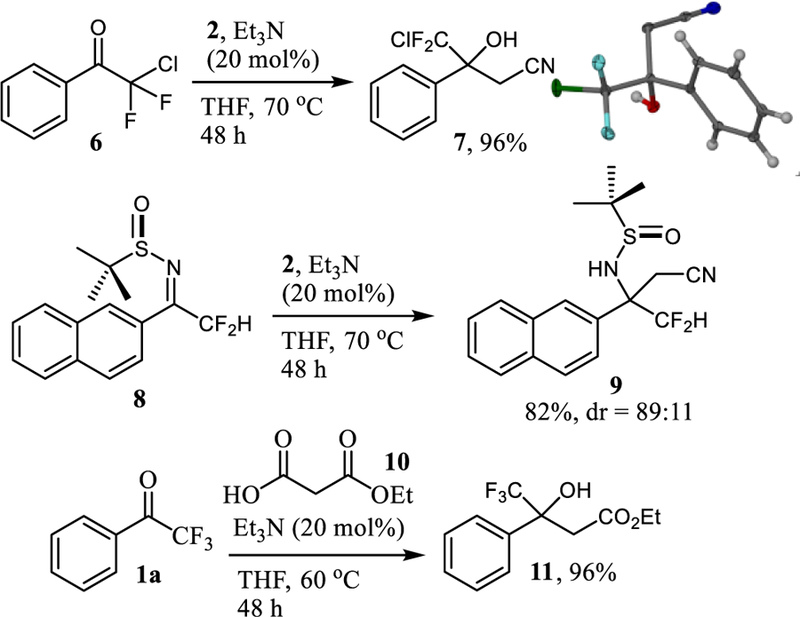
Decarboxylative cyanomethylation of 6 and 8, and conversion of 1a to the ethyl ester 11.
A plausible mechanism of the decarboxylative cyanomethylation is presented in Scheme 5. Initial deprotonation of cyanoacetic acid generates dianionic intermediate I. Nucleophilic addition to a halogenated ketone then forms intermediate II. The carbon-carbon bond formation step is then followed by decarboxylation which affords the β-hydroxy nitrile product and regenerates the amine catalyst. This reaction course is in agreement with literature reports on decarboxylative additions with malonic acids.[19]
Scheme 5.
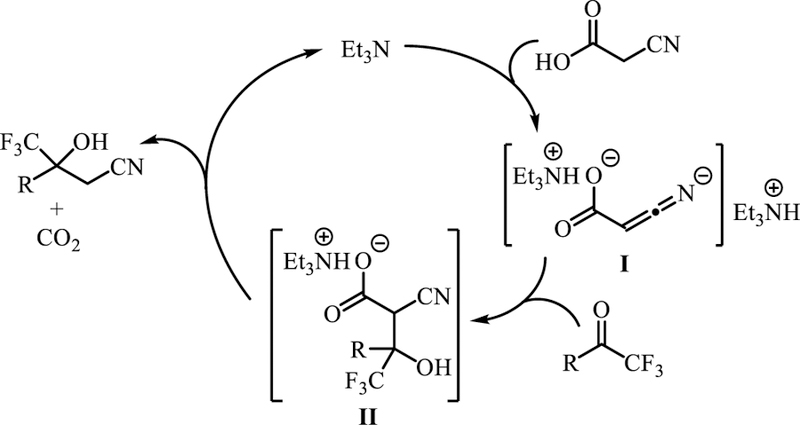
Proposed mechanistic pathway of the organocatalytic decarboxylative cyanomethylation.
In conclusion, we have introduced a practical organocatalytic method for the synthesis of difluoromethyl and trifluoromethyl substituted β-hydroxynitriles. The decarboxylative cyanomethylation of fluorinated ketones with cyanoacetic acid occurs under mild conditions in the presence of 20 mol% of inexpensive triethylamine and affords a wide variety of multifunctional tertiary alcohols in 90–99% yield without concomitant water elimination. The general reaction protocol was successfully upscaled and extended to an asymmetric Mannich reaction with a tert-butylsulfinyl ketimine derivative.
Experimental Section
Representative Procedure for the Organocatalytic Cyanomethylation of Trifluoromethyl Ketone.
To a solution of 2,2,2-trifluoro-1-phenylethan-1-one (52 mg, 0.3 mmol) and cyanoacetic acid (51 mg, 0.6 mmol) in THF (1.0 mL) was added triethylamine (20 mol%). The resulting mixture was stirred at 60 °C for 24 hours and the reaction was monitored by 19F NMR for the disappearance of the trifluoromethyl ketone. The crude product was purified by flash chromatography on silica gel using hexanes-ethyl acetate as mobile phase to give 4,4,4-Trifluoro-3-hydroxy-3-phenylbutanenitrile (3a) as a colorless solid in 98% yield (63 mg, 0.294 mmol). Mp. 75.1–75.9 °C; Rf = 0.2 (hexanes/EtOAc, 8:2); 1H NMR (400 MHz, Chloroform-d) δ = 7.61 – 7.52 (m, 2H), 7.52 – 7.42 (m, 3H), 3.25 – 3.12 (m, 2H), 3.12 (s, 1H); 13C NMR (100 MHz, Chloroform-d) δ = 134.3, 129.8, 128.9, 125.9 (q, JC-F = 1.3 Hz), 124.1 (q, JC-F = 286.0 Hz), 114.8, 75.2 (q, JC-F = 29.7 Hz), 26.9 (q, JC-F = 1.7 Hz); 19F NMR (376 MHz, Chloroform-d) δ = −79.5; HRMS (ESI-TOF) m/z: [M]+ calcd for C10H8F3NO 215.0558, found 215.0553.
Supplementary Material
Acknowledgements
We gratefully acknowledge financial support from NIH (GM106260).
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.201######.
References
- [1].a) Palomo C, Aizpurua JM, Lopez MC, Lecea B, J. Chem. Soc. Perkin Trans 1 1989, 1692–1694; [Google Scholar]; b) Suto Y, Kumagai N, Matsunaga S, Kanai M, Shibasaki M, Org. Lett 2003, 5, 3147–3150; [DOI] [PubMed] [Google Scholar]; c) Kawano Y, Kaneko N, Mukaiyama T, Chem. Lett 2005, 34, 1508–1509; [Google Scholar]; d) Denmark SE, Wilson TW, Burk MT, Heemstra JR Jr, J. Am. Chem. Soc 2007, 129, 14864–14865; [DOI] [PubMed] [Google Scholar]; e) Wadhwa K, Verkade JG, J. Org. Chem 2009, 74, 5683–5686; [DOI] [PubMed] [Google Scholar]; f) Fan Y-C, Du G-F, Sun W-F, Kang W, He L, Tetrahedron Lett 2012, 53, 2231–2233; [Google Scholar]; g) Kondo M, Nishi T, Hatanaka T, Funahashi Y, Nakamura S, Angew. Chem. Int. Ed 2015, 54, 8198–8202; [DOI] [PubMed] [Google Scholar]; h) Kondo M, Kobayashi N, Hatanaka T, Funahashi Y, Nakamura S, Chem. Eur. J 2015, 21, 9066–9070; [DOI] [PubMed] [Google Scholar]; i) Kondo M, Saito H, Nakamura S, Chem. Commun 2017, 53, 6776–6779. [DOI] [PubMed] [Google Scholar]
- [2].a) Kumagai N, Matsunaga S, Shibasaki M, J. Am. Chem. Soc 2004, 126, 13632–13633; [DOI] [PubMed] [Google Scholar]; b) Kumagai N, Matsunaga S, Shibasaki M, Chem. Commun 2005, 3600–3602; [DOI] [PubMed]; c) Suto Y, Tsuji R, Kanai M, Shibasaki M, Org. Lett 2005, 7, 3757–3760; [DOI] [PubMed] [Google Scholar]; d) Fan L, Ozerov OV, Chem. Commun 2005, 4450–4452; [DOI] [PubMed]; e) Kumagai N, Matsunaga S, Shibasaki M, Tetrahedron 2007, 63, 8598–8608; [Google Scholar]; f) Goto A, Endo K, Ukai Y, Irle S, Saito S, Chem. Commun 2008, 2212–2214; [DOI] [PubMed]; g) Chakraborty S, Patel YJ, Krause JA, Guan H, Angew. Chem. Int. Ed 2013, 52, 7523–7526; [DOI] [PubMed] [Google Scholar]; a) Sureshkumar D, Ganesh V, Kumagai N, Shibasaki M, Chem. Eur. J 2014, 20, 15723–15726. [DOI] [PubMed] [Google Scholar]
- [3].a) Kisanga P, McLeod D, D’Sa B, Verkade J, J. Org. Chem 1999, 64, 3090–3094; [DOI] [PubMed] [Google Scholar]; b) Yin L, Kanai M, Shibasaki M, J. Am. Chem. Soc 2009, 131, 9610–9611; [DOI] [PubMed] [Google Scholar]; c) Hyodo K, Kondo M, Funahashi Y, Nakamura S, Chem. Eur. J 2013, 19, 4128–4134; [DOI] [PubMed] [Google Scholar]; d) Ren Q, Huang J, Wang L, Li W, Liu H, Jiang X, Wang J, ACS Catal 2012, 2, 2622–2625; [Google Scholar]; e) Gajulapalli VPR, Vinayagam P, Kesavan V, Org. Biomol. Chem 2014, 12, 4186–4191; [DOI] [PubMed] [Google Scholar]; f) For examples of decarboxylative aldol reactions with trifluoromethyl ketones:Khodakovskiy PV, Volochnyuk DM, Tolmachev AA, Synthesis 2009, 1099–1104;; g) Zheng Y, Xiong H-Y, Nie J, Hua M-Q, Ma J-A, Chem. Commun 2012, 48, 4308–4310. [DOI] [PubMed] [Google Scholar]
- [4].For examples of ethyl isocyanoacetate additions to ketones, see Yuan W-K, Cui T, Liu W, Wen L-R, Li M, Org. Lett 2018, 20, 1513–1516. [DOI] [PubMed] [Google Scholar]
- [5].a) Wang J, Sanchez-Rosello M, Acena JL, del Pozo C, Sorochinsky AE, Fustero S, Soloshonok VA, Liu H, Chem. Rev 2014, 114, 2432–2506; [DOI] [PubMed] [Google Scholar]; b) Smith BR, Eastman CM, Njardarson JT, J. Med. Chem 2014, 57, 9764–9773. [DOI] [PubMed] [Google Scholar]
- [6].a) Liang T, Neumann CN, Ritter T, Angew. Chem. Int. Ed 2013, 52, 8214–8264; [DOI] [PubMed] [Google Scholar]; b) Yang X, Wu T, Phipps RJ, Toste FD, Chem. Rev 2015, 115, 826–870; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhou Y, Wang J, Gu Z, Wang S, Zhu W, Acena JL, Soloshonok VA, Izawa K, Liu H, Chem. Rev 2016, 116, 422–518. [DOI] [PubMed] [Google Scholar]
- [7].Schenck HA, Lenkowski PW, Choudhury-Mukherjee I, Ko S-H, Stables JP, Patel MK, Brown ML, Bioorg. Med. Chem 2004, 12, 979–993. [DOI] [PubMed] [Google Scholar]
- [8].Sani M, Belotti D, Giavazzi R, Panzeri W, Volonterio A, Zanda M, Tetrahedron Lett 2004, 45, 1611–1615. [Google Scholar]
- [9].Caron S, Do NM, Sieser JE, Arpin P, Vazquez E, Org. Process Res. Dev 2007, 11, 1015–1024. [Google Scholar]
- [10].Betageri R, Zhang Y, Zindell RM, Kuzmich D, Kirrane TM, Bentzien J, Cardozo M, Capolino AJ, Fadra TN, Nelson RM, Paw Z, Shih D-T, Shih C-K, Zuvela-Jelaska L, Nabozny G, Thomson DS, Bioorg. Med. Chem. Lett 2005, 15, 4761–4769. [DOI] [PubMed] [Google Scholar]
- [11].Corbett JW, Ko SS, Rodgers JD, Gearhart LA, Magnus NA, Bacheler LT, Diamond S, Jeffrey S, Klabe RM, Cordova BC, Garber S, Logue K, Trainor GL, Anderson PS, Erickson-Viitanen SK, J. Med. Chem 2000, 43, 2019–2030. [DOI] [PubMed] [Google Scholar]
- [12].Silva AM, Cachau RE, Sham HL, Erickson JW, J. Mol. Biol 1996, 255, 321–346. [DOI] [PubMed] [Google Scholar]
- [13].Perrin SR, Hauck W, Ndzie E, Blehaut J, Ludemann-Hombouger O, Nicoud R-M, Pirkle WH, Org. Process Res. Dev 2007, 11, 817–824. [Google Scholar]
- [14].a) Yearick K, Wolf C, Org. Lett 2008, 10, 3915–3918; [DOI] [PubMed] [Google Scholar]; b) Xu H, Wolf C, Chem. Commun 2010, 8026–8028; [DOI] [PubMed]; c) Zhang P, Wolf C, Adv. Synth. Catal 2011, 353, 760–766; [Google Scholar]; d) Zhang P, Wolf C, J. Org. Chem 2012, 77, 8840–8844; [DOI] [PubMed] [Google Scholar]; e) Cook AM, Wolf C, Angew. Chem. Int. Ed 2016, 55, 2929–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Imamura K, Tomita N, Kawakita Y, Ito Y, Ono K, Nii N, Miyazaki T, Yonemori K, Tawada M, Sumi H, Satoh Y, Yamamoto Y, Miyahisa I, Sasaki M, Satomi Y, Hirayama M, Nishigaki R, Maezaki H, Bioorg. Med. Chem 2017, 25, 3768–3779. [DOI] [PubMed] [Google Scholar]
- [16].a) Leng DJ, Black CM, Pattison G, Org. Biomol. Chem 2016, 14, 1531–1535 [DOI] [PubMed] [Google Scholar]; b) Balaraman K, Wolf C, Angew. Chem. Int. Ed 2017, 56, 1390–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17]. Epoxide formation was not observed.
- [18].For Mannich-type reactions with β-phenylsulfonyl nitriles or β-keto acids:a) Gonzalez PB, Lopez R, Palomo C, J. Org. Chem 2010, 75, 3920–3922 [DOI] [PubMed] [Google Scholar]; b) Ohmatsu K, Goto A, Ooi T, Chem. Commun 2012, 48, 7913–7915 [DOI] [PubMed] [Google Scholar]; c) Lahosa A, Soler T, Arrieta A, Cossio FP, Foubelo F, Yus M, J. Org. Chem 2017, 82, 7481–7491. [DOI] [PubMed] [Google Scholar]
- [19].a) Li J, Li Y-L, Jin N, Ma A-L, Y.-N. H., Deng J, Adv. Synth. Catal 2015, 357, 2474–2478. [Google Scholar]; b) Gao H, Luo Z, Ge P, He J, Zhou F, Zheng P, Jiang J, Org. Lett 2015, 17, 5962–5965. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.