

Abstract
Exposure to organophosphorus compounds (OPs) may be fatal if untreated, and a clear and present danger posed by nerve agent OPs has become palpable in recent years. OPs inactivate acetylcholinesterase (AChE) by covalently modifying its catalytic serine. Inhibited AChE cannot hydrolyze the neurotransmitter acetylcholine leading to its build-up at the cholinergic synapses and creating an acute cholinergic crisis. Current antidotes, including oxime reactivators that attack the OP-AChE conjugate to free the active enzyme, are inefficient. Better reactivators are sought, but their design is hampered by a conformationally rigid portrait of AChE extracted exclusively from 100K X-ray crystallography and scarcity of structural knowledge on human AChE (hAChE). Here, we present room temperature X-ray structures of native and VX-phosphonylated hAChE with an imidazole-based oxime reactivator, RS-170B. We discovered that inhibition with VX triggers substantial conformational changes in bound RS-170B from a “nonproductive” pose (the reactive aldoxime group points away from the VX-bound serine) in the reactivator-only complex to a “semi-productive” orientation in the VX-modified complex. This observation, supported by concurrent molecular simulations, suggested that the narrow active-site gorge of hAChE may be significantly more dynamic than previously thought, allowing RS-170B to reorient inside the gorge. Furthermore, we found that small molecules can bind in the choline-binding site hindering approach to the phosphorous of VX-bound serine. Our results provide structural and mechanistic perspectives on the reactivation of OP-inhibited hAChE and demonstrate that structural studies at physiologically relevant temperatures can deliver previously overlooked insights applicable for designing next-generation antidotes.
Keywords: acetylcholinesterase (AChE), X-ray crystallography, molecular modeling, protein chemical modification, structure-function, oxime reactivator, reactivation mechanism, RS-170B, VX-inhibition
Introduction
Acetylcholinesterase (AChE)2 (EC 3.1.1.7), an enzyme of the α/β hydrolase-fold superfamily, has a critical role in synaptic neurotransmission (1). Nerve agent organophosphorus (OP) compounds, the most poisonous substances made by man, are covalent AChE inhibitors (2). The use of these chemicals is banned internationally. However, recent high-profile incidents of OP intoxication, including mass population sarin poisoning in the Syrian Khan Sheikhoun attack in 2017, poisoning of North Korean dissident Kim Jong Nam with VX, and use of a Novichok agent against Sergei and Yulia Skripal in 2018, demonstrate the existence of a real threat from these substances. Chemically, OPs covalently attach to the side chain hydroxyl of the catalytic serine of AChE to form phosphoserine conjugates resulting in the enzyme's inactivation (3, 4). Catalytic function of the OP-inhibited AChE can be restored through a chemical reaction with nucleophiles, which attack the phosphorus atom of the OP-AChE to regenerate the active enzyme (5, 6). Due to the complexity of the reactivation reaction a universal reactivator equally effective against all OP-AChE conjugates has not been identified. In several aspects reactivation efficiency depends on: 1) the chemical and steric nature of the OP agent; 2) the chemical structure of the oxime reactivator determining its nucleophilic and OP-AChE–binding properties; and 3) the spatially constricted active site environment of the AChE active-center gorge, which controls the orientation of the bound phosphonyl moiety as well as oxime access to the phosphorus atom. A structure-based approach is therefore well-justified for the design of improved antidotes against OP intoxication.
Many X-ray structures of AChE:oxime complexes, OP-AChE conjugates, and complexes of OP-AChE with reactivator oxime molecules have been published with mouse (mAChE) (6–11) and electric ray (Torpedo californica, TcAChE) (12–16) and only a few with human AChE (hAChE) (17–21). However, this wealth of structural information has yet to yield improved reactivation efficacy of oxime antidotes (22). Most of the promising antidotes (the bisquaternary oxime HI-6, as well as novel bifunctional oximes) occupy only the upper volume of the TcAChE and hAChE active-center gorges, above the narrow “choke point” defined by the Tyr-124 (Tyr-121 in TcAChE) phenolic side chain and physically separated from the targeted inhibitory P atom. Only some of the longer bispyridinium oximes, HLo7 and ortho 7, extend into the lower part of the gorge, beyond the choke point (9). The environment of the active-site gorge appears to be rigid and unresponsive to both reactivator binding and OP inhibition that is reflected in the limited number of the reported AChE side chain fluctuations. Differences in the side chain conformations were observed for the catalytic histidine in VX-TcAChE, His-440, and tabun-mAChE, His-447 (3, 7). The catalytic histidine alone, or with the help of a water molecule, was suggested (10, 11) to serve as a proton acceptor for the oxime's hydroxyl group, thus boosting an essential reactivator property, its nucleophilicity. Reorientation of the Phe-338 was also captured in the structures of mAChE inhibited with different nerve agents, resulting in a narrower gorge that restricts oxime access to the active site (7, 8). Although there is a large degree of sequence and structural similarity among AChEs from different species (57% sequence identity for hAChE/TcAChE, 88% for hAChE/mAChE, and 59% for TcAChE/mAChE) (18), kinetic data on OP inhibition and oxime reactivation shows substantial differences between human and other species (4, 10, 12, 23, 24). It is thus clear that the most valuable X-ray structural analysis is that of human AChE and OP-hAChE conjugates in complex with oxime reactivators when developing highly efficient antidotes for human OP intoxication.
Nearly all of more than 200 AChE X-ray structures deposited in the Protein Data Bank have been extracted from data collected at nonphysiological cryogenic temperatures (∼100 K). Only one structure of TcAChE was reported as being obtained at room temperature (25, 26). Although there are many indisputable benefits of the cryo-method for macromolecular crystallography, there are several significant disadvantages. For instance, cryocooling has been documented to affect intermolecular interactions, suggesting that specific contacts observed in cryo-structures may not be relevant at room (or physiological) temperatures (27–29). Cryogenic temperatures can hide alternative side chain conformations important for protein function and alter the environment of chemical groups (30), preventing a complete understanding of how enzymes work and/or resulting in incorrect interpretations (31, 32). Cryoprotectants, which are required for most cryo-crystallographic experiments, can bind in active sites and displace lower affinity ligands, thus precluding analysis of the protein-ligand interactions, or can bind to allosteric sites thereby altering conformations and dynamics of the residues at those locations (33). Physiologically critical conformations of small molecule ligands and a macromolecule's amino acid side chains can thus be artificially altered by nonphysiological cryogenic temperatures of an X-ray diffraction experiment.
Here we report room and low temperature (LT, 100 K) X-ray crystal structures of native and VX-phosphonylated recombinant hAChE, both in complex with an imidazole-based oxime reactivator, RS-170B (Fig. 1), which is structurally similar to but less potent than HI-6 (34). Comparison of these structures revealed that covalent inhibition with VX, as well as flash-cooling of hAChE crystals, can trigger changes in how the reactivator molecule binds within the active-site gorge. Specifically, a “nonproductive” pose of RS-170B is observed in the reactivator-only binary complex that is converted into a “semi-productive” pose in the VX-modified ternary complex at room temperature. In contrast, the reactivator is distributed equally across these two orientations in the binary complex at LT. Clearly, the observed conformational diversity of RS-170B, which is distinctly influenced by experimental conditions of the X-ray data collection, illustrates the value of RT X-ray crystallography in identifying productive RS-170B interactions with the VX-hAChE conjugate and guiding structural interpretations regarding the mechanism and efficiency of the nucleophilic reactivation reaction. Furthermore, we found that small molecules can bind in the choline-binding site possibly affecting the reactivation.
Figure 1.
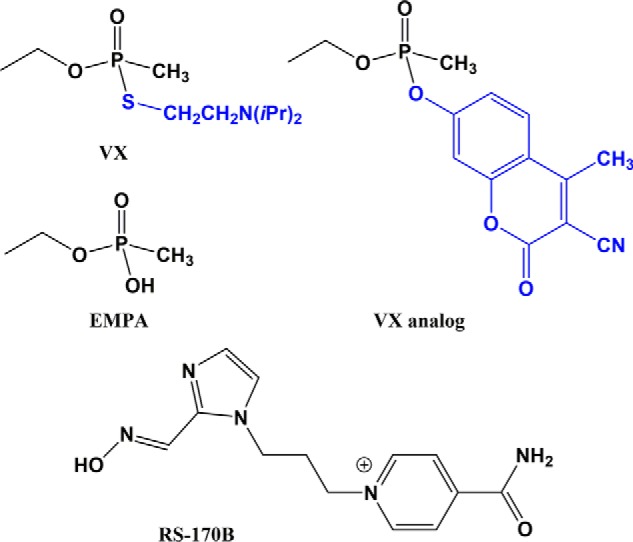
Chemical diagrams of VX nerve agent, nonvolatile VX analog, VX metabolite EMPA, and RS-170B oxime reactivator. The leaving groups of VX and its analog used in this study are colored blue.
Results
New crystal form of AChE
The hAChE protein, expressed in Gnt1-mutant HEK293 mammalian cell culture using a FLAG-tagged construct (35) and eluted from the affinity column by specific proteolysis at the engineered Prescission recognition site, yielded well-defined structures spanning Glu-4 at the N terminus to Thr-543 at the C terminus. This hAChE preparation, when in complex with the oxime RS-170B, was found to crystallize in the P31 unit cell with dimensions of a = b = 125.6, c = 131.4 (Table S1). This unit cell is significantly smaller than those in the previously reported X-ray structures of hAChE(17–21). None of the published AChE structures were from crystals with this unit cell and space group. The asymmetric unit contains two independent molecules that face each other across the active-site gorges, forming face-to-face dimers (Fig. S1). Importantly, this packing does not interfere with binding of long inhibitors that span AChE's catalytic (Trp-86) and peripheral (Trp-286) sites such as donepezil (PDB ID 6O4W) or BW286c51 (PDB ID 6O50) or restrict noticeably the diffusion of small molecules in and out of the active site as demonstrated by our soaking experiments. A brief 4-min soak with a nonvolatile VX analog (Fig. 1) was sufficient to yield well-defined covalent VX-hAChE:RS-170B conjugate starting from crystals of the binary hAChE:RS-170B complex. Examination of the crystal packing also revealed that physiologically relevant four-helix bundle hAChE dimers, normally observed in other AChE structures, are formed through symmetry operations in the current crystal lattice. Thus, both face-to-face and four-helix bundle dimers were concurrently observed in our structures. The lack of glycosylation at Asn-350, one of the three otherwise conserved N-glycosylation sites, is responsible for the new crystal form. Only Asn-265 and Asn-464 are found glycosylated in the current hAChE preparation. The absence of the side chain amide-linked oligosaccharide at Asn-350 frees this site for interactions with another hAChE molecule in the crystal (Fig. S1). This clearly facilitates crystal packing and, perhaps, promotes growth of larger crystals, allowing X-ray data collection at room temperature. The absence of glycosylation at these sites, as was demonstrated by site-directed mutagenesis, has minimal effect on the catalytic activity of hAChE (36). In each complex studied, the two molecules in the asymmetric unit are structurally very similar (root mean square deviation on the main chain atoms of ∼0.3 Å) and exhibit comparable ligand-binding features. To simplify the description and comparison of the structures we henceforth focus on chain A in all hAChE complexes discussed below, unless stated otherwise.
RS-170B orientations observed in the binary complexes
RS-170B belongs to the family of N-substituted imidazole aldoximes whose structure has been optimized for high reactivation efficiency of a potential OP bioscavenger, the Y337A/F338A double mutant hAChE (YAFA) (37). In the binary complex with native hAChE at RT obtained at a resolution of 2.80 Å, RS-170B binds with its pyridinium ring locked at the peripheral anionic site (PAS) through π-π interactions with Trp-286, whereas the rest of the reactivator molecule inserts into the active-site gorge (Figs. 2A and 3, and Fig. S2). The imidazole ring, however, is rotated such that the aldoxime group points away and is ∼11 Å from the catalytic Ser-203 (Fig. 3), instead making a weak hydrogen bond with the main chain amide of Phe-295. In addition, RS-170B makes several weak hydrophobic interactions with the side chains of Trp-286 and Tyr-341. With the aldoxime group turned away from Ser-203, this RS-170B pose can be considered as nonproductive, because the reactivator would not be able to approach the OP-modified Ser-203 and attack the phosphorus atom. In this pose, penetration of RS-170B deeper into the gorge is guarded by Tyr-124, Tyr-337, and Phe-338 residues. Interestingly, our DFT calculations of RS-170B in implicit water also demonstrated that the reactivator molecule favors a similar nonproductive conformation when free in solution (Fig. S3). Therefore, this nonproductive conformation is not imposed on RS-170B by the confines of the active-site gorge, but rather is the most stable geometry of the reactivator in solution before it enters the binding site.
Figure 2.
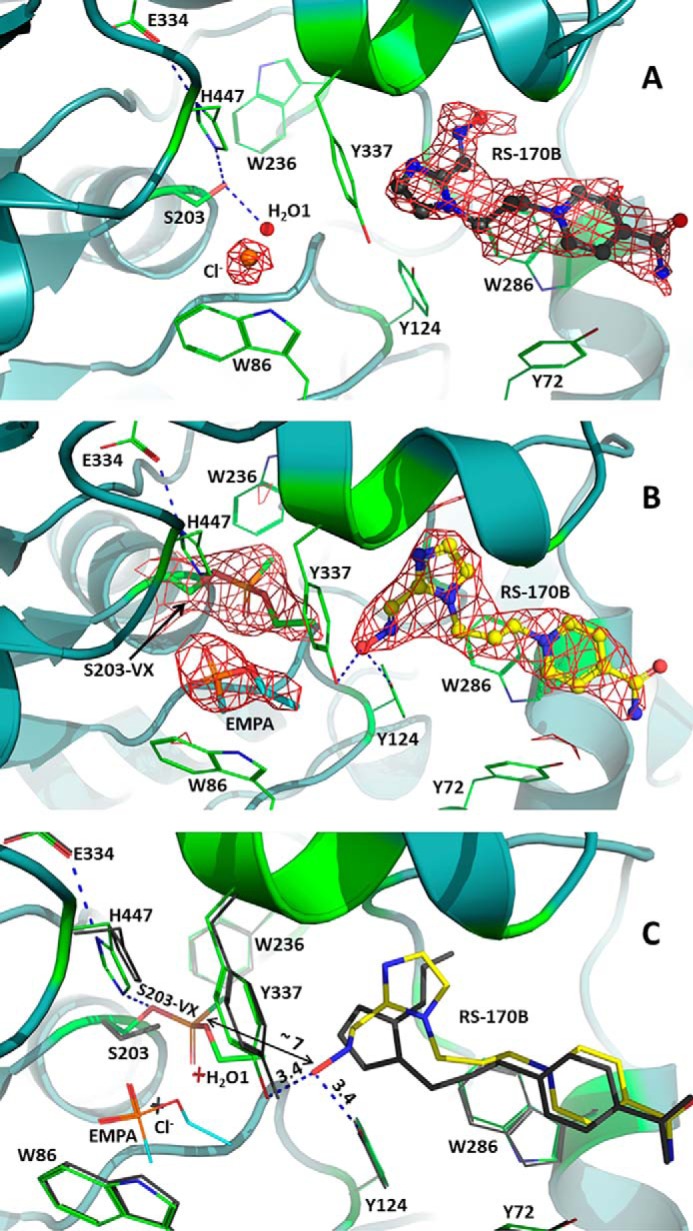
Productive reorientation of the reactivator molecule RS-170B triggered by VX inhibition: RT crystal structures of the native and VX-phosphonylated hAChE in complex with the oxime reactivator RS-170B. A, view of the active-center gorge of the native hAChE with bound nonproductive RS-170B. Here and for panel B, the 2Fo − FC electron density map (red) is contoured at 1σ. RS-170B is shown as ball and stick model with charcoal carbon atoms; spheres represent bound water (red) and chloride anion (orange). Here and in all the panels, blue dashed lines represent possible hydrogen bonding interactions. B, view of the active-center gorge of the VX-hAChE with bound semi-productive RS-170B. RS-170B is colored with yellow carbon atoms; EMPA is in stick model with cyan carbon atoms. C, the overlay of the binary hAChE:RS-170B (carbon colored green for the active-center gorge residues and yellow for RS-170B) and the ternary VX-hAChE:RS-170B (the active-center gorge residues and RS-170B colored charcoal) RT structures. Distances are in Å. Single letter amino acid residues are used in the figure.
Figure 3.

Location of RS-170B in the nonproductive conformation in the RT binary complex structure in relation to the active-center gorge (orange surface). The RS-170B pyridinium ring is anchored by the side chain of Trp-286, whereas the aldoxime oxygen atom is 11 Å away from the hydroxyl of Ser-203 shown by the double-headed arrow. The left and right panels are related by 90° rotation. Single letter amino acid residues are used in the figure.
In the LT hAChE:RS-170B structure at 2.15 Å resolution, the reactivator was found in two equally populated poses (Fig. S4), one being similar to the nonproductive pose seen in the RT structure, and the second having the imidazole ring rotated ∼180° so that the aldoxime group fully extends toward Ser-203 and is only ∼5 Å from its OH group: a pose required for the reactivation reaction. This fully extended pose, therefore, can be considered “productive.” Notably this pose was only observed in the low temperature structure, whereas the more compact, nonproductive, pose is only observed in the room temperature structure. Such temperature-dependent differences in ligand geometry were previously observed in structures of carbonic anhydrase in complex with clinically used inhibitors (38).
Assessment of RS-170B binding poses in the binary complexes with molecular modeling
To quantify the energetics of the two binding poses, we estimated the free energies and individual energy components of RS-170B binding to hAChE in the nonproductive and productive conformations using molecular mechanics-generalized Born/surface area (MM-GB/SA) method. As summarized in Table S2, the MM-GB/SA calculation results indicate that the nonproductive conformation binds more favorably than the productive conformation to the native hAChE, consistent with the potential of mean force (PMF) calculation discussed below. The more favorable binding of the nonproductive conformation arises mainly from the enthalpic term, whereas the entropic contributions are highly comparable between the two different poses. Given a similar amount of entropy decrease and a more negative enthalpic gain upon RS-170B binding in a nonproductive versus productive pose, the enthalpic contribution will dominate the free energy of binding at higher temperatures for the nonproductive conformation more than for the productive one. This means that as the temperature increases the binding energy of the productive conformation becomes unfavorable sooner than that of the nonproductive pose. Thus, whereas both conformations may be equally probable at LT, as observed in the LT binary hAChE:RS-170B complex, the nonproductive binding pose will be preferred at RT, as observed in the RT binary hAChE:RS-170B complex.
To further explore the conformational transition between the two binding poses, we computed the PMF free energy profiles for converting RS-170B from the nonproductive conformation to the productive one both in solution and within hAChE gorge using umbrella sampling molecular dynamics (MD) simulations. The PMF profiles illustrated in Fig. S5 show that the nonproductive conformation is thermodynamically slightly more stable (lower PMF), thus more favorable, than the productive one both in aqueous solution and in the active-site gorge of hAChE. The PMF profiles are broadly consistent with the DFT and MM-GB/SA calculations presented above. It is interesting to note that the protein environment has a significant impact on both the stabilization of the two poses and the interconversion between them. Although the nonproductive conformation is only marginally more stable (<2 kcal/mol, ΔG3) than the productive one in solution, its stabilization inside the protein is more significant with a ∼6 kcal/mol (ΔG2) free energy difference between the two conformations. Conformational ring “flipping” inside the active-site gorge experiences an energy barrier, ΔG1, of ∼12.5 kcal/mol, whereas in solution the energy barrier, ΔG3, for this conformational change is only ∼1.5 kcal/mol. This suggests that the conformational transition of RS-170B inside the native hAChE active-site gorge is more energetically demanding, but possible, and can also be triggered by a chemical driving force provided by OP binding to Ser-203 and the subsequent oxime attack on the phosphorus. The elevated energy barrier to ring flipping could partially account for the slow reactivation kinetics of the inhibited hAChE by RS-170B, but by itself would not be the rate-limiting factor, because the k2 constant (Table S3) suggests a reactivation energy barrier of ∼19–20 kcal/mol.
Ser-203 hydration shell
In both RT and LT structures of hAChE:RS-170B water molecules are present near the unmodified Ser-203 (Fig. 2A and Fig. S4), indicating that it is well-hydrated even though it is buried ∼20 Å below the protein surface. One water molecule is directly hydrogen-bonded to the Ser-203 side chain oxygen, with O … O distances of 2.5 Å in the RT and 2.4 Å in LT structures, and additional interactions made with the main chain amides of the oxyanion hole residues Gly-121 and Gly-122. The other water molecule, clearly visible only in the LT structure, is within hydrogen-bonding distance from the first water. Similarly positioned water molecules were observed previously in the LT structure of hAChE in complex with the inhibitor dihydrotanshinone I (19), and such a water molecule can also be fitted into the unmodeled residual electron density at the active site of apo-hAChE (18). When hAChE is inhibited with VX, these waters are displaced from the active site (Fig. 2, B and C, and Fig. S6). The choline-binding site, proximal to Ser-203, is occupied by a chloride ion in the RT hAChE:RS-170B structure, coming from NaCl present in the buffer. In the LT structure of the binary complex the halide is replaced by a glycerol molecule used as a cryoprotectant (Fig. S4). Affinity of the choline-binding site for various kinds of molecules or ions, such as halides, SO42−, 2-pralidoxime, etc., is well-documented in the literature (20, 39–41). However, significant residual electron density, probably belonging to various cryoprotectant molecules, is not modeled in many published AChE structures.
Inhibition with VX analog
In the RT and LT structures of VX-hAChE:RS-170B obtained at resolutions of 2.80 and 2.45 Å, respectively, the VX moiety is unmistakably observed attached to the side chain of Ser-203, with the highest electron density peak corresponding to the phosphorus atom (Fig. 2B and Figs. S2 and S6). In the VX–Ser-203 adduct, the methyl group of the phosphonyl moiety points toward the acyl-binding pocket made up of residues Trp-231, Phe-295, and Phe-297. The OEt substituent faces, but is not inserted into, the choline site with its oxygen making weak interactions with the catalytic His-447, and the phosphonyl oxygen occupying the oxyanion hole. The covalent modification of hAChE with VX does not alter the secondary structure of the native enzyme (in both RT and LT, binary and ternary structures have root mean square deviation of 0.2 Å); however, the changes in the RS-170B–binding mode in the ternary complex relative to the binary complex at RT are pronounced. In the RT structure of the ternary VX-hAChE:RS-170B complex the reactivator's imidazole ring is flipped about 180° relative to its geometry in the RT structure of the binary complex, placing the oxygen atom of the oxime group ∼7 Å away from the phosphorus atom of the VX moiety (Fig. 2C and Fig. S2) and resulting in a more extended conformation. Because in solution, RS-170B exists predominantly in a more compact conformation, as evidenced by our DFT and MM-GB/SA calculations, we propose that it could reorient from this solution pose (also seen in hAChE:RS-170B at RT) either prior to or after association with the VX-hAChE conjugate active-site gorge. Such a reorientation inside the enzyme may be energetically costly, as it may require movement of Tyr-337, Tyr-341, Tyr-124, and Phe-297 residues to avoid a clash with the aldoxime group of RS-170B but has not been detected in the existing hAChE structural data. MD simulations were therefore performed for 300 ns using the structure of ternary VX-hAChE:RS-170B complex, containing no bound ligand in the choline-binding site, as a starting model. Remarkably, the pose of the reactivator remained the same as in the X-ray structure for several nanoseconds (ns). Subsequently, RS-170B reoriented in concert with subtle changes to the VX–Ser-203 conformation to bring its aldoxime group within 3.4 Å of the P atom of VX–Ser-203 at 11 ns, essentially producing a near-attack configuration (Fig. 4). By 19 ns, the reactivator began translating away from the P atom and, after 22 ns of simulation, RS-170B flipped to a nonproductive orientation, with the aldoxime group now pointing away from the VX–Ser-203 moiety. Following this transition, RS-170B began translating out of the active site until a soft restraint imposed in the MD simulation was activated to bring the oxime back into the hAChE gorge (see “Experimental procedures”). Thus, these MD simulations demonstrate that RS-170B can indeed rotate between productive and nonproductive conformations inside the active-site gorge and only subtle fluctuations in both RS-170B and VX–Ser-203 moiety are required to generate the reactivator pose suitable for the reactivation reaction.
Figure 4.
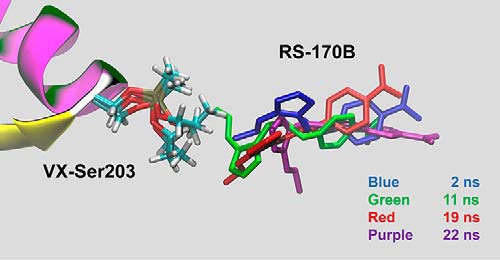
Snapshots from the MD simulation of the VX-hAChE:RS-170B complex revealing translational and conformational changes that bring the oxime within a reactive pose (green) and subsequently to an unproductive one (purple), after which the oxime is able to leave the active site. RS-170B colored in blue corresponds to a snapshot at 2 ns, green at 11 ns, red at 19 ns, and purple at 22 ns.
Although the RS-170B orientation seen in the structure of VX-hAChE:RS-170B complex can be considered semi-productive, as it resembles the fully extended pose found in the LT structure of hAChE:RS-170B, the reactivator is still not primed for an in-line attack on the phosphorus of VX–Ser-203. The oxime group is rotated by 60° out of the plane of the imidazole ring, which is in sharp contrast to an almost coplanar geometry of the two groups (17° out of plane) observed in the extended RS-170B orientation in the binary complex at LT. The deviation in co-planarity between the oxime group and an adjacent aromatic ring is not surprising, as it was previously observed for other reactivators (6, 15) and is always a result of steric constraints. In our RT VX-hAChE:RS-170B structure, the OEt substituent of VX–Ser-203 pushes the aldoxime group of RS-170B away from the VX moiety (Fig. 5). Importantly, no direct or indirect interactions are observed between the aldoxime functional group and the catalytic His-447 residue, whose imidazole side chain is assumed to play a role in the hAChE reactivation as a possible proton acceptor from the aldoxime group of an incoming reactivator (10, 11). Instead, we find that the oxime's oxygen is positioned equidistantly (∼3.4 Å) from the hydroxyl groups of Tyr-124 and Tyr-337 (Fig. 2C), suggesting a possible proton acceptor role for either of these phenolic side chains, assuming RS-170B binds with its aldoxime group protonated. Conversely, if the reactivator molecule is deprotonated when it enters the active site, as was proposed based on earlier reactivation kinetics data (42), Tyr-124 and Tyr-337 may stabilize the negative charge on the aldoxime's oxygen atom. Interestingly, the overlay of the RT VX-hAChE:RS-170B and LT hAChE:RS-170B structures places the nucleophilic oxygen of the extended RS-170B conformer of the LT binary complex only 4.4 Å away from the VX phosphorus atom (Fig. 5).
Figure 5.
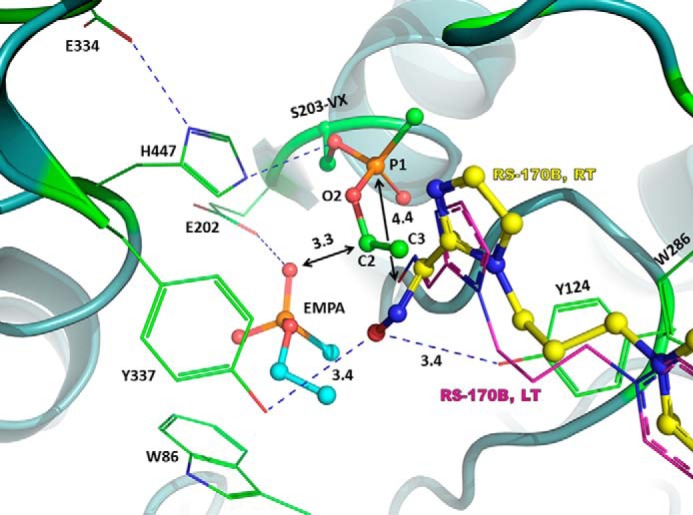
Possible effects of the constricted active-site environment on the RS-170B conformation. Overlay of the RT VX-hAChE:RS-170B (carbons colored green for the VX-hAChE and yellow for RS-170B) and LT hAChE:RS-170B (carbons colored magenta). For the LT hAChE:RS-170B only stick model of the fully extended RS-170B conformer is shown for clarity. EMPA is shown in ball and stick representation with cyan carbon atoms. Distances are in Å. Single letter amino acid residues are used in the figure.
It is possible that the extended conformation of RS-170B seen in the LT hAChE:RS-170B structure represents the reactivator orientation optimal for in-line attack, provided the OEt substituent of VX–Ser-203 moves away and into the choline-binding site. The view along the Oγ-P bond of the VX-phosphonylated Ser-203 clearly shows that the only obstacle for the aldoxime group in the semi-productive RS-170B conformation seen in the RT VX-hAChE:RS-170B structure is the OEt substituent of the phosphonyl group (Fig. 5). The choline-binding site is large enough to allow rotations of VX–Ser-203 around the single bonds of P-O2 and O2-C2. Therefore, if the choline site is not occupied, the OEt substituent would be very dynamic, continuously exposing the phosphorus atom to the nucleophilic attack. However, if the choline-binding site is occupied, the bound molecule's steric hindrance may restrict possible conformations the VX moiety can have.
Remarkably, we discovered a product of VX analog hydrolysis, EMPA, bound at RT in the choline-binding site above the Trp-86 indole side chain. The O3 atom of EMPA makes a 2.8 Å hydrogen bond interaction with the Glu-202 carboxylate Oϵ1, suggesting that one of these oxygen atoms must be protonated (Fig. 5). The EMPA's O3 is also within hydrogen-bonding distance (3.1 Å) to the imidazole of His-447. It is well-known that, in addition to covalent inhibition of hAChE, VX is quickly hydrolyzed into EMPA and diisopropylethyl mercaptoamine (DESH) when absorbed into mammalian tissues (43, 44). Detection of the VX decomposition products is often performed for verification of exposure to the nerve agent; however, little is known about VX metabolism in vivo. Nevertheless, it is unlikely that large amounts of EMPA can be generated in vivo from exposure to VX. In addition, our enzyme kinetics experiments showed that, in solution under physiological conditions, EMPA demonstrates high (∼40 mm) dissociation constants for both native hAChE (Fig. S7) and VX-hAChE conjugate (Fig. S8), which underscores the suggestion that in vivo EMPA would not be a significant factor in oxime reactivation. The presence of EMPA in our RT structure of VX-hAChE:RS-170B complex is most probably due to spontaneous hydrolysis of the VX analog in aqueous solution (45) before or during crystal soaking.
In the RT VX-hAChE:RS-170B structure, EMPA appears to hinder rotation of the OEt group of VX–Ser-203, thus possibly restricting the latter's conformational freedom and keeping VX in a conformation with the OEt blocking access to the phosphorus. In the LT structure of the VX-hAChE:RS-170B complex we detect a molecule of cryoprotectant glycerol bound to the choline-binding site instead of EMPA. We note that the glycerol concentration of 25% used for cryoprotection, corresponding to a concentration of ∼3 m, is very high and can easily out-compete a low-affinity ligand for binding at the choline-binding site. In addition, like EMPA, the glycerol molecule is bulky enough to prevent rotation of the OEt substituent of VX–Ser-203, keeping its conformation the same as in the RT structure. We hypothesize that the presence of a small molecule with high affinity for the choline site could influence the dynamics of the whole VX–Ser-203 moiety and stabilize a conformation that prevents oxime access to the phosphorus of VX–Ser-203. The choline site-bound molecules could thus add to the steric bulk of the active site helping to shield the phosphorous atom from the reactivator's aldoxime group. Similar conformations of the VX group are found in other previously published structures of VX-AChE conjugates (21).
Distinct conformations of RS-170B observed in all studied complexes
Comparison of the alternative RS-170B conformers observed in the noncrystallographic monomers A and B of the hAChE complexes studied here (see Table S1) clearly demonstrates that they can be grouped into two clusters distinguished by the position of the imidazole ring and the reactive aldoxime moiety (Fig. 6 and Fig. S3). Depending on their respective orientations relative to the Ser-203 or VX–Ser-203 moieties we refer to these orientations as nonproductive (Fig. 6, B and E, and Fig. S3) or extended productive (Fig. 6, C and F, and Fig. S3). Although the overall resolutions of these structures (2.15–2.80 Å; Table S1) may not allow for fine analyses of structural differences, the distances between the two clearly identifiable atoms in each oxime molecule, the pyridinium N and oxime O, were either 5.4 ± 0.8 Å for the nonproductive conformation or much longer, 8.3 ± 0.4 Å, for the extended pose. In the context of the active-center gorge of the VX-hAChE conjugate (Fig. 6D) only extended conformers can penetrate with their aldoxime groups into the hAChE catalytic site beyond the choke point of the gorge and assume an orientation that might be productive for the nucleophilic attack on the conjugated phosphorus of VX. This is consistent with the longer distances between the N atoms of their pyridinium rings, making π-π interactions with Trp-286, and their oxime O atoms of ∼8.3 Å. These distances are significantly longer than ∼7 Å distance between Trp-286 and the opening of the catalytic site defined by the choke point (Fig. 6, E and F). Therefore, the nonproductive RS-170B conformers would be ineffective not only because their reactive aldoxime groups point away from the VX's P atom target, but also because they are physically prevented from entering the catalytic site of VX-hAChE by the choke point. It is nonetheless encouraging that in the RT structure of VX-hAChE:RS-170B, obtained at a near-physiological temperature, the reactivator is observed in a semi-productive conformation, consistent with the functional data.
Figure 6.

Clusters of RS-170B conformers created using the α-carbon protein backbone overlay of the eight hAChE monomers from the structures reported in Table S1. A, all 10 conformers. B, four nonproductive conformers. C, six extended conformers. Average distances indicated in B and C are between pyridinium N and aldoxime O for the RS-170B conformers of the respective clusters. D, all conformers in the context of the VX-hAChE active-center gorge stabilized by Trp-286 and Tyr-341. E, nonproductive conformers cluster above the narrow choke point (defined by Trp-124, Tyr-337, and Phe-338 side chains) with aldoxime oxygens pointing away from the conjugated VX. F, only extended conformers can span the ∼7 Å distance between the main RS-170B anchoring point (Trp-286) and the choke point and project their nucleophilic aldoxime groups into the catalytic hAChE site toward conjugated VX.
Discussion
Our findings provide several new mechanistic insights regarding the reactivation of OP-inhibited hAChE by oximes such as RS-170B. The RT X-ray structures and molecular simulations suggest that most RS-170B molecules initially enter the hAChE active-site gorge not poised for the reactivation reaction, but in a compact nonproductive orientation, which is energetically the most stable pose both in solution and within the active-site gorge. Either prior to and/or after association with the active-site gorge of the native hAChE, RS-170B may then flip between nonproductive and productive conformations. Inhibition of hAChE with VX may trigger an additional change in the conformation of RS-170B by providing a chemical driving force for the subsequent attack on the phosphorus by the oxime, resulting in the predominant semi-productive extended reactivator orientation caused by a flip of the imidazole ring. When in the productive conformation, the RS-170B nucleophilic oxygen atom of the aldoxime group could be as close as 4.4 Å to the phosphorus of VX–Ser-203 as indicated by the LT binary complex structure superimposed with the RT VX-hAChE:RS-170B structure or even as close as 3.4 Å as observed in our MD simulations. Additionally, choline-binding site interactions with various kinds of small molecules and ions may affect the conformational flexibility of the OP substituent bound to Ser-203, thus masking the phosphorus atom and making it less accessible to attack by reactivators.
The observed reorientation of RS-170B also allows us to provide a possible explanation for poor reactivation abilities of RS-170B. If the nonproductive orientation binds to VX-hAChE with higher affinity, and RS-170B must reorient in the active site to assume the productive conformation, this would likely impair its ability to reactivate OP-inhibited hAChE. In fact, RS-170B demonstrated superior ability to reactivate soman-inhibited YAFA hAChE compared with the structurally similar but conformationally more rigid bispyridinium aldoxime HI-6 (37). However, RS-170B is about 1 order of magnitude slower and less efficient than HI-6 in reactivation of native hAChE (Table S3). In YAFA, replacement of the bulky aromatic side chains of Tyr-337 and Phe-338 with much smaller methyl substituents creates a substantially larger volume in the active-site gorge (∼345 Å3 compared with ∼170 Å3 in native hAChE), which might allow the imidazole ring of RS-170B to freely rotate.
Finally, it is instructive to compare the overall binding of the RS-170B reactivator in the current structures to that observed previously for other oximes that have similar variable-length alkyl linkers flanked by two heterocyclic rings. Binding of such reactivators was demonstrated to induce a conformational change of Trp-286 (Trp-279 for TcAChE) at the PAS relative to its position in the apo-enzyme, allowing π-π sandwich formation between the nonreactive pyridinium heterocycle of HI-6 (4, 6, 10, 11), HLo-7 (9), ortho-7, or obidoxime (6, 9, 14) and the side chains of Trp-286 and Tyr-72, or Trp-286 and Tyr-124. The sandwiched heterocycle was often characterized by well-defined electron density, most likely signifying tight binding of the ligand within the π-π sandwich. In addition, in all such binary and OP-inhibited ternary AChE structures, the reactivator adopted a conformation in which the two heteroaromatic rings were nearly perpendicular to each other. In hAChE:RS-170B and VX-hAChE:RS-170B complexes, the reactivator does not alter the position of Trp-286, and enters the active-site gorge in a flattened conformation with the pyridinium and imidazole rings roughly in the same plane (Fig. 3). The electron density for RS-170B in our structures is somewhat less distinct compared with that of reactivators in the aforementioned structures. Nevertheless, the positions of the oxime groups could be accurately determined in all our RS-170B conformers. Thus, RS-170B bound in the active-site gorge is conformationally dynamic and only a fraction of the oxime molecules assumes a productive orientation for facile access to the OP-conjugated Ser-203 residue. It is worth noting that improved reactivation of the tabun-inhibited mAChE Trp286–Ala variant relative to the WT enzyme by bis-pyridinium reactivators was observed recently (46). The Trp286–Ala substitution would not only facilitate initial oxime association by increasing the size of the active center opening, but also avert π-π stacking interactions between the PAS Trp residue and a reactivator, thus precluding formation of a π-π sandwich and possibly increasing the reactivator's conformational freedom. The observation with the Trp286–Ala variant supports the hypothesis that more efficient reactivators could benefit from a weaker interaction with the PAS Trp when reactivator is bound in the reversible reactant complex that precedes transition-state formation. Proper stabilization of the transition-state geometry for many bisquaternary oximes would not require stabilizing interactions with Trp-286, thus permitting the oxime to slide deeper into the gorge to reach the phosphorus of OP–Ser-203 thereby avoiding the choke point to allow for quick transphosphorylation and recovery of AChE catalytic activity.
Recently, six new LT crystal structures of hAChE in complex with the HI-6 reactivator in the binary and VX-inhibited ternary forms have been reported by Height et al. (21). Some aspects of the authors' interpretation of the results and their conclusions, however, do not fully align with our observations based on the RT and LT structures reported here. According to our experimental observations, the post-reactivation state of hAChE proposed by Height et al. (21) with noninhibited Ser-203 and bound HI-6 and EMPA molecules, would have been difficult to achieve under the given experimental conditions. To capture this state, hAChE crystals were pre-soaked in ∼4 mm PR/S-VX for 6 min, then incubated for 10 min in a solution of 11 mm HI-6 supplemented with 20–25% ethylene glycol, and then mounted for LT X-ray data collection. A complete absence of covalent inhibitor in hAChE would have been expected with the reactivation constant (k2 = 0.63 ± 0.04 min−1) determined in the same study but in solution. From our experience with RS-170B and with other more efficient bisquaternary reactivators, a complete reactivation under the described experimental conditions would be hard to reach in the crystalline state. An alternative explanation is more plausible: due to the low concentration of the VX analog and its instability in aqueous solution, only a very small fraction of hAChE molecules in the crystal, undetectable by X-ray crystallography, were probably inhibited. Based on our experience with interactions between cryoprotectants and EMPA in the choline-binding site, a disordered ethylene glycol molecule could have been easily mistaken for EMPA, as the cryoprotectant was present at the very high concentration of ∼25% (∼4 m) in the soaking drop. In addition, the proposed mechanism of HI-6 reactivation, in which HI-6 enters the hAChE active site as a deprotonated oxime, was devised from the structure of the PR-VX-hAChE:HI-6 ternary complex. In this structure, HI-6 is drawn into the choline-binding site, with its aldoxime group making a very short (2.5 Å) interaction with the His-447 main chain carbonyl, suggestive of a strong hydrogen bond. Such a short hydrogen bond between two oxygens is only possible if one of them is protonated, in this case, it must be the oxime. Furthermore, this HI-6 binding pose is allowed because the sterically small methyl group of the PR-VX–Ser-203 side chain points to the choline-binding site and the larger ethoxy group is situated in the acyl pocket. In this conformation of the enzyme, the phosphorus atom of the adduct is fully exposed for the nucleophilic attack. Therefore, if the HI-6 reactivator would enter the active site in the deprotonated form, it should have no difficulty attacking the phosphorus; yet, reactivation of PR-VX–hAChE was almost undetectable. Finally, our soaking experiments in which VX analog is added to the oxime:hAChE binary complex clearly contradict the proposed mechanistic model in which bound oxime blocks the flow of small molecules in and out the active site.
In conclusion, we demonstrated here that with RT X-ray diffraction data, we were able to observe RS-170B reorientation due to VX inhibition and to capture the VX hydrolysis product, EMPA, bound in the choline-binding site. This information provides a new structural perspective on the reactivation of OP-inhibited hAChE, offers a new mechanistic hypothesis for the reactivation reaction, and demonstrates the power of combining RT and LT X-ray crystallography for more complete and in-depth structural analyses.
Experimental procedures
Chemicals
Imidazole aldoxime RS-170B was synthesized as described earlier (34) and dissolved in DMSO at 100 mm concentration. The low toxicity VX analog (Fig. 1) was synthesized and kindly provided by Dr. Gabriel Amitai of the IIBR, Ness Ziona, Israel (47). EMPA (Fig. 1) and acetylthiocholine (ATCh) were purchased from Sigma.
Protein expression and purification
Monomeric human AChE expressed in the Gnt1-HEK293 mammalian cell culture deficient in complex N-glycans using a FLAG-tagged construct (35) was eluted from the anti-FLAG affinity column by specific HRV 3C (Sino Biological Inc.) proteolysis, cleaving the FLAG tag off, at the engineered Prescission protease recognition site. The resulting N-terminal sequence of the pure eluted hAChE was G-P-L-E-G-R- …, where the amino acid sequence of the mature hAChE protein starts at E-G-R- … and ends at the truncated C terminus (35) with sequence … -S-A-T-D-T-L-D547. Physiologically relevant hAChE dimers spontaneously and reversibly associate in solution from the expressed monomers.
Enzymatic activity, reversible EMPA inhibition, and reactivation kinetics
Activity of hAChE was measured using a spectrophotometric Ellman assay (48) in 0.1 m phosphate buffer, pH 7.4, containing 0.01% BSA at room temperature. Reversible inhibition of hAChE by EMPA was measured as described before (49) where either (EMPA + ATCh) was pipetted at varying concentrations into buffered solution of hAChE, or hAChE solution was pipetted into varying concentrations of EMPA and ATCh in the buffer. hAChE inhibition reactions with VX analog and oxime reactivation measured at 37 °C in phosphate buffer were done as described before (37).
Crystallization
For crystallization experiments a sample of hAChE was dialyzed in 10 mm NaCl, 10 mm HEPES, pH 7, and concentrated to 6–10 mg/ml. About 1 h prior to crystallization, the solution of hAChE was combined with a 100 mm stock solution of RS-170B reactivator in DMSO at a molar ratio of 1:5 or 1:10 to obtain a binary complex of hAChE:RS-170B. Crystals were grown by vapor diffusion at 10 °C in sitting drop microbridges or 9-well glass plates (Hampton Research, Aliso Viejo, CA) using well solutions containing 10 mm sodium citrate, 100 mm HEPES, pH 7, and 6–8% PEG6000.
Soaking experiments
For the soaking experiments 3 μl of 50 mm VX analog solution in DMSO were diluted with 200 μl of solution of 10 mm sodium citrate, 100 mm HEPES, pH 7, and 7% PEG6000 to give a 0.75 mm solution of the VX analog. This solution was then used to obtain the ternary complex of VX-hAChE:RS-170B through a 4-min soak at RT (∼22 °C) of crystals of the binary hAChE:RS-170B complex. The soaking drop (10 μl of the VX solution) for the crystal intended for the RT X-ray data collection was supplemented with 0.5 μl of the RS-170B stock solution.
X-ray data collection
For both complexes, X-ray crystallographic data were collected at room temperature (∼22 °C) and from frozen crystals at 100 K. Prior to data collection at 100 K crystals were subjected to two very brief consecutive soaks in the cryoprotectant solutions, first in 12% glycerol followed by 25% glycerol, and then flash cooled by plunging into liquid nitrogen. For the RT data collection, crystals were mounted on the Litholoops (Molecular Dimensions, Maumee, OH) directly from the crystallization or soaking drops and kept hydrated using the MiTeGen (Ithaca, NY) room temperature setup. Diffraction data were collected from a single crystal for each complex on the ID19 beamline at SBC-CAT using a Pilatus3 × 6M detector at the Advanced Photon Source (APS). X-ray diffraction data were integrated and scaled using the HKL3000 software suite (50). The structures were solved by molecular replacement using the CCP4 suite (51). The structure of the apo-hAChE (PDB ID 4EY4) (18) was used as a starting model with all waters and the N-linked glycosylated saccharides removed. Refinement was performed using the phenix.refine program in the PHENIX (52) suite and the resulting structure analyzed with molprobity (53). The structures were built and manipulated with the program Coot (54). Figures were generated using PyMol molecular graphics software (version 1.5.0.3; Schrödinger LLC). A summary of the crystallographic data and refinement is given in Table S1. Crystallographic data have been deposited to the PDB with the following codes: 6O5R for RT hAChE:RS-170B, 6O5S for RT VX-hAChE:RS-170B, 6O5V for LT hAChE:RS-170B, and 6O66 for LT VX-hAChE:RS-170B.
DFT quantum chemical calculations
Geometry optimization of RS-170B in implicit solvent was performed using density functional theory with B3LYP (55–58), the def2-TZVPP (59, 60) basis set, the D3 dispersion correction (61, 62), and the COSMO implicit solvent model with the dielectric constant set to 80 (63) using the ORCA (64) software version 3.0.3. Tight convergence parameters were used for both the energy minimization and geometry optimization. The optimized conformation of RS-170B reactivator is shown in Fig. S2.
MM-GB/SA calculations
The MM-GB/SA method (65, 66) was used to estimate the binding free energies of RS-170B with hAChE in the two conformations observed in the LT binary hAChE:RS-170B complex. The total binding energy ΔGbind was defined as ΔGbind = Gcomplex − Greceptor − Gligand. Each free energy term consisted of the gas phase MM energy (ΔEvdW + ΔEele), the solvation-free energy (ΔGsol), and the vibrational entropy contributions (TΔS). ΔGsol was estimated from the GB theory and solvent accessible surface area calculations, which yielded ΔGGB and ΔGSA, respectively. In GB, energies were evaluated at 0.1 m NaCl concentration. A surface tension coefficient (γ) of 0.0072 kcal/(mol Å2) was used to calculate the nonpolar solvation-free energy contribution. The entropy contribution was obtained by performing normal mode analysis on the two complex structures only as entropy contributions from the protein and the ligand alone can be neglected due to RS-170B binding to the same protein. ΔEvdW, ΔEele, ΔGGB, and ΔGSA were computed for 7000 frames, normal mode analysis was performed on 140 frames, both extracted evenly from 30∼100 ns of the MD trajectories. Error bars are mean ± S.E. computed with block averaging.
PMF calculations
MD simulations coupled with umbrella sampling (67, 68) were used to compute the PMF profiles for the interconversion in solution and in hAChE between the two conformations observed in the LT binary hAChE:RS-170B complex. Umbrella windows were generated by driving the intramolecular rotation of RS-170B about the C12-N13 bond by 360°, from which 18 windows (every 20°) were selected for umbrella sampling along the reaction coordinate of the torsional angle C11-C12-N13-C17 (Fig. S5). Umbrella sampling simulations (independent simulations in each window) were performed at T = 298.5 K, with a harmonic potential applied to the torsional angle C11-C12-N13-C17. Post-processing of the umbrella simulations to calculate the PMF profiles was performed using the weighted-histogram analysis method (69, 70), using the last 3 ns of each production umbrella window trajectory, with 50 bins. Error estimates for the weighted-histogram analysis method were obtained from bootstrapping.
MD simulations
MD simulations of the binary hAChE:RS-170B complex, coupled with umbrella sampling, were performed in the NPT ensemble, with P ∼1 bar and T = 298.5 K, using the Amber software suite (71). Temperature was maintained using the Berendsen thermostat and pressure was controlled using the Berendsen barostat (72). To allow for an integration time step of 2 fs for all simulations, all bonds involving a hydrogen atom were constrained using the SHAKE algorithm (73). Amber ff14SB (74) and TIP3P (75, 76) force fields were used for protein and water, respectively. Force field parameters for RS-170B were taken from the Amber general force field (77). MD simulations of the ternary VX-hAChE:RS-170B complex were performed in a similar fashion using CHARMM/OpenMM (78) running on Graphic processing units and using periodic boundary conditions and particle mesh Ewald for the treatment of long-range electrostatics. The force field parameters for RS-170B and the conjugated Ser-203 were obtained from the CGENFF software (79) and DFT calculations. Hydrogen atoms were added to the protein using HBUILD in CHARMM. All protein residues were assigned to their canonical protonation state. His-447 was represented by neutral or protonated states in separate simulations. All tautomeric states of other histidines were assigned based on visualization of surrounding residues. The VX-hAChE:RS-170B complex was surrounded by a cube of water molecules (TIP3P model) (75, 76) with 99.0 Å sides with potassium chloride ions corresponding to 0.15 mmol concentration and sufficient to bring the summed charge of the system to zero. The total atom count was 91,100 atoms. Three simulations were run for 300 ns each using a slightly altered restraint between the middle carbon atom of the reactivator's central alkyl linkage, which connects imidazole and pyridinium rings, and the phosphorus atom on the VX-conjugated Ser-203 in each simulation. This restraint was only active if this distance reached 11.0, 13.0, and 15.0 Å, respectively. In this manner, the substrate would experience a force only if it drifted outside the active site and thus provide an opportunity for the substrate to rebind to the active site.
Author contributions
O. G., X. K., Z. R., and A. K. conceptualization; O. G., X. K., X. C., T. W., D. K. B., P. T., Z. R., and A. K. formal analysis; O. G., X. C., T. W., D. K. B., P. T., Z. R., and A. K. supervision; O. G., D. K. B., P. T., Z. R., and A. K. funding acquisition; O. G., X. K., X. C., T. W., D. K. B., P. T., Z. R., and A. K. investigation; O. G., X. K., X. C., T. W., D. K. B., P. T., Z. R., and A. K. methodology; O. G., X. K., Z. R., and A. K. writing-original draft; O. G. and A. K. project administration; O. G., X. K., X. C., T. W., D. K. B., P. T., Z. R., and A. K. writing-review and editing; X. K., X. C., T. W., D. K. B., P. T., Z. R., and A. K. visualization; D. K. B., P. T., Z. R., and A. K. validation; T. W. molecular modeling.
Supplementary Material
Acknowledgments
X-ray crystallographic data presented here were collected at Argonne National Laboratory using Structural Biology Center (SBC) beamline ID19 at the Advanced Photon Source. Use of the Advanced Photon Source, an Office of Science User Facility operated for the United States Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the United States DOE under Contract number DE-AC02–06CH11357. The Office of Biological and Environmental Research supported research at the Center for Structural Molecular Biology (CSMB) at ORNL using facilities supported by the Scientific User Facilities Division, Office of Basic Energy Sciences, United States Department of Energy.
This work was supported by the CounterACT Program, National Institutes of Health Office of the Director, and National Institutes of Health NINDS Grants U01 NS083451 and R21 NS098998 (to Z. R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 6O5R, 6O5S, 6O5V, and 6O66) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.pdb.org/).
This article contains Figs. S1–S8 and Tables S1–S3.
- AChE
- acetylcholinesterase
- DESH
- diisopropylethyl mercaptoamine
- DFT
- density functional theory
- EMPA
- ethylmethylphosphonic acid
- hAChE
- human acetylcholinesterase
- LT
- low temperature
- MD
- molecular dynamics
- MM-GB/SA
- molecular mechanics-generalized born/surface area
- OP
- organophosphate
- PAS
- peripheral anionic site
- PMF
- potential of mean force
- RT
- room temperature
- TcAChE
- Torpedo californica acetylcholinesterase
- YAFA
- Y337A/F338A double mutant of hAChE
- ATCh
- acetylthiocholine
- PDB
- Protein Data Bank.
References
- 1. Taylor P. (1991) The cholinesterases. J. Biol. Chem. 266, 4025–4028 [PubMed] [Google Scholar]
- 2. Solberg Y., and Belkin M. (1997) The role of excitotoxicity in organophosphorous nerve agents central poisoning. Trends Pharmacol. Sci. 18, 183–185 10.1016/S0165-6147(97)90617-9 [DOI] [PubMed] [Google Scholar]
- 3. Millard C. B., Koellner G., Ordentlich A., Shafferman A., Silman I., and Sussman J. L. (1999) Reaction products of acetylcholinesterase and VX reveal a mobile histidine in the catalytic triad. J. Am. Chem. Soc. 121, 9883–9884 10.1021/ja992704i [DOI] [Google Scholar]
- 4. Allgardsson A., Berg L., Akfur C., Hörnberg A., Worek F., Linusson A., and Ekström J. (2016) Structure of a prereaction complex between the nerve agent sarin, its biological target acetylcholinesterase, and the antidote HI-6. Proc. Natl. Acad. Sci. U.S.A. 113, 5514–5519 10.1073/pnas.1523362113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gorecki L., Korabecny J., Musilek K., Malinak D., Nepovimova E., Dolezal R., Jun D., Soukup O., and Kuca K. (2016) SAR study to find optimal cholinesterase reactivator against organophosphorous nerve agents and pesticides. Arch. Toxicol. 90, 2831–2859 10.1007/s00204-016-1827-3 [DOI] [PubMed] [Google Scholar]
- 6. Ekström F., Pang Y.-P., Boman M., Artursson E., Akfur C., and Börjegren S. (2006) Crystal structures of acetylcholinesterase in complex with HI-6, ortho-7 and obidoxime: structural basis for differences in the ability to reactivate tabun conjugates. Biochem. Pharmacol. 72, 597–607 10.1016/j.bcp.2006.05.027 [DOI] [PubMed] [Google Scholar]
- 7. Ekström F., Akfur C., Tunemalm A.-K., and Lundberg S. (2006) Structural changes of phenylalanine 338 and histidine 447 revealed by the crystal structures of tabun-inhibited murine acetylcholinesterase. Biochemistry 45, 74–81 10.1021/bi051286t [DOI] [PubMed] [Google Scholar]
- 8. Hörnberg A., Tunemalm A.-K., and Ekström F. (2007) Crystal structures of acetylcholinesterase in complex with organophosphorus compounds suggest that the acyl poket modulates the aging reaction by precluding the formation of the trigonal bipyramidal transition state. Biochemistry 46, 4815–4825 10.1021/bi0621361 [DOI] [PubMed] [Google Scholar]
- 9. Ekström F. J., Åstot, Pang C. Y.-P. (2007) Novel nerve-agent antidote desidn based on crystallographic and mass spectrometric analyses of tabun-conjugated acetylcholinesterase in complex with antidotes. Clin. Pharmacol. Ther. 82, 282–293 10.1038/sj.clpt.6100151 [DOI] [PubMed] [Google Scholar]
- 10. Ekström F., Hörnberg A., Artursson E., Hammarström L.-G., Schneider G., and Pang Y.-P. (2009) Structure of HI-6·sarine-acetylcholinesterase determined by X-ray crystallography and molecular dynamics simulation: reactivator mechanism and design. PLoS ONE 4, e5957 10.1371/journal.pone.0005957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Artursson E., Andersson P. O., Akfur C., Linusson A., Börjegren S., and Ekström F. (2013) Catalytic-site conformational equilibrium in nerve-agent adducts of acetylcholinesterase: possible implications for the HI-6 antidote substrate specificity. Biochem. Pharmacol. 85, 1389–1397 10.1016/j.bcp.2013.01.016 [DOI] [PubMed] [Google Scholar]
- 12. Millard C. B., Kryger G., Ordentlich A., Greenblatt H. M., Harel M., Raves M. L., Segall Y., Barak D., Shafferman A., Silman I., and Sussman J. L. (1999) Crystal structures of aged phosphonylated acetylcholinesterase: nerve agent reaction products at the atomic level. Biochemistry 38, 7032–7039 10.1021/bi982678l [DOI] [PubMed] [Google Scholar]
- 13. Sanson B., Nachon F., Colletier J.-P., Froment M.-T., Toker L., Greenblatt H. M., Sussman J. L., Ashani Y., Masson P., Silman I., and Weik M. (2009) Crystallographic snapshots of nonaged and aged conjugates of soman with acetylcholinesterase, and of a ternary complex of the aged conjugate with pralidoxime. J. Med. Chem. 52, 7593–7603 10.1021/jm900433t [DOI] [PubMed] [Google Scholar]
- 14. Legler P. M., Soojhawon I., and Millard C. B. (2015) A conformational change in the peripheral anionic site of Torpedo californica acetylcholinesterase induced by a bis-imidazolium oxime. Acta Crystallogr. D Biol. Crystallogr. 71, 1788–1798 10.1107/S1399004715011281 [DOI] [PubMed] [Google Scholar]
- 15. Santoni G., de Sousa J., de la Mora E., Dias J., Jean L., Sussman J. L., Silman I., Renard P.-Y., Brown R. C. D., Weik M., Baati R., and Nachon F. (2018) Structure-based optimization of non-quaternary reactivators of acetylcholinesterase inhibited by organophosphorus nerve agents. J. Med. Chem. 61, 7630–7639 10.1021/acs.jmedchem.8b00592 [DOI] [PubMed] [Google Scholar]
- 16. Zorbaz T., Braïki A., Maraković N., Renou J., de la Mora E., Maček Hrvat N., Katalinić M., Silman I., Sussman J. L., Mercey G., Gomez C., Mougeot R., Pérez B., Baati R., Nachon F., Weik M., Jean L., Kovarik Z., and Renard P.-Y. (2018) Potent 3-hydroxy-2-pyridine aldoxime reactivators of organophosphate-inhibited cholinesterases with predicted blood-brain barrier penetration. Chem. Eur. J. 24, 1–18 10.1002/chem.201705257 [DOI] [PubMed] [Google Scholar]
- 17. Carletti E., Colletier J.-P., Dupeux F., Trovaslet M., Masson P., and Nachon F. (2010) Structural evidence that human acetylcholinesterase inhibited by tabun ages through O-dealkylation. J. Med. Chem. 53, 4002–4008 10.1021/jm901853b [DOI] [PubMed] [Google Scholar]
- 18. Cheung J., Rudolph M. J., Burshteyn F., Cassidy M. S., Gary E. N., Love J., Franklin M. C., and Height J. J. (2012) Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 55, 10282–10286 10.1021/jm300871x [DOI] [PubMed] [Google Scholar]
- 19. Cheung J., Gary E. N., Shiomi K., and Rosenberry T. L. (2013) Structures of human acetylcholinesterase bound to dihydrotanshinone I and territrem B show peripheral site flexibility. ACS Med. Chem. Lett. 4, 1091–1096 10.1021/ml400304w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Franklin M. C., Rudolph M. J., Ginter C., Cassidy M. S., and Cheung J. (2016) Structures of paraoxon-inhibited human acetylcholinesterase reveal perturbations of the acyl loop and the dimer interface. Proteins 84, 1246–1256 10.1002/prot.25073 [DOI] [PubMed] [Google Scholar]
- 21. Bester S. M., Guelta M. A., Cheung J., Winemiller M. D., Bae S. Y., Myslinski J., Pegan S. D., and Height J. J. (2018) Structural insights of stereospecific inhibition of human acetylcholinesterase by VX and subsequent reactivation by HI-6. Chem. Res. Toxicol. 31, 1405–1417 10.1021/acs.chemrestox.8b00294 [DOI] [PubMed] [Google Scholar]
- 22. Kovalevsky A., Blumenthal D. K., Cheng X., Taylor P., and Radićc Z. (2016) Limitations in current acetylcholinesterase structure-based design of oxime antidotes for organophosphate poisoning. Ann. N.Y. Acad. Sci. 1378, 41–49 10.1111/nyas.13128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Katalinić M., and Kovarik Z. (2012) Reactivation of tabun-inhibited acetylcholinesterase investigated by two oximes and mutagenesis. Croat. Chem. Acta 85, 209–212 10.5562/cca1815 [DOI] [Google Scholar]
- 24. Worek F., Reiter G., Eyer P., and Szinicz L. (2002) Reactivation kinetics of acetylcholinesterase from different species inhibited by highly toxic organophosphates. Arch. Toxicol. 76, 523–529 10.1007/s00204-002-0375-1 [DOI] [PubMed] [Google Scholar]
- 25. Kreimer D. I., Dolginova E. A., Raves M., Sussman J. L., Silman I., and Weiner L. (1994) A metastable state of Torpido californica acetylcholinesterase generated by modification with organomercurials. Biochemistry 33, 14407–14418 10.1021/bi00252a006 [DOI] [PubMed] [Google Scholar]
- 26. Clifford E. F., Harel M., Silman I., and Sussman J. L. (2002) Structure of a complex of the potent and specific infibitor BW184C51 with Torpido californica acetylcholinesterase. Acta Crystallogr. D Biol. Crystallogr. 58, 1765–1771 10.1107/S0907444902011642 [DOI] [PubMed] [Google Scholar]
- 27. Weber I. T., Waltman M. J., Mustyakimov M., Blakeley M. P., Keen D. A., Ghosh A. K., Langan P., and Kovalevsky A. Y. (2013) Joint X-ray/neutron crystallographic study of HIV-1 protease with clinical inhibitor amprenavir-insights for drug design. J. Med. Chem. 56, 5631–5635 10.1021/jm400684f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang G. Y., Gerlits O. O., Blakeley M. P., Sankaran B., Kovalevsky A. Y., and Kim C. (2014) Neutron diffraction reveals hydrogen bonds critical for cGMP-selective activation: insights for cGMP dependent protein kinase agonist design. Biochemistry 53, 6725–6727 10.1021/bi501012v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gerlits O., Keen D. A., Blakeley M. P., Louis J. M., Weber I. T., and Kovalevsky A. (2017) Room temperature neutron crystallography of drug resistant HIV-I protease uncovers limitations of X-ray structural analysis at 100 K. J. Med. Chem. 60, 2018–2025 10.1021/acs.jmedchem.6b01767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vandavasi V. G., Blakeley M. P., Keen D. A., Hu L. R., Huang Z., and Kovalevsky A. (2018) Temperature-induced replacement of phosphate proton with metal ion captured in neutron structure of A-DNA. Structure 26, 1645–1650.e3 10.1016/j.str.2018.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fraser J. S., Clarkson M. W., Degnan S. C., Erion R., Kern D., and Alber T. (2009) Hidden alternative structures of proline isomerase essential for catalysis. Nature 462, 669–673 10.1038/nature08615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fraser J. S., van den Bedem H., Samelson A. J., Lang P. T., Holton J. M., Echols N., and Alber T. (2011) Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proc. Natl. Acad. Sci. U.S.A. 108, 16247–16252 10.1073/pnas.1111325108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dym O., Song W., Felder C., Roth E., Shnyrov V., Ashani Y., Xu Y., Joosten R. P., Weiner L., Sussman J. L., and Silman I. (2016) The impact of crystallization conditions on structure-based drug design: a case study on the methylene blue/acetylcholinesterase complex. Protein Sci. 25, 1096–1114 10.1002/pro.2923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sit R. K., Fokin V. V., Amitai G., Sharpless K. B., Taylor P., and Radić Z. (2014) Imidazole aldoximes effective in assisting butyrylcholinesterase catalysis of organophosphate detoxification. J. Med. Chem. 57, 1378–1389 10.1021/jm401650z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cochran R., Kalisiak J., Küçükkilinç T., Radic Z., Garcia E., Zhang L., Ho K. Y., Amitai G., Kovarik Z., Fokin V. V., Sharpless K. B., and Taylor P. (2011) Oxime-assisted acetylcholinesterase catalytic scavengers of organophosphates that resist aging. J. Biol. Chem. 286, 29718–29724 10.1074/jbc.M111.264739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Velan B., Kronman C., Ordentlich A., Flashner Y., Leitner M., Cohen S., and Shafferman A. (1993) N-Glycosylation of human acetylcholinesterase: effects on activity, stability and biosynthesis. Biochem. J. 296, 649–656 10.1042/bj2960649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kovarik Z., Maček H. N., Katalinić M., Sit R. K., Paradyse A., Žunec S., Musilek K., Fokin V. V., Taylor P., and Radić Z. (2015) Catalytic soman scavenging by the Y337A/F338A acetylcholinesterase mutant assisted with novel site-directed aldoximes. Chem. Res. Toxicol. 28, 1036–1044 10.1021/acs.chemrestox.5b00060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kovalevsky A., Aggarwal M., Velazquez H., Cuneo M. J., Blakeley M. P., Weiss K. L., Smith J. C., Fisher S. Z., and McKenna R. (2018) “To be or not to be” protonated: atomic details of human carbonic anhydrase-clinical drug complexes by neutron crystallography and simulation. Structure 26, 383–390.e3 10.1016/j.str.2018.01.006 [DOI] [PubMed] [Google Scholar]
- 39. Bourne Y., Taylor P., Bougis P. E., and Marchot P. (1999) Crystal structure of mouse acetylcholinesterase. J. Biol. Chem. 274, 2963–2970 10.1074/jbc.274.5.2963 [DOI] [PubMed] [Google Scholar]
- 40. Dvir H., Silman I., Harel M., Rosenberry T. L., and Sussman J. L. (2010) Acetylcholinesterase: from 3D structure to function. Chem.-Biol. Interact. 187, 10–22 10.1016/j.cbi.2010.01.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wandhammer M., de Konig M., van Grol M., Loiodice M., Saurel L., Noort D., Goeldner M., and Nachon F. (2013) A step toward the reactivation of aged cholinesterases: crystal structure of ligands binding to aged human butyrylcholinestarase. Chem.-Biol. Interact. 203, 19–23 10.1016/j.cbi.2012.08.005 [DOI] [PubMed] [Google Scholar]
- 42. Radić Z., Kalisiak J., Fokin V. V., Sharpless K. B., and Taylor P. (2010) Interaction kinetics of oximes with native, phosphylated and aged human acetylcholinesterase. Chem.-Biol. Interact. 187, 163–166 10.1016/j.cbi.2010.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tsuchihashi H., Katagi M., Nishikawa M., and Tatsuno M. (1998) Identification of metabolites of nerve agent VX in serum collected from a victim. J. Anal. Toxicol. 22, 383–388 10.1093/jat/22.5.383 [DOI] [PubMed] [Google Scholar]
- 44. Barr J. R., Driskell W. J., Aston L. S., and Martinez R. A. (2004) Quantitation of metabolites of the nerve agents sarin, soman, cyclohexylsarin, VX, and Russian VX in human urine using isotope-dilution gas chromatography-tandem mass spectrometry. J. Anal. Toxicol. 28, 372–378 10.1093/jat/28.5.372 [DOI] [PubMed] [Google Scholar]
- 45. Yang Y.-C., Szafraniec L. L., Beaudry W. T., Rohrbaugh D. K., Procell L. R., and Samuel J. B. (1996) Autocatalytic hydrolysis of V-type nerve agents. J. Org. Chem. 61, 8407–8413 10.1021/jo9614506 [DOI] [Google Scholar]
- 46. Katalinić M., Šinko, Maček Hrvat G. N., Zorbaz T., Bosak A. Q., and Kovarik Z. (2018) Oxime-assisted reactivation of tabun-inhibited acetylcholinesterase analyzed by active site mutations. Toxicology 406–407, 104–113 [DOI] [PubMed] [Google Scholar]
- 47. Amitai G., Adani R., Yacov G., Yishay S., Teitlboim S., Tveria L., Limanovich O., Kushnir M., and Meshulam H. (2007) Asymmetric fluorogenic organophosphates for the development of active organophosphate hydrolases with reversed stereoselectivity. Toxicology 233, 187–198 10.1016/j.tox.2006.09.020 [DOI] [PubMed] [Google Scholar]
- 48. Ellman G. L., Courtney K. D., Andres V. Jr., and Featherstone R. M. (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 7, 88–95 10.1016/0006-2952(61)90145-9 [DOI] [PubMed] [Google Scholar]
- 49. Radić Z., Pickering N. A., Vellom D. C., Camp S., and Taylor P. (1993) Three distinct domains in the cholinesterase molecule confer selectivity for acetyl- and butyrylcholinesterase inhibitors. Biochemistry 32, 12074–12084 10.1021/bi00096a018 [DOI] [PubMed] [Google Scholar]
- 50. Minor W., Cymborowski M., Otwinowski Z., and Chruszcz M. (2006) HKL-3000: the integration of data reduction and structure solution: from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62, 859–866 10.1107/S0907444906019949 [DOI] [PubMed] [Google Scholar]
- 51. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 10.1107/S0907444994003112 [DOI] [PubMed] [Google Scholar]
- 52. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., Snoeyink J., and Richardson J. S. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 10.1093/nar/gkm216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Becke A. D. (1993) Density-functional thermochemistry: III. the role of exact exchange. J. Chem. Phys. 98, 5648–5652 10.1063/1.464913 [DOI] [Google Scholar]
- 56. Lee C., Yang W., and Parr R. G. (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789 10.1103/PhysRevB.37.785 [DOI] [PubMed] [Google Scholar]
- 57. Vosko S. H., Wilk L., and Nusair M. (1980) Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can. J. Phys. 58, 1200–1211 10.1139/p80-159 [DOI] [Google Scholar]
- 58. Stephens P. J., Devlin F. J., Chabalowski C. F., and Frisch M. J. (1994) Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 98, 11623–11627 10.1021/j100096a001 [DOI] [Google Scholar]
- 59. Schäfer A., Horn H., and Ahlrichs R. (1992) Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 97, 2571–2577 10.1063/1.463096 [DOI] [Google Scholar]
- 60. Weigend F., and Ahlrichs R. (2005) Balanced basis sets of split valence, triple zeta valence and quadruple ζ valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 10.1039/b508541a [DOI] [PubMed] [Google Scholar]
- 61. Grimme S., Ehrlich S., and Goerigk L. (2011) Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 10.1002/jcc.21759 [DOI] [PubMed] [Google Scholar]
- 62. Grimme S., Antony J., Ehrlich S., and Krieg H. (2010) A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 10.1063/1.3382344 [DOI] [PubMed] [Google Scholar]
- 63. Klamt A., and Schüürmann G. (1993) COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2, 799–805 10.1039/P29930000799 [DOI] [Google Scholar]
- 64. Neese F. (2012) The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2, 73–78 10.1002/wcms.81 [DOI] [Google Scholar]
- 65. Kollman P. A., Massova I., Reyes C., Kuhn B., Huo S., Chong L., Lee M., Lee T., Duan Y., Wang W., Donini O., et al. (2000) Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 33, 889–897 10.1021/ar000033j [DOI] [PubMed] [Google Scholar]
- 66. Genheden S., and Ryde U. (2015) The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 10, 449–461 10.1517/17460441.2015.1032936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Torrie G. M., and Valleau J. P. (1977) Nonphysical sampling distributions in Monte Carlo free-energy estimation: umbrella sampling. J. Comput. Phys. 23, 187–199 10.1016/0021-9991(77)90121-8 [DOI] [Google Scholar]
- 68. Kästner J. (2011) Umbrella sampling. Wiley Interdiscip. Rev. Comput. Mol. Sci. 1, 932–942 10.1002/wcms.66 [DOI] [Google Scholar]
- 69. Kumar S., Rosenberg J. M., Bouzida D., Swendsen R. H., and Kollman P. A. (1992) The weighted histogram analysis method for free-energy calculations on biomolecules: I. the method. J. Comput. Chem. 13, 1011–1021 10.1002/jcc.540130812 [DOI] [Google Scholar]
- 70. Souaille M., and Roux B. (2001) Extension to the weighted histogram analysis method: combining umbrella sampling with free energy calculations. Comput. Phys. Commun. 135, 40–57 10.1016/S0010-4655(00)00215-0 [DOI] [Google Scholar]
- 71. Case D. A., Cheatham T. E. 3rd, Darden T., Gohlke H., Luo R., Merz K. M. Jr., Onufriev A., Simmerling C., Wang B., and Woods R. J. (2005) The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 10.1002/jcc.20290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Berendsen H. J., Postma J. V., van Gunsteren W. F., DiNola A. R. H. J., and Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 10.1063/1.448118 [DOI] [Google Scholar]
- 73. Ryckaert J. P., Ciccotti G., and Berendsen H. J. (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 10.1016/0021-9991(77)90098-5 [DOI] [Google Scholar]
- 74. Maier J. A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K. E., and Simmerling C. (2015) ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 10.1021/acs.jctc.5b00255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., and Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 10.1063/1.445869 [DOI] [Google Scholar]
- 76. Jorgensen W. L., and Tirado-Rives J. (2005) Potential energy functions for atomic-level simulations of water and organic and biomolecular systems. Proc. Natl. Acad. Sci. U.S.A. 102, 6665–6670 10.1073/pnas.0408037102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wang J., Wolf R. M., Caldwell J. W., Kollman P. A., and Case D. A. (2004) Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 10.1002/jcc.20035 [DOI] [PubMed] [Google Scholar]
- 78. Brooks B. R., Brooks C. L. 3rd, Mackerell A. D. Jr., Nilsson L., Petrella R. J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., Caflisch A., Caves L., Cui Q., Dinner A. R., Feig M., et al. (2009) CHARMM: the biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 10.1002/jcc.21287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J., Darian E., Guvench O., Lopes P., Vorobyov I., and MacKerell A. D. Jr. (2010) CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 31, 671–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.