

Abstract
The initiation and development of diabetes are mainly ascribed to the loss of functional β-cells. Therapies designed to regenerate β-cells provide great potential for controlling glucose levels and thereby preventing the devastating complications associated with diabetes. This requires detailed knowledge of the molecular events and underlying mechanisms in this disorder. Here, we report that expression of microRNA-223 (miR-223) is up-regulated in islets from diabetic mice and humans, as well as in murine Min6 β-cells exposed to tumor necrosis factor α (TNFα) or high glucose. Interestingly, miR-223 knockout (KO) mice exhibit impaired glucose tolerance and insulin resistance. Further analysis reveals that miR-223 deficiency dramatically suppresses β-cell proliferation and insulin secretion. Mechanistically, using luciferase reporter gene assays, histological analysis, and immunoblotting, we demonstrate that miR-223 inhibits both forkhead box O1 (FOXO1) and SRY-box 6 (SOX6) signaling, a unique bipartite mechanism that modulates expression of several β-cell markers (pancreatic and duodenal homeobox 1 (PDX1), NK6 homeobox 1 (NKX6.1), and urocortin 3 (UCN3)) and cell cycle–related genes (cyclin D1, cyclin E1, and cyclin-dependent kinase inhibitor P27 (P27)). Importantly, miR-223 overexpression in β-cells could promote β-cell proliferation and improve β-cell function. Taken together, our results suggest that miR-223 is a critical factor for maintaining functional β-cell mass and adaptation during metabolic stress.
Keywords: insulin, diabetes, beta cell (B-cell), cell proliferation, microRNA (miRNA), microRNA-223, β-cell proliferation, metabolic disorder, gene regulation, forkhead box O1 (FOXO1), SRY-box 6 (SOX6)
Introduction
Globally, the number of adults with diabetes has quadrupled in the past three decades, and it is projected to increase to 642 million in 2040 (1). Type 1 diabetes (T1D)3 results from notable insulin deficiency caused by autoimmune attack on β-cells, which leads to pronounced β-cell death and dysfunction. Type 2 diabetes (T2D), which accounts for about 90% of all diabetes cases, is believed to be the consequence of insulin resistance in key organs such as liver, adipose tissue, and skeletal muscle (2). However, accumulating evidence suggests that insulin resistance leads to T2D only when accompanied by β-cell dysfunction, in which β-cells can no longer compensate for the increased demand of insulin by increasing functional output and number (3). Therefore, loss of functional β-cells is a critical culprit responsible for both types of diabetes.
Pancreatic β-cell mass is maintained through dynamic balance of neogenesis, proliferation, and apoptosis (4). At embryonic and neonatal stages, β-cells are primarily generated via differentiation from stem or progenitor cells, a process called neogenesis; in adult pancreas, however, cell lineage tracing studies have shown that replication/proliferation of existing β-cells is the dominant mechanism to increase β-cell mass (5). Yet β-cell proliferation rate declines rapidly in early childhood and ultimately reaches to almost zero in adults (4). Interestingly, it has been proposed that β-cell dedifferentiation is needed prior to proliferation (6, 7), followed by redifferentiation into mature β-cells (8, 9). In fact, β-cell dedifferentiation has been repeatedly reported to play a role in the pathogenesis of T2D in both rodent and human studies (10–12), and the redifferentiated cells are capable of correcting hyperglycemia in T1D mice (13). Thus, it is very likely that, during early stage of diabetes, pancreatic β-cells undergo the process of dedifferentiation–proliferation–redifferentiation to expand functional β-cell mass and cope with increasing demand of insulin. Despite years of intensive research in this field, currently available therapies fail to prevent β-cells from their inevitable deterioration. Although transplantation of β-cells has been proposed to address this problem, it becomes implausible because of the scarcity of β-cells from cadaveric donors and transplant rejection (14). Therefore, it is urgently needed to identify novel factors to preserve and restore functional β-cell mass, which can eventually lead to the development of β-cell replacement or regeneration therapies.
MicroRNAs (miRNAs or miRs) are non–protein coding RNAs of ∼22 nucleotides, which can degrade or inhibit the translation of hundreds of target mRNAs by binding to the 3′ untranslated region (UTR) (15). Therefore, miRNAs are able to regulate a broad spectrum of biological processes such as cellular proliferation and survival. Genetic deletion of dicer1, an essential processor of miRNAs, in pancreas or β-cells results in the development of diabetes because of smaller pancreatic size and loss of insulin mRNA and protein (16, 17). This study suggests that miRNAs, as an integrated entity in whole, may act as single positive regulator of β-cell development, survival, and function. Like many other miRNAs, miR-223 is ubiquitously expressed in tissues such as heart, adipose tissues, and liver (18). Consistent with its tissue distribution, miR-223 has been implicated in various physiological and pathological conditions including cardiac hypertrophy (19), systemic cholesterol homeostasis (20), development of atherosclerosis (21), and adipose tissue–associated insulin resistance (18). Furthermore, it has been reported that miR-223 plays a critical role in regulating cell proliferation in diabetic retinopathy and some cancers (22, 23). However, the functional role of miR-223 in pancreatic β-cells and its underlying mechanism have never been investigated. Thus, we performed in vivo loss-of-function and in vitro gain-of-function studies to determine the role of miR-223 in maintaining functional β-cell mass. Our data unveil a unique bipartite molecular mechanism whereby miR-223 positively controls functional β-cell mass through regulating the Foxo1 and Sox6 signaling cascades, providing potential effective targets for diabetes intervention.
Results
MiR-223 is up-regulated in β-cells treated with TNFα or high glucose as well as islets from diabetic mouse model and human patients
To investigate the functional role of miR-223 in pancreatic β-cells, we first characterized the expression levels of miR-223 in diabetic mouse islets and β-cells (Fig. 1, A–E). We observed that, compared with chow diet (CD) control mice, levels of miR-223 were increased by 5.6-fold and 12.8-fold in islets of HFD and db/db mice, respectively (Fig. 1, A and B). Similar results were noticed in islets of Akita mice, a genetic mouse model associated with T1D because of Insulin 2 gene mutation and increased β-cell toxicity (Fig. S1A). Consistent with these in vivo data, the expression levels of miR-223 were increased by 2.5- and 3.4-fold, respectively, in Min6 β-cells treated with TNFα or high glucose (25 mm) medium, compared with BSA or low glucose controls (5 mm) (Fig. 1, C and D). More importantly, expression levels of miR-223 were also enhanced by about 2-fold in islets from human donors with obesity and type 2 diabetes (Fig. 1E). Together, both in vivo and in vitro data clearly indicate that the miR-223 expression in β-cells and islets is dysregulated under various stress conditions.
Figure 1.

miR-223 is up-regulated in β-cells treated with TNFα or high glucose and islets from diabetic mouse model and human patients. A and B, expression of miR-223 in islets of mice with 18 weeks of HFD feeding (A) and db/db mice (B). C and D, mRNA levels of miR-223 in Min6 insulinoma β-cells treated with 25 mm glucose (C) or 5 ng/ml TNFα (D). E, levels of miR-223 were detected in islets from healthy, obese, and type 2 diabetic human donors. Data are shown as mean ± S.E. (error bar).*, p < 0.05; ***, p < 0.001 versus controls by t test.
Ablation of miR-223 exacerbates β-cell dysfunction in diabetic condition
We next went on to assess the potential role of miR-223 in the regulation of functional β-cell mass under metabolic stress. A global miR-223 KO mouse model was used to avoid confounding effects from organ crosstalks, since miR-223 can be transported via exosomes or released into circulation (24, 25). The results of qRT-PCR analysis confirmed that miR-223 was deleted (Fig. 2A and Fig. S1B). After 18 weeks. of HFD feeding, we did not observe any differences in body weight changes between WTs and KOs (Fig. S1C). Interestingly, when compared with chow diet–fed WT mice, miR-223 KO mice showed higher fasting blood glucose but lower insulin levels (Fig. 2, B and C). More importantly, HFD-fed KO mice displayed exacerbated response, with 31% higher fasting glucose levels, whereas insulin levels were 44% lower when compared with WT HFD mice (Fig. 2, B and C). Next, we performed homeostasis model assessment (HOMA) to estimate insulin resistance (HOMA-IR) and steady-state β-cell function (HOMA-%β) as described previously (6). The results showed that KO CD mice exhibited dramatic increase of insulin resistance and 81% decrease in β-cell function, compared with chow diet–fed WTs, and further aggravated upon HFD feeding (Fig. 2, D and E). In accordance with HOMA index, KO HFD mice showed overall significantly higher increase in glucose levels during insulin tolerance test, suggesting more severe systemic insulin resistance (Fig. 2, F and G). Furthermore, we observed higher blood glucose levels during glucose tolerance test in KO HFD mice, when compared with WT HFD controls (Fig. 2, H and I), indicating blunted β-cell function to secrete insulin in response to glucose injection. Taken together, these data reveal that miR-223 ablation provokes insulin resistance and β-cell dysfunction, which are worsened in HFD-induced condition.
Figure 2.

Ablation of miR-223 exacerbates β-cell dysfunction in diabetic condition. A, deletion of miR-223 in KO mice was confirmed by 3% agarose gel electrophoresis. B and C, fasting plasma glucose (B) and insulin levels (C) were measured. D and E, HOMA index for insulin resistance (D) and β-cell function (E) were calculated based on fasting blood glucose and insulin levels. F and G, IPITT was performed after 6-h fasting (F) and quantification (G). H and I, IPGTT was performed after overnight fasting (H) and quantification (I). Data are shown as mean ± S.E. (error bar).*, p < 0.05; ***, p < 0.001 versus controls by t test.
MiR-223 deficiency leads to maladaptive β-cell proliferation and apoptosis
The impaired insulin secretion from β-cells in KO mice could be because of either smaller β-cell mass or β-cell dysfunction. To assess the in vivo relevance of miR-223 in the adaptive β-cell proliferative response upon metabolic stress conditions, we adopted two approaches. First, we fed 5-/6-week-old WT mice and miR-223 KO mice with HFD for 18 weeks and analyzed β-cell proliferation by immunofluorescent staining and flow cytometry. We noticed significant reduction in Ki-67 signal, a marker of cell proliferation, in miR-223 KO mice under basal condition (Fig. 3, A–D). More importantly, we detected a ∼36% decrease in insulin-positive cells (Fig. 3, A and B) and a ∼39% reduction in the number of Ki-67–positive cells (Fig. 3, A and C) in HFD-fed miR-223 KO mice, when compared with age-matched HFD-fed WT mice, suggesting impaired compensatory β-cell proliferation. This was further confirmed by staining with additional proliferation marker pHH3 and decreased cyclin D1 mRNA levels (Fig. S2, A–C). Similarly, H&E staining of pancreas tissue showed reduced islet sizes in KO mice (Fig. 3E). In the second model, we injected WT and miR-223 mice with STZ to deplete β-cells. These mice became and remained diabetic (blood glucose >250 mg/dl) 10 days after injection. Although no spontaneous reversal of diabetes was observed, there was noticeable amount of proliferating β-cells to cope with hyperglycemia in WT mice. However, such adaptive β-cell proliferative response was diminished in miR-223 KO STZ mice, which was supported by a reduction in the number of β-cells that costained positive for insulin and Ki-67 (29% lower in KO STZ compared with WT STZ mice) (Fig. 3, A and C), leading to overall smaller insulin-positive islet size (Fig. 3, A and B). In addition, TUNEL staining revealed more β-cell apoptosis in KO mice when compared to WT controls. A similar trend was observed in mice fed with HFD (Fig. S2, D–F). Together, these data provide evidence for the necessity of miR-223 to preserve β-cell proliferation and survival.
Figure 3.

miR-223 deficiency leads to maladaptive β-cell proliferation. A–C, pancreatic tissues from WT and miR-223 KO mice after HFD or STZ injection were stained with insulin and Ki-67 antibodies. Quantification of insulin-positive area (B), and percent of Ki-67 positive cells within insulin-positive area (C) were shown. D, flow cytometry analysis of pancreatic tissue from WT and KO mice showed decreased Ki-67 signal within insulin-positive population. E, H&E staining and quantification of pancreatic tissue were shown. Scale bar, 100 μm. Data are shown as mean ± S.E. (error bar).*, p < 0.05; **, p < 0.01; ***, p < 0.001 versus controls by t test.
MiR-223 directly targets Foxo1 and Sox6 pathways
To seek potential mechanisms underlying the miR-223 KO–mediated decrease in β-cell mass, we first performed bioinformatics analysis using the database TargetScan. Two potential targets, forkhead box protein O1 (Foxo1) and sex determining region Y-box (Sox6), were identified. As shown in Fig. 4, A and B, the 3′-UTRs of Foxo1 and Sox6 contain miR-223 interacting region that is highly conserved among human, mouse, and rat species. Using luciferase reporter assay, we validated that miR-223 directly recognized both 3′-UTRs of Foxo1 and Sox6 in HEK293 cells (Fig. 4, C and D). Cotransfection of miR-223 strongly inhibited luciferase activity from the reporter constructs harboring 3′-UTR segments of Foxo1 and Sox6, whereas no effect was observed when a control miRNA was cotransfected with either reporter construct (Fig. 4, C and D). Similar results were also obtained when tested using Min6 β-cells (Fig. 4, E and F).
Figure 4.

miR-223 directly targets Foxo1 and Sox6 pathways. A and B, the putative miR-223–binding sites in the 3′-UTR regions of Foxo1 (A) and Sox6 (B) are conserved among mammalian species (human, mouse, and rat). C and D, luciferase reporter assays showed that Foxo1 (C) and Sox6 (D) were authentic targets of miR-223 in HEK293 cells. E and F, similar Luciferase reporter assays performed using Min6 β-cells. β-Gal was used as a transfection control. Similar results were observed in three additional independent experiments. Ctl, control. Data are shown as mean ± S.E. (error bar).*, p < 0.05; **, p < 0.01 versus controls by t test.
Remarkably, immunofluorescence analysis of pancreatic tissue showed significantly increased number of insulin-positive cells that were also expressing Foxo1. Of more interest, islets of miR-223 KO mice showed higher levels of nuclear localization of Foxo1 (Fig. 5A). Consistently, Western blotting results showed that the expression of Foxo1 and Sox6 were increased by 2- and 3.3-fold in pancreas of miR-223 KO mice, respectively (Fig. 5B), when compared with WT controls. Considering that 1) the activity and stability of Foxo1 is regulated by posttranslational modification such as phosphorylation (26); 2) Akt-mediated phosphorylation of Foxo1 results in Foxo1 translocation from nucleus to cytoplasm, thereby inactivating its transcriptional activity (26, 27); and 3) miR-223 increases phosphorylation of Akt in cardiomyocytes (19), we next hypothesized that deletion of miR-223 might reduce Akt phosphorylation, resulting in lower levels of Foxo1 phosphorylation, and subsequently increases its accumulation in nucleus. Indeed, Western blot results revealed that phosphorylation levels of Akt at both Thr-308 and Ser-473 sites were lower in pancreas of KO mice than WT controls, leading to 47% decrease in the phosphorylation levels of Foxo1 protein (Fig. 5B).
Figure 5.

Foxo1 and Sox6 signaling cascades are activated upon miR-223 deletion. A, immunofluorescent analysis of pancreatic tissue exhibited higher Foxo1 signal and localization to nucleus. B, Western blotting results showed that Foxo1 and Sox6 were increased by at least 2-fold in pancreas of KO mice, and protein levels of phosphorylated Akt at Thr(p)308 and Ser(p)473 sites as well as phosphorylated Foxo1 protein levels were significantly decreased. C, KO mice showed decreased Pdx1 and Glut2 protein levels, whereas total p27 levels were increased. D, mRNA levels of β-cell marker (Pdx1, Ucn3, Nkx6.1, Mafa) and progenitor marker (Ngn3) were measured. Scale bar, 100 μm. Data are shown as mean ± S.E. (error bar).*, p < 0.05; **, p < 0.01; ***, p < 0.001 versus controls by t test.
Foxo1 is known to inhibit the expression of Pdx1, a master regulator of β-cell growth and function (28), and a recent publication has shown that impaired β-cell function is mediated by Foxo1-Pdx1-Glut2 pathway (29). On the other hand, it has been reported that Sox6 can control β-cell function and proliferation via repressing Pdx1 and cyclin D1 (30, 31). Therefore, we next determined whether miR-223 deficiency affects the expression levels of Pdx1, a common downstream target of Foxo1 and Sox6. The results of Western blotting and qPCR analysis showed that the expression levels of Pdx1 were markedly decreased in pancreas of miR-223 KO mice compared with WT controls, which are consistent with existing literature (29). As a consequence, levels of Glut2 protein were greatly reduced (Fig. 5, C and D). Additionally, protein levels of cell cycle inhibitor p27, another downstream target of Foxo1, were 2.4-fold higher in pancreas of KO mice than WT controls (Fig. 5C). Overall, these results indicate that the repressed proliferative response of β-cells in miR-223 KO mice is mainly mediated by activated Foxo1 and Sox6 signaling cascades.
To further assess the effects of miR-223 KO on β-cell identity, we performed qRT-PCR analyses to determine the expression levels of various β-cell markers. Surprisingly, the results showed 2.4-fold increase in Ucn3 levels, but no change in the expression of Mafa, whereas levels of Nkx6.1 and neurogenin 3 (Ngn3), were significantly reduced in islets of miR-223 KO mice, compared with WT-controls (Fig. 5D). These data suggest the bidirectional effects of miR-223 on β-cell identity with an overall net effect of reduced functional β-cell mass upon miR-223 ablation.
MiR-223 regulates β-cell growth in a cell-autonomous manner
To further clarify the in vivo findings, we utilized Min6 β-cells, which were infected with adenovirus encoding inhibitory miR-223 sequence (Ad.223off) for 48 h to knock down the expression of miR-223, followed by a series of experiments. qRT-PCR analysis validated that the expression levels of miR-223 were reduced by about 50% in Ad.223off-infected Min6 β-cells, compared with control cells (Fig. 6, A and B). Consistent with our in vivo findings, knockdown of miR-223 in Min6 β-cells significantly suppressed proliferation as indicated by 40% decrease in Ki-67–positive cells after FACS analysis, when compared with Ad.GFP-infected control cells (Fig. 6, C and D). In addition, down-regulation of miR-223 increased Min6 cell death as evidenced by MTS assay and FACS analysis with propidium iodide staining (Fig. 6E and Fig. S3, A and B). Moreover, cell death was exacerbated upon H2O2 and palmitate treatment (Fig. 6E). Similar to the in vivo findings on the molecular change pattern, Western blot results showed that down-regulation of miR-223 resulted in significantly higher levels of Foxo1, Sox6, and p27, whereas phosphorylated Foxo1, Pdx1 and Glut2 protein levels were decreased (Fig. 6, F and G). In addition, the protein levels of phosphorylated Akt were reduced in Ad.223off group, when compared with Ad.GFP controls (Fig. S3C). Furthermore, Ucn3 expression levels were greatly increased, and the expression levels of Nkx6.1, Ngn3, cyclin D1, and cyclin E1 were severely reduced when miR-223 was down-regulated (Fig. 6H). Overall, these data clarify that cell-autonomous mechanisms regulated by miR-223 contribute to the diminished functional β-cell mass exhibited by KO mice.
Figure 6.
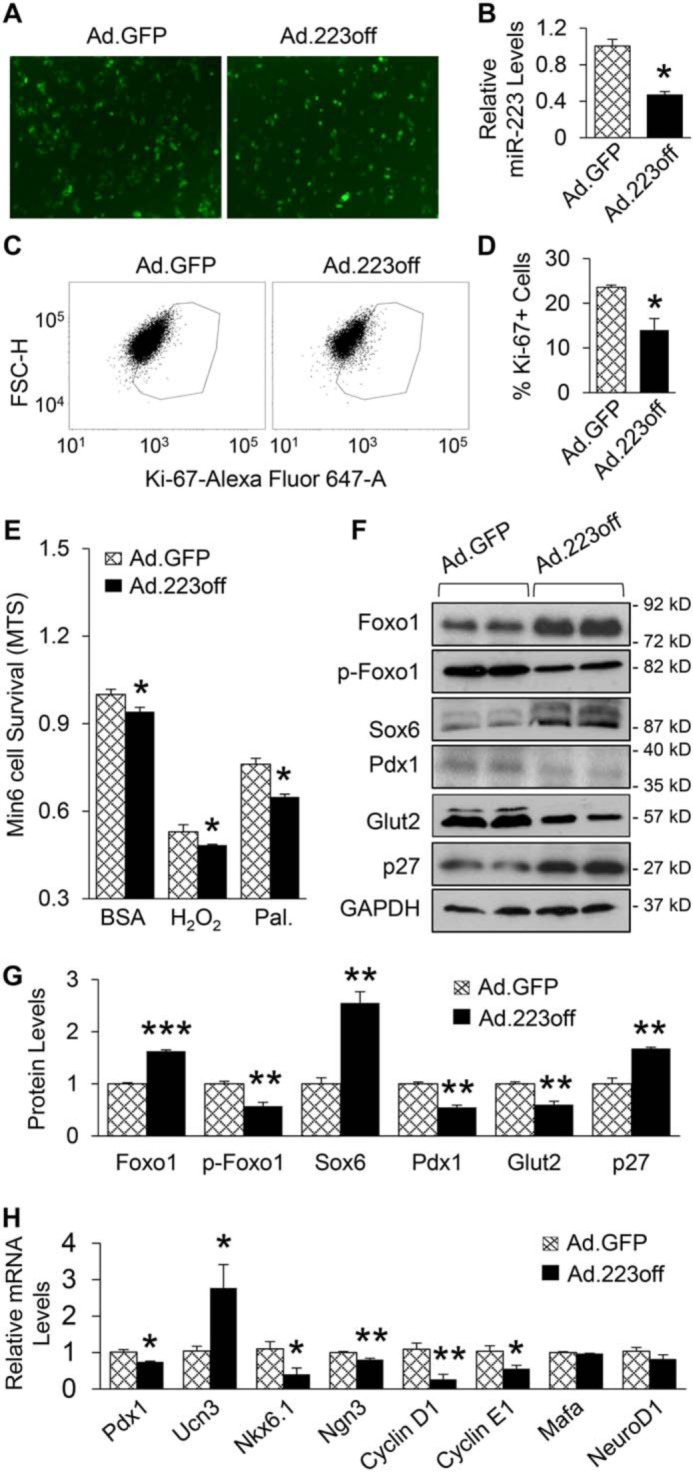
miR-223 regulates β-cell proliferation and identity in cell-autonomous manner. A, representative images of Ad.GFP- or Ad.223off-infected Min6 β-cells. B, expression of miR-223 was decreased after knockdown as measured by qPCR. C and D, representative FACS scatter plots (C) and quantification (D) of Ki-67 positive cells. E, MTS assay of Min6 β-cells after miR-223 knockdown, followed by 1 h H2O2 (200 mm) and 24 h palmitate treatment. F and G, protein levels of Foxo1, Sox6, and p27 were increased, whereas p-Foxo1, Pdx1, and Glut2 levels were reduced 48 h after Ad.223off infection. H, expression of key genes related to β-cell identity and proliferation were measured. Data are shown as mean ± S.E. (error bar).*, p < 0.05; **, p < 0.01; ***, p < 0.001 versus controls by t test
Overexpression of miR-223 rescues Min6 β-cell by improving proliferation and viability
Given that deficiency of miR-223 displayed overall detrimental effect on β-cells, we tested whether up-regulating miR-223 could rescue β-cell growth and function. Using adenovirus encoding miR-223 construct (Ad.miR-223), we overexpressed miR-223 in Min6 cells for 48 h (Fig. 7, A and B). Cell proliferation was increased by about 2-fold in Ad.miR-223-β-cells, compared with control Ad.GFP-cells (Fig. 7, C and D). Importantly, results of MTS assay revealed that elevation of miR-223 could promote Min6 cell viability under basal condition. Such protective effects were also observed when Min6 cells were treated with H2O2 and palmitate (Fig. 7E). In contrast to KO mice, Ad.miR-223 β-cells showed significant decrease in protein levels of Foxo1, Sox6, and p27, whereas p-Foxo1, Pdx1, Glut2, and phosphorylated Akt were dramatically increased (Fig. 7, F and G and Fig. S3D). qRT-PCR results also showed reversed expression pattern of β-cell markers: Ucn3 levels were reduced, whereas the levels of Nkx6.1, Ngn3, cyclin D1, and cyclin E1 were increased. Expression of Mafa and NeuroD1 were unaffected when miR-223 was up-regulated (Fig. 7H). Moreover, as the result from glucose-stimulated insulin secretion assay showed, overexpression of miR-223 could rescue and further augment insulin secretion (Fig. 7I). Taken together, these results demonstrate that up-regulation of miR-223 is able to promote β-cell proliferation, viability, and function.
Figure 7.

Overexpression of miR-223 rescues Min6 β-cell by improving proliferation response and viability. A, representative images showed Ad.GFP- or Ad.miR-223–infected Min6 β-cells. B, expression of miR-223 mRNA was measured by qPCR. C and D, proliferation of Min6 cells was significantly increased upon up-regulation of miR-223 as shown by representative FACS scatter plots (C) and quantification (D) of Ki-67–positive cells. E, MTS assay showed that Min6 cell viability was increased after miR-223 overexpression followed by 24 h palmitate (0.5 mm) or 1 h H2O2 (200 mm) treatments. F, protein levels of Foxo1, Sox6, and p27 were decreased, whereas p-Foxo1, Pdx1 and Glut2 levels were increased 48 h after Ad.miR223 infection. H, key genes related to β-cell identity and proliferation were measured after overexpressing miR-223. I, glucose-stimulated insulin secretion assay showed that miR-223 down-regulation impaired insulin secretion, which could be rescued by overexpression of miR-223. Data are shown as mean ± S.E. (error bar).*, p < 0.05; **, p < 0.01; ***, p < 0.001 versus controls by t test.
Discussion
Our study is the first attempt to elucidate the effects of miR-223 on functional β-cell mass. We observed higher miR-223 levels in islets of diabetic mice and humans as well as in Min6 β-cells treated with TNFα and high glucose. We also demonstrate that deletion of miR-223 interrupted glucose homeostasis. Further investigation revealed that miR-223 deficiency reduced proliferation rate and increased β-cell death under basal condition, and the compensatory response of β-cells to HFD-induced insulin resistance was blunted. Insights into the mechanism involved in the regulation of proliferative response in β-cells have been provided by examining Foxo1 and Sox6, two direct targets of miR-223. Our data showed that ablation of miR-223 caused increased expression and nuclear localization of Foxo1, which suppressed Pdx1 and Glut2 expression but increased p27 protein levels. In addition, increased protein levels of Sox6 also contributed to the down-regulation of Pdx1 and cyclin D1. Furthermore, overexpression of miR-223 in β-cells was able to improve their proliferation and function. Taken together, our data unveil a critical molecular mechanism whereby miR-223 is essential for maintaining functional β-cell mass by controlling Foxo1 and Sox6 signaling cascades (Fig. 8), providing potential effective targets for diabetes intervention.
Figure 8.
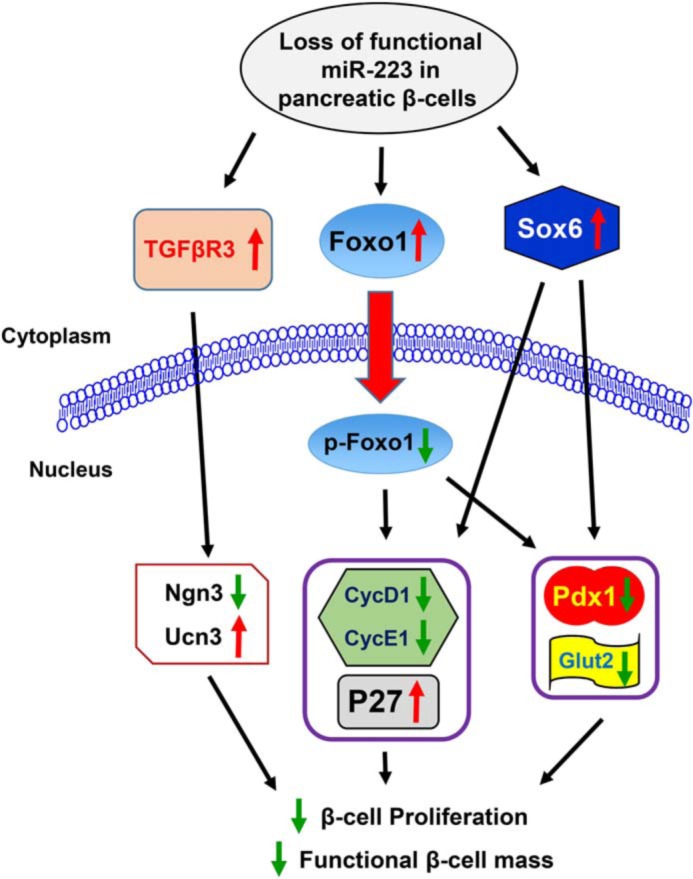
Graphical summary. Proposed scheme for the mechanism illustrating the regulation of β-cell function plasticity by miR-223.
It has been proposed that transient β-cell dedifferentiation is required prior to proliferation, which plays a pivotal role in their in vivo dynamics (6, 7). A recent study reported that TGFβR3 could reverse β-cell dedifferentiation ex vivo, evidenced by restoration of Ucn3 (32). In addition, TGFβ receptor I inhibitor, Alk5 inhibitor II, reversed β-cell dedifferentiation and restored key transcription factors such as Pdx1 and Nkx6.1 (32). However, Alk5 inhibitor II can also inhibit SMAD7-induced β-cell proliferation, which reiterates the intriguing hypothesis that β-cell dedifferentiation proceeds before proliferation. Indeed, as Alk5 inhibitor II reverses β-cell dedifferentiation under cytokine stress condition, it increases gene expression of Foxo1, which has been widely reported as a repressor of β-cell proliferation (26, 33). In contrast, it has been shown that β-cells with Foxo1 deficiency could undergo dedifferentiation (10). In the present study, we also showed that miR-223 could directly target TGFβR3 (Fig. S4, A and B), which was up-regulated in miR-223 knockdown β-cells (Fig. S4C). As shown in Fig. 7H, gene expression of Ngn3 was enhanced when miR-223 was up-regulated, this suggests that miR-223 might be able to induce β-cell dedifferentiation, yet further investigation is required to confirm this hypothesis.
Foxo family proteins play critical roles in the regulation of whole body energy metabolism. Of interest, Foxo1, a key transcription factor in insulin signaling, is highly expressed in pancreatic β-cells, in which Foxo1 is best known for its role in regulating the expression of Pdx1 (26, 28). Nonetheless, numerous studies have suggested that the multifunctional roles of Foxo1 may be attributed to its involvement in crosstalk between various intracellular signaling pathways. For example, Foxo1 inhibition is required for the proliferative effects of GLP-1 on β-cells (34). It has also been reported that ER stress- or lipotoxicity-triggered β-cell apoptosis could be attenuated by overexpressing dominant-negative Foxo1 (27). Furthermore, Foxo1 inhibits cell proliferation via up-regulating cyclin-dependent kinase inhibitor such as p27 (26). In fact, inactivation of Foxo1–p27 cascade mediates the proliferative action of liraglutide in β-cells (33). In accordance with current literature, our study presented here shows that miR-223 is essential for β-cell proliferation via targeting the Foxo1 signaling pathway. Therefore, inhibition of Foxo1 by overexpression of miR-223 may possess great potential to promote β-cell proliferation and alleviate diabetes.
Given that miR-223 directly targets Sox6, a key molecule known to suppress β-cell redifferentiation (8), up-regulation of Sox6 in miR-223 KO β-cells might inhibit the process of their redifferentiation. Nonetheless, further experiments and proper animal model would be needed to validate this conclusion. There is another limitation in this work that needs to be addressed. In vitro evidence of the beneficial effects of miR-223 was based on experiments involving substantial overexpression in Min6 cells; such findings should be evaluated in a more physiological setting, ideally using β-cell–specific overexpression of miR-223 animal model.
It is important to note that previous studies have shown up-regulation of miR-223 in the blood, heart, and adipose tissue of type 2 diabetic patients (36–38). In line with our present findings, these results suggest that increased miR-223 levels in diabetic conditions may represent a compensatory mechanism to protect against T2D, as miR-223 could augment functional β-cell mass to cope with the increasing demand of insulin under obese/T2D conditions. Furthermore, miR-223 has been reported to protect against inflammatory response in adipose tissue and insulin resistance (18). Therefore, increased levels of miR-223 may be the compensatory outcome to mitigate the progression of T2D. Indeed, our data presented here demonstrate that down-regulation of miR-223 resulted in the loss of functional β-cell mass during diabetes. Taken together, the current and prior observations consistently indicate that miR-223 is a critical factor for maintaining functional β-cell mass and adaptation to metabolic stress.
Experimental procedures
Mouse manipulation
Pre–miR-223 knockout mice (B6.Cg-Ptprca MiR223tm1Fcam/J) and WT mice (C57BL6 background) were purchased from The Jackson Laboratory (Bar Harbor, ME). All animal protocols followed the Guidelines for the Care and Use of Laboratory Animals prepared by the National Institutes of Health and were approved by the University of Cincinnati Animal Care and Use Committee.
Diabetic mouse models
To generate type 1 diabetes, male adult mice (5–6 weeks old) were intraperitoneally injected with STZ (50 mg/kg body weight; S0130-1G, Sigma-Aldrich), or citrate buffer as control, daily for 5 days. At days 7 and 10 after the first injection, blood glucose levels were measured. Mice with glucose above 250 mg/dl were considered diabetic and used for experiments. For high-fat diet feeding paradigm, starting at age of 5 or 6 weeks, male mice were given 60% kcal HFD (D12492, Research Diet, New Brunswick, NJ) for 18 weeks; mice fed with normal CD were used as control. In addition, db/db mice (stock no. 000697) were purchased from The Jackson Laboratory; islets were isolated from 14-week-old male mice for experiments.
Islet isolation
Mouse islets were isolated as described previously (35). Briefly, mice were anesthetized and the pancreas was perfused with collagenase and neutral protease (CIzyme RI, catalog no. 005-1030, VitaCyte, Indianapolis, IN) followed by incubation in 37 °C water bath for 15 min with gentle swirling every 3 min. Then HBSS (with 0.3% BSA) was added to pancreas and centrifuged at 290 × g for 1 min. Supernatant was discarded, and cell pellets were resuspended in 10 ml HBSS, which were then filtered through a plastic tea strainer and rinsed with another 10 ml HBSS. After centrifugation at 330 × g for 2 min, supernatant was discarded and cell pellets were resuspended in 10 ml cold Histopaque-1100 and 10 ml HBSS. After centrifugation at 900 × gfor 18 min, entire 20 ml of supernatant was collected and passed through an inverted 70-μm filter, which was rinsed into a Petri dish with 10 ml media. Islets were then handpicked for future experiments. Human islets from both male and female healthy donors (ages 29–57 years old, BMI 21.2–25.9 kg/m2, HbA1c 4.6–5.7%), obese donors (ages 20–62 years old, BMI 31–36.9 kg/m2, HbA1c 5–5.8%), and type 2 diabetic donors (ages 58–66 years old, BMI 30.3–32.7 kg/m2, HbA1c 6.5–6.7%) were purchased from Prodo Laboratories.
Cell culture and treatment
Mouse insulinoma Min6 β-cells were purchased (C0018008, AddexBio, San Diego, CA) and maintained in DMEM supplemented with 10% heat-inactivated FBS, 50 μm β-mercaptoethanol (M3148, Sigma-Aldrich), 10 mm HEPES, and 1% antibiotic antimycotic solution (30-004-Cl, Corning, Corning, NY) in humidified 5% (v/v) CO2, 95% (v/v) air at 37 °C. Min6 cells were seeded on 100-mm dishes with 2 × 106 density and exposed to TNFα (5 ng/ml, 410-MT-050, R&D Systems, Minneapolis, MN) for 24 h. BSA-treated cells were used as control. For glucose treatment, Min6 cells were first incubated in low-glucose medium (5 mm) for 4 h, then treated with low- (as control) or high-glucose (25 mm) medium for 24 h.
MiR-223–overexpressing adenovirus (Ad.miR223) was constructed as described previously (21) and miR-223-inhibitory adenovirus (Ad.223off) was purchased (mr5233, Applied Biological Materials, Richmond, BC, Canada). Plated cells on dishes/wells were infected with Ad.miR-223 or Ad.223off at 10 multiplicity of infection; adenovirus carrying only GFP vector (Ad.GFP) was used as control. After 48–72 h, cells were treated or harvested for further experiments.
RNA isolation and quantitative RT-PCR (qRT-PCR)
Total RNA was extracted from pancreatic islets or Min6 cells with miRNeasy Mini Kit (217004, Qiagen, Venlo, Netherlands) following the manufacturer's protocol, and qRT-PCR was performed using miScript PCR Starter Kit (218193, Qiagen). U6 and GAPDH were used as internal control. The primer sequences are shown in Table S1.
Blood glucose and insulin measurement
Mice were fasted overnight, and blood glucose was measured through tail tip bleeding with the use of Accu-Chek Smartview Nano meter (Roche Diabetes Care). For insulin measurement, whole blood was drawn from submandibular vein, and plasma was separated by centrifugation at 12,000 rpm at 4 °C for 15 min. in microvette tubes coated with EDTA (KMIC-EDTA, Kent Scientific, Torrington, CT). Plasma insulin levels were measured by mouse ELISA kit (cat. no. 90080, Crystal Chem, Elk Grove Village, IL) according to manufacturer's instructions. The HOMA index was used to calculate insulin resistance (HOMA-IR) and β-cell function (HOMA-%β). The following formula was used: HOMA-IR = (fasting glucose × insulin)/22.5; HOMA-%β = (20 × fasting insulin)/(fasting glucose − 3.5)%. Units of glucose and insulin are mm and milliunit/liter, respectively.
Intraperitoneal glucose tolerance testing (IPGTT) and insulin tolerance testing (IPITT)
Mice were fasted overnight (for IPGTT) or for 6 h (for IPITT) prior to injection. Glucose (2 g/kg body weight) or insulin (0.75 units/kg body weight) was injected intraperitoneally. Glucose levels were measured in blood collected from tail tip prior to and at 15, 30, 60, 90, and 120 min after injection.
Histological analysis
Immunohistochemistry staining was performed as described previously (19). For measuring cell apoptosis, TUNEL staining was performed using DeadEnd Fluorometric TUNEL System (catalog no. G3250, Promega) following manufacturer's protocol.
Images of insulin-positive cells were taken from pancreatic sections using Zeiss LSM710 LIVE Duo Confocal Microscope (20× objective, Live Microscopy Core, University of Cincinnati). Zen/ZenLight software or ImageJ were used to quantify all insulin-positive cells or cells costained with insulin and Ki-67 or pHH3 or Foxo1. Images of sections from a minimum of three mice per group, three to five pancreas sections per mouse, were captured.
Luciferase reporter assay for validation of miR-223 targets
Luciferase reporter experiments were performed as described previously (21). Briefly, 3′-UTR segments of Foxo1 and Sox6 and their respective mutants were amplified and validated prior to transfection in HEK293 or Min6 cells. 100 nm mimic miR-223 or mimic miR control (Thermo Fisher Scientific) were added to each well in 12-well plates. Cell lysates were prepared 48 h later, and luciferase activity was measured and expressed as relative light units. All transfections were performed in triplicate from three independent experiments.
Western blotting
Western blotting was performed as described previously (19). The antibodies used are listed in Table S2.
MTS assay
Min6 β-cells were plated on 96-well plates at 104 seeding density and transfected with adenovirus for 48 h followed by 24 h palmitate (0.5 mm) or 1 h H2O2 (200 mm) treatment. Cell viability was assessed using the MTS incorporation assay kit (Promega) according to the manufacturer's protocol.
Flow cytometry
Mice were anesthetized and pancreases were perfused with collagenase and neutral protease followed by incubation in 37 °C for 30 min using 700 agitation. Digestion was stopped by adding excessive volume of cold HBSS, and digested tissue was filtered through 70-μm cell restrainer and washed with HBSS. Cells were then stained with insulin (catalog no. FAB1544P, R&D Systems, Minneapolis, MN) and Ki-67 (catalog no. 61-5698-82, Invitrogen).
For in vitro experiments, after adenovirus infection, cells were fixed with cold 75% ethanol and stored at −20 °C for at least 2 h. Cell proliferation was determined by staining with Alexa Fluor® 647 mouse anti–Ki-67 antibody (catalog no. 558615, BD Biosciences). For cell death analysis, cells were stained with propidium iodide staining solution (00-6990, eBioscience, Waltham, MA). Flow cytometry was performed using LSRII Analyzer (SHC Flow Cytometry Core, Cincinnati) and analyzed with FCSexpress software.
Statistical analysis
Animals were randomly assigned to groups, and sample size estimates were not used. All data were analyzed by two-tailed Student's t test and reported as mean ± S.E. or S.D. as specified. A p < 0.05 was considered statistically significant.
Author contributions
Y. L., Z.-X. M., and G.-C. F. conceptualization; Y. L., Z.-X. M., and G.-C. F. resources; Y. L., S. D., J. P., and X. W. data curation; Y. L., S. D., X. W., K. E., X. M., T. P., and Z.-X. M. formal analysis; Y. L., Z.-X. M., and G.-C. F. supervision; Y. L., Z.-X. M., and G.-C. F. funding acquisition; Y. L. and S. D. validation; Y. L., S. D., J. P., X. W., Z.-X. M., and G.-C. F. investigation; Y. L., S. D., X. W., K. E., X. M., T. P., and Z.-X. M. methodology; Y. L. and S. D. writing-original draft; Y. L., S. D., Z.-X. M., and G.-C. F. project administration; Y. L., S. D., T. P., Z.-X. M., and G.-C. F. writing-review and editing.
Supplementary Material
This work was supported by American Heart Association (AHA) Established Investigator Award 17EIA33400063 and National Institutes of Health Grants GM-112930 and GM-126061 (to G.-C. F.); National Key Research and Development Programme of China 2016YFC1305303, the Fundamental Research Funds for the Central Universities, National Natural Science Foundation of China Grants 81670740 and 81722012, and the Thousand Young Talents Plan of China (to Z. X. M.); and National Natural Science Foundation of China Grant 81600616 (to S. D.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4 and Tables S1 and S2.
- T1D
- Type 1 diabetes
- T2D
- Type 2 diabetes
- miRNA
- microRNA
- miR
- microRNA
- CD
- chow-diet
- HFD
- high-fat diet
- qRT-PCR
- quantitative real-time PCR
- HOMA
- homeostatic model assessment
- STZ
- streptozotocin
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- HBSS
- Hanks' Balanced Salt Solution
- IPGTT
- intraperitoneal glucose tolerance test
- IPITT
- intraperitoneal insulin tolerance test.
References
- 1. Zheng Y., Ley S. H., and Hu F. B. (2018) Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 14, 88–98 10.1038/nrendo.2017.151 [DOI] [PubMed] [Google Scholar]
- 2. Vetere A., Choudhary A., Burns S. M., and Wagner B. K. (2014) Targeting the pancreatic β-cell to treat diabetes. Nat. Rev. Drug Discov. 13, 278–289 10.1038/nrd4231 [DOI] [PubMed] [Google Scholar]
- 3. Cerf M. E. (2013) Beta cell dysfunction and insulin resistance. Front. Endocrinol. 4, 37 10.3389/fendo.2013.00037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang P., Fiaschi-Taesch N. M., Vasavada R. C., Scott D. K., García-Ocaña A., and Stewart A. F. (2015) Diabetes mellitus—advances and challenges in human β-cell proliferation. Nat. Rev. Endocrinol. 11, 201–212 10.1038/nrendo.2015.9 [DOI] [PubMed] [Google Scholar]
- 5. Dor Y., Brown J., Martinez O. I., and Melton D. A. (2004) Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 429, 41–46 10.1038/nature02520 [DOI] [PubMed] [Google Scholar]
- 6. Cheng C. W., Villani V., Buono R., Wei M., Kumar S., Yilmaz O. H., Cohen P., Sneddon J. B., Perin L., and Longo V. D. (2017) Fasting-mimicking diet promotes Ngn3-driven β-cell regeneration to reverse diabetes. Cell 168, 775–788.e12 10.1016/j.cell.2017.01.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim-Muller J. Y., Zhao S., Srivastava S., Mugabo Y., Noh H. L., Kim Y. R., Madiraju S. R., Ferrante A. W., Skolnik E. Y., Prentki M., and Accili D. (2014) Metabolic inflexibility impairs insulin secretion and results in MODY-like diabetes in triple FoxO-deficient mice. Cell Metab. 20, 593–602 10.1016/j.cmet.2014.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sintov E., Nathan G., Knoller S., Pasmanik-Chor M., Russ H. A., and Efrat S. (2015) Inhibition of ZEB1 expression induces redifferentiation of adult human β cells expanded in vitro. Sci. Rep. 5, 13024 10.1038/srep13024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lenz A., Toren-Haritan G., and Efrat S. (2014) Redifferentiation of adult human β cells expanded in vitro by inhibition of the WNT pathway. PLoS One 9, e112914 10.1371/journal.pone.0112914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Talchai C., Xuan S., Lin H. V., Sussel L., and Accili D. (2012) Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 150, 1223–1234 10.1016/j.cell.2012.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. White M. G., Marshall H. L., Rigby R., Huang G. C., Amer A., Booth T., White S., and Shaw J. A. (2013) Expression of mesenchymal and α-cell phenotypic markers in islet β-cells in recently diagnosed diabetes. Diabetes Care 36, 3818–3820 10.2337/dc13-0705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Russ H. A., Ravassard P., Kerr-Conte J., Pattou F., and Efrat S. (2009) Epithelial-mesenchymal transition in cells expanded in vitro from lineage-traced adult human pancreatic beta cells. PLoS One 4, e6417 10.1371/journal.pone.0006417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bar Y., Russ H. A., Sintov E., Anker-Kitai L., Knoller S., and Efrat S. (2012) Redifferentiation of expanded human pancreatic β-cell-derived cells by inhibition of the NOTCH pathway. J. Biol. Chem. 287, 17269–17280 10.1074/jbc.M111.319152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Plaisance V., Waeber G., Regazzi R., and Abderrahmani A. (2014) Role of microRNAs in islet beta-cell compensation and failure during diabetes. J. Diabetes Res. 2014, 618652 10.1155/2014/618652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhu H., and Fan G. C. (2011) Extracellular/circulating microRNAs and their potential role in cardiovascular disease. Am. J. Cardiovasc. Dis. 1, 138–149 [PMC free article] [PubMed] [Google Scholar]
- 16. Filios S. R., and Shalev A. (2015) β-cell microRNAs: Small but powerful. Diabetes 64, 3631–3644 10.2337/db15-0831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Osmai M., Osmai Y., Bang-Berthelsen C. H., Pallesen E. M., Vestergaard A. L., Novotny G. W., Pociot F., and Mandrup-Poulsen T. (2016) MicroRNAs as regulators of beta-cell function and dysfunction. Diabetes Metab. Res. Rev. 32, 334–349 10.1002/dmrr.2719 [DOI] [PubMed] [Google Scholar]
- 18. Zhuang G., Meng C., Guo X., Cheruku P. S., Shi L., Xu H., Li H., Wang G., Evans A. R., Safe S., Wu C., and Zhou B. (2012) A novel regulator of macrophage activation: miR-223 in obesity-associated adipose tissue inflammation. Circulation 125, 2892–2903 10.1161/CIRCULATIONAHA.111.087817 [DOI] [PubMed] [Google Scholar]
- 19. Yang L., Li Y., Wang X., Mu X., Qin D., Huang W., Alshahrani S., Nieman M., Peng J., Essandoh K., Peng T., Wang Y., Lorenz J., Soleimani M., Zhao Z. Q., and Fan G. C. (2016) Overexpression of miR-223 tips the balance of pro- and anti-hypertrophic signaling cascades toward physiologic cardiac hypertrophy. J. Biol. Chem. 291, 15700–15713 10.1074/jbc.M116.715805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vickers K. C., Landstreet S. R., Levin M. G., Shoucri B. M., Toth C. L., Taylor R. C., Palmisano B. T., Tabet F., Cui H. L., Rye K. A., Sethupathy P., and Remaley A. T. (2014) MicroRNA-223 coordinates cholesterol homeostasis. Proc. Natl. Acad. Sci. U.S.A. 111, 14518–14523 10.1073/pnas.1215767111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang J., Bai X., Song Q., Fan F., Hu Z., Cheng G., and Zhang Y. (2015) miR-223 inhibits lipid deposition and inflammation by suppressing toll-like receptor 4 signaling in macrophages. Int. J. Mol. Sci. 16, 24965–24982 10.3390/ijms161024965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhao Y., Li D., Chen D., Ji H., Hu Q., and Zhang J. (2017) Increased miR-223 expression promotes proliferation and migration of retinal endothelial cells and pathogenesis of diabetic retinopathy by targeting EIF4E3 and IGF-1R. Int. J. Clin. Exp. Pathol. 10, 2950–2959 [Google Scholar]
- 23. Johnnidis J. B., Harris M. H., Wheeler R. T., Stehling-Sun S., Lam M. H., Kirak O., Brummelkamp T. R., Fleming M. D., and Camargo F. D. (2008) Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 451, 1125–1129 10.1038/nature06607 [DOI] [PubMed] [Google Scholar]
- 24. Wang X., Gu H., Qin D., Yang L., Huang W., Essandoh K., Wang Y., Caldwell C. C., Peng T., Zingarelli B., and Fan G. C. (2015) Exosomal miR-223 contributes to mesenchymal stem cell-elicited cardioprotection in polymicrobial sepsis. Sci. Rep. 5, 13721 10.1038/srep13721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang J., Li S., Li L., Li M., Guo C., Yao J., and Mi S. (2015) Exosome and exosomal microRNA: Trafficking, sorting, and function. Genomics Proteomics Bioinformatics 13, 17–24 10.1016/j.gpb.2015.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kitamura T. (2013) The role of FOXO1 in β-cell failure and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 9, 615–623 10.1038/nrendo.2013.157 [DOI] [PubMed] [Google Scholar]
- 27. Martinez S. C., Tanabe K., Cras-Méneur C., Abumrad N. A., Bernal-Mizrachi E., and Permutt M. A. (2008) Inhibition of Foxo1 protects pancreatic islet beta-cells against fatty acid and endoplasmic reticulum stress-induced apoptosis. Diabetes 57, 846–859 10.2337/db07-0595 [DOI] [PubMed] [Google Scholar]
- 28. Kitamura T., Nakae J., Kitamura Y., Kido Y., Biggs W. H. 3rd, Wright C. V., White M. F., Arden K. C., and Accili D. (2002) The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J. Clin. Invest. 110, 1839–1847 10.1172/JCI200216857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Song M. Y., Wang J., Ka S. O., Bae E. J., and Park B. H. (2016) Insulin secretion impairment in Sirt6 knockout pancreatic β cells is mediated by suppression of the FoxO1-Pdx1-Glut2 pathway. Sci. Rep. 6, 30321 10.1038/srep30321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Iguchi H., Urashima Y., Inagaki Y., Ikeda Y., Okamura M., Tanaka T., Uchida A., Yamamoto T. T., Kodama T., and Sakai J. (2007) SOX6 suppresses cyclin D1 promoter activity by interacting with beta-catenin and histone deacetylase 1, and its down-regulation induces pancreatic beta-cell proliferation. J. Biol. Chem. 282, 19052–19061 10.1074/jbc.M700460200 [DOI] [PubMed] [Google Scholar]
- 31. Iguchi H., Ikeda Y., Okamura M., Tanaka T., Urashima Y., Ohguchi H., Takayasu S., Kojima N., Iwasaki S., Ohashi R., Jiang S., Hasegawa G., Ioka R. X., Magoori K., Sumi K., et al. (2005) SOX6 attenuates glucose-stimulated insulin secretion by repressing PDX1 transcriptional activity and is down-regulated in hyperinsulinemic obese mice. J. Biol. Chem. 280, 37669–37680 10.1074/jbc.M505392200 [DOI] [PubMed] [Google Scholar]
- 32. Blum B., Roose A. N., Barrandon O., Maehr R., Arvanites A. C., Davidow L. S., Davis J. C., Peterson Q. P., Rubin L. L., and Melton D. A. (2014) Reversal of β cell de-differentiation by a small molecule inhibitor of the TGFβ pathway. Elife 3, e02809 10.7554/eLife.02809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fang D., Huang Z., Guan H., Liu J., Yao B., Xiao H., and Li Y. (2012) The Akt/FoxO1/p27 pathway mediates the proliferative action of liraglutide in β cells. Mol. Med. Rep. 5, 233–238 10.3892/mmr.2011.607 [DOI] [PubMed] [Google Scholar]
- 34. Buteau J., Spatz M. L., and Accili D. (2006) Transcription factor FoxO1 mediates glucagon-like peptide-1 effects on pancreatic beta-cell mass. Diabetes 55, 1190–1196 10.2337/db05-0825 [DOI] [PubMed] [Google Scholar]
- 35. Stull N. D., Breite A., McCarthy R., Tersey S. A., and Mirmira R. G. (2012) Mouse islet of Langerhans isolation using a combination of purified collagenase and neutral protease. J. Vis. Exp. 67, e4137 10.3791/4137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lu H., Buchan R. J., and Cook S. A. (2010) MicroRNA-223 regulates Glut4 expression and cardiomyocyte glucose metabolism. Cardiovasc. Res. 86, 410–420 10.1093/cvr/cvq010 [DOI] [PubMed] [Google Scholar]
- 37. Chuang T. Y., Wu H. L., Chen C. C., Gamboa G. M., Layman L. C., Diamond M. P., Azziz R., and Chen Y-H. (2015) MicroRNA-223 expression is upregulated in insulin resistant human adipose tissue. J. Diabetes Res. 2015, 943659 10.1155/2015/943659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nunez Lopez Y. O., Garufi G., and Seyhan A. A. (2016) Altered levels of circulating cytokines and microRNAs in lean and obese individuals with prediabetes and type 2 diabetes. Mol. Biosyst. 13, 106–121 10.1039/c6mb00596a [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.