

Abstract
The yeast Saccharomyces cerevisiae is the pre-eminent organism for the study of basic functions of eukaryotic cells1. All of the genes of this simple eukaryotic cell have recently been revealed by an international collaborative effort to determine the complete DNA sequence of its nuclear genome. Here we describe some of the features of chromosome XII.
The nucleotide composition of the chromosome, which is 38.48% G+C overall, and gene density vary across the chromosome. This has been observed for other yeast chromosomes2–6 (Fig. 1). There are three main regions deficient in G+C, centred at approximately 150, 685 and 1,043 kilobases; one of these, as expected, coincides with the centromere. There is only one main peak of high G+C content, at approximately 473 kb, centred over the rDNA repeats. There does not seem to be any regularity in the variation in nucleotide composition, as may be the case for some other yeast chromosomes3.
Figure 1.
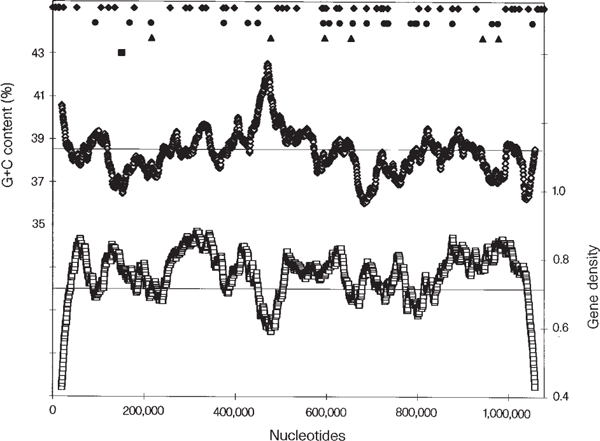
Top, non-coding elements of chromosome XII Including autonomously replicating sequence (ARS) (filled diamonds); tRNA (filled circles); Ty element (filled triangles); centromere (filled square). Middle, G+C content as a percentage (open diamonds). Bottom, gene density (open squares).
Like other yeast chromosomes, 72% of chromosome XII is predicted to code for protein (considering only two copies of the rDNA cluster). The sequence contains 534 open reading frames (ORFs) of 100 or more sense codons (excluding the 13 ORFs contained within yeast transposable elements), distributed roughly equally on the two strands (255 on the Watson (top) strand and 279 on the Crick (bottom) strand. The average ORF size is 485 codons. The largest gene in the chromosome, YLR106c, containing 4,910 codons, is the largest in the yeast genome. The average distance between ORFs is 545 base pairs for the 121 divergently transcribed genes (promoters abutting), 282 bp for the 120 convergently transcribed genes (terminators abutting), and 493 bp for the 208 genes that are transcribed in the same direction (promoter abutting terminator). Of the ORFs, 17 (3.2% of the total) contain introns, all of which are at the extreme 5′ end of the gene (except for YLR464w, a probable pseudogene). Two genes (YLL057c and YLR388W) may contain introns in the 5′-untranslated region of their mRNA. As expected7, about half of the intron-containing genes (9) encode ribosomal proteins.
Only 170 (31.8%) of the genes were previously identified. Of the 364 newly identified genes, 34 (6.4% of the total genes) are obviously similar to proteins of known function, and 54 (10.1%) are weakly similar to proteins of known function. Thus a function is known or can be predicted for 48.3% of the encoded proteins. A further 69 genes (12.9%) encode proteins similar to proteins of unknown function; 207 (38.8%) of the predicted proteins are not similar to other proteins.
Included in the predicted ORFs are 55 that are ‘questionable’, that is, they consist of fewer than 150 codons and have a codon adaptation index (CAI)8 of less than 0.110, or they overlap with another ORF. Of the 40 questionable ORFs that overlap with another ORF, the true gene can be predicted for 27 of these pairs, which include either a gene whose product is known (16 pairs) or whose predicted product is similar to another protein in the databases (11 pairs). There are therefore 13 overlapping ORFs that are suspect, although which of these ORFs is actually a gene awaits experimental determination.
Chromosome XII contains 22 tRNA genes, of which 7 are predicted to contain introns. Most of the tRNA genes are widely separated, although there are two clusters of three tRNA genes, each in a region of 9 kb to 13 kb (725,746–734,874 and 784,352–797,247). As expected9, many (12) of the tRNA genes are near yeast retrotransposons (Ty elements) or their isolated long terminal repeats (LTRs). Three known small nuclear RNAs, SNR6, SNR30 and SNR34 are encoded on chromosome XII. Four of the six retro-transposons on chromosome XII are of the Ty1 type and two are Ty2 elements. There are several complete or partial ‘solo’ retrotransposon LTRs, including nine delta elements, four sigma elements, and a tau element.
The subtelomeric regions of chromosome XII are typical10. The left subtelomeric region contains a ‘core X’ element, and subtelomeric repeats STR-D, C, B and A, along with two tandem Y′ elements (short versions). The right subtelomeric region contains a core X element, the STR elements listed above, and 3–4 tandem Y′ (long version) elements. The sequence of the first 1.5 and the last Y′ elements were fused to give two copies of the Y′ element in the presented sequence. Proximal to the core X are shared homologies with several other telomere regions. As with several other chromosomes, both chromosome ends contain members of the PAU/TIP/SRP gene family11.
Chromosome XII is estimated to contain 100–200 copies of the 9,137 base pair rDNA repeat9,12–14. Only one complete copy (the leftmost repeat in Fig. 2) and one nearly complete copy of the rDNA (the rightmost repeat in Fig. 2) are represented in the assembled sequence. In some strains these repeats seem to be interrupted by non-rDNA sequence14.
Figure 2.

Diagram of the rDNA repeats and surrounding sequence, as assembled for cosmids YSCL9634 (left, GenBank accession no. U53879) and YSCL9362 (right, GenBank accession no. U51921). The numbers shown are the nucleotide coordinates for cosmid YSCL9634. The left rDNA junction (U53879 coordinate 21,811) begins at nucleotide 451,418 of chromosome XII; the right rDNA junction (U53879 coordinate 39,322) is at nucleotide 468,929. The sequence includes 1.92 rDNA repeats, representing the leftmost and rightmost copies in the genome. The remaining 100–200 rDNA repeats in the genome are represented as an insertion at coordinate 30,947. Only one complete 5S rDNA gene (in the left rDNA repeat) is included in this sequence (nucleotides 30,069–30,189); the 5S rDNA genes in the 3.6-kb repeats are variant genes. The 5S rDNA gene in the last rDNA repeat (nucleotides 39,204–39322) includes all 5′ non-translated sequences (like the normal 5S rDNA in the first repeat), but is missing sequences downstream of the 5S rRNA transcript (like the 5S rDNA genes in the 3.6-kb repeats).
The boundaries of the rDNA repeats are in non-transcribed regions downstream of the 35S rDNA (the left, or centromere-proximal, boundary) and 5S rDNA (the right, or centromere-distal, boundary)15. The structure of the left boundary of the rDNA (nucleotide 21,811 in Fig. 2) is straightforward; the right boundary16 is more complicated. Immediately to the right of the rDNA repeats are several copies of a 3.6-kb repeat (one of which is interrupted by a Ty element) that includes the ASP3 gene17 and ends with a nearly complete 5S rDNA gene (5Svar in Fig. 2). The precise number of copies of this 3.6-kb repeat in the genome is not known. The rightmost rDNA repeat ends in a 5S rDNA that adjoins a 3.6-kb repeat. Thus this rightmost rDNA repeat is lacking the 759 bp of sequence between the end of 5S rDNA and the end of the rDNA repeat (equivalent to nucleotides 30,186–30,947 in Fig. 2). The structure of the right rDNA junction differs in other yeast strains15.
The 5S rDNA gene in the 3.6-kb repeats lacks the non-transcribed regions of the gene. It begins two nucleotides upstream of the 5′ end of 5S rRNA, and is missing the last four nucleotides of the 5S rRNA. These genes are labelled ‘5Svar’ in Fig. 2 to indicate that they are incomplete. Immediately downstream of this gene is a run of 10 T residues that is reminiscent of the transcription termination sequence of RNA polymerase III (there are 29 T residues downstream of the 5S rDNA gene in the rDNA repeats). Because this gene seems to be missing the promoter and much of the terminator, it might represent a reverse-transcribed copy of the 5S rRNA that integrated into the genome. Nevertheless these genes produce 5S rRNA transcripts18.
One possible explanation of the structure of the right junction is that a reverse-transcribed copy of 5S rRNA is inserted into the genome near the right border of the rDNA cluster. This gene could then have been part of a 3.6-kb duplication. It is then easy to imagine a recombination event between an intact 5S rDNA gene in one of the rDNA repeats and a 5Svar rDNA in one of the 3.6-kb repeats that generated the right rDNA junction we sequenced. Other explanations for the origin of this junction have been proffered16.
To speed the completion of the sequence of this large chromosome, two groups collaborated on its sequencing. The rDNA repeats on chromosome XII served as a convenient point to divide the effort: the EU sequencing network19 determined the sequence of the chromosome to the left of the rDNA repeats; the sequence to the right was determined at the Washington University Genome Sequencing Center. The sequence of both strands of the entire 1,078,171 base-pair chromosome (but including only two copies of the rDNA repeats) was determined, nearly all the way to the telomeres.
The 781,865 nucleotides determined at Washington University came from 24 partly overlapping cosmid and two lambda clones20. The sequence of each clone was determined by a ‘shotgun’ strategy followed by directed sequencing2. The sequence of each clone was submitted to GenBank, and the entire non-overlapping sequence was assembled, analysed and annotated2–5.
The 460,166 nucleotides to the left of the rDNA were determined by the EU network from a set of cosmid clones constructed from gel-purified chromosome XII DNA and mapped specifically for this purpose21. Sequencing was done by a directed approach that combines the advantages of primer walking (low redundancy) and ‘shotgun’ sequencing (use of a single primer)22. The sequence was determined from ‘shotgun’ sublibraries of 1-kb fragments of the cosmids that were then ordered by hybridization fingerprinting22. The sublibraries were arrayed on high-density filters, and sorted into smaller groups by hybridization with restriction fragments of the cosmids . Detailed mapping information was obtained by hybridizations with both oligodeoxynucleotides and pools of clone inserts amplified by the polymerase chain reaction (PCR).
The sequence of the left telomere region, including the TG1–3 sequence at the very end of the chromosome, was obtained from clones generated by integrating then excising a plasmid at the telomere, with capture of the flanking sequence10. The right telomere sequence was obtained by cycle sequencing of an anchored PCR product of the last Y′ element from a strain whose chromosome end was specifically marked by unique vector sequence23. The sequence of the very end of the Y′ element (about 130 bp short of the end of the chromosome) was not determined.
Only the sequence of the leftmost rDNA repeat (see Fig. 2) and about 300 nucleotides across the junction of the first and second repeat was determined. It was assembled appropriately to give the two rDNA repeats presented in Fig. 2 and in the database (GenBank accession no. U53879). The right junction sequence was not present in the cosmid closest to the rDNA on the right (YSCL9362; GenBank accession no. U51921), nor in two phage lambda clones that were mapped to this region. The structure of the junction was inferred from our ability to obtain a product of the expected size (the size of a 3.6-kb repeat) in PCR using an oligonucleotide primer in the 3.6-kb repeat (lying just to the right of 5Svar) and a primer unique to the rDNA repeat (lying just to the left of 5S rDNA). Our sequence was assembled from these results, and found to match the sequence of the previously determined junction16.
The complete, assembled, non-overlapping sequence of chromosome XII can be obtained at: http://speedy.mips.biochem.mpg.de/mips/yeast/ and http://genome-www.stanford.edu/Saccharomyces/.
Verification of 71,072 bp of sequence determined by the EU network (64,001 bp of overlaps between cosmids sequenced independently, and 7,071 bp of selected region that were resequenced) revealed five mistakes per 10 kb, but most errors were clustered in just a few regions. Only 14 differences were found in 175,891 nucleotides that were sequenced independently by both groups; six of these were sequencing errors, leading to an error frequency of only one mistake per 29 kb. The origins of the remaining eight discrepancies were determined by sequencing PCR products of the genome of the two strains used to generate the clones. Seven of the differences are due to changes that arose in the clones, presumably during propagation in Escherichia coli; only one results from differences between the two yeast strains (which are isogenic, but were propagated separately for many years) used to generate the two sets of clones. Thus the number of errors in the sequence is equivalent to the number of errors resulting from propagation of the DNA in E. coli and yeast.
Acknowledgements.
We thank J. Warner for help with interpreting the rDNA sequence. This work was supported by the NIH National Center for Human Genome Research and the European Commision; the Belgian Federal Services for Science Policy (D.W.T.C.) and the Service Fédéraux des Affaires Scientifiques, Techniques et Culturelles, Pôles d’attraction Inter-universitaire, the Region de Bruxelles-Capitale, Region Wallonne, the Research Fund of the Katholieke Universiteit Leuven and The Wellcome Trust.
References
- 1.Jones EW, Pringle JR & Broach JR The Molecular and Cellular Biology of the Yeast Saccharomyces, Vols 1–3, (Cold Spring Harbor Laboratory Press, NY, 1991–1996). [Google Scholar]
- 2.Johnston M et al. Science 265, 2077–2082 (1994). [DOI] [PubMed] [Google Scholar]
- 3.Dujon B et al. Nature 369, 371–378 (1994). [DOI] [PubMed] [Google Scholar]
- 4.Feldmann H et al. EMBO J. 13, 5795–5809 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galibert A et al. EMBO J. 15, 2031–2049 (1996). [PMC free article] [PubMed] [Google Scholar]
- 6.Sharp P & Lloyd A Nucleic Acids Res. 21, 179–183 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodriguez-Medina JR & Rymond BC Mol. Gen. Genet. 243, 532–539 (1994). [DOI] [PubMed] [Google Scholar]
- 8.Sharp PM & Li WH Nucleic Acids Res. 15, 1281–1295 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olson MV in The Molecular and Cellular Biology of the Yeast Saccharomyces, Vol. 1 (ed. Broach JR, Pringle JR & Jones EW) 1–39 (Cold Spring Harbor Laboratory Press, NY, 1991). [Google Scholar]
- 10.Louis EJ & Borts R Genetics 139, 125–136 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Viswanathan M, Muthukumar G, Lenard J & Cong YS Gene 148, 149–153 (1994). [DOI] [PubMed] [Google Scholar]
- 12.Pasero P & Marilley M Mol. Gen. Genet. 236, 448–452 (1993). [DOI] [PubMed] [Google Scholar]
- 13.Chindamporn A, Iwaguchi S, Nakagawa Y, Homma M & Tanaka KJ Gen. Microbiol. 139, 1409–1415 (1993). [DOI] [PubMed] [Google Scholar]
- 14.Rustchenko EP & Sherman F Yeast 10, 1157–1171 (1994). [DOI] [PubMed] [Google Scholar]
- 15.Zamb T & Petes TD Cell 28, 355–364 (1982). [DOI] [PubMed] [Google Scholar]
- 16.McMahon ME, Stamenkovich D & Petes TD Nucleic Acids Res. 12, 8001–8016 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim KW, Kamerud JQ, Livingston DM & Roon RJ J. Biol. Chem. 263, 11948–11953 (1988). [PubMed] [Google Scholar]
- 18.Piper PW, Lockheart A & Patel N Nucleic Acids Res. 12, 4083–4096 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vassarotti A et al. J. Biotechnol. 41, 131–137 (1995). [DOI] [PubMed] [Google Scholar]
- 20.Riles L et al. Genetics 134, 81–150 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scholler P, Schwarz S & Hoheisel JD Yeast 11, 659–666 (1995). [DOI] [PubMed] [Google Scholar]
- 22.Scholler P et al. Nucleic Acids Res. 23, 3842–3849 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Louis EJ Biochemica 3, 25–26 (1995). [Google Scholar]