

Abstract
The insufficient resolution of conventional methods has long limited the structural elucidation of cellulose and its derivatives, especially for those with relatively low crystallinities or in native cell walls. Recent 2D/3D solid-state NMR studies of 13C uniformly labeled plant biomaterials have initiated a re-investigation of our existing knowledge in cellulose structure and its interactions with matrix polymers but for unlabeled materials, this spectroscopic method becomes impractical due to limitations in sensitivity. Here, we investigate the molecular structure of unlabeled cotton cellulose by combining natural abundance 13C-13C 2D correlation solid-state NMR spectroscopy, as enabled by the sensitivity-enhancing technique of dynamic nuclear polarization (DNP), with statistical analysis of the observed and literature-reported chemical shifts. The atomic resolution allows us to monitor the loss of Iα and Iβ allomorphs and the generation of a novel structure during ball-milling, which reveals the importance of large crystallite size for maintaining the Iα and Iβ model structures. Partial order has been identified in the “disordered” domains, as evidenced by a discrete distribution of well-resolved peaks. This study not only provides heretofore unavailable high-resolution insights into cotton cellulose but also presents a widely applicable strategy for analyzing the structure of cellulose-rich materials without isotope-labeling. This work was part of a multi-technique study of ball-milled cotton described in the previous article in the same issue.
Keywords: Cotton, Cellulose, Solid-state NMR, Dynamic nuclear polarization, Natural abundance
Graphical abstract
Introduction
For decades of studies on cellulose structure and crystallinity, solid-state NMR (ssNMR) has been a standard method that relies primarily on the measurement of 1D 13C spectra followed by analysis of peak multiplicity and intensities (Atalla and Vanderhart 1984; Atalla and Vanderhart 1999; Larsson et al. 1999). Due to limited resolution, ambiguity may exist in the spectral deconvolution and resonance assignment of different components. Recent studies on uniformly 13C-labeled plant materials have substantially improved the resolution by measuring two- and three-dimensional (2D/3D) 13C-13C correlation spectra, which have advanced our understanding of the structure of cellulose and its interactions with matrix polymers (Simmons et al. 2016; Wang and Hong 2016; Wang et al. 2015; Wang et al. 2016a). The atomic resolution of ssNMR allows us to resolve seven types of cellulose allomorphs in plant primary cell walls and directly measure their spatial location and hydroxymethyl conformation (Phyo et al. 2018; Wang et al. 2016b). Two types of glucan chains that primarily adopt the gauche-trans (gt) conformation for the O-6 primary alcohol are found on the cellulose surfaces, while another five types of glucan chains having O-6 in the trans gauche tg conformation form the internal cores of microfibrils. The conformational structures of these cellulose forms in primary plant cell walls are found to differ substantially from the crystallographic structures of Iα and Iβ allomorphs that were determined using purified cellulose microcrystals from tunicate and algae (Kono and Numata 2006; Nishiyama et al. 2002; Nishiyama et al. 2003). This inconsistency is also retained in the secondary cell walls from the mature stems of grasses and woods (unpublished results), indicating that cellulose structures in their native cell walls are far more complicated than in the purified, highly crystalline state.
Therefore, it becomes imperative to establish non-destructive and high-resolution methods to evaluate the consistency and diversity of cellulose structure from a wide range of sources using 2D ssNMR. However, isotope enrichment, a pre-requisite for multidimensional ssNMR, has been a major barrier. Feeding a plant with 13CO2 and/or 13C-glucose as the sole carbon source is typically very expensive and becomes impractical for plants with large size and/or long lifecycles. Without isotope-labeling, we can still rely on the very low natural 13C abundance (1%) for 1D experiments, but the probability for observing a cross peak between two carbons in 2D spectra is only 0.01%, making it unrealistic for conventional NMR. This issue can be addressed by the implementation of the cutting-edge technique of Dynamic Nuclear Polarization (DNP), which enhances NMR sensitivity by tens to hundreds of times by transferring polarization from the electrons in radicals to the NMR-active nuclei in biomacromolecules (Koers et al. 2014; Lee et al. 2015; Ni et al. 2013; Rossini et al. 2013; Saliba et al. 2017). This method has been applied to study many 13C/15N-labeled carbohydrate-rich systems such as the cell walls in plants, bacteria and fungi (Kang et al. 2018; Perras et al. 2017; Takahashi et al. 2013a; Wang et al. 2013). More importantly, it enables structural characterization of unlabeled materials using 2D 13C correlation spectroscopy (Mentink-Vigier et al. 2017; Rossini et al. 2012; Takahashi et al. 2012), thus allowing us to rapidly screen cellulose structures in various systems without worrying about isotope-enrichment.
Here we employ DNP ssNMR on a Wiley-milled cotton sample (the control) and a subsequently ball-milled cotton sample to characterize the structural change of cellulose using unlabeled samples. 2D 13C-13C correlation on these unlabeled samples, assisted by statistical analysis of 13C chemical shifts, allows us to obtain unprecedented molecular information on the structural changes of cellulose. First, the control cotton sample has a good match with the Iα and Iβ structures from model cellulose. Second, these Iα and Iβ structures of Wiley-milled cotton cellulose are fully removed by 2 hours ball-milling. Third, the ball-milled cellulose adopts a new type of chain-arrangement that cannot align with any of the existing structures. Fourth, the disordered domains consist of a collection of discrete conformers instead of a continuous distribution of conformations. Statistical analysis of 13C chemical shifts revealed how these observed sub-forms correlate with literature structures. These data suggest that the crystallographic structures only apply to model cellulose with relatively large crystallite size, and also revise our understanding of the disordered domains that were otherwise difficult to characterize. These methods are widely applicable to other functional cellulose or lignocellulosic materials.
Methods
Wiley-milled and ball-milled cotton samples
Wal-Mart White Cloud cotton balls were chopped in a Wiley mill (Eberbach E3300 mini cutting mill, Eberbach Corp., Belleville, Michigan) until they passed through a 20-mesh screen. Subsequently, the powdered samples were placed in a locally built ball mill (Forziati et al. 1950) with a motor running at 1750 rpm. We used a graduated beaker to measure around 500 mL volume of balls, which are 0.25 in. (~4 mm) stainless steel. The balls are then transferred to a 1 L steel jar (chromium plated). A fan was blown at the mill to minimize heating during the milling for 120 minutes. Samples were processed in an air-conditioned laboratory but without other attention to moisture at this stage.
Matrix-free preparation of DNP samples
The Wiley-milled cotton sample and the 120 min ball-milled material were processed using the matrix-free protocol for DNP experiments (Takahashi et al. 2013b; Takahashi et al. 2012). Briefly, the stock solution of 10 mM AMUPol radical (Sauvee et al. 2013), the DNP matrix, was prepared using D2O and a radical concentration of 10 mM. About 60 mg of the cotton sample was immersed in 150 μL of the AMUPol solution. The sample was stored in an Eppendorf tube and dried in a desiccator under vacuum for about ten hours. The excess radicals that did not mix well with cotton formed orange crystals that were manually removed using a needle. Another 3 μL D2O were added to ensure the moisturization of these cotton samples, previously reported to be essential for ensuring sensitivity enhancement (Takahashi et al. 2012). Around 50 mg of sample was packed into a thin-wall 3.2 mm zirconium rotor for DNP experiments. No silicone soft plug is used so that more space will be created. These thin-wall rotors allow us to pack 10-20 mg more material than the standard sapphire rotors.
Compared with the conventional methods that typically sacrifices a large volume for solvents, such as d8-glycerol, D2O, and H2O, the matrix-free method maximizes the amount of cotton material that can be packed into an NMR rotor, which increases the absolute sensitivity and accelerates the measurement of 2D 13C-13C correlation spectra at natural abundance. This protocol is appropriate for our cotton samples and other cellulose materials that are largely dried in their native state, but it is not suitable for well-hydrated bio-samples, such as plant seedling or stems.
DNP-NMR experiments
The DNP experiments were carried out on a 600MHz/395 GHz MAS-DNP spectrometer (Dubroca et al. 2018). The experiments were conducted using a 3.2 mm probe under 8 kHz MAS frequency. The microwave irradiation power was set to ~12 W and the temperature was 104 K with the microwave on and 98-100 K with the microwave off. The DNP buildup time was 7.2 s for the control cotton and 2.5 s for the material processed with 120 min ball-milling, which has better association with paramagnetic radicals due to enhanced disorder or surface area. 2D 13C-13C INADEQUATE spectra (Lesage et al. 1997) were measured using the SPC5 sequence (Hohwy et al. 1999) under 8 kHz MAS with 5 ms total recoupling time (2.5 ms each for excitation and reconversion). The recycle delays were set to be 1.3 times of the DNP buildup time: 9.4 s for the control cotton and 3.2 s for the ball-milled sample. The acquisition time was 18 ms and 5-6 ms for the direct and indirect dimensions. A spectral width of 60 ppm (130-190 ppm) was used for the indirect dimension of both spectra. This spectral width can effectively cover the double-quantum chemical shifts of cellulose and many other polysaccharides in plants, but it should be extended slightly (130-200 ppm) if arabinose exists in the sample. The number of t1 increment is 110 and 90 for the Wiley-milled and ball-milled samples, respectively. The number of scans was 32 and 64 the Wiley-milled and ball-milled samples, respectively. The experimental time is 9.5 hr for the control sample and 5 hr for the ball-milled sample.
Results and Discussion
DNP enables structural characterization of unlabeled cotton cellulose with atomic resolution
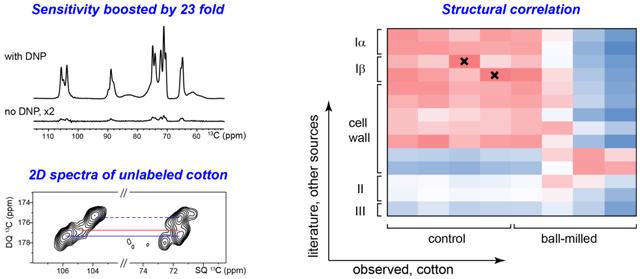
Multidimensional (2D/3D) solid-state spectroscopy is an indispensable method for characterizing the structure of cellulose, especially for those with substantial structural disorder and polymorphism, or those mixed with other biopolymers such as pectin, hemicellulose and lignin (Cosgrove and Jarvis 2012; Wang et al. 2012). This method, however, was not applicable to materials without isotope-enrichment due to the challenging sensitivity until the recent development of Dynamic Nuclear Polarization (DNP) technique. Figure 1a shows the 23-fold enhancement of NMR sensitivity achieved for the Wiley-milled cotton sample, which translates to a saving of NMR experimental time by 529 fold. It is remarkable that the spectral resolution is retained at the low temperature (104 K) of DNP experiment, evidenced by the 0.8-0.9 ppm 13C linewidth of resolved peaks, which could be explained by the high structural order of these cellulose materials. The peak multiplicity is also consistent with the Iα and Iβ allomorphs in literature (Kono et al. 2003b). A slightly lower enhancement, 18 fold, is achieved for the ball-milled sample, providing a time-saving of 324 fold for NMR experiments (Fig. 1b).
Fig. 1.

Sensitivity enhancement from Dynamic Nuclear Polarization (DNP). The DNP-enhancement spectra measured with microwave irradiation provide sensitivity enhancement of 23 and 18-fold for (a) Wiley-milled cotton and (b) ball-milled samples, respectively. The εon/off is the enhancement factor by comparing spectra recorded with microwave on and off. In contrast, the non-DNP spectra have very low intensities even with the same number of scans and measurement time. The representative positions for Iα and Iβ cellulose and their non-equivalent glucose units are labeled in magenta
Compared with room-temperature spectra (Ling et al. 2019), the intensity of the disordered cellulose (relative to the ordered forms) has reduced for both Wiley-milled and ball-milled samples. This can be attributed to the better association of DNP radicals with the surface disordered cellulose, which causes a more pronounced paramagnetic relaxation enhancement that suppresses the intensity of molecules nearby, within a few nanometers. In contrast, the room-temperature samples do not contain DNP radicals and their spectra are reported in an accompanying paper (Ling et al. 2019).
The benefit from the good sensitivity is that these 1D experiments can be finished within 5 mins, with only 8 to 32 scans, and the signal-to-noise ratios are 400-700, which is almost “noiseless.” With this sensitivity boost, we measured 2D 13C-13C correlation spectra that allow us to resolve many unique molecular environments, for both crystalline and non-crystalline domains. The de novo assignment is achieved by first identifying the well-resolved C4-C5 and C3-C4 signals, from which the connectivity to other carbons can be found. Peak intensities are tracked to validate the connectivity-based assignment and carbons from the same cellulose form should have comparable intensities. The control cotton sample mainly contains the crystalline cellulose, with negligible signals for non-crystalline domains, and the representative carbon connectivity is shown in Fig. 2a, without any ambiguity. In contrast, the ball-milled sample shows dominant signals from the disordered cellulose (Fig. 2b). The complete 13C-connectivity and resonance assignments have been identified for all crystalline forms and for two types of disordered cellulose, among the five forms identified (Fig. S1). The 13C-chemical shifts have been documented in Table 1.
Fig. 2.

DNP enables 2D spectroscopy on unlabeled cellulose. All 2D spectra are measured using the natural abundance (1.1%) of 13C. 2D 13C-13C INADEQUATE spectra have been measured on (a) Wiley-milled cotton and (b) ball-milled sample. The two spectra were plotted so that the crystalline domains of cellulose show comparable contour lines. Representative carbon connectivity is shown in magenta. The representative regions of crystalline and disordered cellulose are highlighted using red and blue dashed-line circles, respectively
Table 1.
13C-chemical shifts of cellulose in Wiley-milled and ball-milled cotton samples.
| Sample | Type | C1 (ppm) |
C2 (ppm) |
C3 (ppm) |
C4 (ppm) |
C5 (ppm) |
C6 (ppm) |
|---|---|---|---|---|---|---|---|
| A | 105.0 | 71.1 | 74.1 | 89 | 71.8 | 64.9 | |
| Wiley-milled cotton | A' | 105.0 | 71.4 | 74.9 | 89.8 | 72.5 | 64.9 |
| B | 105.8 | 71.4 | 74.9 | 88.5 | 70.7 | 65.5 | |
| B' | 103.8 | 71.5 | 74.3 | 89.2 | 72.4 | 64.9 | |
| Crystalline, major | 104.5 | 71.7 | 74 | 88.8 | 72 | 64.9 | |
| Ball-milled | Crystalline, minor | 104.5 | 72.5 | 74.9 | 87.1 | 74.1 | 63.6 |
| Disordered, type-1 | 105.1 | 73 | 75.6 | 84.9 | 75.6 | 61.1 | |
| Disordered, type-2 | 105.1 | 73.1 | 75.1 | 82.8 | 75.1 | 59.8 |
The standard Iα and Iβ structures are removed after 120-min ball-milling
The 2D spectra of crystalline cellulose in Wiley-milled cotton allow us to resolve the signals of all carbons in four glucose units (Fig. 3a), The typical signal-to-noise ratio is 10-20 and the full-width at half maximum linewidth (FWHM) is as sharp as 0.9 ppm (Fig. 3a). The 13C chemical shifts of these glucose residues dovetail well with the Iα and Iβ allomorphs that were previously measured on 13C-labeled samples from Cladophora and tunicate (Kono et al. 2003b; Kono and Numata 2006). The four glucose types are better resolved at the C4, C5 and C1 sites rather than the C6 site, suggesting that the C6 hydroxymethyl conformation and the χ’ (C4-C5-C6-O6) torsion angle are relatively focused, primarily at the tg conformational minima as revealed by the C6 chemical shift at 65 ppm.
Fig. 3.

High-resolution structural insights of cellulose in unlabeled cotton. (a) Resonance assignments of Iα and Iβ signals in Wiley-milled cotton. Representative 13C cross section shows a typical 13C linewidth is 0.9-1.0 ppm. The double-quantum and single-quantum chemical shifts are abbreviated as DQ and SQ, respectively. (b) The crystalline region of cellulose is altered after 2 h of ball-milling. The spectral features of Iα and Iβ cellulose in the control Wiley-milled sample are fully removed in the ball-milled sample. (c) The C4 region of disordered cellulose in ball-milled sample resolved five sub-forms as indicated using arrows
The limited resolution of 1D spectra in the many conventional ssNMR studies on cellulose and carbohydrate-rich materials can easily lead to misinterpretations, and here is an example. The C4 region of crystalline cellulose (88-90 ppm) in Wiley-milled and 120-min ball-milled sample shows comparable spectral patterns in 1D spectra except for the substantially reduced intensity (Fig. 1), which naturally misleads us to assume that the remaining crystalline cellulose retains the same molecular structure, but with a considerably decreased amount. However, this is wrong. The well-resolved signals of the crystalline C4 unambiguously differ for these two samples (Fig. 3b). Although both samples show a major signal at 89 ppm for single-quantum (SQ) chemical shift, which will result in similar 1D patterns in the C4 region, their double-quantum (DQ) chemical shift that sums the SQ chemical shifts of two bonded carbons completely differs, revealing a change in the adjacent carbons, C3 and C5. Therefore, the Iα and Iβ structures are not retained in the heavily ball-milled samples.
This substantial change cannot be detected in conventional NMR studies since they only use the well-resolved C4 peaks as the indicators of structure, sometimes with the help of partially resolved C1 and C6 signals, while the C2, C3 and C5 signals are fully ignored. Therefore, any structural change of these spectroscopically “invisible” carbons will be omitted in the conventional 1D work, as shown above.
What is the cause of the structural inconsistency observed here? We speculate that the Iα and Iβ can only exist in large cellulose aggregates with high-crystallinity across a large dimension. After 120 min of ball milling, the remaining material is not bulky enough to support the molecular organization of these model allomorphs. This is supported by the recent observation that Iα and Iβ structures are not present in the native cell walls of plant seedlings, coleoptile, mature stems, woody branches and bark in a variety of plants, from dicot to monocot and from grasses to trees (Wang et al. 2016b; Wang et al. 2014)(unpublished results). Examples include Arabidopsis, Brachypodium, maize, switchgrass, rice, spruce, poplar and Eucalyptus, which collectively indicate that the Iα and Iβ model structures cannot be extended to cellulose from many of the natural sources.
The exact size of the crystallites is difficult to measure directly but could be roughly estimated using NMR-derived crystallinity, a parameter reflecting the ratio between internal and surface chains in cellulose (Fernandes et al. 2011; Wang and Hong 2016). The crystallinity of the control sample (68%) derived from room-temperature 1D 13C spectra best fit a simplified model with 81 crystalline chains arranged as a 9 × 9 matrix, with another 40 disordered chains coating the surface. This will result in large lateral dimensions of 6-9 nm, which is enough to support the presence of Iα and Iβ structures. Because there is rising evidence that plants produce fundamental or elementary fibrils with 18 molecules (Cosgrove 2014; Hill et al. 2014; Newman et al. 2013; Sethaphong et al. 2013; Vandavasi et al. 2016), the averaged cellulose structure of cotton cellulose should contain at least six or seven such fibrils. Note that the real dimension of these crystallites may be substantially larger than the value estimated above because the signals of disordered chains may have contributions from primary cell walls, whose cellulose is much smaller. Also, other processes may introduce structural disorder, for example, the bundling process of multiple elementary microfibrils. If we adopt a previous model that include both accessible and inaccessible surfaces (the aggregated surface between multiple elementary fibrils), a lateral dimension of 11-12 chains is estimated (Verlhac et al. 1990; Larsson et al. 1999). However, we should be aware that large uncertainties may exist due to the ambiguity of these models and our limited understanding of the crystalline and disordered cellulose, as well as their spectral characteristics (see below).
Partial structural order exists in the “disordered” domains
Our contemporary views of the disordered domains of cellulose usually involve a less ordered form in the interior of the fibril (para-crystalline cellulose) and multiple components of fibrillar surface. The inaccessible surface induced by fibril aggregation is often assumed as a dominant disordered form (70-90% of the surface chains), which is typically presented as a very broad component (FWHM linewidth of 4-9 ppm) resonating at ~84 ppm in 1D 13C spectra (Larsson et al. 1999; Larsson and Westlund 2005). The introduction of this component simplified the spectral deconvolution process, and the broad lineshape used in fitting usually suggest a Gaussian distribution of conformations. The disordered C4 region of the ball-milled sample, however, shows several well-resolved peaks, the linewidth of each one is not much broader than the crystalline forms (Fig. 3c). These high-resolution data indicate that conformational disorder happens in a discrete manner instead of a continuous distribution: several energetic minima of cellulose conformation are present even in the heavily ball-milled sample that bears a great degree of disorder. Although the concept of “crystallite surface” was proposed two decades ago (Newman 1998), our 2D data provides, with high-resolution, a far more direct and striking view of this partial order.
Structural comparison of cellulose from various sources
It has long been difficult to extract useful structural information out of the NMR observables, for example, chemical shifts, which sometimes are “indirect” for non-NMR scientists. To begin to understand the 13C chemical shifts for these samples, they can be compared with those for cellulose from other sources. Figure 4 shows “heat maps” of the root-mean-square deviation (RMSD) values for comparisons of the chemical shifts of the corresponding peaks from each source with those of the control and ball-milled samples. A good correlation between two structures will exhibit low RMSD values. The 13C chemical shifts of Iα and Iβ, II and III have been systematically measured by Kono and coworkers using a series of 2D 13C-13C and 13C-1H correlation spectra on uniformly 13C-labeled model cellulose (Kono et al. 2003a; Kono et al. 2003b; Kono et al. 2004). These cellulose materials are typically produced in model algae, bacterial or tunicate for the ease of 13C-labeling, followed by isolation, purification or chemical treatment. Values for cellulose in native cell walls of dicots and grasses are also included for comparison (Wang et al. 2016b).
Fig. 4.

13C chemical shift RMSD map for comparisons between cotton and other cellulose sources. The color scale is shown and the units are ppm. The x-axis contains the observed cellulose forms in Wiley-milled and 2 hr ball-milled cotton. The y-axis contains various cellulose types including Iα, Iβ, II, III and cellulose in native cell walls. The source and treatments are in italic. All good correlations with RMSD of 0.6 ppm or less are highlighted in bold. The crosses indicate very good correlations with RMSD of 0.3 ppm or less.
The crystalline cellulose in Wiley-milled cotton exhibits poor correlation with surface disordered chains (f and g) in plant cell walls or cellulose II/III structures (Fig. 4). Instead, it correlates better with the Iα and Iβ allomorphs and the interior crystalline chains in native cell walls. For the B and B' units in cotton and tunicate, a very low RMSD of 0.2-0.3 ppm is observed, indicating a highly preserved structure of Iβ. For A and A' in cotton, however, reasonably good correlations (0.4-0.6 ppm RMSD) are established with many cellulose subtypes, including the A and A' in Cladophora, the B' in tunicate and the a and e types in plant cell walls. Clearly, in cotton, the structural characteristics of Iα allomorph are more ambiguous than those of Iβ. This finding might be relevant to an unresolved question of how these two major allomorphs are mixed on the molecular level.
A more interesting goal here is to identify similarities between the ball-milled cotton and the known cellulose structures. The major signals of crystalline cellulose in the ball-milled sample have high similarity to tunicate Iβ B', with a low RMSD of 0.4 ppm. In Iβ, B' and B only form B-B or B'-B' repeating units, each forming a different type of glucan chain arranged in alternating sheets (the center and origin chains): one sheet contains only B units and the adjacent sheet only has B' units (Jarvis 2003; Kono and Numata 2006; Nishiyama et al. 2002; Nishiyama et al. 2003). The fact that the remaining crystallites resemble B' rather than B indicate that, after thorough ball-milling, the remaining crystallites adopt a structure that could be viewed as stacked B' sheets. Further computational effort is needed to connect 13C chemical shifts with molecular structure and reveal how this structure is stabilized by inter-sheet C-H O hydrogen bonds (Kubicki et al. 2013; Kubicki et al. 2014; Yang et al. 2018). In contrast, a minor type of crystalline cellulose in the ball-milled sample does not match Iα or Iβ structures. Instead, similarity is found with the type-d cellulose in plants, which is a special type responsible for interacting with matrix polysaccharides, and thus, is considered to bear higher disorder. Therefore, this minor form can be treated as an intermediate between crystalline and disordered cellulose. In contrast, the disordered component of ball-milled cellulose clearly shows better correlations with the surface chains (type-f and g) of cellulose microfibrils in plant cell walls than with any of the crystalline structures.
Conclusions and Future Perspectives
This study shows how atomic-level structural insight can be obtained on unlabeled cellulose samples by integrating DNP ssNMR spectroscopy with chemical shift analysis. The methods presented here can be readily applied to various functional cellulose materials or carbohydrate-rich polymers. This strategy can be substantially facilitated by the implementation of a solid-state NMR database and its auxiliary software that systematically indexes and analyzes the megadata of cellulose and other complex carbohydrates, which are currently being developed by our (TW) lab.
The high-resolution and large data analysis presented in this study also provides novel insights into cellulose structure. A large crystallite size is needed to accommodate the Iα and Iβ crystallographic structures, causing their absence in many types of plants. These model allomorphs will be fully abolished by ball-milling, thus our evaluations of the function-structure relationship of cellulose-based materials need to be cautious under many circumstances, especially for those with mechanical processing, chemical treatment or other structure-perturbing mechanisms. The fact that partial order exists in the “disordered” domains also urges us to revise our thinking of cellulose structure and substantiate it with further molecular evidence from different physical methods.
Supplementary Material
Acknowledgments
This work is supported by the National Science Foundation through NSF OIA-1833040. The National High Magnetic Field Laboratory is supported by the National Science Foundation through NSF/DMR-1644779 and the State of Florida. The MAS-DNP system at NHMFL is funded in part by NIH S10 OD018519 and NSF CHE-1229170.
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s10570-018-2095-6) contains supplementary material, which is available to authorized users.
References
- Atalla RH, Vanderhart DL (1984) Native cellulose: a composite of two distinct crystalline forms. Science 223:283–285. doi: 10.1126/science.223.4633.283 [DOI] [PubMed] [Google Scholar]
- Atalla RH, Vanderhart DL (1999) The role of solid state 13C NMR spectroscopy in studies of the nature of native celluloses. Solid State Nucl Magn Reson 15:1–19 [DOI] [PubMed] [Google Scholar]
- Cosgrove DJ (2014) Re-constructing our models of cellulose and primary cell wall assembly. Curr Opin Plant Biol 22C:122–131. doi: 10.1016/j.pbi.2014.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove DJ, Jarvis MC (2012) Comparative structure and biomechanics of plant primary and secondary cell walls. Front Plant Sci 3. doi: 10.3389/Fpls.2012.00204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubroca T et al. (2018) A quasi-optical and corrugated waveguide microwave transmission system for simultaneous dynamic nuclear polarization NMR on two separate 14.1 T spectrometers. J Magn Reson 289:35–44. doi: 10.1016/j.jmr.2018.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes AN et al. (2011) Nanostructure of cellulose microfibrils in spruce wood. Proc Natl Acad Sci USA 108:E1195–E1203. doi: 10.1073/pnas.1108942108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forziati FH, Stone WK, Rowen JW, Appel WD (1950) Cotton Powder for Infrared Transmission Measurements. J Res Nat Bur Stand 45:109–113. doi: 10.6028/jres.045.009 [DOI] [Google Scholar]
- Hill JL, Hammudi MB, Tien M (2014) The Arabidopsis Cellulose Synthase Complex: A Proposed Hexamer of CESA Trimers in an Equimolar Stoichiometry. Plant Cell 26:4834–4842. doi: 10.1105/tpc.114.131193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohwy M, Rienstra CM, Jaroniec CP, Griffin RG (1999) Fivefold symmetric homonuclear dipolar recoupling in rotating solids: Application to double quantum spectroscopy. J Chem Phys 110:7983–7992. doi: 10.1063/1.478702 [DOI] [Google Scholar]
- Jarvis M (2003) Chemistry: cellulose stacks up. Nature 426:611–612. doi: 10.1038/426611a [DOI] [PubMed] [Google Scholar]
- Kang X et al. (2018) Molecular architecture of fungal cell walls revealed by solid-state NMR. Nat Commun 9, 2747. doi: 10.1038/S41467-018-05199-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koers EJ et al. (2014) NMR-based structural biology enhanced by dynamic nuclear polarization at high magnetic field J Biomol NMR 60:157–168. doi: 10.1007/s10858-014-9865-8 [DOI] [PubMed] [Google Scholar]
- Kono H, Erata T, Takai M (2003a) Complete assignment of the CP/MAS 13C NMR spectrum of cellulose IIII. Macromolecules 36:3589–3592. doi: 10.1021/Ma021015f [DOI] [Google Scholar]
- Kono H, Erata T, Takai M (2003b) Determination of the through-bond carbon-carbon and carbon-proton connectivities of the native celluloses in the solid state. Macromolecules 36:5131–5138. doi: 10.1021/Ma021769u [DOI] [Google Scholar]
- Kono H, Numata Y (2006) Structural investigation of cellulose Ia and Ib by 2D RFDR NMR spectroscopy: determination of sequence of magnetically inequivalent D-glucose units along cellulose chain. Cellulose 13:317–326 [Google Scholar]
- Kono H, Numata Y, Erata T, Takai M (2004) 13C and 1H resonance assignment of mercerized cellulose II by two-dimensional MAS NMR spectroscopies. Macromolecules 37:5310–5316. doi: 10.1021/Ma030465k [DOI] [Google Scholar]
- Kubicki JD, Mohamed MNA, Watts HD (2013) Quantum mechanical modeling of the structures, energetics and spectral properties of I alpha and I beta cellulose. Cellulose 20:9–23. doi: 10.1007/s10570-012-9838-6 [DOI] [Google Scholar]
- Kubicki JD, Watts HD, Zhao Z, Zhong LH (2014) Quantum mechanical calculations on cellulose-water interactions: structures, energetics, vibrational frequencies and NMR chemical shifts for surfaces of I alpha and I beta cellulose. Cellulose 21:909–926. doi: 10.1007/s10570-013-0029-x [DOI] [Google Scholar]
- Larsson PT, Hult EL, Wickholm K, Pettersson E, Iversen T (1999) CP/MAS 13C NMR spectroscopy applied to structure and interaction studies on cellulose I. Solid State Nucl Magn Reson 15:31–40. doi: 10.1016/S0926-2040(99)00044-2 [DOI] [PubMed] [Google Scholar]
- Larsson PT, Westlund PO (2005) Line shapes in CP/MAS 13C NMR spectra of cellulose I. Spectrochim Acta A 62:539–546. doi: 10.1016/j.saa.2005.01.021 [DOI] [PubMed] [Google Scholar]
- Lee D, Hediger S, De Paepe G (2015) Is solid-state NMR enhanced by dynamic nuclear polarization? Solid State Nucl Magn Reson 66-67:6–20. doi: 10.1016/j.ssnmr.2015.01.003 [DOI] [PubMed] [Google Scholar]
- Lesage A, Auger C, Caldarelli S, Emsley L (1997) Determination of through-bond carbon-carbon connectivities in solid-state NMR using the INADEQUATE experiment. J Am Chem Soc 119:7867–7868. doi: 10.1021/Ja971089k [DOI] [Google Scholar]
- Ling Z, Wang T, Makarem M, Cheng HN, Bacher M, Potthast A, Rosenau T, King H, Delhom CD, Nam S, Edwards JV, Kim SH, Xu F, French AD (2019) Effects of ball milling on the structure of cotton cellulose. Cellulose 26:XXXX–XXYY [Google Scholar]
- Mentink-Vigier F et al. (2017) Efficient cross-effect dynamic nuclear polarization without depolarization in high-resolution MAS NMR. Chem Sci 8:8150–8163. doi: 10.1039/c7sc02199b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman RH (1998) Evidence for Assignment of 13C NMR Signals to Cellulose Crystallite Surfaces in Wood, Pulp and Isolated Celluloses. Holzforschung 52:157–159 [Google Scholar]
- Newman RH, Hill SJ, Harris PJ (2013) Wide-angle x-ray scattering and solid-state nuclear magnetic resonance data combined to test models for cellulose microfibrils in mung bean cell walls. Plant Physiol 163:1558–1567. doi: 10.1104/pp.113.228262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni QZ et al. (2013) High Frequency Dynamic Nuclear Polarization. Acc Chem Res 46:1933–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama Y, Langan P, Chanzy H (2002) Crystal structure and hydrogen-bonding system in cellulose Ib from synchrotron X-ray and neutron fiber diffraction. J Am Chem Soc 124:9074–9082. doi: 10.1021/Ja0257319 [DOI] [PubMed] [Google Scholar]
- Nishiyama Y, Sugiyama J, Chanzy H, Langan P (2003) Crystal structure and hydrogen bonding system in cellulose Ia, from synchrotron X-ray and neutron fiber diffraction. J Am Chem Soc 125:14300–14306. doi: 10.1021/Ja037055w [DOI] [PubMed] [Google Scholar]
- Perras FA, Luo H, Zhang X, Mosier NS, Pruski M, Abu-Omar MM (2017) Atomic-Level Structure Characterization of Biomass Pre- and Post-Lignin Treatment by Dynamic Nuclear Polarization-Enhanced Solid-State NMR. J Phys Chem A 121:623–630. doi: 10.1021/acs.jpca.6b11121 [DOI] [PubMed] [Google Scholar]
- Phyo P, Wang T, Yang Y, O'Neill H, Hong M (2018) Direct Determination of Hydroxymethyl Conformations of Plant Cell Wall Cellulose Using 1H Polarization Transfer Solid-State NMR. Biomacromolecules 19:1485–1497. doi: 10.1021/acs.biomac.8b00039 [DOI] [PubMed] [Google Scholar]
- Rossini AJ et al. (2012) Dynamic Nuclear Polarization NMR Spectroscopy of Microcrystalline Solids. J Am Chem Soc 134:16899–16908. doi: 10.1021/ja308135r [DOI] [PubMed] [Google Scholar]
- Rossini AJ, Zagdoun A, Lelli M, Lesage A, Coperet C, Emsley L (2013) Dynamic Nuclear Polarization Surface Enhanced NMR Spectroscopy. Acc Chem Res 46:1942–1951. doi: 10.1021/ar300322x [DOI] [PubMed] [Google Scholar]
- Saliba EP et al. (2017) Electron Decoupling with Dynamic Nuclear Polarization in Rotating Solids. J Am Chem Soc 139:6310–6313. doi: 10.1021/jacs.7b02714 [DOI] [PubMed] [Google Scholar]
- Sauvee C, Rosay M, Casano G, Aussenac F, Weber RT, Ouari O, Tordo P (2013) Highly Efficient, Water-Soluble Polarizing Agents for Dynamic Nuclear Polarization at High Frequency. Angew Chem Int Edit 52:10858–10861. doi: 10.1002/anie.201304657 [DOI] [PubMed] [Google Scholar]
- Sethaphong L, Haigler CH, Kubicki JD, Zimmer J, Bonetta D, DeBolt S, Yingling YG (2013) Tertiary model of a plant cellulose synthase. Proc Natl Acad Sci USA 110:7512–7517. doi: 10.1073/pnas.1301027110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons TJ et al. (2016) Folding of xylan onto cellulose fibrils in plant cell walls revealed by solid-state NMR. Nat Commun 7:13902. doi: 10.1038/ncomms13902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Ayala I, Bardet M, De Paepe G, Simorre JP, Hediger S (2013a) Solid-state NMR on bacterial cells: selective cell wall signal enhancement and resolution improvement using dynamic nuclear polarization. J Am Chem Soc 135:5105–5110. doi: 10.1021/ja312501d [DOI] [PubMed] [Google Scholar]
- Takahashi H, Hediger S, De Paepe G (2013b) Matrix-free dynamic nuclear polarization enables solid-state NMR C-13-C-13 correlation spectroscopy of proteins at natural isotopic abundance. Chem Commun 49:9479–9481. doi: 10.1039/c3cc45195j [DOI] [PubMed] [Google Scholar]
- Takahashi H, Lee D, Dubois L, Bardet M, Hediger S, De Paepe G (2012) Rapid Natural-Abundance 2D 13C-13C Correlation Spectroscopy Using Dynamic Nuclear Polarization Enhanced Solid-State NMR and Matrix-Free Sample Preparation. Angew Chem Int Edit 51:11766–11769. doi : 10.1002/anie.201206102 [DOI] [PubMed] [Google Scholar]
- Vandavasi VG et al. (2016) A Structural Study of CESA1 Catalytic Domain of Arabidopsis Cellulose Synthesis Complex: Evidence for CESA Trimers. Plant Physiol 170:123–135. doi: 10.1104/pp.15.01356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verlhac C, Dedier J, Chanzy H (1990), Availability of surface hydroxyl groups in valonia and bacterial cellulose. Polymer Sci. Part A 28: 1171–1177 [Google Scholar]
- Wang T, Hong M (2016) Solid-state NMR investigations of cellulose structure and interactions with matrix polysaccharides in plant primary cell walls. J Exp Bot 67:503–514. doi: 10.1093/jxb/erv416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Park YB, Caporini MA, Rosay M, Zhong LH, Cosgrove DJ, Hong M (2013) Sensitivity-enhanced solid-state NMR detection of expansin's target in plant cell walls. Proc Natl Acad Sci USA 110:16444–16449. doi: 10.1073/pnas.1316290110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Park YB, Cosgrove DJ, Hong M (2015) Cellulose-Pectin Spatial Contacts Are Inherent to Never-Dried Arabidopsis thaliana Primary Cell Walls: Evidence from Solid-State NMR. Plant Physiol 168:871–884. doi: 10.1104/pp.15.00665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Phyo P, Hong M (2016a) Multidimensional solid-state NMR spectroscopy of plant cell walls. Solid State Nucl Magn Reson 78:56–63. doi: 10.1016/j.ssnmr.2016.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Yang H, Kubicki JD, Hong M (2016b) Cellulose Structural Polymorphism in Plant Primary Cell Walls Investigated by High-Field 2D Solid-State NMR Spectroscopy and Density Functional Theory Calculations. Biomacromolecules 17:2210–2222. doi: 10.1021/acs.biomac.6b00441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Zabotina O, Hong M (2012) Pectin-cellulose interactions in the Arabidopsis primary cell wall from two-dimensional magic-angle-spinning solid-state nuclear magnetic resonance. Biochemistry 51:9846–9856. doi: 10.1021/Bi3015532 [DOI] [PubMed] [Google Scholar]
- Wang T, Salazar A, Zabotina O, Hong M (2014) Structure and dynamics of Brachypodium primary cell walls polysaccharides from two-dimensional 13C Solid-State NMR Spectroscopy. Biochemistry 53: 2840–2854. doi: 10.1021/bi500231b [DOI] [PubMed] [Google Scholar]
- Yang H, Wang T, Oehme D, Petridis L, Hong M, Kubicki JD (2018) Structural factors affecting 13C NMR chemical shifts of cellulose: a computational study. Cellulose. 25:23–36. doi: 10.1007/s10570-017-1549-6 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.