

Abstract
Cyclic di-AMP (c-di-AMP) is the only second messenger known to be essential for bacterial growth. It has been found mainly in Gram-positive bacteria, including pathogenic bacteria like Listeria monocytogenes. CdaA is the sole diadenylate cyclase in L. monocytogenes, making this enzyme an attractive target for the development of novel antibiotic compounds. Here we report crystal structures of CdaA from L. monocytogenes in the apo state, in the post-catalytic state with bound c-di-AMP and catalytic Co2+ ions, as well as in a complex with AMP. These structures reveal the flexibility of a tyrosine side chain involved in locking the adenine ring after ATP binding. The essential role of this tyrosine was confirmed by mutation to Ala, leading to drastic loss of enzymatic activity.
Keywords: second messenger, cyclic di-AMP (c-di-AMP), X-ray crystallography, prokaryotic signal transduction, metal ion–protein interaction
Introduction
Bacteria have the ability to perceive environmental changes, leading to rapid and effective adaptation by utilizing different proteins as well as second messengers to transduce signals in the cell. In response to external stimuli, the intracellular concentration of second messengers, like cyclic dinucleotides and linear mononucleotides, varies to regulate and coordinate cellular processes (1–3). Cyclic di-AMP (c-di-AMP)2 is the most recently discovered bacterial signaling nucleotide and, to date, has been found mostly in Gram-positive bacteria. c-di-AMP is involved in different cellular processes, such as DNA integrity scanning, cell wall metabolism, and osmolyte homeostasis (for a review, see Refs. 4–6). c-di-AMP is the only essential second messenger in bacteria because of its role in potassium homeostasis. It regulates potassium importers at high intracellular K+ concentrations, whereas c-di-AMP is not essential at low K+ concentrations (7). Interestingly, c-di-AMP becomes toxic when its degradation is blocked; hence, a tightly controlled intracellular c-di-AMP concentration is required for bacterial growth (8).
Proteins containing a diadenylate cyclase (DAC) domain have been bioinformatically identified, mainly in Gram-positive bacteria of the phyla Actinobacteria and Firmicutes but also in Gram-negative Cyanobacteria, Chlamydiae, Bacteroidetes, Fusobacteria, and Deltaproteobacteria and even in archaea of the phylum Euryarchaeota (5). Several DAC domain–containing proteins from various bacterial species have also been experimentally proven to produce c-di-AMP. Many of these bacteria are well-known pathogens, e.g. Mycobacterium tuberculosis (9), Staphylococcus aureus (10) and Listeria monocytogenes (11). In total, eight families of diadenylate cyclases have been identified so far, sharing the highly conserved DAC domain (12). However, DACs differ in their additional domains and domain organization, suggesting that DAC enzymes are regulated by different signals (12).
The three-dimensional structure of a DAC domain was first reported for DisA, a multidomain protein with an N-terminal DAC domain (13). This structure revealed that, within the homo-octameric DisA, two adjacent and properly positioned DAC domains, each with one ATP bound, catalyze the synthesis of c-di-AMP. Based on the homology of all DAC domains, it was proposed that DAC domains with bound ATP need to dimerize in a specific arrangement to catalyze c-di-AMP formation.
The importance of c-di-AMP for the growth of several pathogenic bacteria is marked by an increased resistance to cell wall–targeting antibiotics (10, 14). Its absence in humans makes DAC enzymes an interesting target for the development of novel antibiotics by structure-based drug design. Therefore, CdaA, the only DAC of the human pathogen L. monocytogenes, was previously characterized biochemically and structurally. The analysis revealed that CdaA is active with Co2+ or Mn2+ ions as cofactors but inactive in the presence of Mg2+ ions (15). The CdaA crystal structure unveiled the monomeric and catalytically inactive enzyme–substrate complex with bound ATP and Mg2+, leaving the structure of a dimeric and active form with a bound Co2+ or Mn2+ cofactor still to be determined. Such a crystal structure could shed light on the role of the metal ion in the catalytic reaction.
In this study we report two new crystal structures of CdaA from L. monocytogenes at 2.0 Å and 2.8 Å resolution, representing the enzyme in its apo form and the post-catalytic homodimeric enzyme–product complex, respectively. The structure of CdaA with bound c-di-AMP was obtained by co-crystallization of CdaA in the presence of ATP and Co2+ ions. Comparison of the CdaA structure in the apo state with the ligand-bound forms of CdaA (ATP, AMP, or c-di-AMP) revealed conformational changes of a tyrosine residue present in the active site. Mutation of this tyrosine to alanine abolishes c-di-AMP formation and, thus, demonstrates its functional importance. Furthermore, we confirmed that CdaA is active in the presence of Mn2+ or Co2+ ions, with significantly higher activity in the case of Mn2+, but it is inactive in the presence of Mg2+ ions. These new CdaA structures could serve as an important starting point for future rational drug design.
Results
Structure-based development of novel antibiotic drugs requires high-resolution three-dimensional structures of the targeted enzyme and enzyme–inhibitor complexes. CdaA of the human pathogen L. monocytogenes appears to be an attractive target, as c-di-AMP synthesis is essential for bacterial growth and CdaA is the only DAC in this pathogenic bacterium, whereas there are no DACs in humans. For this study, truncated Δ100CdaA, missing the N-terminal transmembrane (TM) helices and the 20 amino acids linking the TM to the DAC domain, was used because the transmembrane helices hamper the solubility of the recombinant full-length protein. We have demonstrated previously that this truncated Δ100CdaA has preserved its enzymatic activity with a higher enzymatic activity for Co2+ compared with Mn2+ but no activity for of Mg2+ (15). Although, in this previous study, the in vitro activity was measured by LC-MS/MS, we now applied a direct fluorescence-based measurement of c-di-AMP formation by its binding to coralyne (16). In contrast to the results obtained with the LC-MS/MS method, more efficient c-di-AMP synthesis was observed in the presence of Mn2+ compared with Co2+ (Fig. 1A).
Figure 1.

In vitro diadenylate cyclase activity of Δ100CdaA. Presented is a histogram displaying three independent measurements. A control measurement was performed using WT Δ100CdaA without addition of any divalent metal cations. The histogram represents the divalent metal cation preferences of WT Δ100CdaA. The highest amount of c-di-AMP was formed in the presence of MnCl2, whereas, in the presence of CoCl2, the amount of the product is significantly reduced. For MgCl2 and CaCl2, production of c-di-AMP could not be confirmed, as it was within the range of the control. Additionally, it represents the importance of Tyr-187 on catalysis. The mutant Y187A causes a significant reduction (5-fold) of diadenylate cyclase activity, confirming its essential role in c-di-AMP synthesis.
Structure of apo CdaA
One approach for identification of potential inhibitors is crystallographic fragment screening, which desires crystals of CdaA in its apo state. Therefore, Δ100CdaA was crystallized in the absence of ATP and divalent metal ions. Crystals of apo-CdaA were obtained and belong to space group P212121, containing two Δ100CdaA molecules per asymmetric unit. The phase problem was solved by means of molecular replacement using the monomeric Δ100CdaA structure of L. monocytogenes (PDB code 4RV7) as a search model. The resulting crystal structure of apo-CdaA was determined at 2.0 Å resolution (Table 1). The CdaA monomer is composed of a slightly twisted central β-sheet made up of seven mixed-parallel and antiparallel β-strands (β1–β7), flanked on both sides by five α-helices (α1–α5) in total (Fig. 2). The two Δ100CdaA molecules in the asymmetric unit are structurally very similar, as indicated by the root mean square deviation (r.m.s.d.) of 1.19 Å between all Cα positions.
Table 1.
Crystallographic data collection and refinement statistics
| Δ100CdaA with AMP and c-di-AMP | Δ100CdaA-APO | Δ100CdaA_Y187A-APO | |
|---|---|---|---|
| Crystallographic data | |||
| Beamline | Petra III-P14, EMBL, Hamburg | Petra III-P14, EMBL, Hamburg | Petra III-P13, EMBL, Hamburg |
| Wavelength (Å) | 0.97620 | 0.97620 | 0.97625 |
| Resolution range (Å)a | 42.27–2.80 (2.90–2.80) | 45.89–2.00 (2.10–2.00) | 46.49–2.23 (2.33–2.23) |
| Unique reflections | 9,435 | 24,884 | 19,512 |
| Redundancy | 5.6 (5.7) | 7.1 (7.0) | 5.8 (4.2) |
| Completeness (%) | 93.0 (95.4) | 99.7 (98.5) | 97.1 (79.3) |
| Space group | H32 | P212121 | P212121 |
| a (Å) | 121.90 | 42.69 | 46.49 |
| b (Å) | 121.90 | 64.67 | 65.13 |
| c (Å) | 141.59 | 129.75 | 131.33 |
| Rmerge (%) | 10.9 (80.5) | 9.4 (119.0) | 8.0 (52.0) |
| I/σ (I) | 12.4 (1.9) | 13.6 (2.0) | 15.6 (2.8) |
| CC1/2 | 99.8 (72.6) | 99.9 (77.2) | 99.8 (80.5) |
| Refinement statistics | |||
| Rwork/Rfree | 0.1875/0.2337 | 0.1858/0.2245 | 0.1837/0.2258 |
| No. of atoms | 2453 | 2610 | 2740 |
| Average B-factor (Å2) | 58.0 | 47.6 | 39.8 |
| Root mean square deviation | |||
| Bonds Å | 0.003 | 0.008 | 0.006 |
| Angles (°) | 0.644 | 1.003 | 1.258 |
| Ramachandran plot | |||
| Favored (%) | 98.05 | 98.11 | 98.79 |
| Allowed (%) | 1.95 | 1.57 | 1.21 |
| Outlier (%) | 0.00 | 0.31 | 0. 00 |
| PDB codes | 6HVL | 6HVM | 6HVN |
a Values for the data in the highest-resolution shell are shown in parentheses.
Figure 2.
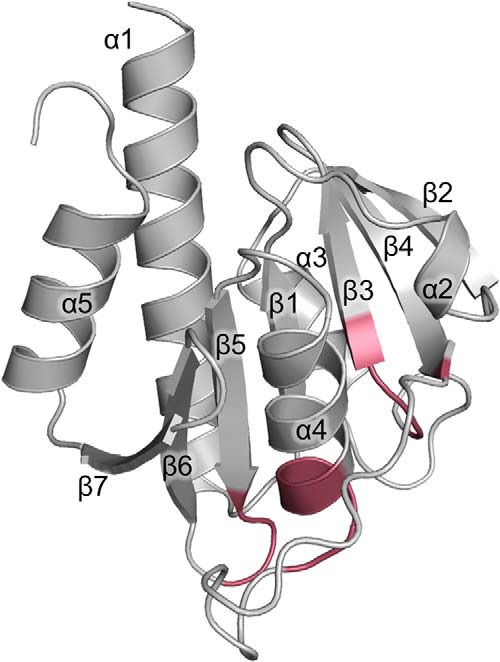
Crystal structure of Δ100CdaA in the apo state, refined at 2 Å resolution. The fold of the CdaA DAC domain consists of seven β-strands forming a central β-sheet surrounded by five α-helices. The positions of residues forming the active site are highlighted in red.
The structure of apo-CdaA closely resembles that of CdaA with bound ATP (PDB code 4RV7), as they exhibit an r.m.s.d. of 1.56 Å, but a few differences are seen in a loop region (residues 137–140) and the C-terminal residues. Careful inspection of the difference electron density map revealed a small molecule bound to the surface of one of two CdaA molecules in the asymmetric unit (Fig. S1). This electron density was interpreted as a sucrose molecule originating from the utilized cryo-protectant solution. In the apo-CdaA crystal structure, the active site is accessible from solvent channels; hence, this crystal form of apo-CdaA appears to be suitable for a fragment screen.
Structure of the CdaA–c-di-AMP complex
To gain more insight into the structure and function of CdaA, we also crystallized Δ100CdaA in the presence of ATP and the cofactor Co2+. The obtained crystals belong to a different space group (H32) than the previously determined structure but also contain two CdaA molecules in the asymmetric unit. The newly obtained crystal structure was determined at 2.8 Å resolution. The two CdaA molecules in the asymmetric unit superpose very well, as the r.m.s.d. calculated between all Cα positions amounts to 0.65 Å. Analysis of the protein contact surfaces in the crystal revealed that one of the two CdaA molecules in the asymmetric unit forms a dimer with a symmetry mate related by a crystallographic two-fold axis (Fig. 3A). This CdaA homodimer corresponds to the catalytically active DAC domain dimers seen in the DisA homo-octamer. The calculated r.m.s.d between superimposed CdaA and DisA dimers amounts to 1.72 Å (198 matched Cα positions, Fig. S4). The CdaA–CdaA dimer interface buries about 605 Å2 of the accessible surface area (7.3%) and is stabilized by six hydrogen bonds and two salt bridges. However, additional interactions between the monomers are mediated by the ligand bound to the active site (see below). Surprisingly, the difference electron density map clearly revealed the presence of a c-di-AMP molecule and two metal ions bound in the active site of the CdaA crystallographic dimer (Fig. S2A). As only ATP and Co2+ were added to the protein right before it was subjected to crystallization, the c-di-AMP must have formed during or after crystallization droplets were set up. It appears very likely that the bound metal ion is a Co2+, as no other catalytic metal cation was present in the crystallization solution. The Co2+ is coordinated by the phosphate moiety and the carboxylate group of Glu-224 as well as the carboxylate group of Asp-171 and the imidazole ring nitrogen of His-170 of the symmetry-related subunit (Fig. 3, B and C). The metal–oxygen distances of 2.1 Å for Asp-171 and Glu-224 and 2.3 Å for phosphate correspond to distances observed in other proteins containing a Co2+ ion (17). Elongated distances observed between Co2+ and the imidazole ring of His-170 (3.1 Å) and the c-di-AMP 3′OH group (3.8 Å) indicate that this complex corresponds to the post-catalytic state. To fulfill its catalytic role, the metal ion must be shifted. Only then it can act as Lewis acid to increase the nucleophilicity of the metal-activated 3′ hydroxyl group of ATP and enhance the electrophilicity of the phosphorus atom of the adjacent ATP molecule.
Figure 3.

The active site of dimeric CdaA with bound c-di-AMP. A, the catalytically active Δ100CdaA homodimer is depicted as a cartoon, and the bound reaction product c-di-AMP is shown as balls and sticks (carbon in pale blue and blue, phosphate in orange, oxygen in red, and nitrogen in dark blue). The two Δ100CdaA monomers are colored according to the c-di-AMP in pale blue and blue, respectively. Co2+ ions are depicted as pale red spheres. B, detailed view of the CdaA active site. Amino acids involved in binding the c-di-AMP molecule (colored and depicted as in A) are shown as sticks (carbon in pale blue and blue, oxygen in red, and nitrogen in dark blue). The Co2+ ions are colored and depicted according to A. For simplicity, only one half of the two-fold symmetric CdaA active site is shown. C, detailed view of the Co2+ binding site and its coordination sphere.
The asymmetric unit of the crystal contains a second CdaA molecule that also accommodates a nucleotide bound in the active site but no bound metal ion (Fig. 4). Based on the observed difference omit electron density map (Fig. S2B), the nucleotide was identified as AMP. Because previous crystal structures of DisA and CdaA with bound ATP or 3′-dATP showed well-defined electron density for the β and γ phosphates, it appears likely that the second CdaA molecule indeed has AMP bound, which must have formed out of ATP during the crystallization process. ATP hydrolysis also explains the presence of another difference electron density map peak, which has been interpreted as a free phosphate. This phosphate ion is bound in the vicinity of the c-di-AMP molecule and could potentially mark an exit route of the pyrophosphate molecule on the surface of CdaA.
Figure 4.

Structure of the CdaA-AMP complex. A, CdaA monomer (cartoon, pale green) with a bound AMP molecule depicted as balls and sticks. B, a detailed view of the active site showing the amino acids (sticks, carbon in pale green, oxygen in red, and nitrogen in blue) involved in AMP binding. The bound AMP is shown as a ball-and-stick model (carbon in wheat, phosphate in orange, oxygen in red, and nitrogen in dark blue).
Conformational rearrangements of the active site induced by ligands
The comparison of CdaA in the apo state to CdaA complexed with AMP or c-di-AMP unveils different orientations of the Tyr-187 side chain, which is located in close proximity to the adenine base. In the CdaA apo state, this tyrosine side chain is rotated outward from the active site, leading to an opening of the binding site for the adenine base (Fig. 5). In the monomeric CdaA–AMP complex, the tyrosine is rotated inward at the active site and stacks on the adenine in an almost coplanar orientation. In contrast, in the dimeric c-di-AMP complex, the tyrosine side chain is flipped outward, as the Thr-202 side chain of the other subunit packs against the adenine ring (Fig. 3B).
Figure 5.
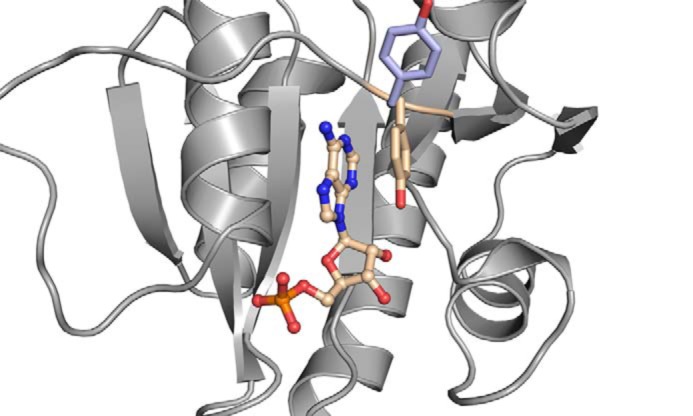
Conformational switch of Tyr-187 during c-di-AMP synthesis. For convenience, only the Δ100CdaA monomer (gray) with bound AMP (carbon in wheat, phosphate in orange, oxygen in red, and nitrogen in dark blue) and Tyr-187 (wheat) is shown. AMP is depicted as a ball-and-stick model. The side chain of Tyr-187 (pale blue) of the Δ100CdaA–c-di-AMP complex structure is superimposed. Upon ATP binding to monomeric CdaA, Tyr-187 stacks parallel on the adenine base (π–π interaction) and stabilizes the protein–substrate complex. Upon homodimer formation, the side chain of Tyr-187 rotates outwards as it is replaced by the Thr-202 side chain of the second monomer in the catalytically active homodimer.
To investigate whether Tyr-187 plays an important role in c-di-AMP formation, a Y187A mutant was generated. This mutation led to a significant reduction (about 80%) in activity, confirming the functional impact of Tyr-187 (Fig. 1B). To exclude that the Y187A mutation perturbed the fold of CdaA, the crystal structure of Δ100CdaA_Y187A was determined as well (Table 1). Comparison with the structure of WT Δ100CdaA demonstrates no structural changes caused by the mutation.
Discussion
Synthesis of c-di-AMP requires dimerization and proper orientation of two DAC domains, each with one ATP bound and accompanied by the metal ion cofactor. In DisA, this is achieved permanently by the homo-octameric oligomerization state (13). The first structure of CdaA of L. monocytogenes showed that the DAC domain crystallized as a monomer even though ATP was bound to the active site (15). However, for the previous study and this one, a truncated CdaA was used. So far, the influence of the missing transmembrane domain on oligomerization and catalytic activity is unknown.
Here, a new crystal form of CdaA was obtained that contains two CdaA molecules with different nucleotides bound. One CdaA molecule forms a catalytically active dimer with a symmetry mate in the crystal. This dimer contains a c-di-AMP molecule and two metal ions in the active site; hence, it closely resembles the dimer arrangement of DAC domains seen in DisA (Fig. S4).
The c-di-AMP must have been formed during crystallization, as only ATP and Co2+ ions were added to CdaA. This complex corresponds to the enzyme–product complex, which is supposed to have a lower stability. However, c-di-AMP mediates multiple contacts between the monomers, increasing the interaction surface area between the monomers, and the catalytically active dimer appears to be caught in the crystalline lattice.
Conserved active-site residues of DisA and CdaA directly involved in substrate binding and catalysis have been identified previously (15, 18). Each of the mutations in DisA (D75N, R130A, RHR (108–110)AAA, T107V+T111V), and in CdaA (D171N, G172A, and T202N) led to a reduction or complete loss of enzymatic activity. However, by analyzing the structure of the monomeric CdaA in the asymmetric unit with bound AMP, we realized that the Tyr-187 side chain might also be involved in substrate binding, as it stacks on the adenine ring, but it is rotated outward in the structure of apo CdaA (Fig. 5). Hence, it appears likely that, upon binding of ATP to monomeric CdaA, Tyr-187 rotates toward the adenine moiety and locks the ATP in the active site by a π–π stacking interaction, as observed for many other ATP binding proteins (19). However, upon CdaA dimerization, the tyrosine side chain is replaced by the side chain of Thr-202 of the other subunit, which then stabilizes the bound ATP. The replacement of the Tyr by a side chain from the other subunit might facilitate product release after catalysis, as, upon dimer dissociation, the product can be released more easily. By mutation of Tyr-187 to Ala, which strongly reduced the activity in vitro, we demonstrate that Tyr-187 indeed plays an essential role in c-di-AMP formation by CdaA. Notably, this Tyr-187 is conserved in most CdaA enzymes but not in other DAC proteins, like DisA, suggesting a slightly different mechanism of substrate binding between different classes of DACs.
A remarkable difference between DisA and CdaA concerns the metal ion specificity in the catalytic center. Although DisA appears to be active with Mg2+ and Mn2+ (20), CdaA is not active in the presence of Mg2+. Such unexpected differences in metal ion preferences have also been observed for other protein families, e.g. the metal-dependent serine/threonine phosphoprotein phosphatase family (21). Because the catalytic mechanism of phosphoprotein phosphatase enzymes as well as that of DAC proteins does not require the redox potential of Mn2+ to carry out the catalyzed reaction, it is not clear why some members of the DAC family would prefer Mn2+ or other divalent cations over Mg2+ (22). Hence, the observed strict dependence of CdaA on Mn2+ or Co2+ ions raised questions concerning its structural basis. The observed metal ion dependence is most likely related to different chemical properties of the cation, e.g. ionic radius, and to the amino acid composition of the active site.
Comparison of the DisA and CdaA structures reveals significant differences in metal ion coordination. In the catalytically active dimer of DisA with bound ATP, the Mg2+ ion is coordinated by three phosphate groups and the Asp carboxylate. In CdaA, more protein residues contribute to metal binding, resulting in a more crowded active site. In addition to Asp-171 (which corresponds to Asp-75 of DisA), the side chains of Glu-224 and His-170 coordinate the Co2+ ion. These two residues are not structurally conserved in DisA, as there is an Arg instead of the Glu and a Met instead of the His. The side chains of both Arg and Met are rotated outward from the metal binding site, making it less crowded. Hence, the major difference appears to be presence of the His. Although Mg2+ strongly prefers coordination by Asp and Glu, the transition metal ions Mn2+ and Co2+ are bound as well by His, as deduced from analysis of all metal binding sites in known protein structures (23).
The reason for this difference between DisA and CdaA is most likely related to the fact that DisA contains stably associated, catalytically active dimers, whereas, for CdaA, the catalytic dimer might just exist transiently. Therefore, in DisA, Asp-171, which belongs to the second DAC domain, is sufficient for binding the substrate ATP and the metal ion. In CdaA, ATP and the metal ion are initially bound to the monomeric DAC domain by the Glu-224 side chain, and solvent molecules complete the metal coordination sphere. Upon formation of the catalytically active dimer, the metal ion needs to be slightly repositioned to be further coordinated by His-170 and Asp-171, provided by the second monomer (Fig. S3).
Recently, CdaA from S. aureus was characterized structurally and biochemically (24). Surprisingly, in contrast to L. monocytogenes CdaA, the S. aureus CdaA homolog shows activity not only in the presence of transition metal ions but also in the presence of Mg2+. Comparison of the active sites of both available CdaA structures unveils identical positioning of the amino acids directly involved in c-di-AMP and metal ion coordination (Fig. S5). The largest structural differences can be observed for N- and C-terminal α-helices and a loop connecting β-strand 4 and α-helix 4. The latter is located in close proximity to the phosphate moiety of ATP and could indirectly alter the metal binding preferences or even metal catalytic efficiency. This could serve as a potential explanation for the metal ion promiscuity of CdaAs showing strict conservation of three residues (His-170, Asp-171, and Glu-224) directly involved in metal binding, as revealed by sequence alignment of ten bacterial CdaAs (Fig. S6). The chemical properties of these three residues are most likely the structural basis for the observed metal ion promiscuity of CdaAs, which has also been observed for other enzymes, e.g. mannosylglycerate synthase (25). The observed capability of utilizing several ions by one enzyme is still one of many not well-understood marvels of enzymology that require further investigation.
Experimental procedures
Bacterial strains and growth conditions
For cloning procedures and protein overexpression, Escherichia coli strains DH5α and BL21(DE3) were used. The E. coli strains were cultivated in 2xYT ((trypton 1.6% (w/v), yeast extract 1.0% (w/v), NaCl 0.5% (w/v)) medium, whereas transformed cells were selected on lysogeny broth-medium plates containing ampicillin (100 μg/ml).
Plasmid construction
For purification, the DAC-type CdaA was equipped with a GST tag. CdaA is known to be a transmembrane protein. The Δ300cdaA allele, which lacks the TM domain, was amplified using the primer pairs JH004 forward (5′-CCGGATCCTATGGATCAAGAATTGAGCG-3′)/JH005 reverse (5′ GGCTCGAG TCATTCGCTTTTGCCTCCTTTCC-3′). As a template, the plasmid pBP33 was used (15). The resulting PCR products were cloned in the pGEX-6P-1 (GE Healthcare) expression vector using the restriction sites XhoI and BamHI, leading to plasmid pGEXpBP33, which encodes for the truncated Δ100CdaA protein with an N-terminal GST tag.
Site-directed mutagenesis
Δ100CdaA mutants were generated with site-directed mutagenesis to identify amino acid residues that have an important function in the catalytic reaction mechanism. The CdaA mutant Y187A was created by PCR using the mutagenesis primer pairs JH_Y187A_forward (5′-CAGCAAGTGCCTTGCCA CTTTCAGATAGCCCGTTCTTATCCAAAGAAC-3′) and JH_Y187A_reverse (5′-GTGGCAAGGCACTTGCTGCCGATGC AATTTCGTTTCCTTTAATAATAACTGC-3′), resulting in the plasmid encoding the truncated mutant variant Δ100CdaA_Y187A.
Protein expression and purification
E. coli BL21(DE3) was used for expression of the fusion protein GST-Δ100CdaA. The cells were grown in 1 liter of 2xYT medium at 37 °C. Protein expression was induced after the culture reached an A600 of ∼ 0.6 by addition of 1 mm isopropyl 1-thio-β-d-galactopyranoside and incubated at 16 °C for 18 h. After harvesting and subsequent to cell disruption with a microfluidizer (M-110S Microfluidizer, Microfluidics) and centrifugation at 15,600 × g for 30 min to remove cell debris, the lysate was loaded onto a GSH-Sepharose column (GE Healthcare) in 300 mm NaCl, 20 mm Tris/HCl (pH 7.5), and 10 mm EDTA. The target protein GST-Δ100CdaA was eluted from the column with 40 mm reduced GSH. The eluate was incubated overnight with PreScission protease (1:100 (w/w)) in cellulose tubing placed in dialysis buffer (100 mm NaCl and 20 mm Tris/HCl (pH 7.5)) at 4 °C to remove the high GSH concentration and to dissect the GST tag from Δ100CdaA. To remove the cleaved-off tag from the truncated CdaA, a second GSH-Sepharose purification step was included.
Crystallization and cryoprotecion
For crystallization, the sitting-drop vapor diffusion method was applied. Initial crystallization trials were performed at 20 °C using Δ100CdaA at a concentration of 4.0 mg/ml supplemented with 500 μm CoCl2 and 500 μm ATP. Rectangular crystals grew after approximately 48 h in a 2-μl droplet composed of the aforementioned protein solution mixed with reservoir in a 1:1 ratio. The reservoir was composed of 0.2 m Ca(CH3COO)2, 0.1 m Na-HEPES (pH 7.5), and 10% (w/v) PEG8000. Crystals were cryoprotected by soaking them in a reservoir solution supplemented with 25% PEG8000.
For crystallization of the apo form and the Y187A variant of Δ100CdaA, a protein concentration of 6.0 mg/ml was used, keeping the 2-μl droplet size and 1:1 protein-to-reservoir ratio. To facilitate crystal growth, microseeding was performed in combination with small alterations of NaCl concertation. Thin crystal plates were obtained after approximately 18 h in a salt concentration ranging between 3.7–4.5 M NaCl and 0.1 m Na-HEPES (pH 8.5). Crystals were cryoprotected by soaking them in a saturated sucrose solution obtained by solubilizing sucrose in reservoir solution.
X-ray data collection and processing
Diffraction images were collected at PETRA III EMBL beamlines P13 and P14 (DESY, Hamburg, Germany) and processed with the XDS package (26, 27). Data collection and processing statistics are summarized in Table 1. A trigonal lattice with unit cell parameters of a = b = 121.90 Å, c = 141.59 Å was determined for the crystals containing the CdaA–c-di-AMP complex. Cell content analysis indicated the presence of two CdaA molecules occupying the asymmetric unit (Vm = 2.89 Å3/Da, corresponding solvent content of 57.4%). The crystals of apo CdaA and the Y187A mutant exhibited an orthorhombic lattice and the unit cell parameters of a = 42.96 Å, b = 64.67 Å, c = 129.75 Å and a = 46.49 Å, b = 65.13 Å, c = 131.33 Å, respectively. The Matthews coefficient (Vm = 2.55 Å3/Da, corresponding solvent content of 51.78%) implicates two molecules occupying the asymmetric unit.
Structure determination and refinement
The crystallographic phase problem was solved by molecular replacement with PHASER (28) using the structure of the DAC Δ100CdaA from L. monocytogenes (PDB code 4RV7) as a search model. Manual model building was preformed with Coot (29), and the structure was refined with Refmac (30) and PHENIX (31). To monitor the refinement progress using Rfree, 5% of the reflections were selected randomly and excluded from refinement. During the refinement process, the coordination distance for the Co2+ ion in the ligand-bound structure was restrained to 2.1 Å for the Glu-224 and Asp-171 side chains and to 2.3 Å for the cyclic di-AMP phosphate. The final structure of the CdaA–c-di-AMP complex was refined at a resolution of 2.8 Å to Rwork of 18.7% and Rfree of 23.4%. The apo-CdaA was refined at a resolution of 2.0 Å to Rwork of 18.6% and Rfree of 22.5%. The structure of the Y187A mutant was determined at 2.32 Å resolution and refined to 17.5% and 22.0% for Rwork and Rfree, respectively. Protein contact areas in the crystals were analyzed using “Protein interfaces, surfaces and assemblies” services at the European Bioinformatics Institute using standard settings (32).
In vitro DAC activity assay
Diadenylate cyclase activity was measured with a quantitative fluorescence assay based on an increased fluorescence signal because of the specific interaction of the fluorescent dye coralyne with c-di-AMP (16). A 200-μl reaction mixture contained 40 mm Tris/HCl (pH 7.5), 100 mm NaCl, 10 mm XCl2 (X = Mg, Co, Mn, or Ca) and 100 μm ATP. The reaction was started by addition of 10 μm Δ100CdaA and incubated for 1 h at 37 °C. To stop the reaction, the reaction mixture was boiled for 5 min and centrifuged for another 5 min at 13,400 × g to remove the precipitated protein. For quantification of the synthesized c-di-AMP, 250 mm KBr and 10 μm coralyne were added to the reaction mixture. After incubating the samples for 30 min in the dark, c-di-AMP concentration was determined by excitation at a wavelength of 420 nm and measuring the fluorescence emission at a wavelength of 475 nm using a microplate reader (Victor Nivo multimode microplate reader, PerkinElmer Life Sciences).
Author contributions
J. L. H. data curation; J. L. H., P. N., A. D., and R. F. formal analysis; J. L. H. and P. N. validation; J. L. H., P. N., and R. F. writing-original draft; J. L. H., P. N., A. D., and R. F. writing-review and editing; R. F. conceptualization; R. F. funding acquisition.
Supplementary Material
Acknowledgments
We thank the EMBL-Outstation Hamburg (DESY PETRA III beamlines P13 and P14, Germany) for allocation of beam time and Isabel Bento, Johanna Hakanpää, as well as Saravanan Panneerselvam for excellent support at the beamline. We also thank Johannes Gibhardt, Fabian Commichau, and Jörg Stülke for fruitful discussions.
This work was supported by Deutsche Forschungsgemeinschaft Priority Programs SPP1879 and INST186/1117. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S6.
- c-di-AMP
- cyclic di-AMP
- DAC
- diadenylate cyclase
- TM
- transmembrane
- r.m.s.d.
- root mean square deviation.
References
- 1. Gomelsky M. (2011) cAMP, c-di-GMP, c-di-AMP and now cGMP: bacteria use them all!. Mol. Microbiol. 79, 562–565 10.1111/j.1365-2958.2010.07514.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kalia D., Merey G., Nakayama S., Zheng Y., Zhou J., Luo Y., Guo M., Roembke B. T., and Sintim H. O. (2013) Nucleotide, c-di-GMP, c-di-AMP, cGMP, cAMP, (p)ppGpp signaling in bacteria and implications in pathogenesis. Chem. Soc. Rev. 42, 305–341 10.1039/C2CS35206K [DOI] [PubMed] [Google Scholar]
- 3. Hengge R., Gründling A., Jenal U., Ryan R., and Yildiz F. (2016) Bacterial signal transduction by cyclic di-GMP and other nucleotide second messengers. J. Bacteriol. 198, 15–26 10.1128/JB.00331-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Commichau F. M., Dickmanns A., Gundlach J., Ficner R., and Stülke J. (2015) A jack of all trades: the multiple roles of the unique essential second messenger cyclic di-AMP. Mol. Microbiol. 97, 189–204 10.1111/mmi.13026 [DOI] [PubMed] [Google Scholar]
- 5. Corrigan R. M., and Gründling A. (2013) Cyclic di-AMP: another second messenger enters the fray. Nat. Rev. Microbiol. 11, 513–524 10.1038/nrmicro3069 [DOI] [PubMed] [Google Scholar]
- 6. Witte C. E., Whiteley A. T., Burke T. P., Sauer J. D., Portnoy D. A., and Woodward J. J. (2013) Cyclic di-AMP is critical for Listeria monocytogenes growth, cell wall homeostasis, and establishment of infection. MBio 4, e00282–00213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gundlach J., Herzberg C., Kaever V., Gunka K., Hoffmann T., Weiß M., Gibhardt J., Thürmer A., Hertel D., Daniel R., Bremer E., Commichau F. M., and Stülke J. (2017) Control of potassium homeostasis is an essential function of the second messenger cyclic di-AMP in Bacillus subtilis. Sci. Signal. 10, eaal3011 10.1126/scisignal.aal3011 [DOI] [PubMed] [Google Scholar]
- 8. Gundlach J., Dickmanns A., Schröder-Tittmann K., Neumann P., Kaesler J., Kampf J., Herzberg C., Hammer E., Schwede F., Kaever V., Tittmann K., Stülke J., and Ficner R. (2015) Identification, characterization, and structure analysis of the cyclic di-AMP-binding PII-like signal transduction protein DarA. J. Biol. Chem. 290, 3069–3080 10.1074/jbc.M114.619619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bai Y., Yang J., Zhou X., Ding X., Eisele L. E., and Bai G. (2012) Mycobacterium tuberculosis Rv3586 (DacA) is a diadenylate cyclase that converts ATP or ADP into c-di-AMP. PLoS ONE 7, e35206 10.1371/journal.pone.0035206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Corrigan R. M., Abbott J. C., Burhenne H., Kaever V., and Gründling A. (2011) c-di-AMP is a new second messenger in Staphylococcus aureus with a role in controlling cell size and envelope stress. PLoS Pathog. 7, e1002217 10.1371/journal.ppat.1002217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Woodward J. J., Iavarone A. T., and Portnoy D. A. (2010) c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328, 1703–1705 10.1126/science.1189801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Commichau F. M., Heidemann J. L., Ficner R., and Stülke J. (2019) Making and breaking of an essential poison: the cyclases and phosphodiesterases that produce and degrade the essential second messenger cyclic di-AMP in bacteria. J. Bacteriol. 201, e00462–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Witte G., Hartung S., Büttner K., and Hopfner K. P. (2008) Structural biochemistry of a bacterial checkpoint protein reveals diadenylate cyclase activity regulated by DNA recombination intermediates. Mol. Cell 30, 167–178 10.1016/j.molcel.2008.02.020 [DOI] [PubMed] [Google Scholar]
- 14. Cho K. H., and Kang S. O. (2013) Streptococcus pyogenes c-di-AMP phosphodiesterase, GdpP, influences SpeB processing and virulence. PLoS ONE 8, e69425 10.1371/journal.pone.0069425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rosenberg J., Dickmanns A., Neumann P., Gunka K., Arens J., Kaever V., Stülke J., Ficner R., and Commichau F. M. (2015) Structural and biochemical analysis of the essential diadenylate cyclase CdaA from Listeria monocytogenes. J. Biol. Chem. 290, 6596–6606 10.1074/jbc.M114.630418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou J., Sayre D. A., Zheng Y., Szmacinski H., and Sintim H. O. (2014) Unexpected complex formation between coralyne and cyclic diadenosine monophosphate providing a simple fluorescent turn-on assay to detect this bacterial second messenger. Anal. Chem. 86, 2412–2420 10.1021/ac403203x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harding M. M. (2006) Small revisions to predicted distances around metal sites in proteins. Acta Crystallogr. D Biol. Crystallogr. 62, 678–682 10.1107/S0907444906014594 [DOI] [PubMed] [Google Scholar]
- 18. Müller M., Deimling T., Hopfner K. P., and Witte G. (2015) Structural analysis of the diadenylate cyclase reaction of DNA-integrity scanning protein A (DisA) and its inhibition by 3′-dATP. Biochem. J. 469, 367–374 10.1042/BJ20150373 [DOI] [PubMed] [Google Scholar]
- 19. Mao L., Wang Y., Liu Y., and Hu X. (2004) Molecular determinants for ATP-binding in proteins: a data mining and quantum chemical analysis. J. Mol. Biol. 336, 787–807 10.1016/j.jmb.2003.12.056 [DOI] [PubMed] [Google Scholar]
- 20. Manikandan K., Sabareesh V., Singh N., Saigal K., Mechold U., and Sinha K. M. (2014) Two-step synthesis and hydrolysis of cyclic di-AMP in Mycobacterium tuberculosis. PLoS ONE 9, e86096 10.1371/journal.pone.0086096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shi L. (2004) Manganese-dependent protein O-phosphatases in prokaryotes and their biological functions. Front. Biosci. 9, 1382–1397 10.2741/1318 [DOI] [PubMed] [Google Scholar]
- 22. Samol I., Shapiguzov A., Ingelsson B., Fucile G., Crèvecoeur M., Vener A. V., Rochaix J. D., and Goldschmidt-Clermont M. (2012) Identification of a photosystem II phosphatase involved in light acclimation in Arabidopsis. Plant Cell 24, 2596–2609 10.1105/tpc.112.095703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Putignano V., Rosato A., Banci L., and Andreini C. (2018) MetalPDB in 2018: a database of metal sites in biological macromolecular structures. Nucleic Acids Res. 46, D459–D464 10.1093/nar/gkx989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tosi T., Hoshiga F., Millership C., Singh R., Eldrid C., Patin D., Mengin-Lecreulx D., Thalassinos K., Freemont P., and Gründling A. (2019) Inhibition of the Staphylococcus aureus c-di-AMP cyclase DacA by direct interaction with the phosphoglucosamine mutase GlmM. PLoS Pathog. 15, e1007537 10.1371/journal.ppat.1007537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nielsen M. M., Suits M. D., Yang M., Barry C. S., Martinez-Fleites C., Tailford L. E., Flint J. E., Dumon C., Davis B. G., Gilbert H. J., and Davies G. J. (2011) Substrate and metal ion promiscuity in mannosylglycerate synthase. J. Biol. Chem. 286, 15155–15164 10.1074/jbc.M110.199844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kabsch W. (2010) Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66, 133–144 10.1107/S0907444909047374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.