

Abstract
Proteases sustain hyperexcitability and pain by cleaving protease-activated receptor-2 (PAR2) on nociceptors through distinct mechanisms. Whereas trypsin induces PAR2 coupling to Gαq, Gαs, and β-arrestins, cathepsin-S (CS) and neutrophil elastase (NE) cleave PAR2 at distinct sites and activate it by biased mechanisms that induce coupling to Gαs, but not to Gαq or β-arrestins. Because proteases activate PAR2 by irreversible cleavage, and activated PAR2 is degraded in lysosomes, sustained extracellular protease-mediated signaling requires mobilization of intact PAR2 from the Golgi apparatus or de novo synthesis of new receptors by incompletely understood mechanisms. We found here that trypsin, CS, and NE stimulate PAR2-dependent activation of protein kinase D (PKD) in the Golgi of HEK293 cells, in which PKD regulates protein trafficking. The proteases stimulated translocation of the PKD activator Gβγ to the Golgi, coinciding with PAR2 mobilization from the Golgi. Proteases also induced translocation of a photoconverted PAR2-Kaede fusion protein from the Golgi to the plasma membrane of KNRK cells. After incubation of HEK293 cells and dorsal root ganglia neurons with CS, NE, or trypsin, PAR2 responsiveness initially declined, consistent with PAR2 cleavage and desensitization, and then gradually recovered. Inhibitors of PKD, Gβγ, and protein translation inhibited recovery of PAR2 responsiveness. PKD and Gβγ inhibitors also attenuated protease-evoked mechanical allodynia in mice. We conclude that proteases that activate PAR2 by canonical and biased mechanisms stimulate PKD in the Golgi; PAR2 mobilization and de novo synthesis repopulate the cell surface with intact receptors and sustain nociceptive signaling by extracellular proteases.
Keywords: G protein–coupled receptor (GPCR), Golgi, pain, protease, protein kinase D (PKD), signal transduction, trafficking, F2R-like trypsin receptor, nociception, proteinase-activated receptor-2, Gβγ
Introduction
Sustained signaling by membrane-impermeant agonists in the extracellular environment requires the presence of functional receptors at the surface of cells. The level of functional receptors at the plasma membrane is a dynamic balance between processes that inactivate or remove receptors and those that reactivate or replenish receptors (1). Desensitization and endocytosis inactivate and remove functional receptors from the plasma membrane and thereby diminish cellular responsiveness. Recovery of responsiveness requires repopulation of the plasma membrane with functional receptors by mechanisms that include resensitization of receptors or membrane insertion of fresh receptors. However, in contrast to the mechanisms of receptor desensitization and endocytosis, which have been extensively studied (2), the mechanisms that repopulate the plasma membrane with functional receptors are not fully understood (1).
G protein–coupled receptors (GPCRs)2 comprise the largest family of transmembrane receptors and are the target of one-third of therapeutic drugs (3). The mechanisms that attenuate plasma membrane signaling of GPCRs have been extensively studied. G protein–coupled receptor kinases and second messenger kinases can phosphorylate activated receptors, which then associate with β-arrestins. By uncoupling GPCRs from G proteins and coupling GPCRs to clathrin and adaptor protein 2, β-arrestins mediate receptor desensitization and endocytosis, which together attenuate plasma membrane signaling (4). Some endocytosed receptors, including the substance P neurokinin 1 receptor (5) and the μ-opioid receptor (6), recycle back to the plasma membrane, which restores their function at the cell surface. Other receptors, exemplified by chemokine receptor type 4 (CXCR4), traffic to lysosomes (7). For these receptors, de novo protein synthesis or translocation of intact receptor from a preexisting pool is required for replenishment of the plasma membrane with functioning receptors.
PARs are a small family of GPCRs that are activated by proteolytic cleavage within the extracellular N-terminal region (8). Several serine and cysteine proteases can cleave and activate PAR2, including trypsins (9, 10), mast cell tryptase (11), kallikreins (12), NE (13, 14) and CS, a cysteine protease from antigen-presenting cells (15). Trypsin, tryptase, and kallikreins activate PAR2 by a canonical mechanism. Trypsin cleaves PAR2 at R36↓S37, which reveals the tethered ligand domain (S37LIGKV for human PAR2). This cleavage results in PAR2 coupling to Gαq, Gαs, and β-arrestins, leading to mobilization of Ca2+, generation of cAMP, and activation of protein kinase C (PKC) and A (PKA) and of extracellular signal-regulated kinases (16, 17). β-Arrestins mediate desensitization and endocytosis of PAR2, which then traffics to lysosomes and is degraded (17). Given the irreversible mechanism of proteolytic activation, cleaved PAR2 cannot be reactivated by proteolysis (16). Recovery of cell surface PAR2 signaling involves mobilization of intact receptors from a preexisting Golgi pool as well as synthesis of fresh receptors (16). NE and CS are activated and released from inflammatory cells at sites of injury and inflammation, retain activity in extracellular fluid, and can cause PAR2-dependent inflammation and pain (14, 15, 18). However, CS and NE activate biased pathways of PAR2 signaling and trafficking. CS cleaves human PAR2 at E56↓T57 to expose the tethered ligand (T57VFSVDEFSA), which promotes PAR2 coupling to Gαs (15). NE cleaves at A66↓S67 and S67↓V68, adjacent to the first transmembrane domain, which activates PAR2 by a nontethered ligand mechanism and induces coupling to Gαs and Gα12/13 (13, 14). CS- and NE-activated PAR2 fails to couple to Gαq and neither recruits β-arrestins nor internalizes. We recently reported that trypsin cleavage of PAR2 at the plasma membrane induces translocation of Gβγ to the Golgi apparatus, where Gβγ activates PKD (19). PKD mediates the mobilization of PAR2 stores from the Golgi apparatus, which replenishes the plasma membrane with fresh receptors that are necessary for sustained trypsin signaling (19). In the present study, we investigated the mechanisms that underlie sustained signaling of proteases that activate PAR2 by biased mechanisms.
Results
Proteases that activate PAR2 by canonical and biased mechanisms induce PAR2-dependent PKD activation
By using immunoblotting and immunofluorescence, we have previously reported that trypsin activation of PAR2 leads to PKD phosphorylation (activation) in the Golgi apparatus (19). To quantitatively assess PKD activation in live cells with high spatial and temporal fidelity, we expressed in human embryonic kidney (HEK293) cells genetically encoded FRET biosensors for PKD that are targeted to the cytosol (Cyto-DKAR) or Golgi apparatus (Golgi-DKAR) (20). We examined whether proteases that activate PAR2 by canonical or biased mechanisms can stimulate PKD activity in the cytosol or plasma membrane of HEK293 cells transiently expressing human PAR2 (hPAR2). To confirm that alterations in FRET were attributable to PKD activation, we expressed a T/A mutated PKD sensor (DKAR-T/A) in which the PKD phosphorylation site in the substrate domain is mutated.
We first confirmed the expected subcellular localization of PKD FRET biosensors expressed in HEK293 cells by confocal microscopy. Cyto-DKAR was uniformly distributed throughout the cytosol, whereas Golgi-DKAR colocalized exclusively with immunoreactive TGN58K, a marker of the Golgi apparatus (Fig. 1A).
Figure 1.

Protease-induced activation of PKD. A, localization of FRET PKD biosensors Cyto-DKAR and Golgi-DKAR with TGN58K in HEK293 cells. B–E, time course of trypsin-induced (B, 10 nm), CS-induced (C, 100 nm), NE-induced (D, 100 nm), and 2-furoyl-LIGRLO-NH2–induced (E, 10 μm) PKD activity in HEK293 cells expressing FRET biosensors for PKD in the cytosol (Cyto-DKAR) and Golgi apparatus (Golgi-DKAR). DKAR-T/A is a PKD control sensor. Agonists were added at the arrow. F–H, AUC for Cyto-DKAR and DKAR-T/A in HEK293 cells treated with trypsin (F), CS (G), or NE (H) and either vehicle or PAR2 antagonist (I-343). I, effects of I-343 on PKD activity in the Golgi apparatus stimulated by phorbol 12,13-dibutyrate (PDBu) or thrombin. n = 3–5 experimental replicates, triplicate observations. *, p < 0.05; **, p < 0.01 to vehicle (one-way ANOVA, Bonferroni multiple comparisons). Error bars, S.E.
Trypsin (10 nm), CS (100 nm), NE (100 nm), and the PAR2-selective agonist 2-furoyl-LIGRLO-NH2, an analogue of the tethered ligand (10 μm), increased PKD activity in the cytosol and Golgi apparatus within 2 min, which was sustained for at least 20 min (Fig. 1, B–E). The PAR2 antagonist I-343 (18) (10 μm) inhibited trypsin-, CS-, and NE-induced PKD activation (Fig. 1, F–H). I-343 did not inhibit activation of PKD in the Golgi apparatus in response to phorbol 12,13-dibutyrate (200 nm) or thrombin (30 units/ml), a PAR1 agonist, and I-343 alone had no effect on PKD activity (Fig. 1I). These results show that proteases that activate PAR2 by canonical and biased mechanisms activate PKD in the cytosol and Golgi apparatus of HEK293 cells. These effects are mediated by PAR2. There was no effect of trypsin or 2-furoyl-LIGRLO-NH2 on DKAR-T/A FRET (Fig. 1, B and E), although CS and NE caused a minor increase in DKAR-T/A FRET (Fig. 1, C and D). One possibility is that kinases other than PKD might phosphorylate DKAR-T/A, such as PKA, which is robustly activated by NE (13, 14) and CS (15). Further studies are necessary to address this question.
Proteases that activate PAR2 by canonical and biased mechanisms induce Gβγ translocation to Golgi
Gβγ subunits can activate PKD in the Golgi apparatus (21). Trypsin activation of PAR2 at the cell surface induces translocation of Gβγ subunits to the Golgi apparatus, where Gβγ activates PKD (19). We used bioluminescence resonance energy transfer (BRET) to examine whether CS and NE, which activate PAR2 by biased mechanisms, also promote Gβγ trafficking to the Golgi apparatus. We transiently expressed in HEK293 cells Gγ2-Venus and Giantin-RLuc8, a Golgi-resident protein (22), which permitted measurement of the proximity between Gγ2-Venus and Giantin-RLuc8 using bystander BRET. We also expressed hPAR2, Gβ1, and either Gαq or Gαs, which allowed assessment of the requirement of different Gα subunits.
We confirmed the expected subcellular localization of BRET biosensors expressed in HEK293 cells by confocal microscopy. In unstimulated cells, Gγ2-Venus was uniformly cytosolic, whereas Giantin-Venus colocalized exclusively with immunoreactive TGN58K in the Golgi apparatus (Fig. 2A). We were unable to localize Giantin-RLuc8 because of the lack of a suitable epitope tag or antibody, but we expect that it would have the identical localization as Giantin-Venus.
Figure 2.

Protease-induced translocation of Gγ to Golgi apparatus. A, localization of BRET biosensors Gγ-Venus and Giantin-Venus with TGN 58K in HEK293 cells. B–J, protease-evoked BRET. Effects of trypsin (B–D), CS (E–G), and NE (H–J) on Gγ-Venus/Giantin-RLuc8 BRET in HEK293 cells. B, E, and H, time course of trypsin-induced (10 nm), CS-induced (100 nm), and NE-induced (100 nm) BRET. Agonists were added at the arrow. C, F, and I, AUC of BRET response. D, G, J, concentration–response analysis. Gβ was coexpressed with either Gαq or Gαs. n = 3–6 experimental replicates, triplicate observations. *, p < 0.05 (Student's t test). Error bars, S.E.
Trypsin increased Gγ2-Venus/Giantin-RLuc8 BRET in the presence of either Gαq or Gαs (Fig. 2, B–D). However, trypsin increased Gγ2-Venus/Giantin-RLuc8 BRET with higher potency in cells expressing Gαq (pEC50 (negative logarithm of the EC50) - 12.74 ± 0.36) compared with Gαs (pEC50 = 9.75 ± 0.65). CS also increased Gγ2-Venus/Giantin-RLuc8 BRET in the presence of either Gαq or Gαs (Fig. 2, E–G). In contrast to trypsin, CS increased BRET with higher potency in cells expressing Gαs (pEC50 = 11.90 ± 0.35) than Gαq (pEC50 = 10.35 ± 0.81). NE increased Gγ2-Venus/Giantin-RLuc8 BRET only in the presence of Gαs (pEC50 = 11.84 ± 0.45), not Gαq (Fig. 2, H–J). Thus, proteases that activate PAR2 at the cell surface by canonical and biased mechanisms may evoke the rapid translocation of Gβγ to the Golgi apparatus. Although Gβγ subunits could originate from the plasma membrane, it is also possible that PAR2 activation evokes translocation of Gβγ from the cytosol to the Golgi apparatus or may reveal Gβγ subunits already present in the Golgi apparatus. The effects of overexpression of Gα subunits on Gγ2-Venus/Giantin-RLuc8 BRET are consistent with the capacity of trypsin-activated PAR2 to couple to Gαq or Gαs and for CS- and NE-activated PAR2 to couple preferentially to Gαs (13–15). Studies with selective Gα inhibitors or with Gα-deficient HEK293 cells will be required to determine the absolute requirement of particular Gα subunits for protease-stimulated Gγ2-Venus/Giantin-RLuc8 BRET.
Proteases that activate PAR2 by canonical and biased mechanisms mobilize PAR2 from the Golgi apparatus
To quantitatively examine whether PAR2 activation at the cell surface mobilizes receptors from the Golgi apparatus, we examined the proximity between PAR2-RLuc8 and TGN38-Venus, a Golgi-resident protein (23). PAR2-RLuc8 was prominently localized to the plasma membrane, and TGN38-Venus was confined to the Golgi apparatus, as confirmed by colocalization with immunoreactive TGN58K (Fig. 3A). Thus, BRET sensors have the expected subcellular localization.
Figure 3.

Protease-induced translocation of PAR2 from the Golgi apparatus. A, localization of BRET biosensors PAR2-RLuc8 and TGN38-Venus with TGN 58K in HEK293 cells. B–J, protease-evoked BRET. B–D, time course of trypsin-, CS-, and NE-induced BRET between PAR2-RLuc8 and TGN38-Venus in HEK293 cells. E–J, effects of inhibitors of Gβγ (gallein), PKD (CRT0066101), PKC (Gö6983), and PKA (H-89) on trypsin-induced (10 nm), CS-induced (100 nm), and NE-induced (100 nm) BRET between PAR2-RLuc8 and TGN38-Venus. E–G, time course. H–J, AUC. n = 3 (A–C) or 3–5 (D–J) experimental replicates, triplicate observations. *, p < 0.05; **, p < 0.01; ****, p < 0.0001 (one-way ANOVA, Bonferroni multiple comparisons). Error bars, S.E.
Trypsin (10 and 100 nm), CS (100 nm), and NE (100 nm) all induced a rapid decrease in PAR2-RLuc8/TGN38-Venus BRET within 2 min that was sustained for 20 min (Fig. 3, B–D). Gallein, which inhibits Gβγ activity (24), suppressed the decrease in PAR2-RLuc8/TGN38-Venus BRET in cells exposed to trypsin, CS, and NE (Fig. 3, E–J). To confirm involvement of Gβγ, we treated cells with a peptide corresponding to the C terminus of GPCR kinase 2 (GRK2i), which inhibits Gβγ activity (25). GRK2i (100 μm) attenuated the decrease in PAR2-RLuc8/TGN38-Venus BRET in cells exposed to trypsin, CS, and NE to a similar degree as gallein (Fig. 3, H–J).
We then evaluated the role of kinases in mobilization of PAR2 from the Golgi apparatus. Surprisingly, in view of the known role of PKD in protein sorting within the Golgi apparatus (19, 21, 26, 27), the PKD inhibitor CRT0066101 (100 nm) (28) did not affect the decrease in PAR2-RLuc8/TGN38-Venus BRET (Fig. 3, E–J). A Gαq/Ca2+/PKC pathway has been reported to control Golgi organization and secretion (29). Gö6983 (1 μm), a nonselective PKC inhibitor (30), did not significantly inhibit trypsin-, CS-, or NE-evoked mobilization of PAR2 from the Golgi apparatus (Fig. 3, E–J). PKA has been shown to be required in the formation of vesicles from the Golgi (31). Given the capacity of CS- and NE-activated PAR2 to couple to Gαs, adenylyl cyclase, cAMP, and PKA, we also evaluated the contribution of PKA to PAR2 mobilization from the Golgi apparatus. H-89 (1 μm), which inhibits PKA and other kinases (32, 33), had no effect on trypsin-, CS-, or NE-induced mobilization of PAR2 (Fig. 3, H–J).
We confirmed that the PKD and PKC inhibitors blocked kinase activity in HEK293 cells by expressing FRET biosensors for cytosolic PKD (Cyto-DKAR) and PKC (Cyto-CKAR). CRT0066101 (100 nm) abolished trypsin (10 nm)-evoked activation of PKD (Fig. 4, A and B), and Gö6983 (1 μm) abolished trypsin-evoked activation of PKC (Fig. 4, C and D). We have previously shown that H-89 inhibits CS-mediated PAR2 signaling events (15).
Figure 4.
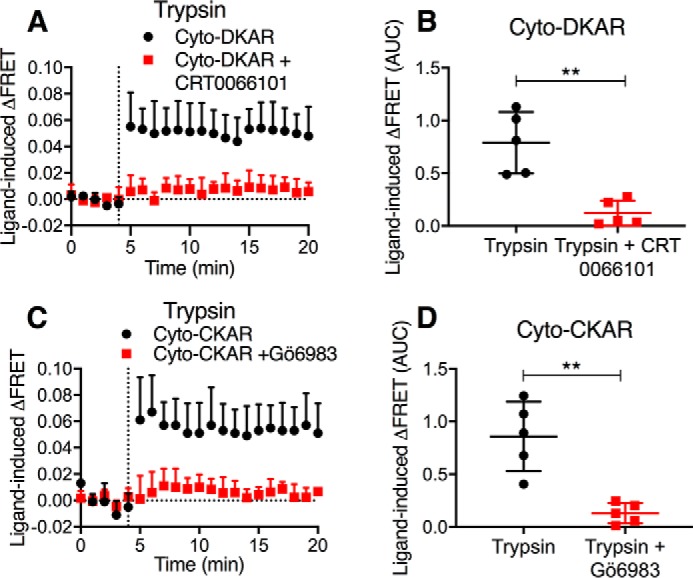
Antagonism of PKD and PKC. Time course (A and C) and AUC (B and D) of trypsin-stimulated activation of cytosolic PKD (A and B) and cytosolic PKC (C and D). The effects of inhibitors of PKD (CRT0066101) and PKC (Gö6983) are shown. n = 5 experimental replicates, triplicate observations. **, p < 0.01 (Student's t test). Error bars, S.E.
These results indicate a major role for Gβγ subunits in mediating the capacity of trypsin, CS, and NE to mobilize PAR2 from the Golgi apparatus of HEK293 cells. Further studies are necessary to understand the unexpected finding that PKD inhibitors did not affect protease-evoked liberation of PAR2 from the Golgi apparatus as determined by measurements of PAR2-RLuc8/TGN38-Venus BRET. However, because TGN38 can shuttle between the Golgi apparatus and plasma membrane, it is possible that proximity between TGN38 and PAR2 in the exocytic and endocytic pathways may confound the results (23).
Proteases that activate PAR2 by canonical and biased mechanisms induce Gβγ and PKD-dependent translocation of PAR2 to the plasma membrane
In light of the paradoxical inhibition of PAR2 mobilization from the Golgi apparatus by Gβγ inhibitors but not by PKD inhibitors, as determined by PAR2-RLuc8/TGN38-Venus BRET, we sought an alternative approach to study depletion of PAR2 from the Golgi apparatus. To localize the redistribution of PAR2 from the Golgi apparatus to the plasma membrane, we expressed in rat kidney epithelial (KNRK) cells PAR2 fused at the C terminus to Kaede, a photoconvertible protein. We have previously shown that PAR2-Kaede is functional and suitable for analysis of intracellular trafficking of PAR2 (19). We selected KNRK cells for these studies rather than HEK293 cells because the Golgi pool of PAR2 is particularly distinct in KNRK cells compared with HEK293 cells, where it is diffuse and more challenging to localize (16). Trypsin induces β-arrestin–mediated endocytosis and endosomal signaling of PAR2 of PAR2 in KNRK cells (17, 34). The prominent Golgi store of PAR2-Kaede was photoconverted from green to red using a confocal microscope laser. Cells were then exposed to proteases and monitored to assess depletion of PAR2-Kaede from the Golgi apparatus and insertion of mobilized PAR2-Kaede into the plasma membrane. Before photoconversion, PAR2-Kaede (green) was detected at the plasma membrane and in the Golgi apparatus (Fig. 5, A and B). Confocal illumination of the Golgi resulted in rapid green to red photoconversion of PAR2-Kaede in the Golgi apparatus but not the plasma membrane. NE (100 nm, 30 min) induced a significant depletion of PAR2-Kaede red from the Golgi apparatus and a corresponding increase in PAR2-Kaede red at the plasma membrane (Fig. 5A). CRT0066101 (Fig. 5, C and D) and gallein (Fig. 5, E and F) both prevented the depletion of PAR2-Kaede red from the Golgi apparatus and insertion into the plasma membrane. Similarly, CS (100 nm, 30 min) induced a significant depletion of PAR2-Kaede red from the Golgi apparatus and a nonsignificant increase in PAR2-Kaede red at the plasma membrane (Fig. 6A). CRT0066101 (Fig. 6B) and gallein (Fig. 6C) both prevented the depletion of PAR2-Kaede red from the Golgi apparatus. These results suggest that NE and CS activate PAR2 at the plasma membrane to induce Gβγ- and PKD-mediated recruitment of intact PAR2 from the Golgi apparatus to the plasma membrane.
Figure 5.
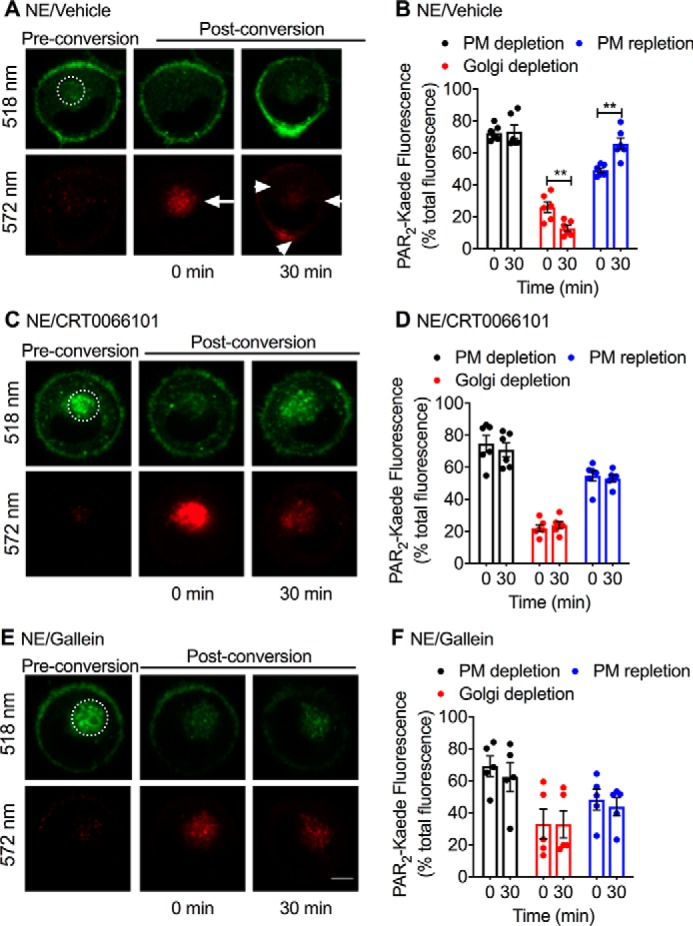
NE-induced trafficking of PAR2-Kaede from the Golgi apparatus to the plasma membrane. PAR2-Kaede located in the region of the perinuclear Golgi apparatus of KNRK-PAR2-Kaede cells was green/red photoconverted using a confocal laser (dashed circle, illuminated region). Cells were then incubated with NE (100 nm) for 0 or 30 min. PAR2-Kaede fluorescence was measured at 518 and 572 nm at the cell surface or Golgi. Cells were pre-incubated with vehicle (A and B), PKD inhibitor (CRT0066101, 100 nm) (C and D), or Gβγ inhibitor (gallein, 10 μm) (E and F). A, C, and E, representative images before and after photoconversion and at 0 or 30 min after protease challenge. B, D, and F, quantification of PAR2-Kaede (518 nm, green) at the plasma membrane and of photoconverted PAR2-Kaede (572 nm, red) in the Golgi region and at the plasma membrane. n = 5–6 experiments, >30 cells analyzed per condition. **, p < 0.01 (Student's t test). Scale bar, 10 μm. Error bars, S.E.
Figure 6.
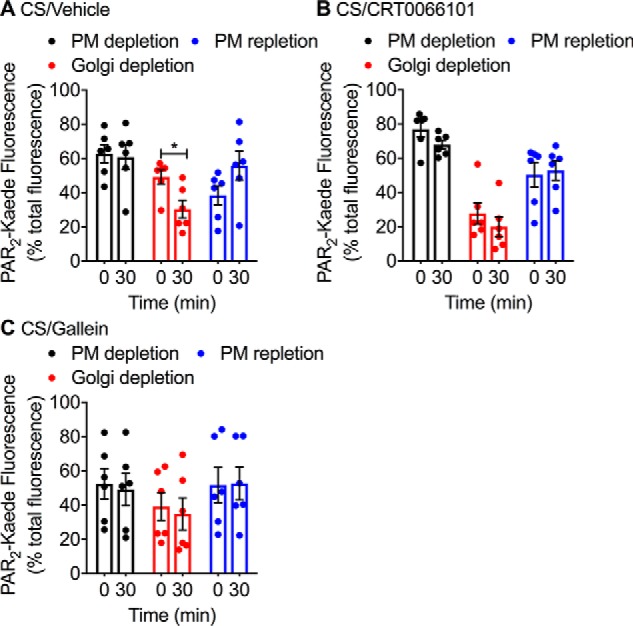
CS-induced trafficking of PAR2-Kaede from the Golgi apparatus to the plasma membrane. PAR2-Kaede located in the region of the perinuclear Golgi apparatus of KNRK-PAR2-Kaede cells was green/red photoconverted. Cells were then incubated with CS (100 nm) for 0 or 30 min. PAR2-Kaede fluorescence was measured at the cell surface or Golgi. Cells were pre-incubated with vehicle (A), PKD inhibitor (CRT0066101, 100 nm) (B), or Gβγ inhibitor (gallein, 10 μm) (C). Quantification of PAR2-Kaede (518 nm, green) at the plasma membrane and of photoconverted PAR2-Kaede (572 nm, red) in the Golgi region and at the plasma membrane is shown. n = 6 experiments, >30 cells analyzed per condition. *, p < 0.05 (Student's t test). Error bars, S.E.
PKD and Gβγ mediate recovery of plasma membrane PAR2 signaling
CS and NE cleave PAR2 distal to the trypsin cleavage site and the trypsin-exposed tethered ligand (14, 15). By removing these domains, CS and NE prevent the capacity of trypsin to activate PAR2. Recovery of responsiveness to trypsin thus requires repopulation of the plasma membrane with intact PAR2. We made use of this paradigm to determine whether Gβγ- and PKD-induced mobilization of intact PAR2 from the Golgi apparatus mediates the recovery of trypsin-responsiveness in HEK293 cells.
HEK293 cells were first exposed to trypsin (10 nm), CS (100 nm), NE (100 nm), or vehicle (control) for 10 min (Fig. 7A). Cells were then washed, recovered for 110 min at 37 °C, and challenged with trypsin (10 nm) at 120 min after the first challenge with proteases. [Ca2+]i was monitored to assess PAR2 activation. We have previously reported that exposure to CS and NE (100 nm, 10 min) prevents the capacity of trypsin to mobilize Ca2+, which indicates that CS and NE can initially cleave and disarm the receptor under these conditions (14, 15). In some experiments, cells were pre-incubated with CTR0066101, gallein, or vehicle to assess the mechanism of recovery. The first challenge with trypsin caused a prompt increase in [Ca2+]i, and this response was unaffected by CRT0066101 or gallein (Figs. 7, B–D and 8A). In contrast, CS and NE did not affect [Ca2+]i, which is in accordance with the inability of CS- and NE-activated PAR2 to couple to Gαq and mobilize intracellular Ca2+ ions (14, 15). When cells were challenged with trypsin at 120 min after the first exposure to trypsin, CS, or NE, the secondary responses to trypsin were almost fully recovered relative to those of cells challenged initially with vehicle (Figs. 7, C and D and 8). These results are consistent with replenishment of the plasma membrane with intact PAR2. CRT0066101 or gallein inhibited recovery of trypsin responsiveness in cells first challenged with trypsin, CS, or NE (Figs. 7, C and D and 8, B–J). The combination of both CTR0066101 and gallein had a further inhibitory effect, which might suggest parallel PKD and Gβγ pathways (Figs. 7, C and D and 8, D, G, and J). These data suggest that Gβγ- and PKD-mediated recruitment of intact PAR2 from the Golgi apparatus to the plasma membrane is required for recovery of trypsin responses in HEK293 cells.
Figure 7.

Recovery of PAR2-mediated signaling. A, experimental design. HEK293 cells were first challenged with trypsin (10 nm), CS (100 nm), NE (100 nm), or vehicle for 10 min. Cells were then washed, recovered for 110 min, and then challenged with trypsin (10 nm) at 120 min after the first challenge with proteases. B–D, measurement of [Ca2+]i in HEK293 cells. B, effects of PKD inhibitor (CRT0066101, CRT, 100 nm) or Gβγ inhibitor (gallein, 10 μm) on responses to a single challenge with trypsin. C and D, responses to a first challenge with trypsin, CS, or vehicle (C) or with trypsin, NE, or vehicle (D) and a second challenge with trypsin after 120 min. n = 4 (B) or 5 (C and D) experimental replicates from triplicate observations. Error bars, S.E.
Figure 8.
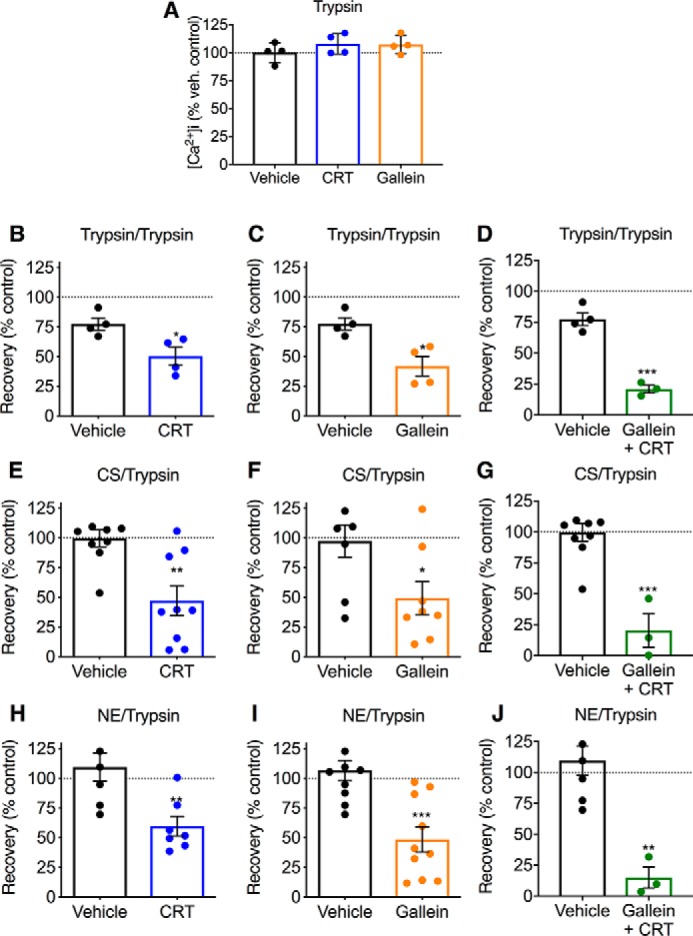
Recovery of PAR2-mediated signaling. A, effects of PKD inhibitor (CRT0066101 (CRT), 100 nm) or Gβγ inhibitor (gallein, 10 μm) on responses of HEK293 cells to a single challenge with trypsin. B–J, recovery of Ca2+ signaling in HEK293 cells. Cells were first challenged with trypsin (10 nm) (B–D), CS (100 nm) (E–G) or NE (H–J). Cells were then washed, recovered for 120 min, and then challenged with trypsin (10 nm). In some experiments, cells were pre-incubated with PKD inhibitor (CRT0066101) (B, E, and H) or Gβγ inhibitor (gallein) (C, F, and I) or both PKD and Gβγ inhibitor (D, G, and J). n = 3–10 experimental replicates from triplicate observations. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (Student's t test). Error bars, S.E.
NE-induced activation of PKD in the Golgi apparatus of nociceptors mediates sustained hyperexcitability
We have recently reported that trypsin, CS, and NE induce sustained nociception in mice by activating PAR2 expressed by a subpopulation of primary sensory neurons (18). Trypsin, CS, and NE also cause a PAR2-dependent hyperexcitability of nociceptors, which is a hallmark of chronic pain (18). PKD activity is necessary for maintaining trypsin-evoked hyperexcitability of nociceptors, consistent with PAR2 mobilization from Golgi stores (19). We examined whether PKD and Gβγ contribute to the maintenance of protease-evoked hyperexcitability. The excitability of nociceptors of mouse dorsal root ganglia (DRG) was determined by measurement of the rheobase (minimum current required to fire a single action potential) using patch-clamp recordings. Neurons were exposed to NE (7.8 nm, 0.2 units/ml, 20 min) or vehicle (control) and washed, and rheobase was measured at 0, 30, or 150 min after washing (Fig. 9A). Immediately after washing (0 min), there was a 32% decrease in rheobase of NE-treated neurons, when compared with vehicle (p < 0.001), consistent with hyperexcitability (Fig. 8B). Hyperexcitability was maintained at 30 min after washing (25% decrease in rheobase compared with vehicle, p < 0.05), but after 150 min, hyperexcitability had declined to control levels (Fig. 9B). To determine whether neurons had recovered their capacity to respond to PAR2 agonists at 150 min after exposure to NE and washing, they were challenged with trypsin (50 nm, 10 min) and washed, and rheobase was immediately measured. In NE-exposed neurons, trypsin induced a 35% decrease in rheobase when compared with vehicle-treated controls (p < 0.01) (Fig. 9B). These results show that NE causes an initial hyperexcitability of nociceptors, which is sustained for 30 min and declines after 150 min. At this time, neurons have regained their capacity to respond to trypsin, which may require mobilization of intact PAR2.
Figure 9.

Recovery of PAR2-mediated hyperexcitability of nociceptors. A, experimental design. DRG neurons from mice were exposed to NE (7.8 nm, 0.2 units/ml) for 20 min, washed, and recovered for up to 150 min. Changes in neuronal excitability were assessed at 0, 30, or 150 min after washing by measurement of rheobase. To assess recovery of excitability, some neurons were challenged with trypsin (Tryp, 50 nm, 10 min). At 150 min after NE exposure, neurons were washed, and rheobase was immediately measured. To examine the mechanism of recovery, neurons were pre-incubated with PKD inhibitor (CRT0066101 (CRT), 10 nm), Gβγ inhibitor (gallein, 10 μm), protein synthesis inhibitor (cycloheximide (CHX), 10 μg/ml), or vehicle for 30 min before NE (inhibitors were included throughout). B–E, measurements of rheobase in protease-treated DRG neurons. B, time course of responses at time 0 min (T = 0), 30 min (T = 30), and 150 min (T = 150) after NE and recovery of trypsin response at 150 min after NE. C, NE responses at time 0 min after NE. D, effects of CRT and gallein on responses to trypsin at 150 min after NE. E, effects of cycloheximide on initial response to trypsin (0 min) and on responses to trypsin at 150 min after NE. Numbers in parentheses indicate numbers of neurons studied from 6–13 mice. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (one-way ANOVA, Bonferroni multiple comparisons). Error bars, S.E.
To determine whether PKD, Gβγ, or new protein synthesis mediates this recovery of hyperexcitability, neurons were pre-incubated with CRT0066101 (10 nm, PKD inhibitor), gallein (10 μm, Gβγ inhibitor), cycloheximide (10 μg/ml, protein synthesis inhibitor), or vehicle 30 min before NE exposure; inhibitors were present throughout the experiment. NE- and trypsin-evoked hyperexcitability was determined. CRT0066101 and gallein had no effect on the capacity of NE to cause an immediate hyperexcitability (0 min after NE) (Fig. 9C). However, both the PKD and Gβγ inhibitors prevented trypsin-induced hyperexcitability at 150 min after NE exposure (Fig. 9D). Similarly, cycloheximide did not affect an initial response to trypsin (0 min), but abolished the recovery of trypsin-induced hyperexcitability at 150 min after NE (Fig. 9D). Thus, after exposure to NE, PKD, Gβγ, and new protein synthesis are necessary for the restoration of protease-evoked hyperexcitability. This result is consistent with the role of PKD and Gβγ in mobilization of newly synthesized PAR2 from the Golgi apparatus and replenishment of the plasma membrane with fresh receptors that are required for recovery of protease signaling.
PKD and Gβγ are required for sustained nociception to proteases that activate PAR2 by canonical and biased mechanisms
The local (intraplantar) injection of trypsin, CS, and NE causes persistent mechanical allodynia in mice by activating PAR2 on nociceptors (18). Whereas PAR2 signaling from endosomes mediates trypsin-induced allodynia, PAR2 signaling from the plasma membrane mediates CS- and NE-induced allodynia. Because cleaved PAR2 cannot be reactivated by proteases and is eventually degraded, the mobilization of fresh PAR2 may be necessary for the continuation of protease-evoked pain. To examine this possibility, we administered to mice inhibitors of PKD and Gβγ. The PKD inhibitor CRT0066101 (80 mg/kg, orally), the Gβγ inhibitor gallein (100 mg/kg, intraperitoneally), or vehicle (control) was administered to mice. After 2 h (CRT0066101) or 30 min (gallein), mice received an intraplantar injection of trypsin (140 nm, 0.04 units/μl), CS (2.5 μm, 0.06 units/μl), or NE (1.18 μm, 0.03 units/μl) (10 μl). Paw withdrawal responses to stimulation of the plantar surface with calibrated von Frey filaments (VFF) were measured every hour for 4 h after protease injection. In vehicle-treated mice, trypsin, CS, and NE caused mechanical allodynia of the ipsilateral (protease-injected) paw after 1 h that was sustained for 4 h (Fig. 10). CRT0066101 significantly blunted responses to trypsin, CS, and NE from 1 to 2 h (Fig. 10, A–C). Gallein also inhibited trypsin-, CS-, and NE-induced allodynia (Fig. 9, E–G). CRT0066101 and gallein did not affect withdrawal responses of the contralateral (noninjected) paw (Fig. 10, D and H). These results reveal a role for PKD and Gβγ in sustained protease-mediated mechanical allodynia in mice.
Figure 10.
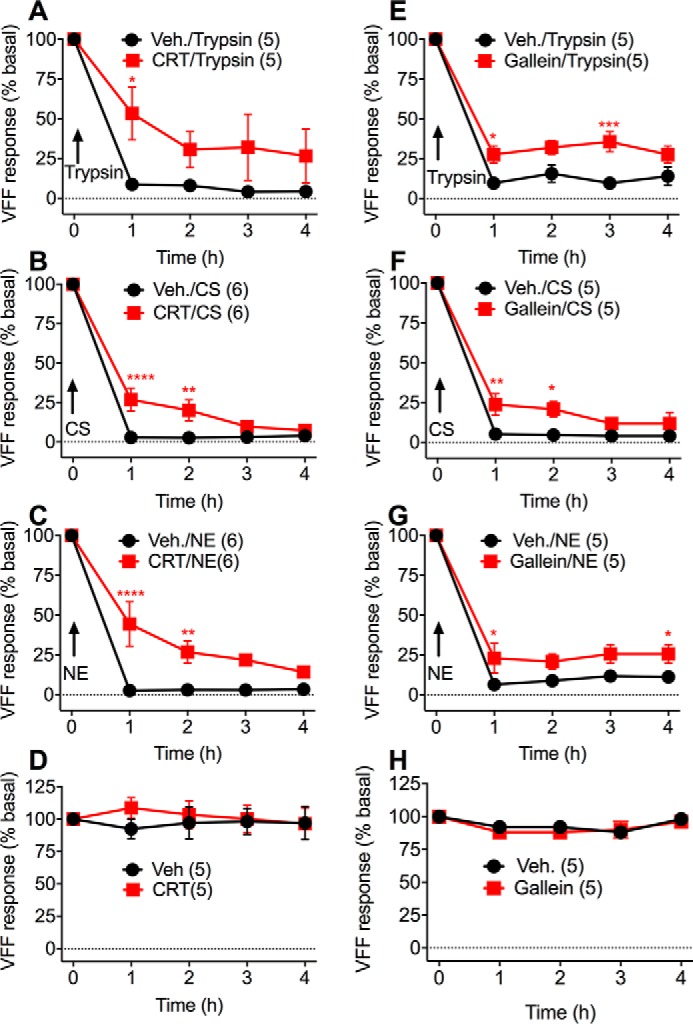
PKD and Gβγ-dependent mechanical hyperalgesia. Mice were pretreated with CRT0066101 (80 mg/kg, orally), gallein (100 mg/kg, intraperitoneally), or vehicle (orally or intraperitoneally) for 2 h (CRT0066101) or 30 min (gallein) before intraplantar injection of trypsin (140 nm) (A, E, D, and H), CS (2.5 μm) (B and F), or NE (1.18 μm) (C and G) (all 10 μl). A–C and E–G, VFF withdrawal responses of the ipsilateral protease-injected paws. D and H, VFF withdrawal responses of the contralateral noninjected paws. Numbers in parentheses indicate mouse numbers. *, p < 0.05; **, p > 0.01; ***, p > 0.001; ****, p < 0.0001 (two-way ANOVA, Bonferroni multiple comparisons). Error bars, S.E.
Discussion
We have recently investigated the mechanisms by which trypsin, CS, and NE initiate pain (18). After intraplantar injection in mice, trypsin, CS and NE cause mechanical allodynia by activating PAR2 on NaV1.8-positive nociceptors. These proteases also evoke hyperexcitability of nociceptors, a characteristic of chronic pain, but by distinct mechanisms (18). Trypsin induces an initial hyperexcitability of nociceptors by a mechanism that entails PAR2 signaling at the plasma and activation of PKC. PAR2 then internalizes and signals from endosomes by β-arrestin– and Gαq–mediated mechanisms that activate extracellular signal–regulated kinase, which contributes to sustained hyperexcitability of nociceptors. In contrast, CS and NE evoke hyperexcitability by PAR2 signaling at the plasma membrane, which activates PKA (14, 15, 18). Accordingly, whereas inhibitors of clathrin- and dynamin-mediated endocytosis block trypsin-evoked allodynia, they have no effect on nociceptive responses to CS and NE, which do not cause PAR2 endocytosis (18). Regardless of the mechanism of PAR2 activation and initial hyperexcitability, the maintenance of pain evoked by extracellular proteases requires mobilization of intracellular stores or de novo synthesis of PAR2. These processes are necessary because trypsin-activated PAR2 traffics to lysosomes and is degraded. Even though CS or NE do not cause endocytosis of PAR2, once cleaved, this receptor cannot be reactivated by a protease (14, 15).
Our current results show that proteases that activate PAR2 by biased mechanisms evoke Gβγ translocation to the Golgi apparatus of HEK293 cells. They also indicate that these proteases activate PKD within the Golgi apparatus. Gβγ and PKD promote mobilization of intact PAR2 from the Golgi apparatus, which replenishes the plasma membrane with intact receptors that allow for a recovery of cellular responsiveness to extracellular proteases. These mechanisms also appear to operate in another cell line (KNRK) as well as in primary sensory neurons, where inhibitors of Gβγ and PKD prevented the recovery of trypsin-evoked hyperexcitability of NE-treated neurons. They may also function in intact mice, because Gβγ and PKD inhibitors blunted trypsin-, CS-, and NE-evoked mechanical allodynia. These results agree with our report that Gβγ and PKD mediate trypsin-evoked mobilization of PAR2 from the Golgi apparatus (19). They also support the role for Gβγ in activating PKD within the Golgi apparatus (21), where PKD controls protein trafficking to secretory pathways (26, 27).
The use of FRET biosensors for PKD that are targeted to the cytosol or Golgi apparatus enabled analysis of PKD activity with high spatial and temporal fidelity and revealed that trypsin, CS, and NE can all activate PKD in the cytosol and Golgi apparatus. These responses are likely mediated by PAR2 because the PAR2-selective agonist 2-furoyl-LIGRLO-NH2 also activated PKD in the Golgi apparatus, and I-343, a PAR2 antagonist (18), abolished responses to trypsin, CS, and NE. Although I-343 can also inhibit PAR1, which is expressed in HEK cells, I-343 had no effect on thrombin-activated PKD.
We observed that trypsin, CS, and NE all stimulated an increase in bystander BRET between Gγ-Venus and Giantin-RLuc8, which is consistent with translocation of Gγ to the Golgi apparatus. Whereas the overexpression of Gαq was required for trypsin-evoked translocation of Gγ-Venus, overexpression of GαS was necessary for maximal CS- and NE-evoked translocation. These results are in accordance with the mechanisms by which these proteases activate PAR2. Thus, the canonical agonist trypsin induces PAR2 coupling to Gαq, and the biased agonists CS and NE evoke PAR2 coupling to Gαs (14, 15).
Our studies of the contribution of PKD to mobilization of PAR2 from the Golgi apparatus and sustained signaling by extracellular proteases were not always consistent between different cell types and experimental approaches. In HEK293 cells, we found that trypsin, CS, and NE all activated PKD in the Golgi apparatus, assessed using a Golgi-targeted FRET biosensor. Trypsin, CS, and NE caused a decrease in bystander BRET between PAR2-RLuc8 and TGN38-Venus, which is consistent with mobilization of PAR2 from the Golgi apparatus. By selective photoconversion of PAR2-Kaede within the Golgi apparatus of KNRK cells, we were able to observe that CS and NE stimulated removal of PAR2 from the Golgi apparatus and insertion into the plasma membrane. Inhibitors of PKD and Gβγ attenuated the mobilization of PAR2-Kaede from the Golgi apparatus of KNRK cells (confocal imaging). They also suppressed recovery of sustained PAR2 signaling in HEK293 cells (Ca2+ assays) and neurons (rheobase assays). Although a PKD inhibitor suppressed removal of PAR2-Kaede from the Golgi apparatus of KNRK cells, it did not affect the decrease in bystander BRET between PAR2-RLuc8 and TGN38-Venus in HEK293 cells. We have no explanation for this discrepancy. One possibility is that PKD promotes mobilization of PAR2 from the Golgi apparatus of KNRK cells but not HEK293 cells. This discrepancy could also be related to differences in assay sensitivity, where an effect of the PKD inhibitor could be assessed in assays of PAR2-Kaede mobilization in individual KNRK cells but not in BRET assays of PAR2-RLuc8 and TGN38-Venus proximity in populations of HEK293 cells. Another explanation could be that TGN38 is not confined to the Golgi apparatus. Although at steady state TGN38 is principally localized to the Golgi apparatus, it constitutively shuttles between the Golgi apparatus and plasma membrane by the exocytic and endocytic pathways (23). It is therefore possible that proximity between TGN38 and PAR2 in these pathways may confound the results. It is also possible that multiple kinases, in addition to PKD, regulate PAR2 trafficking from the Golgi apparatus or that PKD differentially controls trafficking of newly synthesized but not stored PAR2. Further experiments are required to examine these possibilities. However, the observation that Gβγ and PKD inhibitors disrupt the recovery of PAR2 Ca2+ signaling in HEK293 cells pretreated with trypsin, CS, or NE supports their role in maintaining the capacity of extracellular proteases to signal via PAR2.
It is increasingly appreciated that GPCRs can generate signals in subcellular compartments. Compartmentalization of signaling can regulate specific physiological processes, including pain transmission (18, 35, 36). GPCRs can also activate PKD in distinct subcellular compartments, often by different mechanisms. Thus, PAR2 agonists induce PKC-dependent activation of PKD at the plasma membrane, where PKD may regulate the activity of transient receptor potential ion channels (37). Vasopressin, bombesin, and neurokinin 3 receptors rapidly activate PKD by a PKC-dependent mechanism (38–40). Canonical and biased agonists of PAR2 also cause Gβγ-dependent activation of PKD in the Golgi apparatus, where PKD can control PAR2 mobilization (19). Gαq subunits can also control PKD activity and regulate trafficking from the Golgi apparatus to the plasma membrane (29, 41).
Biased agonism describes the phenomenon whereby the binding of different ligands to the same receptor in an identical cellular background results in differential activation of signaling pathways (42). Proteases, which are activated during injury and inflammation, that cleave PAR2 at different sites activate very distinct pathways of receptor signaling and trafficking (14, 15, 18). Our results reveal a common mechanism of recovery of PAR2 signaling capacity at the plasma membrane, regardless of the mechanism of activation. Thus, after activation of PAR2 by trypsin (PAR2 coupling to Gαq, Gαs, β-arrestins, PAR2 endocytosis), CS, or NE (PAR2 coupling to Gαs, retention at cell surface), Gβγ-mediated activation of PKD within the Golgi apparatus mediates the mobilization and plasma membrane trafficking of fresh PAR2 that sustains signaling by extracellular proteases. Other GPCR family members that couple to different Gα subunits also evoke trafficking of Gβγ to the Golgi apparatus. For example, agonists of muscarinic M2 receptors (which couple to Gαi/o) and M3 receptors (which couple to Gαq) both evoke Golgi translocation of Gβγ (43). This may be a general mechanism by which activation of GPCRs at the plasma membrane repopulates the cell surface with nondesensitized receptors.
Our results also provide information about the maintenance of pain. We observed that brief exposure of nociceptors to NE induced an initial period of hyperexcitability that gradually recovered to baseline levels, when neurons recovered their capacity to respond to trypsin. Inhibitors of PKD and Gβγ both impeded this recovery. An inhibitor of protein synthesis also impeded the recovery of trypsin-evoked hyperexcitability of NE-treated neurons. The systemic administration of PKD and Gβγ inhibitors to intact mice also blunted nociception evoked by intraplantar administration of trypsin, CS, or NE. These results highlight the importance of GPCR trafficking for pain transmission. Endocytosis of GPCRs, including PAR2, is important for the initiation and maintenance of pain transmission (18, 35, 36). However, in the case of nonrecycling GPCRs, notably PAR2, synthesis of new receptors and mobilization and exocytosis of intact receptors is necessary for the maintenance of nociceptive signaling by extracellular ligands. These findings have implications for therapy. Inhibitors of clathrin- and dynamin-mediated endocytosis and lipid-conjugated antagonists that target GPCRs in endosomes can provide effective pain relief (18, 35, 36). Inhibitors of exocytosis, including Gβγ and PKD inhibitors, may also provide pain relief.
Limitations to the current study include the use of model HEK293 and KNRK cells, which may not replicate findings in nociceptive neurons and intact animals, possible confounding effects of tagging PAR2 with luciferase or Kaede, and the use of pharmacological inhibitors that can lack selectivity (32, 33). However, we observed that inhibitors of different mediators of Golgi-to-plasma membrane trafficking (PKD and Gβγ) gave consistent results in experiments with model cell lines, primary sensory neurons in short-term culture, and intact mice. Moreover, fusion to Kaede does not affect the function of PAR2 (19).
In summary, this study provides evidence that proteases that activate PAR2 by canonical and biased mechanisms stimulate PKD in the Golgi; PAR2 mobilization and de novo synthesis repopulate the cell surface with intact receptors and sustain nociceptive signaling by extracellular proteases. Antagonism of these pathways may offer another approach to treat chronic pain.
Experimental procedures
Animals
Animal Ethics Committees of Monash University and Queen's University approved the procedures using mice. C57BL/6 mice (8–12 weeks, male) were studied. Mice were maintained in a light-controlled (12-h light/dark cycle) temperature-controlled (22 ± 4 °C) environment with free access to food and water.
Cell culture
HEK293 cells and KNRK cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and containing 1% penicillin and streptomycin (5% CO2, 37 °C). HEK293 and KNRK cells expressing human PAR2 with N-terminal FLAG and C-terminal HA11 epitopes (FLAG-PAR2-HA) have been described previously (19).
FRET PKD and PKC activation assays
HEK293 cells were plated at a density of 4 × 106 cells/10-cm dish. Cells were transiently transfected with the hPAR2 (1 μg) plus FRET PKD biosensors Cyto-DKAR, Golgi-DKAR, or DKAR-T/A (2.5 or 5 μg) or PKC sensor Cyto-CKAR (5 μg) using polyethyleneimine (20). At 24 h after transfection, cells were seeded on poly-d-lysine–coated OptiPlate-96 96-well plates (PerkinElmer Life Sciences). FRET was assessed 72 h post-transfection, following overnight serum restriction. Cells were equilibrated with HBSS, 10 mm HEPES, pH 7.4, for 30 min. Cyan fluorescent protein/yellow fluorescent protein FRET was measured using a PHERAstar plate reader (BMG LabTech) with excitation wavelength 425 ± 5 nm, and emission wavelength 550 ± 25 nm/490 ± 10 nm (18, 35, 36). Cells were challenged with trypsin (10 nm), CS (100 nm), or NE (100 nm), and measurements continued for 20 min at 1-min intervals. In some experiments, cells were pre-incubated with the PAR2-antagonist I-343 (10 μm) for 30 min, and antagonist was present throughout the experiments. For kinetic measurements, data were corrected to baseline and vehicle controls. The area under the curve (AUC, 20 min) was determined to quantify the effects of inhibitors.
BRET Gγ translocation assays
HEK293 cells were plated at a density of 4 × 106 cells/10-cm dish. Cells were transiently transfected with hPAR2 (0.9 μg), Gαq (1.33 μg) or Gαs (1.33 μg), Gβ1 (1.33 μg), Gγ2-Venus (2 μg), and Giantin-RLuc8 (0.5 μg) using polyethyleneimine. At 24 h after transfection, cells were seeded on poly-d-lysine–coated OptiPlate-96 96-well plates (PerkinElmer Life Sciences), and cultured overnight. Cells were equilibrated with HBSS, 10 mm HEPES, pH 7.4, for 30 min before the assay. Coelenterazine h (5 μm, Nanolight Technologies) was added to the cells for 5 min before measurements. RLuc8 luminescence (480 nm) and Venus fluorescence (530 nm) were measured using a LumiSTAR Omega Luminometer (BMG LabTech) before and after exposure to proteases: trypsin, 10−13 to 10−7 m; NE, 10−12 to 10−7 m; CS, 10−12 to 10−7 m; or vehicle (control) (18, 35, 36). Data were corrected to baseline and vehicle-treated conditions to generate the ligand-induced signal, which was normalized to the maximum trypsin response.
BRET PAR2 Golgi trafficking assays
HEK293 cells were transiently transfected with hPAR2-RLuc8 (1 μg) and TGN38-Venus (4 μg). BRET was measured as described above.
[Ca2+]i assays
HEK293 cells were plated (30,000 cells/well) in poly-d-lysine–coated ViewPlate-96 96-well plates (PerkinElmer Life Sciences). Cells were loaded with Fura-2/AM (1 μm; Invitrogen) for 1 h at 37 °C in assay buffer (150 mm NaCl, 2.6 mm KCl, 0.1 mm CaCl2, 1.18 mm MgCl2, 10 mm d-glucose, 10 mm HEPES, pH 7.4) containing 4 mm probenecid and 0.5% BSA. Fluorescence was measured at 340- and 380-nm excitation and 530-nm emission using a FlexStation III Microplate Reader (Molecular Devices) (19). The 340- and 380-nm ratio was measured as an indication of [Ca2+]i. To measure loss and recovery of PAR2-mediated changes in [Ca2+]i, cells were first incubated with trypsin (10 nm), CS (100 nm), NE (100 nm), or vehicle for 10 min, washed three times with assay buffer, recovered at 37 °C for 110 min, and then rechallenged with trypsin (10 nm) at 120 min after the first challenge. For quantification of the recovery of Ca2+ signaling, the maximal responses to the second trypsin challenge were normalized to the vehicle-pretreated control.
Localization of FRET and BRET biosensors
HEK293 cells were transfected as stated with PAR2-HA-RLuc8, Cyto-DKAR, Golgi-DKAR, Giantin-Venus, TGN38-Venus, or Gγ-Venus (2.5 μg). Cells were plated on 12-mm glass coverslips, and 48 h after transfection, cells were fixed in 4% paraformaldehyde (20 min, 4 °C). Cells were washed with PBS and incubated with blocking buffer (PBS + 0.3% saponin + 3% horse serum) (1 h, room temperature). PAR2-HA-RLuc8 was detected using immunofluorescence with HA antibody; other sensors were detected with fluorescent tags. Cells were incubated with rat anti-HA (1:1000; Roche Applied Science, clone 3F10), and mouse anti-TGN58K (1:200; Abcam) in PBS + 0.2% saponin + 1% horse serum (1 h, room temperature). Cells were washed three times in PBS and incubated with goat anti-rat Alexa 488 and donkey anti-mouse Alexa 568 IgG (1:1000; Invitrogen) (1 h, room temperature). Slides were washed three times in PBS, counterstained with 4′,6-diamidino-2-phenylindole (1 μg/ml), and mounted with ProLong Glass antifade mounting medium (Invitrogen). Cells were imaged on a Leica SP8 confocal microscope with a ×63 (numerical aperture 1.4) objective with a digital zoom of 2.5. Images were processed with ImageJ (National Institutes of Health).
Live-cell imaging and quantification of PAR2-Kaede trafficking
KNRK-PAR2-Kaede cells were plated onto glass 8-well chamber slides at a density of 100,000 cells/well and cultured overnight. Cells were equilibrated in HBSS, 10 mm HEPES, pH 7.4, at 37 °C for 30 min before the assay. Live-cell images were collected using a Leica TCS SP8 laser-scanning confocal microscope with a Leica HCX PL APO ×63 oil immersion objective.
The Leica FRAP wizard was used to green/red photoconvert PAR2-Kaede within the perinuclear region of interest with the laser settings of pre-photoconversion image at 400 Hz, UV laser at 10% power, and two passes at 400 Hz, as we have described previously (19). Images were collected for 5 min, cells were challenged with CS (100 nm) or NE (100 nm), and images were collected at 5-min intervals for another 30 min. FIJI software (National Institutes of Health) was used to determine the intensity of PAR2-Kaede signal at the plasma membrane, in the perinuclear region, and throughout the cell. To quantify the depletion of PAR2 from the plasma membrane, the intensities of Kaede green at the plasma membrane and throughout the entire cell were determined and expressed as a ratio. To quantify the depletion of PAR2 from the Golgi apparatus and repletion of the plasma membrane with mobilized PAR2, the intensities of Kaede red were determined at the plasma membrane, in the perinuclear region of the cell, and throughout the cell and were expressed as ratios.
Inhibitors
Cells were pre-incubated with CRT0066101 (100 nm; Tocris Bioscience), gallein (10 μm; Tocris Bioscience), Gö6983 (1 μm; Sigma-Aldrich), H-89 (1 μm; Sigma-Aldrich), GRK2i (100 μm; Tocris), or vehicle (control) for 30 min, and the inhibitors were present throughout the experiments.
Patch-clamp studies of nociceptors
To obtain dispersed neurons, mouse DRG (T9–T13) were incubated in collagenase (1 mg/ml; Worthington) and dispase (4 mg/ml; Roche Applied Science) (10 min, 37 °C) and triturated with a fire-polished Pasteur pipette. Dispersed cells were seeded onto laminin-coated (0.017 mg/ml) and poly-d-lysine–coated (2 mg/ml) glass coverslips in 24-well plates. Neurons were maintained in F12 medium (Sigma-Aldrich) supplemented with 10% fetal calf serum and containing penicillin and streptomycin (5% CO2, 37 °C) for 16 h. Neurons were pre-incubated for 20 min with NE (7.8 nm, 0.2 units/ml) or vehicle (control) and washed three times with F12 medium. Changes in neuronal excitability were assayed immediately (0 min) or at 30 and 150 min after NE application. After 150 min, some cells were challenged with trypsin (50 nm, 10 min) and washed. To evaluate the role of PKD, Gβγ, and new protein synthesis on NE-evoked neural excitability, some cells were pre-incubated with CRT0066101 (10 nm), gallein (10 μm), cycloheximide (10 μg/ml), or vehicle (control) 30 min before NE application, and inhibitors were included throughout the study. The excitability of small-diameter neurons (<30-picofarad capacitance) was assessed using perforated patch-clamp with amphotericin B (240 μg/ml) in current clamp mode at room temperature, as we have described (18, 19). Only those neurons with resting membrane potentials more negative than −40 mV were analyzed. Changes in excitability were quantified by measuring the rheobase. Recordings were made using Axopatch 200B amplifiers, digitized by Digidata 1322A, and stored and processed using pClamp 10.1 software (Molecular Devices). The recording chamber was continuously perfused (2 ml/min) with external solution comprising 140 mm NaCl, 5 mm KCl, 10 mm HEPES, 10 mm d-glucose, 1 mm MgCl2, 2 mm CaCl2, pH to 7.4 with 3 m NaOH. The pipette solution was as follows: 110 mm potassium gluconate, 30 mm KCl, 10 mm HEPES, 1 mm MgCl2, 2 mm CaCl2, pH 7.25, with 1 m KOH.
Mechanical hyperalgesia
Mechanical hyperalgesia was assessed by measuring paw withdrawal responses to stimulation of the plantar surface of the mouse paw with calibrated VFF (14, 15, 18). CRT0066101 (Tocris Bioscience; 80 mg/kg) or vehicle (control, 0.9% NaCl) was administered by gavage (200 μl) 2 h before intraplantar injections of proteases. Gallein (100 mg/kg) or vehicle (control, 0.5% DMSO in 0.9% NaCl) was administered intraperitoneally (200 μl) 30 min before intraplantar injection of proteases. For intraplantar injections, mice were sedated with 5% isoflurane. Trypsin (140 nm, 0.04 units/μl), CS (2.5 μm, 0.06 units/μl), NE (1.18 μm, 0.03 units/μl), or vehicle was injected subcutaneously into the plantar surface of the left hind paw (10 μl). Withdrawal responses to stimulation of the plantar surface of the ipsilateral (injected) and contralateral (noninjected) hind paws with calibrated VFF were measured hourly for 4 h. Investigators were blinded to the experimental treatments, and mice were randomly assigned to treatment groups.
Statistics
Data are presented as mean ± S.E. For studies of cells, triplicate observations from n > 3 experiments were made for each treatment group. For experiments with isolated neurons, n = 7–27 DRG neurons from 6–13 mice were studied per treatment. For studies of nociceptive behavior, n = 5–6 mice were studied per treatment. Student's t tests were used to assess differences between two groups. Differences between multiple groups were assessed by one-way (FRET, BRET, electrophysiology) or two-way (mouse behavior) ANOVAs followed by Bonferroni's correction for multiple comparisons. p < 0.05 was considered significant at the 95% confidence level.
Author contributions
P. Z., D. P. P., S. J. V., B. L. S., and N. W. B. conceptualization; P. Z., L. A. P., D. D. J., N. N. J.-V., R. L., T. L., J. O. J., and C. L.-L. data curation; P. Z., L. A. P., D. D. J., N. N. J.-V., R. L., and T. L. formal analysis; P. Z., D. P. P., S. J. V., B. L. S., and N. W. B. supervision; P. Z., L. A. P., D. D. J., N. N. J.-V., R. L., T. L., S. J. V., B. L. S., and N. W. B. investigation; P. Z., D. D. J., N. N. J.-V., and R. L. visualization; P. Z., L. A. P., D. D. J., N. N. J.-V., R. L., T. L., and N. W. B. methodology; P. Z., L. A. P., D. D. J., R. L., and N. W. B. writing-original draft; D. D. J., N. N. J.-V., R. L., and T. L. validation; D. D. J., D. P. P., S. J. V., B. L. S., and N. W. B. project administration; D. P. P., S. J. V., B. L. S., and N. W. B. funding acquisition; D. P. P., S. J. V., B. L. S., and N. W. B. writing-review and editing; S. J. V., B. L. S., and N. W. B. resources.
This work was supported by National Institutes of Health Grants NS102722, DE026806, and DK118971; Department of Defense Grant W81XWH1810431 (to N. W. B. and B. L. S.); and National Health and Medical Research Council Grants 63303, 1049682, and 1031886) (to N. W. B.). Nigel Bunnett is a founding scientist of Endosome Therapeutics Inc. Research in Nigel Bunnett's laboratory was funded in part by Takeda Pharmaceuticals Inc. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- GPCR
- G protein–coupled receptor
- PAR
- protease-activated receptor
- NE
- neutrophil elastase
- CS
- cathepsin-S
- PKC
- protein kinase C
- PKA
- protein kinase A
- PKD
- protein kinase D
- PAR
- protease-activated receptor
- hPAR
- human PAR
- BRET
- bioluminescence resonance energy transfer
- DRG
- dorsal root ganglia
- VFF
- von Frey filament(s)
- HBSS
- Hanks' balanced salt solution
- AUC
- area under the curve
- HA
- hemagglutinin
- ANOVA
- analysis of variance.
References
- 1. Dong C., Filipeanu C. M., Duvernay M. T., and Wu G. (2007) Regulation of G protein-coupled receptor export trafficking. Biochim. Biophys. Acta 1768, 853–870 10.1016/j.bbamem.2006.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Smith J. S., and Rajagopal S. (2016) The β-arrestins: multifunctional regulators of G protein-coupled receptors. J. Biol. Chem. 291, 8969–8977 10.1074/jbc.R115.713313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hauser A. S., Attwood M. M., Rask-Andersen M., Schiöth H. B., and Gloriam D. E. (2017) Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov. 16, 829–842 10.1038/nrd.2017.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kang D. S., Tian X., and Benovic J. L. (2014) Role of β-arrestins and arrestin domain-containing proteins in G protein-coupled receptor trafficking. Curr. Opin. Cell Biol. 27, 63–71 10.1016/j.ceb.2013.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grady E. F., Garland A. M., Gamp P. D., Lovett M., Payan D. G., and Bunnett N. W. (1995) Delineation of the endocytic pathway of substance P and its seven-transmembrane domain NK1 receptor. Mol. Biol. Cell 6, 509–524 10.1091/mbc.6.5.509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. von Zastrow M., Svingos A., Haberstock-Debic H., and Evans C. (2003) Regulated endocytosis of opioid receptors: cellular mechanisms and proposed roles in physiological adaptation to opiate drugs. Curr. Opin. Neurobiol. 13, 348–353 10.1016/S0959-4388(03)00069-2 [DOI] [PubMed] [Google Scholar]
- 7. Marchese A., and Benovic J. L. (2001) Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR4 mediates lysosomal sorting. J. Biol. Chem. 276, 45509–45512 10.1074/jbc.C100527200 [DOI] [PubMed] [Google Scholar]
- 8. Ossovskaya V. S., and Bunnett N. W. (2004) Protease-activated receptors: contribution to physiology and disease. Physiol. Rev. 84, 579–621 10.1152/physrev.00028.2003 [DOI] [PubMed] [Google Scholar]
- 9. Bohm S. K., Kong W., Bromme D., Smeekens S. P., Anderson D. C., Connolly A., Kahn M., Nelken N. A., Coughlin S. R., Payan D. G., and Bunnett N. W. (1996) Molecular cloning, expression and potential functions of the human proteinase-activated receptor-2. Biochem. J. 314, 1009–1016 10.1042/bj3141009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cottrell G. S., Amadesi S., Grady E. F., and Bunnett N. W. (2004) Trypsin IV, a novel agonist of protease-activated receptors 2 and 4. J. Biol. Chem. 279, 13532–13539 10.1074/jbc.M312090200 [DOI] [PubMed] [Google Scholar]
- 11. Corvera C. U., Déry O., McConalogue K., Böhm S. K., Khitin L. M., Caughey G. H., Payan D. G., and Bunnett N. W. (1997) Mast cell tryptase regulates rat colonic myocytes through proteinase-activated receptor 2. J. Clin. Invest. 100, 1383–1393 10.1172/JCI119658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oikonomopoulou K., Hansen K. K., Saifeddine M., Tea I., Blaber M., Blaber S. I., Scarisbrick I., Andrade-Gordon P., Cottrell G. S., Bunnett N. W., Diamandis E. P., and Hollenberg M. D. (2006) Proteinase-activated receptors, targets for kallikrein signaling. J. Biol. Chem. 281, 32095–32112 10.1074/jbc.M513138200 [DOI] [PubMed] [Google Scholar]
- 13. Ramachandran R., Mihara K., Chung H., Renaux B., Lau C. S., Muruve D. A., DeFea K. A., Bouvier M., and Hollenberg M. D. (2011) Neutrophil elastase acts as a biased agonist for proteinase-activated receptor-2 (PAR2). J. Biol. Chem. 286, 24638–24648 10.1074/jbc.M110.201988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhao P., Lieu T., Barlow N., Sostegni S., Haerteis S., Korbmacher C., Liedtke W., Jimenez-Vargas N. N., Vanner S. J., and Bunnett N. W. (2015) Neutrophil elastase activates protease-activated receptor-2 (PAR2) and transient receptor potential vanilloid 4 (TRPV4) to cause inflammation and pain. J. Biol. Chem. 290, 13875–13887 10.1074/jbc.M115.642736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhao P., Lieu T., Barlow N., Metcalf M., Veldhuis N. A., Jensen D. D., Kocan M., Sostegni S., Haerteis S., Baraznenok V., Henderson I., Lindström E., Guerrero-Alba R., Valdez-Morales E. E., Liedtke W., et al. (2014) Cathepsin S causes inflammatory pain via biased agonism of PAR2 and TRPV4. J. Biol. Chem. 289, 27215–27234 10.1074/jbc.M114.599712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Böhm S. K., Khitin L. M., Grady E. F., Aponte G., Payan D. G., and Bunnett N. W. (1996) Mechanisms of desensitization and resensitization of proteinase-activated receptor-2. J. Biol. Chem. 271, 22003–22016 10.1074/jbc.271.36.22003 [DOI] [PubMed] [Google Scholar]
- 17. Déry O., Thoma M. S., Wong H., Grady E. F., and Bunnett N. W. (1999) Trafficking of proteinase-activated receptor-2 and β-arrestin-1 tagged with green fluorescent protein: β-arrestin-dependent endocytosis of a proteinase receptor. J. Biol. Chem. 274, 18524–18535 10.1074/jbc.274.26.18524 [DOI] [PubMed] [Google Scholar]
- 18. Jimenez-Vargas N. N., Pattison L. A., Zhao P., Lieu T., Latorre R., Jensen D. D., Castro J., Aurelio L., Le G. T., Flynn B., Herenbrink C. K., Yeatman H. R., Edgington-Mitchell L., Porter C. J. H., Halls M. L., et al. (2018) Protease-activated receptor-2 in endosomes signals persistent pain of irritable bowel syndrome. Proc. Natl. Acad. Sci. U.S.A. 115, E7438–E7447 10.1073/pnas.1721891115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jensen D. D., Zhao P., Jimenez-Vargas N. N., Lieu T., Gerges M., Yeatman H. R., Canals M., Vanner S. J., Poole D. P., and Bunnett N. W. (2016) Protein kinase D and Gβγ subunits mediate agonist-evoked translocation of protease-activated receptor-2 from the Golgi apparatus to the plasma membrane. J. Biol. Chem. 291, 11285–11299 10.1074/jbc.M115.710681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kunkel M. T., Toker A., Tsien R. Y., and Newton A. C. (2007) Calcium-dependent regulation of protein kinase D revealed by a genetically encoded kinase activity reporter. J. Biol. Chem. 282, 6733–6742 10.1074/jbc.M608086200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jamora C., Yamanouye N., Van Lint J., Laudenslager J., Vandenheede J. R., Faulkner D. J., and Malhotra V. (1999) Gβγ-mediated regulation of Golgi organization is through the direct activation of protein kinase D. Cell 98, 59–68 10.1016/S0092-8674(00)80606-6 [DOI] [PubMed] [Google Scholar]
- 22. Linstedt A. D., and Hauri H. P. (1993) Giantin, a novel conserved Golgi membrane protein containing a cytoplasmic domain of at least 350 kDa. Mol. Biol. Cell 4, 679–693 10.1091/mbc.4.7.679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Banting G., and Ponnambalam S. (1997) TGN38 and its orthologues: roles in post-TGN vesicle formation and maintenance of TGN morphology. Biochim. Biophys. Acta 1355, 209–217 10.1016/S0167-4889(96)00146-2 [DOI] [PubMed] [Google Scholar]
- 24. Bonacci T. M., Mathews J. L., Yuan C., Lehmann D. M., Malik S., Wu D., Font J. L., Bidlack J. M., and Smrcka A. V. (2006) Differential targeting of Gβγ-subunit signaling with small molecules. Science 312, 443–446 10.1126/science.1120378 [DOI] [PubMed] [Google Scholar]
- 25. Koch W. J., Hawes B. E., Inglese J., Luttrell L. M., and Lefkowitz R. J. (1994) Cellular expression of the carboxyl terminus of a G protein-coupled receptor kinase attenuates Gβγ-mediated signaling. J. Biol. Chem. 269, 6193–6197 [PubMed] [Google Scholar]
- 26. Liljedahl M., Maeda Y., Colanzi A., Ayala I., Van Lint J., and Malhotra V. (2001) Protein kinase D regulates the fission of cell surface destined transport carriers from the trans-Golgi network. Cell 104, 409–420 10.1016/S0092-8674(01)00228-8 [DOI] [PubMed] [Google Scholar]
- 27. Yeaman C., Ayala M. I., Wright J. R., Bard F., Bossard C., Ang A., Maeda Y., Seufferlein T., Mellman I., Nelson W. J., and Malhotra V. (2004) Protein kinase D regulates basolateral membrane protein exit from trans-Golgi network. Nat. Cell Biol. 6, 106–112 10.1038/ncb1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Harikumar K. B., Kunnumakkara A. B., Ochi N., Tong Z., Deorukhkar A., Sung B., Kelland L., Jamieson S., Sutherland R., Raynham T., Charles M., Bagherazadeh A., Foxton C., Boakes A., Farooq M., et al. (2010) A novel small-molecule inhibitor of protein kinase D blocks pancreatic cancer growth in vitro and in vivo. Mol. Cancer Ther. 9, 1136–1146 10.1158/1535-7163.MCT-09-1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Coria A. S., Masseroni M. L., and Díaz Añel A. M. (2014) Regulation of PKD1-mediated Golgi to cell surface trafficking by Gαq subunits. Biol. Cell 106, 30–43 10.1111/boc.201300052 [DOI] [PubMed] [Google Scholar]
- 30. Gschwendt M., Dieterich S., Rennecke J., Kittstein W., Mueller H.-J., and Johannes F.-J. (1996) Inhibition of protein kinase C μ by various inhibitors. Inhibition from protein kinase C isoenzymes. FEBS Lett. 392, 77–80 10.1016/0014-5793(96)00785-5 [DOI] [PubMed] [Google Scholar]
- 31. Muñiz M., Martín M. E., Hidalgo J., and Velasco A. (1997) Protein kinase A activity is required for the budding of constitutive transport vesicles from the trans-Golgi network. Proc. Natl. Acad. Sci. U.S.A. 94, 14461–14466 10.1073/pnas.94.26.14461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bain J., Plater L., Elliott M., Shpiro N., Hastie C. J., McLauchlan H., Klevernic I., Arthur J. S., Alessi D. R., and Cohen P. (2007) The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297–315 10.1042/BJ20070797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Davies S. P., Reddy H., Caivano M., and Cohen P. (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351, 95–105 10.1042/0264-6021:3510095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. DeFea K. A., Zalevsky J., Thoma M. S., Déry O., Mullins R. D., and Bunnett N. W. (2000) β-Arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell Biol. 148, 1267–1281 10.1083/jcb.148.6.1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jensen D. D., Lieu T., Halls M. L., Veldhuis N. A., Imlach W. L., Mai Q. N., Poole D. P., Quach T., Aurelio L., Conner J., Herenbrink C. K., Barlow N., Simpson J. S., Scanlon M. J., Graham B., et al. (2017) Neurokinin 1 receptor signaling in endosomes mediates sustained nociception and is a viable therapeutic target for prolonged pain relief. Sci. Transl. Med. 9, eaal3447 10.1126/scitranslmed.aal3447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yarwood R. E., Imlach W. L., Lieu T., Veldhuis N. A., Jensen D. D., Klein Herenbrink C., Aurelio L., Cai Z., Christie M. J., Poole D. P., Porter C. J. H., McLean P., Hicks G. A., Geppetti P., Halls M. L., et al. (2017) Endosomal signaling of the receptor for calcitonin gene-related peptide mediates pain transmission. Proc. Natl. Acad. Sci. U.S.A. 114, 12309–12314 10.1073/pnas.1706656114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Amadesi S., Grant A. D., Cottrell G. S., Vaksman N., Poole D. P., Rozengurt E., and Bunnett N. W. (2009) Protein kinase D isoforms are expressed in rat and mouse primary sensory neurons and are activated by agonists of protease-activated receptor 2. J. Comp. Neurol. 516, 141–156 10.1002/cne.22104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jacamo R., Sinnett-Smith J., Rey O., Waldron R. T., and Rozengurt E. (2008) Sequential protein kinase C (PKC)-dependent and PKC-independent protein kinase D catalytic activation via Gq-coupled receptors: differential regulation of activation loop Ser744 and Ser748 phosphorylation. J. Biol. Chem. 283, 12877–12887 10.1074/jbc.M800442200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Poole D. P., Amadesi S., Rozengurt E., Thacker M., Bunnett N. W., and Furness J. B. (2008) Stimulation of the neurokinin 3 receptor activates protein kinase C epsilon and protein kinase D in enteric neurons. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G1245–G1256 10.1152/ajpgi.00521.2007 [DOI] [PubMed] [Google Scholar]
- 40. Sinnett-Smith J., Jacamo R., Kui R., Wang Y. M., Young S. H., Rey O., Waldron R. T., and Rozengurt E. (2009) Protein kinase D mediates mitogenic signaling by Gq-coupled receptors through protein kinase C-independent regulation of activation loop Ser744 and Ser748 phosphorylation. J. Biol. Chem. 284, 13434–13445 10.1074/jbc.M806554200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Waldron R. T., Innamorati G., Torres-Marquez M. E., Sinnett-Smith J., and Rozengurt E. (2012) Differential PKC-dependent and -independent PKD activation by G protein α subunits of the Gq family: selective stimulation of PKD Ser748 autophosphorylation by Gαq. Cell. Signal. 24, 914–921 10.1016/j.cellsig.2011.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kenakin T. (2011) Functional selectivity and biased receptor signaling. J. Pharmacol. Exp. Ther. 336, 296–302 10.1124/jpet.110.173948 [DOI] [PubMed] [Google Scholar]
- 43. Akgoz M., Kalyanaraman V., and Gautam N. (2004) Receptor-mediated reversible translocation of the G protein βγ complex from the plasma membrane to the Golgi complex. J. Biol. Chem. 279, 51541–51544 10.1074/jbc.M410639200 [DOI] [PubMed] [Google Scholar]