

Abstract
Human cytosolic serine hydroxymethyltransferase (hcSHMT) is a promising target for anticancer chemotherapy and contains a flexible “flap motif” whose function is yet unknown. Here, using size-exclusion chromatography, analytical ultracentrifugation, small-angle X-ray scattering (SAXS), molecular dynamics (MD) simulations, and ligand-binding and enzyme-kinetic analyses, we studied the functional roles of the flap motif by comparing WT hcSHMT with a flap-deleted variant (hcSHMT/Δflap). We found that deletion of the flap results in a mixture of apo-dimers and holo-tetramers, whereas the WT was mostly in the tetrameric form. MD simulations indicated that the flap stabilizes structural compactness and thereby enhances oligomerization. The hcSHMT/Δflap variant exhibited different catalytic properties in (6S)-tetrahydrofolate (THF)-dependent reactions compared with the WT but had similar activity in THF-independent aldol cleavage of β-hydroxyamino acid. hcSHMT/Δflap was less sensitive to THF inhibition than the WT (Ki of 0.65 and 0.27 mm THF at pH 7.5, respectively), and the THF dissociation constant of the WT was also 3-fold lower than that of hcSHMT/Δflap, indicating that the flap is important for THF binding. hcSHMT/Δflap did not display the burst kinetics observed in the WT. These results indicate that, upon removal of the flap, product release is no longer the rate-limiting step, implying that the flap is important for controlling product release. The findings reported here improve our understanding of the functional roles of the flap motif in hcSHMT and provide fundamental insight into how a flexible loop can be involved in controlling the enzymatic reactions of hcSHMT and other enzymes.
Keywords: structure-function, pyridoxal phosphate, folate, protein structure, enzyme kinetics, molecular dynamics, small-angle X-ray scattering (SAXS), analytical ultracentrifugation, oligomerization, nucleoside/nucleotide biosynthesis, dTMP synthesis cycle, flap motif/flexible loop, PLP-dependent enzymes, serine hydroxymethyl transferase, tetrahydrofolate-dependent reactions
Introduction
Serine hydroxymethyltransferase (SHMT2; EC 2.1.2.1.) is a pyridoxal 5′-phosphate (PLP)-dependent enzyme in which the PLP cofactor facilitates the reversible transfer of a hydroxymethyl group from l-serine to (6S)-tetrahydrofolate (THF) to yield glycine and 5,10-methylenetetrahydrofolate (5,10-CH2-THF) as products (1–3). SHMT is one of the enzymes in the deoxythymidylate (dTMP) synthesis cycle in which the other two enzymes, thymidylate synthase (TS) and dihydrofolate reductase (DHFR), also take part in the recycling of folate compounds and production of 5,10-CH2-THF via the SHMT reaction. 5,10-CH2-THF serves as a methyl donor for the TS reaction to convert dUMP to dTMP, which is a requisite precursor for DNA biosynthesis (4). Due to its important role in DNA biosynthesis, cell proliferation, and cell survival, SHMT is one of the attractive targets for antimalarial (5–12) and anticancer chemotherapy (10, 13–16).
In humans, two isoforms of SHMT, cytosolic (hcSHMT or SHMT 1) and mitochondrial (hmSHMT or SHMT 2), have been proposed as targets for anticancer chemotherapy. Studies to explore the potential of SHMT inhibitors in cancer treatment have been performed at the preclinical level (17). hmSHMT is responsible for maintaining intracellular glycine levels, and the enzyme is expressed at a constant level throughout the cell cycle (18–20). However, in glioma tumor cell proliferation, expression of the mitochondrial isoform was demonstrated to be increased significantly, which was reflected by increased glycine accumulation (21, 22). For hcSHMT, its physiological roles in nucleotide biosynthesis and involvement in cancer proliferation have been reported. Its expression level is increased rapidly during cell proliferation, especially in cancer cells (19, 23, 24). Therefore, a better understanding of the structure and mechanism of SHMT is valuable for the development and discovery of better inhibitors or agents to decrease SHMT activity that may be promising candidates for anticancer drugs.
Recent reports revealed that the human cytosolic SHMT displays distinct biochemical and kinetic properties as compared with homologous Plasmodium enzymes (10, 25). These enzymes possess different quaternary structures, homotetramer for hcSHMT and homodimer for Plasmodium SHMT. The turnover number (kcat) of the THF-dependent hcSHMT reaction is much faster than that of the Plasmodium enzymes by ∼30-fold. The catalytic activity of hcSHMT can be inhibited by high concentrations of THF under low-pH conditions (6.6, 7.1, and 7.5). In contrast, at higher pH values (7.9 and 8.3) THF substrate inhibition was not significant (10, 25). Conversely, substrate inhibition by THF in the reactions of the Plasmodium enzymes is pH-independent and requires much higher concentrations for inhibition (>0.4 mm) (7, 10, 26). Previous studies on the burst kinetics of the reaction indicated that the rate-limiting step of the overall reaction of hcSHMT is glycine product release (10), whereas for the overall reaction of the Plasmodium enzyme, PvSHMT, the rate-limiting step is glycine formation (3).
Structural comparison between human cytosolic (PDB code 1BJ4) (27) and Plasmodium SHMTs (PDB codes 4O6Z and 4PFF) (28, 29) revealed that each subunit of hcSHMT tetramer possesses a unique β-hairpin structure, or “flap motif,” comprising 13 amino acids (273VKSVDPKTGKEIL285) located on top of the THF-binding site (Fig. 1). The flap is presented by two flap-connected regions, β-strand (263–272) and α-helix (286–302), linking to the functional sites of the dimer neighboring subunit. This flap motif is conserved only among the mammalian cytosolic SHMTs, such as those from rabbit (PDB code 1LS3) (28, 30), mouse (PDB code 1EJI) (28, 31), and sheep liver enzymes (30, 32). However, the dimeric structures of Plasmodium and bacterial SHMTs do not contain the flap motif (28, 33, 34). Interestingly, alternative splicing in hcSHMT with flap deletion in some cancer cells was reported, but the role has not been extensively investigated (19, 35).
Figure 1.

Quaternary structure of hcSHMT. hcSHMT (PDB code 1BJ4) is presented as a dimer (AB)–dimer (CD) association to form a native tetrameric quaternary structure with stick models of ligands PLP (orange) and folinic acid (FON; gray; adopted from PvSHMT; PDB code 4OYT) and key amino acid residues. The zoomed-in view is the active site of protomer A (green). The flap motif (blue; residues 273–285; in a red rectangular box) and the flap-connected region (blue; residues 263–272 and 286–302) of protomer B (cyan) link to the active pocket of protomer A. The flap-connected region is located at the dimer interface and is involved in maintaining dimer integrity and THF-binding affinity. Gly-302 and Gly-303 of protomer B form H-bonds with the phosphate group of PLP in protomer A, stabilizing the dimer integrity.
Here, we characterized the functional role of the flap motif in hcSHMT by comparing biochemical and biophysical properties of the WT and the flap-deleted variant (hcSHMT/Δflap). Several analytical techniques, namely size-exclusion chromatography (SEC), analytical ultracentrifugation (AUC), small-angle X-ray scattering (SAXS), molecular dynamics (MD) simulations, and ligand-binding and enzyme kinetic studies were employed. Results revealed that the flap motif is important for the binding affinity of THF and crucial for maintaining the dimer integrity for dimer–dimer assembly into tetramer. Taken together, the flap involves entrapping of the glycine product; thereby the product release is the rate-limiting step.
Results
Expression, purification, and molar absorptivity of hcSHMT/Δflap
The expression and purification of the hcSHMT/Δflap were carried out similarly to procedures described previously for the WT (10, 25) to obtain 220 mg of purified protein/liter of culture with a specific activity of 4.25 units/mg of protein (Fig. S1A and Table S1). The purified hcSHMT/Δflap exhibits a molecular weight of 52 kDa as shown in SDS-PAGE analysis (Fig. S1B) and absorption characteristics typical of a PLP-bound enzyme with a maximum absorption wavelength of 430 nm, similar to the WT (Fig. S1C) (25). The results indicated that removal of the flap motif does not influence PLP absorption. The molar absorption coefficient (ϵ430) of the hcSHMT/Δflap-bound PLP was determined to be 7.2 ± 0.2 mm−1 cm−1, similar to that of the WT (8.0 ± 0.1 mm−1 cm−1) (25), suggesting that the molar absorption coefficient of the PLP cofactor residing within the active site of the hcSHMT is not affected by deletion of the flap motif.
The oligomeric states of the hcSHMT/Δflap
To explore whether the flap motif is necessary for stabilization of the oligomeric structure or intersubunit interactions of hcSHMT, three analytical techniques, including SEC, AUC, and SAXS, were employed to determine the oligomeric species and molecular weights (MWs) of hcSHMT/Δflap and compare them with those of WT. The results obtained from these techniques are summarized in Table 1.
Table 1.
Summary of MWs and oligomeric states as well as thermodynamic and kinetic parameters of the hcSHMT/Δflap compared with those of the wildtype
*, the SAXS analysis for MW estimation typically contains 10–20% errors. ND, non-detectable.
| Parameters | hcSHMT |
|
|---|---|---|
| Δflap variant | Wildtype | |
| MWs (kDa) | ||
| SEC | ||
| Dimer | 96a | ND |
| Tetramer | 206b | 206b |
| AUC | ||
| Dimer | 121a | ND |
| Tetramer | 195b | 220b |
| SAXS | ||
| Dimer | 156c* | ND |
| Tetramer | 173d* | |
| l-Serine and THF as substratese | ||
| kcat (s−1) | 11.3 ± 0.3 | 19.8 ± 0.8f |
| Km (mm) | ||
| l-Serine | 0.32 ± 0.05 | 0.22 ± 0.04f |
| THF (×10−3) | 19 ± 3 | 18 ± 3f |
| kcat/Km (mm−1 s−1) | ||
| l-Serine | 35 | 90f |
| THF | 595 | 1,100f |
| KiTHF (mm)g | 0.65 ± 0.09 | 0.27 ± 0.08f |
| l-allo-Threonine as a substrateh | ||
| kcat (s−1) | 0.42 ± 0.02 | 0.44 ± 0.01 |
| Km (mm) | 0.43 ± 0.07 | 0.63 ± 0.02 |
| kcat/Km (mm−1 s−1) | 0.98 | 0.70 |
| Dissociation constant (Kd) (μm)e | ||
| E·THF | 108 ± 8 | 35 ± 8 |
a Apoenzyme form obtained from SEC.
b Holoenzyme form obtained from SEC.
c The calculated MW of the purified hcSHMT/Δflap (a mixture of dimer and tetramer at 17 and 83%, respectively, from Oligomer program).
d The calculated MW of wildtype at 100% tetrameric form from Oligomer program.
e The reactions were performed in buffer D.
f Data were obtained from previous report (10).
g KiTHF value was determined by Equation 3 using the highest concentration of l-serine at 6.4 and 3.2 mm for the hcSHMT/Δflap and wildtype, respectively.
h The reactions were performed in buffer A.
The SEC revealed that the purified hcSHMT/Δflap was eluted into two peaks with a similar peak area at 12 and 13.5 ml, corresponding to MWs of 206 and 96 kDa (Figs. 2A and S1D). The result indicated that hcSHMT/Δflap coexists in tetrameric and dimeric forms, which clearly differ from the WT showing almost a single peak at 12 ml of 206 kDa for tetramer and a very small fraction of 96-kDa dimer (Fig. 2B). It is noted that only the 206-kDa peak (tetramer) in both enzymes showed absorption at 430 nm (Fig. 2A, dashed line), suggesting that only the tetrameric form contains bound PLP, whereas the dimeric enzyme from hcSHMT/Δflap exists in an apo form. The melting temperature (Tm) of the apo-dimeric hcSHMT/Δflap form was determined to be 57 °C, lower than those (Tm, 65 °C) of the holo-tetrameric form of both hcSHMT/Δflap and WT. The results suggested that holo-tetramer is more stable than the apo-dimeric form.
Figure 2.

Analysis of oligomeric states of hcSHMT/Δflap compared with WT. The elution profile of purified hcSHMT/Δflap (A) and WT enzymes (B) obtained from SEC are shown. The absorptions at 280 and 430 nm are presented in solid and dashed lines, respectively. The peaks in A at 12 and 13.5 ml at 280 nm correspond to MWs of 206 (tetramer) and 96 (dimer) kDa, respectively. The dimer peak lacks a PLP characteristic peak at 430 nm, indicating the presence of apo-dimer. The insets of A (hcSHMT/Δflap) and B (WT) represent the plots of continuous size (c(s)) distribution versus sedimentation coefficient (S) from AUC scans at 280 nm. The observed MWs are 121 and 195 kDa for hcSHMT/Δflap and 220 kDa for WT. The SAXS X-ray scattering patterns and Guinier analysis (insets) at two different concentrations of purified hcSHMT/Δflap (a mixture of apo-dimer and holo-tetramer) (C) and WT (D) indicate no aggregation. E, X-ray scattering patterns of hcSHMT/Δflap (4.5 mg/ml; black) and WT (9 mg/ml; gray) fitted by the Oligomer program. hcSHMT/Δflap (red) contained a mixture of 17 and 83% dimer and tetramer, whereas the WT (blue) showed only tetramer.
The apparent MWs (MWapp) of hcSHMT/Δflap (containing both apo-dimeric and holo-tetrameric forms) and WT were further investigated using AUC scans at 280 nm. The plots between continuous size c(s) distribution and sedimentation coefficient (S) using SEDFIT program (36, 37) revealed two distinct ranges of S values, 6–8 and 9–11 (Fig. 2A, inset), corresponding to the observed MWapp of 121 and 195 kDa, likely consistent with the dimeric and tetrameric states of the hcSHMT/Δflap, respectively. On the contrary, the WT showed S value of 10–12 corresponding to an observed MWapp of 220 kDa (tetrameric form) (Fig. 2B, inset).
For SAXS studies, X-ray scattering data at two different concentrations of the purified hcSHMT/Δflap (4.5 and 9 mg/ml) (Fig. 2C) or WT (9 and 18 mg/ml) (Fig. 2D) were analyzed. Guinier analysis of the X-ray scattering patterns of the hcSHMT/Δflap (Fig. 2C, inset) or WT (Fig. 2D, inset) can be fitted to a linear relationship, indicating no aggregation or interparticle interference. The radius of gyration (Rg) and maximal particle dimensions (Dmax) obtained from the Guinier slope and pair distribution function (P(r)) analysis (Fig. S2A) were similar between hcSHMT/Δflap (Rg, 40.5–41.2 Å; Dmax, 127–128 Å) and WT (Rg, 39.4–39.7 Å; Dmax, 128 Å). Moreover, Kratky analysis suggested similar compactness of the two enzymes (Fig. S2B), suggesting that both enzymes adopt similar compact globular structures. From the above analysis, the size and shape of the two enzymes were similar; however, the calculated MW (see more details under “Experimental procedures”) of the purified hcSHMT/Δflap (156 kDa) was lower than that of the WT (173 kDa). The oligomeric-state analysis of each X-ray scattering curve (Fig. 2E) as fitted by the Oligomer program (38) indicated that the hcSHMT/Δflap (Fig. 2E, red line) contained the mixed forms of dimer and tetramer at 17 and 83%, respectively. The data firmly support that hcSHMT/Δflap exists as mixed oligomers, whereas the WT (Fig. 2E, blue line) showed only a tetramer. Furthermore, the ab initio modeling was carried out by two scattering curves of apo-dimeric and holo-tetrameric forms prepared by SEC to give a visual representation of the SAXS data. The ab initio modeling was done using the DAMMIF server (39) and DAMAVER (40) with 10 independent bead models without imposed symmetry (P1) of each protein. The models illustrated the mixed forms of apo-dimeric and holo-tetrameric hcSHMT/Δflap (Fig. S3). Only the holo-tetrameric form of hcSHMT/Δflap showed the same symmetrical shape as that of the WT (Fig. S3A). Based on the three analytical techniques demonstrated here, the data clearly support the presence of mixed dimeric and tetrameric forms in the hcSHMT/Δflap, whereas the WT exists mainly in a tetrameric form.
Influence of PLP binding to the shift in equilibrium between the dimer and tetramer of hcSHMT/Δflap
Interestingly, the dimeric form of hcSHMT/Δflap lost PLP binding, probably influenced by the absence of the flap motif. However, the tetramer was present with bound PLP. We hypothesized that enzyme tetramerization may require PLP binding. Therefore, the shift in dimer–tetramer equilibrium of the hcSHMT/Δflap in the presence of excess PLP was monitored by SEC, AUC, and SAXS.
The SEC results showed that addition of 5-fold excess PLP could convert apo-dimers to holo-tetramers (Fig. 3, A and B). Consistently, the equilibrium shift from apo-dimer to holo-tetramer upon addition of PLP was observed in AUC analyses. The holo-tetramer to dimer ratio was 1:2 for apo-dimer and 3:1 for PLP-treated apo-dimer (Fig. 3, A and B, insets). Therefore, PLP binding can promote the formation of the holo-tetramer.
Figure 3.

Effects of PLP on oligomerization equilibrium shift. The SEC elution profiles of 20 μm apo-dimer solution in no additional PLP (A) and 100 μm PLP (B), monitored at 280 (solid line) and 430 (dashed line) nm. Insets of A and B show the plots of c(s) distribution versus S value from AUC scans at 430 nm. The observed MWs are 171 and 241 kDa for the samples obtained from A and B. C, SAXS X-ray scattering patterns of apo-dimer (6.25 mg/ml; black), PLP-added apo-dimer (7.8 mg/ml, 5× PLP excess; green), and holo-tetramer (4.4 mg/ml; blue) of hcSHMT/Δflap. The linear fits (red lines) of Guinier analysis of these protein samples are shown in the inset of C accordingly.
Results from SAXS analysis also indicated that the overall X-ray scattering patterns and Guinier analysis shifted more toward the tetrameric form in the presence of PLP (Fig. 3C). The Rg and Dmax values of the apo-dimer were determined from Guinier (Fig. 3C, inset) and P(r) analysis (Fig. S2C) as 34.1 and 109–110 Å, respectively. The addition of PLP into the apo-dimer solution altered the scattering patterns (Fig. 3C, green line) to have higher values of Rg of 43.3 Å and Dmax of 134 Å. These values are similar to the values for the holo-tetramer enzyme separated by SEC (Rg, 39.5 Å; Dmax, 129–130 Å). These data were notably different from the values of the apo-dimer. In addition, Kratky analysis of the apo-dimer also showed a more rigid shape than that of the tetrameric enzyme (Fig. S2D). The ab initio model of the PLP-reconstituted enzyme was similar to that of the holo-tetramer fraction and the WT (Fig. S3D). Taken together, these results emphasize that the binding of PLP to apo-dimer can shift equilibrium toward the tetrameric form of the hcSHMT/Δflap.
Equilibrium binding of PLP to holo-tetramer of hcSHMT/Δflap
With the truncation of the flap motif, the tetrameric form has only been found as a PLP-bound enzyme in hcSHMT/Δflap and not as the apo form. To study the effect of the flap motif on PLP binding, we determined the equilibrium binding of PLP to the tetramer via ultrafiltration by collecting the PLP released from the PLP–tetramer at equilibrium. The results showed that only a small amount of released PLP could be detected in the filtrate fraction (Fig. S4). The calculated Kd for the PLP–Δflap-tetramer was 0.228 ± 0.005 μm, whereas the affinity of PLP to the WT (Kd 0.010 ± 0.005 μm) is 23-fold greater than that to the hcSHMT/Δflap (25). This indicates that the flap motif has an intrinsic function to promote PLP binding. Furthermore, the binding of PLP reconstitutes Δflap-dimer integrity proper for tetrameric formation.
Use of molecular dynamics simulations to investigate the functional role of the flap motif in protein dynamics
The dimeric hcSHMT/Δflap isolated by SEC was mainly obtained in the apo form, whereas the isolated tetrameric hcSHMT/Δflap was present in the holo form (see results in Fig. 2A). Based on SAXS results in Figs. 3C; S2, C and D; and S3, B and C, the X-ray scattering curve, Guinier plot, Kratky, and P(r) analyses as well as the ab initio models, which can differentiate between dimer and tetramer, the data indicated that the isolated apo-dimer mainly existed in that form without any significant formation of the apo-tetramer. Using the same analysis, the isolated holo-tetramer also mainly existed in that form without any significant amount of the holo-dimeric form. Moreover, the experiment in which we probed the effect of PLP on the formation of tetramer confirms the equilibrium between the apo-dimer and holo-tetramer (Figs. 3C and S3D). Therefore, to determine whether the flap motif has a role in controlling protein dynamics, 100-ns MD simulations of hcSHMT/Δflap were carried out for four species, apo-dimer, holo-dimer, apo-tetramer, and holo-tetramer, to assess protein mobility compared with the WT. The output of the simulation is the relationship between the structural integrity and the B-factor, which indicates the thermal motion of the molecule.
The results showed that the obligate tetramer of apo-hcSHMT/Δflap has much greater thermal motion (high B-factor) between dimer–dimer interfaces than the holo forms (Fig. 4). This suggested that loss of the flap motif induced high dynamic disorder or less compact dimers, probably imposed by the high mobility of the flap-connected region (see also Movies S1 and S2), and reduction of tetramer integrity, leading to dissociation into a more thermodynamically stable apo-dimer. Unlike the hcSHMT/Δflap, the MD simulations for the WT showed similar thermal motion for both apo and holo forms of the dimer and tetramer (Fig. S5). Moreover, the B-factors were lower than those of the hcSHMT/Δflap, indicating tightly packed domains in the WT.
Figure 4.

Proposed equilibrium link between dimer and tetramer of the hcSHMT/Δflap. The equilibrium between apo-dimer and holo-tetramer in hcSHMT/Δflap upon addition of PLP can be explained by the diagram. Path A, PLP can bind to the apo-dimer to form a holo-dimer, which is finally assembled to the holo-tetramer. Path B, PLP can be dissociated from the holo-tetramer to give the apo-tetramer with a KdPLP value of 0.228 ± 0.005 μm. High thermal motion (high B-factor) was found in the apo-tetramer, indicating high probability of dissociation of the apo-tetramer to a more stable apo-dimer (low B-factor). Upon increasing the PLP concentration, the binding of PLP promotes a shift toward holo-tetramer stabilization, possibly by more tightly packing the domains (indicated by the relative B-factors). The relative B-factors were calculated from MD simulations as described under “Experimental procedures.”
MD simulations showed interesting results to explain the structural integrity of hcSHMT/Δflap in that, in the absence of bound PLP, the dynamic disorder of the apo-tetramer is much greater, making it difficult to maintain the structural integrity of the tetramer. High motion between the protein–protein interface would lead to worse tetramer packing and therefore promote release to the more stable apo-dimer form (Fig. 4 and see also Movies S1 and S2). Accordingly, a significant portion of the hcSHMT/Δflap existed in the apo-dimeric form. These results agreed very well with the SEC and AUC results (Fig. 2A), which showed that the oligomeric equilibrium is driven toward the apo-dimer when the supply of PLP is not in excess. However, once PLP is added in excess, the equilibrium is shifted toward the holo-tetramer due to the formation of a more tightly packed dimer induced by stabilization of the structure by the bound PLP (Fig. 4). In contrast, the WT does not require PLP for tetrameric stabilization (Fig. S5). The results support that the human flap motif plays a crucial role in stabilizing the obligate tetrameric structure found naturally for hcSHMT.
In addition to stabilization of the tetrameric structure of hcSHMT, MD simulation showed that the flap motif promotes retention of THF in the binding pocket (see Movies S3 and S4). Without the flap, THF can be easily released, thereby lowering the binding affinity of THF (see Movie S3). In contrast, in the WT, the flap motif stabilizes THF retention in the pocket (see Movie S4).
Kinetics of the hcSHMT/Δflap reactions
To identify the catalytic steps for which the flap motif controls, initially, steady-state kinetics of the hcSHMT/Δflap reaction using l-serine (0.1–6.4 mm) and THF (0.025–1.6 mm) as substrates were carried out at pH 7.5, similar to that described previously for the WT (10). The Michaelis–Menten plot revealed that substrate inhibition was apparent when the THF concentration was >0.2 mm (Fig. 5A, red solid lines). This substrate inhibition is less severe than that observed for the WT (>0.08 mm) (Fig. 5A, blue dashed lines) (10). The double-reciprocal plots gave intersecting lines (Fig. S6), indicating that the hcSHMT/Δflap also employs ternary-complex kinetics similar to the reaction catalyzed by the WT (10, 25). Steady-state kinetic parameters were determined and are summarized in Table 1. The kcat value of hcSHMT/Δflap was 11.3 ± 0.3 s−1, about half of that of the WT (19.8 ± 0.8 s−1) (10). Km values for l-serine and THF for the hcSHMT/Δflap were similar to those of WT. The resulting kcat/Km value of the hcSHMT/Δflap was 2–3-fold lower than those of the WT, indicating that hcSHMT/Δflap has lower catalytic efficiency. Therefore, in addition to its effects on protein quaternary structure, structural dynamics, and PLP affinity, deletion of the flap motif also decreases the catalytic efficiency of the enzyme.
Figure 5.

Kinetic studies of the hcSHMT/Δflap reactions. A, two-substrate steady-state kinetics of hcSHMT/Δflap (red) and WT enzyme (blue) reactions at pH 7.5. The plot of ν/e versus THF concentrations (0.025–1.6 mm for hcSHMT/Δflap and 0.005–0.16 mm for WT) at fixed l-serine concentrations (0.1–6.4 mm for hcSHMT/Δflap and 0.1–3.2 mm for WT) is shown. Each fit curve represents low to high concentrations of l-serine with relative increasing velocity. Inset a of A, the apparent kinetics of the hcSHMT/Δflap at pHs 6.5–8.5 using 0.025–1.6 mm THF and 6.4 mm l-serine. Inset b of A, pH-activity profile (pH 6–10) of hcSHMT/Δflap (red filled circles) and WT enzyme (blue filled squares). B, kinetics of glycine product formation of hcSHMT/Δflap (red filled circles) and WT enzyme (blue filled squares) at pH 7.0. C, THF-independent aldol cleavage of l-allo-threonine (0.1–10 mm) of hcSHMT/Δflap (red filled circles) and WT (blue filled squares) enzymes measured by an SHMT–ADH coupled assay. Error bars represent S.D. (or S.E.).
Furthermore, the fact that THF inhibition depended on the pH in the WT prompted us to study this effect in hcSHMT/Δflap. We found that THF inhibition in hcSHMT/Δflap showed a tendency similar to the WT in that the inhibition was more pronounced at low pH (Fig. 5A, inset a) (10). The apparent THF inhibition constants (KiTHF) increased from 0.41 to 1.9 mm upon increasing pH from 6.5 to 8.5. However, the KiTHF values of the hcSHMT/Δflap are larger than those obtained from the WT (10), suggesting that the THF inhibition is less sensitive in the hcSHMT/Δflap. These data indicate that the flap motif plays a role in THF inhibition, previously hypothesized to be involved in the slow release of product that allows THF binding to form the hcSHMT·Gly·THF dead-end species (10).
The activity of hcSHMT/Δflap investigated at various pH values (6.5–8.5) showed a half-bell–shaped plot, similar to the WT (Fig. 5A, inset b) (25). However, the pKa value for the reaction of the hcSHMT/Δflap was 8.8 ± 0.1 (Fig. 5), 1 pH unit higher than that of the WT (7.8 ± 0.2) (25). Shifts in the pKa values of ionizable groups in proteins are typically observed upon mutation or truncation (41). The data imply that, upon the removal of flap motif, a key catalytic residue of SHMT that should be protonated to participate in the catalysis has a raised pKa. In the hcSHMT active site, there are two ionizable residues, Lys-257 and Glu-75. Lys-257 is the active lysine that forms a Schiff base with the PLP cofactor, and it needs to be deprotonated to be active. Thus, the pKa of Lys-257 is unlikely changed. Glu-75 has been proposed to act as the general acid required to protonate the reaction intermediate to facilitate the product formation upon C–C bond cleavage (2). Therefore, Glu-75 may be a candidate residue responsible for the pKa observed in the pH-rate profile and is the residue with the pKa shift in the hcSHMT/Δflap. It is known that pKa values of Glu in enzyme active sites can vary over a wide range from 2 to 9 (41).
A previous study showed that the glycine release is the rate-determining step of hcSHMT (10). To investigate the implication of flap motif on the glycine release step, we compared pre-steady-state kinetics of hcSHMT/Δflap and WT. The kinetics of glycine product formation for the hcSHMT/Δflap (Fig. 5B, red circles) is clearly different from that of WT (Fig. 5B, blue squares). No burst kinetics was observed for hcSHMT/Δflap reaction, whereas the WT showed burst kinetics, and 41 ± 17 μm glycine formed in the burst phase (0–0.5 s), resulting from the first turnover of the WT reaction. Therefore, in the WT enzyme, the product release is the rate-limiting step as shown previously (10). In contrast, the rate-limiting step of hcSHMT/Δflap is not the product release step. Other processes such as substrate binding or the catalytic steps prior to the product release may be slower than the product release and become the rate-limiting step instead. Taken together, the results indicate that the flap controls the glycine release step.
Removal of the flap motif does not interfere with THF-independent SHMT activity
Because hcSHMT can catalyze aldol cleavage reaction of β-hydroxyamino acid (1, 19, 25), whether the flap interferes with the THF-independent reaction, the conversion of l-allo-threonine to acetaldehyde and glycine, was studied in hcSHMT/Δflap and WT. They showed similar kcat and Km (Fig. 5C and Table 1), indicating that the flap does not interfere with THF-independent SHMT activity.
Direct evidence showing that the flap motif is involved in THF binding
The above sections suggest the role of the flap motif in controlling glycine release in THF-dependent catalysis by retaining THF binding. Therefore, the dissociation constant of THF was measured to validate the higher affinity of THF in the WT.
THF can bind to both WT and hcSHMT/Δflap (Fig. S7) to form an E·THF binary complex with a maximum absorbance at 490 nm. The Kd values were calculated to be 108 ± 8 and 35 ± 8 μm for the hcSHMT/Δflap and WT, respectively (Fig. 6). This indicated that the flap enhances the binding affinity of THF by 3-fold, agreeing well with the MD results (see Movies S3 and S4).
Figure 6.
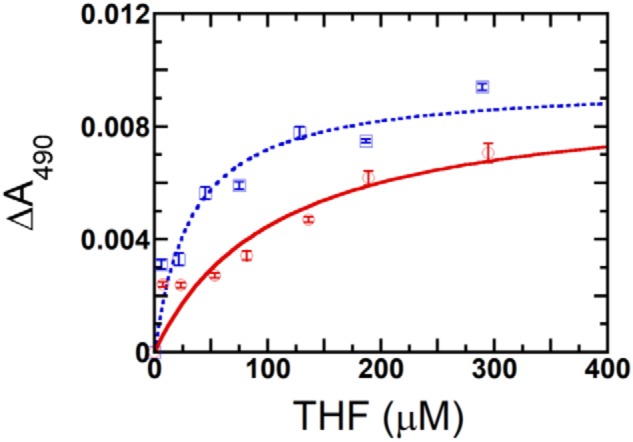
Comparison of THF binding to hcSHMT/Δflap versus WT enzyme. The binding plot of THF with hcSHMT/Δflap (red) and WT enzyme (blue) at 6–290 μm THF is shown. The Kd values were 108 ± 8 and 35 ± 8 μm, respectively, calculated from triplicate experiments. Error bars represent S.D. (or S.E.).
Discussion
This study is the first to elucidate the role of the hcSHMT flap motif, a unique β-hairpin structure, that is conserved among mammalian SHMTs. Although the first structure of human SHMT was elucidated almost two decades ago (19, 27), the key observation that the loop is unique only to mammalian SHMTs was only recognized when the structures of SHMTs from Plasmodium falciparum and Plasmodium vivax were elucidated (28, 29).
The importance of the flap motif in hcSHMT in stabilizing the tetrameric structure and PLP and THF binding has been demonstrated here. These properties are crucial for hcSHMT activity. We showed that the truncation of flap resulted in an apo-dimer, loss of PLP binding, and change of the rate-determining step. Lack of the flap affected the pKa of the ionizable groups, particularly Glu-75, which may be a general acid in the SHMT reaction. Binding of PLP can shift the equilibrium from apo-dimer to holo-tetramer. This situation is similar to human ornithine δ-aminotransferase in which PLP stabilizes the tetrameric structure (42).
The presence of the flexible loop to control oligomerization, cofactor and substrate binding, and product release is crucial for the protein evolution point of view. Within the SHMT family, the lack of the flap motif results in a dimeric structure for SHMTs in prokaryotes such as Escherichia coli (33) and Bacillus stearothermophilus (34) and some eukaryotes such as P. vivax and P. falciparum (3, 7, 26, 28, 29), whereas in higher eukaryotes like human and rabbit the structure is tetrameric with the presence of flap (27, 30). In other enzyme families, a flexible loop/flap region is present in the Plasmodium bifunctional DHFR-TS enzyme from a unique insert (so-called Insert 1) and stabilizes the domain attachment between DHFR and TS in the homodimeric structure of the enzyme (43). In other organisms in which the DHFR lacks this flap region, such as human and E. coli, DHFR and TS exist as separate enzymes (44). A well-known flap motif is also found in HIV protease, which is well-recognized in its functional roles in pocket formation, ligand binding, and importantly enzyme catalysis in the enzyme homodimer (45).
Our findings about the THF inhibition and binding in hcSHMT/Δflap may explain why hcSHMT mRNA splicing takes place in MCF-7 breast and SH-SY5Y neuroblastoma cancer cells. The alternative mRNA splicing in cancer cells was found in exons 2, 9, and 10 (19, 35), and the splicing of exon 9 leads to production of hcSHMT without the flap motif (residues 273–287). The resultant diminished THF affinity likely raises drug resistance against antifolate chemotherapy. Another example of this is the observed overexpression of alternatively spliced variants of folylpolyglutamate synthase mRNAs in leukemia cells, which causes antifolate anticancer drug resistance in acute lymphoblastic leukemia (46, 47).
In conclusion, these findings provide mechanistic insights into how a flap motif can influence the overall structural architecture and catalytic activity of hcSHMT. Understanding the role of the flap motif paves the way for development of more effective antifolate drugs for anticancer chemotherapy in the future. The results reported here also contribute to the understanding of how a flexible loop can take part in stabilization of oligomeric structure and ligand binding in other enzymes in general.
Experimental procedures
Chemicals and reagents
All chemicals and reagents used in this study were analytical grade and of the highest purity commercially available and were prepared as described in previous reports (10, 25). Buffers used throughout this report were (i) buffer A, 10 mm HEPES buffer, pH 7.5, containing 100 mm NaCl, 1 mm DTT, and 0.5 mm EDTA;, (ii) buffer B, 10 mm HEPES buffer, pH 7.5, containing 150 mm NaCl; (iii) buffer C, 10 mm HEPES buffer, pH 7.5, containing 100 mm NaCl; and (iv) buffer D, a three-component buffer system (AME buffer, pH 7.5, composed of 0.05 m acetic acid, 0.05 m MES, and 0.1 m N-ethylmorpholine) containing 1 mm DTT and 0.5 mm EDTA.
Construction of an expression plasmid containing the hcshmt/Δflap gene
An expression plasmid carrying the hcshmt/Δflap gene was generated by site-directed mutagenesis using pET100D/TOPO-hcshmtΔH (25) as a template in the presence of primers 5′-GATCTTCTACAGGAAAGGAGTGAAATACAACCTGGAGTCTCTTATCAATTC-3′ and 5′-GAATTGATAAGAGACTCCAGGTTGTATTTCACTCCTTTCCTGTAGAAGATC-3′. The plasmid was sequenced at the 1st Base DNA sequencing service (Malaysia) to verify the deletion of the flap sequence. The expression plasmid was named pET100D/TOPO-hcshmt/Δflap. The sequences encoding 12 amino acids (274KSVDPKTGKEIL285) of the flap motif was deleted to remove the β-hairpin that makes up for the flap motif. Although the overall flap motif includes residues 273–285, we decided to retain residue 273 in the variant to prevent deterioration in protein folding.
Enzyme expression, purification, and assay
The overexpression of the hcSHMT/Δflap in E. coli BL21(DE3) was carried out at 16 °C in ZY autoinduction medium system (1% (w/v) peptone, 0.5% (w/v) yeast extract, 5 mm Na2SO4, 2 mm MgSO4, 1X NPS (25 mm Na2HPO4, 25 mm KH2PO4, and 50 mm NH4Cl), and 1X 5052 (0.5% (w/v) glycerol, 0.05% (w/v) D-glucose, and 0.2% (w/v) alpha-lactose)) as described previously for the WT (10, 25). The purification protocol for the hcSHMT/Δflap was performed similarly to that for the WT (10, 25) but with slight modifications. In brief, the hcSHMT/Δflap was purified to homogeneity using 1% (w/v) polyethyleneimine and 30–50% (w/v) ammonium sulfate precipitations and diethylaminoethyl (DEAE)-Sepharose and Sephacryl S-200 gel-filtration columns. The buffer system used for enzyme purification and storage was buffer A.
The activity of the hcSHMT/Δflap was measured at 25 °C in buffer A and carried out using an SHMT–MTHFD coupled assay method (10, 25) except that 0.2 mm THF and 0.1 μm hcSHMT/Δflap were added. The reaction progression of NADPH production was monitored at 375 nm by a diode array spectrophotometer (Hewlett Packard). One unit of SHMT activity is defined as the formation of 1 μmol of NADPH/min at pH 7.5 and 25 °C.
Determination of the molar absorption coefficient of hcSHMT/Δflap-bound PLP under denaturing condition
The hcSHMT/Δflap was denatured by SDS, and the amount of PLP cofactor liberated was spectroscopically measured and used to determine the molar absorption coefficient of the hcSHMT/Δflap. The molar absorption coefficients of PLP and the hcSHMT/Δflap-bound PLP were determined in the presence of SDS as described previously (7, 26) except that a final concentration of 2% (w/v) SDS was employed.
Analyses of the MWs, oligomeric states, and quaternary structures
Three analytical techniques, including SEC, AUC, and SAXS, were used to analyze MWs, oligomeric states, and quaternary structures of the hcSHMT/Δflap. By the SEC technique, the native MW and oligomeric states of the hcSHMT/Δflap (77 μm) were determined using a SuperdexTM 200 Increase 10/300 GL (GE Healthcare) gel-filtration column performed with an ÄKTA FPLC system (GE Healthcare). The column was calibrated with the known MWs of protein standards (GE Healthcare). All proteins were eluted with buffer B at 25 °C at a constant flow rate of 0.5 ml/min, and the elution volume (Ve) for each protein was measured. The absorbance at 280 nm (A280) was monitored for the proteins, whereas A430 was monitored for bound PLP of the hcSHMT/Δflap. The oligomeric form of the hcSHMT/Δflap was estimated based on the MW of the subunits of the hcSHMT/Δflap for which the amino acid sequence was calculated using the ProtParam program on the ExPaSy Proteomics Server (https://web.expasy.org/protparam/).
For size-distribution analysis, peaks corresponding to the apo-dimer, holo-tetramer, and mixed oligomers (as purified form) as separated by Superdex gel filtration and detected by optical detectors (Beckman Coulter, ProteomeLab XL-I) were analyzed by AUC. The hcSHMT/Δflap sample (20 μm) was dialyzed overnight and diluted into buffer A. The protein sample (400 μl) and buffer A (425 μl) were injected in each side of a double-sector centerpiece cell. The double-sector cells were placed into an An-60 Ti rotor and then centrifuged at 20,000 rpm at 20 °C for 16 h. The spectrum of each protein sample was monitored continuously at 280 nm using a time interval of 600 s/scan. The sedimentation velocities obtained from multiple scans at 280 nm and at different time intervals were fitted to a continuous c(s) distribution model using the SEDFIT program (36, 37) to obtain the corresponding MW. The partial-specific volume of the protein sample was set as 0.73 ml/g, based on a solvent density of 1.035 g/ml (48).
For SAXS analysis, the experiments were measured using a multipole wiggler source on Beamline 1.3W (BL1.3W:SAXS) of the Synchrotron Light Research Institute (Public Organization), Thailand. A MarCCD Rayonix SX165 detector with a sample-to-detector distance of 2207 mm was used, and at least two different concentrations (3–20 mg/ml) of each protein sample (apo-dimer, holo-tetramer, the as-purified form of hcSHMT/Δflap, and WT in the absence and presence of 5-fold excess PLP) were exposed to X-ray emission (λ = 1.38 Å) at 16 °C for 10 min. This enabled us to capture SAXS data with a q range of 0.022 < q < 0.32 Å−1. The empty sample cell was measured first followed by the matched reference buffer and the protein sample, respectively. The 2D images were reduced and radially averaged by the program SAXSIT, which was developed by Synchrotron Light Research Institute staff, and BioXTAS RAW (49) to obtain 1D scattering curves. Guinier and Kratky analyses were calculated from the scattering patterns using functions of BioXTAS RAW (49). Pair distribution function (P(r)) analysis was calculated from the scattering curves using the GNOM program (50) from ATSAS package version 2.8.2 (51). The MW of each protein was calculated by using a zero extrapolated intensity from a calibration standard protein (lysozyme) with a known concentration (10 mg/ml) and MW (14 kDa) (52). The oligomeric states of the proteins were calculated by the Oligomer program (38). It should be noted that only X-ray scattering patterns at the lowest protein concentration for each protein sample were used for curve-fitting analysis by the Oligomer program to avoid any apparent aggregation scattering. The ab initio envelope reconstructions were carried out by the DAMMIF server (39) and DAMAVER (40) with 10 independent bead models without imposed symmetry (P1) of each protein. The SAXS data at q < 0.25 Å−1 was used for the modeling. For the WT, the SAXS-derived molecular envelope generated was superimposed with the available crystal structure (PDB code 1BJ4). Due to the lack of an X-ray structure, the model of hcSHMT/Δflap was constructed from the WT structure (PDB code 1BJ4) with deletion of residues 274–285 in Coot (53). Both dimeric and tetrameric models of hcSHMT/Δflap were superimposed with the ab initio envelopes generated. The best alignment of any superimposed model was assessed by a normalized spatial discrepancy value that should be generally close to a value of 1 (54).
Measurements of the dissociation constant for the equilibrium binding of PLP with holo-tetrameric form of the hcSHMT/Δflap
Ultrafiltration (Centriprep® YM-30, Amicon) was used to study the equilibrium binding of PLP with the holo-tetrameric hcSHMT/Δflap. An 11-ml holo-tetrameric hcSHMT/Δflap (A430 ∼ 0.09; 12 μm) was placed into a 10-kDa-cutoff ultrafiltration unit and then centrifuged at 1380 × g at 25 °C for 1 min to obtain ∼1 ml of filtrate containing the released free PLP. The concentrations of the holo- and apo-tetrameric enzyme and the released free PLP were calculated, and the Kd value under the equilibrium condition was determined as described (7, 25, 26).
Molecular dynamics simulations
Molecular dynamic simulations were performed for eight modeled systems, WT and hcSHMT/Δflap in dimeric and tetrameric forms with or without PLP bound, using the AMBER 16 package program (55). The starting configurations of SHMT with or without PLP covalently bound to Lys-257 were taken from the available crystal structure in the Protein Data Bank (PDB code 1BJ4). It should be noted that both dimeric and tetrameric models for the hcSHMT/Δflap were derived from the WT (PDB code 1BJ4) enzyme with deletion of flap motif residues 274–285 in Coot (53) and prepared before MD simulation as follows. The protonation state of amino acids was assigned by PDB2PQR 2.0.0 (56). The atomic charges of PLP were developed according to standard procedures (57). The AMBER ff14SB force field (58) was adopted for the protein, whereas the atom types and the other molecular parameters of PLP were assigned by general AMBER force field version 2 (GAFF2) (59). The protonated model was energy-minimized using two steps consisting of 2500 steps of steepest descents followed by 2500 steps of conjugated gradient. The model was then solvated by a TIP3P water in an octahedron box within 10 Å from the protein surface, neutralized by adding Na+ ions, and minimized with the two steps described above under a restraint on the protein atoms with a weight of 10.0 kcal/mol2-Å2 followed by minimization on the whole system. The system was then heated to 298 K at 1-atm pressure and subsequently subjected to MD simulation using the isothermal-isobaric (NPT) ensemble for 100 ns as described previously (60). To indicate protein flexibility and mobility, the B-factor was calculated (Equation 1) for the backbone atoms of all minimized structures at the last 20-ns MD trajectories using the CPPTRAJ module and normalized in a relative scale. Herein, T is a set of frames over the last 5-ns trajectories, ri(tj) is the position of atom i at time tj, and ri is the time-averaged position of the same atom i. Note that B-factor values are normalized in a range of 0 (blue for low fluctuation) to 1 (red for high fluctuation).
| (Eq. 1) |
Steady-state kinetics of two-substrate reactions
The steady-state kinetics of the hcSHMT/Δflap were studied at pH 7.5 and 25 °C by an SHMT–MTHFD coupled assay (10). The assays were performed in buffer D containing 250 μm NADP+, 10 μm MTHFD, 0.1 μm hcSHMT/Δflap, and various concentrations of l-serine (0.1–6.4 mm) and THF (0.025–1.6 mm) using stopped-flow spectrophotometry in single-mixing mode under anaerobic conditions. The kinetics of NADPH production was monitored at 375 nm, and the initial velocities (ν) were calculated. Direct plots of initial velocities versus concentrations of either substrate were fitted to Equation 2 where Vmax is the maximum rate, KA and KB are Michaelis constants for substrates A and B, and KiA is the dissociation constant of substrate A. The double-reciprocal plots of initial velocities versus concentrations of either substrate were plotted to identify the steady-state mechanism as described by Dalziel's equation. Kinetic parameters of the hcSHMT/Δflap were determined without inhibition of THF substrate and compared with those of the WT.
In addition to pH 7.5, the steady-state kinetics at various pH values (6.5–8.5) and THF concentrations (0.025–1.6 mm) at an l-serine concentration 6.4 mm were carried out to determine the magnitude of THF substrate inhibition at different pH values. The inhibition constant (Ki) of the THF substrate was determined using Equation 3, which was analyzed by Marquardt–Levenberg algorithms in KaleidaGraph version 4.0 software (Synergy Software).
| (Eq. 2) |
| (Eq. 3) |
pH-activity profile
The activity of the hcSHMT/Δflap at different pH values was determined by a coupled enzyme assay method as described previously under “Enzyme expression, purification, and assay.” The buffers were as follows: 100 mm HEPES buffer for pH 6–8.5 and 100 mm carbonate buffer for pH 9–10. Initial velocities were plotted as a function of pH and fitted according to Equation 4 to determine the pKa value by using Marquardt–Levenberg algorithms in KaleidaGraph version 4.0 program. Here, Y is the maximum rate, C is the maximum rate that depends on the pH, and pKa is the dissociation constant of the acid.
| (Eq. 4) |
Analyses of glycine formation kinetics using rapid-quench techniques and HPLC-MS
All experiments were performed at pH 7.0 similarly to those reported previously using a rapid-acid quench coupled with HPLC-MS techniques (10) except that the final concentration of THF used for the hcSHMT/Δflap and WT was 0.2 and 0.08 mm, respectively, to avoid THF substrate inhibition. The glycine obtained from the rapid-quench technique was monitored and quantified by HPLC-MS (10). The kinetics of glycine product formation of the hcSHMT/Δflap was then analyzed.
Activity assay of the THF-independent aldol cleavage reaction
The aldol cleavage kinetics was performed using l-allo-threonine as a substrate. The formation of acetaldehyde was detected by coupling the reaction with alcohol dehydrogenase (ADH) (26). The assay reaction contained 100 μm NADH; 3 mg/ml ADH; 1 and 0.5 μm hcSHMT for the hcSHMT/Δflap and WT, respectively; and 0.1–10 mm l-allo-threonine in buffer A. The assay reactions were monitored for an absorbance decrease at 340 nm, and the initial velocities were determined and plotted as a function of l-allo-threonine concentration. The plot was fitted by Michaelis–Menten equation and analyzed using Marquardt–Levenberg algorithms in KaleidaGraph version 4.0 software to determine the kinetic parameters.
Measurement of the equilibrium constants for the binding of enzyme with THF
To measure the Kd value for the binding of THF to the hcSHMT/Δflap and WT, the binding reactions were measured by spectrophotometry (DH-2000-TgK/CCD-204 spectrophotometer, Ocean Optics, UK) under anaerobic conditions in an anaerobic glovebox (<5 ppm O2; Belle Technology, UK) to avoid oxidation of THF. A 1-ml anaerobic solution of the enzyme ([P]total, 28 μm; A430 ≈ 0.2) in buffer D was added into a quartz cuvette, and the absorption spectrum of the enzyme was recorded as a baseline for spectrum subtraction after each THF addition. Absorption spectra were recorded after adding THF, and the spectrum of the enzyme alone was subtracted to obtain the difference spectrum. The absorbance change values at 490 nm (ΔA) for the ligand-bound enzyme were extracted from the difference spectra derived from 16–319 μm THF ([L]total). The ΔAmax was determined from the plot between ΔA and various [L]total and input into Equations 5–7 for calculation of the unbound THF concentrations ([L]free), 6–290 μm. The Kd was determined from the nonlinear least square using Marquardt–Levenberg algorithms in KaleidaGraph version 4.0 software from the plot between the unbound concentrations of THF (6–290 μm) and ΔA.
| (Eq. 5) |
| (Eq. 6) |
| (Eq. 7) |
Dye-binding thermal shift assay
The protein sample (40 μm), apo-dimeric, holo-tetrameric, and mixed oligomeric (as-purified) forms of hcSHMT/Δflap, was mixed with a fluorescent SYPRO Orange dye solution (10×) (Invitrogen). The fluorescence signal was monitored using a real-time PCR machine with a temperature increase from 25 to 95 °C at a constant interval of 1 °C/min (61). The melting curve of temperature versus fluorescence signal was analyzed and used to determine the Tm at which half of the total protein is transitioned to an unfolded state.
Author contributions
S. U., P. Chitnumsub, U. L., P. Chaiyen, and S. M. conceptualization; S. U., W. P., N. K., P. W., P. M.-u., T. R., O. K., P. Chitnumsub, U. L., P. Chaiyen, and S. M. resources; S. U., J. J., N. K., P. W., P. M.-u., T. R., O. K., P. Chitnumsub, U. L., P. Chaiyen, and S. M. data curation; S. U., N. K., P. W., P. M.-u., T. R., O. K., P. Chitnumsub, U. L., P. Chaiyen, and S. M. software; S. U., J. J., N. K., P. W., P. M.-u., T. R., O. K., P. Chitnumsub, U. L., P. Chaiyen, and S. M. formal analysis; S. U., P. Chitnumsub, U. L., P. Chaiyen, and S. M. supervision; S. U., U. L., P. Chaiyen, and S. M. funding acquisition; S. U., J. J., W. P., N. K., P. W., T. R., O. K., P. Chitnumsub, U. L., P. Chaiyen, and S. M. validation; S. U., J. J., W. P., N. K., P. W., P. M.-u., T. R., O. K., P. Chitnumsub, U. L., P. Chaiyen, and S. M. investigation; S. U., J. J., N. K., P. W., P. M.-u., T. R., O. K., P. Chitnumsub, U. L., P. Chaiyen, and S. M. visualization; S. U., J. J., W. P., N. K., P. W., P. M.-u., T. R., O. K., P. Chitnumsub, U. L., P. Chaiyen, and S. M. methodology; S. U., J. J., W. P., N. K., P. Chitnumsub, U. L., P. Chaiyen, and S. M. writing-original draft; S. U., P. Chitnumsub, U. L., P. Chaiyen, and S. M. project administration; S. U., J. J., W. P., N. K., P. W., T. R., O. K., P. Chitnumsub, P. Chaiyen, and S. M. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Professor Martino di Salvo (Università di Roma, Italy) for the plasmid used for overexpressing MTHFD and Merck Eprova AG (Schafhausen Switzerland) for providing high-quality folate compounds. We thank Timothy M. Ryan (Australian Synchrotron, Australia) for useful suggestions of ab initio reconstruction. We also thank staffs at the beamline BL1.3W:SAXS, Synchrotron Light Research Institute (Public Organization, Nakhon Ratchasima, Thailand).
This work was supported by Thailand Research Fund Grants RTA5980001 (to P. Chaiyen), MRG5980001 (to S. M.), and RDG6050101 (to P. Chaiyen and S. M.); the Faculty of Science, Mahidol University and School of Biomolecular Science and Engineering, Vidyasirimedhi Institute of Science and Technology (to P. Chaiyen and J. J.); the Faculty of Science, Burapha University (to S. M.); Cluster Program and Management Office, National Science and Technology Development Agency, Thailand Grant CPMO-P-13-00835 (to U. L., W. P., P. Chitnumsub, P. Chaiyen, and S. M.); Thailand Graduate Institute of Science and Technology Scholarship Grant TG-22-14-59-031M (to S. U.); Synchrotron Light Research Institute–Public Organization Grant 1-2561/Project I.D. 3212 (to S. M. and S. U.); and a Science Achievement Scholarship of Thailand (to P. M.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S7, Movies S1–S4, and Table S1.
- SHMT
- serine hydroxymethyltransferase
- ADH
- alcohol dehydrogenase
- AUC
- analytical ultracentrifugation
- 5,10-CH2-THF
- 5,10-methylenetetrahydrofolate
- DHFR
- dihydrofolate reductase
- hcSHMT
- human cytosolic SHMT
- hcSHMT/Δflap
- flap-deleted hcSHMT
- hmSHMT
- human mitochondrial SHMT
- MD
- molecular dynamics
- MTHFD
- 5,10-methylenetetrahydrofolate dehydrogenase
- MW
- molecular weight
- MWapp
- apparent MW
- PLP
- pyridoxal 5′-phosphate
- SAXS
- small-angle X-ray scattering
- SEC
- size-exclusion chromatography
- THF
- (6S)-tetrahydrofolate
- Tm
- melting temperature
- TS
- thymidylate synthase
- PDB
- Protein Data Bank.
References
- 1. Schirch L. (1982) Serine hydroxymethyltransferase. Adv. Enzymol. Relat. Areas Mol. Biol. 53, 83–112 [DOI] [PubMed] [Google Scholar]
- 2. Schirch V., and Szebenyi D. M. (2005) Serine hydroxymethyltransferase revisited. Curr. Opin. Chem. Biol. 9, 482–487 10.1016/j.cbpa.2005.08.017 [DOI] [PubMed] [Google Scholar]
- 3. Maenpuen S., Amornwatcharapong W., Krasatong P., Sucharitakul J., Palfey B. A., Yuthavong Y., Chitnumsub P., Leartsakulpanich U., and Chaiyen P. (2015) Kinetic mechanism and the rate-limiting step of Plasmodium vivax serine hydroxymethyltransferase. J. Biol. Chem. 290, 8656–8665 10.1074/jbc.M114.612275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alfadhli S., and Rathod P. K. (2000) Gene organization of a Plasmodium falciparum serine hydroxymethyltransferase and its functional expression in Escherichia coli. Mol. Biochem. Parasitol. 110, 283–291 10.1016/S0166-6851(00)00282-6 [DOI] [PubMed] [Google Scholar]
- 5. Nirmalan N., Wang P., Sims P. F., and Hyde J. E. (2002) Transcriptional analysis of genes encoding enzymes of the folate pathway in the human malaria parasite Plasmodium falciparum. Mol. Microbiol. 46, 179–190 10.1046/j.1365-2958.2002.03148.x [DOI] [PubMed] [Google Scholar]
- 6. Leartsakulpanich U., Kongkasuriyachai D., Imwong M., Chotivanich K., and Yuthavong Y. (2008) Cloning and characterization of Plasmodium vivax serine hydroxymethyltransferase. Parasitol. Int. 57, 223–228 10.1016/j.parint.2007.11.001 [DOI] [PubMed] [Google Scholar]
- 7. Maenpuen S., Sopitthummakhun K., Yuthavong Y., Chaiyen P., and Leartsakulpanich U. (2009) Characterization of Plasmodium falciparum serine hydroxymethyltransferase-A potential antimalarial target. Mol. Biochem. Parasitol. 168, 63–73 10.1016/j.molbiopara.2009.06.010 [DOI] [PubMed] [Google Scholar]
- 8. Pornthanakasem W., Kongkasuriyachai D., Uthaipibull C., Yuthavong Y., and Leartsakulpanich U. (2012) Plasmodium serine hydroxymethyltransferase: indispensability and display of distinct localization. Malar. J. 11, 387–395 10.1186/1475-2875-11-387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Witschel M. C., Rottmann M., Schwab A., Leartsakulpanich U., Chitnumsub P., Seet M., Tonazzi S., Schwertz G., Stelzer F., Mietzner T., McNamara C., Thater F., Freymond C., Jaruwat A., Pinthong C., et al. (2015) Inhibitors of plasmodial serine hydroxymethyltransferase (SHMT): cocrystal structures of pyrazolopyrans with potent blood- and liver-stage activities. J. Med. Chem. 58, 3117–3130 10.1021/jm501987h [DOI] [PubMed] [Google Scholar]
- 10. Amornwatcharapong W., Maenpuen S., Chitnumsub P., Leartsakulpanich U., and Chaiyen P. (2017) Human and Plasmodium serine hydroxymethyltransferases differ in rate-limiting steps and pH-dependent substrate inhibition behavior. Arch. Biochem. Biophys. 630, 91–100 10.1016/j.abb.2017.07.017 [DOI] [PubMed] [Google Scholar]
- 11. Schwertz G., Witschel M. C., Rottmann M., Bonnert R., Leartsakulpanich U., Chitnumsub P., Jaruwat A., Ittarat W., Schäfer A., Aponte R. A., Charman S. A., White K. L., Kundu A., Sadhukhan S., Lloyd M., et al. (2017) Antimalarial inhibitors targeting serine hydroxymethyltransferase (SHMT) with in vivo efficacy and analysis of their binding mode based on x-ray cocrystal structures. J. Med. Chem. 60, 4840–4860 10.1021/acs.jmedchem.7b00008 [DOI] [PubMed] [Google Scholar]
- 12. Schwertz G., Frei M. S., Witschel M. C., Rottmann M., Leartsakulpanich U., Chitnumsub P., Jaruwat A., Ittarat W., Schäfer A., Aponte R. A., Trapp N., Mark K., Chaiyen P., and Diederich F. (2017) Conformational aspects in the design of inhibitors for serine hydroxymethyltransferase (SHMT): biphenyl, aryl sulfonamide, and aryl sulfone motifs. Chemistry 23, 14345–14357 10.1002/chem.201703244 [DOI] [PubMed] [Google Scholar]
- 13. Paiardini A., Fiascarelli A., Rinaldo S., Daidone F., Giardina G., Koes D. R., Parroni A., Montini G., Marani M., Paone A., McDermott L. A., Contestabile R., and Cutruzzolà F. (2015) Screening and in vitro testing of antifolate inhibitors of human cytosolic serine hydroxymethyltransferase. ChemMedChem 10, 490–497 10.1002/cmdc.201500028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marani M., Paone A., Fiascarelli A., Macone A., Gargano M., Rinaldo S., Giardina G., Pontecorvi V., Koes D., McDermott L., Yang T., Paiardini A., Contestabile R., and Cutruzzolà F. (2016) A pyrazolopyran derivative preferentially inhibits the activity of human cytosolic serine hydroxymethyltransferase and induces cell death in lung cancer cells. Oncotarget 7, 4570–4583 10.18632/oncotarget.6726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Paiardini A., Tramonti A., Schirch D., Guiducci G., di Salvo M. L., Fiascarelli A., Giorgi A., Maras B., Cutruzzolà F., and Contestabile R. (2016) Differential 3-bromopyruvate inhibition of cytosolic and mitochondrial human serine hydroxymethyltransferase isoforms, key enzymes in cancer metabolic reprogramming. Biochim. Biophys. Acta 1864, 1506–1517 10.1016/j.bbapap.2016.08.010 [DOI] [PubMed] [Google Scholar]
- 16. Ducker G. S., Ghergurovich J. M., Mainolfi N., Suri V., Jeong S. K., Hsin-Jung Li S., Friedman A., Manfredi M. G., Gitai Z., Kim H., and Rabinowitz J. D. (2017) Human SHMT inhibitors reveal defective glycine import as a targetable metabolic vulnerability of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. U.S.A. 114, 11404–11409 10.1073/pnas.1706617114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Amelio I., Cutruzzolá F., Antonov A., Agostini M., and Melino G. (2014) Serine and glycine metabolism in cancer. Trends Biochem. Sci. 39, 191–198 10.1016/j.tibs.2014.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stover P. J., Chen L. H., Suh J. R., Stover D. M., Keyomarsi K., and Shane B. (1997) Molecular cloning, characterization, and regulation of the human mitochondrial serine hydroxymethyltransferase gene. J. Biol. Chem. 272, 1842–1848 10.1074/jbc.272.3.1842 [DOI] [PubMed] [Google Scholar]
- 19. Snell K., Baumann U., Byrne P. C., Chave K. J., Renwick S. B., Sanders P. G., and Whitehouse S. K. (2000) The genetic organization and protein crystallographic structure of human serine hydroxymethyltransferase. Adv. Enzyme Regul. 40, 353–403 10.1016/S0065-2571(99)00035-7 [DOI] [PubMed] [Google Scholar]
- 20. Jain M., Nilsson R., Sharma S., Madhusudhan N., Kitami T., Souza A. L., Kafri R., Kirschner M. W., Clish C. B., and Mootha V. K. (2012) Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 336, 1040–1044 10.1126/science.1218595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim D., Fiske B. P., Birsoy K., Freinkman E., Kami K., Possemato R. L., Chudnovsky Y., Pacold M. E., Chen W. W., Cantor J. R., Shelton L. M., Gui D. Y., Kwon M., Ramkissoon S. H., Ligon K. L., et al. (2015) SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature 520, 363–367 10.1038/nature14363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang B., Wang W., Zhu Z., Zhang X., Tang F., Wang D., Liu X., Yan X., and Zhuang H. (2017) Mitochondrial serine hydroxymethyltransferase 2 is a potential diagnostic and prognostic biomarker for human glioma. Clin. Neurol. Neurosurg. 154, 28–33 10.1016/j.clineuro.2017.01.005 [DOI] [PubMed] [Google Scholar]
- 23. Thorndike J., Pelliniemi T. T., and Beck W. S. (1979) Serine hydroxymethyltransferase activity and serine incorporation in leukocytes. Cancer Res. 39, 3435–3440 [PubMed] [Google Scholar]
- 24. Rao N. A., Talwar R., and Savithri H. S. (2000) Molecular organization, catalytic mechanism and function of serine hydroxymethyltransferase—a potential target for cancer chemotherapy. Int. J. Biochem. Cell Biol. 32, 405–416 10.1016/S1357-2725(99)00126-0 [DOI] [PubMed] [Google Scholar]
- 25. Pinthong C., Maenpuen S., Amornwatcharapong W., Yuthavong Y., Leartsakulpanich U., and Chaiyen P. (2014) Distinct biochemical properties of human serine hydroxymethyltransferase compared with the Plasmodium enzyme: implications for selective inhibition. FEBS J. 281, 2570–2583 10.1111/febs.12803 [DOI] [PubMed] [Google Scholar]
- 26. Sopitthummakhun K., Maenpuen S., Yuthavong Y., Leartsakulpanich U., and Chaiyen P. (2009) Serine hydroxymethyltransferase from Plasmodium vivax is different in substrate specificity from its homologues. FEBS J. 276, 4023–4036 10.1111/j.1742-4658.2009.07111.x [DOI] [PubMed] [Google Scholar]
- 27. Renwick S. B., Snell K., and Baumann U. (1998) The crystal structure of human cytosolic serine hydroxymethyltransferase: a target for cancer chemotherapy. Structure 6, 1105–1116 10.1016/S0969-2126(98)00112-9 [DOI] [PubMed] [Google Scholar]
- 28. Chitnumsub P., Ittarat W., Jaruwat A., Noytanom K., Amornwatcharapong W., Pornthanakasem W., Chaiyen P., Yuthavong Y., and Leartsakulpanich U. (2014) The structure of Plasmodium falciparum serine hydroxymethyltransferase reveals a novel redox switch that regulates its activities. Acta Crystallogr. D Biol. Crystallogr. 70, 1517–1527 10.1107/S1399004714005598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chitnumsub P., Jaruwat A., Riangrungroj P., Ittarat W., Noytanom K., Oonanant W., Vanichthanankul J., Chuankhayan P., Maenpuen S., Chen C. J., Chaiyen P., Yuthavong Y., and Leartsakulpanich U. (2014) Structures of Plasmodium vivax serine hydroxymethyltransferase: implications for ligand-binding specificity and functional control. Acta Crystallogr. D Biol. Crystallogr. 70, 3177–3186 10.1107/S1399004714023128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Scarsdale J. N., Kazanina G., Radaev S., Schirch V., and Wright H. T. (1999) Crystal structure of rabbit cytosolic serine hydroxymethyltransferase at 2.8 Å resolution: mechanistic implications. Biochemistry 38, 8347–8358 10.1021/bi9904151 [DOI] [PubMed] [Google Scholar]
- 31. Szebenyi D. M., Liu X., Kriksunov I. A., Stover P. J., and Thiel D. J. (2000) Structure of a murine cytoplasmic serine hydroxymethyltransferase quinonoid ternary complex: evidence for asymmetric obligate dimers. Biochemistry 39, 13313–13323 10.1021/bi000635a [DOI] [PubMed] [Google Scholar]
- 32. Usha R., Savithri H. S., and Rao N. A. (1992) Arginine residues involved in binding of tetrahydrofolate to sheep liver serine hydroxymethyltransferase. J. Biol. Chem. 267, 9289–9293 [PubMed] [Google Scholar]
- 33. Scarsdale J. N., Radaev S., Kazanina G., Schirch V., and Wright H. T. (2000) Crystal structure at 2.4 Å resolution of E. coli serine hydroxymethyltransferase in complex with glycine substrate and 5-formyl tetrahydrofolate. J. Mol. Biol. 296, 155–168 10.1006/jmbi.1999.3453 [DOI] [PubMed] [Google Scholar]
- 34. Trivedi V., Gupta A., Jala V. R., Saravanan P., Rao G. S., Rao N. A., Savithri H. S., and Subramanya H. S. (2002) Crystal structure of binary and ternary complexes of serine hydroxymethyltransferase from Bacillus stearothermophilus: insights into the catalytic mechanism. J. Biol. Chem. 277, 17161–17169 10.1074/jbc.M111976200 [DOI] [PubMed] [Google Scholar]
- 35. Girgis S., Nasrallah I. M., Suh J. R., Oppenheim E., Zanetti K. A., Mastri M. G., and Stover P. J. (1998) Molecular cloning, characterization and alternative splicing of the human cytoplasmic serine hydroxymethyltransferase gene. Gene 210, 315–324 10.1016/S0378-1119(98)00085-7 [DOI] [PubMed] [Google Scholar]
- 36. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 10.1016/S0006-3495(00)76713-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brown P. H., and Schuck P. (2006) Macromolecular size-and-shape distributions by sedimentation velocity analytical ultracentrifugation. Biophys. J. 90, 4651–4661 10.1529/biophysj.106.081372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., and Svergun D. I. (2003) PRIMUS—a Windows-PC based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 10.1107/S0021889803012779 [DOI] [Google Scholar]
- 39. Franke D., and Svergun D. I. (2009) DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 10.1107/S0021889809000338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Volkov V. V., and Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 10.1107/S0021889803000268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pahari S., Sun L., and Alexov E. (2019) PKAD: a database of experimentally measured pKa values of ionizable groups in proteins. Database 2019, baz024 10.1093/database/baz024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Montioli R., Zamparelli C., Borri Voltattorni C., and Cellini B. (2017) Oligomeric state and thermal stability of apo- and holo-human ornithine δ-aminotransferase. Protein J. 36, 174–185 10.1007/s10930-017-9710-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yuvaniyama J., Chitnumsub P., Kamchonwongpaisan S., Vanichtanankul J., Sirawaraporn W., Taylor P., Walkinshaw M. D., and Yuthavong Y. (2003) Insights into antifolate resistance from malarial DHFR-TS structures. Nat. Struct. Biol. 10, 357–365 10.1038/nsb921 [DOI] [PubMed] [Google Scholar]
- 44. Sawaya M. R., and Kraut J. (1997) Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: crystallographic evidence. Biochemistry 36, 586–603 10.1021/bi962337c [DOI] [PubMed] [Google Scholar]
- 45. McGillewie L., Ramesh M., and Soliman M. E. (2017) Sequence, structural analysis and metrics to define the unique dynamic features of the flap regions among aspartic proteases. Protein J. 36, 385–396 10.1007/s10930-017-9735-9 [DOI] [PubMed] [Google Scholar]
- 46. Stark M., Wichman C., Avivi I., and Assaraf Y. G. (2009) Aberrant splicing of folylpolyglutamate synthase as a novel mechanism of antifolate resistance in leukemia. Blood 113, 4362–4369 10.1182/blood-2008-08-173799 [DOI] [PubMed] [Google Scholar]
- 47. Wojtuszkiewicz A., Raz S., Stark M., Assaraf Y. G., Jansen G., Peters G. J., Sonneveld E., Kaspers G. J., and Cloos J. (2016) Folylpolyglutamate synthetase splicing alterations in acute lymphoblastic leukemia are provoked by methotrexate and other chemotherapeutics and mediate chemoresistance. Int. J. Cancer 138, 1645–1656 10.1002/ijc.29919 [DOI] [PubMed] [Google Scholar]
- 48. Jaturontakul K., Jatuyosporn T., Laohawutthichai P., Kim S. Y., Mori T., Supungul P., Hakoshima T., Tassanakajon A., and Krusong K. (2017) Molecular characterization of viral responsive protein 15 and its possible role in nuclear export of virus in black tiger shrimp Penaeus monodon. Sci. Rep. 7, 6523–6534 10.1038/s41598-017-06653-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hopkins J. B., Gillilan R. E., and Skou S. (2017) BioXTAS RAW: improvements to a free open-source program for small-angle x-ray scattering data reduction and analysis. J. Appl. Crystallogr. 50, 1545–1553 10.1107/S1600576717011438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 10.1107/S0021889892001663 [DOI] [Google Scholar]
- 51. Franke D., Petoukhov M. V., Konarev P. V., Panjkovich A., Tuukkanen A., Mertens H. D. T., Kikhney A. G., Hajizadeh N. R., Franklin J. M., Jeffries C. M., and Svergun D. I. (2017) ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 50, 1212–1225 10.1107/S1600576717007786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mylonas E., Dmitri I., and Svergun D. I. (2007) Accuracy of molecular mass determination of proteins in solution by small-angle X-ray scattering. J. Appl. Crystallogr. 40, s245–s249 10.1107/S002188980700252X [DOI] [Google Scholar]
- 53. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Trewhella J., Duff A. P., Durand D., Gabel F., Guss J. M., Hendrickson W. A., Hura G. L., Jacques D. A., Kirby N. M., Kwan A. H., Pérez J., Pollack L., Ryan T. M., Sali A., Schneidman-Duhovny D., et al. (2017) 2017 publication guidelines for structural modelling of small-angle scattering data from biomolecules in solution: an update. Acta Crystallogr. D Struct. Biol. 73, 710–728 10.1107/S2059798317011597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Case D. A., Betz R. M., Cerutti D. S., Cheatham T. E. III, Darden T. A., Duke R. E., Giese T. J., Gohlke H., Goetz A. W., Homeyer N., Izadi S., Janowski P., Kaus J., Kovalenko A., Lee T. S., et al. (2016) AMBER 2016, University of California, San Francisco [Google Scholar]
- 56. Dolinsky T. J., Nielsen J. E., McCammon J. A., and Baker N. A. (2004) PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667 10.1093/nar/gkh381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kaiyawet N., Rungrotmongkol T., and Hannongbua S. (2013) Effect of halogen substitutions on dUMP to stability of thymidylate synthase/dUMP/mTHF ternary complex using molecular dynamics simulation. J. Chem. Inf. Model. 53, 1315–1323 10.1021/ci400131y [DOI] [PubMed] [Google Scholar]
- 58. Maier J. A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K. E., and Simmerling C. (2015) ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 10.1021/acs.jctc.5b00255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang J., Wolf R. M., Caldwell J. W., Kollamn P. A., and Case D. A. (2004) Development and testing of a general Amber force field. J. Comput. Chem. 25, 1157–1174 10.1002/jcc.20035 [DOI] [PubMed] [Google Scholar]
- 60. Mahalapbutr P., Chusuth P., Kungwan N., Chavasiri W., Wolschann P., and Rungrotmongkol T. (2017) Molecular recognition of naphthoquinone-containing compounds against human DNA topoisomerase IIα ATPase domain: a molecular modeling study. J. Mol. Liq. 247, 374–385 10.1016/j.molliq.2017.10.021 [DOI] [Google Scholar]
- 61. Lavinder J. J., Hari S. B., Sullivan B. J., and Magliery T. J. (2009) High-throughput thermal scanning: a general, rapid dye-binding thermal shift screen for protein engineering. J. Am. Chem. Soc. 131, 3794–3795 10.1021/ja8049063 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.