

Abstract
Polycyclic aromatic hydrocarbons (PAHs) like benzo[a]pyrene (BaP) are ubiquitous environmental contaminants formed during incomplete combustion of organic materials. Our prior work showed that transplacental exposure to BaP depletes ovarian follicles and increases prevalence of epithelial ovarian tumors later in life. We used the MutaMouse transgenic rodent model to address the hypothesis that ovarian mutations play a role in tumorigenesis caused by prenatal exposure to BaP. Pregnant MutaMouse females were treated with 0, 10, 20, or 40 mg/kg/day BaP orally on gestational days 7–16, covering critical windows of ovarian development. Female offspring were euthanized at 10 weeks of age; some ovaries with oviducts were processed for follicle counting; other ovaries/oviducts and bone marrow were processed for determination of lacZ mutant frequency. Mutant plaques were pooled within dose groups and sequenced to determine the mutation spectrum. BaP exposure caused highly significant dose-related decreases in ovarian follicles and increases in ovarian/oviductal and bone marrow mutant frequencies at all doses. Absence of follicles, cell packets, and epithelial tubular structures were observed with 20 and 40 mg/kg/day BaP. Depletion of ovarian germ cells was inversely associated with ovarian mutant frequency. BaP induced primarily G>T and G>C transversions and deletions in ovaries/oviducts and bone marrow cells and produced a mutation signature highly consistent with that of tobacco smoking in human cancers. Overall, our results show that prenatal BaP exposure significantly depletes ovarian germ cells, causes histopathological abnormalities, and increases the burden of ovarian/oviductal mutations, which may be involved in pathogenesis of epithelial ovarian tumors.
Keywords: polycyclic aromatic hydrocarbon, critical window, ovary, mutation, transplacental
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are common environmental pollutants produced during incomplete combustion of wood, fossil fuels, foods, tobacco, and other organic materials [ATSDR 1995]. Many PAHs, such as benzo[a]pyrene (BaP), are metabolized to reactive species that are mutagenic and carcinogenic [Xue and Warshawsky 2005]. Over 50 years ago it was shown that dermal (5 mg applied weekly for 4 weeks or 1 mg applied weekly for 8 weeks) and oral (1 mg weekly by gavage for 8 weeks) treatment with BaP induced ovarian tumors in adult mice [Mody 1960, Biancifiori et al. 1961]. These foundational studies also noted that ovaries were devoid of ovarian follicles at early time points after the onset of treatment and before tumors developed. Subsequent research showed that quiescent primordial follicles in the mouse ovary were dose-dependently destroyed by BaP, with 50% reductions following single or cumulative daily doses in the range of 25 to 50 mg/kg intraperitoneally [Mattison and Thorgeirsson 1979, Mattison et al. 1980, Borman et al. 2000]. Because there is no or minimal neo-oogenesis in the post-natal ovary [Tilly et al. 2009, Zhang et al. 2012, Lei and Spradling 2013], the primordial follicle pool constitutes the ovarian reserve, and its depletion causes irreversible ovarian failure. Thus, the consequences of adult female exposure to BaP include both reduction in fertility and increased probability of ovarian tumors.
BaP exposure during early embryonic development can also have permanent impacts on fertility in adults. In the mouse, oogenesis during embryonic development initiates around 7 days post conception (dpc) with the formation of primordial germ cells that proliferate mitotically to generate the pool of oogonia until entering meiosis and becoming oocytes beginning at 13.5 dpc [McLaren 2003, Pepling 2006]. Before birth, the oocytes arrest in the diplotene stage of the first meiotic prophase. Beginning during the last few days of pregnancy and continuing during the first postnatal week, the oocytes are encapsulated by a single layer of flattened granulosa cells to establish the primordial follicle pool [Pepling 2006]. Transplacental exposures to toxicants that destroy ovarian germ cells before follicle formation can diminish the initial size of the primordial follicle pool. Indeed, female mice exposed to BaP in utero experience premature depletion of the primordial follicle pool and decreased fertility [MacKenzie and Angevine 1981, Lim et al. 2013]. Moreover, doses of BaP that deplete germ cells during in utero exposure also cause increases in ovarian tumors later in life [Lim et al. 2013], suggesting a relationship between these two adverse outcomes.
It is well established that environmental chemicals, including several PAHs [Goerttler et al. 1981, Shorey et al. 2012, Lim et al. 2013], can induce transplacental carcinogenicity [Anderson et al. 2000]. Compelling evidence is also accumulating that genetic changes associated with disease in adult life arise during in utero development [Biesecker and Spinner 2013, Lupski 2013, Lupski 2015]. Mutations occurring during embryogenesis may result in extensive mosaicism in the developing organism and affect a large proportion of cells in the body. Indeed, we recently demonstrated that in utero exposure to BaP significantly enhances the occurrence of mutation fixation as a result of rapid conversion of adducts into mutations and clonal expansion during F1 male organogenesis [Meier et al. 2017]. Furthermore, we showed that mutation induction in the germ cells of F1 males exposed in utero was inversely correlated with reduced sperm count and impaired sperm motility [Meier et al. 2017]. Therefore, in utero development represents a critical window for the induction of mutations caused by xenobiotics that can lead to adverse health effects after birth, including cancer.
Ninety percent of malignant ovarian tumors in women are epithelial and believed to be derived from the ovarian surface epithelium or the fallopian tube epithelium [Mullany and Richards 2012]. Different subtypes of epithelial ovarian cancers have characteristic mutational signatures [Kurman and Shih 2016]. It is not known whether induction of ovarian tumors by transplacental or direct exposure to BaP or other PAHs in mice is consistent with any of the common mutational mechanisms or signatures found in human ovarian cancers. Activating mutations in codon 12 of the KRAS oncogene are the most common mutations in low grade, Type I mucinous epithelial ovarian cancers [Cuatrecasas et al. 1997, Romero and Bast 2012, MacKenzie et al. 2015]. Risk for mucinous epithelial ovarian cancers is increased in women who smoke [Rossing et al. 2008, Tworoger et al. 2008, Gates et al. 2010]. Greater than 90% of KRAS mutations occur in codon 12, and two KRAS codon 12 mutations account for 90% of mucinous ovarian cancer KRAS mutations, KRAS codon 12 GGT>GAT (G12D, 40%) and codon 12 GGT>GTT (G12V, 50%) [Matias-Guiu and Prat 1998, MacKenzie et al. 2015]. We recently reported no significant increase in these Kras codon 12 mutations in mouse ovaries after prenatal exposure to a dose of BaP that resulted in >70% depletion of the primordial follicle pool [Luderer et al. 2017], but we did not analyze any other mutations.
We hypothesize that early developmental exposure to BaP results in ovarian mutations and clonal expansion that increase the risk of ovarian tumors in adulthood. In the present study, we: 1) quantify the dose-dependent effects of in utero exposure to BaP during critical windows of ovarian development on ovarian follicle number to assess potential impacts on adult fertility; 2) determine the impact of in utero BaP exposure on ovarian histopathological lesions that may be precursors to ovarian tumors; and 3) characterize the frequency and spectrum of ovarian mutations induced by transplacental BaP exposure, compared with mutations arising in bone marrow of the same females, as an estimate of mutagenic events with the potential to contribute to ovarian tumorigenesis. Furthermore, we compare the mutation spectra derived from bone marrow and ovarian samples to the COSMIC database of mutational signatures across the spectrum of human cancer types [Forbes et al. 2017] to investigate whether the BaP-induced lacZ mutation signature is consistent with signatures of PAH exposure in human ovarian cancers.
Methods
Animals
Animal treatment, transgene mutation analysis, sequencing, statistical, and bioinformatic analyses performed for scoring and characterizing mutations in the lacZ transgene of MutaMouse females were as described previously [Meier et al. 2017]. The use of animals in these experiments was approved by the Health Canada Ottawa Animal Care Committee. Animals used in this study were humanely treated with regard to the alleviation of suffering following the guidelines of the Canadian Council on Animal Care (http://www.ccac.ca/en_/standards/policies/policy-ethics_animal_investiga-tion ). A total of 37 pregnant MutaMouse females (80% mating success) were dosed by oral gavage with 0, 10, 20, or 40 mg/kg/day BaP (CAS number 50–32-8) dissolved in olive oil on postconception days 7 through 16, spanning several critical windows of ovarian development: primordial germ cell proliferation during and after migration to the gonadal ridge, gonadal differentiation, proliferation of oogonia, and entry and arrest of oogonia in meiosis. The exposures were performed in two rounds, one with 0 and 10 mg/kg/day dose groups and the second with 0, 20, and 40 mg/kg/day dose groups. The goal was to obtain about eight litters per dose group, however, some of the females thought to be pregnant did not produce litters (eight females) and 4 litters were lost within a few days from birth. There was no apparent correlation of these events with the dose of BaP administered, and there was no statistically significant difference in the number of pups born or the number that were weaned among the dose groups (Supplemental Table 1). In total there were 11, 6, 8 and 5 litters for 0, 10, 20 and 40 mg/kg/day dose groups, respectively. One of the litters from the 20 mg/kg/day dose group and two of the litters from the 40 mg/kg/day dose group did not have females that reached weaning at 3 weeks of age, resulting in 7 and 3 litters available for analysis, respectively, in those dose groups. Animals exposed in utero were euthanized 10 weeks after birth in accordance with Health Canada’s ethical guidelines, after which tissues were collected. One ovary with attached oviduct from one mouse per litter was fixed in Bouin’s fixative (Fisher Scientific) for 24 h, rinsed in 50% ethanol, then washed for 30 or more minutes three times in 50% ethanol, and stored in 70% ethanol until processing for ovarian stereology. The other ovary with attached oviduct was flash frozen while the bone marrow was processed as previously described [Meier et al. 2017].
Ovarian stereology and histopathology
One ovary from a randomly chosen female per litter was used for ovarian stereology. The same ovary also underwent independent histopathological evaluation by a board-certified veterinary pathologist (G.W.L.). Bouin’s fixed ovaries were embedded in paraffin, serially sectioned at 5 μm thickness, and stained with Gill’s hematoxylin (Fisher Scientific) and 1% eosin Y (Sigma Aldrich). Primordial, primary, and secondary follicles were enumerated using the physical dissector principle [Miller et al. 1997, Myers et al. 2004]. Primordial and small primary follicles were counted in every 5th serial section if the nucleus was clearly visible; large primary and secondary follicles were counted if the nucleolus was clearly visible. Antral follicles and corpora lutea were followed through every serial section taking care not to count any of these structures twice. Follicles were further classified as healthy or atretic [Lopez and Luderer 2004, Lim et al. 2013]. All counting was performed blind to treatment group by one of the authors (U.L.). The effect of in utero BaP exposure on ovarian follicle numbers was analyzed using linear regression on the log base 10 transformed data and 1-way analysis of variance with post-hoc Dunnett T3 tests for intergroup comparisons. Fisher’s exact test was used to analyze differences in the prevalence of histopathological ovarian abnormalities among groups. Analyses were performed using SPSS Version 23 for Macintosh (IBM).
Mutation analysis
DNA was obtained from bone marrow and ovaries from all females in each litter using phenol/chloroform extraction, although we could not get sufficient DNA to perform the lacZ assay from some of the ovaries. LacZ mutations in the MutaMouse transgene were scored by packaging into lambda phage (Transpack packaging extract, Agilent Technologies) and transfecting the DNA into gale- E. coli as previously described [Gingerich et al. 2014]. As per OECD Test Guideline 488 [OECD 2013], a minimum of 125,000 plaque forming units (PFUs) were analyzed per mouse, with a range of 5–20 animals per dose group. Statistical analysis of the data was performed in R using a generalized linear model with a quasi-Poisson distribution and multiple comparisons corrected by the Bonferroni method. Dose response analysis of mutant frequencies was carried out using PROAST version 38.9 (http://www.rivm.nl/en/Documents_and_publications/Scientific/Models/PROAST ).
Mutant plaques were selected for amplicon sequencing as described [Beal et al. 2015, Meier et al. 2017]. Sequencing was performed on an Ion Torrent Proton using a P1 chip. Sequence data for plaques pooled by dose group (0 mg/kg/day compared to 20 and 40 mg/kg/day BaP dose groups combined) and tissue were screened for mutations by alignment to the lacZ gene with bowtie2 (version 2.1.0). Mutant plaques were not collected from the 10 mg/kg/day BaP group because when the response is small, the mutation spectrum is more affected by the spontaneous mutation spectrum than that obtained at the higher doses and because when only a few mutant plaques per plate are recovered, it would require conducting the lacZ assay an unreasonable number of times to generate a reliable mutation spectrum. The read depth for each mutant was enumerated using samtools (version 0.1.19). Mutations passing the threshold (i.e., those having a read depth equal to at least 1/number of plaques in pool) in two technical replicate libraries were counted and corrected using a limit of detection (LOD) and linear model described in detail previously [Beal et al. 2015]. Briefly, this model was established by sequencing known mutant plaques mixed in different proportions and using the Fitting Linear Models algorithm in R to determine the correlation between expected and observed mutants. This resulted in the following formula: adjusted mutant count = (observed count – y-intercept) / slope for mutant counts above the LOD, where the LOD was 4, y intercept was 0.3123, and slope was 1.1363. Mutations that had an adjusted mutant count >1 were considered the result of clonal expansion, because their read proportions were greater than expected based on the number of plaques sequenced after applying the correction.
Mutation counts for bone marrow and ovary exposed to BaP were imported into R as row counts from a VCF file using the “readVcfAsVRranges” command from the package “SomaticSignatures” [Gehring et al. 2015] with the lacZ coding sequence as the reference FASTA file. For bone marrow, data from the male siblings [Meier et al. 2017] were added to increase the number of mutants available for signature deconstruction, thereby reducing error on the measurement of signatures. The observed lacZ mutation spectra in bone marrow and ovary was compared against COSMIC mutation signatures to identify the signature that best explained the mutation data. In order to do this comparison, the COSMIC mutation signature weights, which are derived from human mutation data, were first normalized to lacZ trinucleotide frequencies. This was done using the ratio of trinucleotide frequencies in lacZ to the trinucleotide frequencies in the human genome. Following normalization, each of the 96 trinucleotide substitutions within each signature were represented as the relative frequency (i.e., all values in a signature sum to 1) by dividing each normalized value by the sum of all values for that signature. The trinucleotide mutation context (the nucleotide immediately upstream and downstream of the mutation) was obtained with the “mutationContext” command and converted to a motif matrix using the “motifMatrix” command (both in the “Somatic Signatures” package). The motif matrix was then transposed to obtain the required format and was finally decomposed into the constituent lacZ-normalized signatures using the “whichSignature” command from “deconstructSigs” [Rosenthal et al. 2016]. Pearson correlation was then used to identify the signature that best explained the BaP-induced mutations in utero. The best matching signature and mutation profiles were plotted with the “plotSignatures” command from ‘deconstructSigs”.
Results
In utero exposure to BaP depletes ovarian germ cells and induces ovarian histopathological changes
Analysis of the ovaries of F1 females at 10 weeks of age showed that in utero exposure to BaP during critical windows of ovarian development dose-dependently depleted germ cells, resulting in dramatic reductions in the numbers of healthy and atretic follicles of all stages of development (Table 1 and Figure 1A). Only 3% as many healthy follicles as were observed in control mice remained in the ovaries of 10 mg/kg/day exposed females. Only one of seven ovaries in the 20 mg/kg/day group had any follicles, and no follicles were found in the ovaries from the 40 mg/kg/day group (Figure 1 and Table 1). The numbers of corpora lutea, which are derived from preovulatory follicles that ovulated during prior estrous cycles, were similarly decreased (Table 1). The ED50 for follicle depletion in MutaMouse females exposed during this developmental window was therefore below 10 mg/kg/day.
Table 1:
Ovarian follicle counts in F1 MutaMouse females exposed to BaP in utero
| Follicles/ovary ± SEM | |||||||
|---|---|---|---|---|---|---|---|
| BaP Dose (mg/kg/day)a |
N | Healthy Primordial |
Healthy Primary |
Healthy Secondary |
Healthy Antral |
Atreticb | Corpora lutea |
| 0 | 11 | 2220 ± 238 | 1140 ± 77 | 186 ±13 | 22.6 ± 3.1 | 110 ± 16 | 8.3 ± 2.4 |
| 10 | 6 | 19.2 ± 29.2* | 55.0±25.3* | 17.0 ± 6.6* | 2.5 ± 1.1* | 6.0 ± 2.4* | 2.8 ± 1.1† |
| 20c | 7 | 0.3 ± 0.3* | 0.3 ± 0.3* | 0* | 0* | 0.6 ± 0.6* | 0* |
| 40 | 3 | 0* | 0* | 0* | 0* | 0* | 0* |
Gestational days 7–16
All stages combined
Only one ovary in this group had any follicles
P<0.001 versus respective 0 mg/kg/day, Dunnett T3 test;
P=0.011 versus 0 mg/kg/day
Figure 1: Effects of in utero exposure to BaP on F1 ovarian follicle numbers and histopathology at 10 weeks of age.
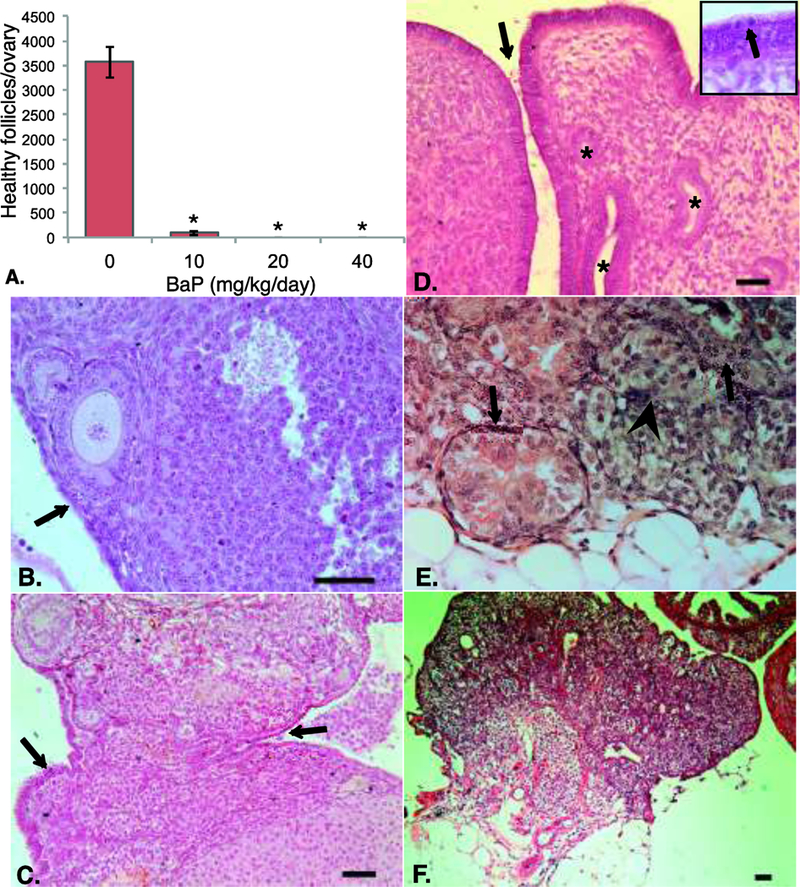
Ovaries from F1 females exposed to the indicated doses of BaP on gestation days 7 to 16 were processed for ovarian histology and stereology as described in Methods. (A) Mean ± SEM of healthy follicles of all developmental stages per ovary. *P<0.001 versus 0 mg/kg/day BaP. (B) Ovary from control mouse shows follicles at various stages of development and single layer of ovarian surface epithelial cells (arrow). (C) Multilayered and invaginated areas of ovarian surface epithelium (OSE; arrows) in ovary from 10 mg/kg/day BaP exposed mouse. (D) Abnormal multi-layered, highly invaginated OSE (arrow) and inclusion cysts (asterisks) in ovary of 20 mg/kg/day BaP-exposed mouse. Inset shows mitotic cell in epithelium (arrow). (E) Representative packet of cells surrounded by fibroblasts (arrowhead) and tubules (black arrows) observed in ovaries from 20 and 40 mg/kg/day exposed mice. (F) Ovary from 40 mg/kg/day BaP exposed mouse shows absence of ovarian follicles and multiple tubular, epithelial-lined structures within ovarian parenchyma. Structure at upper right is portion of oviduct. Scale bars are 50 μm throughout.
The ovarian surface epithelium normally consists of a single layer of flattened to cuboidal cells (Figure 1B). Small areas of surface epithelium more than one layer thick or epithelial invaginations were occasionally noted at the 10 mg/kg/day dose (Figure 1C). One of seven ovaries from the 20 mg/kg/day dose group exhibited multi-layered ovarian surface epithelium with multiple invaginations, tubules extending from the surface into the parenchyma, epithelial inclusions, and mitotic figures (Figure 1D). All of the ovaries from the two high dose groups had areas of fibroblast-like cells separating cell packets (Figure 1E). In some areas where fibroblasts were prominent around packeted cells, packeted cells appeared to degenerate, forming tubular structures lined by cuboidal epithelium in 5 of 7 ovaries in the 20 mg/kg/day group and 3 of 3 in the 40 mg/kg/day group (Figure 1E,F). We propose that these histological changes represent precursor lesions to epithelial ovarian tumors in view of our prior study in which we observed 83% prevalence of epithelial ovarian tumors in wild type F1 mice exposed to 10 mg/kg/day BaP during the same developmental window [Lim et al. 2013]. None of the control ovaries exhibited any of these changes. Overall, the incidence of epithelial tubular structures increased significantly with prenatal BaP dose (p < 0.001, Supplemental Table 2).
In utero exposure to BaP induces mutations in ovaries/oviducts and bone marrow
In utero exposure to BaP during the critical period of organogenesis induced significant dose-related increases (p < 0.0001) in mutations in the bone marrow and ovaries/oviducts of F1 female mice (Figure 2, top panel, Table 2). The increase in mutations was especially pronounced in the ovaries/oviducts, where a 3-fold increase in mutant frequency over controls was observed at the 10 mg/kg/day dose (p = 0.003). At the 20 and 40 mg/kg/day doses, 23-fold and 24-fold increases in mutant frequency was observed in the bone marrow and ovaries/oviducts, respectively (p < 0.0001). Dose-response modelling using PROAST revealed a benchmark dose (BMD; the dose at which a 10% increase in response occurs) of 1.3 mg/kg/day, with a BMDU (upper 95% confidence interval on the BMD) of 1.4 (Supplemental Table 3). This was lower than the BMDL (lower 95% confidence interval on the BMD) of 2.1 mg/kg/day in bone marrow.
Figure 2. Effects of transplacental BaP exposure on ovarian/oviductal mutant frequencies.
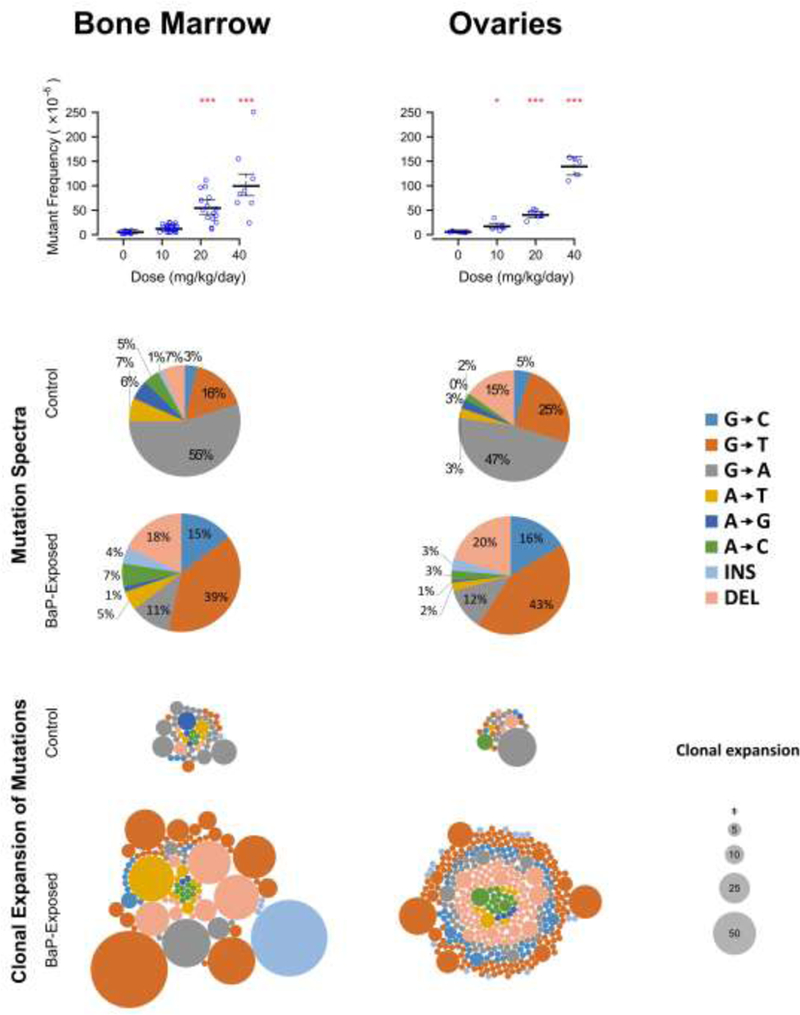
The lacZ mutant frequency of female mice exposed to benzo[a]pyrene (BaP) in utero is significantly elevated in the bone marrow and ovaries/oviducts (top panel). The mutation spectra of the 20 and 40 mg/kg BaP-exposed animals show the characteristic increase in G→T, G→C, and deletion mutations that is associated with BaP-induced mutagenesis (middle panel). An increased proportion of BaP-induced mutations are clonally expanded (bottom panel). Each circle within a group corresponds to a different mutation and the area of each circle represent the number of times that the mutation was observed within an animal. *P<0.05 versus 0 mg/kg/day BaP.
Table 2.
Summary of lacZ mutant frequency in bone marrow and ovaries of MutaMouse females exposed to BaP in utero during the period of organogenesis.
| Tissue | Dose administered to dam (mg/kg/d) |
No of F1 animals |
Total Mutants |
Total Plaque Forming Units |
Avga MF (×105) |
SEb | P-valuec |
|---|---|---|---|---|---|---|---|
| Bone | 0 | 11 | 327 | 6,223,765 | 4.5 | 1.89 | - |
| 10 | 20 | 983 | 8,154,942 | 13.6 | 2.50 | 0.1530 | |
| marrow | 20 | 15 | 2,449 | 4,502,489 | 54.4 | 7.15 | <0.0001 |
| 40 | 8 | 3,471 | 3,493,579 | 105.9 | 10.97 | <0.0001 | |
|
Ovaries |
0 | 8 | 76 | 1,326,824 | 5.7 | 1.37 | - |
| 10 | 5 | 176 | 1,032,932 | 17.5 | 2.67 | 0.0030 | |
| 20 | 7 | 704 | 1,743,973 | 41.9 | 3.16 | <0.0001 | |
| 40 | 5 | 1,059 | 760,410 | 138.9 | 8.90 | <0.0001 |
Arithmetic mean of mutant frequency (MF) of all animals in group
Standard error (SE) calculated using the esticon function in the doBy R package
Generalized linear model in R with quasi-Poisson distribution and Bonferonni correction
High-throughput next-generation sequencing of the mutant plaques recovered from the MutaMouse lacZ transgene revealed that the BaP-induced mutation spectrum was similar between the two tissues and that they both contained an increase in G>T and G>C transversions as well as deletions (Figure 2, middle panel). In addition, the proportion of clonally expanded G-based transversions increased following in utero exposure in both tissues, consistent with the early developmental origin of such mutations (Figure 2, lower panel). Finally, the spontaneous mutation spectra of bone marrow and ovary/oviduct are also comparable, with both being comprised of roughly 50% G>A transitions.
We next analyzed our lacZ sequencing data to determine if BaP-induced mutational signatures are found in human cancers. The lacZ transgene in MutaMouse consists of a ~3000 bp sequence of bacterial origin, which is heavily biased towards GC rich sequence composition [Beal et al. 2015]. Thus, the deconstruction of signatures was performed using signature weights that were normalized to the ratio of lacZ trinucleotide frequencies to human trinucleotide frequencies. When adjusted in this manner, decomposition of known mutation signatures from the lacZ mutants of BaP exposed mice and Pearson Correlation revealed Signature 4, which is enriched for C>A mutations, as the best matching signature to explain bone marrow (Pearson Coefficient: 0.74) and ovary/oviduct (Pearson Coefficient: 0.61) mutations (Figure 3). Note that substitutions in the COSMIC signatures are displayed with respect to the mutated pyrimidine in the base pair nd, therefore, C>A in Figure 3 is equivalent to G>T in Figure 2. Signature 4 is associated with tobacco smoking and is also the main signature observed after exposure of mouse embryonic fibroblasts (MEF) to BaP [Nik-Zainal et al. 2015, Alexandrov et al. 2016]. Comparison of the BaP signature in MEF with our signature obtained with the lacZ gene shows an improved Pearson Coefficient for both bone marrow (0.81) and ovary (0.68). Thus, these results further support that the observed mutations in our study are the direct consequence of BaP exposure and that this is a relevant mutational mechanism in human cancer.
Figure 3. Tissue-specific lacZ transgene mutation signatures after transplacental exposure to BaP.
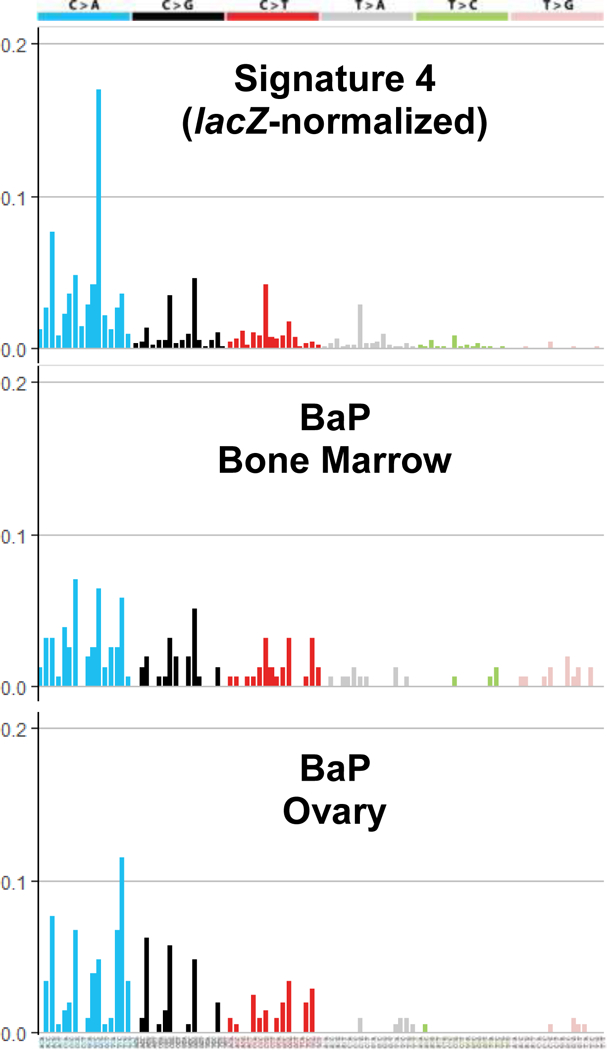
Top to botton: lacZ normalized Signature 4; BaP signature in bone marrow; BaP signature in the ovary. Note that mutation signatures are represented using the pyrimidine context, and therefore C>A mutations here are equivalent to G>T mutations in Figure 2. Each bar represents the relative frequency of the mutation in the trinucleotide contexts shown on the x-axis.
Discussion
We examined the potential relationship between ovarian germ cell depletion and induction of ovarian/oviductal mutations following in utero exposure to BaP. We observed strong, inverse dose-response relationships for ovarian follicle counts and mutant frequencies, with follicle numbers decreasing and mutant frequencies increasing with dose. Notably, ovarian primordial follicle numbers were decreased by 97% in the lowest dose group, and essentially no ovarian follicles remained in the ovaries of mice exposed to the two higher doses. Ovaries in the higher dose groups showed histological changes that are likely precursor lesions of epithelial ovarian tumors. The mutation spectra were very similar between ovaries/oviducts and bone marrow of F1 females exposed to BaP in utero and were highly correlated with a human cancer signature associated with tobacco smoking.
Although environmental mutagens such as BaP are well-established causative agents of tumor initiation in adults, the role that environmental mutagens play in the formation of tumors following transplacental exposures is not as well studied. We previously showed that treatment of pregnant C57BL/6J mice with 10 mg/kg/day BaP during the same dosing window as in the present study resulted in 50% prevalence of bilateral and 33% prevalence of unilateral epithelial ovarian tumors in wild type F1 offspring at 7.5 months of age, with no tumors noted in other tissues [Lim et al. 2013]. Two other PAHs, 7,12-dimethylbenz[a]anthracene (DMBA) and dibenz[def,p]chrysene, are transplacental ovarian tumorigens at doses that cause tumors in other organs [Goerttler et al. 1981, Shorey et al. 2012]. Like BaP, DMBA depletes ovarian germ cells in fetal and neonatal ovaries, and for DMBA this has been shown to be aryl hydrocarbon receptor (AHR)-dependent [Matikainen et al. 2001, Matikainen et al. 2002]. To our knowledge no studies have investigated the impact of Ahr deletion on the ovarian toxicity of prenatal BaP exposure. However, one previous study reported that Ahr non-responsive genotype of the fetus was protective against transplacental carcinogenesis by the methylated PAHs DMBA and 3-methylcholanthrene, but had no effect on transplacental carcinogenesis by BaP [Anderson et al. 1995]. Antral follicles produce steroid hormones (estrogens and progestins) and peptide hormones, such as inhibin, that feed back to the hypothalamus to inhibit gonadotropin releasing hormone secretion and to the pituitary to inhibit luteinizing hormone and follicle stimulating hormone secretion (LH and FSH). Germ cell/ovarian follicle depletion results in lack of antral follicles with resultant loss of ovarian negative feedback and high levels of circulating LH and FSH. These high gonadotropin levels are thought to play an ovarian tumor-promoting role [Salehi et al. 2008]. In BaP-exposed ovaries that were devoid of follicles, we observed multiple areas of cell packets surrounded by fibroblast-like cells, which in areas were degenerating and forming tubular, epithelial-lined structures, likely representing the early stages of ovarian tumorigenesis. These findings are similar to surface epithelial papillomatosis, intraovarian epithelial inclusions and invaginations, and epithelial pseudostratification, all of which are observed in ovaries removed prophylactically from women with familial ovarian cancer, and tubular structures observed in ovaries of aged rats, which are thought to be precursor lesions to epithelial ovarian tumors [Engle 1946, Salazar et al. 1996, Romero and Bast 2012, Ng and Barker 2015].
In this study, we observed statistically significantly increased mutant frequencies in the lacZ transgene at all doses evaluated in the ovaries/oviducts. Bone marrow mutant frequencies were also elevated at all doses in these females and closely matched those observed in the male siblings in our previous work [Meier et al. 2017]. Based on dose-response modelling of the lacZ mutant frequencies, the ovaries/oviducts accumulated mutations at lower doses than the bone marrow (benchmark doses of 1.3 and 3.6 mg/kg/d, respectively, with non-overlapping confidence intervals). The mutation spectra of the exposed bone marrow and ovaries both show a prominent increase in G>T and G>C mutations, and a relative decrease in G>A mutations, consistent with previous studies that examined the mutation spectrum of B[a]P [Beal et al. 2015]. The presence of B[a]P induced mutations and the low BMD observed in this study suggest that developing ovaries may be more sensitive to environmental mutagens than other tissues, and because the mutations induced by environmental agents can occur randomly throughout the genome, any cancer driver gene may be affected. Furthermore, as it was observed in males [Meier et al. 2017], the number of clonally expanded mutants was higher in BaP-exposed animals (Figure 2), consistent with observations that in utero exposure is a sensitive window for the induction of germline and somatic mosaicism.
Mutations in the KRAS GTPase are the second most common mutation in epithelial ovarian cancers in humans, and 32–50% of KRAS mutations in these cancers are G>T transversions in codon 12 (G12V) [Cuatrecasas et al. 1997, Matias-Guiu and Prat 1998, MacKenzie et al. 2015]. The mutation spectrum in the lacZ gene was consistent with these being the predominant mutation types. We also observed increased deletions in the ovaries, which is consistent with what was observed in somatic tissues, but not in sperm, from males exposed in utero [Meier et al. 2017]. It should be noted that since there were very few follicles in the treated groups, the ovarian mutation spectra and frequency presented here essentially represent the contribution of somatic cells only. Indeed, the mutation spectra in the somatic tissues of control animals and those exposed to BaP in utero are highly consistent between females in this study and males in Meier et al. [Meier et al. 2017], suggesting that there are few sex-related differences in mutation mechanisms.
The mutations sequenced in this study, when considered in their trinucleotide context, contain the previously described Signature 4 that has been associated with cigarette smoking (Figure 3) [Alexandrov et al. 2013, Alexandrov et al. 2016]. In addition, we observed an even better correlation between our lacZ signature and the BaP signature found in MEF [Nik-Zainal et al. 2015]. These findings are to be expected given that Signature 4 is a composite of: (1) the initial mutations induced by an exposure (as detected by the lacZ signature and the BaP signature) and the mutations accumulated during the carcinogenesis process; and (2) the mutation signatures induced by the other PAHs and carcinogenic compounds that are found in tobacco smoke [US Department of Health and Human Services 2010]. These results suggest that transgenic rodent models can provide important information on the mutational mechanisms and signatures underlying cancer formation.
Comparison of the mutations found in 441 ovarian cancers in the TCGA mutation dataset indicates that a subset of these cancers (at least 12 cases) were driven primarily by the smoking signature (Supplementary Figure 1), which is congruent with epidemiological studies demonstrating an increased risk for mucinous epithelial ovarian cancers is in women who smoke [Rossing et al. 2008, Tworoger et al. 2008, Gates et al. 2010]. Our study adds experimental support to such a molecular basis. It would be of interest to sequence ovarian tumors induced by transplacental BaP exposure to identify cancer-driving mutations. It would also be of interest in future studies to analyze ovarian and oviductal mutations separately because ovarian tumors may arise from either of these tissues [Kim et al. 2012, Mullany and Richards 2012, Kim et al. 2015]. We were not able to do this in the present study because of the small size of these tissues, particularly the atrophic ovaries from the BaP-exposed females.
To our knowledge, this is the first report of ovarian follicle numbers in MutaMouse. The MutaMouse transgenic strain was generated on a (BALB/c x DBA/2)F1 genetic background [Gossen et al. 1989]. The total number of ovarian follicles in 10 week old MutaMouse controls in the present study is about 1.5-fold higher than the follicle number in 6-week old C57BL/6J mice [Luderer et al. 2017]. MutaMouse females appear to be similarly sensitive to in utero germ cell depletion by BaP as C57BL/6J females. The ED50 for follicle depletion in MutaMouse in the present study is clearly below 10 mg/kg/day and is consistent with the ED50 of about 2 mg/kg/day previously observed in C57BL/6J with the same dosing period [Lim et al. 2013, Luderer et al. 2017]. When comparing the effects of in utero exposure to BaP in MutaMouse on the ovaries in the present study to those on the testis, statistically significant ovarian follicle depletion is observed at 10 mg/kg/day BaP, while no statistically significant effects on testis weights, epididymal sperm counts, or sperm motility were observed in the male siblings at that dose [Meier et al. 2017]. Similarly, greater sensitivity of the developing ovary than the developing testis was observed in C57BL/6J mice exposed in utero to BaP during the same developmental window [Nakamura et al. 2012, Lim et al. 2013], as well as in cultured embryonic gonads of C57BL/6J mice [Lim et al. 2016]. MutaMouse males appear to be less sensitive to the reproductive toxicity of in utero exposure to BaP than C57BL/6J males. Using the same dosing regimen, epididymal sperm counts in C57BL/6J mice exposed to 10 mg/kg/day were 68% decreased compared to oil controls [Nakamura et al. 2012], while no significant decreases were observed in MutaMouse males at that dose [Meier et al. 2017]. Overall, the data in this and other papers indicate that although in utero BaP exposure adversely impacts both male and female fertility, ovarian impairment occurs at lower doses than testicular effects.
Biomonitoring studies show that essentially all people are exposed to PAHs [NHANES 2009, Aquilina et al. 2010, Health Canada 2015], and BaP has been measured in human follicular fluid [Neal et al. 2008]. Exposure of pregnant rats to BaP via oral or inhalation routes results in BaP-dose-dependent concentrations of BaP metabolites in blood and tissues of offspring on the day of birth, with levels declining over the subsequent postnatal weeks [Wu et al. 2003, Jules et al. 2012]. Smoking related DNA and albumin adducts, including BaP-specific adducts, have been detected in human fetal tissues [Hansen et al. 1993, Autrup and Vestergaard 1996]. Humans are exposed to dozens of mutagenic PAHs in addition to BaP. The estimated daily intake of mutagenic PAHs by a hypothetical non-occupationally exposed 60 kg woman from inhalation of polluted urban air (3 μg/day), smoking cigarettes (10 μg/day) and consuming grilled and smoked foods (17 μg/day) would be on the order of 0.5 μg/kg/day [Menzie et al. 1992, ATSDR 1995, Shopland et al. 2001, Lodovici et al. 2004] or about 3.5 μg/kg/day in mice after an allometric interspecies correction [USEPA 2011]. The estimated daily dose of BaP alone in a highly exposed woman from air (0.04 μg/day), tobacco smoke (3 μg/day), and food (1.6 μg/day) would be about 0.08 μg/kg/day, equivalent to 0.5 μg/kg/day in mice. Thus, the daily doses of BaP used in this study are three to four orders of magnitude higher than estimated human exposure to total mutagenic PAHs and four to five orders of magnitude higher than estimated human exposure to BaP. However, gestation in humans is much longer than in rodents, resulting in an estimated cumulative dose during the course of pregnancy in our hypothetical woman of about 150 μg/kg BaP after allometric interspecies correction, which is 1333-fold less than the cumulative dose of 200 mg/kg BaP in the 10 mg/kg/day dose group in the present study. Given that the 10 mg/kg/day dose in the present study resulted in depletion of 97% of ovarian follicles and statistically significant increases in ovarian mutations, future studies should examine the ovarian effects of lower transplacental doses of BaP.
In summary, depletion of ovarian germ cells by transplacental BaP exposure is inversely associated with ovarian/oviductal mutant frequency and increases in G>T and G>C transversions and deletions. The similar mutation spectra in the ovaries and bone marrow of females and in various somatic tissues in transplacentally exposed males [Meier et al. 2017] suggest that there are few sex-related differences in mechanisms by which in utero BaP exposure induces mutations. Together with ovarian lesions in the two highest dose groups that are similar to lesions observed in ovaries from women at high risk of ovarian cancer, our data support a role for ovarian/oviductal somatic mutations in the pathogenesis of epithelial ovarian tumors caused by prenatal BaP exposure.
Supplementary Material
Acknowledgments:
We thank J. O’Brien, M. Rosales, B Allen, J. Gingerich, and L. Soper for help with tissue collections and conduct of the lacZ assay. We thank J. Kim for embedding and sectioning and S. Meharda for help with hematoxylin and eosin staining of ovarian serial sections.
Grant Sponsors:
Supported by NIH R01ES020454 to U.L; NIH grant P30CA062203, the University of California Irvine (UC Irvine) Chao Family Comprehensive Cancer Center; UC Irvine Center for Occupational and Environmental Health; Health Canada Intramural Funding.
Footnotes
Conflict of Interest Statement: The authors declare that they have no actual or potential competing financial interests.
References
- Alexandrov LB, Ju YS, Haase K, Van Lo P, Martincorena I, Nik-Zainal S, Totoki Y, Fujimoto A, Nakagawa H, Shibata T, et al. (2016). Mutational Signatures Associated with Tobacco Smoking in Human Cancer. Science 354(6312): 618–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. (2013). Signatures of mutational processes in human cancer. Nature 500(7463): 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson LM, Diwan BA, Fear NT, Roman E (2000). Critical Windows of Exposure for Children’s Health: Cancer in Human Epidemiological Studies and Neoplasms in Experimental Animals. Environ. Health Perspect. 108 (Suppl 3): 573–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson LM, Rushkie S, Carter J, Pittinger S, Kovatch RM, Riggs CW (1995). Fetal Mouse Susceptibility to Transplacental Carcinogenesis: Differential Influence of Ah Receptor Phenotype on Effects of 3-methylcholanthrene, 12-dimethylbenz[a]anthracene, and Benzo[a]pyrene. Pharmacogenetics 5: 364–372. [DOI] [PubMed] [Google Scholar]
- Aquilina NJ, Delgado-Saborit JM, Meddings C, Baker S, Harrison RM, Jacob PI, Wilson M, Yu L, Duan M, Benowitz NL (2010). Environmental and Biological Monitoring of Exposures to PAHs and ETS in the General Population. Environ. Int. 36: 763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ATSDR (1995). Toxicological Profile for Polycyclic Aromatic Hydrocarbons. Atlanta, GA, US Department of Health and Human Services, Public Health Service, Agency for Toxic Substances and Disease Registry. [PubMed] [Google Scholar]
- Autrup H, Vestergaard AB (1996). Transplacental Transfer of Environmental Genotoxins-Polycyclic Aromatic Hydrocarbon-Albumin in Nonsmoking Women. Environ. Health Perspect. 104(Suppl 3): 625–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MA, Gagné R, Williams A, Marchetti F, Yauk CL (2015). Characterizing Benzo[a]pyrene-Induced lacZ Mutation Spectrum in Transgenic Mice Using Next-Generation Sequencing. BMC Genomics 16: 812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biancifiori C, Bonser GM, Caschera F (1961). Ovarian and Mammary Tumours in Intact C3Hb Virgin Mice Following a Limited Dose of Four Carcinogenic Chemicals. Br. J. Cancer 15: 270–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesecker LG, Spinner NB (2013). A Genomic View of Mosaicism and Human Disease. Nat. Rev. Genet. 14: 307–320. [DOI] [PubMed] [Google Scholar]
- Borman SM, Christian PJ, Sipes IG, Hoyer PB (2000). Ovotoxicity in Female Fischer Rats and B6 Mice Induced by Low-Dose Exposure to Three Polycyclic Aromatic Hydrocarbons: Comparison through Calculation of an Ovotoxic Index. Toxicol. Appl. Pharmacol. 167: 191–198. [DOI] [PubMed] [Google Scholar]
- Cuatrecasas M, Villanueva A, Matias-Guiu X, Prat J (1997). K-ras Mutations in Mucinous Ovarian Tumors: A Clinicopathologic and Molecular Study of 95 Cases. Cancer 79: 1581–1586. [DOI] [PubMed] [Google Scholar]
- Engle ET (1946). Tubular Adenomas and Testis-Like Tubules of the Ovaries of Aged Rats. Cancer Res. 6: 578–582. [PubMed] [Google Scholar]
- Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole C,G, Ward S, Dawson E, Ponting L, et al. (2017). COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 45(D1): D777–D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gates MA, Rosner BA, Hecht JL, Tworoger SS (2010). Risk Factors for Epithelial Ovarian Cancer by Histologic Subtype. Am. J. Epidemiol. 171(1): 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring JS, Fischer B, Lawrence M, Huber W (2015). SomaticSignatures: inferring mutational signatures from single-nucleotide variants. Bioinformatics 31(22): 3673–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingerich JD, Soper L, Lemieux C, Marchetti F, Douglas GR (2014). Transgenic rodent gene mutation assay in somatic cells Genotoxicity and DNA repair. A practical approach. Sierra ML and Gaivao I. Humana Press: 305–321. [Google Scholar]
- Goerttler K, Loehrke H, Hesse B, Milz A, Schweizer J (1981). Diaplacental Initiation of NMRI Mice with 7,12-dimethylbenz[a]anthracene during Gestation Days 6–20 and Postnatal Treatment of the F1-generation with the Phorbol Ester 12-O-tetradecanoylphorbol-13-acetate: Tumor Incidence in Organs Other than Skin. Carcinogenesis 2(11): 1087–1094. [DOI] [PubMed] [Google Scholar]
- Gossen JA, de Leeuw WJF, Tan CHT, Zwarthoff EC, Berends F, Lohman PHM, Knook DL, Vijg J (1989). Efficient Rescue of Integrated Shuttle Vectors from Transgenic Mice: A Model for Studying Mutations in Vivo. Proc. Natl. Acad. Sci. U. S. A. 86(20): 7971–7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen C, Asmussen I, Autrup H (1993). Detection of Carcinogen-DNA Adducts in Human Fetal Tissues by the 32P-Postlabeling Procedure. Environ. Health Perspect. 99: 229–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Health Canada (2015). Third Report on Human Biomonitoring of Environmental Chemicals in Canada. Ottawa, CA, Health Canada. [Google Scholar]
- Jules GE, Pratap S, Ramesh A, Hood DB (2012). In Utero Exposure to Benzo[a]pyrene Predisposes Offspring to Cardiovascular Dysfunction in Later-Life. Toxicology 295: 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Coffey DM, Creighton CJ, Yu Z, Hawkins SM, Matzuk MM (2012). High-Grade Serous Ovarian Cancer Arises from Fallopian Tube in a Mouse Model. Proc. Natl. Acad. Sci. U. S. A. 109(10): 3921–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Coffey DM, Ma L, Matzuk MM (2015). The Ovary Is an Alternative Site of Origin for High-Grade Serous Ovarian Cancer in Mice. Endocrinology 156(6): 1975–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurman RJ, Shih I-M (2016). The Dualistic Model of Ovarian Carcinogenesis. Revisited, Revised, and Expanded. Am. J. Pathol 186(4): 733–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei L, Spradling AC (2013). Female Mice Lack Adult Germ-Line Stem Cells but Sustain Oogenesis Using Stable Primordial Follicles. Proc. Natl. Acad. Sci. U. S. A. 110(21): 8585–8590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Kong W, Lu M, Luderer U (2016). The Mouse Fetal Ovary Has Greater Sensitivity than the Fetal Testis to Benzo[a]pyrene-Induced Germ Cell Death. Toxicol. Sci. 152(2): 372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Lawson GW, Nakamura BN, Ortiz L, Hur JA, Kavanagh TJ, Luderer U (2013). Glutathione-Deficient Mice Have Increased Sensitivity to Transplacental Benzo[a]pyrene-Induced Premature Ovarian Failure and Ovarian Tumorigenesis. Cancer Res. 73(2): 908–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodovici M, Akpan V, Evangelisti C, Dolara P (2004). Sidestream Tobacco Smoke as the Main Predictor of Exposure to Polycyclic Aromatic Hydrocarbons. J. Appl. Toxicol. 24: 277–281. [DOI] [PubMed] [Google Scholar]
- Lopez SG, Luderer U (2004). Effects of Cyclophosphamide and Buthionine Sulfoximine on Ovarian Glutathione and Apoptosis. Free Radic. Biol. Med. 36: 1366–1377. [DOI] [PubMed] [Google Scholar]
- Luderer U, Myers MB, Banda M, McKim KL, Ortiz L, Parsons BL (2017). Ovarian Effects of Prenatal Exposure to Benzo[a]pyrene: Roles of Embryonic and Maternal Glutathione Status. Reprod. Toxicol. 69: 187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR (2013). Genome Mosaicism - One Human, Multiple Genomes. Science 341: 358–359. [DOI] [PubMed] [Google Scholar]
- Lupski JR (2015). Structural Variation Mutagenesis of the Human Genome: Impact on Disease and Evolution. Environ. Mol. Mutagen. 56(5): 419–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie KM, Angevine DM (1981). Infertility in Mice Exposed in Utero to Benzo(a)pyrene. Biol. Reprod. 24: 183–191. [DOI] [PubMed] [Google Scholar]
- MacKenzie R, Kommoss S, Winterhoff BJ, Kipp BR, Garcia JJ, Voss J, Halling K, A K, Senz J, Yang W, et al. (2015). Targeted Deep Sequencing of Mucinous Ovarian Tumors Reveals Multiple Overlapping RAS-Pathway Activating Mutations in Borderline and Cancerous Neoplasms. BMC Cancer 15: 415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matias-Guiu X, Prat J (1998). Molecular Pathology of Ovarian Carcinomas. Virchows Arch. 433: 103–111. [DOI] [PubMed] [Google Scholar]
- Matikainen T, Moriyama T, Morita Y, Perez GI, Korsmayer SJ, Sherr DH, Tilly JL (2002). Ligand Activation of the Fetal Aromatic Hydrocarbon Receptor Transcription Factor Drives Bax-Dependent Apoptosis in Developing Fetal Ovarian Germ Cells. Endocrinology 143(2): 615–620. [DOI] [PubMed] [Google Scholar]
- Matikainen T, Perez GI, Jurisicova A, Pru JK, Schlezinger JJ, Ryu H-Y, Laine J, Sakai T, Korsmeyer SJ, Casper RF, et al. (2001). Aromatic Hydrocarbon Receptor-Driven Bax Gene Expression is Required for Premature Ovarian Failure Caused by Biohazardous Environmental Chemicals. Nat. Genet. 28: 355–360. [DOI] [PubMed] [Google Scholar]
- Mattison DR, Thorgeirsson SS (1979). Ovarian Aryl Hydrocarbon Hydroxylase Activity and Primordial Oocyte Toxicity of Polycyclic Aromatic Hydrocarbons in Mice. Cancer Res. 39: 3471–3475. [PubMed] [Google Scholar]
- Mattison DR, White NB, Nightingale MR (1980). The Effect of Benzo(a)pyrene on Fertility, Primordial Oocyte Number, and Ovarian Response to Pregnant Mare’s Serum Gonadotropin. Pediatr Pharmacol 1: 143–151. [PubMed] [Google Scholar]
- McLaren A (2003). Primordial Germ Cells in the Mouse. Dev. Biol. 262: 1–15. [DOI] [PubMed] [Google Scholar]
- Meier MJ, O’Brien JM, Beal MA, Allan B, Yauk CL, Marchetti F (2017). In Utero Exposure to Benzo[a]pyrene Increases Mutation Burden in the Soma and Sperm of Adult Mice. Environ. Health Perspect. 125(1): 82–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzie CA, Potocki BB, Santodonato J (1992). Ambient Concentrations and Exposure to Carcinogenic PAHs in the Environment. Environ. Sci. Technol. 26(7): 1278–1284. [Google Scholar]
- Miller PB, Charleston JS, Battaglia DE, Klein NA, Soules MR (1997). An Accurate, Simple Method for Unbiased Determination of Primordial Follicle Number in the Primate Ovary. Biol. Reprod. 56: 909–915. [DOI] [PubMed] [Google Scholar]
- Mody JK (1960). The Action of Four Carcinogenic Hydrocarbons on the Ovaries of IF Mice and the Histogenensis of Induced Tumors. Br. J. Cancer 14: 256–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullany LK, Richards JS (2012). Minireview: Animal Models and Mechanisms of Ovarian Cancer Development. Endocrinology 153(4): 1585–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers M, Britt KL, Wreford NGM, Ebling FJP, Kerr JB (2004). Methods for quantifying follicular numbers within the mouse ovary. Reproduction 127(5): 569–580. [DOI] [PubMed] [Google Scholar]
- Nakamura BN, Mohar I, Lawson GW, Hoang YD, Cortés MM, Ortiz L, Patel R, Rau BR, McConnachie L,Kavanagh TJ, et al. (2012). Increased Sensitivity to Testicular Toxicity of Transplacental Benzo[a]pyrene Exposure in Male Glutamate Cysteine Ligase Modifier Subunit Gclm−/−) Knockout Mice. Toxicol. Sci. 126(1): 227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal MS, Zhu J, Foster WG (2008). Quantification of Benzo[a]pyrene and Other PAHs in the Serum and Follicular Fluid of Smokers Versus Non-smokers. Reprod. Toxicol. 25: 100–106. [DOI] [PubMed] [Google Scholar]
- Ng A, Barker N (2015). Ovary and Fimbrial Stem Cells: Biology, Niche and Cancer Origins. Nat Rev Molec Cell Biol 16: 625–638. [DOI] [PubMed] [Google Scholar]
- NHANES (2009). Fourth National Report on Human Exposure to Environmental Chemicals, Department of Health and Human Services, Centers for Disease Control and Prevention. [Google Scholar]
- Nik-Zainal S, Kucab JE, Morganella S, Glodzik D, Alexandrov LB, Arlt VM, Weninger A, Hollstein M, Stratton MR, Phillips DH (2015). The genome as a record of environmental exposure. Mutagenesis 30(6): 763–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OECD (2013). Test No. 488: Transgenic Rodent Somatic and Germ Cell Gene Mutation Assays. Paris, France, Office for Economic Cooperation and Development (OECD) Publishing. [Google Scholar]
- Pepling ME (2006). From Primordial Germ Cell to Primordial Follicle: Mammalian Female Germ Cell Development. Genesis 44: 622–632. [DOI] [PubMed] [Google Scholar]
- Romero I, Bast RC (2012). Minireview: Human Ovarian Cancer: Biology, Current Management, and Paths to Personalizing Therapy. Endocrinology 153(4): 1593–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C (2016). deconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 17(1): 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossing MA, Cushing-Haugen KL, Wicklund KG, Weiss NS (2008). Cigarette Smoking and Risk of Epithelial Ovarian Cancer. Cancer Causes Control 19: 413–420. [DOI] [PubMed] [Google Scholar]
- Salazar H, Godwin AK, Daly MB, Laub PB, Hogan WM, Rosenblum N, Boente MP, Lynch HT, Hamilton TC (1996). Microscopic Benign and Invasive Malignant Neoplasms and a Cancer-Prone Phenotype in Prophylactic Oophorectomies. J. Natl. Cancer Inst. 88: 1810–1820. [DOI] [PubMed] [Google Scholar]
- Salehi F, Dunfield L, Phillips KP, Krewski D, Vanderhyden BC (2008). Risk Factors for Ovarian Cancer: An Overview with Emphasis on Hormonal Factors. J. Toxicol. Environ. Health. B. Crit. Rev. 11(3–4): 301–321. [DOI] [PubMed] [Google Scholar]
- Shopland DR, Burns DM, Benowitz NL, Amacher RH, Eds. (2001). Risks Associated with Smoking Cigarettes with Low Machine-Measured Yields of Tar and Nicotine. Smoking and Tobacco Control Monographs, U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health, National Cancer Institute. [Google Scholar]
- Shorey LE, Castro DJ, Baird WM, Siddens LK, Löhr CV, Matzke MM, Waters KM, Corley RA, Williams DE(2012). Transplacental Carcinogenesis with Dibenz[def,p]chrysene (DBC): Timing of Maternal Exposures Determines Target Tissue Response in Offspring. Cancer Lett. 317: 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilly JC, Niikura Y, Rueda BR (2009). The Current Status of Evidence for and Against Postnatal Oogenesis in Mammals: A Case of Ovarian Optimism Versus Pessimism? Biol. Reprod. 80(1): 2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tworoger SS, Gertig DM, Gates MA, Hecht JL, Hankinson SE (2008). Caffeine, Alcohol, Smoking, and the Risk of Incident Epithelial Ovarian Cancer. Cancer 112: 1169–1177. [DOI] [PubMed] [Google Scholar]
- US Department of Health and Human Services (2010). How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-Attributable Disease: A Report of the Surgeon General. Atlanta, GA, Centers for Disease Control and Prevention. [PubMed] [Google Scholar]
- USEPA (2011). Recommended Use of Body Weight3/4 as the Default Method in Derivation of the Oral Reference Dose. O. o. t. S. A. R. A. Forum. Washington, DC, U.S: Environmental Protection Agency. [Google Scholar]
- Wu J, Ramesh A, Nayyar T, Hood DB (2003). Assessment of Metabolites and AhR and CYP1A1 mRNA Expression Subsequent to Prenatal Exposure to Inhaled Benzo(a)pyrene. Int. J. Dev. Neurosci. 21: 333–346. [DOI] [PubMed] [Google Scholar]
- Xue W, Warshawsky D (2005). Metabolic Activation of Polycyclic Aromatic Hydrocarbon and Heterocyclic Aromatic Hydrocarbons and DNA Damage: A Review. Toxicol. Appl. Pharmacol. 206: 73–93. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zheng W, Shen Y, Adhikari D, Ueno H, Liu K (2012). Experimental Evidence Showing that No Mitotically Active Female Germline Progenitors Exist in Postnatal Mouse Ovaries. Proc. Natl. Acad. Sci. U. S. A. 109(31): 12580–12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.