

Abstract
DNA–protein cross-links can interfere with chromatin architecture, block DNA replication and transcription, and interfere with DNA repair. Here we synthesized a DNA 23-mer containing a site-specific DNA–peptide cross-link (DpC) by cross-linking an 11-mer peptide to the DNA epigenetic mark 5-formylcytosine in synthetic DNA and used it to generate a DpC-containing plasmid construct. Upon replication of the DpC-containing plasmid in HEK 293T cells, approximately 9% of progeny plasmids contained targeted mutations and 5% semitargeted mutations. Targeted mutations included C→T transitions and C deletions, whereas semitargeted mutations included several base substitutions and deletions near the DpC lesion. To identify DNA polymerases involved in DpC bypass, we comparatively studied translesion synthesis (TLS) efficiency and mutagenesis of the DpC in a series of cell lines with TLS polymerase knockouts or knockdowns. Knockdown of either hPol ι or hPol ζ reduced the mutation frequency by nearly 50%. However, the most significant reduction in mutation frequency (50%–70%) was observed upon simultaneous knockout of hPol η and hPol κ with knockdown of hPol ζ, suggesting that these TLS polymerases play a critical role in error-prone DpC bypass. Because TLS efficiency of the DpC construct was not significantly affected in TLS polymerase–deficient cells, we examined a possible role of replicative DNA polymerases in their bypass and determined that hPol δ and hPol ϵ can accurately bypass the DpC. We conclude that both replicative and TLS polymerases can bypass this DpC lesion in human cells but that mutations are induced mainly by TLS polymerases.
Keywords: DNA damage, DNA damage response, DNA polymerase, mutagenesis, mutagenesis mechanism, 5-formylcytosine, DNA–peptide cross-link, translesion synthesis
Introduction
DNA is continuously exposed to a wide range of exogenous and endogenous DNA-damaging agents (1–3). DNA–protein cross-links (DPCs)2 are formed when proteins become covalently bound to DNA (4, 5). Although DPC formation in cells was first reported as early as in the 1960s (6), their effects on biological processes have not been recognized until recently. DPCs can be induced by exposure to various anti-tumor drugs, transition metals, and UV light or can form endogenously as a result of normal cellular processes such as lipid peroxidation, histone demethylation, DNA replication, transcription, and DNA repair (4, 5, 7–11).
It was recently discovered that the endogenously occurring DNA epigenetic mark 5-formylcytosine forms reversible Schiff base conjugates with histone proteins; these can be reductively stabilized to form stable amino conjugates (12, 13). 5-Formylcytosine (5fC) is one of the four epigenetically modified cytosine bases endogenously present in mammalian genomes (14–17). Low levels of 5fC bases, formed by oxidation of 5-methylcytosine, have been detected in all mammalian tissues (14–16). 5fC forms reversible Schiff base cross-links with histone proteins both in vitro and in human cells, a process that is thought to influence gene expression levels (12, 13).
DPCs have the ability to interfere with chromatin architecture (18), block DNA replication and transcription, interfere with DNA repair, and induce mutations and toxicity (4, 5, 7, 8, 19–21). As a result, DPCs contribute to a number of human ailments, including cancer and aging (22–25). Initial steps of DPC repair are thought to involve proteolytic digestion of the cross-linked protein to a smaller peptide fragment, which is subsequently bypassed by translesion synthesis (TLS) polymerases or repaired by nucleotide excision repair (NER) proteins (20, 26, 27).
Many previous studies of DPCs have been focused on their repair (reviewed in Ref. 28). Notably, the recent discovery of a mammalian protease, SPRTN (SprT-like N-terminal domain), required for resolving DNA-protein cross-links in vivo, initiated many published studies, as its function is compromised in Ruijs–Aalfs syndrome patients suffering from accelerated aging and an increased incidence of liver cancer (29). Several canonical DNA repair pathways, including the NER, homologous recombination, and Fanconi anemia pathways, are thought to be involved in DPC repair either with or without proteolytic processing to smaller peptide lesions (19, 27, 30).
In comparison, relatively little is known about the interactions of DPCs with DNA replication machinery. Although in the early 1990s DPC formation and mutagenesis in mammalian cells were shown in one study (31), the majority of the reports published subsequently either failed to link formation of DPCs with mutations (32, 33) or suggested that DPCs may not induce mutagenesis in mammalian cells (34). More recent research using advanced tools, however, established that DPCs containing large proteins completely block replicative bypass but that peptide-containing DpCs allow TLS (19, 35–41).
Our previous studies with a model DpC have established a key role of TLS polymerases, such as human Pol κ and η, in catalyzing DNA replication past DpC lesions (39). Unlike replicative polymerases, whose tight active sites undergo conformational changes upon binding the correct dNTP, TLS polymerases of the Y family have more open active sites that enable them to bypass bulky DNA lesions (42, 43). We found that 5fC conjugated to histones H2A or H4 completely inhibited DNA replication in vitro but that translesion synthesis (TLS) by Y family polymerases occurred when the proteins were subjected to proteolytic digestion (40). 5fC-mediated cross-links to 11-mer or 31-mer peptides were bypassed by human DNA polymerase η and κ (hPol η and κ) in an error-prone manner, inducing targeted C→T transitions and −1 deletions. Similar mutations also occurred when a plasmid containing the 11-mer DNA–peptide cross-link (DpC) is replicated in HEK 293T cells (40).
The main purpose of this work was to establish the roles of the TLS DNA polymerases in both error-free and error-prone bypass of DpCs in living cells. We examined replication of a plasmid containing a site-specific, structurally defined cross-link between 5fC and an 11-mer peptide in HEK 293T cells subjected to knockout and/or knockdown of one or more TLS polymerase(s). To focus on replication rather than repair, we employed a single-stranded plasmid containing the 5fC–cross-linked DpC, as DNA damages in single-stranded DNA are refractory to repair by most common types of DNA repair pathways. We have also investigated the ability of human replicative DNA polymerases δ and ϵ (hPol δ and ϵ) to bypass this DpC lesion in vitro.
Results
Replication of 5fC-linked DpC in human cells
The DNA 23-mer (5′-AGG GTT TTC CXA GTC ACG ACG TT-3′) containing 5fC at the 11th position from the 5′ end (X) was site-specifically conjugated to the lysine residue of the 11-mer peptide (RPKPQQFFGLM-CONH2) in the presence of a reducing agent (NaCNBH3), as reported previously (Scheme 1 and Fig. S1) (12). This particular peptide sequence had been used in previous studies that focused on in vitro replication by TLS polymerases (40). Our methodology generates stable DNA–polypeptide cross-links structurally analogous to Schiff base conjugates formed between 5fC and histone proteins in human cells (12, 13).
Scheme 1.

General procedure for the preparation of a DpC conjugated to the 5fC in DNA.
The resulting DpC-containing 23-mer oligonucleotide (5fC-DpC in Scheme 1) was ligated into a gapped pMS2 plasmid (Scheme 2). The DpC construct and an unmodified plasmid (containing a different sequence at the ligation site) were co-transfected into HEK 293T cells. The unmodified vector served as an internal control of transfection efficiency. Following 24-h incubation to allow for one round of replication, plasmid DNA was isolated and used to transform Escherichia coli DH10B cells (Scheme 2). The resultant colonies were analyzed by oligonucleotide hybridization followed by DNA sequencing. TLS efficiency was determined as the percentages of the colonies originating from the lesion-containing plasmid relative to the internal plasmid control.
Scheme 2.

Schematic representation of plasmid construction, replication, and progeny analysis.
In HEK 293T cells, the replication efficiency of the DpC-containing plasmid was 98.5% ± 1% compared with an unmodified control plasmid (set as 100%) (Fig. 1). We next examined the effects of 5fC DpCs on DNA replication in cells deficient in various TLS polymerases (hPol η, hPol κ, hPol ι, and hPol ζ). Surprisingly, the TLS efficiency did not change upon knockout of hPol η and hPol κ or knockdown of hPol ι and hPol ζ (Fig. 1). TLS remained above 93% even following double and triple knockout/knockdown of TLS polymerases (Fig. 1). This indicates that TLS polymerases are not required for bypass of 5fC DpCs in human cells and that it can be bypassed by replicative DNA polymerases.
Figure 1.
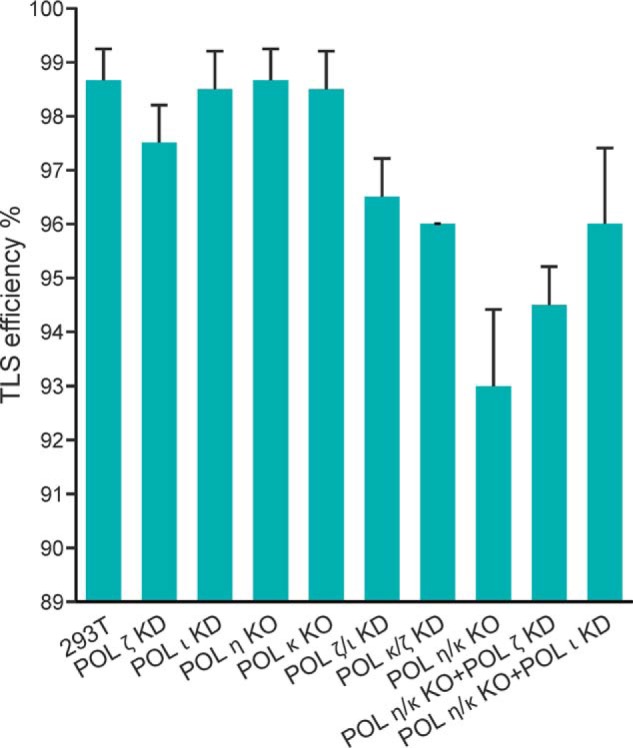
The extent of replicative bypass (or TLS efficiency) of 5fC-linked DpC in HEK 293T cells with or without siRNA knockdowns of various TLS polymerases. Percent TLS of the DpC construct in different polymerase knockout or knockdowns was measured relative to an internal control DNA. The data represent the mean and standard deviation of results from three independent experiments. HEK 293T cells were treated with negative control siRNA (293T), whereas the other single or double polymerase knockouts or knockdowns (KD) are indicated below on the x axis.
Mutations induced by this DpC and the role of TLS polymerases
Nearly 14% of plasmid progeny recovered from HEK 293T cells transfected with the DpC-bearing vector were found to be mutants, which included 9% targeted and 5% semitargeted base changes and frameshift mutations (Fig. 2). We define semitargeted mutations as base substitutions and deletions detected near the peptide-conjugated 5fC even though the lesion might have been replicated correctly as a template cytosine. Targeted mutations were comprised of 6.7% C→T transitions, 0.6% C→G transversions, and 1.3% targeted C deletions (Fig. 3). Knockout or knockdown of TLS polymerases resulted in reduced mutation frequency (MF) upon replication past DpCs, which was most pronounced upon knockdown of either hPol ι or hPol ζ (Figs. 2 and 3). Targeted mutations were reduced from 8.6% ± 1.9% in HEK 293T cells to 2.9% ± 0.4% in hpol ι knockdown cells, a reduction of 50%–80%. Likewise, targeted mutations dropped from 8.6% ± 1.9% in HEK 293T cells to 3.2% ± 0.6% in hpol ζ knockdown cells, a reduction of 45%–75%. Similarly, C→T mutations were reduced by 40%–80% and 30%–70% upon single knockdown of hPol ι and hPol ζ, respectively (Fig. 3). However, further reduction in MF was not achieved upon simultaneous knockdown of hPol ι and hPol ζ (Figs. 2 and 3). Of the various double and triple knockout/knockdown experiments, the greatest reduction in MF occurred upon simultaneous knockout of hPol η and hPol κ along with knockdown of hPol ζ. In the latter case, the total MF was reduced from 13.5% ± 1.7% in HEK 293T cells to 4.8% ± 0.7% in cells carrying hPol η/κ knockout plus hPol ζ knockdown, and the incidence of C→T transitions dropped from 5.9% ± 1.4% to 1.4% ± 0.2% for the same (Fig. 3). Taken together, the results from TLS polymerase knockout/knockdown experiments are indicative of an involvement of TLS polymerases hPol ι, hPol ζ, hPol η, and hPol κ in error-prone bypass of 5fC-mediated DpC lesions in human cells.
Figure 2.
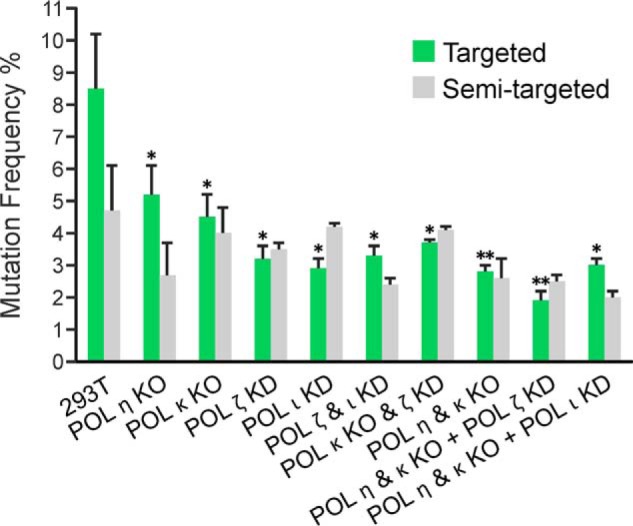
Targeted and semitargeted mutations induced in the progeny from the 5fC-linked DpC construct in HEK 293T (293T) and various polymerase KO or knockdown cells (as indicated). The data represent the mean and standard deviation (of the total MF) from two to five independent experiments. The statistical significance of the difference in MFs between HEK 293T and TLS Pol knockouts and/or knockdowns (KD) was calculated using two-tailed, unpaired Student's t test (*, p < 0.05; **, p < 0.005).
Figure 3.
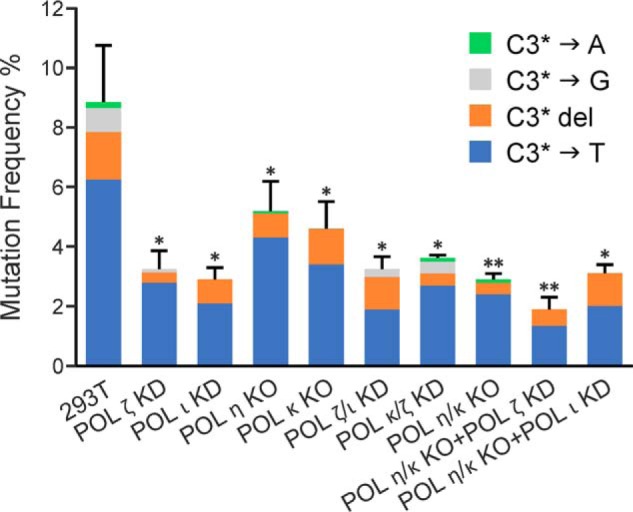
The types and frequencies of targeted mutations induced in the progeny from the 5fC-linked DpC construct in HEK 293T cells and various polymerase KO or knockdown cells (as indicated). The data represent the mean and standard deviation (of the total targeted MF) from two to five independent experiments. The statistical significance of the difference in targeted MFs between HEK 293T and TLS Pol knockouts and/or knockdowns (KD) was calculated using two-tailed, unpaired Student's t test (*, p < 0.05; **, p < 0.005).
Semitargeted base substitutions and one to six base deletions around the DpC site comprised ∼33% of total mutations in HEK 293T cells (Fig. 2). Nearly half of the semitargeted mutations were deletions of one to six bases, whereas insertion mutations did not occur. Of the various semitargeted base substitutions, C→T at several different cytosine sites were predominant. Upon knockout and knockdown of TLS polymerases, the frequency of semitargeted mutations decreased in some cases, but no clear pattern emerged, and the changes were not statistically significantly different (Fig. 2 and Fig. S4). In some knockout/knockdown experiments, the percentage of semitargeted mutations increased to more than 50% of the total mutations (Fig. S4).
In vitro replication of a DpC lesion by replicative DNA polymerases δ and ϵ
Our observation of efficient replicative bypass of the 5fC-linked DpC lesion in HEK 293T cells with radically reduced levels of TLS polymerases (Fig. 1) suggests that these specialized polymerases are not required for DpC lesion bypass in human cells. Therefore, we investigated the possibility that this bulky adduct can be bypassed by replicative DNA polymerases δ and ϵ. Standing-start in vitro primer extension assays were conducted in the presence of hPol ϵ (Fig. 4, left panel) and hPol δ (Fig. 4, right panel). Primer–template complexes were created by annealing 32P-labeled 12-mer primers to the 23-mer template containing 5fC (Fig. 4, A and C) or the 11-mer peptide DpC (Fig. 4, B and D). In vitro replication was initiated by the addition of the corresponding DNA polymerase and a mixture of dNTPs. Primer extension reactions were quenched at selected time points prior to loading onto 20% denaturing polyacrylamide gel.
Figure 4.

Primer extension assays for replication bypass of 5fC-mediated DNA–peptide cross-links by hPol ϵ and hPol δ. A–D, the 32P-labeled 12-mer primer was annealed with a 23-mer template containing 5-formyl-dC (A and C) or the 11-mer peptide cross-link RPKPQQFFGLM (B and D). Polymerase reactions were initiated by addition of DNA polymerases and a mixture of dNTP and quenched at selected time points prior to loading onto 20% denaturing PAGE.
As shown in Fig. 4, left panel, recombinant hPol ϵ was able to extend the primer past unconjugated 5fC to form a full-length 23-mer product, although a significant fraction of the primer remained unchanged, even after a 30-min reaction. Primer extension using the DpC template was even less efficient, but a small part of the full-length product was formed in 5 min, which gradually increased over a 30-min time period. In comparison, hPol δ–catalyzed replication of the control template was significantly more efficient, with more than 95% of the primer extended to the full-length product in 5 min (Fig. 4, right panel). Primer extension using the DpC-containing template was much slower, but the band corresponding to the full-length product continued to increase during the 30-min incubation. This suggests that both hPol ϵ and hPol δ can bypass the 5fC-conjugated DpC, albeit more slowly than the 5fC control. In addition, both replicative polymerases exhibited exonuclease activity, so a significant portion of the primer was converted to smaller products at longer incubation times (results not shown).
Next, single-nucleotide insertion experiments were conducted to investigate the fidelity of hPol ϵ and hPol δ upon in vitro replication of the DpC-containing template. For hPol ϵ, the correct nucleotide (dGTP) was preferentially incorporated opposite both DpC and the 5fC control (Fig. 5). Gel electrophoresis analyses revealed that three successive dGs were incorporated because of the 3′-XCC sequence of the template strand.
Figure 5.

Single-nucleotide insertion opposite 5fC (control) and the 5fC–11-mer peptide cross-links by hPol ϵ. Template–primer complexes were incubated with hPol ϵ in the presence of individual dNTP and then quenched at preselected time points. X = 5-formylcytosine conjugate to RPKPQQFFGLM.
Although low levels of dTTP and dATP misincorporation were noted opposite 5fC, no noticeable misincorporation opposite DpC was detected. Similar results were obtained for hPol δ; dGTP was the only nucleotide inserted opposite DpC, whereas dGTP and low levels of dATP were incorporated opposite the 5fC control (Fig. 6). To determine the catalytic efficiency of dGTP incorporation by hPol δ opposite DpC and 5fC, steady-state kinetic experiments were conducted (Table 1). The same primer–substrate complexes were used, and the primer extension reactions were conducted in the presence of hPol δ with increasing concentrations of dGTP (5–500 μm) (Fig. S2). To ensure steady-state conditions, a 9–90 molar excess of DNA over hPol δ was employed. Steady-state kinetic parameters were determined from Michaelis–Menten curves (Fig. S3). kcat/Km values for dGTP incorporation opposite 5fC and DpC were calculated as 0.056 and 0.008 μm−1 min−1, respectively, indicating that the presence of the peptide lesion significantly inhibits DNA polymerase activity. Overall, these results reveal the ability of replicative DNA polymerases to catalyze error-free nucleotide addition opposite 5fC derived DpC lesions. Perhaps this is not surprising, as such lesions routinely form at epigenetic marks in human cells (12) and, thus, must be tolerated by the cellular machinery.
Figure 6.

Single-nucleotide insertion opposite 5fC (control) and the 5fC–11-mer peptide cross-link by hPol δ. Template–primer complexes were incubated with hPol δ in the presence of individual dNTP and then quenched at preselected time points. X = 5-formylcytosine conjugate to RPKPQQFFGLM.
Table 1.
Steady-state kinetics parameters for single-nucleotide incorporation opposite 5fC or the 5fC–11-mer peptide (RPKPQQFFGLM) conjugated to the C5 position of cytosine by hPol δ
| Polymerase | Template | Incoming nucleotide | kcat (min−1) | Km (μM) | kcat/Km (μM−1 min−1) |
|---|---|---|---|---|---|
| hPol δ | 5fC | dGTP | 1.30 ± 0.08 | 23 ± 5 | 0.056 ± 0.013 |
| 11-mer peptide | dGTP | 0.13 ± 0.007 | 17 ± 4 | 0.008 ± 0.002 |
Discussion
DPC formation in cells was first reported as early as in the 1960s (6). In the early 1990s, it was shown that vanadate (VO3−) produces DPCs and causes mutations in mammalian cells (31). Most other reports in the latter part of the 20th century, however, either failed to associate formation of DPCs with mutations (32, 33) or indicated that DPCs cause cytotoxicity and clastogenicity but may not induce mutagenesis in mammalian cells (34). Research in the last two decades, in contrast, unambiguously established that DPCs containing large proteins pose strong blocks of DNA replication, whereas certain peptide-containing DpCs allow error-prone replicative bypass both in vitro and in vivo (19, 35–40). For example, Lloyd and co-workers (36, 45) have shown that DpCs covalently linked to the N2 position of dG blocked DNA replication, whereas the same peptide conjugated to the N6 position of dA was bypassed and induced small number of mutations in both E. coli and simian kidney cells. In vitro studies showed that E. coli Pol II, Pol III, and Pol V cannot replicate past these DpCs but that pol IV is reasonably proficient in their bypass (36). Moreover, Pol κ is highly efficient and accurate in TLS of the dG-N2-DpCs, as it is with other dG-N2 adducts (36). Indeed, the types and frequencies of DPC-induced mutations appear to be strongly dependent on the site and structure of the cross-links as well as the types of cells in which they are replicated. We have shown that site-specifically modified templates containing 10- to 20-mer peptides conjugated to uracil, 7-deazaguanine, or 5fC, unlike full-length protein DPCs, are subject to bypass by Y family DNA polymerases. Although proteins and large peptides (>20 amino acids) conjugated to the C5 position of dT completely blocked Pol η and κ, smaller C5-dT DpC conjugates (<10-mer peptides) were bypassed in an error-prone manner, giving rise to large numbers of deletions and point mutations (37, 46).
Depletion of one or more TLS DNA polymerases has become an attractive approach to determine their role in lesion bypass, which we employed in this study. A limitation, however, was that we were able to generate only a limited number of knockout cell lines and used the siRNA knockdown approach for the rest. A strict comparative quantitative analysis, therefore, is unwarranted. Even so, the critical role of TLS polymerases in the mutagenic outcome was evident.
In one of our earlier reports, TLS efficiency of a model DpC containing a 10-mer Myc peptide covalently linked to C7 of 7-deaza-dG was 76% in HEK 293T cells compared with 100% progeny derived from the control DNA, suggesting that this lesion slowed down DNA replication (39). In contrast, DpCs conjugated to the C5 position of 5fC examined in this study (Scheme 1) allowed a TLS efficiency comparable with that of the 5fC control in HEK 293T cells (Fig. 1). Additionally, TLS efficiency was barely altered in cells with knockout or knockdown of TLS polymerases, suggesting that none of these polymerases were essential for TLS of the C5–5fC–conjugated DpC lesion (Fig. 1). This led us to hypothesize that C5–5fC DpC–containing DNA may be bypassed by replicative DNA polymerases. Indeed, in vitro replication experiments with recombinant hPol δ and hPol ϵ revealed that both polymerases can catalyze DNA synthesis past the 5fC-linked 11-mer peptide lesion, albeit more slowly than 5fC. Remarkably, nucleotide incorporation opposite the DpC by hPol δ and hPol ϵ was accurate, and no nucleotide misincorporation opposite the lesion was detected (Figs. 5 and 6 and Table 1). Overall, our results indicate that hPol δ and hPol ϵ can bypass the C5–5fC–conjugated DpC in an error-free manner. However, because of the low efficiency of bypass (Table 1), TLS polymerases are likely to be recruited to the replication fork, leading to nucleotide misincorporations. The mechanism of nucleotide misincorporation can be rationalized from our recent molecular dynamics simulations of the hPol η-DNA ternary complexes, which revealed that the 11-mer peptide can be accommodated in the DNA major groove side. It also showed that the adducted 5fC forms a stable pair with the incoming dATP via wobble base-pairing in the polymerase active site, thereby causing C→T transitions (40). The targeted C deletion, on the other hand, can be explained by a slippage mechanism, when the incoming dG opposite the adducted 5fC realigns to the 5′-adjacent C, thereby causing the one-base targeted deletion (40). The cellular mutagenesis results for 5fC-induced DpCs differ from our earlier findings for DpCs conjugated to the C7 of 7-deaza-dG (39). In that study, replication of DpC-containing plasmids produced both targeted (20% targeted G→A and G→T) and semitargeted (15%) mutations in HEK 293T cells (39). TLS efficiency and targeted mutations were reduced upon siRNA knockdown of pol η, pol κ, or pol ζ, indicating that they participated in error-prone bypass of 7-deaza-dG-linked DpC lesions (39). However, the semitargeted mutations at G5 were only reduced upon knockdown of pol ζ, suggesting its critical role in this type of mutations (39). Even though targeted and semi-targeted mutations were observed in both studies, each structurally distinct DpC exhibited a unique spectrum of mutations. We postulate that the semitargeted mutations induced by DpCs arise from local destabilization of the DNA helix in the presence of bulky DpC lesions and from interference of nucleotide incorporations near the lesion site by the long arm of the peptide, which can adopt many different conformations. Future structural studies will likely shed light on the validity of this hypothesis and the mechanistic details of semitargeted mutagenesis by the DpCs.
Although this study was conducted with the 5fC-DpC situated in single-stranded DNA, which is not repaired by most DNA repair systems, immediately after TLS, the DpC-containing plasmid may become a substrate for DNA repair. 5fC itself is repaired by base excision repair (44), whereas the 5fC-DpC conjugate is likely to be an NER substrate (20, 26, 27). In this study, we did not attempt to determine the role of DNA repair in mutagenesis, but, in the future, it would be of interest to determine the interplay of DNA repair with mutagenesis induced by this DpC.
In conclusion, our results indicate that polymerase bypass of 5fC-conjugated DpC lesions by TLS polymerases gives rise to mutations in human cells. Even for such bulky DNA adducts, the specialized bypass polymerases of the Y and B families are not required for bypass of the 5fC–polypeptide cross-link. Replicative hPol δ and hPol ϵ could bypass the 5fC-linked 11-mer peptide lesion in an error-free manner. However, because of the low efficiency of DpC bypass by hPol δ and hPol ϵ, TLS polymerases are recruited to the replication fork, leading to nucleotide misincorporations and mutagenesis in human cells.
Experimental procedures
All materials, including reagents and solvents, were of commercial grade. [γ-32P]ATP was from PerkinElmer Life Sciences (Shelton, CT). The enzymes were purchased from New England Biolabs (Beverly, MA). hPol ϵ was purchased from Enzymax (Lexington, KY). hPol δ was prepared as described previously (47).
Synthesis and characterization of 23-mer DpC with an 11-mer peptide cross-linked to 5fC
A 23-mer oligodeoxynucleotide (5′-AGG GTT TTC CXA GTC ACG ACG TT-3′), where X represents 5fC covalently attached to an 11-mer peptide, RPKPQQFFGLM-CONH2, was synthesized and characterized as described previously (12). The procedure for preparation is outlined briefly in Scheme 1, and its characterization and purity are shown in Fig. S1. The aldehyde group of 5fC forms a reversible Schiff base with the side chain of the third amino acid, Lys, of the polypeptide, which was stabilized by NaCNBH3 reduction. The oligonucleotide–polypeptide conjugate was purified by electrophoresis on a 20% polyacrylamide gel containing 7 m urea (denaturing PAGE), followed by Sep-Pac C18 SPE desalting and characterized by MS.
Construction of TLS polymerase knockout cell lines
For Pol η and Pol κ gene knockouts in HEK 293T cells, commercially available CRISPR/Cas9 knockout plasmids were used (Santa Cruz Biotechnology, sc-406518 for Pol η and sc-405052 for Pol κ). Following plasmid transfection, single cells were sorted by FACS into 96-well plates using a BD FACSAria II instrument. After clonal expansion, the resulting monoclonal cultures were screened by Western blotting for protein levels (Fig. S2). We also evaluated two different clones of Pol η knockout as well as Pol η/Pol κ double knockout cell lines by comparing the MF induced by 5fC-DpC, which was found statistically to be the same. This suggests that the CRISPR/Cas9 approach did not induce any off-target effects in the knockout cells lines that would influence the TLS and mutagenesis results.
Construction and characterization of a pMS2 vector containing a single DpC and its replication in HEK 293T cells
The DpC-containing plasmid pMS2, which carries neomycin and ampicillin resistance genes, was constructed according to published reports (39, 40) The pMS2 construct (50 ng) in 6 μl of Lipofectamine cationic lipid reagent (Invitrogen) was used to transfect HEK 293T cells after they were grown to ∼90% confluency. Following transfection, the cells were grown at 37 °C in 5% CO2 for 48 h. The plasmid DNA was collected, purified, and used to transform E. coli DH10B. The transformants were analyzed by oligonucleotide hybridization followed by DNA sequence analysis (39).
Determination of TLS efficiency
DpC-containing or control pMS2 constructs were mixed with equal amounts of a single-stranded pMS2 DNA construct containing a 23-nt sequence different from the DpC (or control) DNA sequence. After transfection in HEK 293T cells followed by amplification in E. coli, oligonucleotide probes for both types of DNA were used to analyze the progeny. The altered DNA was used as an internal control, and it gave a number of progeny equal to the control construct. TLS efficiency was determined as the percentages of the colonies originating from the DpC-containing plasmid relative to the internal control. Mutational analyses of TLS products from human cells with polymerase knockdowns were performed as described previously. RT-PCR analysis and the Western blot procedure have been reported in our earlier publication (48).
Primer extension assays using human replicative DNA polymerases
32P-labeled primer–template complexes containing 5fC or 5fC–11-mer peptide cross-links (RPKPQQFFGLM) were prepared as described previously (40). These primer–template complexes (1 pmol) were incubated at 37 °C with all four dNTPs (1 mm final concentration) in a buffer containing 25 mm Tris/HCl (pH 7.5), 8 mm MgCl2, 8 mm NaCl, 5 mm DTT, 100 μg/ml BSA, and 10% glycerol (v/v) (total reaction volume, 30 μl). The polymerization reaction was initiated by addition of human DNA polymerases δ (0.56 pmol) or ϵ (0.6 pmol). Aliquots (4 μl) of the reaction mixtures were quenched with a gel loading buffer (20 mm EDTA in 95% formamide, including 0.05% bromphenol blue and xylene cyanol) at the 0-, 5-, 15-, and 30-min time points. The primer extension products were then resolved using a 20% (w/v) denaturing PAGE containing 7 m urea at 80 W for 2 h in 1× Tris borate-EDTA buffer and visualized using the Typhoon FLA 7000 PhosphorImager (GH Healthcare).
Single-nucleotide incorporation assays
32P-labeled primer–template complexes containing 5f-dC or 5fC–11-mer peptide cross-links (RPKPQQFFGLM, 1 pmol) were subjected to primer extension reactions at the same conditions as described above, except for addition of a single dNTP (dGTP, dCTP, dTTP, or dATP; 1 mm final concentration). The reaction mixtures were quenched, denatured, and imaged under the same conditions as described above.
Steady-state kinetics analysis
Steady-state kinetics for incorporation of dGTP opposite 5fC or the 5fC–11-mer peptide cross-link (RPKPQQFFGLM) were examined by performing single-nucleotide insertion assays in the presence of hPol δ and increasing concentrations of dGTP. Primer–template complexes (2.25 pmol) were incubated with hPol δ (0.025–0.25 pmol) in the presence of dGTP (5, 10, 25, 50, 100, 150, 250, or 500 μm) for specified time periods (0–30 min). Extension products were visualized using the Typhoon FLA 7000 system and quantified by volume analysis using the ImageQuant TL 8.0 software (GE Healthcare). Steady-state kinetic parameters were calculated by nonlinear regression analysis using one-site hyperbolic fits in Prism 4.0 (GraphPad Software, La Jolla, CA).
Author contributions
S. N., S. J., N. Y. T., and A. K. B. conceptualization; S. N., S. J., J. T., C. M. N., Z. Z., G.-L. M., N. Y. T., and A. K. B. data curation; S. N., S. J., J. T., C. M. N., M. L., Z. Z., G.-L. M., N. Y. T., and A. K. B. formal analysis; S. N., S. J., J. T., C. M. N., M. L., Z. Z., G.-L. M., N. Y. T., and A. K. B. investigation; S. N., S. J., J. T., C. M. N., M. L., Z. Z., G.-L. M., N. Y. T., and A. K. B. methodology; S. N., S. J., J. T., N. Y. T., and A. K. B. writing-original draft; S. N., Z. Z., G.-L. M., N. Y. T., and A. K. B. project administration; S. J., J. T., C. M. N., M. L., Z. Z., G.-L. M., N. Y. T., and A. K. B. validation; Z. Z., G.-L. M., N. Y. T., and A. K. B. resources; Z. Z., G.-L. M., N. Y. T., and A. K. B. supervision; N. Y. T. and A. K. B. funding acquisition.
Supplementary Material
Acknowledgment
We thank Robert Carlson (University of Minnesota) for help with the figures.
This work was supported by the NIEHS, National Institutes of Health Grants R01 ES-023350 (to N. Y. T. and A. K. B.) and R01 ES-026184 (to G-L.M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4.
- DPC
- DNA–protein cross-link
- 5fC
- 5-formylcytosine
- TLS
- translesion synthesis
- NER
- nucleotide excision repair
- hPol
- human DNA polymerase
- DpC
- DNA–peptide cross-link
- MF
- mutation frequency.
References
- 1. Singer B., and Grunberger D. (1981) Molecular Biology of Mutagens and Carcinogens, Plenum Press, New York [Google Scholar]
- 2. Basu A. K. (2018) DNA damage, mutagenesis and cancer. Int. J. Mol. Sci. 19, E970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wogan G. N., Hecht S. S., Felton J. S., Conney A. H., and Loeb L. A. (2004) Environmental and chemical carcinogenesis. Semin. Cancer Biol. 14, 473–486 10.1016/j.semcancer.2004.06.010 [DOI] [PubMed] [Google Scholar]
- 4. Barker S., Weinfeld M., and Murray D. (2005) DNA-protein crosslinks: their induction, repair, and biological consequences. Mutat. Res. 589, 111–135 10.1016/j.mrrev.2004.11.003 [DOI] [PubMed] [Google Scholar]
- 5. Tretyakova N. Y., Groehler A. 4th, and Ji S. (2015) DNA-protein cross-links: formation, structural identities, and biological outcomes. Acc. Chem. Res. 48, 1631–1644 10.1021/acs.accounts.5b00056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Smith K. C., Hodgkins B., and O'Leary M. E. (1966) The biological importance of ultraviolet light induced DNA-protein crosslinks in Escherichia coli 15 TAU. Biochim. Biophys. Acta 114, 1–15 10.1016/0005-2787(66)90248-6 [DOI] [PubMed] [Google Scholar]
- 7. Speit G., Schütz P., and Merk O. (2000) Induction and repair of formaldehyde-induced DNA-protein crosslinks in repair-deficient human cell lines. Mutagenesis 15, 85–90 10.1093/mutage/15.1.85 [DOI] [PubMed] [Google Scholar]
- 8. Barker S., Weinfeld M., Zheng J., Li L., and Murray D. (2005) Identification of mammalian proteins cross-linked to DNA by ionizing radiation. J. Biol. Chem. 280, 33826–33838 10.1074/jbc.M502477200 [DOI] [PubMed] [Google Scholar]
- 9. Wu F. Y., Chang P. W., Wu C. C., and Kuo H. W. (2002) Correlations of blood lead with DNA-protein cross-links and sister chromatid exchanges in lead workers. Cancer Epidemiol. Biomarkers Prev. 11, 287–290 [PubMed] [Google Scholar]
- 10. Chválová K., Brabec V., and Kaspárková J. (2007) Mechanism of the formation of DNA-protein cross-links by antitumor cisplatin. Nucleic Acids Res. 35, 1812–1821 10.1093/nar/gkm032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Voitkun V., and Zhitkovich A. (1999) Analysis of DNA-protein crosslinking activity of malondialdehyde in vitro. Mutat. Res. 424, 97–106 10.1016/S0027-5107(99)00011-1 [DOI] [PubMed] [Google Scholar]
- 12. Ji S., Shao H., Han Q., Seiler C. L., and Tretyakova N. Y. (2017) Reversible DNA-protein cross-linking at epigenetic DNA marks. Angew. Chem. Int. Ed. Engl. 56, 14130–14134 10.1002/anie.201708286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li F., Zhang Y., Bai J., Greenberg M. M., Xi Z., and Zhou C. (2017) 5-Formylcytosine yields DNA-protein cross-links in nucleosome core particles. J. Am. Chem. Soc. 139, 10617–10620 10.1021/jacs.7b05495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ito S., Shen L., Dai Q., Wu S. C., Collins L. B., Swenberg J. A., He C., and Zhang Y. (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303 10.1126/science.1210597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pfaffeneder T., Hackner B., Truss M., Münzel M., Münzel M., Deiml C. A., Hagemeier C., and Carell T. (2011) The discovery of 5-formylcytosine in embryonic stem cell DNA. Angew. Chem. Int. Ed. Engl. 50, 7008–7012 10.1002/anie.201103899 [DOI] [PubMed] [Google Scholar]
- 16. Bachman M., Uribe-Lewis S., Yang X., Burgess H. E., Iurlaro M., Reik W., Murrell A., and Balasubramanian S. (2015) 5-Formylcytosine can be a stable DNA modification in mammals. Nat. Chem. Biol. 11, 555–557 10.1038/nchembio.1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Song C. X., Szulwach K. E., Dai Q., Fu Y., Mao S. Q., Lin L., Street C., Li Y., Poidevin M., Wu H., Gao J., Liu P., Li L., Xu G. L., Jin P., and He C. (2013) Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell 153, 678–691 10.1016/j.cell.2013.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nackerdien Z., Rao G., Cacciuttolo M. A., Gajewski E., and Dizdaroglu M. (1991) Chemical nature of DNA-protein cross-links produced in mammalian chromatin by hydrogen peroxide in the presence of iron or copper ions. Biochemistry 30, 4873–4879 10.1021/bi00234a006 [DOI] [PubMed] [Google Scholar]
- 19. Ide H., Shoulkamy M. I., Nakano T., Miyamoto-Matsubara M., and Salem A. M. (2011) Repair and biochemical effects of DNA-protein crosslinks. Mutat. Res. 711, 113–122 10.1016/j.mrfmmm.2010.12.007 [DOI] [PubMed] [Google Scholar]
- 20. Baker D. J., Wuenschell G., Xia L., Termini J., Bates S. E., Riggs A. D., and O'Connor T. R. (2007) Nucleotide excision repair eliminates unique DNA-protein cross-links from mammalian cells. J. Biol. Chem. 282, 22592–22604 10.1074/jbc.M702856200 [DOI] [PubMed] [Google Scholar]
- 21. Tretyakova N. Y., Michaelson-Richie E. D., Gherezghiher T. B., Kurtz J., Ming X., Wickramaratne S., Campion M., Kanugula S., Pegg A. E., and Campbell C. (2013) DNA-reactive protein monoepoxides induce cell death and mutagenesis in mammalian cells. Biochemistry 52, 3171–3181 10.1021/bi400273m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dizdaroglu M., and Gajewski E. (1989) Structure and mechanism of hydroxyl radical-induced formation of a DNA-protein cross-link involving thymine and lysine in nucleohistone. Cancer Res. 49, 3463–3467 [PubMed] [Google Scholar]
- 23. Dizdaroglu M., Gajewski E., Reddy P., and Margolis S. A. (1989) Structure of a hydroxyl radical induced DNA-protein cross-link involving thymine and tyrosine in nucleohistone. Biochemistry 28, 3625–3628 10.1021/bi00434a071 [DOI] [PubMed] [Google Scholar]
- 24. Izzotti A., Cartiglia C., Taningher M., De Flora S., and Balansky R. (1999) Age-related increases of 8-hydroxy-2′-deoxyguanosine and DNA-protein crosslinks in mouse organs. Mutat. Res. 446, 215–223 10.1016/S1383-5718(99)00189-8 [DOI] [PubMed] [Google Scholar]
- 25. Wu F. Y., Lee Y. J., Chen D. R., and Kuo H. W. (2002) Association of DNA-protein crosslinks and breast cancer. Mutat. Res. 501, 69–78 10.1016/S0027-5107(02)00006-4 [DOI] [PubMed] [Google Scholar]
- 26. Quievryn G., and Zhitkovich A. (2000) Loss of DNA-protein crosslinks from formaldehyde-exposed cells occurs through spontaneous hydrolysis and an active repair process linked to proteosome function. Carcinogenesis 21, 1573–1580 10.1093/carcin/21.5.573 [DOI] [PubMed] [Google Scholar]
- 27. Reardon J. T., Cheng Y., and Sancar A. (2006) Repair of DNA-protein cross-links in mammalian cells. Cell Cycle 5, 1366–1370 10.4161/cc.5.13.2892 [DOI] [PubMed] [Google Scholar]
- 28. Stingele J., Bellelli R., and Boulton S. J. (2017) Mechanisms of DNA-protein crosslink repair. Nat. Rev. Mol. Cell Biol. 18, 563–573 10.1038/nrm.2017.56 [DOI] [PubMed] [Google Scholar]
- 29. Stingele J., Bellelli R., Alte F., Hewitt G., Sarek G., Maslen S. L., Tsutakawa S. E., Borg A., Kjær S., Tainer J. A., Skehel J. M., Groll M., and Boulton S. J. (2016) Mechanism and regulation of DNA-protein crosslink repair by the DNA-dependent metalloprotease SPRTN. Mol. Cell 64, 688–703 10.1016/j.molcel.2016.09.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chesner L. N., and Campbell C. (2018) A quantitative PCR-based assay reveals that nucleotide excision repair plays a predominant role in the removal of DNA-protein crosslinks from plasmids transfected into mammalian cells. DNA Repair 62, 18–27 10.1016/j.dnarep.2018.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cohen M. D., Klein C. B., and Costa M. (1992) Forward mutations and DNA-protein crosslinks induced by ammonium metavanadate in cultured mammalian cells. Mutat. Res. 269, 141–148 10.1016/0027-5107(92)90169-3 [DOI] [PubMed] [Google Scholar]
- 32. Craft T. R., Bermudez E., and Skopek T. R. (1987) Formaldehyde mutagenesis and formation of DNA-protein crosslinks in human lymphoblasts in vitro. Mutat. Res. 176, 147–155 10.1016/0027-5107(87)90262-4 [DOI] [PubMed] [Google Scholar]
- 33. Zwelling L. A., Bradley M. O., Sharkey N. A., Anderson T., and Kohn K. W. (1979) Mutagenicity, cytotoxicity and DNA crosslinking in V79 Chinese hamster cells treated with cis- and trans-Pt(II) diamminedichloride. Mutat. Res. 67, 271–280 10.1016/0165-1218(79)90021-1 [DOI] [PubMed] [Google Scholar]
- 34. Merk O., and Speit G. (1998) Significance of formaldehyde-induced DNA-protein crosslinks for mutagenesis. Environ. Mol. Mutagen. 32, 260–268 [DOI] [PubMed] [Google Scholar]
- 35. Kuo H. K., Griffith J. D., and Kreuzer K. N. (2007) 5-Azacytidine induced methyltransferase-DNA adducts block DNA replication in vivo. Cancer Res. 67, 8248–8254 10.1158/0008-5472.CAN-07-1038 [DOI] [PubMed] [Google Scholar]
- 36. Minko I. G., Yamanaka K., Kozekov I. D., Kozekova A., Indiani C., O'Donnell M. E., Jiang Q., Goodman M. F., Rizzo C. J., and Lloyd R. S. (2008) Replication bypass of the acrolein-mediated deoxyguanine DNA-peptide cross-links by DNA polymerases of the DinB family. Chem. Res. Toxicol. 21, 1983–1990 10.1021/tx800174a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wickramaratne S., Boldry E. J., Buehler C., Wang Y. C., Distefano M. D., and Tretyakova N. Y. (2015) Error-prone translesion synthesis past DNA-peptide cross-links conjugated to the major groove of DNA via C5 of thymidine. J. Biol. Chem. 290, 775–787 10.1074/jbc.M114.613638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wickramaratne S., Ji S., Mukherjee S., Su Y., Pence M. G., Lior-Hoffmann L., Fu I., Broyde S., Guengerich F. P., Distefano M., Schärer O. D., Sham Y. Y., and Tretyakova N. (2016) Bypass of DNA-protein cross-links conjugated to the 7-deazaguanine position of DNA by translesion synthesis polymerases. J. Biol. Chem. 291, 23589–23603 10.1074/jbc.M116.745257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pande P., Ji S., Mukherjee S., Schärer O. D., Tretyakova N. Y., and Basu A. K. (2017) Mutagenicity of a model DNA-peptide cross-link in human cells: roles of translesion synthesis DNA polymerases. Chem. Res. Toxicol. 30, 669–677 10.1021/acs.chemrestox.6b00397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ji S., Fu I., Naldiga S., Shao H., Basu A. K., Broyde S., and Tretyakova N. Y. (2018) 5-Formylcytosine mediated DNA-protein cross-links block DNA replication and induce mutations in human cells. Nucleic Acids Res. 46, 6455–6469 10.1093/nar/gky444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sanchez A. M., Minko I. G., Kurtz A. J., Kanuri M., Moriya M., and Lloyd R. S. (2003) Comparative evaluation of the bioreactivity and mutagenic spectra of acrolein-derived α-HOPdG and γ-HOPdG regioisomeric deoxyguanosine adducts. Chem. Res. Toxicol. 16, 1019–1028 10.1021/tx034066u [DOI] [PubMed] [Google Scholar]
- 42. Yang W., and Woodgate R. (2007) What a difference a decade makes: insights into translesion DNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 104, 15591–15598 10.1073/pnas.0704219104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vaisman A., and Woodgate R. (2017) Translesion DNA polymerases in eukaryotes: what makes them tick? Crit. Rev. Biochem. Mol. Biol. 52, 274–303 10.1080/10409238.2017.1291576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maiti A., and Drohat A. C. (2011) Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J. Biol. Chem. 286, 35334–35338 10.1074/jbc.C111.284620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Minko I. G., Kozekov I. D., Kozekova A., Harris T. M., Rizzo C. J., and Lloyd R. S. (2008) Mutagenic potential of DNA-peptide crosslinks mediated by acrolein-derived DNA adducts. Mutat Res. 637, 161–172 10.1016/j.mrfmmm.2007.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yeo J. E., Wickramaratne S., Khatwani S., Wang Y. C., Vervacke J., Distefano M. D., and Tretyakova N. Y. (2014) Synthesis of site-specific DNA-protein conjugates and their effects on DNA replication. ACS Chem. Biol. 9, 1860–1868 10.1021/cb5001795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li H., Xie B., Zhou Y., Rahmeh A., Trusa S., Zhang S., Gao Y., Lee E. Y., and Lee M. Y. (2006) Functional roles of p12, the fourth subunit of human DNA polymerase δ. J. Biol. Chem. 281, 14748–14755 10.1074/jbc.M600322200 [DOI] [PubMed] [Google Scholar]
- 48. Pande P., Malik C. K., Bose A., Jasti V. P., and Basu A. K. (2014) Mutational analysis of the C8-guanine adduct of the environmental carcinogen 3-nitrobenzanthrone in human cells: critical roles of DNA polymerases η and κ and Rev1 in error-prone translesion synthesis. Biochemistry 53, 5323–5331 10.1021/bi5007805 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.